

Abstract
Background
Programmed death-ligand 1 (PD-L1) is upregulated in glioblastoma and supports immunosuppression. We evaluated PD-L1 blockade with durvalumab among glioblastoma cohorts and investigated potential biomarkers.
Methods
MGMT unmethylated newly diagnosed patients received radiotherapy plus durvalumab (cohort A; n=40). Bevacizumab-naïve, recurrent patients received durvalumab alone (cohort B; n=31), or in combination with standard bevacizumab (cohort B2; n=33), or low-dose bevacizumab (cohort B3; n=33). Bevacizumab-refractory patients received durvalumab plus bevacizumab (cohort C; n=22). Primary endpoints were: OS-12 (A); PFS-6 (B, B2, B3); and OS-6 (C). Exploratory biomarkers included: a systematic, quantitative and phenotypic evaluation of circulating immune cells; tumor mutational burden (TMB); and tumor immune activation signature (IAS).
Results
No cohort achieved the primary efficacy endpoint. Outcome was comparable among recurrent, bevacizumab-naive cohorts. No unexpected toxicities were observed. A widespread reduction of effector immune cell subsets was noted among recurrent patients compared to newly diagnosed that was partially due to dexamethasone use. A trend of increased CD8+Ki67+ T cells at day 15 was noted among patients who achieved the primary endpoint and were not on dexamethasone. Neither TMB nor IAS predicted outcome.
Conclusion
Recurrent glioblastoma patients have markedly lower baseline levels of multiple circulating immune cell subsets compared to newly diagnosed patients. An early increase in systemic Ki67+CD8+ cells may warrant further evaluation as a potential biomarker of therapeutic benefit among glioblastoma patients undergoing checkpoint therapy. Dexamethasone decreased immune cell subsets. PD-L1 blockade and combination with standard or reduced dose bevacizumab was ineffective.
Keywords: PD-L1, glioblastoma, PD-1, VEGF, bevacizumab, biomarker
Introduction
PD-L1, which is upregulated by glioblastoma tumor cells and infiltrating myeloid cells, contributes to tumor mediated immunosuppression.1 Although PD-1 receptor blockade in glioblastoma has failed to improve OS among recurrent2 and newly diagnosed patients (BMS press releases), the role of PD-L1 blockade has not been effectively studied.3,4
We performed a multicenter, open-label phase 2 study to evaluate the safety and efficacy of durvalumab (MEDI4736), a selective, high-affinity human IgG1 monoclonal antibody that blocks PD-L1 but not PD-L2 binding to PD-1 and CD80 among separate cohorts of newly diagnosed and recurrent glioblastoma patients. Immunocorrelative biomarkers included a detailed quantitative and functional evaluation of circulating immune effector cell subsets as well as tumor mutational burden (TMB) and immune activation mRNA signature. Prospective evaluation of neurologic function was performed using the Neurologic Assessment in Neuro-Oncology (NANO) scale.5
The initial study design included three independent cohorts of glioblastoma patients which were enrolled concurrently. Each cohort included a different population of glioblastoma patients and had its own sample size considerations. For newly-diagnosed patients, durvalumab was administered during and following conventional radiation therapy without temozolomide because temozolomide has nominal benefit in MGMT unmethylated patients.6 Initially there were two cohorts of recurrent glioblastoma patients including those who were either bevacizumab-naïve or bevacizumab-refractory. The bevacizumab-naïve cohort received durvalumab monotherapy while the refractory cohort was treated with durvalumab plus bevacizumab continuation. Bevacizumab was continued in the latter cohort based on its anti-permeability effect as a strategy to mitigate rebound worsening of cerebral edema if discontinued abruptly. While ongoing, the study was amended to include two additional bevacizumab-naïve, recurrent cohorts who received durvalumab plus bevacizumab. They were added to test the hypothesis of whether VEGF blockade enhances the anti-tumor activity of PD-1/PD-L1 blockade based on promising preclinical7 and clinical data; the latter leading to FDA approval of five such combinatorial regimens for solid tumor indications.8–11 One added cohort received standard bevacizumab dosing while the other received a reduced bevacizumab dosing schedule. Reduced versus standard bevacizumab dosing was investigated based on preclinical data demonstrating a greater additive effect with reduced-dose VEGF inhibitor therapy.12,13 In addition, a retrospective review of 219 glioblastoma patients treated with bevacizumab showed that patients treated with lower dose intensity (< 5 mg/kg per week) of bevacizumab had longer PFS and OS when compared with those treated with standard 10 mg/kg biweekly dosing.14
PATIENTS AND METHODS
Study design and participants
This multinational, open-label, phase 2 study (ClinicalTrials.gov identifier: NCT02336165) initially enrolled adults with histologically confirmed glioblastoma to one of three separate cohorts that accrued independently including newly diagnosed, untreated patients with an unmethylated MGMT promotor (cohort A, n=40), and recurrent patients who were either bevacizumab naïve (cohort B, n=31) or bevacizumab refractory (cohort C, n= 22). Following completion of accrual to cohort B, two additional cohorts of bevacizumab-naïve, recurrent patients were added who received durvalumab with bevacizumab at either standard (B2, n=33) or reduced dosing (B3, n=33). These cohorts were open at the same time and patients were randomly assigned into either of these two cohorts at a 1:1 ratio using a randomization list.
Key eligibility criteria for all patients included: ≥ 18 years of age; an ECOG ≤ 1; adequate organ function; and ≤4 mg/day of dexamethasone. Patients with IDH mutant tumors were allowed to enroll. Although the presence of an IDH mutation now precludes a diagnosis of glioblastoma,15 at the time this study was designed and conducted IDH mutant glioblastoma was a recognized entity although such patients were known to have a better outcome than IDH wild-type patients.16 Remaining eligibility criteria are detailed in Supplemental Materials.
The study was compliant with the Declaration of Helsinki and guidelines on Good Clinical Practice. Ethics approval was obtained at all participating centers and all patients provided informed written consent prior to participation.
Study Procedures
All patients received durvalumab at 10 mg/kg biweekly until progression, unacceptable toxicity or consent withdrawal. For cohort A, durvalumab began with the initiation of standard radiation therapy (2 Gy/fraction daily for 30 fractions).
Durvalumab was administered as monotherapy for cohort B and with bevacizumab administered at standard (10 mg/kg biweekly) or reduced (3 mg/kg biweekly) dosing for patients randomized to cohorts B2 or B3, respectively. For cohort C, durvalumab was administered with bevacizumab continuation (10 mg/kg biweekly).
Toxicity was graded using Common Terminology Criteria for Adverse Events version 4.03 and investigator assessed response was performed every eight weeks using the Radiologic Assessment in Neuro-Oncology (RANO) criteria.17 Clinically stable patients with progression on MRI could continue study therapy pending progression confirmation on follow-up imaging as per the Immunotherapy Response Assessment Criteria in Neuro-Oncology (iRANO) criteria.18 Patient neurologic function was assessed at baseline and at MRI assessments using the NANO scale.5
Biomarker Analyses
All biomarker analyses were performed at an AstraZeneca affiliated laboratory. Analysis of TMB and tumor immune activation mRNA signature were assessed from paraffin-embedded tumor material as previously described.19 Briefly, tumor genomic variants were called using VarDict-Java (version 1.7.0), annotated with SnpEff (version 4.3.1t) and selected using the application of custom variant filters with a minimum depth of 50 reads. The variants were put through additional germline filtering using dbSNP, gnomAD and COSMIC and only somatic, non-synonymous and exonic mutations were used to evaluate tumor mutational burden (TMB). TMB was calculated by a custom program where the number of mutations were counted over the total of whole exome regions covered by more than 50 reads.
Peripheral blood populations before and after treatment, were evaluated for any associations with treatment and patient responses. Four bioanalytically-validated flow cytometry-based assays were implemented to quantify T, B or NK cells, regulatory T cells, proliferating T cell subsets, and naïve/memory or activated T cells. In brief, whole blood collected in optimal blood collections tubes were shipped overnight to local testing laboratories (Q2 Solutions, Inc.). Upon sample receipt, optimum quantities of fluorochrome-labelled monoclonal antibodies were added to 50 or 100 μl of whole blood, dependent on assay, and incubated for indicated time prior to erythrocyte lysis.20 Samples were analyzed on BD FACSCanto™ II flow cytometers with 405, 488 and 633 nm lasers running a standard 4–2-2 optical configuration. Lymphocyte populations were gated based on results from isotypic, isoclonal or fluorescence-minus-one treatments. Absolute count values for T cell subpopulations were calculated by multiplying population percentage values by absolute count values from the total CD4+ or CD8+ T cell quantities derived from the T, B and NK cell assay.
