

Abstract
Allogeneic blood or marrow transplantation (BMT) is currently considered the standard of care for patients with specific inborn errors of metabolism (IEM). However, there is a paucity of studies describing long-term survival and cause-specific late mortality after BMT in these patients with individual types of IEM. We studied 273 patients who had survived ≥2 years after allogeneic BMT for IEM performed between 1974 and 2014. The most prevalent IEM in our cohort were X-linked adrenoleukodystrophy (ALD; 37.3%), Hurler syndrome (35.1%), and metachromatic leukodystrophy (MLD; 10.2%). Conditional on surviving ≥2 years after BMT, the overall survival for the entire cohort was 85.5 ± 2.4% at 10 years and 73.5 ± 3.7% at 20 years. The cohort had a 29-fold increased risk of late death compared with an age- and sex-matched cohort from the general US population (95% CI, 22- to 38-fold). The increased relative mortality was highest in the 2- to 5-year period after BMT (standardized mortality ratio [SMR], 207; 95% confidence interval [CI], 130 to 308) and declined with increasing time from BMT, but remained elevated for ≥21 years after BMT (SMR, 9; 95% CI, 4 to 18). Sequelae from the progression of primary disease were the most common causes of late mortality in this cohort (76%). The use of T cell-depleted grafts in patients with ALD and Hurler syndrome was a risk factor for late mortality. Younger age at BMT and use of busulfan and cyclosporine were protective in patients with Hurler syndrome. Our findings demonstrate relatively favorable overall survival in ≥2-year survivors of allogeneic BMT for IEM, although primary disease progression continues to be responsible for the majority of late deaths.
Keywords: Blood or marrow transplantation, Inborn errors of metabolism, Late mortality
INTRODUCTION
Inborn errors of metabolism (IEM) are a group of inherited metabolic disorders characterized by accumulation of toxic substrates that interfere with normal organ function [1]. Patients present with a wide range of clinical symptoms. For example, mucopolysaccharide (MPS) disorders, such as Hurler syndrome (MPS IH), are characterized by coarse facial features, dysostosis multiplex, hydrocephalus, organomegaly, corneal clouding, cardiac abnormalities, and loss of developmental milestones [2–4], whereas peroxisomal disorders, such as X-linked adrenoleukodystrophy (ALD), are primarily neurologic disorders marked by progressive loss of function [5–7]. Although the severity of the phenotype varies, in most cases, if left untreated, these disorders are progressive and ultimately result in premature death [8–10].
Gene therapy for IEM is currently experimental [11,12], and enzyme replacement therapy has been ineffective in treating central nervous system deterioration, owing to its inability to penetrate the blood-brain barrier [13]. Given its potential to correct the underlying defect and traverse the blood-brain barrier, allogeneic blood or marrow transplantation (BMT) has been used with the goal of halting or slowing disease progression [5–7,10,14–20]. Transplantation remains the standard of care in younger patients with Hurler syndrome, early cerebral ALD, and asymptomatic late infantile or early juvenile metachromatic leukodystrophy (MLD) and globoid cell leukodystrophy. BMT has also been used in other types of IEM, including, but not limited to, Hunter syndrome (MPS II), Maroteaux-Lamy syndrome (MPS VI), alpha-mannosidosis, and Wolman syndrome [21–27].
Previous studies have described survival outcomes after allogeneic BMT for a heterogeneous population of patients with IEM [28,29], and most of these studies relied on passive ascertainment of vital status, with no attempt to link with death registries, such as the National Death Index (NDI) [2,3,10,30]. A comprehensive assessment of overall and cause-specific late mortality in patients with specific types of IEM treated with allogeneic BMT is needed to inform both the transplantation physicians as well as the patients and their families. In the present study, we addressed these gaps using the resources provided by the Blood or Marrow Transplant Survivor Study-2 (BMTSS-2).
PATIENTS AND METHODS
BMTSS-2 is a collaboration between City of Hope, University of Minnesota, and University of Alabama at Birmingham, examining the long-term outcomes of individuals treated with BMT. To be included in this analysis, patients had to have undergone allogeneic BMT between 1974 and 2014 for a diagnosis of IEM and have survived for ≥2 years after BMT. The vast majority of the patients underwent transplantation at the University of Minnesota. The Human Subjects Committee at each participating institution approved the BMTSS-2 protocol. Informed consent was obtained in accordance with the Declaration of Helsinki.
Data Collection
Information on demographic characteristics, primary diagnosis, preparative regimens, stem cell source, type of donor (sibling versus unrelated), agents used for graft-versus-host disease (GVHD) prophylaxis and presence of chronic GVHD was obtained from the institutional transplantation databases and supplemented by medical records. Chronic GVHD was classified as none, limited, or extensive based on the Seattle Classification [31]. NDI Plus and/or medical records provided information regarding the date and cause of death through December 31, 2015. Additional information from the Accurint database [32] was used to extend the vital status information through December 31, 2016. All patients were assigned a primary and, if present, secondary cause of death, independently by 2 investigators (A.W. and J.W.). Cause of death assignments were further verified by a third investigator (P.J. O.). In the event of discrepant assignments, a fourth investigator (S.B.) provided adjudication. Causes of death (primary or secondary) attributable to the underlying IEM were classified as disease-related mortality. Causes of death due to therapeutic exposures and the transplantation procedure were collectively classified as transplantation-related mortality.
Statistical Analysis
Kaplan-Meier techniques were used to describe overall survival, conditional on surviving for ≥2 years after allogeneic BMT. The cumulative incidence of cause-specific mortality was calculated using competing-risk methods [33]. The standardized mortality ratio (SMR), a ratio of observed to expected number of deaths, was used to compare the mortality experienced by this cohort to the age- (5-year intervals), sex-, and calendar-specific (5-year intervals) mortality of the general US population, using data obtained from the Centers for Disease Control and Prevention [34]. The 95% confidence intervals (CIs) of the SMR were calculated using the Poisson regression method described by Vandenbroucke [35]. SMRs were calculated for the entire cohort, as well as separately by sex, year of transplantation (1983 to 1988, 1989 to 1994, 1995 to 2000, 2001 to 2006, and 2007 to 2014), primary IEM diagnosis, and presence or absence of chronic GVHD.
