

Abstract
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant multisystemic vascular dysplasia, characterized by arteriovenous malformations (AVMs), mucocutaneous telangiectasia and nosebleeds. HHT is caused by a heterozygous null allele in ACVRL1, ENG, or SMAD4, which encode proteins mediating bone morphogenetic protein (BMP) signaling. Several missense and stop-gain variants identified in GDF2 (encoding BMP9) have been reported to cause a vascular anomaly syndrome similar to HHT, however none of these patients met diagnostic criteria for HHT. HHT families from UK NHS Genomic Medicine Centres were recruited to the Genomics England 100,000 Genomes Project. Whole genome sequencing and tiering protocols identified a novel, heterozygous GDF2 sequence variant in all three affected members of one HHT family who had previously screened negative for ACVRL1, ENG, and SMAD4. All three had nosebleeds and typical HHT telangiectasia, and the proband also had severe pulmonary AVMs from childhood. In vitro studies showed the mutant construct expressed the proprotein but lacked active mature BMP9 dimer, suggesting the mutation disrupts correct cleavage of the protein. Plasma BMP9 levels in the patients were significantly lower than controls. In conclusion, we propose that this heterozygous GDF2 variant is a rare cause of HHT associated with pulmonary AVMs.
Keywords: arteriovenous malformations, bone morphogenetic protein 9, hereditary hemorrhagic telangiectasia
1 |. INTRODUCTION
Hereditary hemorrhagic telangiectasia (HHT; syn. Osler–Weber–Rendu syndrome) is a multisystemic vascular dysplasia inherited as an autosomal dominant trait. It is diagnosed clinically by the presence of at least three diagnostic criteria, known as the Curaçao criteria: spontaneous, recurrent nose bleeds; multiple telangiectases at characteristic sites (lips, oral cavity, fingers, nose); visceral lesions such as gastrointestinal telangiectasia (with or without bleeding); arteriovenous malformations (AVMs) in the lung, liver, brain or spine; and a first degree relative with HHT according to these criteria (Shovlin et al., 2000). Nosebleeds are the most common symptom in HHT and frequently lead to iron deficiency anemia. Pulmonary AVMs (PAVMs) affect at least 50% of HHT patients, are usually silent, but cause low blood oxygen levels (hypoxemia), paradoxical ischemic strokes, and cerebral abscess (Shovlin, 2014). The majority of genotyped HHT patients have a heterozygous pathogenic DNA sequence variant in ENG, encoding endoglin (ENG, HHT1) (McAllister et al., 1994), or ACVRL1, encoding activin receptor-like kinase (ALK1, HHT2) (Johnson et al., 1996). A smaller proportion have HHT due to a heterozygous pathogenic variant in SMAD4, which also cause other pathologies, including juvenile polyposis and aortopathy (Gallione et al., 2004; Heald et al., 2015). Current sequencing strategies do not detect causal variants in ~10% of genotyped HHT patients (Shovlin et al., 2020).
BMP9 is a specific ALK1 ligand encoded by GDF2 (David et al., 2007). BMP9/10 inhibition in neonatal mice leads to hyper-vascularization and induces retinal AVMs (Ruiz et al., 2016). Heterozygous missense substitutions in GDF2 were first identified in three of 191 unrelated individuals with nosebleeds and skin telangiectasia, who did not have a pathogenic variant in any of the established HHT genes (ACVRL1, ENG, or SMAD4) (Wooderchak-Donahue et al., 2013). However, the telangiectases were atypical for HHT and considered more consistent with a capillary-malformation AVM syndrome (Wooderchak-Donahue et al., 2013). With the recent expansion of population-scale whole genome sequencing (WGS), one of these variants (p.Arg333Trp) is found in 0.1% of control chromosomes, and thus its pathogenicity is now uncertain (Table S1). More recently, two children with PAVMs and other features suggestive of HHT were identified with homozygous GDF2 mutations (one frameshift, one nonsense) (Hodgson et al., 2021; Liu et al., 2020), while a third patient with a homozygous nonsense mutation initially presented with pulmonary arterial hypertension (PAH) (Wang et al., 2016), and was later found to have multiple spider-like telangiectases on the face (Hodgson et al., 2021). Again, the telangiectases in these three cases were not typical of HHT (Hodgson et al., 2021; Liu et al., 2020). Remarkably, all of the heterozygous parents and one homozygous sibling had few or no HHT features. In summary, none of the patients previously reported with GDF2 variants met Curaçao criteria (Hodgson et al., 2021; Liu et al., 2020; Wooderchak-Donahue et al., 2013).
