

Abstract
A tumour microenvironment abundant in regulatory T (Treg) cells aids solid tumours to evade clearance by effector T cells. Systemic strategies to suppress Treg cells or to augment immunity can elicit autoimmune side effects, cytokine storms and other toxicities. Here we report the design, fabrication and therapeutic performance of a biodegradable macroporous scaffold, implanted peritumourally, that releases a small-molecule inhibitor of transforming growth factor β to suppress Treg cells, chemokines to attract effector T cells and antibodies to stimulate them. In two mouse models of aggressive tumours, the implant boosted the recruitment and activation of effector T cells into the tumour and depleted it of Treg cells, which resulted in an ‘immunological abscopal effect’ on distant metastases and in the establishment of long-term memory that impeded tumour recurrence. We also show that the scaffold can be used to deliver tumour-antigen-specific T cells into the tumour. Peritumourally implanted immunomodulatory scaffolds may represent a general strategy to enhance T-cell immunity and avoid the toxicities of systemic therapies.
Solid tumours effectively escape immune clearance by modifying their local microenvironment and promoting the development of regulatory T (Treg) cells. Treg cells dramatically accumulate in tumours, making up 20% of T cells or more in many solid tumours, whereas they comprise ~1–3% of total T cells in the blood. Many blockbuster immunotherapies in clinical use, including inhibitors of the cytotoxic T-lymphocyte-associated antigen 4 and programmed death-1 (PD-1) pathways, enact their antitumour roles by both reducing the proportions of Treg cells in tumours1,2 and reducing inhibitory pathways in effector T cells. These therapies are a double-edged sword, however, inflicting in some patients systemic autoimmunity with dermatologic (20–60%), gastrointestinal (30–40%) and endocrine (1–5%) consequences3–5. Reducing Treg cells alone, moreover, is not sufficient, as these therapies do not work well in tumours that lack an appreciable number of effector T cells6. Indeed, decreased ratios of tumour-infiltrating CD8+ effector T cells to Treg cells correlate with poor prognoses in many cancers7–10. Taken together, what is needed is a way to reduce Treg cells—but not at the expense of systemic autoimmunity—combined with a way to recruit and augment the activity of effector T cells. In this Article, we present a biodegradable scaffold (sponge-like biomaterial) that can be placed locally adjacent to the tumour bed that addresses both these concerns, depleting Treg cells locally while robustly activating endogenous effector T cells against tumours globally.
Transforming growth factor beta (TGFβ) is a potent growth factor of the tumour microenvironment that induces Treg cells from the helper T cells drawn to the tumour11. Beyond these effects, TGFβ promotes cancer proliferation and metastasis while inhibiting the effector functions and the migration of cytotoxic T cells in the tumour12,13. TGFβ has thus become a popular target for immunotherapy14. However, systemic TGFβ inhibition in preclinical models and humans has shown major adverse effects on the cardiovascular, gastrointestinal and skeletal systems15,16. The release of TGFβ inhibitors by injected nanoliposomes reduced metastases17 but did not reduce Treg cells in tumours. Several other approaches have been tried to deplete Treg cells in cancer18,19. For example, anti-CD25 antibodies have been used for over 20 years20, but these approaches failed when practiced globally because they delete CD25-expressing effector T cells21. To more specifically target Treg-cell depletion to a particular tumour, a recent effort showed selective depletion of tumour-associated Treg cells with photoimmunotherapy after intravenous (IV) infusion of anti-CD25 antibodies conjugated to a near infrared dye22 or by phototherapy after intratumoural injection of anti-CD25 antibodies conjugated to a photosensitizer dye. However, these approaches are not practical for anything other than superficial tumours (for example, they would not work for breast cancer). These alternative approaches localize Treg-cell suppression, but they are not practical in clinical settings. Moreover, because of the low residence time of the therapeutic compounds in the tumour environment, these techniques must be repeated several times, making these efforts less useful than the one proposed here. In this work, we tested whether sustained blockade of TGFβ signalling locally could suppress Treg cells in the tumour without global harm.
