

Abstract
Cell-surface proteins (SPs) are a rich source of immune and targeted therapies. By systematically integrating single-cell and bulk genomics, functional studies and target actionability, in the present study we comprehensively identify and annotate genes encoding SPs (GESPs) pan-cancer. We characterize GESP expression patterns, recurrent genomic alterations, essentiality, receptor–ligand interactions and therapeutic potential. We also find that mRNA expression of GESPs is cancer-type specific and positively correlates with protein expression, and that certain GESP subgroups function as common or specific essential genes for tumor cell growth. We also predict receptor–ligand interactions substantially deregulated in cancer and, using systems biology approaches, we identify cancer-specific GESPs with therapeutic potential. We have made this resource available through the Cancer Surfaceome Atlas (http://fcgportal.org/TCSA) within the Functional Cancer Genome data portal.
SPs are proteins that span or are anchored/embedded in the surface plasma membrane of cells, controlling communications between cells and the extracellular environment1–5. Along the cell membrane, SPs facilitate fundamental and distinct functions, such as nutrient and ion transport, intercellular interactions, receptor-mediated signaling transduction, enzymatic reactions and immune recognition. Due to their critical biological function and unique subcellular location, SPs have been proposed as a rich source for the identification of targets for immune and targeted therapy5,6. Indeed, SPs serve as targets for >60% of approved drugs for human diseases. In addition, SPs (especially in biological fluids) have been utilized as informative biomarkers for assays of early detection, diagnosis and prediction of diseases.
Targeting SPs that are highly or specifically expressed in the membrane of tumor cells by antibodies or chimeric antigen receptor T cells (CAR-Ts) has become a powerful treatment strategy for cancer patients7–12. More than a dozen antibody drugs against tumor SPs, including naked and conjugated monoclonal antibodies, as well as bispecific monoclonal antibodies, have been developed and used in the clinic for treatment of certain cancers. Most recently, the US Food and Drug Administration (FDA) approved CAR-T therapy to treat selected hematological malignancies. Meanwhile, targeting SPs that function as oncogenic drivers by small molecule inhibitors has also led to a paradigm shift in the treatment of cancer. Multiple receptor tyrosine kinase inhibitors have been developed and applied in oncology, especially for patients with receptor tyrosine kinase genomic alterations such as mutations, copy number alterations (CNAs) or fusions13,14. Taken together, among 151 drug-target genes with FDA-approved therapies in oncology, 90 were in the cell-surface membrane. However, most patients with cancer still do not benefit from these kinds of therapies due to the challenge in identification and prioritization of targetable proteins on the tumor cell surface5,6. To fill this gap, high-throughput transcriptomic and proteomic approaches have been successfully applied to study SPs in select cancer types15–20. Nevertheless, current anticancer drug discovery efforts are still focused on a small fraction of SPs, predominantly due to challenges in systematic characterization of the surfaceome across healthy and tumor tissues. Advances in large-scale and multidimensional studies, such as the Genotype-Tissue Expression (GTEx)21, The Cancer Genome Atlas (TCGA)22, the Dependency Map (DepMap) and the Project Score23–27, and the Open Targets projects6,28 have provided powerful resources for characterizing the GESPs (that is, the surfaceome) in cancer and identifying potential therapeutic targets. The overall goal of the present study is to systematically characterize the surfaceome across cancers, and to develop a comprehensive surfaceome database for research community.
Results
Definition of the human surfaceome on a genome-wide scale.
Both experimental and computational approaches have been applied to identify and predict the proteins located on cell-surface membra nes1–5,29–35, although each strategy has its own limitations, leading to incomplete coverage and false positives. To overcome this problem and comprehensively define the human surfaceome in the genome (Fig. 1a), we integrated GESP candidates from nine independent resources, in which the SPs were identified or predicted by distinct strategies (Fig. 1b and Supplementary Table 1). We estimated a core GESP score for each candidate based on a weighted vote approach, that is, each resource has a different voting power due to its identification/prediction principle. Then, using known GESPs and non-GESPs as positive and negative controls, we established a cutoff (core GESP score ≥4) to define the potential GESPs (Fig. 1c,d). In this setting, both false-negative and false-positive rates were <5%. To evaluate this cutoff, we collected the targets (FDA approved or in clinical development) of CAR-Ts, ADCs (antibody–drug conjugates) and antibody drugs that target SPs of tumor cells, and found that 97.0% of them had a core GESP score ≥4 and many of them showed high core GESP scores (Fig. 1c,d). Next, information from COMPARTMENT, gene ontology (GO) and manual literature searches was used to remove genes encoding proteins in intracellular membranes such as the nuclear membrane and mitochondrial membrane (Fig. 1b). Finally, other features, such as literature evidence, protein structure and evolutionary conservation, were used as additional score-driven factors to finalize the GESP list and estimate a final GESP score (Fig. 1b). Notably, 100 GESPs were defined as integral monotopic proteins on the inner surface of the cell membrane, which were excluded from certain downstream analyses in our study (for example, identification of immunotherapy targets). Taken together, we generated a comprehensive human GESP gene list (n = 3,567; Supplementary Table 2), representing high confidence surfaceome candidates from nine complementary surfaceome resources. Among them, more than half of the GESPs fell into druggable gene families that were predicted as potential targets for small molecules, such as G-protein-coupled receptors (23.13%), ion channels (7.40%), kinases (3.36%), enzymes (9.81%) and transporters (8.83%; Fig. 1e). Importantly, 17.21% of the GESPs were functionally defined as membrane-bound immunological accessory molecules (mIAMs) that modulate immune response in physiological and pathological conditions (Fig. 1f).
Fig. 1 |. Definition of the human surfaceome on a genome-wide scale.
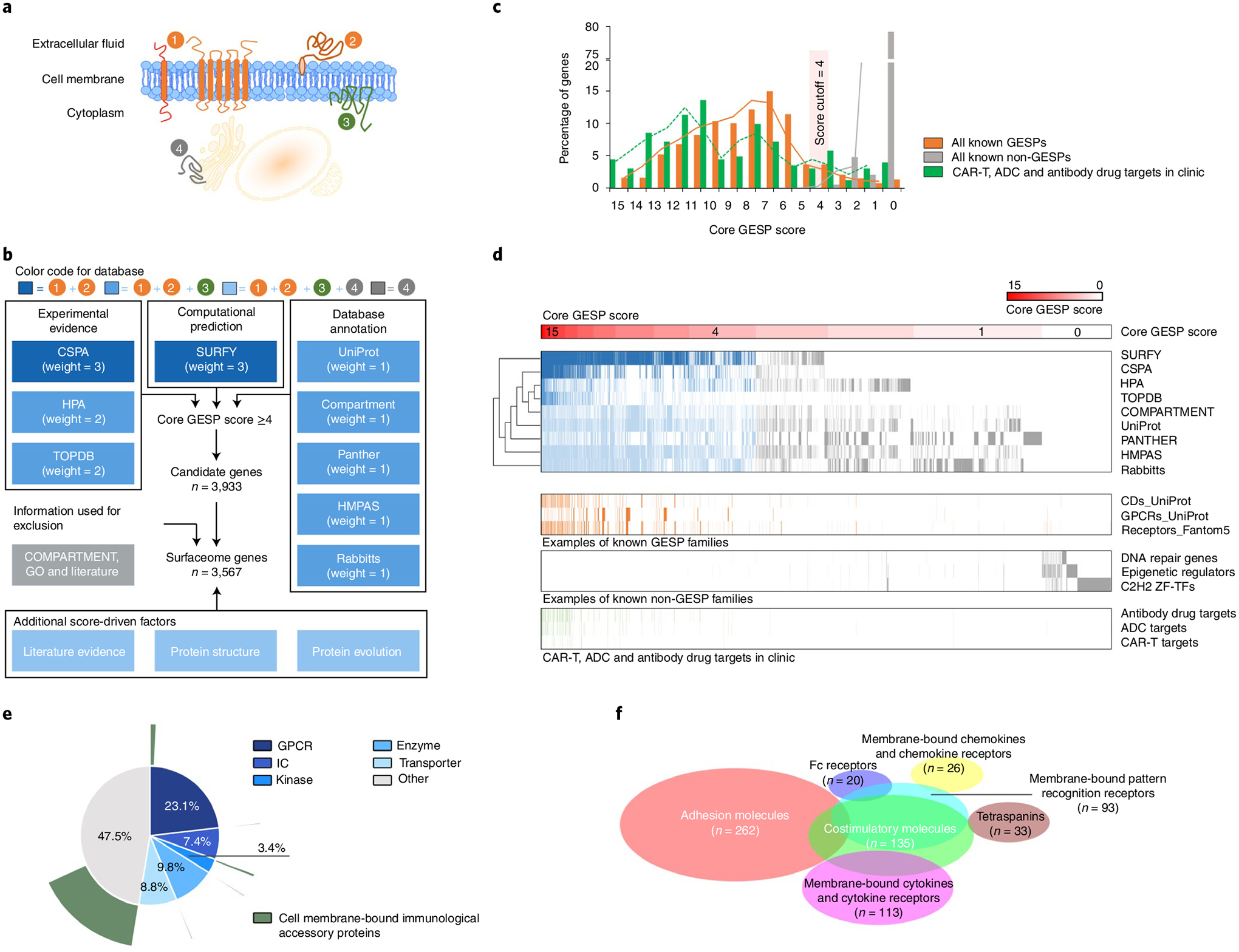
a, Schematic illustration of membrane proteins in cells. 1, Integral bitopic and polytopic proteins on the cell-surface membrane. 2, Integral monotopic proteins on the outer surface of the cell membrane. 3, Integral monotopic proteins on the inner surface of the cell membrane. 4, Membrane proteins on other intracellular membranes. b, The workflow to estimate final GESP scores for candidates of the GESPs. Nine independent and complementary resources were used to initially establish a core GESP score based on a weighted vote approach. Then COMPARTMENT, GO and literature searches were used to remove genes encoding proteins in intracellular membranes. Finally, other information and features were used as additional score-driven factors to estimate a final GESP score for each candidate. c, The distribution of the core GESP scores for known GESPs (orange) and non-GESPs (gray), as well as the target genes of CAR-Ts, ADCs and antibody drugs that are FDA approved or in clinical development (green). A core GESP score ≥4 was chosen as the cutoff to define GESPs. d, Heatmap showing genes across the nine surfaceome resources used in the present study, the examples of known GESPs and non-GESPs, and the target genes of CAR-Ts, ADCs and antibody drugs. Genes were ranked based on their core GESP scores. e, Classification of the GESPs based on gene superfamily category (inner circle), and the cell membrane-bound immunological accessory proteins highlighted in green (outer circle). f, Scaled Venn diagram showing the functional families among the cell mIAMs.
Expression of the GESPs across normal and tumor specimens.
To characterize messenger RNA expression of the GESPs, the RNA-sequencing (RNA-seq) profiles from the GTEx (n = 7,429; Fig. 2a and Supplementary Table 3) and The Cancer Genome Atlas (TCGA; n = 9,807; Fig. 2b and Supplementary Table 4) projects were analyzed. We found that only 22.1% of GESPs were ubiquitously expressed across all cancer types; in contrast, 48.4% of non-GESPs (8,100 of 16,723) were detectable. For each gene, we analyzed the numbers of cancer types in which its mRNA was detectable and found that indeed GESPs were expressed in significantly fewer cancer types compared with non-GESPs (Fig. 2c, odds ratio (OR) = 2.1, P = 1.3 × 10−81). Tissue specificity index36 analysis consistently demonstrated that a larger fraction of GESPs exhibited cancer type specificity compared with non-GESPs (44.5% versus 24.1%; OR = 2.5, P = 7.4 × 10−98; Fig. 2d). These results were further confirmed by enrichment analysis for each subgroup of genes classified by protein subcellular location (Fig. 2e,f). A consistent result was also observed across the normal tissues from GTEx (Extended Data Fig. 1). Supporting these results, t-distributed stochastic neighbor embedding (t-SNE) analysis37 indicated that expression levels of GESP mRNAs could distinguish the tumor specimens from different tumor types, and tumor specimens with related tissue origins were clustered closely (Fig. 2g). Importantly, tumor specimens were closely clustered together with their corresponding normal healthy tissues from GTEx and tumor-adjacent tissues from TCGA (Fig. 2h). Furthermore, when we used samples only from the same lineage to perform the t-SNE analysis, normal tissues were clearly distinct from cancers and their adjacent tissues (Fig. 2h). Collectively, GESP mRNA expression patterns were largely tumor-type specific, and their expression spectrum in cancers reflected tissue lineages. To further characterize GESP expression in the cancer microenvironment, single-cell RNA-seq (scRNA-seq) profiles from 13 cancer types (Supplementary Table 5) were collected and processed by a unified computational pipeline. Among 3,031 GESPs that were detectable by bulk RNA-seq in at least one cancer type from TCGA, 29.7% (n = 899) could be detected by scRNA-seq in at least 50% cells of one cell population in a cancer type (Supplementary Table 6). Importantly, most GESPs showed specific expression patterns among different cell populations within the cancer microenvironment (Supplementary Table 6 and Extended Data Fig. 2). Finally, to examine whether the mRNA expression levels of GESPs were representative of their protein levels in cancers, we analyzed the correlations of mRNA and protein in five cancer types with matched proteomic profiles generated by the Clinical Proteomic Tumor Analysis Consortium (CPTAC; Supplementary Table 7). Consistent with previous reports, significant and positive correlations were observed in an average of 79.3% (69.2–85.6%) of the protein-coding genes (Fig. 2i and Extended Data Fig. 3). Comparing with non-GESPs, a larger fraction of GESPs exhibited significantly and strongly positive correlation between mRNA expression and protein abundance (41.0% versus 24.2%, OR = 2.2, P = 1.2 × 10−19; Fig. 2j). Consistently, a Gene Set Enrichment Analysis (GSEA) indicated that the positively correlated genes were significantly enriched in the GESPs and in the set of genes located in the cytoplasm and peroxisomes (Fig. 2k). This result demonstrated that mRNA expression of GESPs can be used to predict protein expression across cancers.
Fig. 2 |. Expression of GESPs across healthy normal tissues and primary tumor specimens.
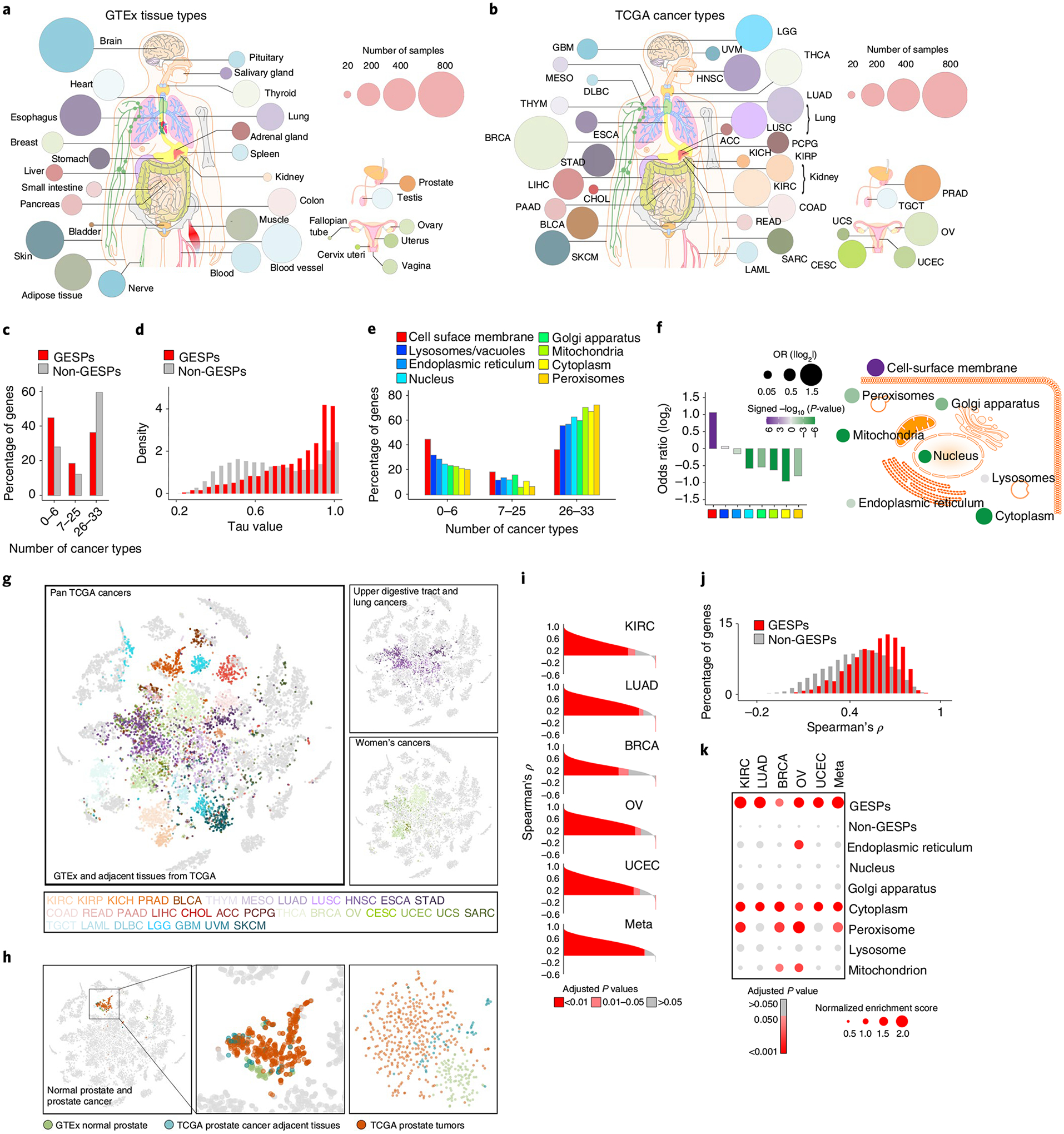
a,b, Summary of the tissue/cancer types and numbers of RNA-seq specimens of GTEx (a) and TCGA (b) cohorts. The size of each circle corresponds to the number of samples of a tissue/cancer type. c, Percentages of genes that were detectable by RNA-seq in 0–6, 7–25 and 26–33 cancer types. d, Histogram of relative frequency distributions of tau values in GESPs and non-GESPs. e, The percentages of genes detectable in 0–6, 7–25 and 26–33 cancer types, stratified by subcellular location of gene products. f, Bar plot (left) and bubble plot (right) showing enrichment of cancer-type-specific genes in the corresponding subgroups based on subcellular location of gene products. P values were calculated using two-sided Fisher’s exact test. Purple, enriched; green, depleted. The size of the bubble: absolute value of log2(OR). g, Based on GESP expression similarity, normal and tumor specimens were presented by t-SNE analysis. Normal and tumor-adjacent tissues: gray; tumor specimens: color coded (color key is listed at the bottom and based on tissue origin). Right: only the cancers derived from the upper digestive tract and lung epithelium (top right) or the women’s cancers (bottom right) are color coded; normal tissues and specimens from other cancer types are shown in gray. h, Left: specimens from normal prostate (GTEx, green), prostate tumor adjacent (TCGA; blue) and prostate tumors (TCGA, red) are highlighted. Other normal tissues and cancer specimens are shown in gray. Right: t-SNE analysis was performed only in the prostate specimens (normal, adjacent and tumors). i, Histogram of Spearman’s correlation coefficients between mRNA and protein expression levels of all genes across five cancer types. P values for Spearman’s rank correlation were calculated and adjusted using the Benjamini–Hochberg method. j, Histogram of frequency distributions of Spearman’s correlation coefficients between mRNA and protein expression levels in GESPs and non-GESPs. k, Bubble plot showing enrichment of positively correlated genes in the corresponding subgroups based on subcellular location of gene products. P values for the GSEA test were based on 1,000 permutations, and adjusted for gene set size and multiple hypotheses testing.