2 |. CLINICAL DESCRIPTION
The pedigree for the family is indicated in Figure 1a. The proband (III.1) was diagnosed with HHT during childhood due to frequent nosebleeds, mucocutaneous telangiectasia, and PAVMs. He had presented at the age of 8 years with cyanosis, and was diagnosed with multiple PAVMs that were too diffuse for treatment. He initially deteriorated steadily, with oxygen saturations (SaO2) less than 80% at rest, and by the age of 11 years was only able to walk a few steps. A possible heart–lung transplant was declined by the family. He was commenced on nifedipine, based on theoretical considerations by his pediatrician, and improved functionally, though remained cyanosed and clubbed. At the time of recruitment to the 100,000 Genomes Project when classical HHT telangiectasia were confirmed, he was 38 years old, working full time, with a low normal exercise capacity (Medical Research Council dyspnea scale 1 [normal], New York Heart Association Class I [normal], VSAQ varying between 8 and 9 corresponding to metabolic equivalents of ~10–11 kcal/kg/min) (Fletcher, 1960; Myers et al., 1994; New York Heart Association, 1964). This corresponded to the upper tertile of a recent PAVM patient cohort (range 1.33–15.55 kcal/kg/h [median 8.84, interquartile range 6.64–11.86 kcal/kg/h]) (Gawecki et al., 2019). The SaO2 was 85% standing at rest, and he was compensating for this by polycythemia (hemoglobin 201 g/L, hematocrit 0.558). On cardiology workup, he was considered to have no significant pulmonary hypertension. His DNA had been sequenced for ACVRL1, ENG, and SMAD4 with no variants identified.
FIGURE 1.
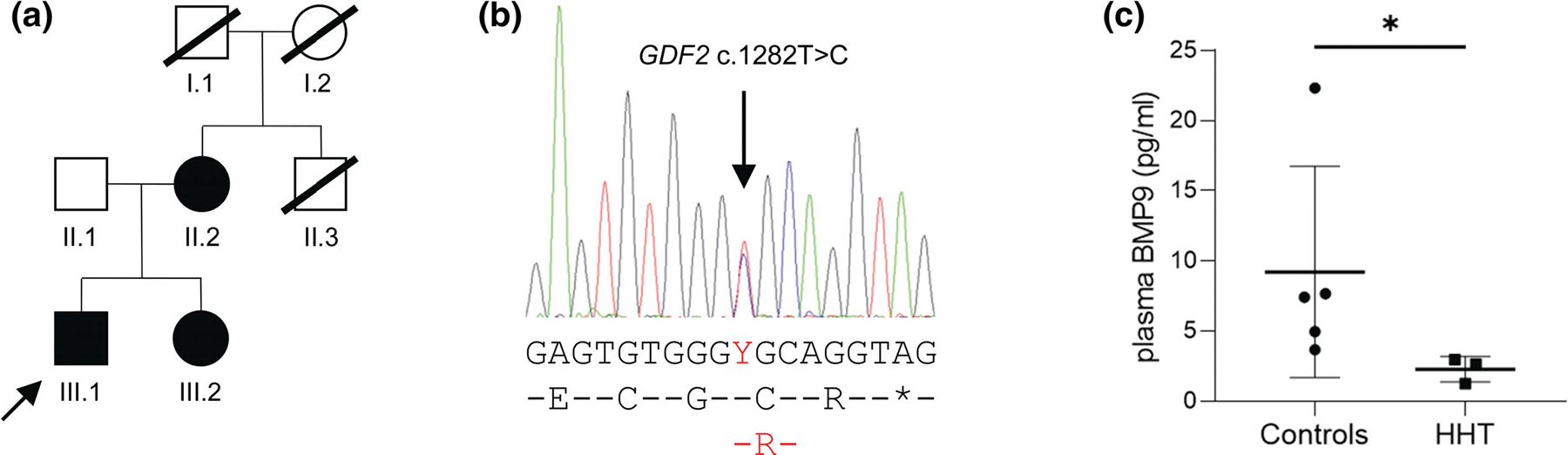
HHT family with GDF2 missense variant and reduced plasma BMP9 levels. (a) Pedigree at the time of family recruitment. The proband (III.1) is arrowed. (b) Sanger sequencing trace from individual III.2 showing the heterozygous GDF2 variant. Nucleotide sequence and protein translation are depicted below, with variants denoted in red font. (c) Circulating BMP9 levels in human plasma from the three individuals carrying the c.1282T>C variant were significantly lower than in five unaffected individuals collected and analyzed under the same conditions (*p = 0.036, Mann–Whitney two-tailed test)
The proband’s sister (III.2) realized she was likely to be affected with HHT when nosebleeds developed in her 30s. She received a definite clinical diagnosis aged 41 years due to the affected sibling, nosebleeds, and typical tiny HHT telangiectasia over the lips and face (none on finger pads, mouth, ears, or conjunctiva). A thoracic CT scan identified no pulmonary AVMs, and SaO2 was normal. The proband’s mother (II.2) received a positive clinical diagnosis at the age of 76 years due to severe nosebleeds as a child, subtle telangiectasia on the lips, and affected children. Again SaO2 was normal. An uncle (II.3) had died during a childhood tonsillectomy due to excessive bleeding. The proband’s maternal grandmother (I.2) died of an unusual stroke in her 50s.
