

Abstract
Metabolic reprogramming is a hallmark of cancer, including lung cancer. However, the exact underlying mechanism and therapeutic potential are largely unknown. Here we report that protein arginine methyltransferase 6 (PRMT6) is highly expressed in lung cancer and is required for cell metabolism, tumorigenicity, and cisplatin response of lung cancer. PRMT6 regulated the oxidative pentose phosphate pathway (PPP) flux and glycolysis pathway in human lung cancer by increasing the activity of 6-phospho-gluconate dehydrogenase (6PGD) and α-enolase (ENO1). Furthermore, PRMT6 methylated R324 of 6PGD to enhancing its activity; while methylation at R9 and R372 of ENO1 promotes formation of active ENO1 dimers and 2-phosphoglycerate (2-PG) binding to ENO1, respectively. Lastly, targeting PRMT6 blocked the oxidative PPP flux, glycolysis pathway, and tumor growth, as well as enhanced the anti-tumor effects of cisplatin in lung cancer. Together, this study demonstrates that PRMT6 acts as a post-translational modification (PTM) regulator of glucose metabolism, which leads to the pathogenesis of lung cancer. It was proven that the PRMT6-6PGD/ENO1 regulatory axis is an important determinant of carcinogenesis and may become a promising cancer therapeutic strategy.
KEY WORDS: Lung cancer, Metabolic reprogramming, Post-translational modification, PRMT6, Pentose phosphate pathway flux, Glycolysis, 6-Phospho-gluconate dehydrogenase, α-enolase, ENO1
Graphical abstract
PRMT6 promotes lung cancer growth and cisplatin response through enhancing oxidative pentose phosphate pathway flux and glycolysis pathway, which mediated by regulating the asymmetric arginine demethylation modification of 6PGD/ENO1.
1. Introduction
Lung cancer remains the second leading cause of cancer mortality, which ranks as the second cancer-related mortality worldwide according to the latest global cancer data1. The reprogramming of energy metabolism has been accepted as a hallmark of cancer, including lung cancer. There have been a number of studies and cancer therapeutic agents targeting the cancer metabolism that are being developed2,3. However, the molecular mechanisms that underlying lung cancer progression are not fully clarified. Thus, there is a critical need not only to elucidate the molecular mechanisms underlying lung cancer carcinogenesis and chemotherapy responses, but also identify molecular targets in lung cancer can be cured and enhance chemotherapy sensitivity.
Several studies reported that the post-translational modification (PTM) participates in essential biological processes, such as cell metabolism4. Protein arginine methylation, which is catalyzed by protein arginine methyltransferases (PRMTs), is one of the most common PTMs. These PRMTs regulate a broad array of cellular processes and are increasingly being recognized as drivers for human diseases, including cancer5, 6, 7. Numerous studies showed that several metabolic enzymes are methylated by PRMTs. Zhong et al.8 reported that coactivator-associated arginine methyltransferase 1 (CARM1) methylates glyceraldehyde-3-phosphate dehydrogenase (GAPDH) at arginine 234 (R234), which is a key regulatory mechanism of glucose metabolism in liver cancer. Studies by Guo et al.9 revealed that CARM1 methylates ribose-5-phosphate isomerase A (RPIA), an enzyme in oxidative pentose phosphate pathway (PPP), at arginine 42 (R42) and enhances its activity, which connects the glucose availability to nucleotide synthesis and redox homeostasis. Yamamoto et al.10 suggested that a carbon monoxide (CO)/cystathionine β-synthase (CBS)-dependent regulation of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB3) methylation by arginine methyltransferase 1 (PRMT1) determines the directional glucose utilization to ensure cancer cell survival under oxidative stress. The findings of Wang et al.11 revealed that CARM1 methylates malate dehydrogenase 1 (MDH1) at arginine 248 (R248), which negatively regulates cellular redox homeostasis and suppresses the glutamine metabolism of pancreatic cancer. Protein arginine methyltransferase 6 (PRMT6) is a type I PRMT that asymmetrically dimethylates histones or non-histones protein substrates on arginine residues5. PRMT6 is frequently overexpressed in human cancers, and contributes in tumor malignancy12, 13, 14. Wong et al.15 revealed that PRMT6 regulates aerobic glycolysis in an extracellular signal-regulated kinase (ERK)-dependent pyruvate kinase M2 (PKM2) nuclear re-localization manner mediated by methylated CRAF. However, the role of PRMT6 in cell metabolism, tumorigenicity, and chemotherapy responses of lung cancer remains largely unknown.
This study aimed to further uncover the role and mechanism of PRMT6 in cell metabolism, tumorigenicity, and explore its potential as a therapeutic target. In this study, it was shown that PRMT6 is highly expressed in lung cancer and is required for lung cancer cell metabolism, progression, and cisplatin response. Furthermore, PRMT6 coordinated the oxidative PPP flux and glycolysis pathway, providing an overall advantage to lung cancer pathogenesis mediated by methylating 6-phospho-gluconate dehydrogenase (6PGD) and α-enolase (ENO1). Lastly, targeting PRMT6 blocked cell metabolism and tumor growth, as well as enhanced the anti-tumor effects of cisplatin on lung cancer. In summary, we show that the PRMT6–6PGD/ENO1 axis is critical for lung cancer progression and inhibiting this axis signaling enhances the anti-tumor effects of cisplatin for treatment of lung cancer preclinical models.
2. Materials and methods
The more detailed information of materials and methods are provided in Supporting Information.
2.1. Reagents and biological resources
The reagents and biological resources are listed in the Supporting Information Table S1.
2.2. Cell culture
Lung cancer H1299, H226, H157, H1944, A549, H460, H2122, and H1437 cells were cultured in RPMI-1640 medium (Thermo Fisher Scientific, MA, USA) supplemented with 10% fetal bovine serum (FBS, ExCell Bio, Shanghai, China). HEK293T cell was maintained in Dulbecco's modified Eagle's medium (DMEM, Thermo Fisher Scientific, MA, USA) supplemented with 10% FBS. Normal proliferating Human Bronchial Epithelial Cell Line (BEAS-2B) was cultured in RPMI 1640 medium with 10% FBS.
2.3. Clinical samples
Twenty-one paired clinical lung tumor tissues and adjacent non-tumor lung tissues (Cohort 1, n = 21) were collected from The First Affiliated Hospital of Wenzhou Medical University (Wenzhou, China) after surgical resection. The clinical lung cancer specimens all have the written consent approving the use of the samples for research purposes from patients. The lung cancer tissue microarrays (Cohort 2, n = 41) were purchased from Shanghai Outdo Biotechnology Co., Ltd. (Shanghai Outdo Biotechnology, Shanghai, China). These microarrays included 41 lung cancer tissues and 41 adjacent non-tumor lung tissues. The relevant characteristics were shown in Supporting Information Table S2. The study protocol was approved by the Institute Research Ethics Committee at Nankai University, Tianjin, China.
2.4. EdU assay
Lung cancer cells that were knocked down for genes or treated with inhibitors were seeded in 96-well plates. On the second day of seeding, the EdU assay was performed using EdU imaging detection kit (RiboBio, Guangzhou, China) according to the manufacturer's instructions.
2.5. Lactate production assay
Cellular lactate production was measured by a colorimetric-based lactic acid assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer's instructions. In brief, we seeded cells in a 6 well-plate and incubated them at 37 °C for 12 h. Media on cells was replaced with phenol red-free RPMI medium without FBS when the cells were 50% confluent. The plate was then incubated for 1 h at 37 °C. After incubation, 1 mL of media from each well was assessed using the lactate assay kit on a Microplate Photometer (Thermo Fisher Scientific, MA, USA) at 570 nm. Cell numbers were counted by a microscope, and the lactate content was normalized by cell numbers.
2.6. NADPH/NADP+ ratio
Intracellular NADPH/NADP+ levels were assayed using NADPH/NADP+ assay kit (Beyotime, Shanghai, China) according to the manufacturer's instructions. Briefly, 2 × 106 cells were washed with cold PBS and pelleted. Homogenized samples were treated with 200 μL counter NADPH (or NADP+) extraction buffer, which were incubated at 60 °C for 5 min. The 20 μL of assay buffer and 200 μL of the counter NADPH (or NADP+) extraction buffer were added to neutralize the extracts. Then, these extracts were centrifuged at 12,000 rpm for 5 min on a Legend Micro 21R Centrifuge (Thermo Fisher Scientific, MA, USA), and the NADP+ or NADPH was measured in the supernatant by reading the fluorescence at 450 nm according to the manufacturer's protocol on a microplate photometer. NADPH/NADP+ ratios were calculated according to the manufacturer's protocol.