Identification of GESPs specifically expressed in cancers.
To systematically identify cancer-specific GESPs (caGESPs), we estimated an expression specificity score for each GESP by comparing its expression level in a given cancer type (TCGA) to all normal tissues (GTEx) using five principally different computational strategies (Fig. 3a and Supplementary Table 8). Given that cancer-testis genes often encode immunogenic antigens for cancer immunotherapy38,39, the RNA-seq profiles of normal testis tissues were excluded from the normal tissue pools (except for analysis on testicular germ-cell tumor (TGCT)). To reduce the expressional interference from tumor-infiltrating immune cells in tumor specimens, GESPs that are highly expressed in immune cells were excluded (except for analysis on hematopoietic malignancies), based on the RNA-seq profiles from 30 distinct types of hematopoietic cells and 6 lymphatic tissues12,40,41. We identified a total of 409 unique caGESPs, which were specifically expressed in at least 1 cancer type (a median of 16 caGESPs for each cancer type; Fig. 3b,c and Supplementary Table 9). Based on their specificity scores, the genes we identified as caGESPs were further classified into three tiers of confidence (Fig. 3c,d, Extended Data Fig. 4 and Supplementary Table 9). In each tier, we were able to find caGESPs that are in current clinical development for CAR-Ts, ADCs or antibody drug treatment in cancer (Fig. 3e). This indicates that caGESP candidates in all three tiers have therapeutic potential for further clinical application. Indeed, 13.4% (55/409) of the caGESPs identified by our systematic approaches have been previously reported as being in advanced clinical development for cancer immune therapy (Fig. 3f). Notably, although most of the caGESPs were identified in a single cancer type, we found that 26.4% (128/409) of caGESPs were shared by more than 1 cancer type (Fig. 3b), suggesting that these caGESPs may be regulated by common oncogenic signals during tumorigenesis.
Fig. 3 |. Identification of GESPs that are specifically expressed in cancers.
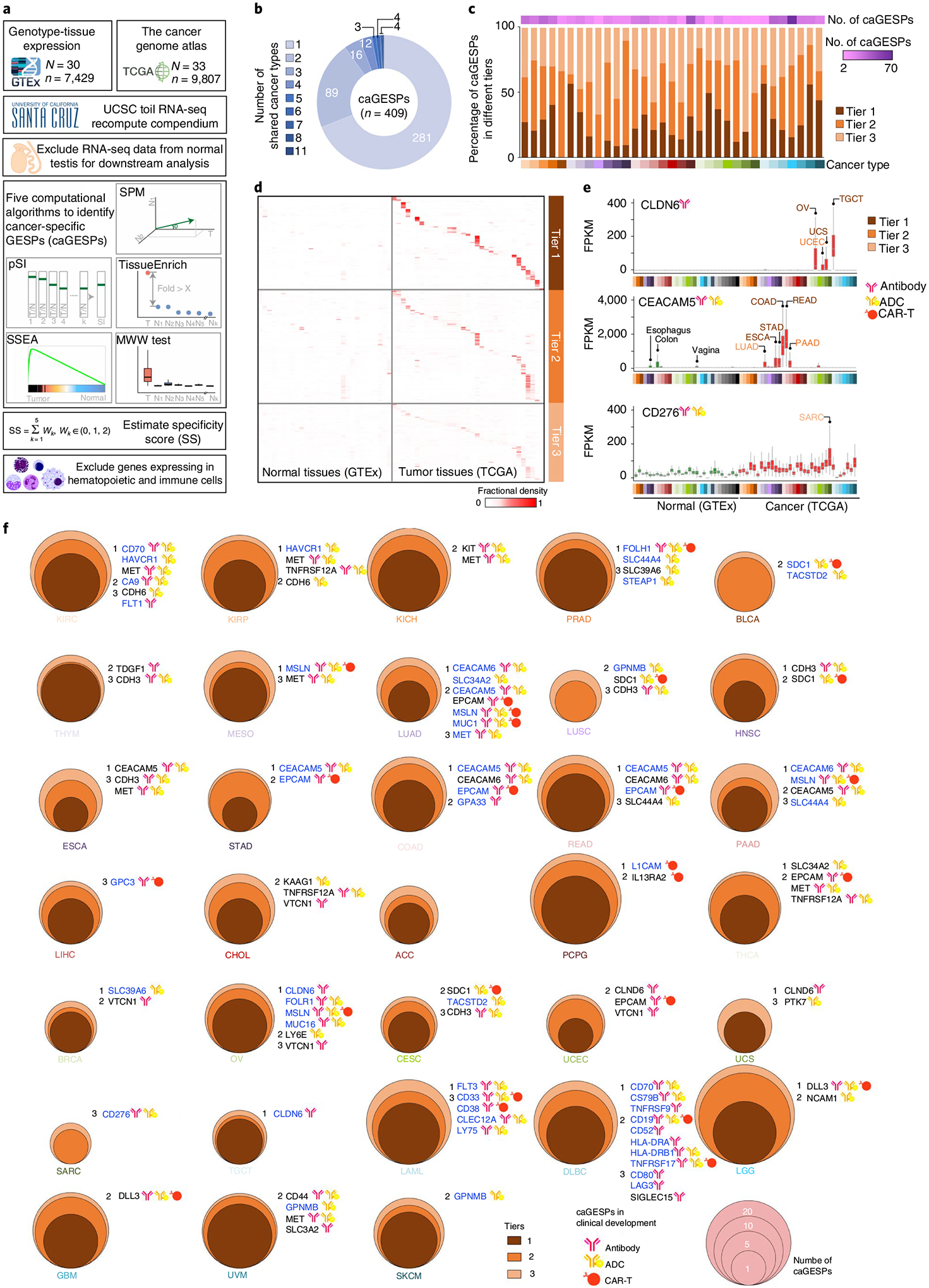
a, The workflow to identify GESPs that are specifically expressed in cancer (caGESPs). b, Numbers of caGESPs that are shared by different cancer types. c, Numbers of caGESPs identified in each cancer type (top), the percentages of caGESPs in each tier (middle) and the cancer-type color code (bottom). d, Heatmap showing expression abundance of identified caGESPs across normal tissues (GTEx) and cancers (TCGA), stratified by tiers. The color intensity represents the fractional density across FPKM values. e, Expression levels of typical examples of identified caGESPs across normal and tumor specimens. Cancer types in which the caGESPs were identified are labeled by color. Based on specificity scores, the identified potential caGESPs were classified into three tiers: tier 1, high confidence (maroon); tier 2, moderate confidence (sienna); and tier 3, low confidence (sandy brown). Note: the CAR-Ts, ADCs or antibody drugs targeting these three caGESPs are currently being evaluated in clinical trials. The horizontal line in the box plot indicates the median, and the whiskers indicate 1.5× IQR of the first and third quartiles. The sample size used to derive statistics is reported in Supplementary Tables 3 and 4. f, Numbers and tiers of caGESPs identified in each cancer type. Note: many caGESPs identified by the present study are being evaluated in the clinic, and those caGESPs are highlighted for each cancer type.
After systematically reviewing each caGESP, we observed that, in most cases, it is still challenging to use an individual caGESP to ‘uniquely’ define tumors and ‘completely’ spare normal cells. For example, low-level expression of MSLN (mesothelin), a widely used caGESP target in CAR-T and ADC therapy development, was also detected in lung, fallopian tube and salivary gland tissue, although the tumor specimens from mesothelioma, pancreatic adenocarcinoma (PAAD), ovarian serous cystadenocarcinoma and lung adenocarcinoma showed specifically higher expression levels. This may lead to potential ‘on-target–off-tumor’ toxic side effects for immunotherapy. To overcome this problem, a combination of multiple caGESPs has been proposed as a promising strategy for more precise and adaptable tumor recognition11,42–46. For example, CAR-T can be designed with a ‘Boolean A AND B’ SP recognition logic gate that is activated only when both proteins (A and B) are expressed in tumor cells (Fig. 4a)47,48. In this regard, we developed a computational approach to identify and prioritize caGESP combinations for design of logic-gated CAR-T (Fig. 4b). To identify candidate pairs of caGESPs for the ‘AND CAR-T’ strategy, mutual exclusivity of caGESP expression in normal tissues was analyzed. We defined caGESP pairs, in which both caGESPs were identified from the same cancer type and showed significantly and mutually exclusive expression patterns across normal tissues, as potential candidates for this strategy (Fig. 4c). An average of 12 pairs was found in 15 cancer types, and a total of 179 unique pairs were identified (Fig. 4d and Supplementary Table 10). In addition, an inhibitory CAR (iCAR) can be designed with an antigen-specific inhibitory signaling domain that recognizes surface proteins expressed only in normal tissues to limit CAR-T activity49. Thus, the iCAR-T is activated only in tumors when the inhibitory signal is absent (Fig. 4e). To identify candidate pairs of GESPs for ‘iCAR-T’ strategy, the expression co-occurrence of the GESPs in normal tissues was analyzed (Fig. 4f). We defined GESP pairs, in which the caGESP and its paired GESP were coexpressed in the same normal tissues, and the paired GESP was not detectable in the cancer type in which the caGESP was identified, as the potential candidates for ‘iCAR-T’ strategy (Fig. 4g). An average of 25 pairs was found in 21 cancer types, and a total of 443 unique pairs were identified (Fig. 4h and Supplementary Table 11).
Fig. 4 |. Evaluation of GESP combinations for logic-gated CAR-T design.
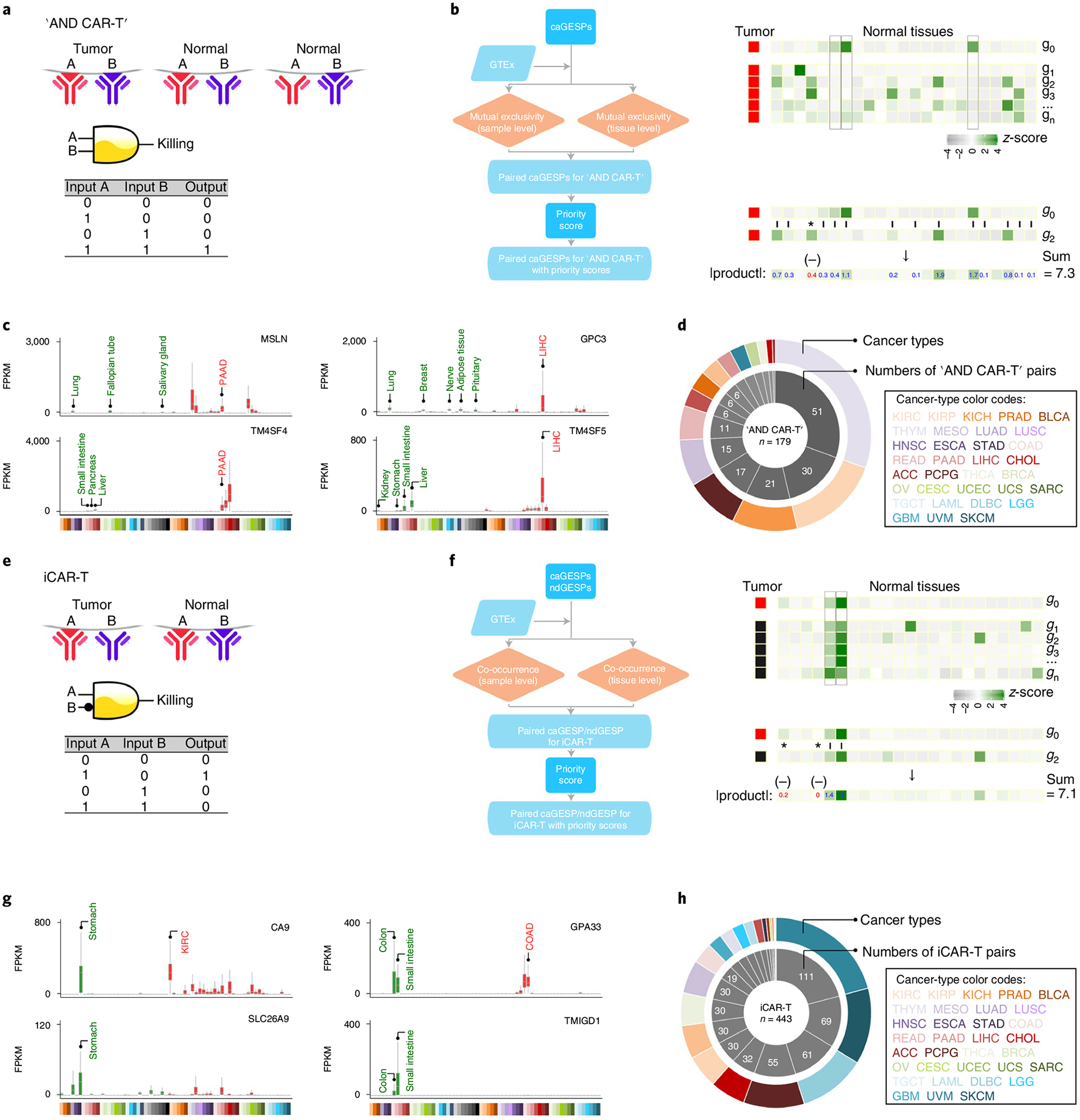
a, Schematic illustration of the logic-gated ‘AND CAR-T’ design. b, The workflow to identify paired GESPs for ‘AND CAR-T’ design. c, Examples of identified caGESP pairs for ‘AND CAR-Ts’: MSLN-TM4SF4 for PAAD (left), and GPC3-TM4SF5 for liver hepatocellular carcinoma (right). Red text: the cancer type targeted by ‘AND CAR-T’, green text: the normal tissues in which the caGESP is expressed at low levels (that is, the tissue types with potential ‘on-target–off-tumor’ effects). Identified caGESP pairs are coexpressed in a select cancer type (red), and are mutually exclusively expressed in normal tissues (green). The horizontal line in the box plot indicates the median, and the whiskers indicate 1.5× IQR of the first and third quartiles. The sample size used to derive statistics is reported in Supplementary Tables 3 and 4. d, Number of the identified ‘AND CAR-T’ pairs for each cancer type. e, Schematic illustration of the iCAR-T design. f, The workflow to identify paired GESPs for iCAR-T design. g, Examples of identified caGESP–GESP pairs for iCAR-Ts: CA9-SLC26A9 for kidney renal clear cell carcinoma (left) and GPA33-TMIGD1 for chronic obstructive airway disease (right). Red text: the cancer type targeted by iCAR-T, green text: the normal tissues in which the caGESP is expressed at low levels (that is, the tissue types with potential ‘on-target–off-tumor’ effects). The identified caGESP–GESP pairs are mutually exclusively expressed in a select cancer type (red) and are coexpressed in normal tissues (green). The horizontal line in the box plot indicates the median, and the whiskers indicate 1.5× IQR of the first and third quartiles. The sample size used to derive statistics is reported in Supplementary Tables 3 and 4. h, Number of the identified iCAR-T pairs for each cancer type.
Characterization of recurrent genomic alterations of GESPs.