3 |. METHODS
3.1 |. Editorial policies and ethical considerations
This study was approved by the East of Scotland Research Ethics Service (16/ES/0095). All participants provided written informed consent.
3.2 |. Sequencing and analysis pipelines
Detailed methodology is provided in the Supporting Information. Due to the absence of a variant in the known HHT genes, the family was identified through the UK National Health Service (NHS) as suitable for the 100,000 Genomes Project. For HHT, Tier 1 variants were defined as clear loss of function variants in ENG, ACVRL1, and SMAD4, Tier 2 as other variants in these genes, and Tier 3 as protein truncating, de novo or protein altering variants in other genes. Genomics England performed all WGS, data alignments, and initial variant tiering using Illumina pipeline alignments and variant calling. Separately, a Respiratory GeCIP (Clinical Interpretation Partnership) was established to analyze full WGS/phenotypic datasets. Customized scripts were written in Python to extract all variants in HHT patients’ variant call format files for single nucleotide variants, small indels and larger copy number variants. Data were exported under Project Code RR42.
3.3 |. In vitro functional analysis
Functional consequences of the GDF2 variant were studied by in vitro expression of mutant and wild-type plasmids. Briefly, site-directed mutagenesis was used to introduce the variant into a full-length human GDF2 cDNA clone. Plasmids were transfected into 293T cells and cultured for 5 days. Western blot analysis was performed on cell lysates and conditioned media, using antibodies that specifically target the mature BMP9 protein or the proprotein. Enzyme-linked immunosorbent assay (ELISA) was performed on conditioned media samples using the DuoSet Human BMP9 kit (R&D Systems) according to the manufacturer’s instructions. To determine if the secreted BMP9 was biologically active, control pulmonary artery endothelial cells were incubated with conditioned medium for 4 h, then harvested for RNA extraction and analyzed by realtime quantitative RT-PCR for expression of the BMP-responsive genes ID1 and pre-miR-21.
3.4 |. BMP9 concentration in human plasma
Plasma samples were obtained from the three affected family members, one unaffected relative (II.1), and four unrelated healthy controls. Circulating BMP9 levels were measured by ELISA as described above, with the addition of 0.1% heat inactivated goat serum to avoid nonspecific binding.
