

Human proteins are assembled from 20 amino acids, nine of which are considered “essential” because they cannot be synthesized from other metabolites in the human body. Among these are the three branched-chain amino acids (BCAAs): leucine (Leu), isoleucine (Ile) and valine (Val), so named because of their branched rather than linear aliphatic side chains. The food sources most enriched in BCAAs are meat, fish, dairy products, and eggs. BCAA have been studied for decades as agents for enhancing muscle protein synthesis and mass during exercise training, in syndromes of cachexia (muscle wasting), and in aging. In this context, Leu is known to activate anabolic signaling molecular mTORC1 (mammalian target of rapamycin complex 1), as well as other factors involved in protein synthesis.
In contrast to the potential health-promoting effects of BCAA in conditions of negative energy balance, chronic elevations in BCAAs are observed in blood from humans with obesity-related conditions such as insulin resistance, type 2 diabetes (T2D), and cardiovascular diseases (collectively referred to as cardiometabolic disease) (1). In 1969, Cahill, et al. (2) reported that the concentrations of the three BCAA and the aromatic amino acids, phenylalanine (Phe), and tyrosine (Tyr) were increased, and glycine (Gly) was decreased, in the blood from obese compared to age- and sex-matched lean individuals (2). The increased BCAAs correlated with circulating insulin levels, suggesting that the rise was a manifestation of insulin resistance. Subsequently, the obesity-associated elevations in circulating lipid concentrations (dyslipidemia) became a focus, fueled by studies suggesting that lipids could function as mechanistic drivers of insulin resistance and T2D. These efforts largely eclipsed investigation of a role of BCAAs in disease pathogenesis.
Broader recognition of the links between BCAAs and chronic human diseases has coincided with the advent of metabolomics as a tool for human disease research. The application of mass spectrometry-based metabolomics exactly replicated the findings of increased BCAA, Tyr and Phe, and decreased Gly in obese, insulin-resistant individuals compared to lean insulin-sensitive individuals (3). But in addition, principal components analysis of the metabolomics data identified a BCAA-related metabolite cluster as more significantly insulin resistance than several lipid-related clusters--a finding that has since been confirmed in multiple human studies (1). Increased circulating amounts of BCAAs, Phe and Tyr were also associated with up to a 5-fold increase in risk for future development of T2D (4), and predicted improved insulin sensitivity in response to a dietary/behavioral weight loss intervention in obese individuals (1). Additionally, obese individuals who underwent gastric bypass surgery had a more dramatic decline in circulating BCAAs than individuals who undertook dietary intervention, despite equivalent weight loss. This was correlated with better improvement in glucose homeostasis in response to the surgical intervention (1). These recent examples of associations of BCAAs with cardiometabolic disease phenotypes raise the important question of whether circulating BCAAs serve as a biomarker, a causal agent, or both in these disorders.
Increases in circulating BCAAs in obesity result in part from decreased rates of their oxidation in adipose tissue, due to coordinated transcriptional suppression of all of the BCAA catabolic enzymes, and also from increased phosphorylation and inactivation of the branched-chain ketoacid dehydrogenase (BCKDH) complex in liver, such that fewer BCAAs are taken up from the blood (see Figure 1). Rodent models of obesity exhibit increased expression of the inhibitory BCKDH kinase, BDK, and reduced expression of the activating BCKDH phosphatase, PPM1K (5). Although mammalian tissues lack the enzymes required for de novo BCAA synthesis, but bacteria can perform these reactions, and the entire BCAA biosynthetic pathway is induced in the gut microbiota of obese compared to lean humans. Moreover, transplantation of microbiota from obese humans into germ free mice (which lack gut microbiota) causes a significant increase circulating BCAAs (6). Additional studies are needed to define the quantitative contribution of bacterially-derived BCAAs to the circulating pool of these metabolites.
Figure 1. BCAAs: Biomarkers and causal agents of cardiometabolic disease.