Cancer-associated GESPs driven by recurrent focal somatic CNAs (SCNAs) were identified by four criteria (Extended Data Fig. 5a), and a G-score50,51 at the individual cancer level was established for each GESP. We initially identified 989 GESPs that met all these criteria in at least one tumor type (Fig. 5a,b, Extended Data Fig. 5b,c and Supplementary Table 12). For instance, the well-established therapeutic target GESP, ERBB2, was recurrently amplified in 11 tumor types. Importantly, SCNAs of GESPs were tumor-type specific: 497 of 989 (50.2%) GESPs with recurrent SCNAs were identified only in a single tumor type, and no GESP recurrent CNA was observed in >11 tumor types. We also estimated a pan-cancer overall G-score50,51 (Fig. 5a,b, Extended Data Fig. 5b,c and Supplementary Table 13), and found that 19.8% (200/989; 113 with amplification and 81 with deletion) showed an overall G-score above a cutoff determined using the elbow method52,53 (0.62 and 0.73 for amplification and deletion, respectively). Cancer-associated GESPs driven by mutations were identified by a combination of five complementary approaches (Extended Data Fig. 6a), and an M-score50,51 at the individual cancer level was established for each GESP. We initially identified 143 GESPs that had recurrent mutations in at least 1 tumor type (Fig. 5c, Extended Data Fig. 6b and Supplementary Table 14). Although CTNNB1 (Catenin Beta 1) and ERBB2 were widely mutated across different tumor types (ten and six tumor types, respectively), recurrent mutations of GESPs were remarkably tumor-type specific: 105 of 143 (73.4%) GESPs with recurrent mutations were found only in a single tumor type, and no GESP recurrent mutation was observed in >10 tumor types. Notably, except for GNAQ (G Protein Subunit Alpha Q), which had a substantially high frequency of mutation in uveal melanoma (50%), most GESPs had mutation frequencies <5% in a certain tumor type (Supplementary Table 15), and rarely those carrying hotspot mutations (Extended Data Fig. 6c). We also estimated a pan-cancer overall M-score50,51 (Fig. 5c and Supplementary Table 16), and found that 37.8% (54/143) of GESPs showed an overall M-score above a cutoff determined using the elbow method (that is, M-score ≥0.10). CTNNB1, EGFR (Epidermal Growth Factor Receptor), GNAQ and FAT1 (FAT Atypical Cadherin 1) had the highest overall M-scores across all tumor types (Fig. 5c). Except for B2M (β2-microglobulin), the most common type of recurrent mutation of GESPs at a pan-cancer level was a missense mutation (37.3–85.7%; Supplementary Table 17) and the dominant type of recurrent mutation was heterozygous (56.9–92.8%; Supplementary Table 18). In contrast, β2-microglobulin was most commonly altered by truncating mutations (72.0%; Supplementary Table 17). Finally, the timing and clonal statuses of the GESP mutations were also determined using the ABSOLUTE algorithm54. More than 41.5% of recurrent mutations in GESPs were early events (Supplementary Table 19) and >52.5% were clonal alterations (Supplementary Table 20). TCGA gene fusion profiles were retrieved from the TumorFusions database55, and 6,280 fusion transcripts (including 5,512 fusion pairs) of 1,771 GESPs were initially identified in 9,799 tumor specimens (Fig. 5d and Supplementary Tables 21 and 22). Only 484 of 6,280 (7.7%) GESP fusion transcripts, representing 104 of 5,512 (1.9%) fusion pairs, were considered as recurrent fusions, indicating that recurrent transcript fusion in GESPs is a rare genetic event. Across all cancer types, 4.9% (86/1,771) of GESPs showed fusion events above a cutoff determined using the elbow method (that is, ≥12; Extended Data Fig. 7a). TMPRSS2–ERG (n = 177), FGFR3–TACC3 (n = 36), NCOR2–SCARB1 (n = 12), CCDC6– RET (n = 12) and ETV6–NTRK3 (n = 10) were the most frequent fusions among TCGA tumor cohort (Extended Data Fig. 7b and Supplementary Table 23).
Fig. 5 |. Characterization of recurrent genomic alterations of GESPs across cancers.
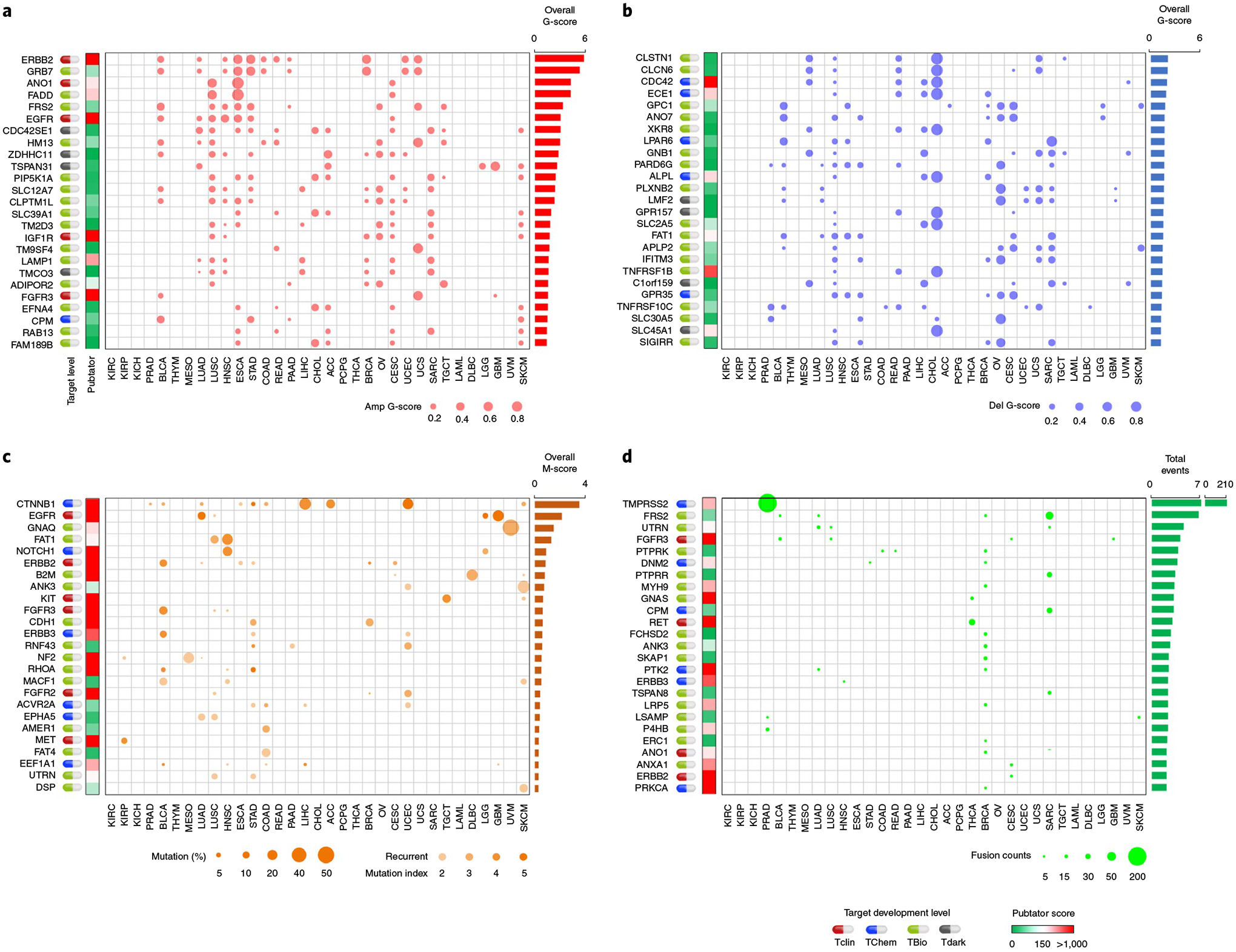
a,b, Bubble plot showing the G-scores (copy number gain (a) and copy number loss (b)) of cancer-associated GESPs driven by SCNAs, plotted for each cancer type. Bubble size: G-score; red: gain; blue: loss. c, Bubble plot showing the mutation indices and mutation frequencies of caGESPs driven by somatic mutations, plotted in each cancer type. Bubble size: mutation frequency; intensity of color: mutation index. d, Bubble plot showing the number of transcript fusions of cancer-associated GESPs driven by fusion, plotted in each cancer type. Bubble size: fusion events. Note: genes are ranked by overall G-scores (a,b), overall M-scores (c) or total fusion events (d), which are shown on the right of each panel. Target development levels of each gene (PHAROS database) are shown on the left of each panel. Red: Tclin; blue: TChem; green: TBio; gray: Tdark. Pubtator scores (Pubtator database) are shown next to the target development levels. Green: 1–150 (understudied genes); red: >150.
Characterization of GESP dependence in cancer cell growth.
Genome-wide CRISPR (clustered regularly interspaced short palindromic repeats) screening data from the DepMap and the Project Score23–26 (Supplementary Tables 24 and 25) were integrated by Pacini et al.27, and the effects of knocking out a given GESP on cell growth were analyzed. As expected, the percentages of GESPs that were defined as essential genes (‘common essential’ or ‘strongly selective’) for cell growth (Supplementary Table 26) were substantially less than those of non-GESPs (4.1% versus 14.0%; Extended Data Fig. 8a). Consistently, after we grouped genes based on the subcellular locations of their protein products, GESPs showed significantly less enrichment for the genes with functions considered as essential for cell growth, whereas the genes encoding products located in the nucleus, mitochondria and cytoplasm showed significant enrichment (Extended Data Fig. 8b). Similar results were also observed at individual cancer-type levels (Supplementary Table 27). These results indicate that most GESPs are not essential for cell growth in vitro. Among the essential GESPs (both common essential and strongly selective) expressed in cancer cell lines, 43 (36.8%) showed a significant and positive correlation between mRNA expression levels and dependence, including 7 GESPs that were recurrently amplified in cancer and with copy numbers that were positively correlated with dependence (Extended Data Fig. 8c and Supplementary Table 28). Notably, among them, three GESPs have already been used as anticancer drug targets for FDA-approved cancer treatment (Extended Data Fig. 8d).
Characterization of receptor–ligand interactions of GESPs.
A large proportion of GESPs functions as cell-surface receptors, directly interacting with soluble or cell membrane-associated ligands (Fig. 6a)56,57. To characterize the receptor–ligand network of GESPs in cancers, we systematically identified receptor–ligand interaction pairs using a computational approach (Fig. 6b). After combining both known and inferred receptor–ligand databases (Supplementary Table 29), we predicted 1,278 pairs of receptor–ligand interactions (Supplementary Table 30; note: non-GESP receptors, such as nuclear receptors, were excluded from our analysis). Among them, 1 receptor binds to 2.8 ligands on average (range: 1–22), whereas 1 ligand interacts with 2.7 receptors on average (range: 1–13; Fig. 6c,d). The numbers of pairs in which both receptors and ligands were detectable by RNA-seq varied widely across tissue and tumor types (Fig. 6e). We next analyzed the expression correlation of each identified receptor–ligand pair in a given tissue or cancer type using Pearson’s test, and found that the expression of 99.1% (1,267/1,278) and 99.2% (1,268/1,278) of receptor–ligand pairs significantly and positively correlated in at least one tissue of GTEx and TCGA, respectively (sample size ≥10, Pearson’s test P value <0.05; Fig. 6f). Importantly, unsupervised cluster analysis on the correlations of receptor–ligand pairs showed that the normal (GTEx) and tumor-adjacent (TCGA) tissues were clustered together, and were largely separated from tumor tissues (Fig. 6f), suggesting that the receptor–ligand interaction networks during tumorigenesis are dramatically dysregulated. As expected, normal and tumor-adjacent tissues from the same lineage were clustered together; a similar pattern was also observed across cancers (Fig. 6f). Given that both soluble and membrane-associated ligands that specifically bind to GESPs have been used to design CAR-Ts or ADCs for cancer treatment42,44,58, we next identified the ligands that bind to caGESPs. Among 166 caGESPs associated with receptor–ligand pairs, 29 ligands bind to limited numbers of GESPs (that is, <2 GESPs; Fig. 6g and Supplementary Table 31), including CD27–CD70 and KLRK1-ULBP2, which are two targets currently being evaluated for CAR-T design in clinical trials (Fig. 6h).
Fig. 6 |. Characterization of receptor–ligand interactions of the GESPs in cancers.
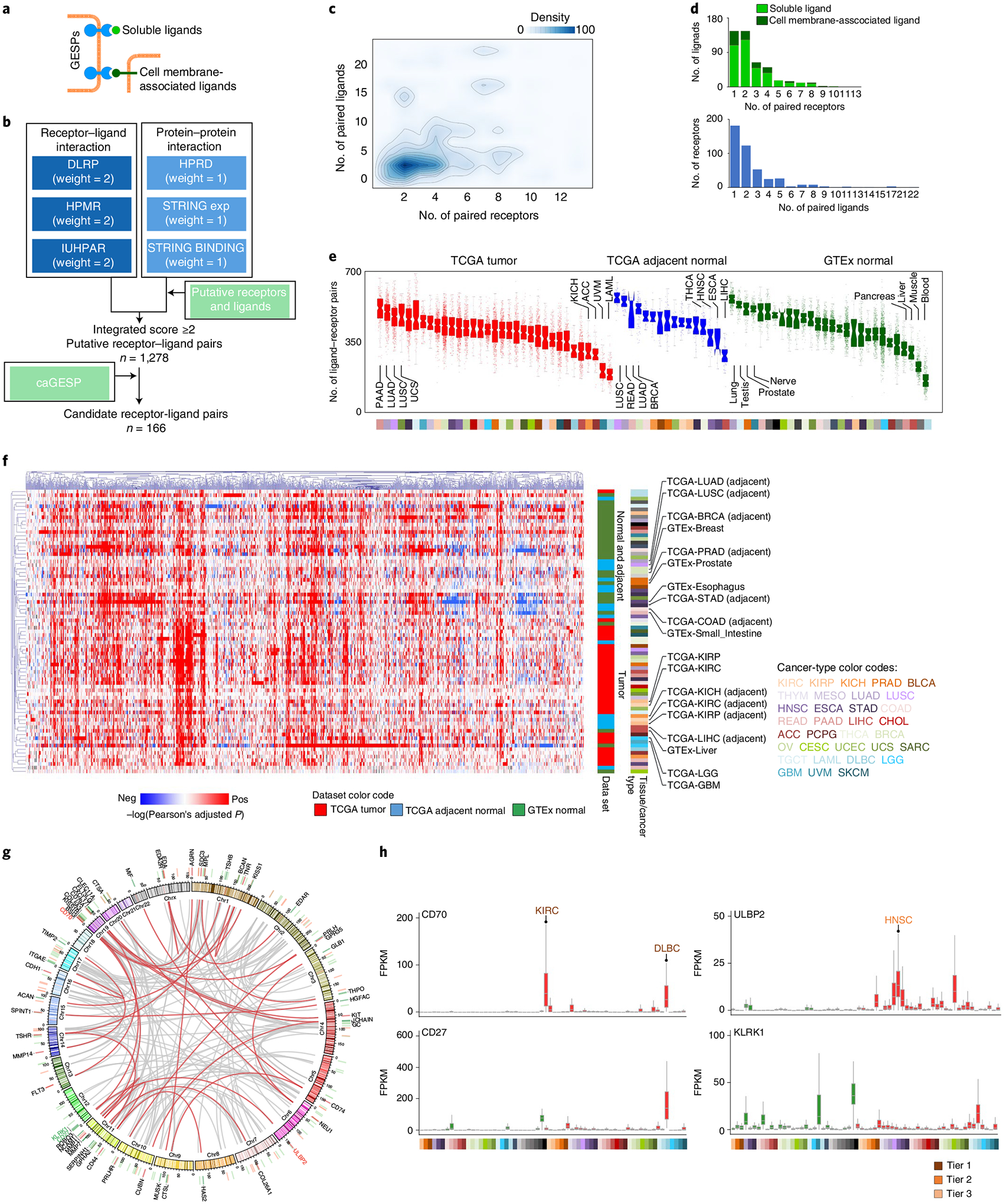
a, Schematic illustration of receptor GESPs and their soluble and membrane-associated ligands. b, The workflow to identify receptor–ligand interaction pairs. c, Density cloud plot showing the binding patterns of receptors and ligands. A given receptor (x axis) is plotted against the number of its corresponding ligands (y axis), and a given ligand (y axis) is plotted against the number of its corresponding receptors (x axis). d, Top: bar plots showing the numbers of ligands that bind to different numbers of their corresponding receptors. Bottom: bar plots showing the numbers of receptors that bind to different numbers of their corresponding ligands. e, Number of expressed receptor–ligand pairs in TCGA tumors, TCGA adjacent normal tissues and GTEx normal tissues. On the violin plot, points represent estimates for individual samples, and the colored areas are estimated density distributions. The sample size used to derive statistics is reported in Supplementary Tables 3 and 4. f, Unsupervised hierarchical cluster heatmap based on the correlations (log(transferred adjusted P value) of Pearson’s test) of expression levels of receptor–ligand pairs across TCGA tumors, TCGA adjacent normal tissues and GTEx normal tissues. Selected tissue/cancer types are highlighted on the right. g, Circle plot showing the interactions of the caGESP-associated receptor–ligand pairs. The receptor–ligand interactions are highlighted by a red color when a receptor is paired with at least one unique ligand. Red and green bars indicate genomic locations of the receptors and ligands, respectively. The names of two identified receptor–ligand pairs that have been used for CAR-T development in the clinic are highlighted by color. h, Examples of identified caGESP–ligand pairs that have been used for CAR-T development in the clinic: CD70–CD27 (left) and ULBP2–KLRK1 (right). Red text: the cancer type in which the caGESPs are highly expressed; green text: the normal tissues in which the ligands are expressed. The horizontal line in the box plot indicates the median, and the whiskers indicate 1.5× IQR of the first and third quartiles. The sample size used to derive statistics is reported in Supplementary Tables 3 and 4.
Characterization of mIAMs across cancers.
The mIAMs, one of the major functional groups of the GESPs that we identified from the human genome (Fig. 1f), play crucial roles in tumorigenesis by modulating immune responses59. To further characterize their expression in the cancer microenvironment, we compared expression similarity of mIAMs within each cell population in the tumor microenvironment across cancers using the scRNA-seq profiles from 13 cancer types (Supplementary Table 5). As expected, mIAM expression was largely distinct among different stromal cell populations, even between differentially related cell types (for example, macrophages and dendritic cells (DCs); Fig. 7a). However, mIAM expression signatures were similar for stromal cell-type populations isolated from different cancer types (Fig. 7b), indicating that their expression is relatively consistent in the same stromal cell population across different cancer types. In contrast, mIAM expression patterns were highly heterogeneous among the tumor cells from different cancer types, reflecting cancer lineage: epithelial tumors were clustered together and separated from neurological and hematological malignancies (Fig. 7c). Heterogeneous expression of mIAMs in tumor cells may lead to intrinsic differences in tumor-immune interactions among different cancers because the expression of mIAMs in stromal cells was relatively homogeneous. Supporting this observation, Spearman’s correlation analysis showed remarkably lower correlation coefficients of mIAM expression among tumor cells compared with those among each stromal cell population (Fig. 7d). To overcome the limitation of low coverage of scRNA-seq, we further analyzed intrinsic mIAM expression in tumor cells (without extrinsic stromal signals) across a large collection of established cancer cell lines (n = 1,200 from 28 tumor types)60. Positive expression of an mIAM in cancer was defined as having mRNA expression that was reliably detected for >5% of cancer cell lines; 488 (79.5% of all) mIAMs were expressed in tumor cells and 126 (20.5%) were defined as undetectable genes (Fig. 7e and Supplementary Table 32).
Fig. 7 |. Characterization of mIAMs in cancers.