2.7. Intracellular reactive oxygen species (ROS) production
Intracellular ROS was measured by the fluorescence probe DCFDA (Beyotime, Shanghai, China) following the manufacturer's instructions. In brief, 2 × 105 cells were seeded in 6-well plates. Twenty-four hours later, the cells were rinsed with PBS and loaded with 5 μmol/L carboxy-H2DCFDA for 1 h in 37 °C. The cells were then harvested, re-suspended in PBS and analyzed using a Microplate Photometer at 488/525 nm.
2.8. 6PGD activity assay
The 6PGD enzyme activity was determined by the NADPH production rate in assay buffer (1 mmol/L MgCl2 and 50 mmol/L Tris–HCl, pH 8.1) with 0.2 mmol/L 6-phosphogluconate (6PG) for 6PGD activity. 10 μg cell lysates or 1 μg recombinant protein was added and the reaction was then initiated by adding 0.1 mmol/L NADP+. The increase in 340 nm absorbance (OD340) as a measure of NADPH production was obtained every minute for 1 h on a microplate photometer (Thermo Fisher Scientific, MA, USA). The 6PGD enzyme activity was calculated by the changes of NADPH production rate from the cure of a steady OD340 increase.
2.9. ENO1 activity assay
ENO1 activity was determined by the NADH reduction rate in reaction buffer containing 50 mmol/L Tris–HCl (pH 7.5), 25 mmol/L MgSO4, 100 mmol/L KCl, 1.3 mmol/L ADP, 1.9 mmol/L 2-phosphoglycerate (2-PG), 0.5 U of pyruvate kinase 2 (PKM2, Sigma, USA) and 0.67 U of lactate dehydrogenase A (LDHA, Sigma, MO, USA). Since a product of ENO1, phosphoenolpyruvate (PEP) is rapidly hydrolyzed to a substrate of PKM2/LDHA in cells. 10 μg cell lysates or 1 μg recombinant protein was added and the reaction was then initiated by adding 0.15 mmol/L NADH. The decrease in 340 nm absorbance (OD340) as measures of NADH reduce was obtained every minute for 1 h on a microplate photometer. The ENO1 enzyme activity was calculated by the changes of NADH reduction rate from the cure of a steady OD340 decrease.
2.10. Kinase assay for PRMT6
For in vivo arginine methylation, HEK293T cells were co-transfected with GST-6PGD/GST-ENO1/GST-aldolase A (ALDOA) and Flag-PRMT6 plasmids. Thirty-six hours later, the cells were lysed with NP40 lysis buffer and IP using an anti-GST antibody was performed to pull down the GST-6PGD/GST-ENO1/GST-ALDOA protein. The input and eluted protein lysates were resolved by SDS-PAGE and analyzed by Western blotting with indicated antibodies. The level of asymmetric dimethylated arginine (ADMA) in GST-6PGD/GST-ENO1/GST-ALDOA was assessed by using an anti-ADMA antibody (Cell signaling, MA, USA).
For in vitro arginine methylation assay, recombination Flag-PRMT6 (or Flag-PRMT6 purified from HEK293T cells) and recombination GST-6PGD/recombination GST-ENO1 (rGST-6PGD/rGST-ENO1) proteins were incubated in methylation rea-ction buffer (50 mmol/L Tris, 150 mmol/L NaCl), with 16 μmol/L methyl donor S-(5′-adenosyl)-l-methionine iodide (SAM, sigma, MO, USA), to a total volume of 60 μL and incubated at 30 °C for 120 min.
2.11. Tumor formation in nude mice
Nude mice (nu/nu, female, 4–6-week-old) were subcutaneously injected with 2 × 107 H1299 cells harboring empty vector on the left flank, and the cells with stable knockdown of endogenous PRMT6 on the right flank. They were performed according to the institutional ethical guidelines for animal experiments. Cells were harvested by trypsinization, washed twice with sterile PBS and re-suspended at 1 × 108 cells/mL. And then 0.2 mL aliquots were injected subcutaneously into female nude mice. Tumor growth was recorded by measurement of two perpendicular diameters using Eq. (1):
| Tumor growth = 4π/3 × (Length/2) × (Width/2)2 | (1) |
The tumors were harvested and weighed at the experimental endpoint, and the masses of tumors (g) derived from cells with and without stable knockdown of endogenous PRMT6. Statistical analyses have been done by comparison in relation to the control group with a two-tailed paired Student's t test.
To explore the effect of DCPR049_12 on tumorigenicity of lung cancer, mice were received either vehicle control or DCPR049_12 (50 mg/kg), and they were administered every two days by intraperitoneal (i.p.) injection (50 mg/kg). The mice were sacrificed, and the tumors were excised, imaged and weighed after inhibitor treatments. Statistical analyses have been done by comparison in relation to the control group with a two-tailed unpaired Student's t test.
To explore the effect of DCPR049_12 in combination with cisplatin on tumorigenicity in lung cancer. Mice were received either vehicle control, DCPR049_12 (30 mg/kg) alone, cisplatin (2 mg/kg) alone, and DCPR049_12 and cisplatin together, and they were administered every two days by i.p. injection. The mice were sacrificed, and the tumors were excised, imaged and weighed after inhibitor treatments. Statistical analyses have been done by comparison in relation to the control group with a two-tailed unpaired Student's t test.
2.12. Cell-derived xenograft (CDX) models for tumor formation
For CDX model, nude mice (nu/nu, female, 4–6-week-old) were subcutaneously injected with 2 × 107 H460 cells on the left and right flank. When tumor volumes reached approximately 20 mm3, control shRNA lentivirus was injected precisely into the center of the xenograft tumors first three days for three times on the left flank. PRMT6 shRNA virus was injected precisely into the center of the xenograft tumors first three days for three times on the right flank. Statistical analyses have been done by comparison in relation to the control group with a two-tailed paired Student's t test.
2.13. Patient-derived xenograft (PDX) models for tumor formation
For PDX model, the PDX samples were from the Jackson Laboratory. The model ID is TM00192/LG0567F. The tumor samples were cut into approximately 1 mm3, and the tissues were pushed under skin of mice by TROCHAR. When tumor volumes reached approximately 20 mm3, mice were randomly distributed into groups of 7 mice. To investigate the effect of PRMT6 on tumorigenicity of lung cancer, PRMT6 shRNA or control shRNA virus were injected precisely into the center of the xenograft tumors first three days for three times. Statistical analyses have been done by comparison in relation to the control group with a two-tailed paired Student's t test.
To explore the combination effect of DCPR049_12 and cisplatin on tumorigenicity of lung cancer, mice were received either vehicle control, DCPR049_12 (30 mg/kg) alone, cisplatin (2 mg/kg) alone or DCPR049_12 and cisplatin together, and they were administered every two days by i.p. injection. The mice were sacrificed, and the tumors were excised, imaged and weighed after inhibitor treatments. Statistical analyses have been done by comparison in relation to the control group with a two-tailed unpaired Student's t test.
2.14. Ethics approval and consent to participate
This study was carried out in accordance with the recommendations of Requirements of the Ethical Review System of Biomedical Research Involving Human by National Health and Family Planning Commission of China, Nankai University and Wenzhou Medical University Ethics Committee with written informed consent from all subjects. All subjects were given a written informed consent in accordance with the Declaration of Helsinki.
2.15. Bioinformatics analysis
The public datasets TCGA and GSE19804 dataset were used for bioinformatics analysis. Kaplan–Meier Plotter (http://kmplot.com/analysis/index.php?p=background) was used for overall survival.
2.16. Statistical analysis
The data are expressed as mean ± standard deviation (SD). The differences between two groups with similar variances were analyzed using a two-tailed Student's t test. All statistical analyses were performed using GraphPad Prism 8 software (GraphPad Software Inc., CA, USA). The P value lower than 0.05 was considered as significant.