To accomplish the goals of suppressing Treg cells while recruiting and activating effector T cells, we combined a variety of biomaterial design elements into a three-dimensional (3D) scaffold. First, the scaffold continually secretes a small-molecule inhibitor that blocks TGFβ. Second, it entices effector T cells through the microporous structure of our scaffold and into the tumour microenvironment by releasing chemokines. Third, it super-activates effector T cells through stimulatory antibodies. Fourth, we tuned the mechanical stiffness of the scaffold to maximize mechanosensing pathways in T cells23,24. Fifth, the scaffold secretes stimulatory cytokines to promote T-cell activation and the development of memory T cells. Combining all five strategies into a single, biodegradable biomaterial allowed us to obtain a highly effective antitumour response.
Results
Scaffold fabrication
To enhance local immunity against tumours, our alginate-based scaffold combines multiple design considerations (Supplementary Fig. 1). The scaffold facilities traffic of T cells throughout the structure by virtue of its microporous design24 (Fig. 1a). Naive T cells pipetted onto the surface of the scaffold infiltrate throughout (Fig. 1b). We assessed pore sizes of the scaffold in vivo 7 days after surgical implantation of scaffolds in wild-type, immunologically intact, non-tumour-bearing mice. As shown here, the pore size of the scaffolds after in vivo implantation (Fig. 1c) is quite similar to the pore size before implantation. We showed using fluorescence microscopy that primary T cells crawled within the scaffold (Fig. 1d). The surfaces of the scaffold were covalently conjugated with stimulatory antibodies anti-CD3 and anti-CD28. To test the activation of T cells, we placed naive T cells onto either our immunoactive scaffolds or control scaffolds that lacked all functionalization and measured proliferation and cytokine production by flow cytometry after 3 days. The T cells were quite activated, showing enhanced proliferation and cytokine production (Fig. 1e,f), confirming that covalent conjugation of stimulatory antibodies to the surface area of the scaffold produced robust T-cell activation. Over days, the scaffold allowed for a dramatic 20-fold increase in proliferated T cells (Fig. 1g). Because mechanical stiffness of the substrate can promote T-cell activation24, we tuned the elastic modulus of the alginate 3D scaffold based on Ca2+ concentration (Fig. 1h) and found the resulting T-cell activation was maximized when the elastic modulus was ~50 kPa (Fig. 1i). Our design allowed for TGFβ inhibitor (TGFβi) to gradually release from embedded poly(lactic-co-glycolic acid) (PLGA) nanoparticles (NPs) (Fig. 1j), which suppressed the development of Treg cells from naive CD4+ T cells being cultured with anti-CD3, anti-CD28 and TGFβ (Fig. 1k,l). The interleukin-2 (IL-2) cytokine was gradually eluted from engineered mesoporous silica microparticles (Supplementary Fig. 2) and from the scaffolds (Supplementary Fig. 4). Finally, chemokines were gradually released from alginate scaffolds resulting in robust recruitment of T cells (Supplementary Fig. 7). Taken together, these results show our scaffold orchestrates suppression of Treg-cell development and promotion of effector-T-cell activation.