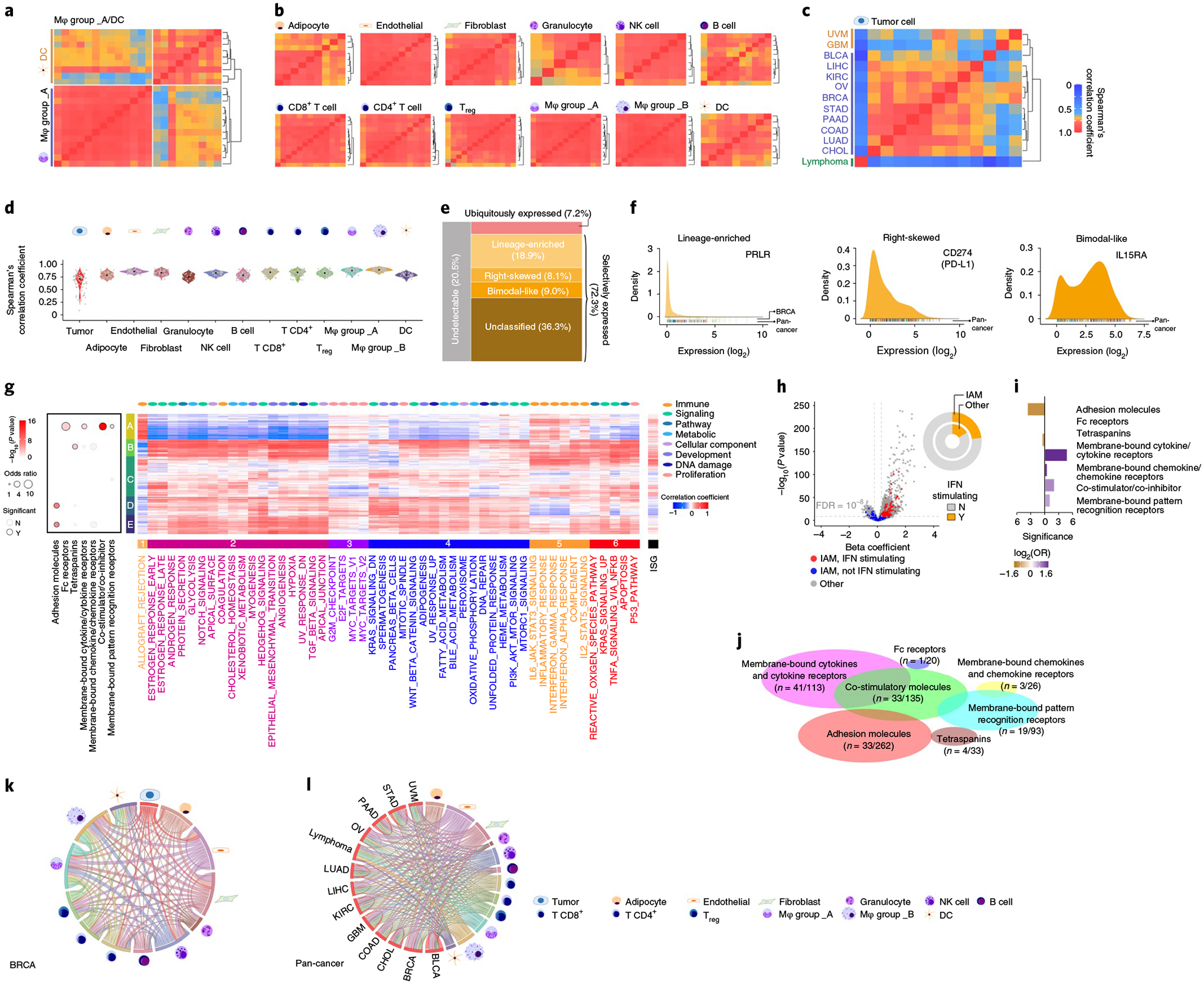
a–c, Heatmaps showing Spearman’s correlation coefficients between the fractional expression profiles of 614 mIAMs in macrophages and DCs (a), 12 stromal cell types (b) and tumor cells (c) from 13 cancer types profiled by scRNA-seq. Rows and columns were ordered by unsupervised hierarchical clustering. d, Violin plot showing distribution of Spearman’s correlation coefficients in tumor cells and 12 stromal cell types. e, Mosaic plot showing the classification of mIAMs based on their expression patterns across cancer cell lines. f, Density plots showing the expression distribution of typical examples of selectively expressed mIAMs across cancers. Left: lineage enriched (PRLR); middle: right skewed (CD274/PD-L1); right: bimodal like (IL15RA). g, Heatmap showing unsupervised hierarchical clustering of Pearson’s correlation coefficients between expressed mIAMs and signature scores of 50 ‘hallmark’ gene sets. The numbered bars indicate mIAMs (left) and gene sets (bottom) sharing similar correlation patterns. Bubble plot (left) shows the enrichment for seven categories of mIAMs within each clustered gene group. P values were calculated using two-sided Fisher’s exact test. h, Volcano plot shows gene expression association with interferon score (ISG). Each dot represents one protein-coding gene. The x axis represents the effect of each gene, reported as the β coefficient. The y axis represents −log10(P) values from the Bioconductor Limma package. The Benjamini–Hochberg method was used to adjust the P values. The mIAMs are highlighted in red if they show significant (adjusted P < 10−8) and positive (β coefficient > 0) association with ISG or blue otherwise. Circle plot shows proportion of ISG positively correlated genes among mIAMs and other genes. i, Bar plot showing enrichment of ISG positively correlated genes in the corresponding mIAM categories. Purple: enriched; orange: depleted. j, Scaled Venn diagram showing the functional families among the cell mIAMs that are positively correlated with ISG. k,l, Circos plots showing the number of mIAM-associated interactions between cell types in breast cancer (k) and pan-cancer (l). Paired cell types with significant cell–cell interactions identified by CellPhoneDB were connected by lines. The width of the lines indicates the normalized number of mIAM-associated interactions between two cell types. Mφ, macrophage; NK, natural killer; Treg, regulatory T cell.
Consistent with observations from scRNA-seq, most mIAMs (72.3%; n = 444) were selectively detectable in a portion of cell lines (defined as selectively expressed), whereas only 7.2% (n = 44) of mIAMs exhibited a ubiquitous expression pattern. The selectively expressed mIAMs were further classified into four categories based on their expression distribution (Fig. 7e,f and Supplementary Table 32). Similar results were observed when we excluded the cell lines from hematological malignancies (Extended Data Fig. 9a). To further characterize intrinsic signaling pathways that may regulate expression of mIAMs in tumor cells, expression correlations between detectable mIAMs and 50 ‘hallmark’ gene sets61 were estimated. Based on expressional correlations with these core signaling pathways in cancer, mIAMs were clustered into five groups (Fig. 7g and Supplementary Table 33). For example, group A mIAMs, in which costimulator/co-inhibitor molecules were significantly enriched, were positively correlated with immune and proliferation pathways and negatively correlated with most cancer-associated pathways. In contrast, group D mIAMs, in which adhesion molecules were significantly enriched, were negatively correlated with immune pathways. Using a signature score for interferon-stimulated gene (ISG) expression62, intrinsic interferon activity was estimated for each cancer cell line. We found that the percentage of mIAMs that was positively correlated with interferon signal was significantly higher than other genes at a genome-wide level (OR = 1.5, P =1.9 × 10−4; Fig. 7h). Among 112 mIAMs with expression levels that were positively associated with interferon score (Fig. 7i and Supplementary Table 34), membrane-bound cytokines/cytokine receptors and costimulator/co-inhibitor molecules were significantly enriched compared with other mIAM groups (Fig. 7j). Finally, potential tumor–stroma interactions mediated by mIAMs were predicted by the CellPhoneDB algorithm63 using scRNA-seq profiles. After normalizing overall interactions, potential mIAM-mediated interactions between tumor cells and individual stromal cell populations were estimated in each tumor type (Fig. 7k,l, Extended Data Fig. 9b and Supplementary Table 35). Consistently, across all cancer types examined, higher numbers of interactions were found between tumor cells and T cells, as well as tumor cells and myeloid cells, whereas B cells and granulocytes showed fewer interactions with tumor cells (Fig. 7l). It is interesting that remarkable numbers of mIAM-mediated interactions were also found between tumor cells and nonimmune stromal cell populations such as fibroblasts and endothelial cells, suggesting that they may also be involved in immune regulation in the tumor microenvironment.
Evaluation of GESPs as therapeutic targets in oncology.
GESPs have been proposed as a major source for druggable targets, given that the vast majority of GESPs can be recognized by antibodies, ADCs or CAR-Ts. Beyond immunotherapy approaches, a considerably large number of GESPs may also be targeted by small molecules6. To this end, we analyzed the druggability of GESPs based on prediction from the Open Target project6,28, and found that 10.5%, 14.0% and 11.0% of GESPs are classified as ‘Clinical_Precedence’, ‘Discovery_Precedence’ and ‘Predicted_Tractable’, respectively, suggesting that ~36.0% of GESPs may be targeted by small molecules (Fig. 8a and Supplementary Table 36). Consistent results were also observed from the PHAROS database64 (Fig. 8a and Supplementary Table 36). However, >66.8% of GESPs are defined as understudied genes (Pubtator score <150; Supplementary Table 36), indicating that functional characterization of GESPs is urgently needed for potential drug development for human diseases. Next, we analyzed current applications of GESPs in cancer treatment. Among 162 FDA-approved anticancer-targeted (small molecules) or immune therapy drugs (Supplementary Table 37), we found that 64.8% (105/162) of them are reported to directly target or bind to GESPs, especially CAR-Ts (100%), ADCs (100%) and antibody drugs (82.8%) for cancer treatment (Fig. 8b).
Fig. 8 |. Evaluation of GESPs as therapeutic targets in anticancer drug development.
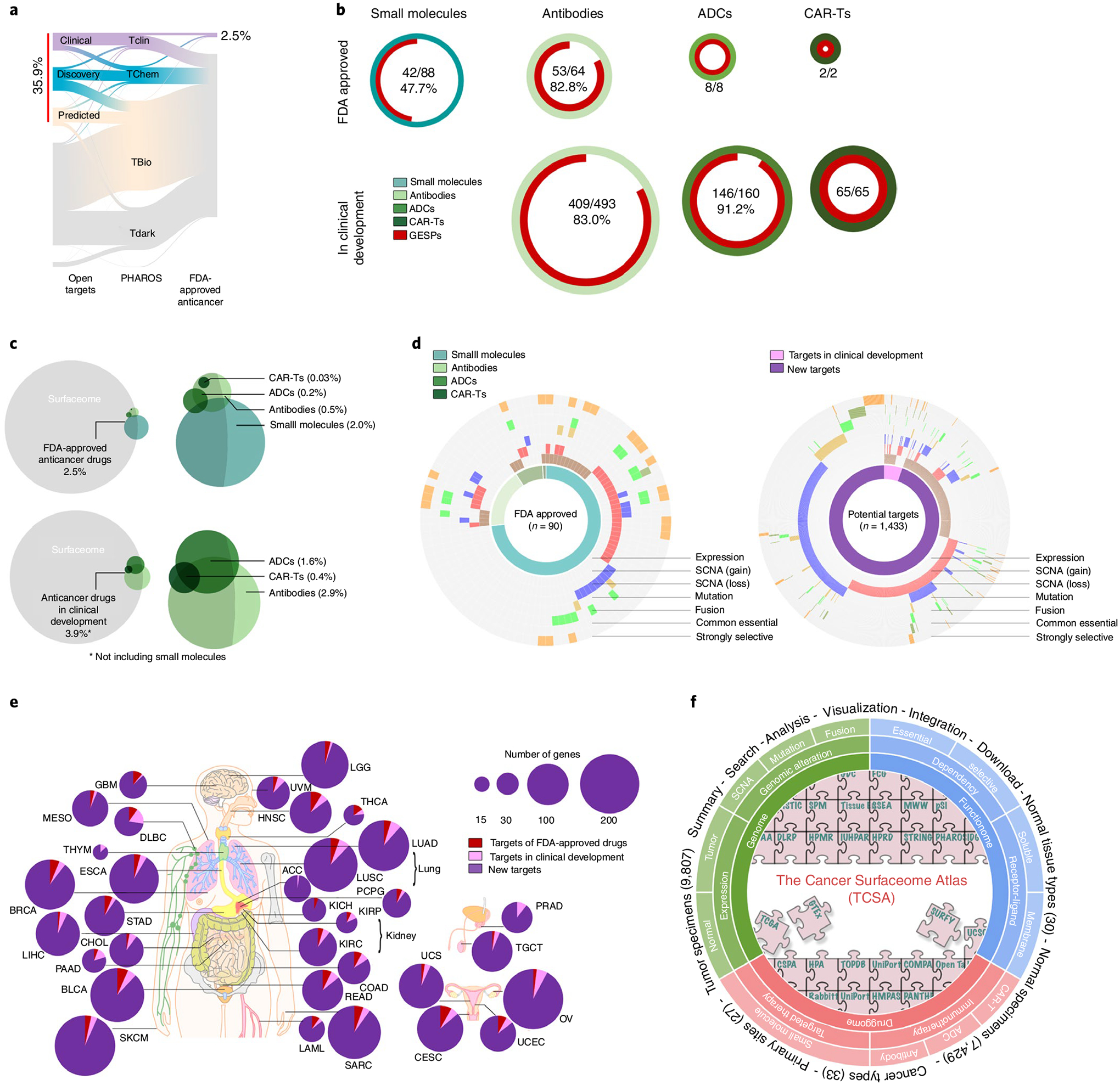
a, River plot showing druggability of GESPs as small-molecule drug targets based on definition by the Open Target and PHAROS projects. The width of the bar is proportional to the number of GESPs at each druggability level. b, Proportion of GESPs (red) serving as direct targets for small molecule drugs (cyan), antibody drugs (light green), ADCs (green) and CAR-Ts (dark green) that have been approved (top) or are in clinical development (bottom) for cancer treatment. Ring size represents the number of drugs. c, Venn diagram showing the overlap between GESPs (gray) and target genes of small molecule drugs (cyan), antibody drugs (light green), ADCs (green) and CAR-Ts (dark green). Top: FDA-approved anticancer drugs; bottom: anticancer drugs in clinical development. d, Genomic and functional features identified in GESPs that are targets of FDA-approved anticancer drugs (left) or genes with therapeutic potential (right). The caGESPs: brown, recurrent SCNA gain: red; recurrent SCNA loss: blue; recurrent mutation: gold; recurrent fusion: green; common essential: olive; strongly selective: orange. e, Summary of GESPs that may serve as therapeutic targets for cancer treatment across 33 cancer types. The size of each circle corresponds to the number of identified GESPs in a given cancer type. Red: targets of FDA-approved anticancer drugs; pink: targets in clinical development; purple: targets identified in the present study. f, Overview of TCSA data portal. TCSA database integrates genomic, functional and pharmacological information on the human surfaceome across 33 cancer types.
Similar results were also observed for drugs in clinical development (Fig. 8b and Supplementary Table 38). Even so, however, only a small percentage of GESPs serve as targets for anticancer drugs that have been approved by the FDA or are in clinical development for oncology applications (2.5% and 3.9%, respectively; Fig. 8c). Among them, ERBB2/Her2, EGFR, MS4A1/CD20, PDCD1/PD1 and CD19 are the top five target proteins and are targeted by >30.9% of approved drugs and 23.1% of drugs in clinical development. This suggests that identification and prioritization of GESP targets are still challenges in anticancer drug development, and that most studies are limited to a small fraction of GESPs. To test whether genomic and functional characterization of GESPs may be used for identification and prioritization of drug targets, we analyzed clinically approved anticancer drugs as an example and found that >68.9% of them have at least one genomic or functional feature identified by our systematic analysis, including specific expression in cancer, recurrent SCNA, mutation, fusion and fitness/essential (Fig. 8d). In this regard, we evaluated GESPs that have such features but have not yet been used as targets for FDA-approved anticancer drugs, and identified a total of 1,433 potential targets across 33 cancer types (Fig. 8d and Supplementary Table 39). For each cancer type, an average of 86 potential targets (range: 15–205) were found (Fig. 8e and Supplementary Table 40). Finally, a publicly accessible data resource, The Cancer Surfaceome Atlas (TCSA), was developed through the Functional Cancer Genome data portal (Fig. 8f; http://fcgportal.org/TCSA).
Discussion
Due to their unique protein subcellular location and crucial biological functions, GESPs have been proposed as a major source for identification of druggable targets for human disease5,6. Indeed, we found that 64.8% of FDA-approved anticancer immune and targeted therapy drugs directly target GESPs. However, the currently drugged GESPs in oncology represent only 2.5% of the surfaceome, and preclinical anticancer drug discovery efforts are also focused on a relatively small fraction of the surfaceome due to challenges in target identification and prioritization. Furthermore, biological functions of 66.8% of GESPs are still understudied, leading to additional difficulties in designing drugs targeting GESPs. Excitingly, recent advances in large-scale and high-throughput studies have provided powerful resources for comprehensive identification and prioritization of GESPs with therapeutic potential in cancers. Thus, systems biology efforts to integrate the above omics resources are urgently needed. In the present study, we combined multiple computational approaches and systematically characterized the human surfaceome across 33 adult cancers. A publicly accessible surfaceome database (TCSA) was developed to assist researchers to explore GESPs in cancer genomes.
Consistent with their functions, the expression of the GESPs is more lineage- and cancer-type specific compared with proteins located in other subcellular locations. We identified 409 unique GESPs that are ‘specifically’ expressed in certain cancer types (caGESPs), providing a genome-wide view of the potential GESP targets for immunotherapy. Supporting our unbiased systematic discovery, 13.4% (55/409) of identified caGESPs have already been used for CAR-T, ADC or antibody drug development in the clinic. Even so, it is still challenging to use an individual caGESP to ‘uniquely’ define tumors and ‘completely’ spare normal cells. The ‘ideal’ targets (that is, highly and specifically expressed in a given cancer type) are still limited, especially for solid tumors. Given that ‘on-target–off-tumor’ toxic side effects are one of the major clinical problems of CAR-T and antibody therapies8,9,11, strategies that can more specifically recognize cancer cells are critically important to the development of effective and safe immunotherapy. Thus, technologies that use a combination of multiple GESPs, such as logic-gated CAR-Ts and bispecific antibodies11,42–46, can further increase drug specificity and thus create more precise treatments for patients with cancer. In this regard, after systematically evaluating potential combinations on a genome-wide scale, 179 and 443 unique pairs were identified for ‘AND CAR-T’ and ‘iCAR-T’ strategies, respectively.