3. Results
3.1. PRMT6 expression is elevated in lung cancer
The human genome encodes at least nine different PRMTs, which catalyze mono-methylation or di-methylation reactions on arginine residues5. To identify PRMTs that are overexpressed in lung cancer and potential drivers for tumorigenicity and cisplatin response, the expression profiling of catalytically active PRMTs and their prognostic significance were analyzed in lung cancer samples. It was found that the expression levels of PRMT1, PRMT3, PRMT4, PRMT5, PRMT6, and PRMT7 were highly expression in lung cancer tissues in TCGA datasets (Supporting Information Fig. S1A). Secondly, we found that PRMT3, PRMT4, PRMT5, PRMT6, and PRMT7 were also highly expressed in lung cancer tissues based on GEO databases (Fig. S1B). In addition, the elevated expression of PRMT4, PRMT5, and PRMT6 predicted the poor prognosis in lung cancer (Fig. S1C). Thus, these findings suggest that the high expression levels of PRMT4, PRMT5, and PRMT6 in lung cancer may indicate worse outcomes for patients with lung cancer.
Thus, to better understand which of the PRMT family members are required for the tumorigenicity of lung cancer, PRMT4, PRMT5, and PRMT6 were efficiently knocked down in H1299 cells. Cell number counting assays showed that the knockdown of PRMT6 dramatically inhibited the growth of H1299 cells (Fig. 1A). Thus, the following investigations were focused on the role of PRMT6 in lung cancer pathogenesis. To validate the clinical relevance of our findings from public databases, the expression of PRMT6 in samples taken from 21 patients with lung cancer was analyzed (Cohort 1: n = 21, lung cancer tissues [T], paired adjacent normal tissues [N]). Western blotting consistently showed that the PRMT6 protein levels were significantly higher in lung tumor tissues than in adjacent non-tumor lung tissues (Fig. 1B and C). To validate these findings, immunohistochemistry (IHC) staining was performed to determine the PRMT6 protein expression levels in lung cancer tissue microarrays (Cohort 2: n = 41, lung cancer tissues [T], paired adjacent normal tissues [N]). IHC staining showed that the expression of PRMT6 was positive in 39 (95.1%) and strongly positive in 21 (51.2%) of the 41 patients with lung cancer (Table S2), which was significantly higher in lung cancer tissues than in paired adjacent normal tissues (Fig. 1D and E). No correlation was observed between tumor grade and PRMT6 levels (Supporting Information Table S3). Lastly, the expression of PRMT6 in various human lung cancer cells, including H1299, H226, H157, H1944, A549, H460, H2122, and H1437 cells, were also checked and compared to that in normal proliferating Human Bronchial Epithelial Cell Line (BEAS-2B). It was found that PRMT6 was highly expressed in most lung cancer cells (Fig. 1F). Together, these data demonstrate that PRMT6 was highly expressed in lung cancer and was correlated with the overall survival, suggesting that PRMT6 is a promising anticancer target.
Figure 1.

PRMT6 expression is elevated in lung cancer. (A) The cell proliferation was determined by cell number counting at third day in PRMT4, PRMT5, and PRMT6-knockdown human lung cancer H1299 cells (Data represent mean values ± SD from three independent experiments). (B) The PRMT6 protein levels in lung cancer tissues (T) and matched adjacent normal tissues (N) were determined by Western blotting. (C) Quantification of PRMT6 expression by Image J software from Western blotting (n = 21). (D) The expression of PRMT6 was determined by IHC assay in lung cancer tissues microarray (TMA), which containing matched adjacent normal tissues (N) and lung cancer tissues (T). (E) Quantification of PRMT6 expression by Image J software from IHC data (n = 41). (F) PRMT6 protein levels were analyzed in a panel of diverse human lung cancer cells, including H1299, H226, H157, H1944, A549, H460, H2122, H1437 cells and normal proliferating human bronchial epithelial cell line (BEAS-2B). ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001.
3.2. PRMT6 promotes lung cancer cell growth
To carefully elucidate the role of PRMT6 upregulation in lung cancer, stable PRMT6 knockdown cells were generated by specific short hairpin RNAs (shRNAs) (Supporting Information Fig. S2A). It was found that PRMT6 knockdown decreased the proliferation of H1299 and H460 cells based on cell number counting assay and colony formation assay (Fig. 2A and B). In contrast, the exogenous expression of PRMT6 in H1299 and A549 cells promoted the cell proliferation (Fig. S2B–S2E). These findings show that PRMT6 promotes lung cancer cell proliferation in vitro. Next, the impact of PRMT6 on lung cancer growth in vivo was explored. As indicated in an H460 cell-derived xenograft (CDX) mouse model, we found that H460 tumor cells with PRMT6 shRNA lentivirus showed a slower growth rate compared to control cells (Fig. 2C–E). The same results were found in an H1299 xenograft model (Fig. S2F–S2H). Finally, Western blotting assay was done to confirm the PRMT6 knockdown efficiency (Figs. 2F and S2I). As PRMT6 enzyme carries out the formation of monomethylarginine (MMA) as an intermediate before the establishment of ADMA5, it was also found that PRMT6 knockdown decreased the ADMA levels in tumors (Figs. 2F and S2I).
Figure 2.

PRMT6 promotes lung cancer cell growth. (A) The cell proliferation was determined by cell number counting in H1299 and H460 cells with stable knockdown of PRMT6 (Data represent mean values ± SD from three independent experiments). (B) The colony formation was determined in H1299 and H460 cells with stable knockdown of PRMT6. (C) Tumor growth was compared between xenograft nude mice with H460 cells injected with PRMT6 shRNA virus and control shRNA virus (n = 4). (D) All tumors from nude mouse are shown. (E) Tumor mass in xenograft nude mice with H460 cells injected with PRMT6 shRNA virus and control shRNA virus (n = 4). (F) The arginine methylation levels of 6PGD/ENO1 and 6PGD/ENO1 were analyzed by immunoprecipitation (IP) assay in a representative H460 tumor cells injected with PRMT6 shRNA virus and control shRNA virus. (G) Tumor growth was compared between xenograft nude mice bearing with lung cancer PDX injected with PRMT6 shRNA virus and control shRNA virus (n = 7). (H) All tumors in xenograft nude mouse are shown. (I) Tumor mass in xenograft nude mice bearing with lung PDX tumor injected with PRMT6 shRNA virus and control shRNA virus (n = 7). (J) The arginine methylation levels of 6PGD/ENO1 and 6PGD/ENO1 were analyzed by immunoprecipitation (IP) assay in presentative PRMT6 knockdown lung PDX tumor. ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001.
Next, a patient-derived xenograft (PDX) mouse model was used to validate the effects of PRMT6 on tumor growth through intratumoral PRMT6 shRNA lentivirus injection. The growth rate of tumors with PRMT6 shRNA lentivirus was significantly slower than that of tumors in the control group (Fig. 2G). Moreover, the xenograft tumor size was smaller than that in the control group (Fig. 2H), and its tumor weight was also reduced (Fig. 2I). Western blotting assay was done to confirm the PRMT6 knockdown efficiency; it was found that PRMT6 knockdown decreased the PRMT6 levels in tumors injected with PRMT6 shRNA lentivirus (Fig. 2J). And we found that ADMA levels were decreased in tumors injected with PRMT6 shRNA lentivirus (Fig. 2J). Collectively, these data indicate that PRMT6 promotes lung cancer cell growth in vitro and in vivo.
3.3. PRMT6 promotes the oxidative PPP flux and glycolysis pathway
Numerous evidence showed that the oxidative PPP flux and glycolysis pathway contribute to tumor development3,4,15, 16, 17. To identify PRMT6 downstream effectors in promoting lung cancer development, the effects of PRMT6 on the oxidative PPP flux and glycolysis pathway were explored. PRMT6 knockdown resulted in a reduced lactate production (Fig. 3A), glycolytic rate (Supporting Information Fig. S3A and S3B), DNA biosynthesis (Figs. 3B and S3C), and NADPH/NADP+ ratio (Fig. 3C), as well as the increased ROS levels (Fig. 3D). To validate the regulation of PRMT6 on oxidative PPP flux and glycolysis pathway, we analyzed the role of PRMT6 in regulating the incorporation of glucose in the oxidative PPP flux and glycolysis pathway by measuring metabolic fluxes via isotopomer analysis. We used [1,2]-13C-glucose as a tracer to probe this issue. As shown in Fig. 3E, the oxidative PPP flux was determined by measuring M1 ribose. The glycolysis pathway was determined by measuring M2 intermediate metabolites. By tracking the carbon atoms of glucose, the fractional contributions of the intermediate metabolites of oxidative PPP flux and glycolysis pathway derived from glucose displayed reduced conversion rates due to PRMT6 knockdown in H1299 cells (Fig. 3F). These data together suggest that PRMT6 promotes oxidative PPP flux and glycolysis pathway in lung cancer cells.