Outcomes
The primary endpoint for each cohort was based on the intent-to-treat population and included overall survival at 12 months (OS-12) for cohort A, progression-free survival at 6 months (PFS-6) for cohorts B, B2 and B3, and OS at 6 months (OS-6) for cohort C. Secondary endpoints were overall response rate (ORR), median PFS and OS as well as safety. Exploratory endpoints included association of outcome with TMB and immune activation mRNA signature, circulating immune effector cell levels and functional status as well as changes in NANO scale.5 Data cut-offs for overall survival and other analyses were 20Nov2019 and 06Sep2019, respectively.
Statistical Analysis
In the study protocol, a primary efficacy endpoint based of OS or PFS was identified for each cohort and pre-specified testing was conducted using a selected alpha level. After completion of the clinical study report, multivariable analysis of the association of baseline factors and OS was performed as exploratory analysis. The hypothesized associations were documented in a brief statistical analysis plan (SAP) which was signed off prior to conducting these exploratory analyses. P-values were considered descriptive in nature and were not adjusted for multiplicity. The exploratory analyses presented to describe the characteristics of the NANO assessment and to examine the associations between NANO scores and clinical outcomes were not pre-specified in the SAP. All p-values from the NANO analysis were also considered descriptive.
Samples size consideration for cohort A was based on the EORTC 26981/NCIC CE3 study.6 Thirty-seven patients were required to detect a 20% increase in OS at 12 months from 50% (historical benchmark) to 70% using a one-sided binomial test (based on normal approximation) at 0.05 significance level with 80% statistical power. Sample size consideration for cohort B was based on three large meta-analyses of clinical trials performed among recurrent glioblastoma patients.21–23 Thirty patients were required to detect a 20% increase in PFS-6 from 10% (historical benchmark) to 30% using a one-sided binomial test (based on normal approximation) at 0.05 significance level with 90% statistical power. Sample size considerations for cohorts B2 and B3 were based on the BRAIN study24 and were done separately for each cohort. Thirty-two patients in each cohort were required to detect a 20% increase in PFS-6 from 42% (historical benchmark) to 62% using a one-sided binomial test (based on normal approximation) at 0.10 significance level with 84% power. Sample size consideration for cohort C was based on several published studies.25–28 Seventeen patients were required to detect 30% increase in OS-6 from 35% (historical benchmark) to 65% using a one-sided binomial test (based on normal approximation) at 0.10 significance level with 90% power.
Time to event analyses for OS and PFS used the Kaplan-Meier method from the time of initial glioblastoma diagnosis for cohort A and from the start of study therapy for cohorts B, B2, B3 and C. Patients who withdrew from the study or were lost to follow-up (survival status unknown) were censored at the date of last contact. Median follow-up was determined using the reverse Kaplan-Meier method. Progression was defined by iRANO criteria18 or date of death due to any cause. Patients without documentation of progression or death during the on-study period were censored at the date of the last disease assessment while on study or date of start of alternate therapy, whichever came first. Patients with no disease assessment were censored at the start date of the treatment. Patients who discontinued treatment or withdrew from the study for other than documented progressive disease or death were censored at the date of last response assessment prior to discontinuation or withdrawal. ORR was defined as the percentage of patients meeting criteria of complete or partial response that was confirmed at least 4 weeks later as per RANO.17 Median durability of ORR was determined as the median duration from start of complete response or partial response to progression/death or last disease assessment.
After completion of the primary analyses for this study, additional explorative multivariable analyses of associations between baseline patient characteristics and overall survival were performed. The association between baseline factors and OS was assessed using multivariable Cox proportional hazard models for cohorts A and B/B2/B3. Associations between baseline predictor variables were examined using Pearson’s correlation coefficient, Fisher’s exact test, or t-test. P-values were considered descriptive in nature and were not adjusted for multiplicity. Frequencies and percentages were calculated for categories of baseline characteristics. Further detail on these analyses is provided in the Supplemental Materials.
Comparisons of baseline or day 15 immune cell counts (cells/mm3) or percent changes from baseline between cohorts or steroid-treatment groups were performed by Mann-Whitney U tests (α=0.01). The Wilcoxon signed-rank test was used to assess elevation magnitudes of baseline-normalized cell populations on day 15 (hypothetical median value of 0, α=0.01).
Descriptive summaries of NANO assessments were calculated including the number and frequency of completed baseline NANO and end of treatment NANO measures. Further detail on NANO assessment parameters and their association with outcome is summarized in the Supplemental Materials. The data generated in this study are available within the article and its supplementary data files. Any additional data are available from the corresponding author upon request.
RESULTS
Patient Characteristics and Disposition
The study enrolled 162 patients between March 2015 and January 2017, and 159 were treated (Table 1; Supplemental Figure 1). Study therapy has discontinued among 155 patients (97.5%) due primarily to progressive disease (79%), while 14 (8.8%) patients discontinued due to toxicity. One hundred forty-five (145) patients (91.2%) have died and 14 (8.8%) were alive at date of last contact including 4 (2.5%) who continued study therapy.
Table 1.
Patient Characteristics at Baseline and Study Dispositiona
| Characteristic | Cohort A (n=40) | Cohort B (n=31) | Cohort B2 (n=33) | Cohort B3 (n=33) | Cohort C (n=22) | TOTAL (n=159) |
|---|---|---|---|---|---|---|
| Median age, years (range) | 57.0 (22 to 77) | 54.0 (24 to 77) | 57.0 (40 to 74) | 54.0 (23 to 73) | 56.5 (37 to 77) | 56.0 (22 to 77) |
| < 60 years, n (%) | 22 (55.0) | 18 (58.1) | 22 (66.7) | 22 (66.7) | 15 (68.2) | 99 (62.3) |
| ≥ 60 years, n (%) | 18 (45.0) | 13 (41.9) | 11 (33.3) | 11 (33.3) | 7 (31.8) | 60 (37.7) |
| Gender, n (%) | ||||||
| Male | 28 (70.0) | 26 (83.9) | 18 (54.5) | 20 (60.6) | 14 (63.6) | 106 (66.7) |
| Female | 12 (30.0) | 5 (16.1) | 15 (45.5) | 13 (39.4) | 8 (36.4) | 53 (33.3) |
| ECOG Performance status, n (%) | ||||||
| 0 | 24 (60.0) | 16 (51.6) | 9 (27.3) | 10 (30.3) | 6 (27.3) | 65 (40.9) |
| 1 | 16 (40.0) | 15 (48.4) | 24 (72.7) | 23 (69.7) | 16 (72.7) | 94 (59.1) |
| # prior PD (%) | ||||||
| 0 | 40 (100.0) | 0 | 0 | 0 | 0 | 40 (25.2) |
| 1 | 0 | 25 (80.6) | 28 (84.8) | 25 (75.8) | 7 (31.8) | 85 (53.5) |
| 2 | 0 | 6 (19.4) | 5 (15.2 | 8 (24.2) | 15 (68.2) | 34 (21.4) |
| Resection prior to study (%) | ||||||
| Gross total | 8 (20.0) | 5 (16.1) | 3 (9.1) | 4 (12.1) | 1 (4.5) | 21 (13.2) |
| Subtotal | 29 (72.5) | 5 (16.1) | 8 (24.2) | 11 (33.3) | 2 (9.1) | 55 (34.6) |
| Biopsy | 3 (7.5) | 1 (3.2) | 0 | 1 (3.1) | 0 | 5 (3.1) |
| None | 0 | 20 (64.5) | 22 (66.7) | 17 (51.5) | 19 (86.4) | 78 (49.1) |
| Initial glioma diagnosis, n (%) | ||||||
| Grade II | 0 | 1 (3.2) | 0 | 1 (3.0) | 0 | 2 (1.3) |
| Grade III | 0 | 3 (9.7) | 1 (3.0) | 1 (3.0) | 0 | 5 (3.1) |
| Grade IV | 40 (100.0) | 27 (87.1) | 32 (97.0) | 31 (93.9) | 22 (100.0) | 152 (95.6) |
| Dexamethasone use at study entry, n (%) | 14 (35.0) | 11 (35.5) | 17 (51.5) | 17 (51.5) | 7 (31.8) | 66 (41.5) |
| Mean time from initial GBM diagnosis to enrollment, mean weeks (SD) | 4.7 (0.99) | 43.9 (28.31) | 71.0 (65.43) | 75.2 (60.42) | 80.3 (49.93) | 51.2 (54.47) |
| MGMT status, n (%) | ||||||
| Methylated | 0 | 9 (29.0) | 12 (36.4) | 12 (36.4) | 9 (40.9) | 42 (26.4) |
| Unmethylated | 40 (100.0) | 15 (48.4) | 18 (54.5) | 18 (54.5) | 11 (50.0) | 102 (64.2) |
| Unknown | 0 | 7 (22.6) | 3 (9.1) | 3 (9.1) | 2 (9.1) | 15 (9.4) |
| IDH1 Status (%) | ||||||
| Mutant | 5 (12.5) | 4 (12.9) | 4 (12.1) | 2 (6.1) | 1 (4.5) | 16 (10.1) |
| Wild-type | 35 (87.5) | 22 (71.0) | 28 (84.8) | 30 (90.9) | 19 (86.4) | 134 (84.3) |
| Unknown | 0 | 5 (16.1) | 1 (3.0) | 1 (3.0) | 2 (9.1) | 9 (5.7) |
| Tumor source for immunocorrelatives | ||||||
| Original diagnosis | 40 (100) | 22 (71.0) | 25 (75.8) | 26 (78.8) | 19 (86.4) | 132 (83.0) |
| Relapse | 0 | 6 (19.4) | 6 (18.2) | 7 (21.2) | 2 (9.1) | 21 (13.2) |
| Unknown | 0 | 3 (9.7) | 2 (6.1) | 0 | 1 (4.5) | 6 (3.8) |
| # study cycles completed, median (range) | 7.6 (1.2, 16.6) |
3.2 (0.8 to 37.0) |
4.1 (1.5 to 37.4) |
4.0 (2.0 to 17.9) |
2.1 (0.2 to 6.1) |
4.0 (0.2 to 37.4) |
| Reason off study, n (%) | ||||||
| PDb | 27 (67.5) | 26 (83.9) | 28 (84.8) | 24 (72.7) | 20 (90.9) | 125 (78.6) |
| Toxicityc | 4 (10.0) | 2 (6.5) | 2 (6.1) | 5 (15.2) | 1 (4.5) | 14 (8.8) |
| Elective withdrawal | 3 (7.5) | 2 (6.5) | 2 (6.1) | 4 (12.1) | 1 (4.5) | 12 (7.5) |
| Death (unrelated to tumor progression) | 3 (7.5) | 0 | 1 (3.0) | 0 | 0 | 4 (2.5) |
| Status at last contact, n (%) | ||||||
| Dead | 36 (90.0) | 25 (80.6) | 31 (93.9) | 31 (93.9) | 22 (100.0) | 145 (91.2) |
| Alive | 4 (10.0) | 6 (19.4) | 2 (6.1) | 2 (6.1) | 0 | 14 (8.8) |
| On study therapy | 3 (7.5) | 1 (3.2) | 0 | 0 | 0 | 4 (2.5) |
Percents are correct for denominators in each cell.