4 |. RESULTS
In keeping with the earlier clinical sequencing studies, WGS and tiering protocols identified no Tier 1 or Tier 2 variants in the family. The family had 76 Tier 3 variants, that is, protein truncating or protein altering variants in other genes, present in the affected mother and children, but absent in the unaffected father. Inspection of these Tier 3 variants identified a novel, heterozygous GDF2 missense variant (c.1282T>C, p.Cys428Arg). This variant is predicted to be deleterious (SIFT) and probably damaging (PolyPhen), with a Combined Annotation Dependent Depletion (CADD) score of 28.8 (Rentzsch et al., 2019). It has been reported once in 152,092 WGS alleles (population frequency of 6.58 × 10−6) in gnomAD v3.1.1 (https://gnomad.broadinstitute.org). Sanger sequencing confirmed the presence of this variant in all three HHT-affected family members (Figure 1b). Six other Tier 3 variants were identified that probably modify function, but were not likely candidate genes for HHT on the basis of their known biology (Table S2). Through the 100,000 Genomes Project Data Release 6.0, WGS data were available for 160 HHT participants from 126 families. No other GDF2 variants were identified in these families.
Given previous reports of GDF2 variants in patients with HHT-like features, this variant was prioritized for further analysis. The concentration of circulating mature BMP9 protein measured in plasma was significantly lower for the three HHT patients who carry the variant than in samples from five unaffected individuals (the father (II.1) and four unrelated controls), which were collected and processed in parallel (median 2.65 vs. 7.43 pg/ml, p = 0.036) (Figure 1c). Notably, the severely affected proband had the lowest BMP9 level of all. He had become increasingly unwell in the months preceding plasma collection, and had been diagnosed with decompensated cirrhosis without evidence of hepatic AVMs. It is unclear whether the very low circulating BMP9 and his liver failure are related.
BMP9 is synthesized as a proprotein, which then dimerizes and is cleaved to produce the mature secreted protein. Western blotting of cells transfected with the wild-type or mutant plasmid demonstrated expression of the proprotein in all cells, whereas the mature protein was not detected from the mutant construct, suggesting the c.1282T>C variant disrupts processing of BMP9 (Figure 2a). This was supported by analysis with the proprotein antibody, where two species were detected in wild-type cells—the uncleaved and cleaved proprotein—whereas in mutant cells, the uncleaved protein predominated (Figure S1). Western blotting of conditioned media confirmed secretion of the mature BMP9 dimer from the wild-type plasmid (20 kDa), together with a larger partially cleaved form detected at about 57 kDa, whereas neither were detected in medium from the mutant construct (Figure S2). ELISA confirmed that mature BMP9 was undetectable in conditioned media from the mutant construct, even at a 1 in 10 dilution, whereas the concentration in wild-type medium was 973 ng/ml (Figure S3). Furthermore, whereas wild-type medium induced a fivefold increase in ID1 expression in cultured endothelial cells and a threefold increase in pre-miR-21, conditioned media from the mutant construct failed to upregulate these BMP-responsive target genes (Figure 2b). ID1 is a target of canonical BMP signaling, whereas miR-21 is upregulated via a noncanonical SMAD4-independent pathway that regulates microRNA maturation (Davis et al., 2008).
FIGURE 2.