Characterization of recurrent genomic alterations is a useful strategy to identify functional GESPs with therapeutic potential13,14. For example, discovery of the recurrent amplification of ERBB2/Her2 in cancer led to the development of anti-Her2 antibody therapy for patients with breast cancer, and identification of the EGFR mutation in cancers provided a strong rationale for small molecule inhibitor-based targeted therapy for lung cancer and CAR-T therapy for glioblastoma. We systematically analyzed recurrent genomic alterations for GESPs across 33 cancer types and identified 1,433 potential targets recurrently altered in at least one cancer type. Notably, 35.5% of GESPs were defined as ‘druggable’ genes for small molecule compounds based on their protein structural and pharmacological properties. This suggests promising opportunities to design small molecule compounds to specifically target cancer driver GESPs.
Recent high-throughput functional studies, such as CRISPR-based genetic screening and protein–protein interaction prediction, have provided additional resources to functionally characterize the surfaceome in cancer. Based on large-scale CRISPR screening in cancer cell lines, we identified 65 and 80 GESPs that are defined as ‘common essential’ and ‘strongly selective’ genes for cancer cell growth in vitro. Most importantly, 38.6% of such essential GESPs showed strong and positive correlations between their cell growth dependence and mRNA expression/CNAs. Thus, targeting these essential GESPs (especially the strongly selective GESPs) by small molecule inhibitors or neutralizing antibodies may serve as strategies to treat cancers. Finally, advances in both experimental and computational protein–protein interaction screens (the protein interactome) allowed us to systematically identify receptor–ligand interactions of GESPs on a genome-wide scale; 1,278 receptor–ligand pairs were identified in our analyses, which not only serve as a rich resource for design of ligand-based CAR-T therapies, but also provide insight into regulations of GESPs. Based on receptor–ligand coexpression patterns, most tumor specimens clustered together and were completely separated from normal tissues. This strongly indicates that intercellular communications in the tumor microenvironment play critical roles during tumorigenesis. However, most current functional studies are largely based on in vitro two-dimensional cell culture assays. High-throughput in vivo functional screening is still urgently needed for further characterization of GESP functions in cancer.
Methods
Definition of the GESPs.
To comprehensively define the human surfaceome at the whole-genome level, we integrated GESP candidates from nine independent resources, in which SPs were identified or predicted by distinct strategies (Fig. 1b and Supplementary Table 1). After converting the protein/gene names to the ENSEMBL gene annotation (GENCODE v.23), we estimated a core GESP score for each candidate based on a weighted vote approach, that is, each resource had a different voting power due to its identification/prediction principle. The resources that can define GESPs with at least one amino acid exposed to extracellular space (that is, the part of a protein located on the outer surface of the cell membrane) were given a weight of 3, the resources that define GESPs based on experimental evidence had a weight of 2 and other resources had a weight of 1. Then, using known GESPs and non-GESPs as positive and negative controls, a cutoff was established to define potential GESPs (that is, core GESP score ≥4). Next, information from COMPARTMENT, GO65 and manual literature searches were used to remove genes encoding proteins in intracellular membranes such as the nuclear membrane and mitochondrial membrane. Finally, other features such as literature evidence, protein structure and evolutionary conservation collected from PubMed and related databases (Supplementary Table 1) were used as additional score-driven factors to finalize the GESP list and estimate a final GESP score, increasing the confidence level for each GESP.
RNA-seq data processing and gene expression analysis.
The RNA-seq data, which were retrieved from TCGA, GTEx, Human Protein Atlas (HPA), Illumina’s Human BodyMap 2.0, Encyclopedia of DNA Elements (ENCODE) project and Sequence Read Archive (SRA, accession no. SRP125125, RNA-seq data for hematopoietic cells), were processed using a standard pipeline that was developed by the University of California Santa Cruz Toil RNAseq Recompute Compendium66, which was able to consistently process large-scale RNA-seq data and analyze gene expression without computational batch effects.
Proteomic data processing and protein expression analysis.
Extensive mass spectrometry-based proteomics data using isobaric tagging approaches (iTRAQ or TMT) for selected cancer types were generated by the National Cancer Institute’s (NCI’s) CPTAC. Protein-level processed data consisting of iTRAQ or TMT log(ratios) were downloaded from the CPTAC data portal (https://cptac-data-portal.georgetown.edu). Proteins that were detectable in at least half the tumor specimens in a given cancer type were subjected to further analysis. Only tumor specimens with both RNA-seq and proteomic data were used for correlation analysis. Spearman’s correlation coefficient was calculated for each mRNA–protein pair (log(FPKM + 0.001) versus log(ratios of iTRAQ or TMT) where FPKM is fagments per kilobase million). False discovery rate (FDR) correction was applied to P values assessing the statistical significance.
Identification of GESPs specifically expressed in cancers (caGESPs).
The caGESPs were identified independently for each individual cancer type by comparing mRNA expression levels of GESPs between a given cancer type (TCGA) and normal tissues from 29 organs (GTEx). As cancer-testis genes often encode immunogenic antigens for cancer immunotherapy38,67, normal testis tissues were excluded from the normal tissue pools (except for analysis on TGCT). To reduce false positives, we applied five independent computational algorithms to identify cancer-specific genes: specificity measure (SPM)68, TissueEnrich40,69, specificity index probability (pSI)70, sample set enrichment analysis (SSEA)71 and differential expression analysis by the Mann–Whitney–Wilcoxon test (MWW) (Fig. 3a). These algorithms were categorized into two groups based on their principles: group I, including TissueEnrich and SPM, which calculated a metric to assess the specificity of each gene independently; and group II, including pSI, SSEA and MWW test, which required an additional step to calculate a rank for each gene across all genes based on the specificity metrics. Notably, distinct input data matrices were used by these algorithms: for the pSI, SPM and TissueEnrich, median FPKM values of a given gene in each tissue or cancer type were used to represent the expression levels; for SSEA and MWW test, FPKM values of a given gene in each individual sample were used for analysis. For each method, both stringent and less stringent criteria were applied to define caGESPs with high and moderate confidence, respectively. SPM. SPM was adopted from TiSGeD68, by which the specificity measure for each gene in a given cancer type was calculated as the cosine value of the intersection angle between the gene’s observed expression pattern and a predefined artificial expression pattern. The observed expression pattern was represented as a vector of expression values of the gene corresponding to the given cancer type and each normal tissue type. An artificial expression pattern was predefined, representing the extreme case in which the gene was expressed in the given cancer type whereas its expression level was zero in all normal tissue types. Genes with SPM values >0.99 and 0.9 were considered to be highly confident (stringent criteria) and moderately confident (less stringent criteria), respectively.
TissueEnrich.
The function GeneRetrieval of TissueEnrich R package69 was used to classify genes into six different groups according to pairwise expression fold-change among tissue types. Genes classified as ‘Tissue-Enriched’ in a given cancer type (that is, its expression level in a given cancer type was at least fivefold higher than all normal tissue types) were considered to be highly confident (stringent criteria). Genes classified as ‘Tissue-Enhanced’ in a given cancer type (that is, its expression level in a given cancer type was at least fivefold higher than the average of all normal tissue types) were considered to be moderately confident (less stringent criteria).
The pSI statistic.
The R package pSI, developed by Dougherty et al.70, was applied to calculate a pSI value for each gene in a given cancer type. Genes with pSI values <0.001 and 0.01 in a given cancer type were considered highly confident (stringent criteria) and moderately confident (less stringent criteria), respectively.
SSEA.
SSEA was adopted from the GSEA71: the R package fgsea was applied for testing differential expression between a given cancer type and each normal tissue type. For each pairwise comparison (a given cancer type versus a given normal tissue type), all samples were ranked according to the expression level of a specific gene. Querying the sample set of cancer against the ranked sample list yielded a normalized enrichment score (higher score means stronger enrichment of expression in cancer). We ranked genes within each pairwise comparison by normalized enrichment score and assigned percentile ranks (for example, a percentile rank of 0.95 implies that the gene ranked in the top 5th percentile of all genes analyzed). Each of the percentile ranks obtained from comparisons against different normal tissue types was then combined. The genes with an average percentile rank >0.99 were considered to be highly confident (stringent criteria); the genes with a minimum percentile rank >0.9 were considered to be moderately confident (less stringent criteria).
MWW test.
Differential expression of a gene between a given cancer type and each normal tissue type was estimated by the function Wilcox_test of the R package coin72. For each pairwise comparison (a given cancer type versus a given normal tissue type), the difference in rank position of expression levels of the two groups was estimated (a higher positive value means a stronger enrichment of expression in cancer). We ranked genes within each pairwise comparison by difference in rank position and assigned percentile ranks (for example, a percentile rank of 0.95 implies that the gene ranked in the top 5th percentile of all genes analyzed). Each percentile rank obtained from comparisons against different normal tissue types was then combined. The genes with an average percentile rank >0.99 were considered to be highly confident (stringent criteria); the genes with a minimum percentile rank >0.9 were considered to be moderately confident (less stringent criteria).
To integrate the results generated by different methods, we summed the potential caGESP lists from all five algorithms based on the confidence levels, then estimated a specificity score for each potential caGESP. For each algorithm: 2 = positive by stringent criteria; 1 = positive by less stringent criteria; and 0 = negative:
where:
After a cutoff (specificity score ≥3) was estimated to define the caGESPs in a given cancer type, the caGESPs were further divided into three tiers: tier 1 (high confident caGESPs)—the caGESPs were identified by at least two algorithms with stringent criteria; tier 2 (moderately confident caGESPs)—the caGESPs were identified by at least one algorithm with stringent criteria and one algorithm with less stringent criteria; and tier 3 (low confident caGESPs)—the caGESPs were identified by at least three algorithms with less stringent criteria. Finally, to reduce the expression interference from tumor-infiltrating immune cells in tumor specimens, GESPs that are highly expressed in immune cells were excluded (except for analysis on hematopoietic malignancies) based on the RNA-seq profiles from 30 distinct types of hematopoietic cells and 6 lymphatic tissues.
Evaluation of GESP combinations for logic-gated CAR-T cell design.
The teGeneRetrieval function of the TissueEnrich R package69 was used to classify expression specificity of GESPs across normal healthy tissues from the GTEx. Among six categories defined by the TissueEnrich algorithm, the GESPs in the categories ‘Tissue Enriched’, ‘Tissue Enhanced’ and ‘Group Enriched’ were considered to have relatively specific expression in normal tissues and used for downstream analysis. GESPs in other categories, such as ‘Expressed in all’, ‘Not Expressed’ and ‘Mixed’, were excluded from our analysis. As with the caGESP analysis, normal testis tissues were excluded from the normal tissue pools (except for analysis on TGCT). Logic-gated GESP pairs were identified independently for each individual cancer type by evaluating all potential combinations between the caGESPs in a given cancer type and the GESPs that were defined as relatively specific to normal tissues.
Identification of caGESP combinations for ‘AND CAR-T’ design: we defined caGESP pairs, in which both caGESPs were identified from the same cancer type (for example, coexpressed in a given cancer type) and showed significantly and mutually exclusive expressional patterns across normal tissues, as potential candidates for the ‘AND CAR-T’ strategy (Fig. 4a, b). To minimize ‘on-target–off-tumor’ toxicity, we evaluated the mutual exclusivity of the caGESPs at both tissue-type and individual levels across normal tissues from the GTEx. At the tissue-type level, caGESP expression in each tissue type was estimated as the median measurement across all samples of the corresponding tissue type. The z-scores were converted from log(transformed FPKM values). The tissue types with potential ‘on-target–off-tumor’ toxicity for a given caGESP (that is, the caGESP is expressed at relatively high level) were defined as having z-scores >1. For each potential combination of caGESPs, the OR of both caGESPs sharing ‘on-target–off-tumor’ tissue types was calculated:
| caGESP A | |||
|---|---|---|---|
| Not expected | Expected | ||
| Not expected | a | b | |
| Expected | c | d | |
where
a = number of normal tissue types in which neither caGESPs were expressed;
b = number of normal tissue types in which only caGESP A was expressed;
c = number of normal tissue types in which only caGESP B was expressed;
d = number of normal tissue types in which both caGESPs were expressed.
The combination pairs of caGESPs that were mutually exclusive at the tissue-type level were defined as those with OR of 0 (the number of normal tissue types in which both caGESPs were expressed was 0). At the individual-sample level, ‘on-target–off-tumor’ samples (that is, a caGESP expressed at a relatively high level) were determined by a similar approach with z-scores >1. Mutually exclusive pairs at the individual-sample level were identified using the CoMEt algorithm73. CoMEt was used to overcome the challenge of low-frequency occurrence combinations because a considerable portion of tissue-specific caGESPs had ‘on-target–off-tumor’ toxicity for a relatively small subset of normal tissues. Pairs of caGESPs that were mutually exclusive at the individual-sample level were defined as those with FDR < 1%. Finally, we further calculated a priority score for each of the candidate pairs:
in which z1k and z2k represented tissue-type-level z-scores of the pair of caGESPs in tissue k. Candidate pairs with higher priority score were considered to have better performance.
Identification of caGESP combinations for iCAR-T design: we defined the GESP pairs, in which the caGESP and its paired GESP were coexpressed in the same normal tissues, but the paired GESP was not detectable in the cancer type in which the caGESP was identified, as potential candidates for the iCAR-T strategy (Fig. 4e,f). The challenge of ‘iCAR’ strategy was identification of GESPs that were absent in a given cancer type but coexpressed with a caGSEP in normal tissues. We considered GESPs with FPKM < 1 in a given cancer type (hereafter called nondetectable GESPs in cancer (ndGESPs)) as potential partners of caGESPs for the ‘iCAR-T’ strategy. We evaluated the co-occurrence of caGESPs and ndGESPs at both the tissue-type and the individual levels across normal tissues from the GTEx. At the tissue-type level, expression of caGESPs and ndGESPs in each tissue type was estimated as the median measurement across all samples of the corresponding tissue type. The z-scores were converted from log(transformed FPKM values). The tissue types with potential ‘on-target–off-tumor’ toxicity for a given caGESP (that is, the caGESP is expressed at a relatively high level) were defined as having z-scores >1. The same criterion was applied to define the tissue types in which the iCAR-T activation may be blocked by select ndGESPs (that is, the ndGESP expressed at a high level). For each potential combination pair (caGESP and ndGESP), the OR of blocking ‘on-target–off-tumor’ toxicity was calculated as:
| ‘on-target/off-tumor’ toxicity | |||
|---|---|---|---|
| Not expected | Expected | ||
| Not expected | a | b | |
| CAR activation inhibited | Expected | c | d |
where
a = number of normal tissue types in which neither caGESP A nor ndGESP B was expressed.
b = number of normal tissue types in which only caGESP A was expressed;
c = number of normal tissue types in which only ndGESP B was expressed;
d = number of normal tissue types in which both caGESP A and ndGESP B were expressed.
The combination pairs (caGESPs and ndGESPs) were considered to be mutually exclusive at the tissue-type level if the OR was infinity (there were no normal tissue types in which caGESP A was expressed whereas ndGESP B was not). In addition, we required that the ndGESPs had a FPKM value >10 in the dominant ‘on-target–off-tumor’ tissue types of the paired caGESPs. At the individual-sample level, ‘on-target–off-tumor’ samples for caGESPs or ‘iCAR-T activation inhibited’ samples for ndGESPs (that is, in these individual samples, caGESP or ndGESP is expressed at relatively high levels across specimens from the GTEx) were determined using a similar approach (z-scores >1). Coexpressing pairs at the level of individual normal samples were identified by one-sided Fisher’s exact test (right tailed), with FDR < 1%. Finally, we further calculated a priority score for each candidate pair:
in which z1k and z2k represented tissue-type level z-scores of the caGESP and ndGESP in each pair, respectively, in tissue k. The candidate pairs with higher priority score were considered to have better performance.
TCGA genomic profile processing and analysis.
TCGA genomic profiles for CNAs, mutations and fusions were retrieved, processed and analyzed through a standard pipeline developed by the Functional Cancer Genome (FCG) project50,51. Recurrent SCNAs, mutations and fusions, as well as G-score and M-score, were estimated at both individual and pan-cancer levels as described in our previous publications50,51.
Characterization of dependence of the GESPs.
The integrated CRISPR–Cas9 dependency profile was retrieved from Pacini et al.27, in which two independent screen profiles from the DepMap (https://depmap.org) and Score (https://score.depmap.sanger.ac.uk) were integrated by a computational approach. Criteria for definition of the common essential and strongly selective genes have been described previously by the DepMap team23,74 and the Score team25. Briefly, common essential genes were defined by two methods: the 90th percentile method75 and the Adaptive Daisy Model25. A strongly selective gene (that is, a gene with dependence observed in a subset of cancer cells in a large pan-cancer screen) was defined as a given gene with a skewed-likelihood ratio test (LRT) value >100. Both common essential and strongly selective genes were considered as essential genes for cancer cell viability. Assessment of enrichment for essential genes for cancer cell viability was performed using Fisher’s exact test across the genes anchored to different subcellular locations. For the GESPs that were defined as either common essential or strongly selective, we used the Bioconductor Limma package76 to estimate the correlation between their dependence (dependence scores) and mRNA expression or DNA copy number levels. The processed mRNA expression (RNA-seq) and DNA copy number (whole-exome sequencing or SNP array) profiles of the cancer cell lines were retrieved from the DepMap portal. Cohen’s effect size was scaled so that it measured the change in dependence across the interquartile range (IQR) of mRNA expression or the DNA copy number. For DNA copy number, log2(relative to ploidy + 1) was used. For mRNA expression, log2(transformed TPM) values using a pseudo-count of 1 were used.
Characterization of receptor–ligand interactions of GESPs.
