

Die Livedovaskulopathie ist eine eher seltene, chronisch-rezidivierende Erkrankung der kutanen Gefäßversorgung, die für die Betroffenen mit starken Schmerzen und deutlich reduzierter Lebensqualität verbunden ist. Daher wäre eine rasche Diagnose wichtig - allerdings dauert es im Schnitt sechs Jahre, bis eine Livedovaskulopathie erkannt wird. Hier erfahren Sie, wie Sie diese Zeitspanne verkürzen und die Erkrankung sicher diagnostizieren können und welche Therapieoptionen Sie haben.
Die Livedovaskulopathie (LV) ist eine seltene, eminent chronische, regelmäßig rezidivierende, thromboembolische Verschlusskrankheit des kutanen Gefäßplexus, die vor allem die unteren Extremitäten betrifft [1]. Die LV zählt zu den seltenen Erkrankungen mit einer Inzidenz von geschätzt 1:100.000, die mehr das weibliche Geschlecht mit einem Verhältnis von 3:1 betrifft und häufiger in jüngeren Lebensjahren (Median 32 Jahre) auftritt, wobei das Durchschnittsalter der Patienten regional sehr unterschiedlich sein kann. So finden sich in asiatischen Ländern zum Beispiel eher jüngere Patienten [1, 2]. Auch wenn die LV bei manchen Patienten in den Sommermonaten verstärkt auftritt und daher initial als "Livedo reticularis mit Sommerulzerationen" [3] beschrieben wurde, können die Hautveränderungen ganzjährig auftreten [4]. Diese initiale Benennung ist nur ein Beispiel für zum Teil verwirrende Synonyme, die in der Dermatologie für die LV kursieren. Teils wird die LV auch als PURPLE ("painful purpuric ulcers with reticular patterning on the lower extremities"), Livedo-Vaskulitis, segmental hyalinisierende Vaskulitis oder einfach nur Atrophie blanche bezeichnet. Dieses Begriffswirrwarr könnte einer der Gründe dafür sein, dass die Inzidenzangabe der LV sehr niedrig ausfällt und es im Schnitt sechs Jahre benötigt, bis die Diagnose LV gestellt wird.
Klinik
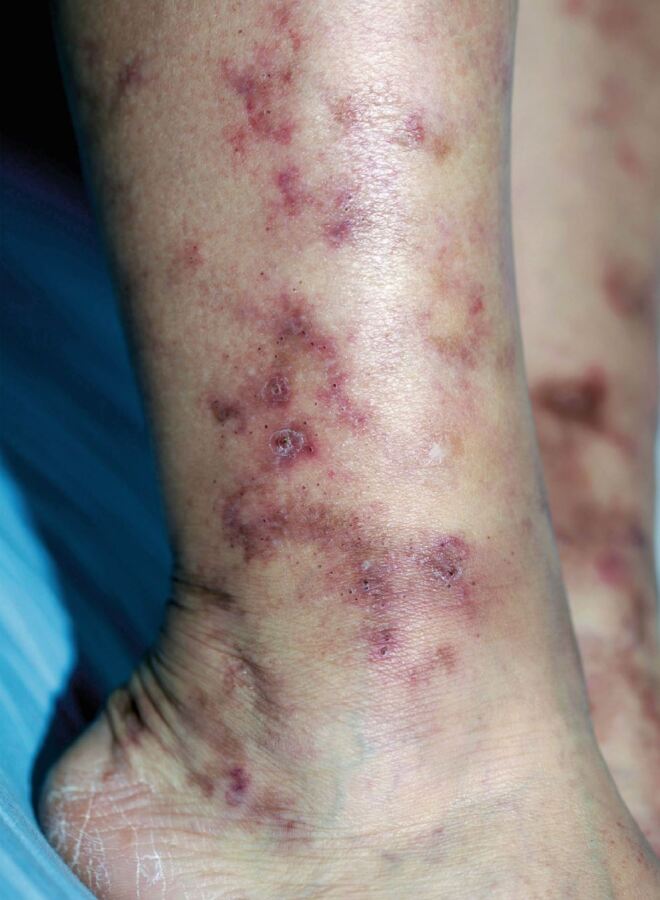
Klinisch imponiert die Trias aus Livedo racemosa, sehr schmerzhaften Ulzera und deren narbiger Abheilung, der Atrophie blanche [5]. Ursache der Hautveränderungen sind thrombotische Gefäßverschlüsse in den Kapillaren und kleinen Gefäßen der oberen und mittleren Dermis. Die somit einhergehende lokale Sauerstoffunterversorgung führt zu Schmerzen (Angina cutis), Erythem, Blasen bis zu Nekrosen und Ulzerationen mit nachfolgender narbiger Abheilung [6]. Sehr häufig besteht eine erythematös-livide Verfärbung der Haut (Abb. 1), die blitzfigurenartig und netzförmig mit diskontinuierlichen Kreisen auftritt. Diese Livedo racemosa ist ein Resultat der lokalen Sauerstoffunterversorgung. Sie tritt nicht nur an den unteren Extremitäten