Cox regression analysis was used to identify predictors of all-cause mortality for the entire cohort, as well as for the most prevalent IEM diagnoses considered individually. Owing to the small number of subjects and deaths in each model and the associated collinearity among the variables, a parsimonious model was created using the variables with associated P values <.10 in the multivariable model. Furthermore, owing to the varied clinical presentations of different IEM and the significant differences in median age at BMT by type of IEM, we used the median age at BMT of each primary disease to evaluate its effect on late mortality. Multivariable regression models could not be constructed for MLD because of the small number of patients and associated deaths. All analyses were performed with SAS version 9.4 (SAS Institute, Cary, NC).
RESULTS
Entire Cohort
A total of 273 patients underwent allogeneic BMT for IEM between 1983 and 2014 and survived for ≥2 years after BMT at 1 of the 3 participating centers (University of Minnesota, n = 272; City of Hope, n = 1; University of Alabama at Birmingham, n = 0). The IEM diagnoses included ALD in 102 patients (37.4%), Hurler syndrome in 96 (35.2%), MLD in 28 (10.2%), and other miscellaneous IEM in 47 (17.2%). Table 1 summarizes the demographic and clinical characteristics of the entire cohort, as well as for the patients with ALD, Hurler syndrome, and MLD. The median age at BMT was 4.9 years (range, .10 to 44.2 years). Overall, 189 patients (69.2%) were male, 233 (85.3%) were non-Hispanic white, 192 (70.3%) underwent an unrelated donor BMT, and 121 (44.3%) received a cord blood transplant. The majority of transplantations (n = 172; 63%) were performed after 2000. Cyclophosphamide was used in the preparative regimen in 228 patients (83.5%), busulfan in 194 (71.1%), and total body irradiation (TBI) in 111 (40.7%). Cyclosporine was the most commonly used agent for GVHD prophylaxis (n = 255; 93.4%); other prophylaxis strategies included mycophenolate mofetil (MMF; n = 103; 37.7%) and T cell depletion (n = 64; 23.4%). T cell depletion for GVHD prophylaxis declined with progressing transplant era (before 2000, n = 46 [72%]; 2000 to 2010, n = 18 [28%]; after 2010, n = 0; P < .0001).
Table 1.
Demographic and Clinical Characteristics of 2-Year Survivors of Allogeneic BMT for IEM and of Patients with the 3 Most Prevalent IEM Diagnoses
| Variable | All (n = 273) | ALD (n = 102) | Hurler Syndrome (n = 96) | MLD (n = 28) |
|---|---|---|---|---|
|
| ||||
| Age at BMT, yr, median (range) | 4.9 (0.1–44.2) | 8.6 (4.0–23.3) | 1.5 (0.4–6.0) | 9.8 (0.4–44.2) |
| Time since BMT, yr, median (range) | 10.5 (1.6–34.0) | 7.8 (1.7–25.2) | 13.2 (1.6–31.4) | 15.6 (2.2–32.0) |
| Sex, n (%) | ||||
| Male | 189 (69.2) | 102 (100) | 57 (59.4) | 6 (21.4) |
| Female | 84 (30.8) | – | 39 (40.6) | 22 (78.6) |
| Race/ethnicity, n (%) | ||||
| Non-Hispanic white | 233 (85.3) | 81 (79.4) | 93 (96.9) | 23 (82.1) |
| Hispanic | 15 (5.5) | 8 (7.8) | 2 (2.1) | 1 (3.6) |
| Black | 18 (6.6) | 8 (7.8) | 1 (1.0) | 4 (14.3) |
| Other | 1 (0.4) | 1 (1.0) | – | – |
| Missing | 6 (2.2) | 4 (3.9) | – | – |
| Type of donor, n (%) | ||||
| Sibling | 77 (28.2) | 30 (29.4) | 24 (25) | 8 (28.6) |
| Unrelated | 192 (70.3) | 72 (70.6) | 70 (72.1) | 20 (71.4) |
| Parent | 4 (1.5) | – | 2 (2.1) | – |
| Stem cell source, n (%) | ||||
| Bone marrow | 151 (55.3) | 49 (48.0) | 53 (55.2) | 17 (60.7) |
| Cord blood | 121 (44.3) | 52 (51.0) | 43 (44.8) | 11 (39.3) |
| Peripheral blood stem cells | 1 (0.4) | 1 (1.0) | – | – |
| Year of BMT, n(%) | ||||
| <2000 | 101 (37.0) | 17 (16.7) | 41 (42.7) | 15 (53.6) |
| 2000–2010 | 112 (41.0) | 50 (49.0) | 31 (32.3) | 12 (42.9) |
| >2010 | 60 (22.0) | 35 (34.3) | 24 (25.0) | 1 (3.6) |
| TBI, n (%) | ||||
| Yes | 111 (40.7) | 44 (43.1) | 31 (32.3) | 17 (60.7) |
| No | 162 (59.3) | 58 (56.9) | 65 (67.7) | 11 (39.3) |
| Cyclophosphamide, n (%) | ||||
| Yes | 228 (83.5) | 77 (75.5) | 86 (89.6) | 22 (78.6) |
| No | 45 (16.5) | 25 (24.5) | 10 (10.4) | 6 (21.4) |
| Busulfan, n (%) | ||||
| Yes | 194 (71.1) | 58 (56.9) | 83 (86.5) | 14 (50.0) |
| No | 79 (28.9) | 44 (43.1) | 13 (13.5) | 14 (50.0) |
| Cyclosporine, n (%) | ||||
| Yes | 255 (93.4) | 100 (98) | 86 (89.6) | 27 (96.4) |
| No | 18 (6.6) | 2 (2.0) | 10 (10.4) | 1 (3.6) |
| Mycophenolate mofetil, n (%) | ||||
| Yes | 103 (37.7) | 59 (57.8) | 29 (30.2) | 5 (17.9) |
| No | 170 (62.3) | 43 (42.2) | 67 (69.8) | 23 (82.1) |
| T cell depletion, n (%) | ||||
| Yes | 64 (23.4) | 11 (10.8) | 25 (26.0) | 12 (42.9) |
| No | 209 (76.6) | 91 (89.2) | 71 (74.0) | 16 (57.1) |
| cGVHD, n (%) | ||||
| Yes | 40 (14.7) | 10 (9.8) | 14 (14.6) | 5 (17.9) |
| No | 233 (85.3) | 92 (90.2) | 82 (85.4) | 23 (82.1) |
| Deceased, n (%) | 54 (19.8) | 13 (12.7) | 14 (14.6) | 6 (21.4) |
Conditional on surviving the first 2 years, 54 patients (19.8%) had died after a median follow-up of 10.5 years (range, 1.6 to 34 years), yielding overall survival rates of 92.8 § 1.7% at 5 years, 85.5 ± 2.4% at 19 years, and 73.5 ± 3.7% at 20 years post-BMT (Figure 1A). The median age at death was 13.8 years (range, 2.7 to 51.4 years). The mortality rate declined with time from BMT (2 to 5 years, n = 22 [40.7%]; 6 to 10 years, n = 12 [22.2%]; 11 to 15 years, n = 7 [12.9%]; 16 to 20 years, n = 6 [11.1%]; and ≥21 years, n = 7 [12.9%)). Causes of death were available for 47 patients (87%), the majority of which were directly attributable to the primary disease (76%). Other causes of death included infection (2%), subsequent malignant neoplasms (4%), cardiac disease (2%), pulmonary disease (2%), and other miscellaneous causes (2%) (supplementary Table S1 and Figure S1). The 20-year cumulative incidence of disease-related mortality (19.7%; 95% CI, 13.7% to 26.5%) was higher than that of transplantation-related mortality (3.9%; 95% CI, 1.4% to 8.5%) (Figure 1B).