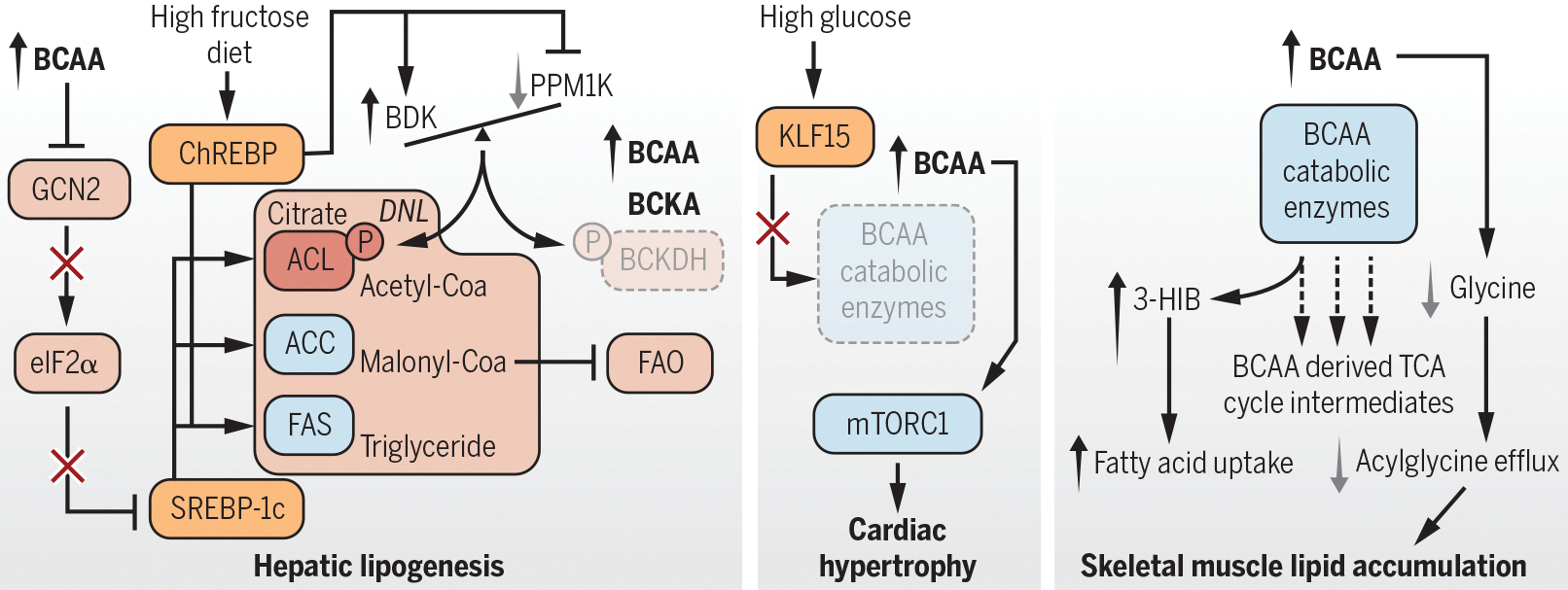
Diet, obesity, the gut microbiota, and genetics can increase circulating BCAAs. BCAAs are proposed to promote increased hepatic lipid storage, cardiac hypertrophy, and lipid accumulation contributing to muscle insulin resistance.
Recent studies show that human genetic variants associated with dyslipidemia and insulin resistance are strongly associated with increased circulating BCAAs. However, variants that specifically raise BCAA, including one near the PPM1K gene, do not associate with insulin resistance (7,8). Nonetheless, in an analysis of large cohorts, increases of 1 standard deviation in Ile, Leu or Val in circulation were associated with a clear increase in risk for development of T2D (7). A unifying model suggests that obesity and insulin resistance are the primary drivers of the obesity-associated rise in BCAAs (8), consistent with conclusions of Cahill, et al. in 1969, but also holds that when chronically elevated in the obese mileu, BCAAs may contribute to T2D pathogenesis (8).
Numerous studies of the effects of dietary supplementation or restriction of BCAAs have been performed in rodent models, yielding inconsistent results. The inconsistency is due in part to whether the study involved manipulation of all three BCAAs or only Leu. Supplementation with Leu alone causes the concentrations of the other two BCAA to decrease in the circulation, and thus does not mimic human cardiometabolic disease states, in which all three BCAA are elevated.