auf, sondern manchmal auch an anderen Lokalisationen oberhalb der Gürtellinie [5]. Initiale Läsionen sind flache bis erhabene Purpura, die im Verlauf ulzerieren [7]. Diese Ulzerationen (Abb. 2) sind typischerweise flach und klein (4-6 mm) und an den Knöcheln, den Fußrücken sowie den Unterschenkeln lokalisiert, jedoch nicht oberhalb der Knie [5, 7, 8]. Nachdem die Ulzerationen abgeheilt sind, bleiben atrophe, sternförmige, narbenähnliche weiße Plaques mit Teleangiektasien und peripherer Hyperpigmentierung (Abb. 3) zurück (Atrophie blanche) [8]. Eine Seitenpräferenz tritt dabei nicht auf [5].


Die Thrombosierung von Gefäßen in den oberen Hautschichten bei Patienten mit LV ist mit starken Schmerzen und somit mit einer dramatischen Verschlechterung der Lebensqualität verbunden [9, 10]. Bei einer sehr schweren Verlaufsform der Erkrankung werden von den Patienten auch Symptome einer Polyneuropathie beschrieben, wie Dys- oder Hypästhesien. Diese können häufig einer Mononeuritis multiplex zugeordnet werden, einer peripheren Neuropathie, die unter anderem auch mit einer Vaskulitis, Diabetes, Neoplasmen und Infektionen assoziiert sein kann. Im Gegensatz zu anderen Erkrankungen, die mit einer Mononeuritis multiplex einhergehen, kommt es bei der LV nicht zu einer Vaskulitis, sondern zu einer Thrombosierung in den Vasa nervorum, ähnlich wie in den kutanen Gefäßen bei der LV [11, 12].
Pathophysiologie
Die Pathophysiologie der LV ist bislang noch nicht vollständig geklärt. Ursprünglich wurde die Erkrankung für eine Vaskulitis gehalten [13]. Zum heutigen Zeitpunkt geht man bei der LV jedoch von einer Gefäßerkrankung aus, bei der es durch ein Übergewicht prokoagulatorischer Faktoren zu einem Status der Hyperkoagulabilität kommt [7]. Der thrombotische Effekt resultiert möglicherweise aus einer Endothelzelldysfunktion, Defekten in der Plasminogen-Aktivierung am Endothel, einer Plättchendysfunktion, einer erhöhten Fibrinbildung oder einer gestörten Fibrinolyse. Fibrinablagerungen und Thrombusbildung fungieren als Diffusionsbarriere und führen zu einer verminderten Sauerstoffversorgung [7, 14] mit anschließendem Gewebeuntergang (Hautinfarkt). Geringe Gewebsperfusion führt wiederum zu schlechter Wundheilung - ein Teufelskreislauf entsteht [15]. Hyperkoagulabilität, Stasis und Endothelschaden - auch Virchow-Trias genannt - sind auch bei der LV Risikofaktoren für eine Thrombose [14, 16]. Als Ursache für die Manifestation der LV an den Beinen wird eine geringere Konzentration thrombolytischer Faktoren sowie Unterschiede im Perfusionsdruck und der Temperatur an der unteren Extremität vermutet [16, 17].