Figure 3.

PRMT6 promotes the oxidative PPP flux and glycolysis pathway. (A–D) PRMT6 knockdown and control vector cells were tested for lactate production (A), DNA biosynthesis (B), and NADPH/NADP+ ratio (C), as well as ROS level (D). (E) Schematic of oxidative PPP flux and glycolysis pathway flux, illustrating labeling from d-[1,2]-13C-glucose. (F) Fractional labeling of metabolites of glycolysis pathway and oxidative PPP flux in PRMT6 knockdown and control vector cells cultured in medium containing d-[1,2]-13C-glucose for 15 min. Data represent mean values ± SD from three independent experiments. ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001.
3.4. PRMT6 methylates 6PGD/ENO1 and enhances their activities
To determine how PRMT6 altered the oxidative PPP flux and glycolysis pathway in lung cancer cells, 12 genes that were involved in oxidative PPP flux and glycolysis pathway were further analyzed. The expression of metabolic enzymes was not changed in PRMT6 knockdown H1299 and H460 cells relative to the control based on Western blotting (Fig. 4A). Thus, we wondered whether PRMT6 regulates oxidative PPP flux and glycolysis pathway through its interaction with metabolic enzymes. Next, a Flag-pull down assay was performed to analyze the metabolic enzymes co-precipitated with Flag-tagged PRMT6 in HEK293T cells. It was found that ALDOA, ENO1, and 6PGD were associated with PRMT6 based on the Flag-pull down assay (Fig. 4B). As PRMT6 not only methylates histone proteins, it also methylates non-histone proteins. Thus, to gain insight on the interaction between PRMT6 and metabolic enzymes, the effects of PRMT6 on 6PGD, ENO1, and ALDOA were determined. GST-tagged 6PGD, ENO1, and ALDOA with or without Flag-PRMT6 were transfected into HEK293T cells. Then, a GST-pull down assay was performed to purify GST-tagged 6PGD, ENO1, and ALDOA. After that, the methylation levels of 6PGD, ENO1, and ALDOA were examined by Western blotting using ADMA antibodies. It was found that the methylation levels of 6PGD and ENO1, not ALDOA, were increased in HEK293T cells, with an exogenous expression of PRMT6 (Supporting Information Fig. S4A, S4C and S4E). Lastly, the activities of 6PGD and ENO1 were also determined, and found these were increased in HEK293T cells, with an exogenous expression of PRMT6 (Fig. S4B and S4D). Additionally, the activity of 6PGD/ENO1 was also decreased in H1299 cell xenograft tumors with PRMT6 knockdown (Fig. S2J and S2K). To rule out the nonspecific pull down by GST-vector, we transfected HEK293T cells with GST-tagged vector or GST-tagged-6PGD (or GST-ENO1) with or without Flag-PRMT6. And then the GST-pull down assay was applied to determine the interaction between 6PGD/ENO1 and PRMT6. It was found that PRMT6 was pulled down by GST-6PGD (or GST-ENO1), not by GST (Fig. S4F). Thus, we focused on the arginine methylation regulation of PRMT6 on 6PGD/ENO1.
Figure 4.

PRMT6 methylates 6PGD/ENO1 and enhances their activities. (A) Analysis of glycolysis-, and oxidative PPP-related gene expression in PRMT6 knockdown cells and control vector cells. (B) The expression of glycolysis-, and oxidative PPP-related enzymes were determined by Western blotting in HEK293T cells by Flag-pull down assay. (C) 6PGD or ENO1 associates with PRMT6 in vitro. The rFlag-PRMT6 was incubated with recombination GST-taged 6PGD (rGST-6PGD or rGST-ENO1). After GST pull down assay, the interaction of rFlag-PRMT6 protein with rGST-6PGD (or rGST-ENO1) were analyzed by Western blotting. (D) Purified rGST-6PGD was incubated with purified Flag-tagged PRMT6 from HEK293T cells, followed by 6PGD enzyme activity assay. (E) Purified rGST-6PGD was incubated with purified Flag-tagged PRMT6 from HEK293T cells, followed by Western blotting. (F) Purified rGST-ENO1 was incubated with purified Flag-tagged PRMT6 from HEK293T cells, followed by ENO1 enzyme activity assay. (G) Purified rGST-ENO1 was incubated with purified Flag-tagged PRMT6 from HEK293T cells, followed by Western blotting. Data represent mean values ± SD from three replicates of each sample (∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001).
First, whether PRMT6 directly binds 6PGD/ENO1 was answered by an in vitro GST-pull down assay, which further demonstrated that 6PGD/ENO1 directly bound to PRMT6 in vitro (Fig. 4C). Besides, an in vitro co-immunoprecipitation (Co-IP) assay demonstrated that 6PGD/ENO1 directly bound to PRMT6 (Fig. S4G–S4H). At same time, a Co-IP assay demonstrated that 6PGD also bound to ENO1 in H460 cells (Fig. S4I); however, an in vitro Co-IP assay using bacterially expressed recombinant GST-tagged 6PGD and ENO1 proven that they are not directly bound, which mediated by PRMT6 (Fig. S4J). These results suggest that PRMT6 directly binds to 6PGD/ENO1.
Then, whether PRMT6 directly methylates 6PGD/ENO1 was tested. First, bacterially expressed recombinant GST-tagged 6PGD and ENO1 with Flag-PRMT6 were incubated, which were purified from HEK239T cells. It was found that recombinant GST-tagged 6PGD and ENO1 were methylated by Flag-PRMT6 with increased enzyme activities (Fig. 4D–G). In addition, bacterially expressed PRMT6 and 6PGD/ENO1 proteins were purified, and an in vitro methylation assay was performed. After the incubation of recombinant 6PGD/ENO1 with recombinant PRMT6, 6PGD/ENO1 showed strongly increased methylation levels (Fig. S4K–S4L). These results suggest that PRMT6 methylates 6PGD and ENO1 directly and enhances their activities.
3.5. PRMT6 regulates 6PGD and ENO1 activities by methylating their R324 and R9/372 sites
Proteomics analysis (Cell Signaling Technology) revealed that 6PGD and ENO1 were arginine methylated at one or more groups of arginine methylation residues (6PGD: http://www.phosphosite.org/proteinAction.do?id=15053&showAllSites=true; ENO1: https://www.phosphosite.org/siteTableNewAction?id=2610&showAllSites=true). To examine the effects of arginine methylation on the activity of 6PGD and ENO1, mutational analysis was performed, and diverse arginine-deficient (R→K) mutants of 6PGD and ENO1 were generated in which arginine residues were substituted by lysine residues. First, it was found that the substitution of R324 with lysine alone abolished the methylation level and activity of 6PGD compared to what was observed in the wild-type (WT) (Supporting Information Fig. S5A and S5B). Moreover, the activity (Fig. 5A) and methylation level (Fig. 5B) of 6PGD R324K mutant were not increased by the PRMT6 treatment. These results suggest that R324 is a key methylation site to control the 6PGD activity mediated by PRMT6.
Figure 5.

PRMT6 regulates 6PGD and ENO1 activities by methylating their R324 and R9/372 sites. (A) Purified rGST-6PGD WT and rGST-6PGD R324K were incubated with purified Flag-tagged PRMT6 from HEK293T cells, followed by 6PGD enzyme activity assay. (B) Purified rGST-6PGD WT and rGST-6PGD R324K were incubated with purified Flag-tagged PRMT6 from HEK293T cells, followed by Western blotting. (C) Structural locations of R324 (PDB ID 2JKV). (D) Purified rGST-ENO1 WT and R9/372K were incubated with purified Flag-PRMT6 from HEK293T cells, followed by ENO1 enzyme activity assay. (E) Purified rGST-ENO1 WT and R9/372K were incubated with purified Flag-PRMT6 from HEK293T cells, followed by Western blotting. (F) Structural locations of R9 and R372 (PDB ID 2PSN). Lower right exploded view shows that R9 forms part of the dimerization interface, with the surface of the partner molecule shown in tan. Upper right exploded view shows that R372 abuts 2-phosphoglycerate (2-PG). (G) Purified rGST-ENO1 WT and R9K were followed by crosslinking and Western blotting experiments to detect dimeric and monomeric ENO1 in the upper panel. ENO1 protein input assessed by Western blotting is shown in the lower panel. (H) An ENO1 mutant R372K that cannot be methylated had reduced enzymatic activity when exposure to the increased concentrations of substrate 2-PG. The data represent mean values ± SD from three replicates of each sample (∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001).