PD includes deaths attributed to progressive tumor
Includes events that occurred at the time of tumor progression but possible attribution to study therapy could not be excluded
Abbreviations: ECOG, Eastern Cooperative Oncology Group; GBM, glioblastoma; IDH1, isocitrate dehydrogenase 1; MGMT, methylguanine methyltransferase; PD, progressive disease; SD, standard deviation
Efficacy
Outcome per cohort is summarized in Figure 1. With a median follow-up for cohort A of 36.8 months, median OS, OS-12 and median PFS for all patients were 15.1 months, 60.0% and 4.6 months, respectively and for the IDH wild-type patients (n=35) were 12.8 months, 54.3%, 4.5 months, respectively. The median PFS and OS for the five IDH mutant patients were 14.7 months and 26.5 months, respectively and two of these patients remain alive without progression at 51.7 and 52.0 months. Median follow-up for cohorts B and B2 was over 36 months and non-evaluable for cohort B3, while the PFS-6 rates were 19.4%, 15.2% and 17.2 %, respectively. Median OS was modestly increased for cohorts B2 and B3 compared to cohort B, but the rate of survival at the 12-, 18-, and 24-month time points favored cohort B. Although the objective response rate (ORR) for cohorts B, B2 and B3 patients was low at 9–13%, the median durability of response per cohort was 9.0, 18.3 and 8.5 months, respectively. Patients in cohort C exhibited the poorest outcome and achieved an OS-6 of 36.4%.
Figure 1. Progression-Free and Overall Survival in All Patients.
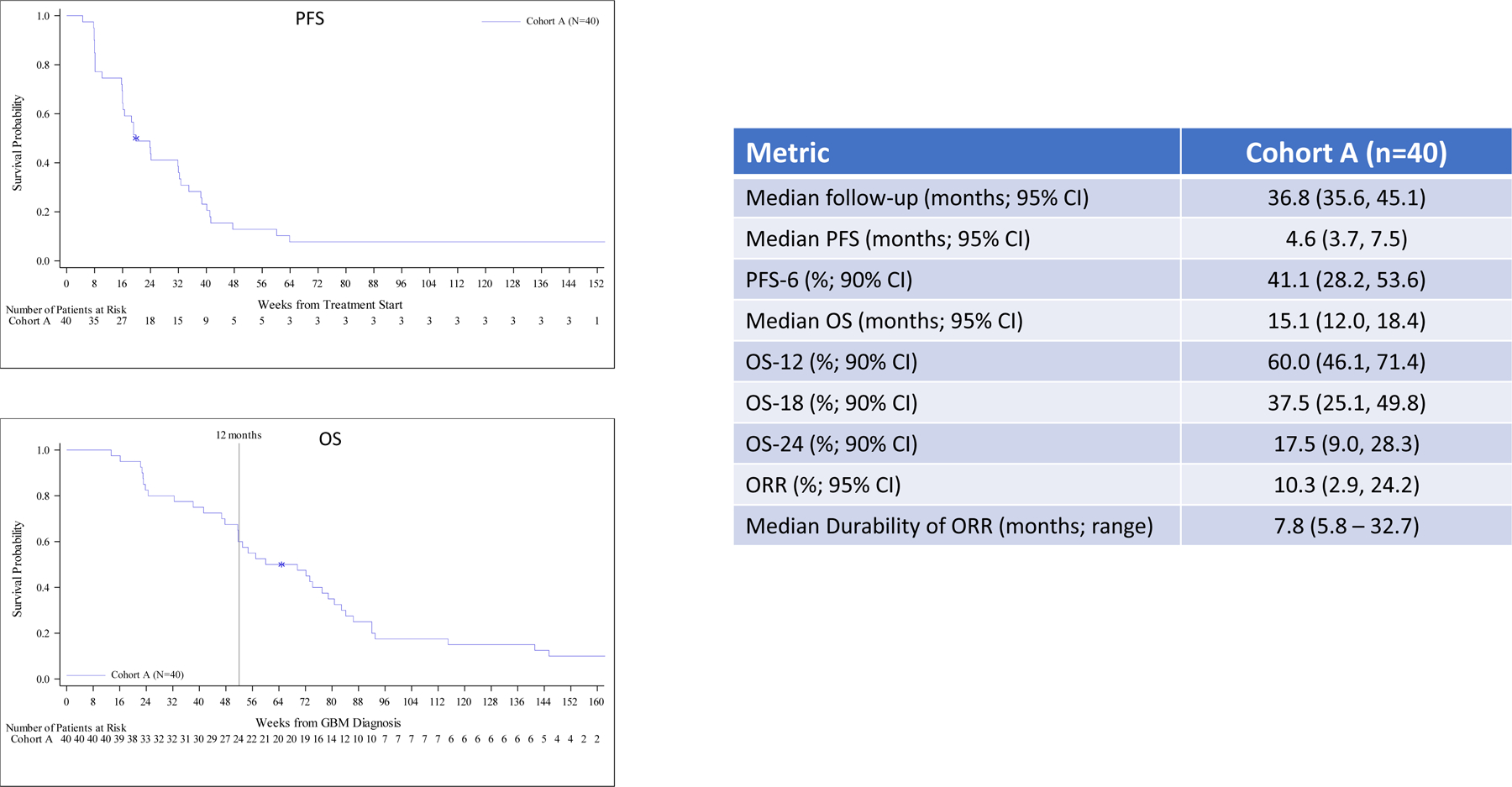
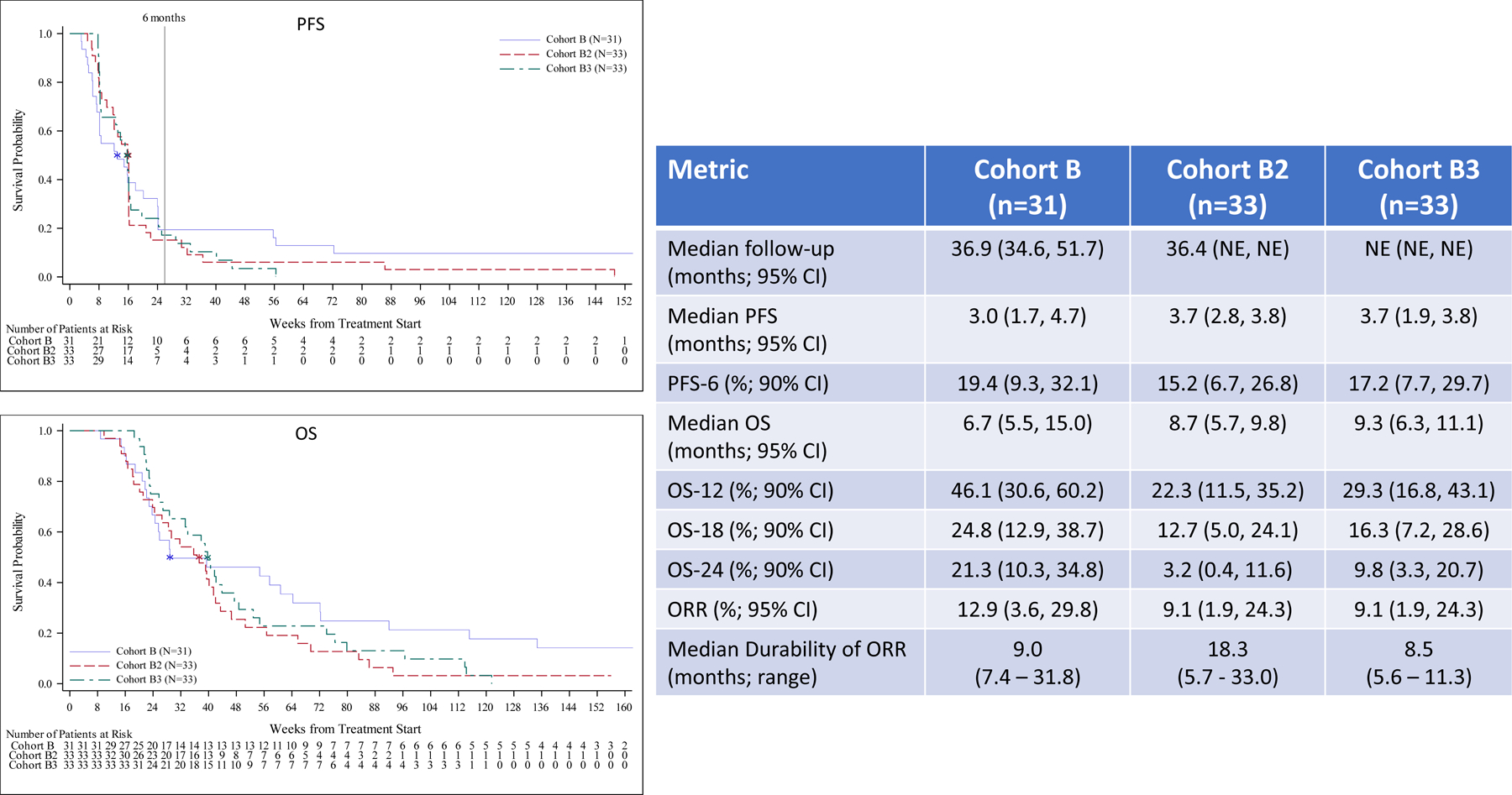
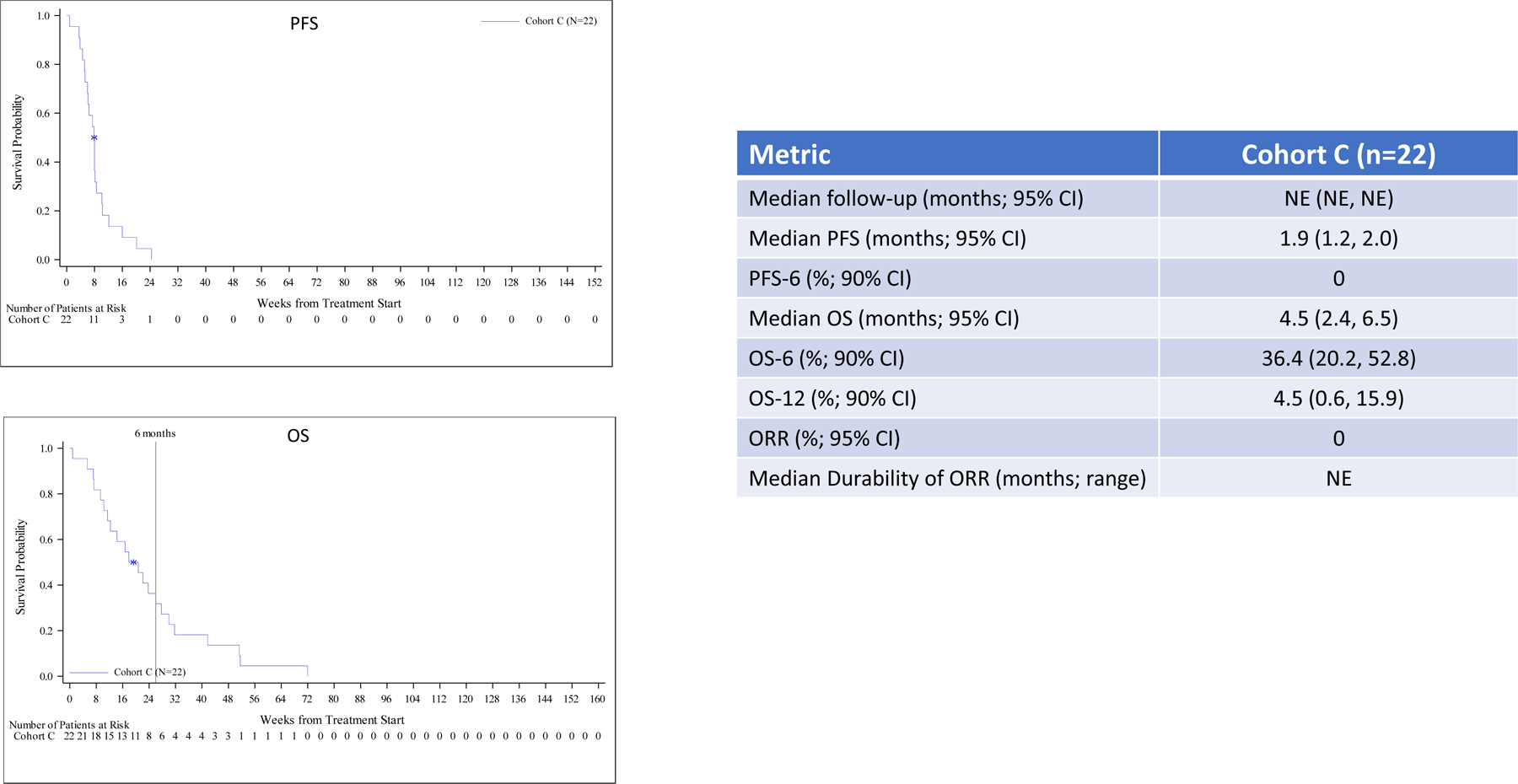
Efficacy for all patients including Kaplan-Meier plots and tabular PFS and OS data by cohort. Outcome for newly diagnosed, MGMT unmethylated patients (cohort A), bevacizumab naïve, recurrent patients (cohorts B, B2 and B3) and bevacizumab refractory, recurrent patients (cohort C) are summarized in panels A, B and C, respectively including Kaplan-Meier plots of PFS (upper) and OS (lower).