Assoziationen zu verschiedenen Erkrankungen, die einen Status der Hyperkoagulabilität bedingen, wurden bei der LV beschrieben [7, 18], darunter
angeborene und erworbene Thrombophilien, darunter unter anderem Faktor-V-Leiden-Mutation (F5LMT), Protein-C- und Protein-S-Mangel, Antithrombin-III-Mangel, Prothrombin-G20210A-Mutation, Plasminogen-Aktivator-Inhibitor(PAI)-1-Promoter-Mutation, Lipoprotein(a), Methylen-Tetrahydrofolat(MTHF)-Genmutation, Homocysteinämie und Antiphospholipid-Antikörper
Autoimmunerkrankungen, unter anderem systemischer Lupus erythematodes
Malignome
Beispielsweise ist Lipoprotein(a) (Lp[a]) ein unabhängiger, genetisch determinierter und nicht durch den Lebensstil beeinflussbarer Risikofaktor für kardiovaskuläre Erkrankungen und wurde gehäuft bei Patienten mit LV nachgewiesen. In einer Studie wurde bei 42 % der insgesamt 75 LV-Patienten ein erhöhtes Lp(a) nachgewiesen [19]. Lp(a) hat strukturelle Ähnlichkeit mit Plasminogen und besitzt hierdurch antifibrinolytische Eigenschaften, was die Thromboseneigung erhöht [20, 21]. Weiterhin kann Lp(a) die Expression des Gewebefaktors ("tissue factor", TF) erhöhen und die Expression vom "tissue factor pathway inhibitor" (TFPI) hemmen. Zuletzt verstärkt die oxidierte Form, ox-Lp(a), die PAI-1-Aktivität. Umfangreiche Untersuchungen an größeren Patientenkollektiven haben jedoch gezeigt, dass sich in weniger als der Hälfte der Fälle pathologische Veränderungen bekannter prokoagulatorischer Faktoren identifizieren lassen [8, 22].
In einem kürzlich publizierten Review wurden genetische Varianten der Thrombophiliefaktoren und deren Nachweis bei LV-Patienten untersucht [18]. So konnte bei 81 von 95 LV-Patienten eine PAI-1-675 4G/5G-Variante gefunden werden. Weitere genetische Varianten waren PAI-1 A-844G (bei 56 % von 18 untersuchten LV Patienten), MTHF-Reduktase (MTHFR) C677T und MTHFR A1298C (55 % von 122 beziehungsweise 44 % von 82 LV-Patienten). Weniger häufig wurden Polymorphismen im Faktor V und Prothrombin festgestellt.
Aufgrund dieser starken Heterogenität der zugrunde liegenden pathogenetischen Faktoren lässt sich die LV unterteilen in eine primäre (idiopathische) und eine sekundäre Form, bei der andere Erkrankungen involviert sind [1, 23].
Interessant ist auch, dass im Krankheitsverlauf Moleküle wie Interleukine (IL; hier IL-2 und löslicher IL-2-Rezeptor) und P-Selektin vom Endothel freigesetzt werden und so entzündliche Prozesse in der Haut fördern, indem sie Leukozyten rekrutieren [13]. Auch wenn bei der LV keine primäre Vaskulitis vorliegt, lässt die Assoziation zu Autoimmunerkrankungen sowie das therapeutische Ansprechen auf Immunmodulatoren zumindest eine sekundäre Inflammation vermuten [7].
Einige Mechanismen in der Ätiopathogenese der LV und COVID-19-assoziierten livedoartigen Läsionen erscheinen ähnlich, jedoch gibt es bisher noch keine Fallberichte über eine COVID-19-induzierte LV [24, 25]. Allerdings wurde von einer Verschlimmerung beziehungsweise einem Rezidiv einer LV nach einer COVID-19-Infektion berichtet [26, 27].
Diagnostik
Auch wenn das klinische Erscheinungsbild der Erkrankung sehr typisch ist [28], sollte die Diagnose einer LV in Zusammenschau von Klinik und Histologie zum Ausschluss anderer Differenzialdiagnosen gestellt werden [29, 30]. Histologisch können Fibrinablagerungen in den Gefäßwänden, eine endotheliale Proliferation und intraluminale hyaline Thromben in Blutgefäßen der oberen und mittleren Dermis im akuten Stadium nachgewiesen werden. Im Verlauf kann es zu einer Reorganisation der Thromben mit subintimaler Proliferation und segmentaler Hyalinisierung der Gefäßwände und der Dermis kommen [23, 31].