Figure 1.
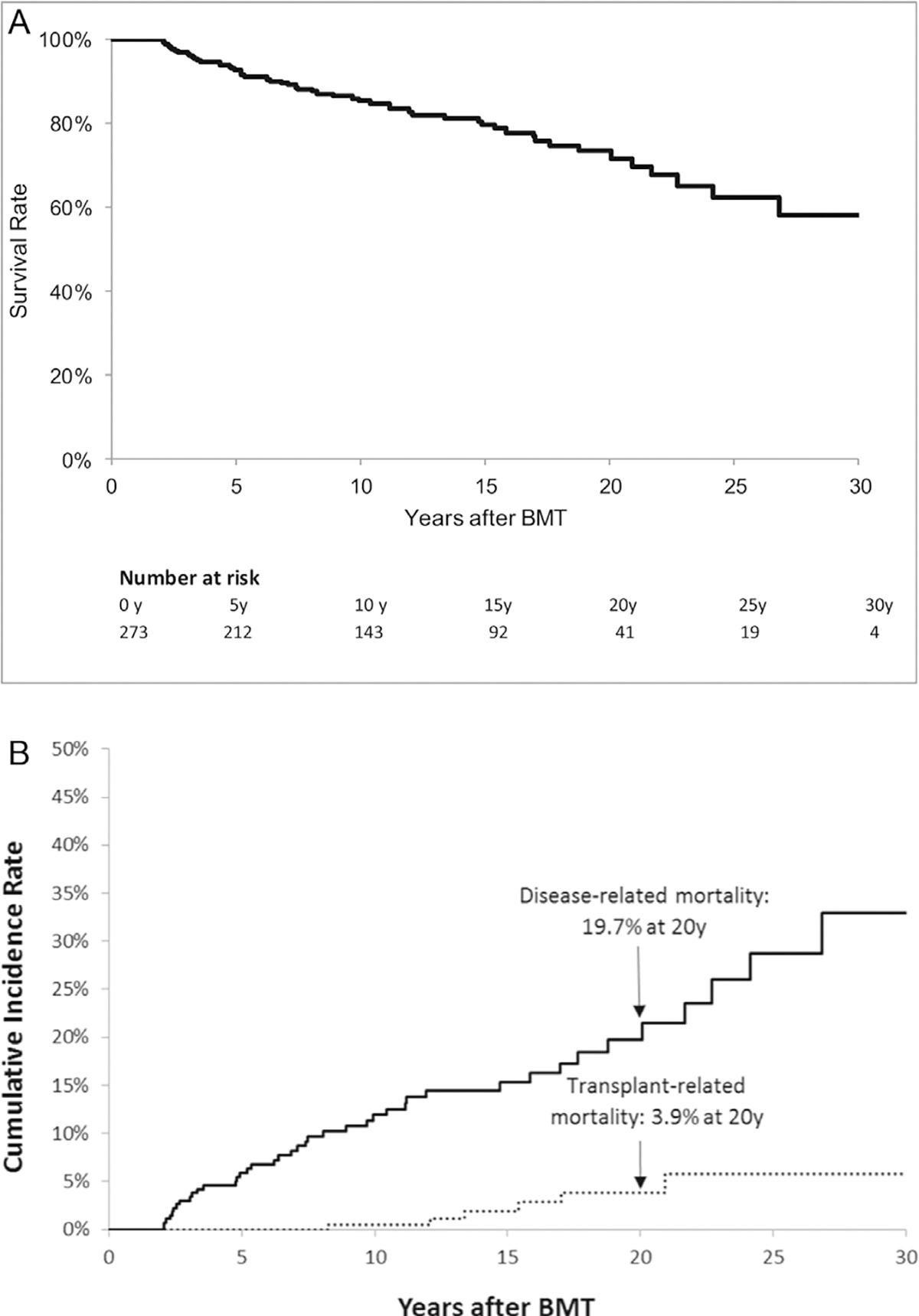
(A) Overall survival for entire cohort of 273 2-year survivors of allogeneic BMT for IEM. (B) Cumulative incidence of cause-specific mortality.