The substitution of R9 or R372 with lysine alone also resulted in decreased methylation levels of purified rGST-ENO1 compared to those of WT (Fig. S5C), whereas the substitution of both R9 and R372 in the double mutant R9/372K abolished the methylation level and activity of ENO1 (Fig. S5D–S5E). This further blocked the PRMT6 treatment-induced activation (Fig. 5D) and methylation level (Fig. 5E) of ENO1, suggesting that R9 and R372 methylation were required to activate ENO1. Interestingly, structural analysis revealed that R9 was located at the dimeric interface between the two sister molecules of an ENO1 homodimer (Fig. 5F), and R372 was directly proximal (2.7 Å) to the substrate 2-PG (Fig. 5F). Thus, we hypothesized that R9 and R372 methylation may contribute to ENO1 activation by affecting the homodimer formation and substrate 2-PG binding, respectively. To examine whether R9 methylation promotes homodimer formation, a crosslinking experiment was performed. As shown in Fig. 5G, R9 arginine residue was replaced by lysine residue resulting in a decreased formation of dimeric ENO1. These results reveal that ENO1 exists as a dimeric ENO1 with an active dimeric state. Lastly, a ‘‘2-PG’’ binding compete experiment was performed using ENO1 activity assay, and it was found that ENO1 WT not R372K enzyme activities were increased with increasing 2-PG concentrations (Fig. 5H). The increased enzyme activity indicated that the binding between ENO1 WT and 2-PG was increased with increasing 2-PG concentrations. In contrast, the substitution at R372 of ENO1 significantly abolished the 2-PG binding (Fig. 5H). These results suggest that R9 and R372 methylation enhances ENO1 activity by affecting homodimer formation and substrate binding, respectively.
3.6. PRMT6 enhances the oxidative PPP flux, glycolysis pathway, and cell proliferation by activating 6PGD and ENO1
It was determined whether PRMT6 promotes oxidative PPP flux, glycolysis pathway, and cell proliferation by regulating 6PGD and ENO1 activities. 6PGD WT or 6PGD R324K was expressed in PRMT6 knockdown cells (Supporting Information Fig. S6A and S6B), and it was found that the exogenous expression of 6PGD WT, not 6PGD R324K, in PRMT6 knockdown cells rescued the decreased cell proliferation and colony formation (Fig. 6A and B). Similarly, the exogenous expression of ENO1 WT, not ENO1 R9/372K, in PRMT6 knockdown cells rescued the decreased cell proliferation and colony formation (Figs. 6C, D, S6C, and S6D). Moreover, the exogenous expression of 6PGD WT, not 6PGD R324K, in PRMT6 knockdown cells rescued the decreased DNA synthesis and the increased ROS (Figs. 6E, F and S6E). Similarly, the exogenous expression of ENO1 WT, not ENO1 R9/372K, in PRMT6 knockdown cells also rescued the decreased lactate production (Fig. 6G). Taken together, these data suggest that PRMT6 enhances oxidative PPP flux, glycolysis pathway, and cell proliferation by regulating the arginine methylation levels and activities of 6PGD and ENO1.
Figure 6.

PRMT6 enhances the oxidative PPP flux, glycolysis pathway, and cell proliferation by activating 6PGD and ENO1. (A) Cell proliferation was determined by cell number counting assay in stable knockdown of PRMT6 cells with or without exogenously expressing 6PGD WT or R324K. (B) Colony formation was determined in stable knockdown of PRMT6 cells with or without exogenously expressing 6PGD WT or R324K. (C) Cell proliferation was determined by cell number counting assay in stable knockdown of PRMT6 cells with or without exogenously expressing ENO1 WT or R9/372K. (D) Colony formation was determined in stable knockdown of PRMT6 cells with or without exogenously expressing ENO1 WT or R9/372K. (E) DNA synthesis was determined by using EdU incorporation assay in stable knockdown of PRMT6 cells with or without exogenously expressing 6PGD WT or R324K. (F) The intracellular ROS was determined in stable knockdown of PRMT6 cells with or without exogenously expressing 6PGD WT or R324K. (G) The lactate production was determined in stable knockdown of PRMT6 cells with or without exogenously expressing ENO1 WT or R9/372K. The data represent mean values ± SD from three independent experiments (∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001).
3.7. DCPR049_12 inhibits the oxidative PPP and glycolysis pathway by regulating activities of 6PGD and ENO1
In our previous study, DCPR049_12 was designed and synthesized as a highly potent inhibitor of type I PRMTs and the IC50 of DCPR049_12 against PRMT1, PRMT3, PRMT4, PRMT5, PRMT6, and PRMT8 was 1.1, 22, 63 nmol/L, >100 μmol/L, 1.2 and 1.1 nmol/L, respectively. It was found that DCPR049_12 effectively inhibited cell proliferation in several leukemia cell lines and reduced the cellular asymmetric arginine demethylation levels18. In this study, we explored whether DCPR049_12 could be a potent inhibitor of PRMT6 in lung cancer. First, it was found that the DCPR049_12 treatment resulted in a reduced lactate production (Fig. 7A), glycolytic rate (Supporting Information Fig. S7A and S7B), DNA biosynthesis (Figs. 7B and S7C), and NADPH/NADP+ ratio (Fig. 7C), as well as increased ROS levels (Fig. 7D). The effect of DCPR049_12 on oxidative PPP flux and glycolysis pathway by affecting the methylation levels and activities of 6PGD/ENO1 mediated by PRMT6 were also confirmed. Then, the effects of DCPR049_12 on 6PGD/ENO1 were explored. Indeed, the DCPR049_12 treatment resulted in a decreased activities and methylation levels of 6PGD and ENO1 (Fig. 7E–H). Because DCPR049_12 is a highly potent inhibitor of type I PRMTs, thus, to rule out the effects of DCPR049_12 on oxidative PPP flux and glycolysis pathway were mediated by targeting PRMT1, PRMT3, and PRMT4. First, 6PGD and ENO1 expressions were decreased with the PRMT1 depletion (Fig. S7D), but these were not affected by PRMT3 or PRMT4 knockdown. However, the 6PGD activity was increased by PRMT3 or PRMT4 depletion (Fig. S7E and S7F), while the ENO1 activity was only increased by PRMT4 depletion (Fig. S7E and S7F). The DCPR049_12 treatment had no effect on the expression of the identified 12 glucose metabolic enzymes (Fig. 7I). The bindings between PRMT1, or PRMT3 or PRMT4 and 12 glucose metabolic enzymes were also analyzed. There was no interaction between PRMT1, or PRMT3, or PRMT4 and the identified 12 glucose metabolic enzymes based on Flag-pull down assay (Fig. S7G). Thus, these data demonstrate that DCPR049_12 inhibits oxidative PPP flux and glycolysis pathway by regulating the 6PGD and ENO1 activities by mainly targeting PRMT6 in lung cancer.
Figure 7.

DCPR049_12 inhibits the oxidative PPP flux and glycolysis pathway by regulating activities of 6PGD and ENO1. (A–D) DCPR049_12 treated and control cells were tested for lactate production (A), DNA biosynthesis (B), and NADPH/NADP+ ratio (C), as well as ROS level (D) (Data represent mean values ± SD from three independent experiments). (E) Flag-6PGD was purified from H1299 cells treated with or without DCPR049_12, followed by 6PGD enzyme activity assay (The data represent mean values ± SD from three replicates of each sample). (F) Flag-6PGD was purified from H1299 cells treated with or without DCPR049_12, followed by Western blotting. (G) Flag-ENO1 was purified from H1299 cells treated with or without DCPR049_12, followed by ENO1 enzyme activity assay (The data represent mean values ± SD from three replicates of each sample). (H) Flag-ENO1 was purified from H1299 cells treated with or without DCPR049_12, followed by Western blotting. (I) The expression of glycolysis-, and oxidative PPP-related enzymes were determined by Western blotting in H1299 and H460 cells treated with DCPR049_12. ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001.