Zu beachten gilt, dass sich das histologische Bild in unterschiedlichen Stadien der Erkrankung unterscheidet [5]. Perivaskulär findet sich ein spärliches Infiltrat aus Lymphozyten, jedoch das für eine primäre Vaskulitis typische Infiltrat aus Neutrophilen oder eine Leukozytoklasie lässt sich bei der LV nicht nachweisen. Neutrophile finden sich höchstens sekundär im Bereich der Ulzera [5]. In der direkten Immunfluoreszenz können granuläre Ablagerungen insbesondere von C3, Fibrinogen und Immunglobulin(Ig)M - weniger hingegen IgA und IgG - gefunden werden, auch wenn diese Ergebnisse als nicht spezifisch und nicht diagnostisch gewertet werden [8, 32, 33, 34]. Positive Ergebnisse der direkten Immunfluoreszenz kommen statistisch signifikant häufiger bei älteren Patienten und frischeren Hautveränderungen (< 6 Monate) vor [33]. Normwertige Serum-Komplement-Spiegel, ein diffuses homogenes statt granuläres Verteilungsmuster, ein geringes perivaskuläres Infiltrat von Leukozyten und das Fehlen von Kernstaub sprechen jedoch gegen eine Immunkomplex-vermittelte Erkrankung [33, 35].
Die Veränderungen bei der LV treten häufig in der oberflächlichen bis mittleren Dermis, seltener auch in der tiefen Dermis auf. Insbesondere zum Ausschluss einer differenzialdiagnostisch abzugrenzenden Panarteriitis nodosa (PAN), einer Vaskulitis der mittelgroßen Arterien, ist eine tiefe Spindelbiopsie zur Diagnose erforderlich; eine oberflächliche Probebiopsie reicht nicht aus [1, 23]. Eine Gewebeentnahme direkt aus einer Ulzeration sollte vermieden werden, da sonst nur Granulationsgewebe und eine sekundäre Inflammationsreaktion im Rahmen der Wundheilung nachgewiesen werden können [8]. Häufig sind mehrere Gewebeproben notwendig, um die bei der LV typischen vaskulären Veränderungen anzutreffen, da diese nicht ubiquitär, sondern nur abschnittsweise auftreten [8].
Die Bestimmung verschiedener Laborparameter dient insbesondere der Abgrenzung zu Differenzialdiagnosen und hat bei deren Nachweis nur mäßig relevante therapeutische Konsequenzen für die betroffenen Patienten, beispielsweise in Hinblick auf eine genetische Beratung oder Vitaminsubstitution bei Hyperhomocysteinämie [23]. Eine generelle Bestimmung jeglicher mit Gerinnungsstörungen assoziierter Laborparameter wird und kann nicht empfohlen werden [36].
Um Differenzialdiagnosen mit laborchemischen Untersuchungen auszuschließen, sollten folgende Parameter bestimmt werden:
IgG
antinukleäre Antikörper (ANA), vor allem wenn Hinweise für eine Kollagenose vorligen
extrahierbare nukleäre Antigene (ENA), um das Sjögren-Syndrom auszuschließen
Antiphospholipid-Antikörper wie Anti-β-2-Glykoprotein-Antikörper, Antikardiolipin-Antikörper und Lupus-Antikoagulans, um ein Antiphospholipid-Syndrom auszuschließen
Paraproteine, gegebenenfalls mit Bestimmung der Kryoglobuline und Kryofibrinogen, um eine Kryoglobulinämie Typ I auszuschließen
Weitere mögliche relevante Thrombophiliefaktoren können untersucht werden [23], wie
Faktor-V-Leiden,
Protein-C- oder -S-Mangel,
Antithrombin-III-Mangel,
Hyperhomocysteinämie.