Overall, the cohort exhibited a 29-fold (95% CI, 22- to 38-fold) increased risk of premature death when compared with age-, sex-, and calendar-specific rates observed in the general US population (Table 2). SMR values were higher in females than in males (47; 95% CI, 31 to 68 versus 22; 95% CI, 15 to 31; P = .01), even after exclusion of the all-male ALD cohort (SMRnon-ALD males, 21; 95% CI, 12 to 33). The excess risk of late mortality remained fairly stable over the transplantation eras. The SMR values declined with time after BMT (2 to 5 years post-BMT: SMR, 207; 95% CI, 130 to 308; ≥21 years post-BMT: SMR, 9; 95% CI, 4 to 18). Patients with chronic GVHD and without chronic GVHD had comparable SMR values (33; 95% CI, 17 to 56 versus 28; 95% CI, 20 to 38; P = .70). As shown in Table 3, multivariable regression analysis of the entire cohort did not identify any statistically significant associations between demographic or clinical characteristics and all-cause late mortality, although use of busulfan appeared to have a protective effect on the risk of late mortality (HR, 0.6; 95% CI, 0.3 to 1; P = .05), and the association trended toward significance.
Table 2.
SMR among 273 2-Year Survivors of Allogeneic BMT for IEM
| Variables | Deaths, n | SMR | 95% CI |
|---|---|---|---|
|
| |||
| All patients | 53 | 28.9 | 21.9–37.5 |
| Sex | |||
| Male | 28 | 21.6 | 14.6–30.6 |
| Female | 25 | 46.9 | 30.9–67.8 |
| Year of BMT | |||
| 1983–1988 | 11 | 36.8 | 19.1–63 |
| 1989–1994 | 10 | 23.1 | 11.6–40.5 |
| 1995–2000 | 12 | 24.8 | 13.3–41.6 |
| 2001–2006 | 14 | 35.2 | 19.9–57.1 |
| 2007–2014 | 6 | 27.7 | 11–56.1 |
| Years after BMT | |||
| 2–5 | 21 | 206.5 | 130.3–307.9 |
| 6–10 | 12 | 52 | 27.8–87.2 |
| 11–15 | 7 | 32.4 | 13.9–62.6 |
| 16–20 | 6 | 11.2 | 4.5–22.7 |
| 21+ | 7 | 9.4 | 4.0–18.2 |
| Chronic GVHD | |||
| Yes | 11 | 32.9 | 17–56.3 |
| No | 42 | 28.0 | 20.4–37.5 |
| Primary diagnosis | |||
| ALD | 14 | 27.8 | 15.7–45 |
| Hurler syndrome | 12 | 22.2 | 11.9–37.3 |
| MLD | 6 | 16.1 | 6.4–32.7 |
Table 3.
Risk of Late Mortality for the Entire Cohort and Patients with ALD or Hurler Syndrome
| Variable | HR (95% CI) | P Value |
|---|---|---|
|
| ||
| Entire cohort | ||
| Race/ethnicity | ||
| Other | 1 | - |
| Non-Hispanic white | 2.8 (.90–8.9) | .09 |
| Busulfan as conditioning agent | ||
| No | 1 | - |
| Yes | .60 (.30–1) | .05 |
| ALD | ||
| Age at BMT | ||
| At or older than the median age of the cohort | 1 | - |
| Younger than the median age of the cohort | .10 (.02–1.1) | .06 |
| Donor type | ||
| Related | 1 | - |
| Unrelated | .20 (.04–1) | .05 |
| Year of BMT | ||
| <2000 | 1 | - |
| 2000–2009 | .40 (.10–1.6) | .20 |
| >2010 | 12.6 (1.3–118.7) .03 | |
| T cell depletion for GVHD prophylaxis | ||
| No | 1 | - |
| Yes | 18.8 (2.8–126) | .003 |
| Hurler syndrome | ||
| Age at BMT | ||
| At or older than median age of cohort | 1 | - |
| Younger than the median age of cohort | .20 (.05–.90) | .03 |
| Chronic GVHD | ||
| No | 1 | - |
| Yes | 3.9 (.80–18.8) | .08 |
| Busulfan as conditioning agent | ||
| No | 1 | - |
| Yes | .30 (.07–.90) | .04 |
| T cell depletion for GVHD prophylaxis | ||
| No | 1 | - |
| Yes | 5.3 (1.4–20.3) | .02 |
| Cyclosporine for GVHD prophylaxis | ||
| No | 1 | - |
| Yes | .10 (.04–.50) | .004 |
X-Linked ALD
ALD was the most prevalent IEM in our cohort of ≥2-year BMT survivors, affecting 102 patients. The median age at transplantation was 8.6 years (range, 4 to 23.3 years) (Table 1). Overall, the majority of patients received stem cells from an unrelated donor (n = 72; 70.6%), and one-half (n = 52; 51%) received a cord blood transplant. Cyclophosphamide was included in the preparative regimen in 75.5% of the patients, and almost all patients (n = 100; 98%) received cyclosporine for chronic GVHD prophylaxis. The prevalence of chronic GVHD in this cohort was 9.8% (n = 10). T cell depletion was used in 10.8% of the patients but was not used in any patients after 2000.
After a median follow-up of 7.8 years (range, 1.7 to 25.2 years), 13 (12.7%) of the 2-year survivors of allogeneic BMT for ALD had died, yielding an overall survival of 94.3 ± 2.5% at 5 years and 86.6 ± 4.4% at 10 years post-BMT (Figure 2A). The ALD cohort was at a 28-fold greater risk of late death (95% CI, 16- to 45-fold) compared with the age- and sex-matched general US population (Table 2). As shown in Table 3, the use of T cell-depleted grafts (HR, 18.8; 95% CI, 2.8 to 126; P = .003) and BMT performed after 2010 (HR, 12.6; 95% CI, 1.3 to 118.7; P = .03) were associated with a significantly greater risk of all-cause late mortality in patients with ALD. Younger age at BMT (ie, less than the median age of the cohort) (HR, .10; 95% CI, .02 to 1.1; P = .06) and unrelated donor BMT compared with related donor BMT (HR, .20; 95% CI, .04 to 1.0; P = .05) were suggestive of having a protective effect on all-cause late mortality. Causes of death were available for 8 patients (62%); all were directly attributable to progression of primary disease, with pulmonary disease the most common secondary cause of death for these patients (n = 5; 63%) (Supplementary Table S1).