3.8. DCPR049_12 suppresses the lung cancer cell growth
To examine the effects of DCPR049_12 on lung cancer cell proliferation, lung cancer cells were treated with various concentrations of DCPR049_12, and the effect of DCPR049_12 on cell proliferation was determined. Cell proliferation and colony formation assays showed that DCPR049_12 exerted a strong inhibition efficacy on lung cancer cell proliferation and colony formation in time- and dose-dependent manner. However, it did not significantly affect the proliferation of normal BEAS-2B cells (Fig. 8A–C, and Supporting Information Fig. S8A). These data demonstrate that DCPR049_12 has a significant inhibitory effect on lung cancer cell proliferation in vitro.
Figure 8.

DCPR049_12 suppresses the lung cancer cell growth. (A) The cell proliferation was determined by cell number counting assay in lung cancer cells treated with DCPR049_12 (Data represent mean values ± SD from three independent experiments). (B) The colony formation was determined in lung cancer cells treated with DCPR049_12 (Data represent mean values ± SD from three independent experiments). (C) The cell proliferation was determined by cell number counting assay in BEAS-2B cells treated with DCPR049_12 (Data represent mean values ± SD from three independent experiments). (D) Tumor growth was compared between xenograft nude mice injected with H460 cells treated with DCPR049_12 (n = 7). (E) All tumors from nude mouse were shown. (F) Tumor mass of xenograft nude mice injected with H460 cells and treated with DCPR049_12 (n = 7). (G) Ki67, 6PGD, ENO1, and PRMT6 were analyzed in a representative DCPR049_12 treated H460 tumor by IHC. (H) The arginine methylation levels of 6PGD/ENO1 and 6PGD/ENO1 were analyzed by immunoprecipitation (IP) assay in a representative DCPR049_12 treated and control H460 cells tumor. (I) Tumor growth was compared between xenograft nude mice injected with H1299 cells and treated with DCPR049_12 (n = 6). (J) All tumors from nude mouse were shown. (K) Tumor mass in xenograft nude mice injected with H1299 cells and treated with DCPR049_12 (n = 6). (L) Ki67, 6PGD, ENO1, and PRMT6 were analyzed in a representative DCPR049_12 treated H1299 cell xenograft tumor by IHC. (M) The arginine methylation levels of 6PGD/ENO1 and 6PGD/ENO1 were analyzed by IP assay in are presentative DCPR049_12 treated and control H1299 cells tumor. ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001.
Next, the anti-tumor effects of DCPR049_12 were determined in vivo using a CDX mouse model. An initial toxicity studies on the chronic injection of DCPR049_12 to nude mice for 12 days revealed that 30 or 50 mg/kg/day of i.p. administered DCPR049_12 was a well-tolerated dose. In addition, the chronic treatment with DCPR049_12 to nude mice did not affect the body weight, serum chemistry, or complete blood count (CBC) and hematopoietic properties of the nude mice compared to the DMSO-treated group (Fig. S8B–S8D). These results demonstrate the DCPR049_12 treatment did not cause obvious toxicities in vivo. Then, 50 mg/kg of DCPR049_12 was administrated i.p. every two days into xenograft mice with subcutaneously injected H460 cells. DCPR049_12 significantly decreased the tumor growth (Fig. 8D and E), tumor masses (Fig. 8F), Ki67 expression (Fig. 8G), and methylation levels of 6PGD/ENO1 (Fig. 8H) in DCPR049_12 treated mice compared to mice receiving DMSO. While, the levels of 6PGD/ENO1 and PRMT6 were not affected by DCPR049_12 treatment (Fig. 8G and H). Similar results were obtained using an H1299 CDX treated with DCPR049_12 for 16 days (Fig. 8I–M). These data suggest that DCPR049_12 significantly suppresses excessive tumor growth in vivo partly by targeting PRMT6.
3.9. DCPR049_12 enhances the anti-tumor effects of cisplatin
To further determine the therapeutic benefit of targeting PRMT6 in combination with cisplatin, the sensitivity of cisplatin in PRMT6 knockdown cells was tested. It was found that endogenous PRMT6 knockdown significantly enhanced the sensitivity to cisplatin treatment based on MTT assay and colony formation (Fig. 9A and B), while the overexpression of PRMT6 significantly decreased the sensitivity to cisplatin treatment (Supporting Information Fig. S9A). A xenograft model of H460 cells in nude mice was established to analyze the combination effect of cisplatin and DCPR049_12. Cisplatin showed a slight inhibitory effect on the tumor growth of H460 by ip administered at a dose of 2 mg/kg every two days. While the co-administration of DCPR049_12 at 30 mg/kg significantly suppressed the xenograft tumor growth (Fig. S9B and S9C). H460 xenograft tumors were weighed at end points (Fig. S9D). The combination of cisplatin and DCPR049_12 showed about 60% tumor growth inhibition, which was much higher than that of cisplatin or DCPR049_12 alone (Fig. S9D), Furthermore, the expression of Ki67 was examined by IHC. The level of Ki67 was significantly suppressed in the combination treatment group (Fig. S9E). However, it did not affect nude the body weight of nude mice (Fig. S9F). Moreover, in a PDX model, the treatment combination also showed a strong inhibition of lung cancer growth (Fig. 9C–F), it also did not affect the body weight of nude mice (Fig. S9G). These data suggest that targeting PRMT6 significantly enhances the anti-tumor effects of cisplatin on lung cancer xenografts.
Figure 9.

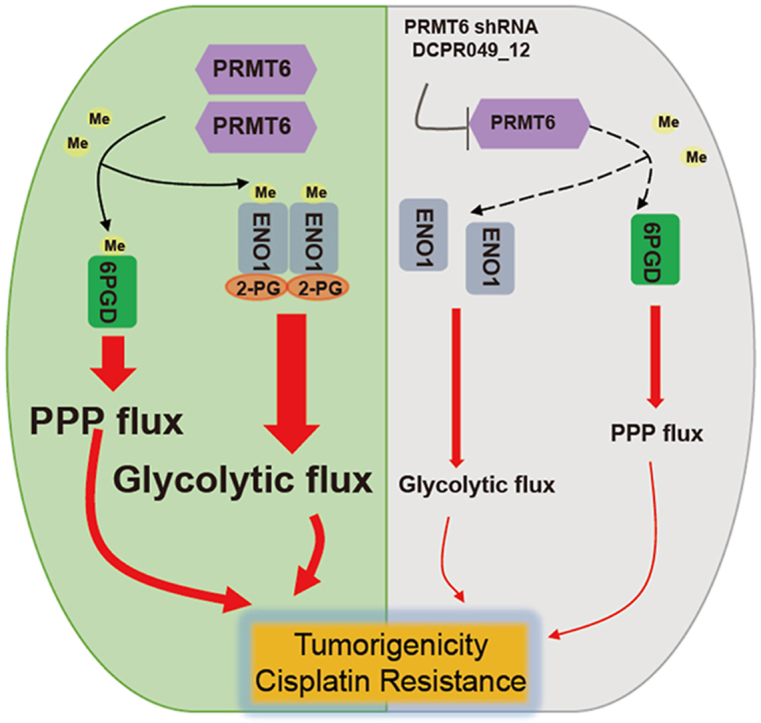
DCPR049_12 enhances the anti-tumor effects of cisplatin. (A) Cell viability determined by MTT in H1299 and H460 cells with stable PRMT6 knockdown when treated with or without cisplatin. Each bar represents mean ± SD (H1299: n = 8; H460: n = 10). (B) Cell proliferation rates were determined by colony formation in H1299 and H460 cells with stable PRMT6 knockdown when treated with or without cisplatin. (C) Tumor growth was compared between xenograft nude mice injected with PDX tumor treated with treated with DCPR049_12 alone, cisplatin alone, and combination (n = 8). (D) All tumors from nude mouse were shown. (E) Tumor mass of xenograft nude mice injected with PDX tumor treated with treated with DCPR049_12 alone, cisplatin alone, and combination (n = 8). (F) Ki67 was analyzed in a representative PDX tumor treated with treated with DCPR049_12 alone, cisplatin alone, and combination by IHC. (G) A schematic model shows that PRMT6 regulates cell metabolism, tumorigenicity, and cisplatin response of lung cancer by methylating 6PGD and ENO1. ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001.