D-Dimere sind zwar unspezifisch für die Erkrankung, stellen jedoch auch bei der LV einen schnellen Test für thrombotische Ereignisse dar [37]. Gerinnungstests sollten nicht während eines thromboembolischen Ereignisses oder unter antikoagulatorischer Therapie durchgeführt werden, da dies die Testergebnisse beeinflusst. Zudem sollten sekundäre Gründe für einen Status der Hyperkoagulabilität bedacht werden, wie Immobilisation durch Hospitalisierung, Trauma oder Operation, konsumierende Erkrankungen (u. a. Krebserkrankungen), Schwangerschaft, Medikamente (u. a. Antikoagulationen oder orale Kontrazeptiva), Adipositas oder fortgeschrittenes Lebensalter [14, 36]. Da sie leicht zu substituieren sind, können auch Vitamin B6, B12 und Folsäure bestimmt werden. Zusätzlich wird Lp(a) bestimmt, da es einen Risikofaktor für kardiovaskuläre Erkrankungen ist und eine entsprechende Vorsorge betrieben werden muss.
Differenzialdiagnosen
Die Differenzialdiagnosen umfassen [23]
kutane PAN
kutane Kleingefäßvaskulitis
Antiphospholipidsyndrom
Pyoderma gangraenosum
Kryoglobulinämie Typ 1
Sjögren-Syndrom
Warfarin-induzierte kutane Nekrose
Die wichtigste Differenzialdiagnose ist die kutane PAN, deren Klinik teilweise genauso wie bei der LV imponieren kann. Als Hinweise für eine PAN dienen subkutane, erythematöse Knoten und eine bereits von Erkrankungsbeginn an bestehende Mononeuritis multiplex, die sich bei der LV erst im späteren Verlauf manifestieren kann [23]. Die histopathologische Begutachtung sollte schließlich beide Erkrankungen differenzieren.
Die als Livedo racemosa bezeichnete livid-erythematöse Verfärbung der Haut mit unregelmäßigem und unterbrochenem Netzwerk kommt durch eine fokale Unterbrechung der Blutzufuhr in den kutanen Gefäßen zustande. Sie ist leicht zu erkennen und sollte stets abgeklärt werden. Eine Livedozeichnung findet sich jedoch nicht nur bei der LV, sondern bei diversen anderen Erkrankungen, darunter PAN, Sneddon-Syndrom, Antiphospholipid-Syndrom, Calciphylaxie, Kryoglobulinämie, verschiedene hämatologische Erkrankungen, Autoimmunerkrankungen wie systemischer Lupus erythematodes, Infektionen, Malignome oder sekundär nach bestimmten Medikamenten, zum Beispiel Cumarinen [38].
Auch wenn der Begriff Atrophie blanche teilweise synonym zur LV verwendet wird, finden sich Atrophie-blanche-artige Narben auch bei anderen Erkrankungen wie chronisch venöser Insuffizienz, Antiphospholipid-Syndrom, kutanen Vaskulitiden oder systemischem Lupus erythematodes. Sie sind somit nicht pathognomonisch für die LV [1, 29, 39].
Therapie
Bei der LV gibt es verschiedenste Therapieansätze, aufgrund der Seltenheit der Erkrankung jedoch keine einheitliche und evidenzbasierte Behandlungsstrategie. Die Therapie zielt darauf ab, die Hautläsionen zu verbessern, Rezidive zu verhindern und insbesondere die Schmerzen zu reduzieren, um die Lebensqualität der Patienten zu verbessern [40]. Ein einzelner Therapieansatz ist nicht für alle Patienten gleich effektiv, sodass oft mehrere Behandlungsoptionen in Kombination zum Einsatz kommen [15].
Aufgrund der pathophysiologisch im Vordergrund stehenden Mikrothrombosierung ist eine antikoagulatorische Therapie, zum Beispiel mit niedermolekularem Heparin oder direkten oralen Antikoagulantien (DOAK), die erste Wahl [22, 40]. Eine antikoagulatorische Therapie ist nicht nur sehr effektiv und schnell wirksam, sie ist im Vergleich zu anderen Therapieoptionen auch relativ kostengünstig und nebenwirkungsarm [5]. Über eine erfolgreiche und prompte Therapie mit niedermolekularen Heparinen wurde nicht nur bei Erwachsenen [41], sondern ebenso bei einer jugendlichen Patientin berichtet [42]. Eine Thrombozytenkontrolle in den ersten vier Wochen ist laut Leitlinie nicht notwendig, aber dennoch empfehlenswert.