Known receptor–ligand pairs were retrieved from the Database of Interacting Proteins (http://dip.doe-mbi.ucla.edu/dip/dlrp/dlrp.txt), the International Union of Basic and Clinical Pharmacology (http://www.guidetopharmacology.org/DATA/interactions.csv) and the Human Plasma Membrane Receptome (http://receptome.stanford.edu; retrieved 23 October 2019). To exclude intracellular receptor–ligand interactions, only surface–surface and surface–extracellular interactions were used for analysis. Computationally inferred receptor–ligand pairs were estimated through searching for experimentally validated the protein–protein interaction Human Protein Reference Database (HPRD) (http://www.hprd.org/RELEASE9/HPRD_Release9_041310.tar.gz) and the STRING database (https://stringdb-static.org/download/protein.links.full.v11.0/9606.protein.links.full.v11.0.txt.gz) between a set of putative receptors and putative ligands. For HPRD, we requested that protein–protein interactions be supported by at least one source (in vitro evidence, in vivo evidence or yeast two-hybrid evidence). For STRING, physical-binding interactions (score ≥ 700) and experimental interactions (score ≥ 700) were included in our analysis. Putative receptors were compiled from known interacting receptors and GESPs. Putative ligands were compiled from known interacting ligands and a set of secreted proteins predicted by DeepLoc (http://www.cbs.dtu.dk/services/DeepLoc). Then, we estimated a confidence score for each receptor–ligand pair based on a weighted vote approach: known receptor–ligand pairs had a weight of 2 and inferred receptor–ligand pairs had a weight of 1. Receptor–ligand pairs with a confidence score ≥2 were considered as potential candidates for further analysis.
ScRNA-seq profile processing and analysis.
ScRNA-seq profiles from 13 cancer types were retrieved and processed by a unified computational pipeline. Unique molecular identifier counts were normalized to transcripts per million (TPM) and transferred into log2(TPM/10 + 1). The identity of individual cells in each dataset was annotated by scMatch algorithm (https://github.com/asrhou/scMatch)77. For tumor-infiltrating stromal cells, reference gene expression data were collected from FANTOM5, and SingleR (https://figshare.com/s/efd2969ce20fae5c118f). For tumor cells, reference gene expression profiles were collected from the CCLE project (https://ndownloader.figshare.com/files/24613349) and the Xena Cancer browser (https://xenabrowser.net). Stromal cells were classified into 12 cell types (adipocyte, B cell, DC, endothelial, fibroblast, granulocyte, macrophage group_A, macrophage group_B, natural killer cell, T CD4, T CD8 and regulatory T cell). To evaluate diversity of the gene expression states of scRNA-seq profiles, gene expression states were estimated using a left truncated mixture algorithm (https://github.com/zy26/LTMGSCA)78. For each cell population that has at least 50 cells from a cancer type, the expression level for a gene was quantified as the fraction of cells defined as having active expression states. Spearman’s correlation coefficients were calculated to assess the similarities between expression profiles across cell types from different cancer types.
Classification of expression distribution across cell lines.
Gene expression data of cancer cell lines were retrieved from the DepMap data portal (https://depmap.org/portal)60. Genes were classified into six categories according to their mRNA expression levels across the cancer cell lines: (1) undetectable genes: genes that showed undetectable RNA expression (FPKM < 1) for >95% of cancer cell lines; (2) ubiquitously expressed genes: genes that were expressed (FPKM > 1) for the majority of tumor samples (95%); (3) lineage-enriched genes: genes with elevated (fivefold) RNA expression levels in an individual cancer type or a group of cancer types (a maximum of seven cancer types) compared with all other cancer types; (4) right-skewed genes: genes with expression levels that had skewedness >0.5 and skewed-LRT values >125 (that is, 125 times more likely to have been sampled from a right-skewed distribution than a normal distribution); (5) bimodal-like genes: genes with expression levels that had a bimodal index79 >1.2 and bimodal-LRT values >125 (that is, 125 times more likely to have been sampled from a bimodal distribution than a normal distribution); and (6) unclassified: genes that were not assigned to any of the above five groups. The hierarchy of groups used to classify genes was: undetectable > ubiquitously expressed > lineage enriched > right skewed > bimodal like > unclassified. Genes from the ‘lineage-enriched’, ‘right-skewed’, ‘bimodal-like’ and ‘unclassified’ groups were considered to be selectively expressed genes.
Association of mIAM expression with signaling pathways.
A collection of 50 hallmark gene sets were downloaded from the Broad/UCSD Molecular Signatures Database (MSigDB)61. The gene set of ISGs was described by Liu et al.62. For each gene from a specific gene set, the mean absolute deviation-modified z-score (ZMAD)-normalized RNA expression was calculated across cancer cell lines. The signature score of the specific gene set in each sample was then defined as the mean ZMAD value of all genes included in the gene set. Finally, the associations between expression of mIAMs and the signature scores of hallmark gene sets were assessed using Pearson’s correlation coefficients. Unsupervised hierarchical clustering was performed to split mIAMs into groups, each of which consisted of mIAMs demonstrating similar association patterns with the core signaling pathways. For each group, enrichment for mIAMs of a specific category was assessed using Fisher’s exact test.
Cell–cell communication network.
Using CellPhoneDB (www.cellphonedb.org)63, the mIAM-associated receptor–ligand pairs were mapped on to different cell types for each cancer type to identify cell–cell interactions. CellPhoneDB was used to infer the potential interaction strength between subsets of cells based on mRNA expression levels. The ligands and receptors, which were expressed in >10% of cells in a given cell type, were considered in our analysis. We iterated through all the cells for 1,000 permutations to determine the receptor/ligand expression levels. Significant interactions (P value (one-sided) for a permutation test <0.05) were identified between cell types in each cancer type. The numbers of mIAM-associated interactions between cell types, as identified by the CellphoneDB algorithm, were normalized to the overall interactions of each cancer type accordingly.
Statistics and reproducibility.
Large-scale and multidimensional profiling data generated by the publicly accessible databases (TCGA, GTEx, CPTAC and DepMap) were used, so statistical analysis was not used to predetermine sample size in the present study. For TCGA analysis, if more than one profiling file existed for a patient in TCGA, only one single file would be selected and used, and detailed methods for exclusion of duplicated profiling files are included in the Reporting Summary. The number of samples in each data cohort is reported in Supplementary Tables 3, 4, 7 and 25. The computational analyses were not randomized and the investigators were not blinded during data analyses of the present study.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Tissue specificity of GESPs across normal tissues.
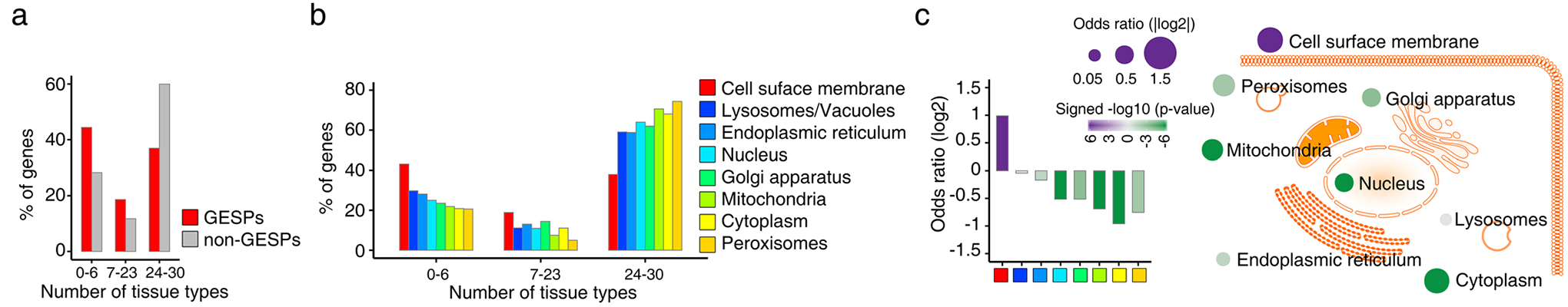
a, Percentages of genes which were detectable (median FPKM value >1) by RNA-seq analysis in 0–6, 7–23, and 24–30 tissue types. Red: GESPs; and gray: non-GESPs. b, The percentages of genes detectable in 0–6, 7–23, and 24–30 tissue types, stratified by subcellular location of gene products. c, Bar plot (left) and bubble plot (right) show enrichment of tissue type-specific genes in the corresponding subgroups based on subcellular location of gene products. P-values were calculated by two-sided Fisher’s exact test. Purple, enriched; green, depleted. The size of the bubble: absolute value of log2(odds ratio).
Extended Data Fig. 2 |. GESPs specifically express in distinct cell populations in tumor microenvironment.

a, Bubble plots show expression levels and percentages of the cells expressing ERBB3, EGFR, MET, PDGFRB, KDR, CTLA4, CSF1R, or IL2RB in each cell population across 13 cancer types. Bubble size: percentage of positive cells; intensity of color: expression level. b, Violin plots show gene expression levels of ERBB3, EGFR, MET, PDGFRB, KDR, CTLA4, CSF1R, and IL2RB in each cell population at single cell. Each plot presents expression level in one cell.
Extended Data Fig. 3 |. Distribution of correlation coefficient between protein and mRNA expression levels.
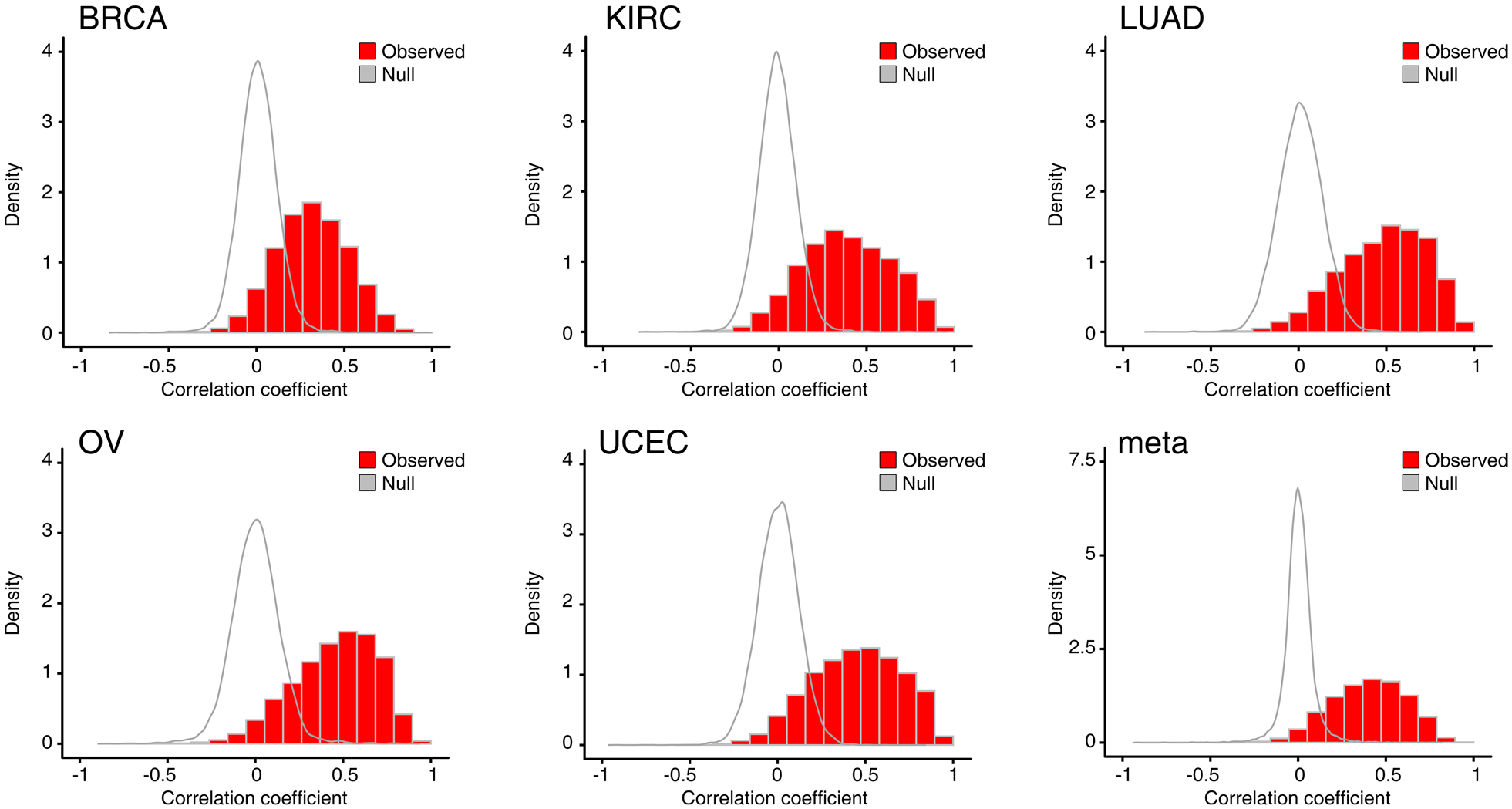
The empirical null distribution for correlation of mRNA and protein generated by permutating samples is shown for comparison (all p-values < 2.2 × 10–16, two-sided Wilcoxon rank-sum test).
Extended Data Fig. 4 |. Characteristic of caGESPs stratified by specificity score.
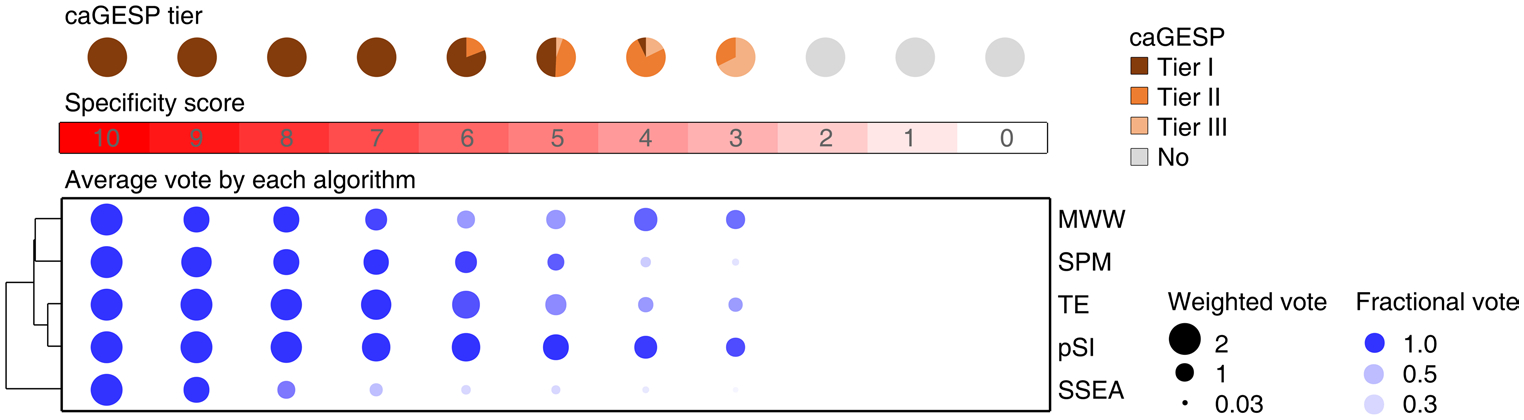
Up panel: pie charts showing percentages of caGESPs in each tier stratified by specificity score. All caGESPs have specificity score ≥ 3. Bottom panel: dot plot showing the relative contribution of each algorithm to identification of caGESPs stratified by specificity score. Size, weighted vote; color intensity, fractional vote.
Extended Data Fig. 5 |. Somatic copy number alterations of the GESPs across cancers.
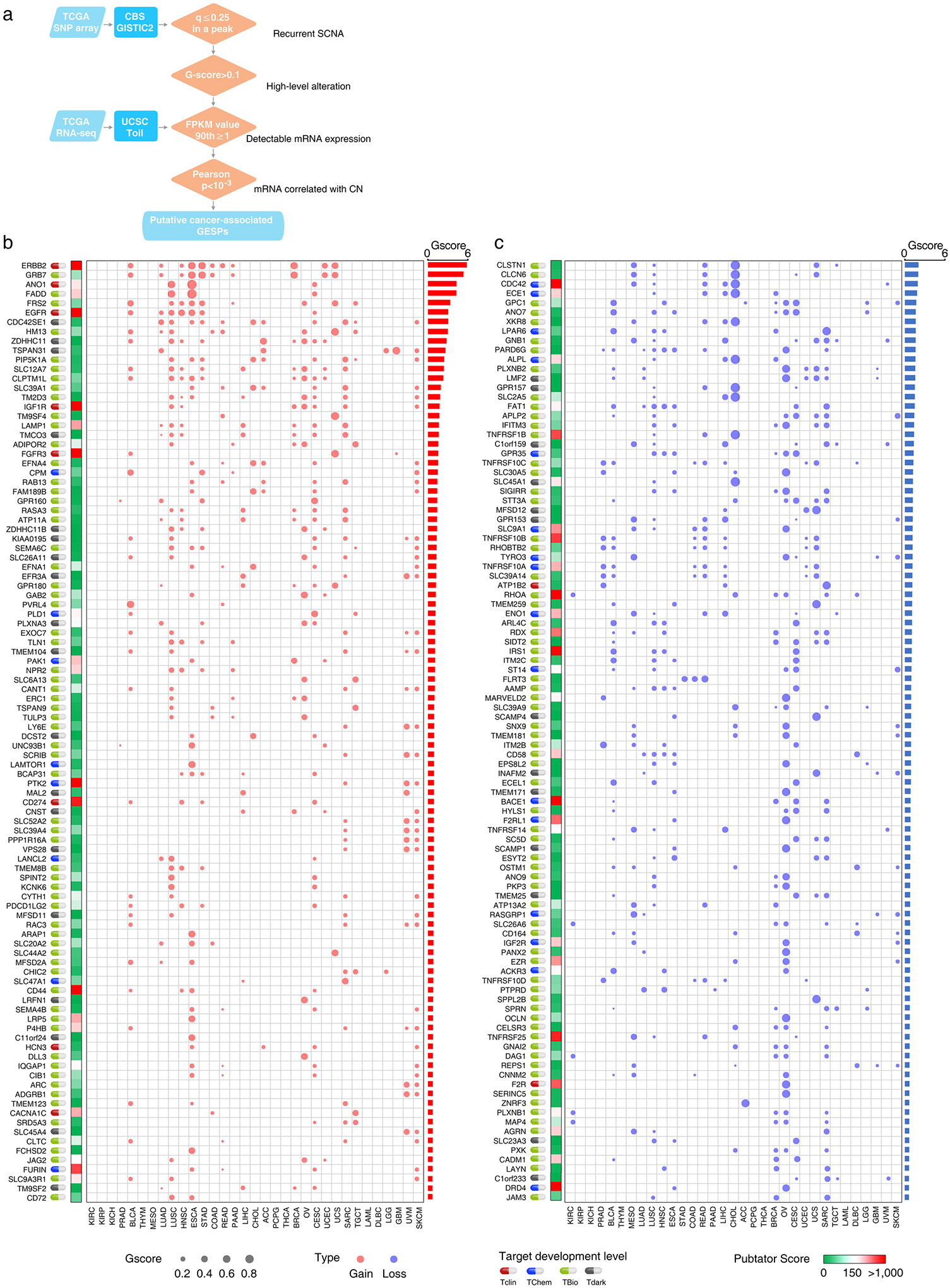
a, The workflow of somatic copy number alteration analysis. Four criteria were used to identify the putative cancer-causing GESPs driven by SCNAs in each cancer type. b and c, Bubble plot shows the SCNA G-scores, which consider both the amplitudes of the aberrations and the frequencies of their occurrence across samples, of the putative cancer-causing GESPs driven by SCNAs in each cancer type. b, copy number gain; c, copy number loss. The size of the bubble: G-score; red: gain; blue: loss. Pubtator scores, which represent the number of publications for a given gene and were retrieved from Pubtator database, are shown next to G-score plot. Green: 1–150 (understudied genes); Red: >150. Target development levels of each gene, which were retrieved from PHAROS database, are shown in the left. Red: Tclin; blue: TChem; green: Tbio; grey: Tdark. Genes are ordered according to overall G-score (from largest values to smallest values). Top 100 GESPs with highest overall G-score were shown.