Analysis of the association between specified factors and OS for cohorts A and B/B2/B3 are summarized in Supplemental Tables 1A and 1B, respectively. For cohort A, univariate variables that associated with poorer OS were presence of measurable disease (hazard ratio [HR] = 2.82; p = 0.024) and ECOG 1 (HR = 2.13; p = 0.028) relative to ECOG 0, whereas IDH mutation (HR = 0.26; p = 0.027) was associated with improved OS. For cohort A, gender was not associated with survival while trends favoring improved survival were noted for age ( <60 vs. >/= 60 years old), lack of baseline steroid use and shorter time from glioblastoma diagnosis to study initiation. In a multivariable model for cohort A which adjusted for age at study baseline as a continuous variable, measurable disease at screening was associated with a higher rate of death (HR = 9.42; 95% CI = 2.98, 29.79; p = 0.0001) as was baseline ECOG status of 1 compared to ECOG status of 0 (HR = 2.81; 95% CI = 1.36, 5.83 p = 0.0054), while IDH mutant patients had a decreased rate of death compared to IDH wild-type patients (HR = 0.12; 95% CI = 0.02, 0.66; p=0.0147). Baseline predictors were not associated with each other, except for IDH mutation status and age (mean age = 60.6 years for IDH wild-type and mean age = 34.2 years for IDH mutant; t-test p-value = <0.0001) and baseline ECOG status and age (mean age = 61.3 years for ECOG status of 1 and mean age = 54.7 for ECOG status of 0; t-test p-value = 0.0620). Within the cohort A IDH wild-type subgroup (n=35), tumor burden at screening was associated with a higher rate of death (HR = 7.54; 95% CI = 2.36, 24.06; p = 0.0006), as was baseline ECOG status of 1 compared to 0 (HR = 2.65; 95% CI = 1.25, 5.61; p = 0.0108) after adjustment for age at study baseline as a continuous variable.