Reducing the dietary supply of all three BCAAs improves insulin sensitivity and glucose homeostasis in rodent models of obesity (9,10), coincident with normalization of multiple lipid-derived metabolites in muscle and lowering of the respiratory exchange ratio (RER), signifying increased efficiency of fatty acid oxidation (FAO) (9). Conversely, feeding of rats with a high-fat diet supplemented with BCAAs (HF/BCAA) reduces food intake and body weight compared to rats fed a HF diet alone, but still promotes insulin resistance and accumulation of lipids in skeletal muscle (3). Rats pair-fed with a HF diet to match the lower rate of HF/BCAA consumption, or fed a low-fat diet supplemented with BCAAs, do not develop insulin resistance. Muscle Gly concentrations are reduced in obese compared to lean rats, but are normalized in obese rats fed a BCAA-restricted diet (9), demonstrating a direct interaction of BCAA and Gly metabolism that may explain the inverse association of their levels in human studies (1). Muscle Gly levels are correlated with urinary acetylglycine, which implies that formation of acylglycine adducts could facilitate removal of excess lipid-derived metabolites from muscle to improve insulin sensitivity. Consistent with the idea that relief of obesity-related impairment in BCAA catabolism could promote enhanced efficiency of FAO and metabolic health, administration of a small molecule inhibitor of BDK, 3,6-dichlorobenzo[b]thiophene-2-carboxylic acid (BT2) or a recombinant adenovirus expressing PPM1K (AdCMV-PPM1K) to Zucker-obese rats lowered circulating BCAA, improved glucose tolerance and insulin sensitivity, and reduced RER and excess storage of fat in the liver (11).
Why does manipulation of the BDK:PPM1K ratio have such broad metabolic effects? The answer may lie, at least in part, with newly identified alternate substrates of this kinase/phosphatase pair, including ATP-citrate lyase (ACL), a critical enzyme of de novo lipogenesis (DNL), which functions in concert with acetyl-coenzyme A (CoA) carboxylase (ACC) and fatty acid synthase (11). Whereas phosphorylation of BCKDH by BDK inhibits its activity, phosphorylation activates ACL. ACL generates cytosolic acetyl-CoA to form malonyl-CoA, the immediate substrate for DNL and an allosteric inhibitor of FAO. Therefore, BT2 treatment or PPM1K overexpression is predicted to inactivate ACL, leading to reduced DNL, increased FAO, and reduced fat accumulation in liver, consistent with the findings in obese rats (11).
Consumption of sugar-sweetened foods and beverages has long been implicated as a risk factor for metabolic diseases, with fructose ascribed a particularly prominent role via its capacity to induce DNL via the Carbohydrate Response Element Binding Protein (ChREBP) transcription factor (12). Feeding of a high fructose diet or hepatic overexpression of ChREBP-β causes an increase in BDK and a decrease in PPM1K expression in liver of lean rats (11). Additionally, the amounts of BDK and ChREBP transcripts are tightly correlated in liver samples from humans with non-alcoholic fatty liver disease (NAFLD). These observations suggest a model wherein overnutrition, particularly involving diets high in fructose, activates ChREBP to increase the BDK/PPM1K ratio, thereby suppressing BCKDH activity and increase circulating BCAAs, while also promoting DNL and accumulation of liver fat.
What are direct mechanisms by which BCAA may drive cardiometabolic disease phenotypes? In cardiomyocytes, glucose suppresses BCAA catabolism via inhibition of cyclic adenosine monophosphate response element binding protein (CREB)-mediated expression of the Kruppel-like factor 15 (KLF15) transcription factor, a global activator of genes encoding BCAA catabolic enzymes (13). The resultant accumulation of BCAAs may activate mTORC1 to drive protein synthesis and cardiac hypertrophy, consistent with studies demonstrating that inhibition of BDK improves, whereas PPM1K deletion impairs, cardiac function in mouse models of heart failure (14). In liver, BCAA restriction activates the eIF2α kinase to engage programs of amino acid conservation, in concert with suppression of lipogenic genes, effects that are lost in GCN2 eIF2α knockout mice (15). Also, a valine-derived metabolite, 3-hydroxyisobutryate (3-HIB) is generated skeletal muscle in response to forced expression of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), resulting in stimulation of trans-endothelial fatty acid transport (16). Circulating 3-HIB levels are positively correlated with blood glucose in diabetic individuals (16), which suggests that this metabolite could promote excessive lipid storage and impaired insulin action in skeletal muscle. However, in studies using BT2 to activate BCKDH--a treatment that would be expected to raise 3-HIB levels--insulin resistance was improved rather than exacerbated in Zucker-obese rats, with no changes in lipid-derived metabolites in skeletal muscle (11). This does not exclude the possibility that 3-HIB contributed to lipid accumulation and the development of insulin resistance in the rats before therapeutic intervention. Further studies of this mechanism are warranted.