Figure 2.
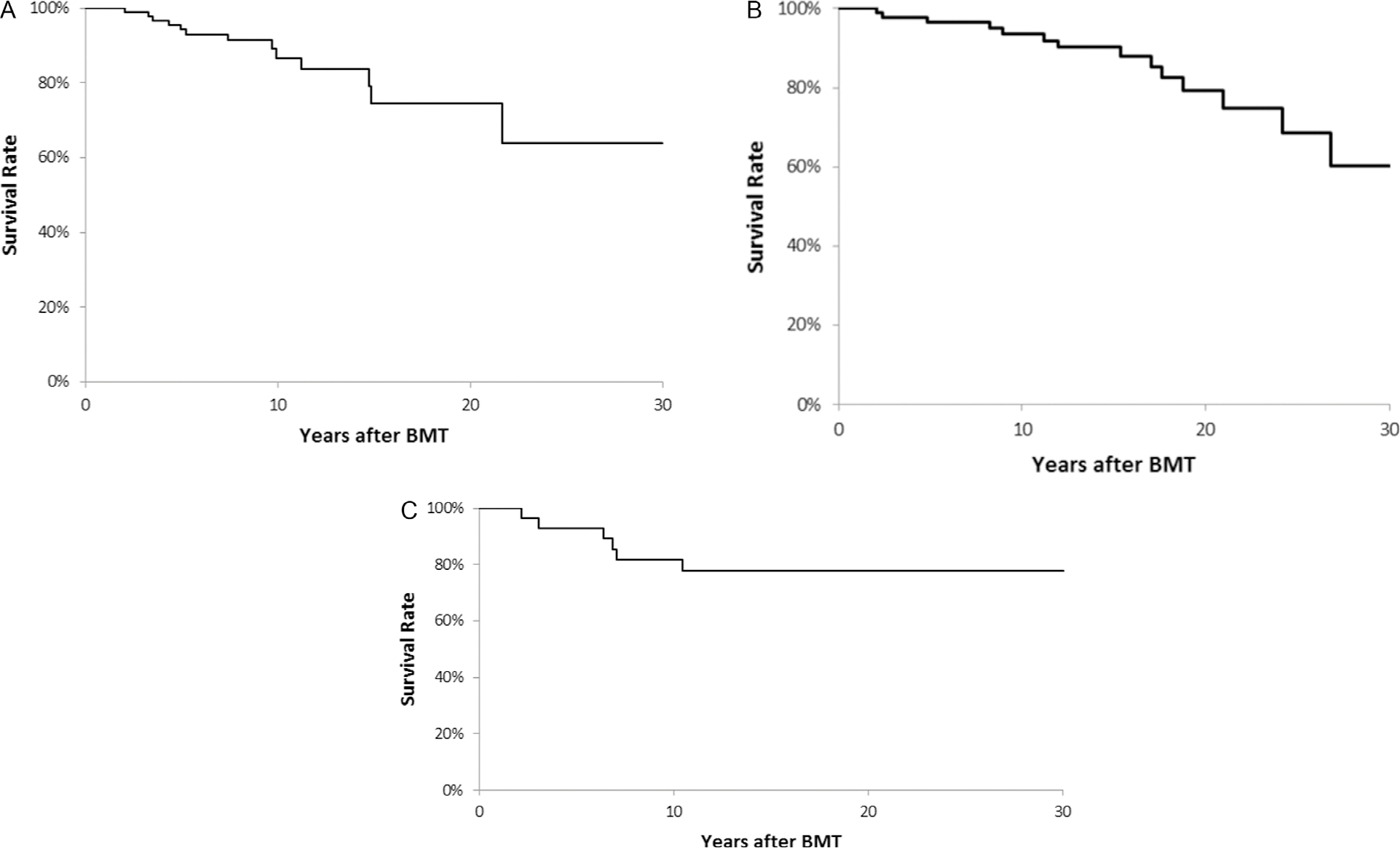
Overall survival for patients with ALD (A), Hurler syndrome (B), and MLD (C).
Hurler Syndrome
A total of 96 patients with Hurler syndrome underwent allogeneic BMT during the study period and survived for ≥2 years. These patients were younger at the time of transplantation (median age at BMT, 1.5 years; range, .40 to 6 years) compared with patients with ALD or MLD (8.6 years [range, 4 to 23.3 years] and 9.8 years [range, .40 to 44.2 years], respectively; P < .001). The majority received an unrelated donor transplant (n = 70; 72%), and bone marrow was the most common source of stem cells (n = 53; 55%). Cyclophosphamide (n = 86; 90%) and busulfan (n = 83; 87%) were the 2 most commonly used agents in the preparative regimen, with only 32% (n = 31) receiving TBI. The use of TBI as part of preparative regimen for patients with Hurler syndrome declined over time (before 2000, n = 21 [67.7%]; 2000 to 2010, n = 7 [22.6%]; after 2010, n = 3 [9.7%]; P = .002). Cyclosporine was the most commonly used agent for GVHD prophylaxis (n = 86; 89.6%). T cell depletion was used in 26% of patients as part of GVHD prophylaxis, and its use declined with transplantation era (before 2000, n = 18 [72%]; 2000 to 2010, n = 7 [28%]; after 2010, n = 0; P < .0001). The prevalence of chronic GVHD in patients with Hurler syndrome was 14.6% (n = 14).