In a multivariable model for cohort B, IDH mutation was marginally associated with a lower rate of death (HR = 0.11; 95% CI = (0.01, 1.02; p = 0.0524) while a trend associated with worse survival that did not achieve statistical significance was noted for increased days from initial glioblastoma diagnosis to start of treatment (>365 days vs. <=365 days) (HR = 2.45; 95% CI = 0.90, 6.67; p = 0.0807), with age at study baseline as a continuous variable included in the model as an adjustment factor (HR = 1.01; 95% CI = 0.97, 1.05; p = 0.6295). Baseline dexamethasone use (HR 1.47; p = 0.3449) was marginally associated with worse survival (without adjustments for other covariates). In cohorts B2/B3, IDH mutation was associated with a higher rate of death (HR = 2.50; 95% CI = 1.05, 5.97; p = 0.0386). This unexpected finding may warrant further investigation in future studies.
Given the overall poor survival for cohort C, evaluation of potential factors associated with outcome was not performed.
Safety
There were no dose limiting toxicities observed during the safety lead-in for cohorts A or C. Table 2 summarizes treatment-related adverse events (TRAEs) grade ≥ 2 that occurred in at least 10% of patients per cohort. Most TRAEs were low-grade including immune related AEs. Fatigue was the most common grade ≥ 2 TRAE, affecting 29 (18.2%) patients across all cohorts, and was grade 2 in all but 1 patient who had grade 3 fatigue. Two patients experienced grade 4 lipase elevation which was asymptomatic and reversible. Other grade 4 TRAEs occurred in single patients and included ALT elevation, altered cognition, and lymphopenia. One potentially treatment-related grade 5 event occurred; this event was an intracranial hemorrhage in a cohort C patient that occurred at tumor progression. On study deaths that were felt to be unlikely related to either study therapy or underlying tumor progression included single patients in cohort A who accidentally drowned or suffered a cardiac arrest. Two patients died after protracted seizures. Overall, 14 patients (8.8%) discontinued study therapy due to adverse events (Supplemental Table 2), including 9 patients (5.7%) with adverse events that were at least possibly related to study therapy.
Table 2.
Grade ≥ 2 treatment related adverse events in ≥ 10% patients by cohort
| Cohort | A (n=40) | B (n=31) | B2 (n=33) | B3 (n=33) | C (n=22) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Grade | 2 | 3 | 4 | 2 | 3 | 4 | 2 | 3 | 4 | 2 | 3 | 4 | 2 | 3 | 4 | |
| AE (n, %) a | ||||||||||||||||
| Amylase elevation | 4 (10.0) | 2 (5.0) | 0 | 0 | 0 | 0 | 0 | 2 (6.1) | 0 | 0 | 1 (3.0) | 0 | 0 | 0 | 0 | |
| Fatigue | 5 (12.5) | 0 | 0 | 6 (19.4) | 0 | 0 | 6 (18.2) | 0 | 0 | 9 (27.3) | 0 | 0 | 2 (9.1) | 1 (4.5) | 0 | |
| Headache | 4 (10.0) | 0 | 0 | 1 (3.2) | 1 (3.2) | 0 | 0 | 0 | 0 | 1 (3.0) | 0 | 0 | 1 (4.5) | 0 | 0 | |
| Hemiparesis | 2 (5.0) | 0 | 0 | 4 (12.9) | 0 | 0 | 0 | 0 | 0 | 1 (3.0) | 0 | 0 | 0 | 0 | 0 | |
| Hypertension | 0 | 0 | 0 | 1 (3.2) | 0 | 0 | 1 (3.0) | 4 (12.1) | 0 | 2 (6.1) | 0 | 0 | 1 (4.5) | 0 | 0 | |
| Lipase elevation | 3 (7.5) | 5 (12.5) | 1 (2.5) | 0 | 0 | 1 (3.2) | 1 (3.0) | 0 | 0 | 0 | 3 (9.1) | 0 | 0 | 0 | 0 | |
Percents are per cohort
Peripheral Blood Immune Cell Populations
Analysis of circulating lymphocytes from fresh, whole blood specimens at baseline revealed that recurrent patients (cohorts B, B2 and B3) had lower levels of total CD3+, CD4+ and CD8+ T cells, CD19+ B cells and CD16+/56+ NK cells compared to newly diagnosed patients (cohort A; Figure 2A; p<0.01 by Mann Whitney U test). The deficit in CD4+ T cells among recurrent patients reflected global reductions in most subsets including naïve as well as central and effector memory cell populations (Figure 2B). Similarly, naïve CD8+ T cells were also significantly reduced among recurrent patients (Figure 2B). In general, activated CD4+ and CD8+ T cell levels, as defined by CD38+, HLA-DR+ or ICOS+ expression, trended lower among recurrent patients including CD4+ICOS+ cells which achieved statistical significance (Figure 2C). Proliferating (Ki67+) CD4+ were also lower among recurrent patients (Figure 2D).
Figure 2. Circulating Immune Cell Subsets Comparing Newly Diagnosed and Recurrent Patients.
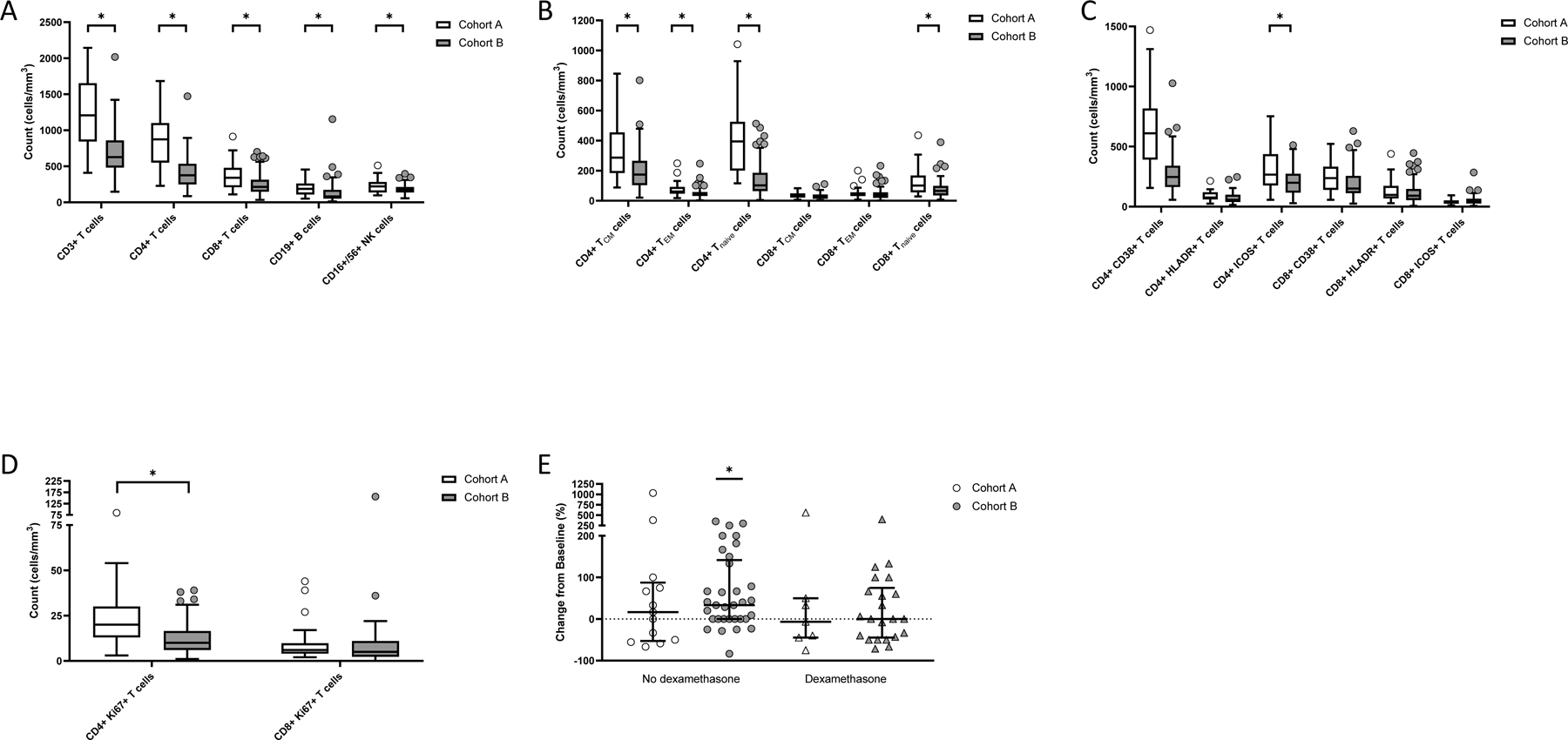
Measurement of baseline immune cell subsets detected by flow cytometry among newly diagnosed patients (cohort A; open bars and symbols) compared to bevacizumab-naïve, recurrent patients (cohorts B, B2 and B3; gray bars and symbols) including major immune cell subsets (panel A), and CD4+ and CD8+ T cells distinguished by memory or naïve (panel B), activation (panel C) or proliferation (panel D) markers. Asterisks indicate p<0.01 by Mann-Whitney U test comparing cohort A to cohorts B, B2 and B3. Panel E: Change in baseline CD8+Ki67+ T cells at day 15 for newly diagnosed (cohort A, open symbols) and bevacizumab-naïve, recurrent patients (cohorts B, B2 and B3, gray symbols) on baseline dexamethasone use (right side triangles) compared to those not on dexamethasone (left side circles). Bars indicate median value and error bars denote 25th and 75th percentiles. Asterisk indicates p ≤ 0.01. P value was determined by Wilcoxon Signed Rank test.