Eine multizentrische, einarmige, prospektive Studie (RILIVA) zeigte den effektiven Einsatz von Rivaroxaban 10-20 mg ein- bis zweimal täglich an einem Patientenkollektiv von 25 Patienten [10]. Die orale Gabe von Rivaroxaban statt der subkutanen Gabe von Enoxaparin sowie die fehlende Notwendigkeit eines Monitorings der Blutwerte zu Beginn der Therapie erhöhen die Compliance beim Patienten. Allerdings muss dieser über die möglichen Nebenwirkungen einer erhöhten Blutungsneigung aufgeklärt werden [10, 43]. Weiterhin ist die Gabe intravenöser Immunglobuline (2 g/kg Körpergewicht über 2-5 Tage, in vierwöchigem Rhythmus) eine sehr wirkungsvolle Therapieoption, deren Einsatz jedoch in Hinblick auf die Wirtschaftlichkeit bei sehr hohen Therapiekosten und mittlerweile begrenzter Verfügbarkeit zunächst geprüft werden sollte [40, 44, 45, 46, 47]. Bei ineffektiver antikoagulatorischer Therapie kann als zweite Wahl das Prostazyklin-Analogon Iloprost eingesetzt werden [48].
Weitere Therapieoptionen umfassen Vitamin-K-Antagonisten, Thrombozytenaggregationshemmer wie Acetylsalicylsäure, Dipyridiamol oder Pentoxifyllin sowie Fibrinolytika wie "tissue-type plasminogen activator" (tPA), Danazol oder Stanozolol, die zwar häufig eingesetzt werden, jedoch aufgrund der pathophysiologischen Überlegungen weniger sinnvoll erscheinen [23, 40]. Darüber hinaus existieren Fallberichte über vorteilhafte Effekte von hyperbarer Sauerstofftherapie sowie Psoralen plus UV-A, Sulfasalazin, Nicotinsäure, Doxycyclin und Ciclosporin [23, 40]. Zudem wird vermutet, dass sich eine Raucherentwöhnung positiv auf die Erkrankung auswirken könnte [49].
Bei häufig koexistenter venöser Insuffizienz ist, nachdem eine arterielle Störung ausgeschlossen wurde, zudem eine Kompressionstherapie sinnvoll, um Ödeme zu reduzieren und die Fibrinolyse zu stimulieren [23, 31].
Eine adäquate Schmerztherapie ist entscheidend, um die Lebensqualität zu verbessern [9], wobei Schmerzen häufig durch den Einsatz antikoagulatorischer Medikamente [50] sowie durch eine angepasste Wundversorgung reduziert werden können [23]. Eine antiinflammatorische Therapie mit nicht steroidalen Antirheumatika wird zwar häufig eingesetzt, zeigt jedoch keine gute Wirkung [5].
In einer retrospektiven Analyse berichteten Forschende kürzlich über fünf Patienten, die nach erfolgloser Vortherapie mit Glukokortikosteroiden, Plättchenhemmern und Danazol gut auf eine Therapie mit dem Tumornekrosefaktor-Inhibitor Etanercept (25-50 mg, einmal pro Woche) ansprachen [51]. In einer weiteren Studie derselben Arbeitsgruppe wurde auch Adalimumab erfolgreich bei zehn Patienten mit LV eingesetzt [52].
Zuletzt wurden in einigen Studien auch gute Behandlungserfolge mit oralen Januskinase-Inhibitoren wie Tofacitinib [53] und Baricitinib [54] beschrieben. Allerdings sind hierzu weitere Untersuchungen nötig.
Fazit
Die LV ist eine okklusive kutane Gefäßerkrankung mit Assoziation zu verschiedenen Gerinnungsstörungen, hauptsächlich angeborenen und erworbenen Thrombophilien, Autoimmunerkrankungen und Malignomen.