Fig. 1 |. Immunoactive scaffold activates T cells and suppresses Treg cells.
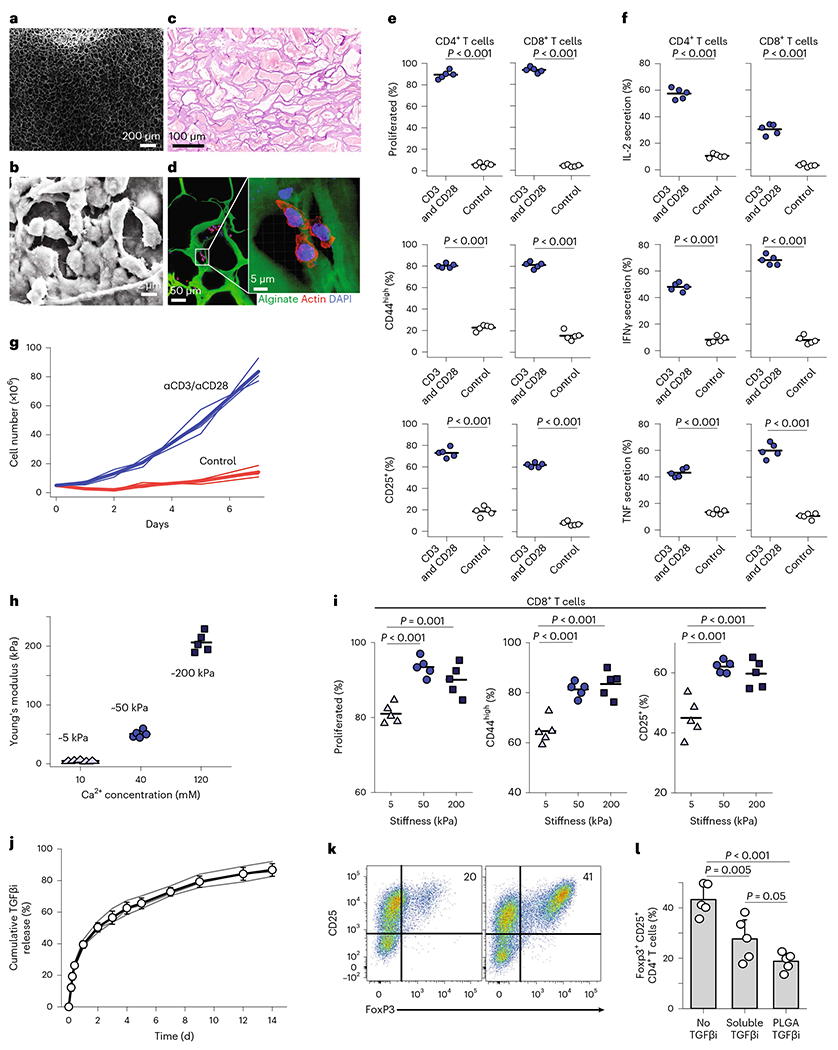
a, SEM images of macroporous scaffolds. Images were taken from a region within the bulk of the scaffold. b, Enlarged SEM images showing the association of T cells within the alginate-based scaffolds. Images were taken from an internal wall of the scaffold where T cells were aligned. c, To better assess the pore sizes of the scaffold in vivo, scaffolds were implanted in wild-type non-tumour-bearing mice for 7 days, with H&E staining showing connective tissue-like scaffolds. d, Confocal fluorescence images showing interaction of primary T cells with the scaffold. e–g, Activation and proliferation. e, Naive CD4+ and CD8+ primary mouse T cells co-cultured with 3D scaffolds. Scaffolds formulated by surface coating with stimulatory antibodies (anti-CD3 and anti-CD28) resulted in robust responses. Flow cytometry analysis of cell division and expression of activation markers CD25 or CD44 assayed 3 days after the introduction of T cells into the biomaterial. Proliferated (%) is the percentage of T cells that divided at least once. The percentage of T cells with high expression of CD44 and percentage of T cells upregulating CD25. f, Naive CD4+ and CD8+ primary mouse T cells co-cultured with 3D scaffolds. Scaffolds formulated by surface coating with stimulatory antibodies (anti-CD3 and anti-CD28) resulted in robust responses. Percentage of T cells expressing the effector cytokines IL-2, interferon gamma (IFNγ) or tumour necrosis factor (TNF). Each dot represents one independent experiment. Flow cytometry analysis of Treg-cell formation using antigen-presenting scaffolds in the absence (left) and presence (right) of TGFβi releasing NPs 4 days into culture. g, Naive CD4+ and CD8+ T cells co-cultured with various formulations of 3D scaffolds. The starting number of T cells was 5 × 106 cells. Absolute counts of viable T cells in scaffolds fabricated with different formulations shows the robust expansion of T cells. Each line represents an independent experiment. h,i, Mechanical stiffness. h, Elastic modulus of the 3D alginate scaffold fabricated with three different concentrations of Ca2+. i, Proliferation and activation markers of T cells in scaffolds of various mechanical stiffness. j–l, TGFβ inhibition. j, Release of TGFβi encapsulated PLGA NPs embedded in the 3D alginate scaffold. The released TGFβi was measured over time under gentle shaking (50 rpm) at 37°C. k, Flow cytometry analysis of Treg-cell formation using antigen-presenting scaffolds in the presence (left) and absence (right) of TGFβi releasing NPs 4 days into culture. l, Inhibition of Treg-cell development in 3D scaffold by soluble TGFβi versus PLGA-encapsulated TGFβi was quantified. The individual data are presented (n = 5). The results were statistically analysed using one-way ANOVA with post hoc analysis. Error bars indicate standard deviation (s.d.). In e, f and i, horizontal bars indicate means.