4. Discussion
The aberrant expression of PRMT6 was reported in various cancers and has been well-recognized to play crucial roles in the regulation of tumorigenesis15,19,20. Also, the expression of PRMT6 is increased in lung cancer and is correlated with poor prognostic significance for patients with lung cancer12,21. Indeed, our study confirmed these observations using two different lung cancer patient tissues. It was found that PRMT6 protein levels were higher in lung cancer tissues than in adjacent non-tumor lung tissues. As we know, lung cancers have concurrent mutations of tumor suppressors and oncogenes, which determinants of the molecular, clinical heterogeneity, and the expression of downstream genes in oncogene-driven lung cancer22, 23, 24. Indeed, we observed that PRMT6 was higher in various human lung cancer cells, while, some lung cancer cells with low PRMT6 expression. However, the exactly mechanism of PRMT6 expression varied among lung cancer cells should be further explored. Moreover, suppressing PRMT6 expression using shRNA or inhibitor DCPR049_12 significantly reduced lung cancer cell proliferation, colony formation, and tumor growth, as well as enhanced anti-tumor effect of cisplatin. However, whether PRMT6 can influence lung cancer tumorigenesis or cisplatin response through the regulation of cell metabolism remains largely unknown.
In this study, we reported a crucial role of PRMT6 in promoting lung cancer tumorigenesis by modulating oxidative PPP flux and glycolysis pathway. Moreover, we demonstrated that PRMT6 is a critical modification enzyme for 6PGD/ENO1 arginine methylation modification, coordinating both oxidative PPP flux and glycolysis pathway, leading to lung cancer growth (Fig. 9G). Several cancer-associated mechanisms have been proposed for PRMT6 in promoting the lung cancer progression from its transcriptional regulation role or non-catalytic role12,15,21,25. For example, PRMT6 may serve as an oncogene in the progression of lung adenocarcinoma (LUAD) through transcriptionally represses the p18 expression21. PRMT6 disrupts the association between p16 and Cyclin-dependent kinase 4 (CDK4), and also weakens the function of p16 in preventing cell proliferation by increasing the methylation level of p1625. PRMT6/interleukin-enhancer binding protein 2 (ILF2)/macrophage migration inhibitory factor (MIF) signaling axis in promoting lung tumor growth by its non-catalytic role12. However, the mechanisms controlling lung cancer progression mediated by PRMT6 have not yet to be fully determined. Energy metabolism reprogramming is one of the main hallmarks of cancer2. Multiple studies reported that metabolic enzymes play an important role in cancer cell metabolism and tumorigenesis9, 10, 11, 12. Herein, we demonstrate that PRMT6 is critically involved in the regulation of 6PGD/ENO1 activities to coordinate both oxidative PPP flux and glycolysis pathway.
Furthermore, the mechanism of arginine methylation on 6PGD/ENO1 was explored. We found that methylation at R324 of 6PGD enhances its activity, and structural analysis revealed that R324 was directly proximal to the E227 and E321 (Fig. 5C). Thus we hypothesized that R324 methylation may contribute to 6PGD activation by affecting the binding between R324 and E227, or E321, which affect 6PGD enzyme activity by changing the overall conformation of 6PGD. To test this hypothesis, an in vitro thermal shift assay was performed to analyze the stability of 6PGD proteins to reflect the changing of 6PGD conformation. And we found that R324 methylation levels affect the stability of 6PGD under higher temperature condition (data not shown). In all, these results suggest that R324 methylation enhances the 6PGD activity maybe by controlling the binding between R324 and E227, or E321, which leading to the changes of 6PGD conformation. However, the exactly mechanism of R324 methylation in regulating 6PGD activity should be further explored. Next, we also explored the mechanism of methylation in regulating ENO1, and found that methylation at arginine 9 and 372 promotes ENO1 dimer formation and 2-PG binding to ENO1, respectively. Thus, these data suggest that PRMT6 could exert its methyltransferase activity in driving lung cancer progression by methylating 6PGD/ENO1, which are involved in oxidative PPP flux and glycolysis, respectively. Although our results analyses excluded histone methylation in regulating the expression of metabolic enzymes, it is possible that there are other mechanisms or protein substrates are responsible for PRMT6 in promoting PPP flux and glycolysis. Indeed, studies by Wong et al.15 indicated that PRMT6 contributes to human hepatocellular carcinoma (HCC) progression by rewriting glucose metabolic activities, demonstrating that the mechanism of PRMT6 regulates glycolysis pathway is through the PRMT6–ERK–PKM2 signaling axis. Moreover, a protein–protein interaction between PRMT6 and ALDOA was also found in our study, but this, including its biological functions, was not explored. Hence, the biological functions of the interaction between PRMT6 and ALDOA are worth exploring, and this will hopefully help to understand the mechanism of tumor progression. Therefore, these studies may have broader implications for targeting PRMT6 to suppress lung cancer growth.
Indeed, targeting PRMT6 activity using DCPR049_12 significantly suppressed tumor growth by regulating oxidative PPP flux and glycolysis. Moreover, the DCPR049_12 treatment also enhanced the anti-tumor effects of cisplatin on lung cancer. In our previous study, a potent type I PRMT inhibitor (DCPR049_12) was designed and synthesized. This is a highly potent inhibitor of type I PRMTs that has a good selectivity against a panel of other methyltransferases. To rule out the possible of DCPR049_12 that regulates oxidative PPP flux and glycolysis pathway by targeting other methyltransferases. Thus we analyzed the effect of type I PRMT (PRMT1, PRMT3 and PRMT4) on oxidative PPP flux and glycolysis pathway. We found that PRMT1 knockdown decreased the levels of 6PGD, ENO1, PFKP, PKM2, and LDHA. Moreover, the knockdown of PRMT3 and PRMT4 had no effect on the expression of oxidative PPP flux and glycolysis pathway enzymes. Lastly, there was no interaction between PRMT1/PRMT3/PRMT4 and oxidative PPP flux/glycolysis enzymes. The DCPR049_12 treatment also had no effect on the expression of oxidative PPP flux/glycolysis pathway enzymes, but it decreased the enzyme activities and asymmetric arginine dimethylation levels of 6PGD and ENO1, as the effect of PRMT6. Thus, this study demonstrated that DCPR049_12 could inhibit oxidative PPP flux/glycolysis pathway, leading to the suppression of tumor growth mainly by partly targeting PRMT6 activity in lung cancer. Even, we rule out the effect of DCPR049_12 by targeting other type I PRMT, there may still have other mechanism or proteins involving in regulating oxidative PPP flux and glycolysis. So, the specificity and activity of DCPR049_12 for targeting PRMT6 should be improved in the future.
Cisplatin is currently one of the most common effective anticancer drugs used for treating lung cancer3. However, the therapeutic activity of cisplatin becomes limited because many patients acquire resistance. Therefore, whether DCPR049_12 could enhance the anti-tumor effects of cisplatin on lung cancer was further investigated. First, the knockdown of endogenous PRMT6 significantly enhanced the sensitivity of lung cancer to cisplatin. DCPR049_12 also enhanced this sensitivity in vitro and in vivo. It was suggested that DCPR049_12 might enhance the sensitivity of lung cancer cells to cisplatin by partly inhibiting PRMT6 activation.
5. Conclusions
In this study, a unique role for PRMT6 in promoting lung cancer growth was identified. The molecular mechanisms on how PRMT6 linked cell metabolism to tumor growth were also explored by regulating the enzyme activity of 6PGD/ENO1 mediated by asymmetric arginine demethylation modification. These findings shed light on new molecular mechanisms of PRMT6 in promoting lung cancer progression, and PRMT6 could be a novel therapeutic target for lung cancer therapy.