Patients were stratified based on baseline dexamethasone use to assess the contribution of corticosteroid use to lymphocyte levels. Overall, patients on dexamethasone at baseline had lower median lymphocyte counts compared to patients not on dexamethasone (Table 3). Moreover, recurrent patients on dexamethasone exhibited more profound reductions in multiple populations compared to newly diagnosed patients; nonetheless, dexamethasone treatment did not solely account for the observed cellular deficits in recurrent patients.
Table 3.
Immune Cell Subsets By Cohort and Stratified by Baseline Dexamethasone (Dex) Use
| Cohort | A | B/B2/B3 | |||||
|---|---|---|---|---|---|---|---|
| Baseline Dex | Yes | No | P value | Yes | No | P value | |
| Lymphocyte Subset | |||||||
| CD3+ | # Patients | 11 | 21 | 39 | 50 | ||
| Median | 1004 | 1240 | NS | 523 | 696 | 0.0178 | |
| CD4+ | # Patients | 11 | 21 | 39 | 50 | ||
| Median | 762 | 910 | NS | 323 | 417 | 0.01404 | |
| CD8+ | # Patients | 11 | 21 | 39 | 50 | ||
| Median | 344 | 336 | NS | 192 | 238 | NS | |
| CD4+ Central Memory | # Patients | 9 | 20 | 36 | 45 | ||
| Median | 197 | 291 | NS | 152 | 199 | 0.0465 | |
| CD4+ Effector Memory | # Patients | 9 | 20 | 36 | 45 | ||
| Median | 72 | 61 | NS | 39 | 43 | NS | |
| CD4+ Naïve | # Patients | 9 | 20 | 36 | 45 | ||
| Median | 281 | 440 | NS | 102 | 103 | NS | |
| CD4+CD38+ | # Patients | 9 | 20 | 36 | 45 | ||
| Median | 459 | 703 | NS | 211 | 273 | 0.0235 | |
| CD4+HLADR+ | # Patients | 9 | 20 | 35 | 45 | ||
| Median | 73 | 85 | NS | 53 | 68 | 0.0130 | |
| CD4+ICOS+ | # Patients | 9 | 20 | 35 | 45 | ||
| Median | 209 | 324 | 0.0477 | 131 | 212 | 0.0015 | |
| CD8+ Central Memory | # Patients | 9 | 20 | 36 | 45 | ||
| Median | 27 | 32 | NS | 21 | 28 | NS | |
| CD8+ Effector Memory | # Patients | 9 | 20 | 36 | 45 | ||
| Median | 33 | 43 | NS | 33 | 35 | NS | |
| CD8+ Naive | # Patients | 9 | 20 | 36 | 45 | ||
| Median | 96 | 125 | NS | 49 | 73 | NS | |
| CD8+ CD38+ | # Patients | 9 | 20 | 35 | 45 | ||
| Median | 168 | 243 | NS | 129 | 154 | NS | |
| CD8+ HLADR+ | # Patients | 9 | 20 | 35 | 45 | ||
| Median | 152 | 97 | NS | 81 | 105 | NS | |
| CD8+ ICOS+ | # Patients | 9 | 20 | 35 | 45 | ||
| Median | 35 | 41 | NS | 35 | 48 | 0.0151 | |
| CD4+ Ki67+ | # Patients | 8 | 19 | 35 | 43 | ||
| Median | 13 | 24 | 0.0407 | 8 | 12 | 0.0147 | |
| CD8+Ki67+ | # Patients | 8 | 18 | 35 | 42 | ||
| Median | 7 | 6 | NS | 5 | 7 | NS | |
| Day 15 CD4+Ki67+ | # Patients | 7 | 14 | 24 | 36 | ||
| Median | Neg 31 | Neg 12 | NS | 24 | 23 | NS | |
| Day 15 CD8+Ki67+ | # Patients | 7 | 13 | 22 | 33 | ||
| Median | Neg 7 | 17 | NS | 0 | 33 | 0.0703 | |
Abbreviations: Dex, dexamethasone; Neg, negative; NS not significant
We then sought to determine if absolute levels of subclasses of immune effector cells at baseline or day 15 correlated with therapeutic benefit. We observed no association between absolute levels of circulating immune cell subsets at either baseline or at day 15 and OS-12 for cohort A or PFS-6 for cohorts B/B2/B3 (Supplemental Table 3; p>0.05 for all comparisons), including upon stratification for baseline dexamethasone use (data not shown). However, for newly-diagnosed patients on dexamethasone, those who achieved OS-12 had a non-significant trend of higher baseline median CD8+ T cell counts compared to patients who did not achieve OS-12, that included subsets expressing HLA-DR, CD38 or naïve cell markers as well as higher median baseline CD4+ICOS+ T cells (Supplemental Figure 2). These findings require cautious interpretation due to small sample size, wide confidence intervals and lack of a study control arm but suggest that therapeutic benefit may require higher levels of baseline T cell subsets, including those expressing markers of activation, to overcome the suppressive activity of concurrent dexamethasone use among glioblastoma patients undergoing immunotherapy.
It has been previously reported that durvalumab induces an average 50–100% increase above baseline in circulating proliferating (Ki67+) CD8+ T cells within 10–15 days following the first dose.29,30 We assessed the elevation magnitudes of this cell population and observed that newly diagnosed and recurrent patients who were not on dexamethasone demonstrated an increase in baseline-normalized CD8+Ki67+ T cells on day 15 that achieved statistical significance among recurrent patients (p<0.01 by Wilcoxon Signed Rank test). In contrast, no day 15 increase was observed among patients who were on baseline dexamethasone (Figure 2E). We then explored the association of changes in baseline-normalized CD8+Ki67+ T cells on day 15 with clinical outcome. As baseline dexamethasone use affected lymphocyte quantities, we stratified patients based on dexamethasone use. Among patients not on dexamethasone, a two-fold or greater median increase in day 15 CD8+Ki67+ T cells was noted among newly diagnosed patients who achieved OS-12 and among recurrent patients who achieved PFS-6 compared to patients who did not achieve these endpoints (Table 4). In contrast, the day 15 CD8+Ki67 T cell count showed little to no increase compared to day 0 for newly diagnosed and recurrent patients who were on dexamethasone (data not shown). These analyses also require cautious interpretation and validation but suggest that an early on-treatment increase in CD8+Ki67+ T cells may indicate a higher likelihood of therapeutic benefit to immune checkpoint blockade and that concurrent dexamethasone may be detrimental to this increase.
Table 4.
Percent change in baseline-normalized CD8+Ki67+ T cells among patients not on dexamethasone.
| Cohort | A | B | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Endpoint | <OS12 | ≥OS12 | <PFS6 | ≥PFS6 | ||||||
| n | Median (Q1, Q3) | n | Median (Q1, Q3) | p | n | Median (Q1, Q3) | n | Median (Q1, Q3) | p | |
| % change CD8+Ki67+ | 5 | −50 (−55.5, −33.3) | 8 | 50 (12.5, 81.3) | 0.22 | 25 | 33.33 (0, 66.7) | 8 | 83.3 (−23.6, 186.4) | 0.98 |
Tumor Immune Biomarker Analyses
Analyzed tumor samples for cohort A patients were collected immediately prior to study enrollment while those from cohort B were primarily archival given that only 35% of these patients had surgery after recurrence and prior to initiation of study therapy. These analyses were not performed for cohorts B2 and B3 due to expected similar findings relative to cohort B. As summarized in Supplemental Table 4, TMB was quite low (< 2.6/Mb) among cohort A and B patients. For cohort B, IAS was also low. Insufficient tumor material prevented analysis for cohort A. There was no apparent association of TMB with PFS or OS for either cohort A or B nor an apparent association of IAS with PFS or OS for cohort B (Supplemental Figure 3).