4T1 breast cancer model
To determine whether local depletion of Treg cells could facilitate clearance of tumours, we tested our scaffolds in a mouse breast cancer model. As described above, the mechanically tuned, 3D, macroporous, alginate scaffolds were coated with stimulatory antibodies and embedded with chemokine (C–C motif) ligand 21 (CCL21), IL-2-eluting mesoporous silicon microparticles and NPs that released TGFβi (Fig. 2a). All mice received an injection of 4T1 breast cancer cells into the mammary fat pad (Fig. 2b). After 5 days, the tumours became palpable, and we implanted the scaffolds adjacent to the tumour in the intramammary fat space (Fig. 2b). Control mice were untreated. Tumour sizes were monitored twice weekly by luciferase imaging for the first 3 weeks. We then followed all mice for survival and ended the experiment on day 70. Four of five mice showed suppression of the growth of tumours in the mice treated with functionalized scaffolds, while all control mice showed growth of the tumours (Fig. 2c,d). Survival was significantly improved in treated mice (Fig. 2e). These results show that our functionalized scaffolds controlled the growth of an aggressive breast cancer model in mice.
Fig. 2 |. Engineered scaffolds that curb breast cancer.
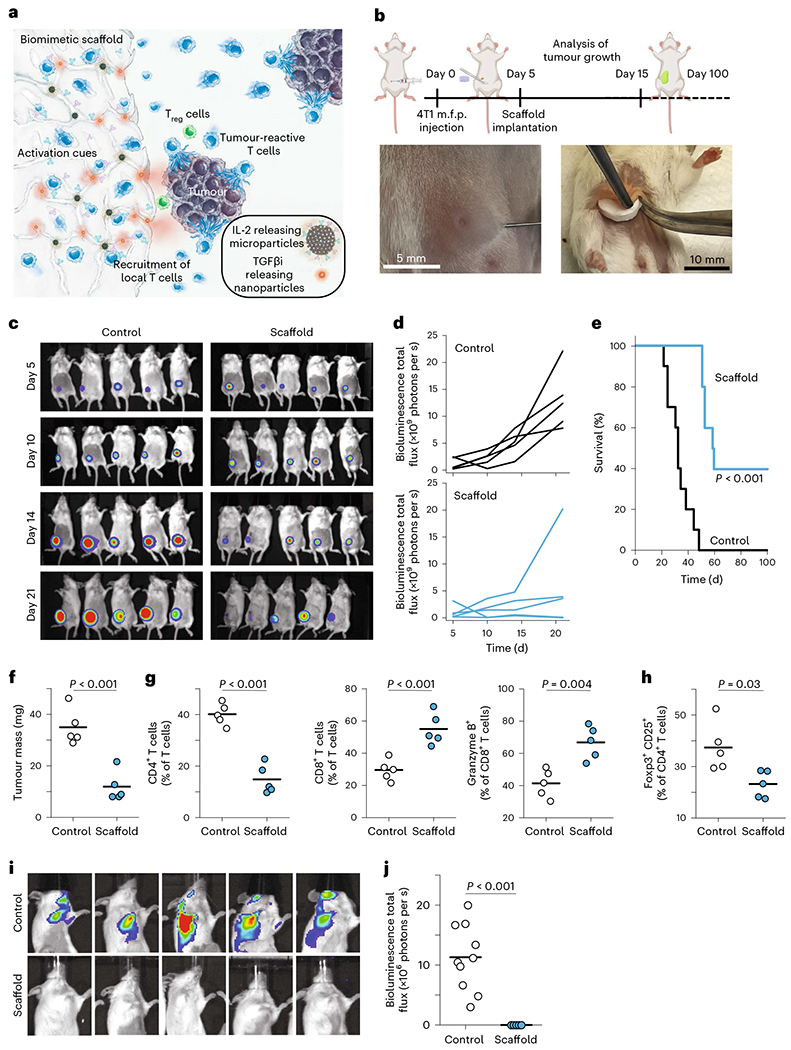
a, Schematic representation of the scaffold components. Sustained release of CCL21 helps to recruit endogenous T cells, while the presentation of surface-conjugated activation cues (anti-CD3 and anti-CD28) and the sustained release of IL-2 activates recruited T cells. Sustained release of TGFβi depletes Treg cells in tumours. b, Schematic of 4T1 breast cancer experiment, along with photos of the injection of tumour cells into the mammary fat pad and the implantation of scaffolds into the mammary fat space. m.f.p., mammary fat pad. c, Bioluminescence imaging of luciferase-producing cancer cells in mice treated with immunoactive scaffolds or controls. d, Bioluminescence quantified over time across mice. e, Survival of mice after introduction of 4T1 breast cancer cells in treated and control groups. f, Tumour mass on day 15. g, T cells are more activated and enriched for cytotoxic T cells. h, Treg cells are suppressed in the tumors adjacent to the immunoactive scaffolds. i, Bioluminescence imaging on day 30 of head and upper limbs reveals metastatic disease distant from the primary site. j, Quantified results of bioluminescence imaging of distant sites. Each point represents one mouse. Horizontal bars indicate means. The results were statistically analysed using one-way ANOVA.