Extended Data Fig. 6 |. Somatic mutations of the GESPs across cancers.
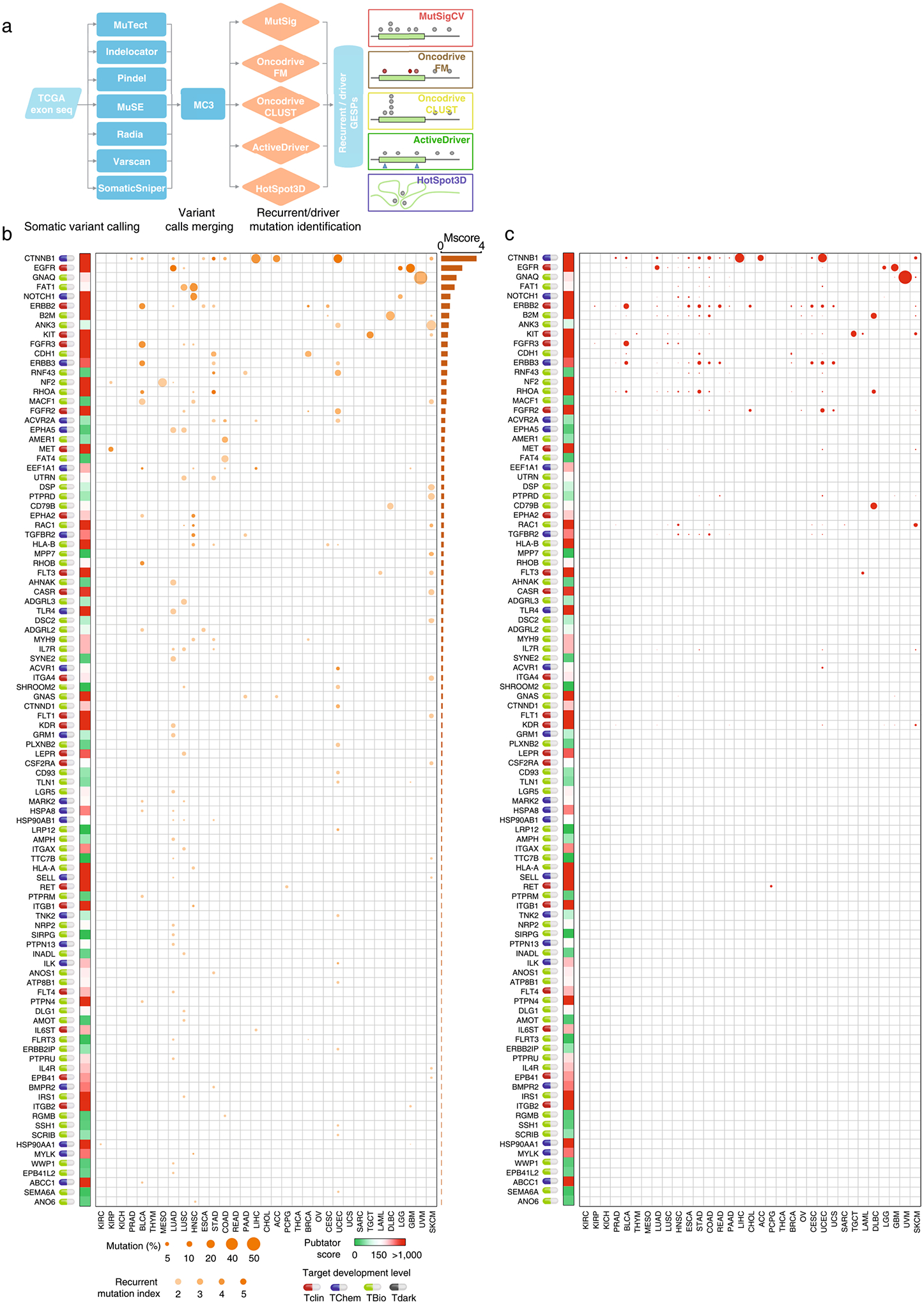
a, The workflow of recurrent somatic mutation analysis. Five complementary methods were integrated to identify the putative cancer-causing GESPs driven by mutatons in each cancer type. b, The bubble plot shows the mutation frequencies and mutation indexes of the putative cancer-causing GESPs driven by somatic mutations in each cancer type. The size of the bubble: mutation frequency; intensity of color: mutation index. c, The bubble plot shows the frequencies of hotspot mutation of GESPs in each cancer type. The size of the bubble: hotspot mutation frequency. The locations of hotspot mutated regions were retrieved from cancerhotspots.org. Pubtator scores, which represent the number of publications for a given gene and were retrieved from Pubtator database, are shown the next to bubble plot. Green: 1–150 (understudied genes); Red: >150. Target development levels of each gene, which were retrieved from PHAROS database, are shown in the left. Red: Tclin; blue: TChem; green: Tbio; grey: Tdark. Genes are ordered according to overall M-score (from largest values to smallest values). Top 100 GESPs with highest overall M-score were shown.
Extended Data Fig. 7 |. Transcript fusions of the GESPs across cancers.
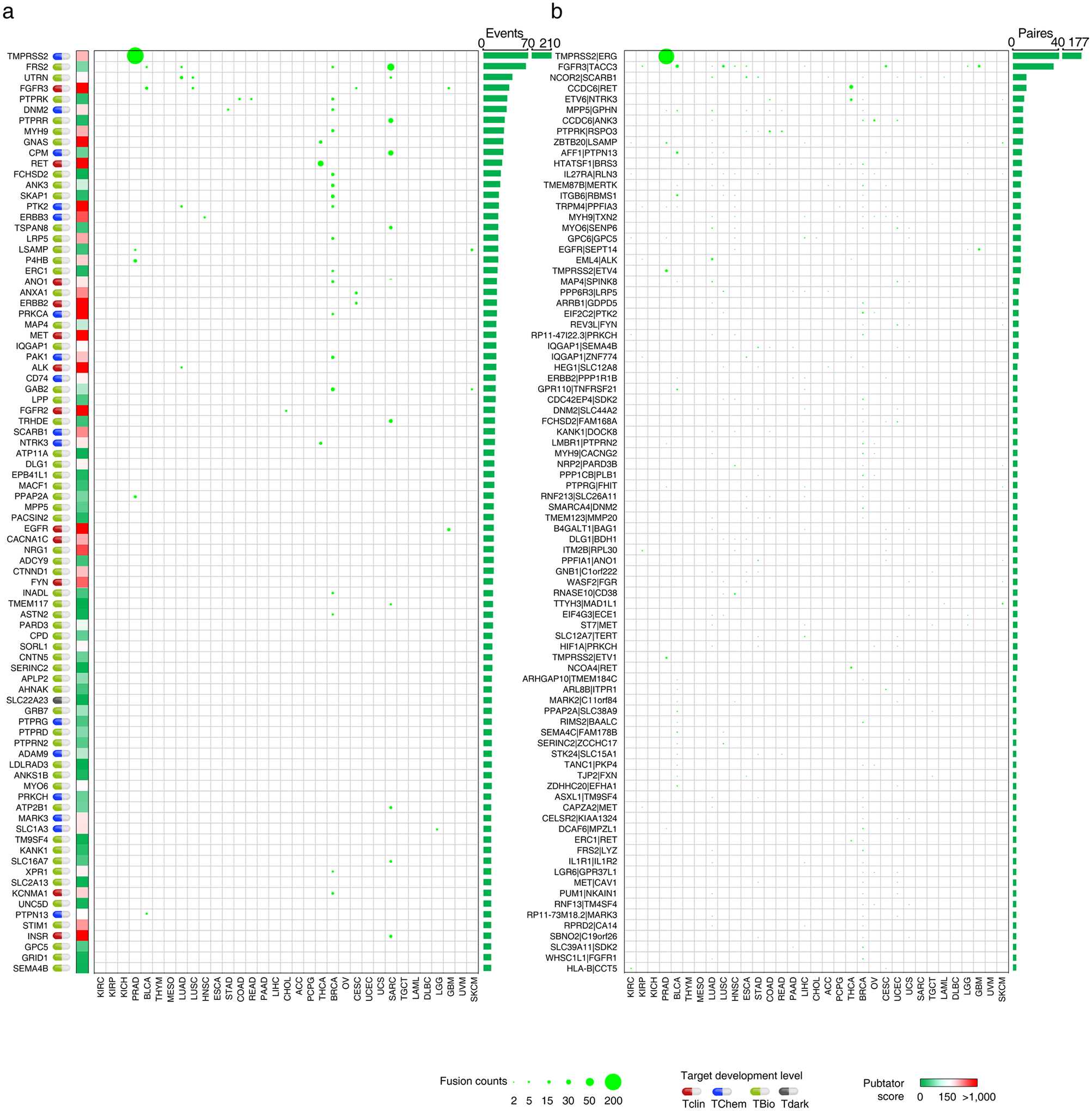
a, Summary of the GESP transcript fusion events across cancers. The size of the bubble: number of the GESP transcript fusion events across 33 cancer types. Pubtator scores, which represent the number of publications for a given gene and were retrieved from Pubtator database, are shown the next to bubble plot. Green: 1–150 (understudied genes); Red: >150. Target development levels of each gene, which were retrieved from PHAROS database, are shown in the left. Red: Tclin; blue: TChem; green: Tbio; grey: Tdark. Genes are ordered according to the overall number of the fusion events (from largest values to smallest values). Top 86 GESPs with highest number of fusion events (≥12) were shown. b, Summary of the GESP transcript fusion pairs across cancers. The size of the bubble: number of the GESP transcript fusion pairs across 33 cancer types. Fusion pairs are ordered according to the overall recurrent pairs number (from largest values to smallest values). Top 86 GESP transcript fusion pairs were shown.
Extended Data Fig. 8 |. Characterization of dependencies of the GESPs in cancer cell growth.
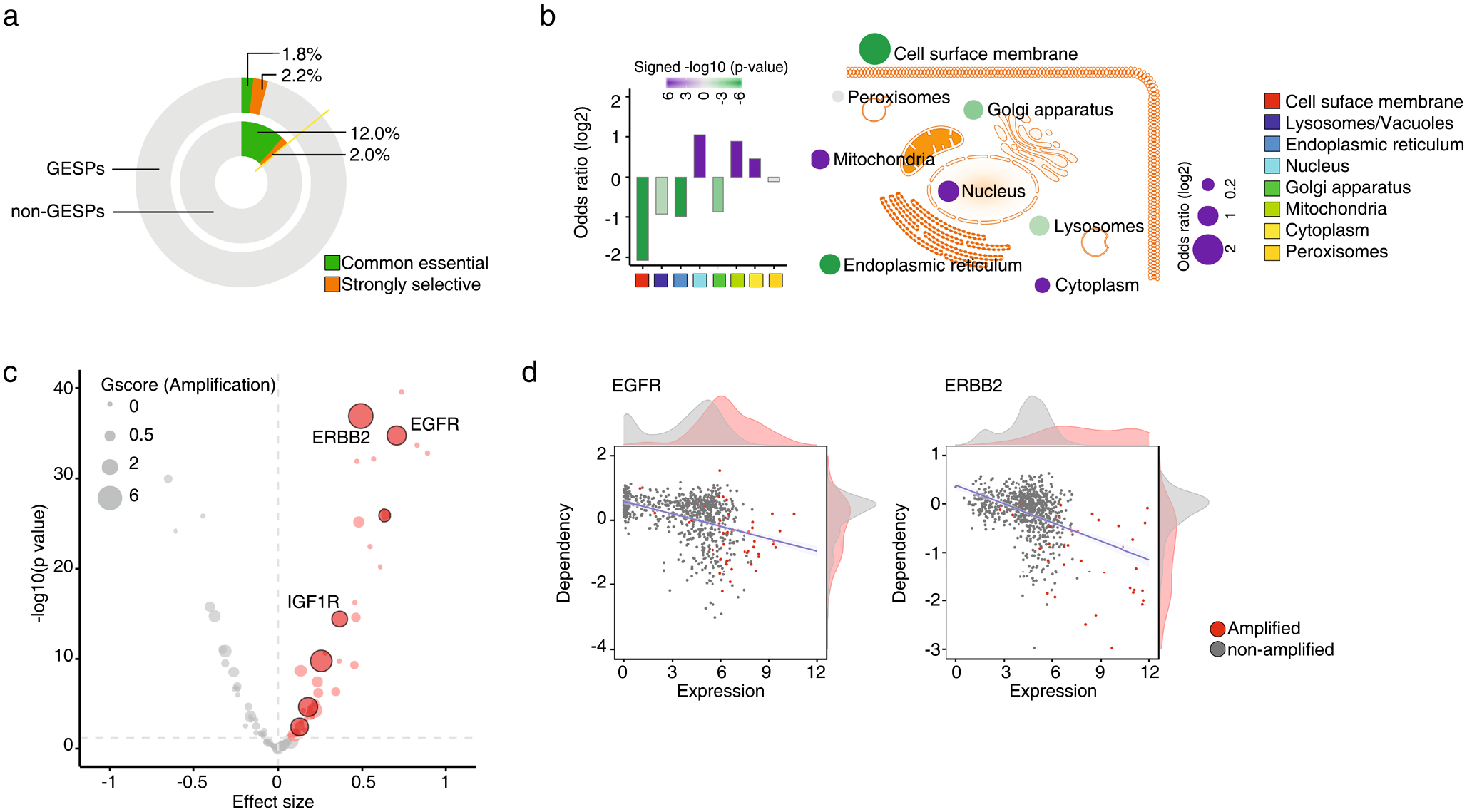
a, Proportional doughnut graph showing the frequency of common essential and strongly selective genes among GESPs (outer layer) and non-GESPs (inner layer). b, Summary of enrichment for essential genes (common essential and strongly selective) in the corresponding subgroups based on subcellular locations. Bar plot (left) and bubble plot (right) show the odds ratios on a log scale for each subgroup. Purple and green bars indicate that essential genes are enriched and depleted in the corresponding subgroups, respectively. The color intensity of the bars and bubbles indicates the enrichment significance calculated by two-sided Fisher’s exact test. c, Summary of association between mRNA expression levels and dependencies for essential GESPs. The x-axis represents the effect size of each gene. Positive effect size values represent higher dependency in cells expressing higher level of mRNA. The y-axis represents the negative logarithm (base 10) of the p-values from the Bioconductor Limma package. Benjamini-Hochberg (BH) method was used to adjust the p-values. Each circle corresponds to a GESP with size proportional to overall G-score (gain). Red circles represent GESPs whose dependencies are significantly and positively correlated with their mRNA expression levels. GESPs which are recurrently amplified in tumors and whose dependencies are significantly and positively correlated with both copy number and mRNA expression levels are outlined with black border. d, Association between mRNA expression levels and dependencies for EGFR (left) and ERBB2 (right). Red dots represent cells with gene copy number amplification. Density plots of gene expression and gene dependencies are stratified by gene amplification status.
Extended Data Fig. 9 |. Characterization of membrane-bound immunological accessory molecules (mIAMs) in cancers.