Conditional on surviving for ≥2 years after BMT and after a median follow-up of 13.2 years (range, 1.6 to 31.4 years), 14 patients (14.6%) had died, yielding an overall survival of 96.6 ± 1.9% at 5 years and 93.6 ± 2.8% at 10 years post-BMT (Figure 2B). This cohort exhibited a 22-fold increased risk of relative mortality (95% CI, 12- to 37-fold) compared with an age- and sex-matched cohort from the general US population (Table 2). BMT at younger age (ie, less than the median age at BMT of 1.5 years) was associated with a lower risk of all-cause late mortality (HR, .20; 95% CI, .05 to .90; P = .03) compared with BMT performed at older than the median age (Table 3). Other factors found to be protective included the use of busulfan in the preparative regimen (HR, .30; 95% CI, .07 to .90; P = .04) and the use of cyclosporine for GVHD prophylaxis (HR, .10; 95% CI, .04 to .50; P = .004). The use of T cell-depleted grafts was associated with an elevated risk of late mortality (HR, 5.3; 95% CI, 1.4 to 20.3; P = .02) compared with non-T cell-depleted grafts. Cause of death data were available for all patients who had died; the most prevalent cause was primary disease progression (n = 10; 71%), with cardiac diseases (n = 4; 29%) and pulmonary diseases (n = 4; 29%) the most common secondary causes of death in this cohort (Supplementary Table S1). Transplantation-related disorders as the primary cause of death included infection, subsequent malignant neoplasm, cardiac disease, and pulmonary disease in 1 patient each.
MLD
A total of 28 patients with MLD underwent allogeneic BMT during the study period and survived for ≥2 years (Table 1). These patients had the longest follow-up in our study, with a median duration since transplantation of 15.6 years (range, 2.2 to 31.9 years). Cyclosporine (n = 27; 96.4%) and T cell depletion (n = 12; 42.9%) were the most commonly used approaches for GVHD prophylaxis, and the prevalence of chronic GVHD approached 18% (n = 5). Six deaths occurred during the study period, yielding an overall survival of 92.9 ± 4.9% at 5 years and 81.7 ± 7.4% at 10 years (Figure 2C). This cohort experienced a 16-fold increased relative mortality (95% CI, 6- to 32-fold) compared with the age- and sex-matched general US population (Table 2). All deaths were attributable to the primary disease (Supplementary Table S1).
DISCUSSION
In the present study, 2-year survivors of allogeneic BMT for IEM had a 10-year overall survival of 86%. These findings are comparable to those in a previous study assessing late mortality in 2-year survivors of BMT for IEM, which reported a 9-year survival rate of 88% [29]. With the availability of longer follow-up in our cohort, we were able to show that the overall survival declined to 79% at 15 years and to 74% at 20 years after BMT. Nonetheless, given that these disorders can have a debilitating and rapidly fatal course if untreated, our study demonstrates relatively favorable long-term survival in 2-year survivors of allogeneic BMT for IEM.
Nonetheless, the excess mortality in these 2-year survivors of allogeneic BMT for IEM is high compared with the low baseline mortality in the age- and sex-matched general population. Our study population had exceptionally high rates of premature death compared with the age- and sex-matched general population in the 2- to 5-year post-BMT period. Thereafter, the relative mortality declined with time but remained significantly elevated beyond 20 years, at a 9-fold greater risk compared with the general population. Previous studies have reported similarly high relative mortality for all IEM considered together [28,29]; we and we found significantly elevated all-cause late mortality in our patients with ALD, Hurler syndrome, and MLD. We also show that the high relative mortality in these patients has remained unchanged over time and is likely attributable to the underlying disease.
An analysis of registry data from the Center for International Bone Marrow Transplant registry [29] found active chronic GVHD and unrelated donor BMT to be associated with increased risk of all-cause late mortality in 2-year survivors of allogeneic BMT for IEM. Our study did not identify these factors as influencing late mortality. We found that use of busulfan was associated with a lower risk of all-cause late mortality when assessed for all patients; however, given the heterogeneity in demographic and clinical characteristics of the various IEM, it is more important to understand the predictors of all-cause mortality and causes of death in these diseases individually.
Age at BMT had a significant impact on survival in patients with Hurler syndrome. Similar to previous studies [2,14], we observed that allogeneic BMT at a younger age for patients with Hurler syndrome was protective against late mortality, likely due to its role in preventing or slowing the progression of the disease when performed at an earlier stage. Although we did not have data on busulfan pharmacokinetic targeting, we observed that the use of busulfan was protective against late mortality in patients with Hurler syndrome. Busulfan pharmacokinetic targeting has been shown to improve engraftment in patients with Hurler syndrome [36], and its use in preparative regimens has been shown to improve engraftment of donor-derived microglial cells post-transplantation in animal models, which potentially could improve disease phenotype [37]. We also found a lower risk of late mortality in patients with Hurler syndrome associated with the use of cyclosporine as GVHD prophylaxis compared with the use of T cell depletion and MMF for prophylaxis. Finally, patients with ALD and Hurler syndrome who received a T cell-depleted graft were at increased risk for late mortality. T cell depletion has been previously identified as a risk factor for graft failure in patients with Hurler syndrome and possibly could have played a role here [36].
We have demonstrated that 2-year survivors of allogeneic BMT for IEM continue to be at increased risk of death from primary disease-related complications. In our cohort, the risk of death at 20 years post-BMT from progression of the primary disease was higher than the risk of death from other transplantation-related causes (19.7% versus 3.9%). It is known that despite the intent of BMT for IEM to correct the underlying deficiency and prevent further worsening of the clinical course, transplantation is insufficient to eradicate disease or to repair damaged tissue, most notably in the brain or skeleton, and patients continue to experience significant disease-related morbidity [2,7,10,19]. We did not have data on the functional status of these patients; however, Aldenhoven et al [2] have shown that although survivors of BMT for Hurler syndrome had an improved clinical course, these survivors faced significant disease-related complications. Similarly, other reports on long-term outcomes following BMT in patients with ALD [6,7,19,38] and MLD [10,39,40] have shown disease stabilization. Nevertheless, patients continue to suffer from disease-related morbidity. In our cohort, 76% of deaths were attributable to primary disease. For patients with Hurler syndrome, cardiac and pulmonary causes were the most common secondary causes of death. The majority of patients with ALD died from pulmonary causes, likely secondary to neurologic impairment and the resultant aspiration. Although these numbers are too small to allow us to draw statistical inferences, these results suggest that progression of primary disease continues to be the leading cause of death for these patients.