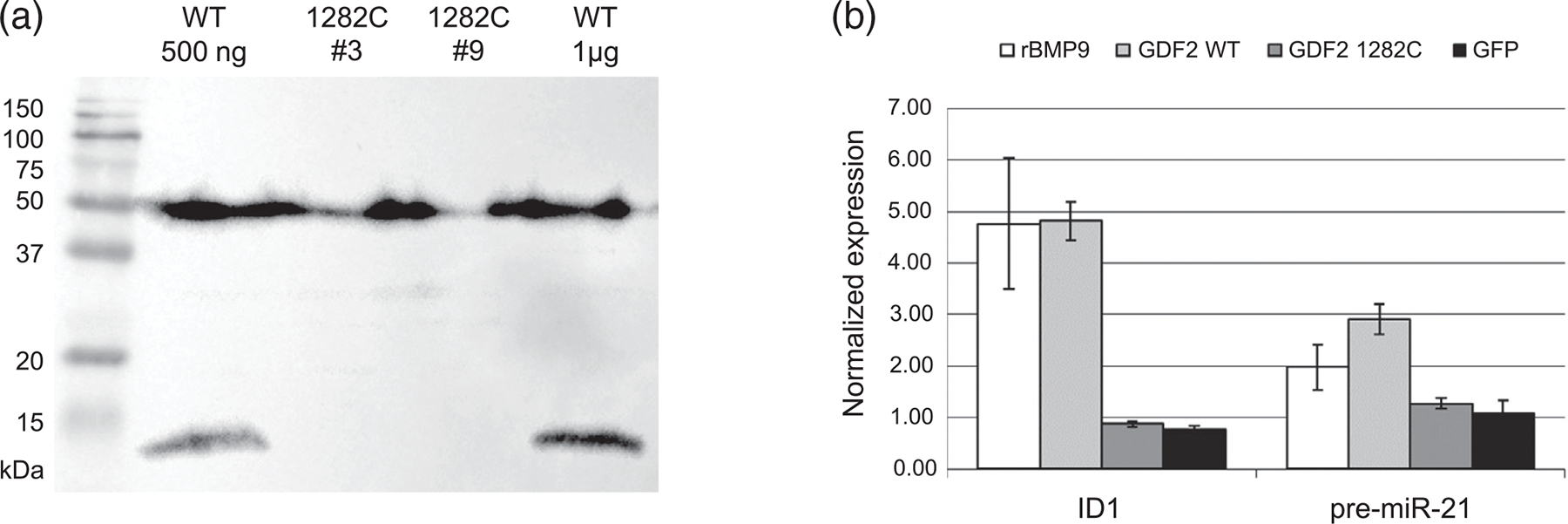
GDF2 c.1282T>C disrupts processing of BMP9 protein. (a) Western blot of lysates from 293T cells transfected with wild-type (WT; 500 ng or 1 μg of DNA) or mutant (1282C, clones #3 and #9; 1 μg) GDF2 plasmids, run under reducing conditions and probed with antibody to human BMP9. Full length protein (50 kDa) is expressed from the mutant plasmids, but is not correctly processed, leading to absence of the mature monomer (12 kDa). (b) Conditioned medium from these cells was applied to cultures of human pulmonary artery endothelial cells (PAEC). Medium from the wild-type (WT) but not mutant (c.1282C) plasmid induces mRNA expression of BMP-responsive genes, ID1 and pre-miR-21. Recombinant BMP9 (rBMP9) was used as a positive control, and medium from GFP transfection as a negative control. Expression was normalized to GAPDH, and fold change was calculated relative to untreated PAEC
5 |. DISCUSSION
We identified a missense GDF2 variant in three affected members of a family with HHT. Results of our in vitro studies suggest this mutation disrupts normal cleavage of BMP9 proprotein, leading to a reduction in the active mature dimer form. Consistent with this, the level of mature protein detectable in plasma from the three mutation-positive patients was more than 2.5-fold lower than controls. Although GDF2 mutations have previously been identified in individuals with a vascular anomaly syndrome similar to HHT, this is the first family that meets Curaçao criteria. Table S1 shows the characteristics of missense GDF2 variants reported in patients with features of HHT. Of note, the c.1282T>C variant in this family has the highest CADD pathogenicity score, whereas some variants now appear likely benign on the basis of low pathogenicity scores, and population frequencies greater than 1/10,000 (Table S1).
Highly variable phenotypes between HHT family members affected by the same pathogenic variant are the norm in HHT, although the discrepancy in HHT phenotype between the proband and other two cases in this pedigree is unusually marked. With the exception of the proband, the clinical phenotype in the other HHT-affected individuals was mild, and in the absence of the proband’s history it is unlikely that their diagnosis would have been suspected. Nevertheless, they both had three Curaçao Criteria, unlike other ACVRL1+/− and ENG+/− confirmed HHT cases in our cohort (Sharma et al., 2021), and were not “atypical” as seen by us for patients with EPHB4+/− and other vasculopathies (Shovlin et al., 2021). WGS identified the scale of genetic variation between the family members, and studies to identify variants potentially relevant to the pulmonary AVM and cirrhotic phenotypes are ongoing. Cirrhosis has been recognized in HHT patients but is not considered part of the direct HHT phenotype (Silvain et al., 2020; VASCERN, 2019), instead usually ascribed to infective or toxin-based etiologies not relevant to individual III.1. Notably, although BMP9 is synthesized in the liver, this is predominantly from nonparenchymal cells (Miller et al., 2000), and therefore it is not currently known if the cirrhosis in the proband was specifically related to the GDF2 mutation.