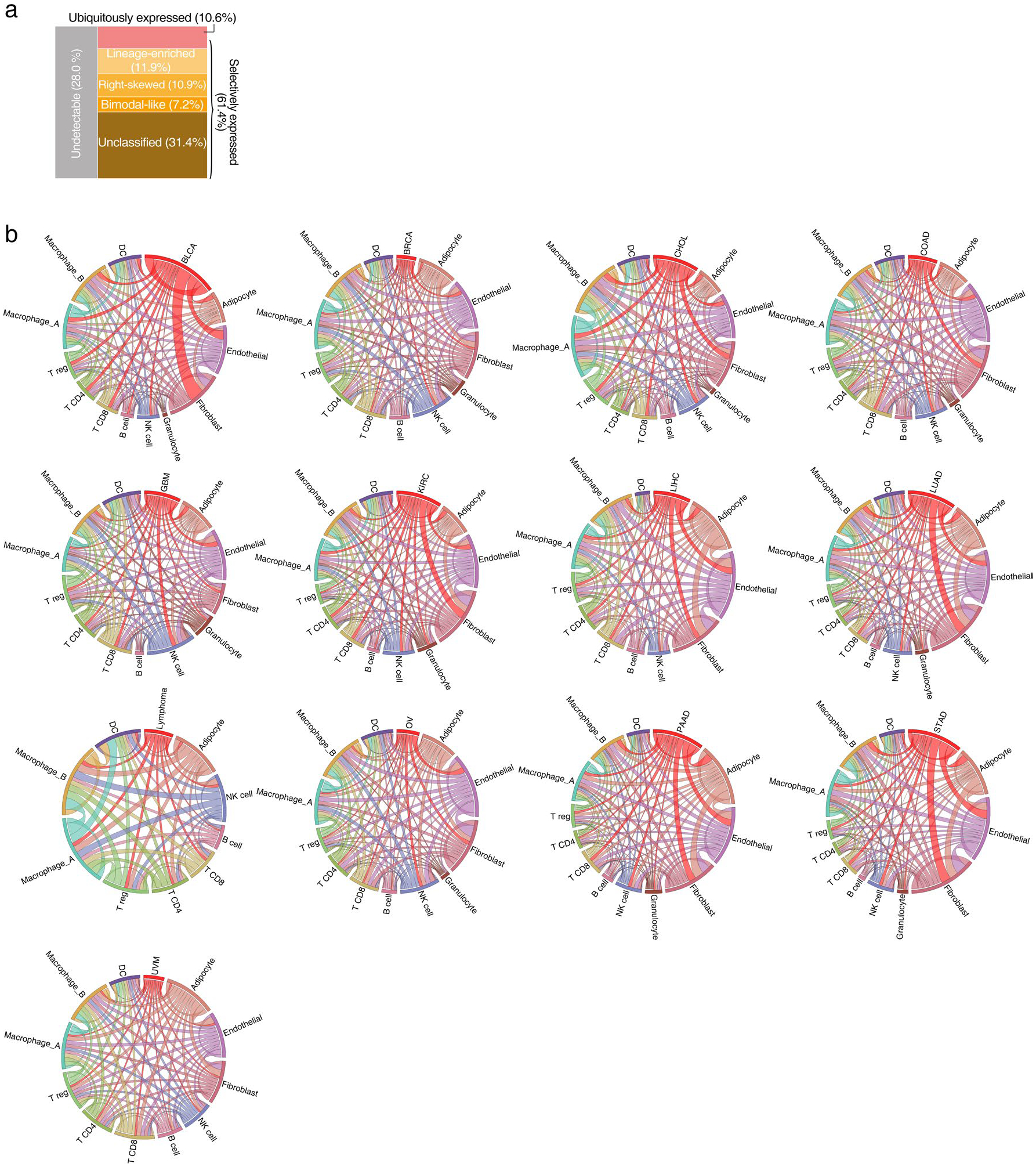
a, Mosaic plot shows the classification of mIAMs based on their expression patterns across cancer cell lines from non-hematological malignancies. b, Circos plot shows the number of mIAMs-associated interactions between cell types across 13 cancer types. Paired cell types with significant cell-cell interactions identified by CellPhoneDB were connected by lines. The width of the lines indicates normalized number of mIAMs-associated interactions between two cell types.
Supplementary Material
Acknowledgements
The present study was supported, in whole or in part, by the grants from the Pennsylvania Department of Health, Harry Fields Professorship and Abramson Cancer Center. L.Z. was supported by the Basser Center for BRCA and US National Institutes for Health (NIH) grants (nos. R01CA142776, R01CA190415, R01CA225929, R01CA262070, P50CA083638 and P50CA174523). R.H.V. was supported by NIH grants (nos. P01CA210944 and R01CA229803). X.H. was supported by the Ovarian Cancer Research Alliance. X.H. and Y.Z. were supported by the Foundation for Women’s Cancer. Support of the core facilities was provided by an NIH Cancer Centre support grant (no. P30CA016520) to Abramson Cancer Center.
Competing interests
L.Z. and X.H. report having received research funding from AstraZeneca, Bristol-Myers Squibb/Celgene and Prelude Therapeutics. R.H.V. is an inventor on a licensed patent relating to cancer cellular immunotherapy and receives royalties from Children’s Hospital Boston for a licensed research-only monoclonal antibody. O.T. and H.M.C. are employees of AstraZeneca. The remaining authors declare no competing interests.
Footnotes
Code availability
The code for analysis of TCSA is available at https://github.com/fcgportal/TCSA.
Extended data are available for this paper at https://doi.org/10.1038/s43018-021-00282-w.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s43018-021-00282-w.
Data availability
The present study is based on genomic profiles generated by TCGA project, which was supported by the NCI and the National Human Genome Research Institute (http://cancergenome.nih.gov). TCGA profiling data are publicly available through TCGA data portal (https://tcga-data.nci.nih.gov/tcga), the Genomic Data Commons portal (GDC, https://gdc-portal.nci.nih.gov), the GDAC Firehose of the Broad Institute (http://gdac.broadinstitute.org), the UCSC Toil RNAseq Recompute Compendium (https://xenabrowser.net/datapages/?hub=https://toil.xenahubs.net:443), TCGA Multi-Center Mutation Calling in Multiple Cancers (MC3) project (https://doi.org/10.7303/syn7214402) and TumorFusions data portal (http://tumorfusions.org/). Proteomics profiles were generated by the NCI’s CPTAC (https://proteomics.cancer.gov/programs/cptac). The CPTAC profiling data are publicly available through the CPTAC data portal (https://cptac-data-portal.georgetown.edu). CRISPR–Cas9 screening profiles in human cancer cell lines are publicly available through the DepMap portal (https://depmap.org/portal) and the Score projects (https://doi.org/10.6084/m9.figshare.c.5289226.v1). ScRNA-seq data are available through http://blueprint.lambrechtslab.org (breast invasive carcinoma, colon adenocarcinoma and ovarian serous cystadenocarcinoma), http://ureca-singlecell.kr (bladder urothelial carcinoma), https://bigd.big.ac.cn/bioproject/browse/PRJCA001063 (pancreatic adenocarcinoma), https://dna-discovery.stanford.edu/research/datasets (follicular lymphoma, and stomach adenocarcinoma), https://science.sciencemag.org/highwire/filestream/713964/field_highwire_adjunct_files/6/aat1699_DataS1.gz.zip (kidney renal clear cell carcinoma) and Gene Expression Omnibus (accession nos. GSE125449, GSE131907, GSE131928 and GSE139829) (cholangiocarcinoma and liver hepatocellular carcinoma, lung adenocarcinoma, glioblastoma multiforme and uveal melanoma), respectively. The data generated by the present study are publicly available through the FCG data portal (http://fcgportal.org/fcgtcsa). All other data supporting the findings of the present study are available from the corresponding author on reasonable request. Source data are provided with this paper.
References
- 1.Wu CC & Yates JR 3rd The application of mass spectrometry to membrane proteomics. Nat. Biotechnol 21, 262–267 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Daley DO et al. Global topology analysis of the Escherichia coli inner membrane proteome. Science 308, 1321–1323 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Almen MS, Nordstrom KJ, Fredriksson R & Schioth HB Mapping the human membrane proteome: a majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol 7, 50 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.da Cunha JP et al. Bioinformatics construction of the human cell surfaceome. Proc. Natl Acad. Sci. USA 106, 16752–16757 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bausch-Fluck D et al. The in silico human surfaceome. Proc. Natl Acad. Sci. USA 115, E10988–E10997 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown KK et al. Approaches to target tractability assessment—a practical perspective. Medchemcomm 9, 606–613 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adams GP & Weiner LM Monoclonal antibody therapy of cancer. Nat. Biotechnol 23, 1147–1157 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Lim WA & June CH The principles of engineering immune cells to treat cancer. Cell 168, 724–740 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadelain M, Riviere I & Riddell S Therapeutic T cell engineering. Nature 545, 423–431 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carter PJ & Lazar GA Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov 17, 197–223 (2018). [DOI] [PubMed] [Google Scholar]
- 11.MacKay M et al. The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat. Biotechnol 38, 233–244 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Weber EW, Maus MV & Mackall CL The emerging landscape of immune cell therapies. Cell 181, 46–62 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garraway LA & Lander ES Lessons from the cancer genome. Cell 153, 17–37 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Vogelstein B et al. Cancer genome landscapes. Science 339, 1546–1558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Town J et al. Exploring the surfaceome of Ewing sarcoma identifies a new and unique therapeutic target. Proc. Natl Acad. Sci. USA 113, 3603–3608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh D et al. A cell-surface membrane protein signature for glioblastoma. Cell Syst 4, 516–529 e517 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perna F et al. Integrating proteomics and transcriptomics for systematic combinatorial chimeric antigen receptor therapy of AML. Cancer Cell 32, 506–519.e505 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JK et al. Systemic surfaceome profiling identifies target antigens for immune-based therapy in subtypes of advanced prostate cancer. Proc. Natl Acad. Sci. USA 115, E4473–E4482 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coscia F et al. Multi-level proteomics identifies CT45 as a chemosensitivity mediator and immunotherapy target in ovarian cancer. Cell 175, 159–170 e116 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yao W et al. Syndecan 1 is a critical mediator of macropinocytosis in pancreatic cancer. Nature 568, 410–414 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Consortium GT et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hutter C & Zenklusen JC The cancer genome atlas: creating lasting value beyond its data. Cell 173, 283–285 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Meyers RM et al. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet 49, 1779–1784 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsherniak A et al. Defining a cancer dependency map. Cell 170, 564–576 e516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Behan FM et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 568, 511–516 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Dwane L et al. Project Score database: a resource for investigating cancer cell dependencies and prioritizing therapeutic targets. Nucleic Acids Res 49, D1365–D1372 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pacini C et al. Integrated cross-study datasets of genetic dependencies in cancer. Nat. Commun 12, 1661 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carvalho-Silva D et al. Open targets platform: new developments and updates two years on. Nucleic Acids Res 47, D1056–D1065 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim MS & Yi GS HMPAS: human membrane protein analysis system. Proteome Sci 11, S7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Binder JX et al. COMPARTMENTS: unification and visualization of protein subcellular localization evidence. Database 2014, bau012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bausch-Fluck D et al. A mass spectrometric-derived cell surface protein atlas. PLoS ONE 10, e0121314 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dobson L, Lango T, Remenyi I & Tusnady GE Expediting topology data gathering for the TOPDB database. Nucleic Acids Res 43, D283–D289 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fonseca AL et al. Bioinformatics analysis of the human surfaceome reveals new targets for a variety of tumor types. Int. J. Genom 2016, 8346198 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thul PJ et al. A subcellular map of the human proteome. Science 10.1126/science.aal3321 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Pais H et al. Surfaceome interrogation using an RNA-seq approach highlights leukemia initiating cell biomarkers in an LMO2 T cell transgenic model. Sci. Rep 9, 5760 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yanai I et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21, 650–659 (2005). [DOI] [PubMed] [Google Scholar]
- 37.van der Maaten L & Hinton G Visualizing data using t-SNE. J. Mach. Learn. Res 9, 2579–2605 (2008). [Google Scholar]
- 38.Hofmann O et al. Genome-wide analysis of cancer/testis gene expression. Proc. Natl Acad. Sci. USA 105, 20422–20427 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang C et al. Systematic identification of genes with a cancer-testis expression pattern in 19 cancer types. Nat. Commun 7, 10499 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uhlen M et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Davis CA et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res 46, D794–D801 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Labanieh L, Majzner RG & Mackall CL Programming CAR-T cells to kill cancer. Nat. Biomed. Eng 2, 377–391 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Wu MR, Jusiak B & Lu TK Engineering advanced cancer therapies with synthetic biology. Nat. Rev. Cancer 19, 187–195 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Rafiq S, Hackett CS & Brentjens RJ Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol 17, 147–167 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dannenfelser R et al. Discriminatory power of combinatorial antigen recognition in cancer T cell therapies. Cell Syst 11, 215–228 e215 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams JZ et al. Precise T cell recognition programs designed by transcriptionally linking multiple receptors. Science 370, 1099–1104 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kloss CC, Condomines M, Cartellieri M, Bachmann M & Sadelain M Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol 31, 71–75 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roybal KT et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 164, 770–779 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fedorov VD, Themeli M & Sadelain M PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med 5, 215ra172 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu Z et al. Genomic characterization of genes encoding histone acetylation modulator proteins identifies therapeutic targets for cancer treatment. Nat. Commun 10, 733 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shan W et al. Systematic characterization of recurrent genomic alterations in cyclin-dependent kinases reveals potential therapeutic strategies for cancer treatment. Cell Rep 32, 107884 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thorndike RL Who belongs in the family? Psychometrika 18, 267–276 (1953). [Google Scholar]
- 53.Goutte C, Toft P, Rostrup E, Nielsen F & Hansen LK On clustering fMRI time series. NeuroImage 9, 298–310 (1999). [DOI] [PubMed] [Google Scholar]
- 54.Carter SL et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol 30, 413–421 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hu X et al. TumorFusions: an integrative resource for cancer-associated transcript fusions. Nucleic Acids Res 46, D1144–D1149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Graeber TG & Eisenberg D Bioinformatic identification of potential autocrine signaling loops in cancers from gene expression profiles. Nat. Genet 29, 295–300 (2001). [DOI] [PubMed] [Google Scholar]
- 57.Ben-Shlomo I, Yu Hsu S, Rauch R, Kowalski HW & Hsueh AJ Signaling receptome: a genomic and evolutionary perspective of plasma membrane receptors involved in signal transduction. Sci. STKE 2003, RE9 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Kahlon KS et al. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res 64, 9160–9166 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Benedict SH, Cool KM, Dotson AL & Chan MA in Encyclopedia of Life Sciences (ed John Wiley & Sons Ltd) 10.1002/9780470015902.a0000923.pub2 (John Wiley & Sons Ltd, 2007). [DOI] [Google Scholar]
- 60.Ghandi M et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liberzon A et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu H et al. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat. Med 25, 95–102 (2019). [DOI] [PubMed] [Google Scholar]
- 63.Efremova M, Vento-Tormo M, Teichmann SA & Vento-Tormo R CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc 15, 1484–1506 (2020). [DOI] [PubMed] [Google Scholar]
- 64.Oprea TI et al. Unexplored therapeutic opportunities in the human genome. Nat. Rev. Drug Discov 17, 317–332 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rhee SY, Wood V, Dolinski K & Draghici S Use and misuse of the gene ontology annotations. Nat. Rev. Genet 9, 509–515 (2008). [DOI] [PubMed] [Google Scholar]
- 66.Vivian J et al. Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol 35, 314–316 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang L et al. MNX1 Is oncogenically upregulated in African-American prostate cancer. Cancer Res 76, 6290–6298 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xiao SJ, Zhang C, Zou Q & Ji ZL TiSGeD: a database for tissue-specific genes. Bioinformatics 26, 1273–1275 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jain A & Tuteja G TissueEnrich: tissue-specific gene enrichment analysis. Bioinformatics 35, 1966–1967 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dougherty JD, Schmidt EF, Nakajima M & Heintz N Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic Acids Res 38, 4218–4230 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Torsten H, Kurt H, Mark A v. d. W & Achim Z A lego system for conditional inference. Am. Stat 60, 257–263 (2006). [Google Scholar]
- 73.Leiserson MD, Wu HT, Vandin F & Raphael BJ CoMEt: a statistical approach to identify combinations of mutually exclusive alterations in cancer. Genome Biol 16, 160 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dempster JM et al. Extracting biological insights from the project achilles genome-Scale CRISPR screens in cancer cell lines. Preprint at bioRxiv 10.1101/720243 (2019). [DOI] [Google Scholar]
- 75.Dempster JM et al. Agreement between two large pan-cancer CRISPR–Cas9 gene dependency data sets. Nat. Commun 10, 5817 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hou R, Denisenko E & Forrest ARR scMatch: a single-cell gene expression profile annotation tool using reference datasets. Bioinformatics 35, 4688–4695 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wan C et al. LTMG: a novel statistical modeling of transcriptional expression states in single-cell RNA-Seq data. Nucleic Acids Res 47, e111 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang J, Wen S, Symmans WF, Pusztai L & Coombes KR The bimodality index: a criterion for discovering and ranking bimodal signatures from cancer gene expression profiling data. Cancer Inform 7, 199–216 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The present study is based on genomic profiles generated by TCGA project, which was supported by the NCI and the National Human Genome Research Institute (http://cancergenome.nih.gov). TCGA profiling data are publicly available through TCGA data portal (https://tcga-data.nci.nih.gov/tcga), the Genomic Data Commons portal (GDC, https://gdc-portal.nci.nih.gov), the GDAC Firehose of the Broad Institute (http://gdac.broadinstitute.org), the UCSC Toil RNAseq Recompute Compendium (https://xenabrowser.net/datapages/?hub=https://toil.xenahubs.net:443), TCGA Multi-Center Mutation Calling in Multiple Cancers (MC3) project (https://doi.org/10.7303/syn7214402) and TumorFusions data portal (http://tumorfusions.org/). Proteomics profiles were generated by the NCI’s CPTAC (https://proteomics.cancer.gov/programs/cptac). The CPTAC profiling data are publicly available through the CPTAC data portal (https://cptac-data-portal.georgetown.edu). CRISPR–Cas9 screening profiles in human cancer cell lines are publicly available through the DepMap portal (https://depmap.org/portal) and the Score projects (https://doi.org/10.6084/m9.figshare.c.5289226.v1). ScRNA-seq data are available through http://blueprint.lambrechtslab.org (breast invasive carcinoma, colon adenocarcinoma and ovarian serous cystadenocarcinoma), http://ureca-singlecell.kr (bladder urothelial carcinoma), https://bigd.big.ac.cn/bioproject/browse/PRJCA001063 (pancreatic adenocarcinoma), https://dna-discovery.stanford.edu/research/datasets (follicular lymphoma, and stomach adenocarcinoma), https://science.sciencemag.org/highwire/filestream/713964/field_highwire_adjunct_files/6/aat1699_DataS1.gz.zip (kidney renal clear cell carcinoma) and Gene Expression Omnibus (accession nos. GSE125449, GSE131907, GSE131928 and GSE139829) (cholangiocarcinoma and liver hepatocellular carcinoma, lung adenocarcinoma, glioblastoma multiforme and uveal melanoma), respectively. The data generated by the present study are publicly available through the FCG data portal (http://fcgportal.org/fcgtcsa). All other data supporting the findings of the present study are available from the corresponding author on reasonable request. Source data are provided with this paper.