Despite the small number of certain IEM in our cohort, limiting the statistical analyses for individual disorders, our study illuminates characteristics and outcomes of one of the largest cohorts of ALD patients reported to date. In addition to medical records, this analysis relied on active ascertainment of vital status by using the NDI Plus Program and Accurint databases, providing comprehensive and complete ascertainment of vital status and cause of death. However, although causes of death were assigned independently by 2 investigators and then verified by a physician who specializes in BMT for IEM, limitations of the causes of death recorded on death certificates could result in misclassification of causes of death [41]. We also lacked information regarding cause of death in 13% of cases. In addition, our study lacks information on pretransplantation enzyme replacement therapy for patients with Hurler syndrome, which is now widely used [42,43]. We did not have the pretransplantation magnetic resonance imaging severity score [44] for patients with ALD, which has been shown to be an important prognostic indicator for outcomes after BMT [5,6]. In addition, information on the phenotype of MLD (late infantile, juvenile, or adult forms) and the extent of disease at transplantation was not available. Despite the multicenter nature of the BMTSS-2 study, almost all patients in this cohort underwent transplantation at 1 center, which possibly could limit the generalizability of our findings. Finally, data on post-BMT donor chimerism and enzyme levels were not available.
These limitations notwithstanding, our data show that patients with IEM who undergo allogeneic BMT and survive for at least 2 years have a relatively favorable overall survival even at 20 years after BMT. These patients continue to be at risk for primary disease-related complications for many years after transplantation, however. These findings regarding the late mortality experience in patients with ALD, Hurler syndrome and MLD provide important information for the families and the healthcare providers caring for these patients. Future research should include collaborative efforts between various transplantation working groups to collectively study outcomes for these rare diseases, in addition to developing transplantation practices that can effectively reduce the disease burden in these patients.
Supplementary Material
ACKNOWLEDGMENTS
Financial disclosure: This study was supported in parts by grants from the National Cancer Institute (R01 CA078938), the Leukemia Lymphoma Society (R6502–16), and the Swedish Childhood Cancer Foundation (TJ2016–0014).
Footnotes
Conflict of interest statement: W.M. is a medical director at Sangamo Therapeutics. The other authors have no conflicts of interest to report.
SUPPLEMENTARY DATA
Supplementary data related to this article can be found on line at doi:10.1016/j.bbmt.2018.09.035.
REFERENCES
- 1.Burrow TA Grabowski GA, Leslie ND, Prada CE. Lysosomal storage diseases. In: Orkin SH, ed. Nathan and Oski’s Hematology and Oncology of Infancy and Childhood. Philadelphia, PA: Elsevier/Saunders; 2015. [Google Scholar]
- 2.Aldenhoven M, Wynn RF, Orchard PJ, et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood. 2015;125:2164–2172. [DOI] [PubMed] [Google Scholar]
- 3.Coletti HY, Aldenhoven M, Yelin K, Poe MD, Kurtzberg J, Escolar ML. Long-term functional outcomes of children with Hurler syndrome treated with unrelated umbilical cord blood transplantation. JIMD Rep. 2015;20:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shapiro EG, Nestrasil I, Rudser K, et al. Neurocognition across the spectrum of mucopolysaccharidosis type I: Age, severity, and treatment. Mol Genet Metab. 2015;116:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peters C, Charnas LR, Tan Y, et al. Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999. Blood. 2004;104:881–888. [DOI] [PubMed] [Google Scholar]
- 6.Miller WP, Rothman SM, Nascene D, et al. Outcomes after allogeneic hematopoietic cell transplantation for childhood cerebral adrenoleukodystrophy: the largest single-institution cohort report. Blood. 2011;118:1971–1978. [DOI] [PubMed] [Google Scholar]
- 7.Shapiro E, Krivit W, Lockman L, et al. Long-term effect of bone-marrow transplantation for childhood-onset cerebral X-linked adrenoleukodystrophy. Lancet. 2000;356:713–718. [DOI] [PubMed] [Google Scholar]
- 8.Boelens JJ, Orchard PJ, Wynn RF. Transplantation in inborn errors of metabolism: current considerations and future perspectives. Br J Haematol. 2014;167:293–303. [DOI] [PubMed] [Google Scholar]
- 9.Eisengart JB, Rudser KD, Xue Y, et al. Long-term outcomes of systemic therapies for Hurler syndrome: an international multicenter comparison [e-pub ahead of print]. Genet Med 2018. 10.1038/gim.2018.29. accessed March 26, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Groeschel S, Kühl JS, Bley AE, et al. Long-term outcome of allogeneic hematopoietic stem cell transplantation in patients with juvenile metachromatic leukodystrophy compared with nontransplanted control patients. JAMA Neurol. 2016;73:1133–1140. [DOI] [PubMed] [Google Scholar]
- 11.Eichler F, Duncan C, Musolino PL, et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N Engl J Med. 2017;377:1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sessa M, Lorioli L, Fumagalli F, et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet. 2016;388:476–487. [DOI] [PubMed] [Google Scholar]
- 13.Shull RM, Kakkis ED, McEntee MF, Kania SA, Jonas AJ, Neufeld EF. Enzyme replacement in a canine model of Hurler syndrome. Proc Natl Acad Sci U S A. 1994;91:12937–12941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boelens JJ, Aldenhoven M, Purtill D, et al. Outcomes of transplantation using various hematopoietic cell sources in children with Hurler syndrome after myeloablative conditioning. Blood. 2013;121:3981–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Staba SL, Escolar ML, Poe M, et al. Cord-blood transplants from unrelated donors in patients with Hurler’s syndrome. N Engl J Med. 2004;350:1960–1969. [DOI] [PubMed] [Google Scholar]
- 16.Peters C, Balthazor M, Shapiro EG, et al. Outcome of unrelated donor bone marrow transplantation in 40 children with Hurler syndrome. Blood. 1996;87:4894–4902. [PubMed] [Google Scholar]