The majority of GDF2 variants reported to date have been found in patients with PAH (Gräf et al., 2018; Wang et al., 2019), a severe progressive lung vasculopathy that is also associated with mutations of the BMP pathway, especially the type-II BMP receptor (BMPR2), but has a distinct clinical spectrum. Many of the GDF2 variants in PAH are also missense and have similar effects in reducing the secretion of mature BMP9 protein (Hodgson et al., 2020; Wang et al., 2019). Thus at present, there is no obvious genotype–phenotype correlation. Some phenotypic crossover between the two diseases is already known, since a small subset of HHT patients develop PAH, while at least two PAH patients with BMPR2 mutations have been reported with PAVM (Rigelsky et al., 2008; Soon et al., 2014). GDF2 now adds to this enigma, highlighting the importance of understanding potential modifier genes and second hits in the pathogenesis of both HHT and PAH.
Supplementary Material
ACKNOWLEDGMENTS
The authors are indebted to the family for their participation in this study. This research was made possible through access to the data and findings generated by the 100,000 Genomes Project. The 100,000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The 100,000 Genomes Project is funded by the National Institute for Health Research (NIHR) and NHS England. The Wellcome Trust, Cancer Research UK, and the Medical Research Council have also funded research infrastructure. The 100,000 Genomes Project uses data provided by patients and collected by the National Health Service as part of their care and support. The authors thank the National Health Service staff of the UK Genomic Medicine Centre and the families for their willing participation. This research was co-funded by the NIHR Imperial Biomedical Research Centre, and by NHLBI grant R35HL140019 (to M.A.A.). The views expressed are those of the authors and not necessarily those of funders, the NHS, the NIHR, or the Department of Health and Social Care. This research was co-funded by the NIHR Imperial Biomedical Research Centre, and Medical Research Council – Northern Ireland Executive support of the Northern Ireland Genomic Medicine Centre (MC_PC_16018). K.K. was supported by a Department for the Economy Co-operative Awards in Science and Technology (DfE-CAST) PhD studentship award with Belfast Health and Social Care Trust.
Funding information
National Institute for Health Research (NIHR) and NHS England, Grant/Award Number: The 100,000 Genomes Project; National Heart, Lung, and Blood Institute, Grant/Award Number: R35HL140019; National Institute for Health Research, Grant/Award Number: Imperial Biomedical Research Centre 2017–2022; Medical Research Council – Northern Ireland Executive: Northern Ireland Genomic Medicine Centre, Grant/Award Number: MC_PC_16018; Department for the Economy Co-operative Awards in Science and Technology (DfE-CAST)
Footnotes
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
DATA AVAILABILITY STATEMENT
Primary data from the 100,000 Genomes Project, which are held in a secure Research Environment, are available to registered users. Please see https://www.genomicsengland.co.uk/about-gecip/for-gecip-members/data-and-data-access for further information.