Die LV sollte immer als Differenzialdiagnose bei chronisch rezidivierenden (kleinen) Ulzerationen der unteren Extremitäten insbesondere bei starken Schmerzen und einer Livedo racemosa in Betracht gezogen werden. Empfohlen werden eine histologische Sicherung sowie eine Laboruntersuchung mit Bestimmung der Antiphospholipid-Antikörper, der ANA, von Antithrombin-III, Lp(a), Protein C und S, Homocystein und der Vitamine B6 und B12.
Therapie der ersten Wahl ist niedermolekulares Heparin. Nach deutlicher und persistierender Besserung kann auf Rivaroxaban oder eine halbtherapeutische Heparin-Dosis umgestellt werden. Bei Therapieresistenz oder einem nicht ausreichenden Ansprechen können nach Genehmigung durch die zuständige Krankenkasse zusätzlich Immunglobuline gegeben werden. Diese haben sich in kleinen Fallserien als sehr wirkungsvolle Therapieoption erwiesen, mit Abheilungsraten von mindestens 90 %. Zu neuen Therapieoptionen wie Tumornekrosefaktor-Antikörpern und Januskinase-Inhibitoren wurden erst in jüngerer Zeit kleine Studien publiziert, daher müssen diese Behandlungsmöglichkeiten ihren Stellenwert erst noch beweisen.
Prof. Dr. med. Stefan W. Schneider.
Universitätsklinikum Hamburg-Eppendorf (UKE)
Klinik und Poliklinik für Dermatologie und Venerologie
Martinistraße 52
20246 Hamburg
st.schneider@uke.de
Ausgabe verpasst?
Jetzt als ePaper lesen!
Lesen Sie hautnah dermatologie jetzt auch digital auf Ihrem Tablet oder Smartphone - jederzeit und überall. SpringerMedizin.de hält für Sie alle Ausgaben der letzten drei Jahre als ePaper bereit, auf die Sie kostenfrei zugreifen können.

Highlights der letzten Ausgabe:
Geriatrische Dermatologie: typische Hauterkrankungen älterer Menschen erkennen und behandeln
Stress und Haut
Nahrungsmittelallergien mithilfe von Laborwerten, Pricktests, Endoskopie und Co. diagnostizieren
Das gesetzliche Hautkrebsscreening auf dem Prüfstand
Bericht vom 16. Dermatologie-Update-Seminar
www.springermedizin.de/hautnah-dermatologie

CME-Fragebogen.
Livedovaskulopathie
Teilnehmen und Punkte sammeln können Sie
als e.Med-Abonnent*in von SpringerMedizin.de
als registrierte*r Abonnent*in dieser Fachzeitschrift
zeitlich begrenzt unter Verwendung der abgedruckten FIN.
Dieser CME-Kurs ist auf SpringerMedizin.de/CME zwölf Monate verfügbar. Sie finden ihn, wenn Sie die FIN oder den Titel in das Suchfeld eingeben. Alternativ können Sie auch mit der Option "Kurse nach Zeitschriften" zum Ziel navigieren oder den QR-Code links scannen.

Dieser CME-Kurs wurde von der Bayerischen Landesärztekammer mit zwei Punkten in der Kategorie I (tutoriell unterstützte Online- Maßnahme) zur zertifizierten Fortbildung freigegeben und ist damit auch für andere Ärztekammern anerkennungsfähig.
Für eine erfolgreiche Teilnahme müssen 70 % der Fragen richtig beantwortet werden. Pro Frage ist jeweils nur eine Antwortmöglichkeit zutreffend. Bitte beachten Sie, dass Fragen wie auch Antwortoptionen online abweichend vom Heft in zufälliger Reihenfolge ausgespielt werden.
Bei inhaltlichen Fragen erhalten Sie beim Kurs auf SpringerMedizin.de/CME tutorielle Unterstützung. Bei technischen Problemen erreichen Sie unseren Kundenservice kostenfrei unter der Nummer 0800 7780777 oder per Mail unter kundenservice@springermedizin.de.
Welche Aussage zur Livedovaskulopathie ist korrekt?
Sie betrifft vor allem die oberen Extremitäten.
Sie tritt erst im höheren Alter auf.
Sie tritt nur in den Sommermonaten auf.
Sie betrifft häufiger Frauen als Männer.