Acknowledgments
This work was supported by grants from the Natural Science Foundation of Tianjin (21JCZDJC00060, China), the National Nature Science Foundation of China (81973356, 91957120, 81902826, and 81672781), the Fundamental Research Funds for the Central Universities of Nankai University (3206054, 91923101, 63213082 and 92122017, China), the State Key Laboratory of Drug Research (SIMM2105KF-08, China), the National Key R&D Program of China (No. 2018YFC2002000), the Innovative S&T Projects for Young Researchers of Tianjin Academy of Agricultural Science (grant No. 201918, China), and the Natural Science Foundation of Tianjin (19JCYBJC29600 and 21JCYBJC00180, China). We also thanks for Lu Zhou (Fudan University, Shanghai, China) for suggestion on analyzing the structural for 6PGD R324 site.
Author contributions
Mingming Sun, Leilei Li: drafting of the manuscript; Mingming Sun, Leilei Li, Yujia Niu, Yingzhi Wang, Qi Yan, Fei Xie, Yaya Qiao, Jiaqi Song, Huanran Sun, Zhen Li, Sizhen Lai, Hongkai Chang, Han Zhang, Jiyan Wang, Chenxin Yang, Huifang Zhao: acquisition of data, analysis and interpretation of data; Junzhen Tan, Yanping Li, Shuangping Liu, Bin Lu, Min Liu, Yujun Zhao, Shuhai Lin, Cheng Luo: technical and/or material support; Chunze Zhang, Shuhai Lin, Shuai Zhang, Changliang Shan: funding acquisition; Shuhai Lin, Cheng Luo, Shuai Zhang, Changliang Shan: study concept and design, study supervision; Changliang Shan: writing-reviewing and editing.
Conflicts of interest
The authors declare no conflicts of interests.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2022.05.019.
Contributor Information
Shu-Hai Lin, Email: shuhai@xmu.edu.cn.
Cheng Luo, Email: cluo@simm.ac.cn.
Shuai Zhang, Email: shuaizhang@tjutcm.edu.cn.
Changliang Shan, Email: changliangshan@nankai.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Du D., Liu C., Qin M., Zhang X., Xi T., Yuan S., et al. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm Sin B. 2022;12:558–580. doi: 10.1016/j.apsb.2021.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zheng W., Feng Q., Liu J., Guo Y., Gao L., Li R., et al. Inhibition of 6-phosphogluconate dehydrogenase reverses cisplatin resistance in ovarian and lung cancer. Front Pharmacol. 2017;8:421. doi: 10.3389/fphar.2017.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hitosugi T., Chen J. Post-translational modifications and the Warburg effect. Oncogene. 2014;33:4279–4285. doi: 10.1038/onc.2013.406. [DOI] [PubMed] [Google Scholar]
- 5.Blanc R.S., Richard S. Arginine methylation: the coming of age. Mol Cell. 2017;65:8–24. doi: 10.1016/j.molcel.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Hwang J.W., Cho Y., Bae G.U., Kim S.N., Kim Y.K. Protein arginine methyltransferases: promising targets for cancer therapy. Exp Mol Med. 2021;53:788–808. doi: 10.1038/s12276-021-00613-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Z., Ding S., Wang Z., Zhu X., Zhou Z., Zhang W., et al. Prmt1 upregulated by Hdc deficiency aggravates acute myocardial infarction via NETosis. Acta Pharm Sin B. 2022;12:1840–1855. doi: 10.1016/j.apsb.2021.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhong X.Y., Yuan X.M., Xu Y.Y., Yin M., Yan W.W., Zou S.W., et al. CARM1 methylates GAPDH to regulate glucose metabolism and is suppressed in liver cancer. Cell Rep. 2018;24:3207–3223. doi: 10.1016/j.celrep.2018.08.066. [DOI] [PubMed] [Google Scholar]
- 9.Guo J., Zhang Q., Su Y., Lu X., Wang Y., Yin M., et al. Arginine methylation of ribose-5-phosphate isomerase A senses glucose to promote human colorectal cancer cell survival. Sci China Life Sci. 2020;63:1394–1405. doi: 10.1007/s11427-019-1562-y. [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto T., Takano N., Ishiwata K., Ohmura M., Nagahata Y., Matsuura T., et al. Reduced methylation of PFKFB3 in cancer cells shunts glucose towards the pentose phosphate pathway. Nat Commun. 2014;5:3480. doi: 10.1038/ncomms4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y.P., Zhou W., Wang J., Huang X., Zuo Y., Wang T.S., et al. Arginine methylation of MDH1 by CARM1 inhibits glutamine metabolism and suppresses pancreatic cancer. Mol Cell. 2016;64:673–687. doi: 10.1016/j.molcel.2016.09.028. [DOI] [PubMed] [Google Scholar]
- 12.Avasarala S., Wu P.Y., Khan S.Q., Yanlin S., Van Scoyk M., Bao J., et al. PRMT6 promotes lung tumor progression via the alternate activation of tumor-associated macrophages. Mol Cancer Res. 2020;18:166–178. doi: 10.1158/1541-7786.MCR-19-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jarrold J., Davies C.C. PRMTs and arginine methylation: cancer’s best-kept secret?. Trends Mol Med. 2019;25:993–1009. doi: 10.1016/j.molmed.2019.05.007. [DOI] [PubMed] [Google Scholar]
- 14.Yang Y., Bedford M.T. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13:37–50. doi: 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- 15.Wong T.L., Ng K.Y., Tan K.V., Chan L.H., Zhou L., Che N., et al. CRAF methylation by PRMT6 regulates aerobic glycolysis-driven hepatocarcinogenesis via ERK-dependent PKM2 nuclear relocalization and activation. Hepatology. 2020;71:1279–1296. doi: 10.1002/hep.30923. [DOI] [PubMed] [Google Scholar]
- 16.Li Y., Liang R., Sun M., Li Z., Sheng H., Wang J., et al. AMPK-dependent phosphorylation of HDAC8 triggers PGM1 expression to promote lung cancer cell survival under glucose starvation. Cancer Lett. 2020;478:82–92. doi: 10.1016/j.canlet.2020.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Shan C., Lu Z., Li Z., Sheng H., Fan J., Qi Q., et al. 4-Hydroxyphenylpyruvate dioxygenase promotes lung cancer growth via pentose phosphate pathway (PPP) flux mediated by LKB1–AMPK/HDAC10/G6PD axis. Cell Death Dis. 2019;10:525. doi: 10.1038/s41419-019-1756-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang C., Jiang H., Jin J., Xie Y., Chen Z., Zhang H., et al. Development of potent type I protein arginine methyltransferase (PRMT) inhibitors of leukemia cell proliferation. J Med Chem. 2017;60:8888–8905. doi: 10.1021/acs.jmedchem.7b01134. [DOI] [PubMed] [Google Scholar]
- 19.Huang T., Yang Y., Song X., Wan X., Wu B., Sastry N., et al. PRMT6 methylation of RCC1 regulates mitosis, tumorigenicity, and radiation response of glioblastoma stem cells. Mol Cell. 2021;81 doi: 10.1016/j.molcel.2021.01.015. 1276–1291.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshimatsu M., Toyokawa G., Hayami S., Unoki M., Tsunoda T., Field H.I., et al. Dysregulation of PRMT1 and PRMT6, type I arginine methyltransferases, is involved in various types of human cancers. Int J Cancer. 2011;128:562–573. doi: 10.1002/ijc.25366. [DOI] [PubMed] [Google Scholar]
- 21.Tang J., Meng Q., Shi R., Xu Y. PRMT6 serves an oncogenic role in lung adenocarcinoma via regulating p18. Mol Med Rep. 2020;22:3161–3172. doi: 10.3892/mmr.2020.11402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim J., Lee H.M., Cai F., Ko B., Yang C., Lieu E.L., et al. The hexosamine biosynthesis pathway is a targetable liability in KRAS/LKB1 mutant lung cancer. Nat Metab. 2020;2:1401–1412. doi: 10.1038/s42255-020-00316-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitajima S., Ivanova E., Guo S., Yoshida R., Campisi M., Sundararaman S.K. Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov. 2019;9:34–45. doi: 10.1158/2159-8290.CD-18-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Skoulidis F., Heymach J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer. 2019;19:495–509. doi: 10.1038/s41568-019-0179-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma W.L., Wang L., Liu L.X., Wang X.L. Effect of phosphorylation and methylation on the function of the p16INK4a protein in non-small cell lung cancer A549 cells. Oncol Lett. 2015;10:2277–2282. doi: 10.3892/ol.2015.3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.