REFERENCES
- Criteria Committee New York Heart Association Inc. (1964). Diseases of the heart and blood vessels: Nomenclature and criteria for diagnosis (6th ed.) p. 114. Boston: Little, Brown & Co. [Google Scholar]
- David L, Mallet C, Mazerbourg S, Feige JJ, & Bailly S (2007). Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood, 109, 1953–1961. [DOI] [PubMed] [Google Scholar]
- Davis BN, Hilyard AC, Lagna G, & Hata A (2008). SMAD proteins control DROSHA-mediated microRNA maturation. Nature, 454, 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher CM (1960). Standardised questionnaire on respiratory symptoms: A statement prepared and approved by the MRC Committee on the aetiology of chronic bronchitis (MRC breathlessness score). BMJ, 2, 1665.13688719 [Google Scholar]
- Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, Mitchell G, Drouin E, Westermann CJJ, & Marchuk DA (2004). A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet, 363, 852–859. [DOI] [PubMed] [Google Scholar]
- Gawecki F, Myers J, & Shovlin CL (2019). Veterans specific activity questionnaire (VSAQ): A new and efficient method of assessing exercise capacity in patients with pulmonary arteriovenous malformations. BMJ Open Respiratory Research, 6(1), e000351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gräf S, Haimel M, Bleda M, Hadinnapola C, Southgate L, Li W, Hodgson J, Liu B, Salmon RM, Southwood M, Machado RD, Martin JM, Treacy CM, Yates K, Daugherty LC, Shamardina O, Whitehorn D, Holden S, Aldred M, … Morrell NW (2018). Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nature Communications, 9, 1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heald B, Rigelsky C, Moran R, LaGuardia L, O’Malley M, Burke CA, & Zahka K (2015). Prevalence of thoracic aortopathy in patients with juvenile polyposis syndrome-hereditary hemorrhagic telangiectasia due to SMAD4. American Journal of Medical Genetics. Part A, 167, 1758–1762. [DOI] [PubMed] [Google Scholar]
- Hodgson J, Ruiz-Llorente L, McDonald J, Quarrell O, Ugonna K, Bentham J, Mason R, Martin JM, Moore D, Bergstrom K, Bayrak-Toydemir P, Wooderchak-Donahue WL, Morrell NW, Condliffe R, Bernabéu C, & Upton PD (2021). Homozygous GDF2 nonsense mutations result in a loss of circulating BMP9 and BMP10 and are associated with either PAH or an “HHT-like” syndrome in children. Molecular Genetics & Genomic Medicine. 10.1002/mgg3.1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson J, Swietlik EM, Salmon RM, Hadinnapola C, Nikolic I, Wharton J, Guo J, Liley J, Haimel M, Bleda M, Southgate L, Machado RD, Martin JM, Treacy CM, Yates KP, Daugherty LC, Shamardina O, Whitehorn D, Holden S, … Morrell NW (2020). Characterization of GDF2 mutations and levels of BMP9 and BMP10 in pulmonary arterial hypertension. American Journal of Respiratory and Critical Care Medicine, 201, 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer MC, Pericak-Vance MA, Diamond AG, Guttmacher A, Jackson CE, Attisano L, Kucherlapati R, Porteous ME, & Marchuk DA (1996). Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nature Genetics, 13, 189–195. [DOI] [PubMed] [Google Scholar]
- Liu J, Yang J, Tang X, Li H, Shen Y, Gu W, & Zhao S (2020). Homozygous GDF2-related hereditary hemorrhagic telangiectasia in a Chinese family. Pediatrics, 146, e20191970. [DOI] [PubMed] [Google Scholar]
- McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrel JC, McCormick MK, Pericak-Vance MA, Heutink P, Oostra BA, Haitjema TJ, Westerman CJ, Porteous ME, Guttmacher A, Letarte M, & Marchuk DA (1994). Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nature Genetics, 8, 345–351. [DOI] [PubMed] [Google Scholar]
- Miller AF, Harvey SA, Thies RS, & Olson MS (2000). Bone morphogenetic protein-9. An autocrine/paracrine cytokine in the liver. The Journal of Biological Chemistry, 275, 17937–17945. [DOI] [PubMed] [Google Scholar]
- Myers J, Do D, Herbert W, Ribisl P, & Froelicher VF (1994). A nomogram to predict exercise capacity from a specific activity questionnaire and clinical data. The American Journal of Cardiology, 73, 591–596. [DOI] [PubMed] [Google Scholar]