Sie tritt meist akut auf und rezidiviert nur selten.
Was ist charakteristisch für die Livedovaskulopathie?
Sie ist selbstlimitierend und heilt rückstandslos ab.
Sie wird meist innerhalb weniger Wochen diagnostiziert.
Sie ist durch ein entzündliches Geschehen gekennzeichnet.
Sie betrifft vorwiegend die größeren Gefäße der tiefen Dermis.
Sie ist in der Regel mit starken Schmerzen verbunden.
Mit welcher anderen Erkrankung ist die Livedovaskulopathie assoziiert?
Polyzythämie
Homocysteinämie
Thrombozythämie
Anämie
Hämophilie
Durch welche Trias ist das klinische Bild der Livedovaskulopathie typischerweise gekennzeichnet?
Livedo racemosa, Papeln, Atrophie blanche
Livedo racemosa, Hyperpigmentierungen, Ulzera
Livedo racemosa, Ulzera, Atrophie blanche
Livedo racemosa, Bullae, Teleangiektasien
Livedo racemosa, Teleangiektasien, Hyperpigmentierungen
Was trifft auf die Livedo racemosa zu?
Sie tritt zwar häufig aber nicht nur an den unteren Extremitäten auf.
Sie ist eine bläulich-livide Verfärbung der Haut.
Sie ist ein Resultat der gesteigerten Hautdurchblutung.
Sie kann im Erkrankungsverlauf wandern.
Sie sieht wie ein gleichmäßiges, ununterbrochenes Netzwerk aus.
Bei Livedovaskulopathie zeigen sich in der direkten Immunfluoreszenz insbesondere granuläre Ablagerungen von C3, Fibrinogen und einem Immunglobulin (Ig). Um welches Ig handelt es sich primär?
IgA
IgD
IgE
IgG
IgM
Was gilt für die Histologie bei Livedovaskulopathie?
Intravaskulär findet sich ein spärliches Infiltrat aus Lymphozyten.
Perivaskulär findet sich ein Infiltrat aus Neutrophilen.
Im Bereich der Ulzera lässt sich eine Leukozytoklasie nachweisen.
In den Gefäßwänden lassen sich Fibrinablagerungen nachweisen.
In der Subcutis lassen sich intraluminale hyaline Thromben in den Blutgefäßen nachweisen.
Welche Aussage zur Biopsie bei einer Livedovaskulopathie ist richtig?
Für die Diagnose ist sie nicht nötig und sollte vermieden werden, da sie schlecht abheilt.
Für die Diagnose ist eine oberflächliche Probebiopsie ausreichend.
Für die Diagnose ist eine tiefe Spindelbiopsie erforderlich.
Für die Diagnose reicht eine Gewebeprobe in der Regel aus.
Für die Diagnose sollte das Gewebe direkt aus einer Ulzeration entnommen werden.
Welches ist die wichtigste Differenzialdiagnose einer Livedovaskulopathie?
kutane Kleingefäßvaskulitis
kutane Panarteriitis nodosa
Pyoderma gangraenosum
Warfarin-induzierte kutane Nekrose
Sjögren-Syndrom
Was ist bei Livedovaskulopathie Therapie der ersten Wahl?
niedermolekulares Heparin
Rivaroxaban
Immunglobuline
Iloprost
Fibrinolytika
Supplementary Information
Interessenkonflikt
Der Autor erklärt, dass er sich bei der Erstellung des Beitrages von keinen wirtschaftlichen Interessen leiten ließ. Er legt folgende potenzielle Interessenkonflikte offen: Novartis, Almirall, Leo Pharma, GSK, Sanofi, Pfizer.
Der Verlag erklärt, dass die inhaltliche Qualität des Beitrags durch zwei unabhängige Gutachten bestätigt wurde. Werbung in dieser Zeitschriftenausgabe hat keinen Bezug zur CME-Fortbildung. Der Verlag garantiert, dass die CME-Fortbildung sowie die CME-Fragen frei sind von werblichen Aussagen und keinerlei Produktempfehlungen enthalten. Dies gilt insbesondere für Präparate, die zur Therapie des dargestellten Krankheitsbildes geeignet sind.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.