Neurologic Assessment in Neuro-Oncology (NANO)
NANO compliance rate at baseline and across all visits were 91% (145 of 159 treated patients) and 60% (462 out of 767), respectively. Fourteen patients (9%) lacked a baseline NANO evaluation and one was deemed non-evaluable; these patients were excluded leaving 144 patients for NANO analyses. One hundred and seven patients (67%) lacked an end of treatment NANO evaluation. At baseline, 62 patients (43%) had a normal NANO neurologic examination. Gait, strength and language were the domains that accounted for the most common changes in neurologic function during the study. Nine patients were assessed as progressive by NANO criteria prior to radiographic progression which was established on subsequent MRI evaluations. Three patients had neurologic response by NANO criteria and concurrent stable imaging findings.
In order to further assess the utility of NANO, we assessed its score relative to other measures of clinical status and outcome at specified time points. First, median and overall survival for those with a normal baseline NANO score was better than those with an abnormal score (11.1 vs 6.7 months, log-rank p=0.0014). Second, at a 2-month landmark, patients with NANO progression had shorter median OS compared to those without NANO progression (3.2 vs 7.7 months, log-rank p=0.04) and shorter median PFS (1.3 vs 1.8 months, log-rank p=0.0022) in cohort B, B2 and B3. Third, a decline in ECOG performance status was associated with NANO progression (p=0.0427) among all patients at the 2-month time point. These data demonstrate that NANO score associates with other measures of outcome among glioblastoma patients.
DISCUSSION
In this multi-arm study, blockade of PD-L1 with durvalumab did not improve outcome among newly diagnosed or recurrent glioblastoma patients relative to appropriate historical benchmarks. Similarly negative results have been previously reported following PD-1 blockade (BMS press releases),2 providing further evidence that solely targeting the PD(L)-1 axis is insufficient to benefit glioblastoma patients. In our study, for newly diagnosed patients with MGMT unmethylated tumors, median survival (15.1 months), OS-12 (60%) and OS-24 (17.5%) were similar to standard of care temozolomide chemoradiotherapy.6,31 As expected from the literature, improved survival was noted among patients without measurable disease, normal performance status and IDH1 mutation, respectively. Among recurrent patients, single agent durvalumab achieved a PFS-6 of 19.4% and a median OS of 6.7 months which are similar to outcomes from meta-analyses of non-immunotherapy, salvage therapy trials,22 as well as phase 3 studies evaluating lomustine or bevacizumab at recurrence.2,32–34
We also noted that the combination of durvalumab with bevacizumab, administered at either standard or reduced dosing schedule, did not appear to improve outcome compared to durvalumab monotherapy or historical benchmarks of bevacizumab monotherapy.33 Our study findings indicate that combinatorial benefit of VEGF blockade to immune checkpoint therapy observed in some cancers is not applicable to glioblastoma. Our data also do not support preclinical data in non-glioblastoma models demonstrating that reduced dose anti-angiogenic therapy can enhance the therapeutic benefit of immunotherapy,12,13 however the reduced bevacizumab dosing schedule we utilized was empirically chosen and may not have induced relative vascular normalization.
Although only 9–13% of bevacizumab-naïve, recurrent patients achieved an ORR, the median duration of response was 9–18 months per cohort and surpassed that of bevacizumab monotherapy which is typically 4–6 months.2 A similar durability of ORR was reported with PD-1 blockade with or without bevacizumab in two recent studies among recurrent glioblastoma patients.2,35
Bevacizumab refractory recurrent glioblastoma patients have a particularly poor outcome with subsequent salvage therapies and such patients on our trial treated with durvalumab plus bevacizumab continuation achieved similarly poor PFS and OS outcomes.36
Administration of durvalumab was well tolerated across study cohorts. The spectrum, severity and frequency of immune-related adverse events was comparable to data reported from anti-PD-1 studies for glioblastoma patients2 and other cancer subtypes. There were also no unexpected safety issues observed for the combination of durvalumab with radiation therapy for newly diagnosed patients and with bevacizumab among recurrent patients.
Our study design incorporated a comprehensive flow cytometry analysis of circulating immune cell subsets measured at baseline and early (day 15) after durvalumab initiation that allowed us to directly compare newly diagnosed and recurrent patients in the same clinical trial setting. We noted significantly greater lymphopenia among recurrent patients which aligns with prior studies showing that standard radiation and temozolomide can induce prolonged lymphopenia.37 Our analysis expanded these observations by including a systematic assessment of immune cell subclasses as well as markers of function and proliferation. We noted that recurrent patients had striking reductions of immune effector cells relative to newly diagnosed patients that included circulating CD3+, CD4+ and CD8+ T cell subsets as well as B cells and NK cells, with reductions in naïve, central memory and effector memory subsets, activated T cells as well as proliferating Ki67+ cells primarily accounting for the deficit in circulating CD4+ cells. The etiology of the observed broad immune cell deficiency among recurrent patients requires further study but is likely multifactorial. These exploratory findings require validation in a larger, dedicated dataset but suggest that relative immune effector cell deficiency may represent an additional challenge for immunotherapy approaches among recurrent glioblastoma patients.
Dexamethasone use was associated with lower immune cell populations among both newly diagnosed and recurrent patients consistent with recently reported data from a retrospective clinical review and preclinical studies in syngeneic murine glioblastoma models.38 Although more substantial reductions were observed with dexamethasone among recurrent compared to newly diagnosed patients, dexamethasone did not solely account for observed differences in immune effector cell subsets between newly diagnosed and recurrent patients.
Additional exploratory analyses suggested that dexamethasone use may have influenced associations between immune cell population levels and outcome. For newly diagnosed patients on dexamethasone, a non-statistical trend was noted between likelihood of achieving OS-12 and higher baseline CD8+ levels including those expressing Ki67 or markers of either a naïve or activated phenotype. This observation was not noted among recurrent patients, possibly reflecting their lower levels of these cell subsets compared to newly diagnosed patients.
An early increase in circulating proliferating CD8+ T cells was noted among both newly diagnosed and recurrent patients not on dexamethasone that achieved statistical significance among recurrent patients. This finding has been observed in previously reported melanoma and NSCLC trials of immune checkpoint blockade but has not been previously assessed in glioblastoma studies. An early increase in these cells was noted to be more frequent among patients who achieved OS-12 or PFS-6, respectively providing they were not on dexamethasone. Cautious interpretation of this observation is warranted due to the small sample size, but an early increase in Ki67+CD8+ T cells may warrant further evaluation as a potential biomarker of benefit to immune checkpoint blockade in future studies.
Although tumor mutational burden and immune activation mRNA signature have been linked with improved benefit to immune checkpoint inhibitors in other cancers,19,39 their utility in this context among glioblastoma patients has not been clarified. In our study, we noted that the quantitative values of both of these measures were low relative to other tumor types19,39 and that neither parameter correlated with either PFS or OS. A limitation of these analyses among the recurrent patients is that archival tumor samples obtained at original diagnosis were primarily used rather than samples obtained at study enrollment and thus, may have not accurately reflected the tumor microenvironment at the time of study therapy.
Our study incorporated the NANO scale to provide an objective, user-friendly measure of neurologic function for CNS tumor patients based on a rapidly performed and simplified neurologic examination.5 Limited data exist describing the utility of NANO to assess neurologic function prospectively as well as its association with outcome for glioblastoma patients in clinical trials. We demonstrated that NANO can be performed efficiently in a multicenter trial with most missed assessments due to lack of clinic follow-up at progression. We noted that NANO appeared to effectively track neurologic function of patients receiving study therapy. Baseline NANO neurologic examination correlated with overall survival. In addition, preservation of baseline neurologic status as measured by NANO was observed among patients without radiographic progression, while neurologic decline was noted at the time of radiographic progression in general, although this was impacted by tumor location relative to functional cortex as expected.
Our study findings are affected by several important limitations. First, the number of patients and associated biospecimens per cohort is small and our circulating immune cell findings require further investigation in larger, dedicated datasets. Second, historical benchmarks were used to assess efficacy and a contemporaneous control arm was not included. Third, our findings may have been affected by potentially confounding factors and multiple comparison analyses, particularly regarding the circulating immune cell flow cytometry analyses. Fourth, our study included use of archival tumor material for immunocorrelative analyses among recurrent patients rather than tumor samples obtained immediately prior to study initiation. Fifth, it is not known whether an anti-PD-L1 Mab requires intratumoral delivery for activity; in this study we did not incorporate an assessment of intra-tumoral durvalumab penetration. Finally, our study only evaluated changes in the peripheral blood compartment of circulating immune cell subsets at day 15, and future studies may consider evaluating additional later time points.