- 17.Peters C, Shapiro EG, Anderson J, et al. Hurler syndrome: II. Outcome of HLA-genotypically identical sibling and HLA-haploidentical related donor bone marrow transplantation in fifty-four children. The Storage Disease Collaborative Study Group. Blood. 1998;91:2601–2608. [PubMed] [Google Scholar]
- 18.Mitchell R, Nivison-Smith I, Anazodo A, et al. Outcomes of haematopoietic stem cell transplantation for inherited metabolic disorders: a report from the Australian and New Zealand Children’s Haematology Oncology Group and the Australasian Bone Marrow Transplant Recipient Registry. Pediatr Transplant. 2013;17:582–588. [DOI] [PubMed] [Google Scholar]
- 19.Kühl JS, Suarez F, Gillett GT, et al. Long-term outcomes of allogeneic haematopoietic stem cell transplantation for adult cerebral X-linked adrenoleukodystrophy. Brain. 2017;140:953–966. [DOI] [PubMed] [Google Scholar]
- 20.van den Broek BTA, Page K, Paviglianiti A, et al. Early and late outcomes after cord blood transplantation for pediatric patients with inherited leukodystrophies. Blood Adv. 2018;2:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peters C, Steward CG. Hematopoietic cell transplantation for inherited metabolic diseases: an overview of outcomes and practice guidelines. Bone Marrow Transplant. 2003;31:229–239. [DOI] [PubMed] [Google Scholar]
- 22.Mynarek M, Tolar J, Albert MH, et al. Allogeneic hematopoietic SCT for alpha-mannosidosis: an analysis of 17 patients. Bone Marrow Transplant. 2012;47:352–359. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka A, Okuyama T, Suzuki Y, et al. Long-term efficacy of hematopoietic stem cell transplantation on brain involvement in patients with mucopolysaccharidosis type II: a nationwide survey in Japan. Mol Genet Metab. 2012;107:513–520. [DOI] [PubMed] [Google Scholar]
- 24.Borgwardt L, Lund AM, Dali CI. Alpha-mannosidosis - a review of genetic, clinical findings and options of treatment. Pediatr Endocrinol Rev. 2014;12(1):185–191. Suppl. [PubMed] [Google Scholar]
- 25.Harmatz P, Shediac R. Mucopolysaccharidosis VI: pathophysiology, diagnosis and treatment. Front Biosci (Landmark Ed). 2017;22:385–406. [DOI] [PubMed] [Google Scholar]
- 26.Kubaski F, Yabe H, Suzuki Y, et al. Hematopoietic stem cell transplantation for patients with mucopolysaccharidosis II. Biol Blood Marrow Transplant. 2017;23:1795–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tolar J, Petryk A, Khan K, et al. Long-term metabolic, endocrine, and neuropsychological outcome of hematopoietic cell transplantation for Wolman disease. Bone Marrow Transplant. 2009;43:21–27. [DOI] [PubMed] [Google Scholar]
- 28.Bhatia S, Francisco L, Carter A, et al. Late mortality after allogeneic hematopoietic cell transplantation and functional status of long-term survivors: report from the Bone Marrow Transplant Survivor Study. Blood. 2007;110:3784–3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eapen M, Ahn KW, Orchard PJ, et al. Long-term survival and late deaths after hematopoietic cell transplantation for primary immunodeficiency diseases and inborn errors of metabolism. Biol Blood Marrow Transplant. 2012;18:1438–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodgers NJ, Kaizer AM, Miller WP, Rudser KD, Orchard PJ, Braunlin EA. Mortality after hematopoietic stem cell transplantation for severe mucopolysaccharidosis type I: the 30-year University of Minnesota experience. J Inherit Metab Dis. 2017;40:271–280. [DOI] [PubMed] [Google Scholar]
- 31.Shulman HM, Sullivan KM, Weiden PL, et al. Chronic graft-versus-host syndrome in man. A long-term clinicopathologic study of 20 Seattle patients. Am J Med. 1980;69:204–217. [DOI] [PubMed] [Google Scholar]
- 32.LexisNexis. Accurint. Available at: www.accurint.com. Accessed December 20, 2016.
- 33.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94:496–509. [Google Scholar]
- 34.Center for Disease Control and Prevention; 2017. https://wonder.cdc.gov/mortSQL.html,https://www.cdc.gov/nchs/ndi/index.htm. Accessed January 19, 2018.
- 35.Vandenbroucke JP. A shortcut method for calculating the 95 per cent confidence interval of the standardized mortality ratio. Am J Epidemiol. 1982;115:303–304. [Google Scholar]
- 36.Boelens JJ, Wynn RF, O’Meara A, et al. Outcomes of hematopoietic stem cell transplantation for Hurler’s syndrome in Europe: a risk factor analysis for graft failure. Bone Marrow Transplant. 2007;40:225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilkinson FL, Sergijenko A, Langford-Smith KJ, Malinowska M, Wynn RF, Bigger BW. Busulfan conditioning enhances engraftment of hematopoietic donor-derived cells in the brain compared with irradiation. Mol Ther. 2013;21:868–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beam D, Poe MD, Provenzale JM, et al. Outcomes of unrelated umbilical cord blood transplantation for X-linked adrenoleukodystrophy. Biol Blood Marrow Transplant. 2007;13:665–674. [DOI] [PubMed] [Google Scholar]
- 39.Boucher AA, Miller W, Shanley R, et al. Long-term outcomes after allogeneic hematopoietic stem cell transplantation for metachromatic leukodystrophy: the largest single-institution cohort report. Orphanet J Rare Dis. 2015;10:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin HR, Poe MD, Provenzale JM, Kurtzberg J, Mendizabal A, Escolar ML. Neurodevelopmental outcomes of umbilical cord blood transplantation in metachromatic leukodystrophy. Biol Blood Marrow Transplant. 2013;19:616–624. [DOI] [PubMed] [Google Scholar]
- 41.Smith Sehdev AE, Hutchins GM. Problems with proper completion and accuracy of the cause-of-death statement. Arch Intern Med. 2001;161:277–284. [DOI] [PubMed] [Google Scholar]
- 42.de Ru MH, Boelens JJ, Das AM, et al. Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: results of a European consensus procedure. Orphanet J Rare Dis. 2011;6:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kakkis ED, Muenzer J, Tiller GE, et al. Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med. 2001;344:182–188. [DOI] [PubMed] [Google Scholar]
- 44.Loes DJ, Hite S, Moser H, et al. Adrenoleukodystrophy: a scoring method for brain MR observations. AJNR Am J Neuroradiol. 1994;15:1761–1766. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.