- Rentzsch P, Witten D, Cooper GM, Shendure J, & Kircher M (2019). CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research, 47, D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigelsky CM, Jennings C, Lehtonen R, Minai OA, Eng C, & Aldred MA (2008). BMPR2 mutation in a patient with pulmonary arterial hypertension and suspected hereditary hemorrhagic telangiectasia. American Journal of Medical Genetics. Part A, 146A, 2551–2556. [DOI] [PubMed] [Google Scholar]
- Ruiz S, Zhao H, Chandakkar P, Chatterjee PK, Papoin J, Blanc L, Metz CN, Campagne F, & Marambaud P (2016). A mouse model of hereditary hemorrhagic telangiectasia generated by transmammary-delivered immunoblocking of BMP9 and BMP10. Scientific Reports, 5, 37366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma L, Alsafi A, Ferguson T, Redhead J, West London Genomic Medicine Centre, & Shovlin CL (2021). Pulmonary arteriovenous malformations – Genetic versus clinical evidence of underlying hereditary haemorrhagic telangiectasia. Thorax, 76(Suppl 2), A62. https://thorax.bmj.com/content/76/Suppl_2/A62.2 [Google Scholar]
- Shovlin CL (2014). Pulmonary arteriovenous malformations. American Journal of Respiratory and Critical Care Medicine, 190, 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, Kjeldsen AD, & Plauchu H (2000). Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-weber syndrome). American Journal of Medical Genetics, 91, 66–67. [DOI] [PubMed] [Google Scholar]
- Shovlin CL, Simeoni I, Downes K, Frazer ZC, Megy K, BernabeuHerrero ME, Shurr A, Brimley J, Patel D, Kell L, Stephens J, Turbin IG, Aldred MA, Penkett CJ, Ouwehand WH, Jovine L, & Turro E (2020). Mutational and phenotypic characterization of hereditary hemorrhagic telangiectasia. Blood, 136, 1907–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shovlin CL, Buscarini E, Sabbà C, Mager HJ, Kjeldsen AD, Pagella F, Sure U, Ugolini S, Toerring PM, Suppressa P, Rennie C, Post MC, Patel MC, Nielsen TH, Manfredi G, Lenato G, Lefroy DC, Kariholu U, Jones B, … Dupuis-Girod S (2021). The European rare disease network for HHT frameworks for management of hereditary haemorrhagic telangiectasia in general and speciality care. European Journal of Medical Genetics. 10.1016/j.ejmg.2021.104370 [DOI] [PubMed] [Google Scholar]
- Silvain C, Thévenot T, Colle I, Vilgrain V, Dupuis-Girod S, Buscarini E, Valla D, Hillaire S, Dutheil D, Sitbon O, Bureau C, & Plessier A (2020). Hereditary hemorrhagic telangiectasia and liver involvement: Vascular liver diseases: Position papers from the francophone network for vascular liver diseases, the French Association for the Study of the liver (AFEF), and ERN-rare liver. Clinics and Research in Hepatology and Gastroenterology, 44(4), 426–432. [DOI] [PubMed] [Google Scholar]
- Soon E, Southwood M, Sheares K, Pepke-Zaba J, & Morrell NW (2014). Better off blue: BMPR-2 mutation, arteriovenous malformation, and pulmonary arterial hypertension. American Journal of Respiratory and Critical Care Medicine, 189, 1435–1436. [DOI] [PubMed] [Google Scholar]
- VASCERN. (2019). Orphanet Definition of Hereditary haemorrhagic telangiectasia. www.orpha.net/consor/www/cgi-bin/OC_Exp.php?lng=EN&Expert=774
- Wang G, Fan R, Ji R, Zou W, Penny DJ, Varghese NP, & Fan Y (2016). Novel homozygous BMP9 nonsense mutation causes pulmonary arterial hypertension: A case report. BMC Pulmonary Medicine, 16, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ, Lian TY, Jiang X, Liu SF, Li SQ, Jiang R, Wu W, Ye JL, Cheng C, Du Y, Xu X, Wu YM, Peng F, Sun K, Mao YM, Yu H, Liang C, Shyy JY, Zhang S, … Jing ZC (2019). Germline BMP9 mutation causes idiopathic pulmonary arterial hypertension. The European Respiratory Journal, 53, 1801609. [DOI] [PubMed] [Google Scholar]
- Wooderchak-Donahue WL, McDonald J, O’Fallon B, Upton PD, Li W, Roman BL, Young S, Plant P, Fülöp GT, Langa C, Morrell NW, Botella LM, Bernabéu C, Stevenson DA, Runo JR, & Bayrak-Toydemir P (2013). BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. American Journal of Human Genetics, 93, 530–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Primary data from the 100,000 Genomes Project, which are held in a secure Research Environment, are available to registered users. Please see https://www.genomicsengland.co.uk/about-gecip/for-gecip-members/data-and-data-access for further information.