In conclusion, PD-L1 blockade with durvalumab was well tolerated but failed to improve outcome among recurrent and newly diagnosed glioblastoma patients. When combined with radiation, durvalumab achieved similar outcome for newly diagnosed, MGMT unmethylated patients as standard temozolomide chemoradiotherapy. Durvalumab as single agent or in combination with standard or reduced dosing schedules of bevacizumab was comparable but not superior to established salvage therapies for recurrent glioblastoma patients. TMB and immune activation signature were lower in general than values reported for other solid tumors and did not associate with outcome. A comprehensive evaluation of circulating immune cell subsets measured at baseline and early after initiation of study therapy revealed that a broad spectrum of peripheral blood lymphocyte populations was reduced among recurrent compared to newly diagnosed patients which was partly attributed to concurrent dexamethasone use. Baseline dexamethasone use also limited the ability of some effector cell populations to increase after durvalumab initiation. An early increase in circulating Ki67+CD8+ T cells may warrant further study as a potential biomarker of PD(L)-1 benefit.
Supplementary Material
Translational Relevance.
Although our study demonstrated that PD-L1 blockade was ineffective among newly diagnosed and recurrent glioblastoma patients, including when combined with reduced or standard bevacizumab dosing, systematic evaluation of circulating immune cell subsets revealed a striking reduction of most cell types among recurrent compared to newly diagnosed patients. We also noted an early increase in circulating, Ki67+CD8+ T cells among newly diagnosed and recurrent patients not on dexamethasone who achieved OS-12 or PFS-6, respectively. These findings require validation in larger datasets but suggest that: 1) recurrent glioblastoma patients may have more difficulty responding to immunotherapy due to decreased levels of relevant circulating immune cells compared to newly diagnosed patients; and 2) an early increase in Ki67+CD8+ T cells may provide a potential biomarker of benefit from immune checkpoint therapy. Our study also further highlights the detrimental effect of dexamethasone and the role of the NANO scale for glioblastoma trials.
Acknowledgments:
We thank the study patients, their families and all the participating investigators and research staff. We also thank the Ludwig Institute for Cancer Research, the Cancer Research Institute, AstraZeneca, the Cure Brain Cancer Foundation and the Jennifer Oppenheimer Cancer Research Initiative for their support of this trial. Durvalumab was provided by AstraZeneca. This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748.
Funding
Ludwig Institute for Cancer Research, Cancer Research Institute, AstraZeneca, the Cure Brain Cancer Foundation and the Jennifer Oppenheimer Cancer Research Initiative. This research was also funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748.
Durvalumab was provided by AstraZeneca.
References
- 1.Nduom EK et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol 18, 195–205, doi: 10.1093/neuonc/nov172 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reardon DA et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol 6, 1003–1010, doi: 10.1001/jamaoncol.2020.1024 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lukas RV et al. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. J Neurooncol 140, 317–328, doi: 10.1007/s11060-018-2955-9 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Awada G et al. Axitinib plus avelumab in the treatment of recurrent glioblastoma: a stratified, open-label, single-center phase 2 clinical trial (GliAvAx). J Immunother Cancer 8, doi: 10.1136/jitc-2020-001146 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nayak L et al. The Neurologic Assessment in Neuro-Oncology (NANO) scale: a tool to assess neurologic function for integration into the Response Assessment in Neuro-Oncology (RANO) criteria. Neuro Oncol 19, 625–635, doi: 10.1093/neuonc/nox029 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stupp R et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10, 459–466 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Allen E et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med 9, doi: 10.1126/scitranslmed.aak9679 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hodi FS et al. Bevacizumab plus Ipilimumab in Patients with Metastatic Melanoma. Cancer Immunol Res 2, 632–642, doi: 10.1158/2326-6066.CIR-14-0053 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wallin JJ et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun 7, 12624, doi: 10.1038/ncomms12624 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Socinski MA et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N Engl J Med 378, 2288–2301, doi: 10.1056/NEJMoa1716948 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Lee MS et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): an open-label, multicentre, phase 1b study. Lancet Oncol 21, 808–820, doi: 10.1016/S1470-2045(20)30156-X (2020). [DOI] [PubMed] [Google Scholar]
- 12.Huang Y et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A 109, 17561–17566, doi: 10.1073/pnas.1215397109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Q et al. Low-Dose Anti-Angiogenic Therapy Sensitizes Breast Cancer to PD-1 Blockade. Clin Cancer Res 26, 1712–1724, doi: 10.1158/1078-0432.CCR-19-2179 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Lorgis V et al. Relation between bevacizumab dose intensity and high-grade glioma survival: a retrospective study in two large cohorts. J Neurooncol 107, 351–358, doi: 10.1007/s11060-011-0748-5 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Louis DN et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol, doi: 10.1093/neuonc/noab106 (2021). [DOI] [PMC free article] [PubMed]
- 16.Brat DJ et al. cIMPACT-NOW update 3: recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol 136, 805–810, doi: 10.1007/s00401-018-1913-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wen PY et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol 28, 1963–1972 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Okada H et al. Immunotherapy response assessment in neuro-oncology: a report of the RANO working group. Lancet Oncol 16, e534–542, doi: 10.1016/S1470-2045(15)00088-1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Higgs BW et al. Interferon Gamma Messenger RNA Signature in Tumor Biopsies Predicts Outcomes in Patients with Non-Small Cell Lung Carcinoma or Urothelial Cancer Treated with Durvalumab. Clin Cancer Res 24, 3857–3866, doi: 10.1158/1078-0432.CCR-17-3451 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Kelley RK et al. Safety, Efficacy, and Pharmacodynamics of Tremelimumab Plus Durvalumab for Patients With Unresectable Hepatocellular Carcinoma: Randomized Expansion of a Phase I/II Study. J Clin Oncol, JCO2003555, doi: 10.1200/JCO.20.03555 (2021). [DOI] [PMC free article] [PubMed]
- 21.Ballman KV et al. The relationship between six-month progression-free survival and 12-month overall survival end points for phase II trials in patients with glioblastoma multiforme. Neuro Oncol 9, 29–38 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamborn KR et al. Progression-free survival: An important end point in evaluating therapy for recurrent high-grade gliomas. Neuro Oncol 10, 162–170 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu W et al. Joint NCCTG and NABTC prognostic factors analysis for high-grade recurrent glioma. Neuro Oncol 12, 164–172, doi:nop019 [pii] 10.1093/neuonc/nop019 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedman HS et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 27, 4733–4740 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Quant EC et al. Role of a second chemotherapy in recurrent malignant glioma patients who progress on bevacizumab. Neuro Oncol 11, 550–555 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Torcuator RG et al. The role of salvage reirradiation for malignant gliomas that progress on bevacizumab. J Neurooncol 97, 401–407, doi: 10.1007/s11060-009-0034-y (2010). [DOI] [PubMed] [Google Scholar]
- 27.Reardon DA et al. Phase II study of metronomic chemotherapy with bevacizumab for recurrent glioblastoma after progression on bevacizumab therapy. Journal of neuro-oncology 103, 371–379, doi: 10.1007/s11060-010-0403-6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu-Emerson C et al. Retrospective study of dasatinib for recurrent glioblastoma after bevacizumab failure. J Neurooncol 104, 287–291, doi: 10.1007/s11060-010-0489-x (2011). [DOI] [PubMed] [Google Scholar]
- 29.Kamphorst AO et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A 114, 4993–4998, doi: 10.1073/pnas.1705327114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang AC et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65, doi: 10.1038/nature22079 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilbert MR et al. in 2103 ASCO Annual Meeting (ASCO, 2013). [Google Scholar]
- 32.Wick W et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 28, 1168–1174, doi: 10.1200/JCO.2009.23.2595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wick W et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N Engl J Med 377, 1954–1963, doi: 10.1056/NEJMoa1707358 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Batchelor TT et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol 31, 3212–3218, doi: 10.1200/JCO.2012.47.2464 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nayak L et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin Cancer Res, doi: 10.1158/1078-0432.CCR-20-2500 (2020). [DOI] [PMC free article] [PubMed]
- 36.Quant E et al. in Proc Am Soc Clin Oncol 91s [Google Scholar]
- 37.Grossman SA et al. Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clinical cancer research : an official journal of the American Association for Cancer Research 17, 5473–5480, doi: 10.1158/1078-0432.CCR-11-0774 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iorgulescu JB et al. Concurrent Dexamethasone Limits the Clinical Benefit of Immune Checkpoint Blockade in Glioblastoma. Clin Cancer Res 27, 276–287, doi: 10.1158/1078-0432.CCR-20-2291 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samstein RM et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 51, 202–206, doi: 10.1038/s41588-018-0312-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.