Therefore, BCAA are clearly a biomarker of cardiometabolic diseases. There is also growing evidence that they participate in disease pathogenesis, primarily in the context of obesity. In diet-induced obesity, dietary sugars stimulate ChREBP to increase the BDK/PPM1K ratio, simultaneously suppressing BCAA catabolism, activating DNL, and suppressing FAO. Because BDK and PPM1K regulate ACL, BCAAs serve as biomarkers of dyslipidemia, which itself contributes to cardiometabolic diseases. Molecular or pharmacologic manipulation of the BDK:PPM1K ratio is sufficient to reverse disease phenotypes in obese rodents. Moreover, supplementation of BCAA in obesogenic, but not low fat diets, or their restriction in diets fed to obese rodents, impacts metabolic health. Finally, the strong association of genetic variants that raise BCAA levels with risk of future T2D is consistent with a causal role for BCAAs in T2D pathogenesis. Ongoing studies of mechanisms by which BCAAs and related metabolites affect cardiometabolic disease pathogenesis are needed and will contribute to this burgeoning field.
References
- 1).Newgard CB. 2017. Metabolomics and metabolic diseases: where do we stand? Cell Metabolism 25: 43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Felig P, Marliss E, Cahill GF. 1969. Plasma amino acid levels and insulin secretion in obesity. New Engl J Med. 281:811–816. [DOI] [PubMed] [Google Scholar]
- 3).Newgard CB, An J. Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, et al. 2009. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 9:311–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, et al. 2011. Metabolic profiles and the risk of developing diabetes. Nat Med. 17:448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).She P, Van Horn C, Reid T, Hutson SM, Cooney RN, and Lynch CJ 2007. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am. J. Physiol. Endocrinol. Metab. 293, E1552–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. 2013. Cultured gut bacterial consortia from twins discordant for obesity modulate adiposity and metabolic phenotypes in gnotobiotic mice. Science. 341:1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Lotta LA, Scott RA, Sharp SJ, Burgess S, Luan J, Tillin T, et al. 2016. Genetic predisposition to an impaired metabolism of the branched-chain amino acids and risk of type 2 diabetes: a Mendelian randomization analysis. PLoS Med. 13:e1002179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Wang Q, Holmes MV, Smith GD, Ala-Korpela M. 2017. Genetic support for a causal role of insulin resistance on circulating branched-chain amino acids and inflammation. Diabetes Care 40: 1779–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).White PJ, Lapworth AL, An J, Wang L, McGarrah RW, Stevens RD, et al. 2016. Branched-chain amino acid restriction in Zucker-obese rats improves muscle insulin sensitivity by enhancing efficiency of fatty acid oxidation and acyl-glycine export. Mol Metab. 22:538–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Cummings NE, Williams EM, Kasza I, Konon EN, Schaid MD, Schmidt BA, Poudel C, Sherman DS, Yu D, Arriola Apelo SI, Cottrell SE, Geiger G, Barnes ME, Wisinski JA, Fenske RJ, Matkowskyj KA, Kimple ME, Alexander CM, Merrins MJ, Lamming DW. 2018. Restoration of metabolic health by decreased consumption of branched-chain amino acids. J. Physiology 596: 623–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).White PJ, McGarrah RW, Grimsrud PA, Tso S-C, Yang W-H, Haldeman J, Grenier-Larouche T, An J, Lapworth AL, Astapova I, Hannou SA, George T, Arlotto M, Lai M, Zhang G, Ilkayeva O, Herman MA, Wynn RM, Chuang DT, Newgard CB. 2018. The BCKDH kinase and phosphatase integrate BCAA and lipid metabolism via regulation of ATP-citrate lyase. Cell Metabolism 27: 1281–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Hannou SA, Haslam DE, McKeown NM, Herman MA. 2018. Fructose metabolism and metabolic disease. J. Clin Invest 128: 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Shao D, Villet O, Zhang Z, Choi SW, Yan J, Ritterhoff J, Gu H, Djukovic D, Christodoulou D, Kolwicz SC Jr, Raftery D, Tian R. 2018. Glucose promotes cell growth by suppressing branched-chain amino acid degradation. Nature Communications 9: 2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, Jeyaraj D, Youn JY, Ren S, Liu Y, Rau CD, Shah S, Ilkayeva O, Gui WJ, William NS, Wynn RM, Newgard CB, Cai H, Xiao X, Chuang DT, Schulze PC, Lynch C, Jain MK, Wang Y. 2016. Catabolic defects of branched-chain amino acids promotes heart failure. Circulation 133: 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Guo F, Cavener DR. 2007. The GCN2 eIF2α kinase regulates fatty acid homeostasis in the liver during deprivation of an essential amino acid. Cell Metabolism 5: 103–114. [DOI] [PubMed] [Google Scholar]
- 16).Jang C, Oh SF, Rowe GC, Liu L, Chan MC, Rhee J, Hoshino A, Kim B, Ibrahim A, Baca LG, et al. 2016. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nature Med. 22, 421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]