To assess the mechanism by which the scaffold allowed mice to control tumours, some mice were killed on day 15 (10 days after scaffold implantation), and the tumours were examined by flow cytometry. The tumours in treated mice were much smaller (Fig. 2f). T cells were examined in the tumours, and there was a significant increase in infiltrating CD8+ T cells with a concomitant decrease in CD4+ T cells (Fig. 2g). Infiltrating CD8+ T cells showed an increase in their killing capability (Fig. 2g). We saw that the proportion of Treg cells in the tumour was reduced by half (Fig. 2h). These results confirm that the immunoactive scaffold depletes Treg cells and enriches the presence of activated, cytotoxic T cells.
To highlight the contribution of each component of our scaffold, mice were injected with 4T1 breast cancer cells as above and were implanted with alginate scaffolds comprising the individual ingredients. These experiments allowed us to examine the effects on tumour growth and on responding T cells within the tumour (Supplementary Fig. 10). Scaffolds included ones with only: surface functionalization with anti-CD3 and anti-CD28, PLGA NPs releasing TGFβi, release of the chemokine CCL21 and release of the cytokine IL-2. We also tested an injection of all these components intratumourally and peritumourally. To control for the surgical implantation of the scaffold, we used unfunctionalized scaffolds. We found that release of either TGFβi or IL-2 significantly reduced tumour growth. The ‘full’ scaffold combining all the components was the most potent at reducing tumour growth (see Supplementary Table 1 for statistical analysis). The reduction of Treg cells in the tumour was only observed in the scaffold that secreted TGFβi. The degree of Treg-cell depletion by about 35% was comparable to the fully functionalized scaffolds. To understand which component(s) participated in the recruitment of cytotoxic T cells into the tumour, we found that CD3/CD28 antibody coating of the scaffolds was the single most effective, even more than the CCL21 chemokine alone. Thus, local activation and expansion of cytotoxic T cells is important for their recruitment into the tumour. To understand which component(s) contributed to the elevated cytotoxicity of T cells (as measured by granzyme B expression), we found that CD3/CD28 antibody coating, release of IL-2 and release of TGFβi were comparably potent. We did expect that offering more stimulation of the T cell receptor (TCR) to T cells would drive more cytotoxic function, and IL-2 is well known to potentiate cytotoxicity as well. We can understand the finding that TGFβ inhibition increased cytotoxicity of effector T cells through both direct and indirect mechanisms. It is known that TGFβ signalling in T cells directly inhibits proliferation and cytotoxicity12. In addition, global diminishment of Treg cells was shown to indirectly potentiate the cytotoxicity of intratumoural CD8+ T effectors25,26. Our work extended this finding to reveal that local diminishment of intratumoural Treg cells evokes greater cytotoxicity in T effectors. On the basis of these observations, reducing intratumoural Treg cells while activating and recruiting T effectors makes for the most potent therapy.
We found a dramatic extension of survival in our treated mice. To better understand the therapeutic impact by which survival was enabled, we noted that in the 4T1 breast cancer model metastases develop spontaneously from the primary tumour, and the aggressive spread of metastases to the draining lymph nodes and other organs resembles human breast cancer27. We found that metastases in the brain and axillary lymph nodes were utterly eliminated in treated mice, whereas they were universally found in untreated mice (Fig. 2i,j). These results show that our immunoactive scaffold reduces both primary tumours and metastatic spread.
We sought to better understand whether the long-term survivors, which resisted both the primary tumour and metastases, had developed immunological memory that could protect them from recurrence. When the tumour-free mice were seen to survive to 100 days (~40 days beyond the last non-surviving mouse) the survivors were rechallenged with an IV injection of 4T1 tumour cells, which deposits tumour cells into the lungs and emulates new metastases (Fig. 3a). To be sure this lung-metastatic model was performing as intended, and to directly compare the impact of immunological memory versus that of naive T-cell responses, we compared this IV injection of tumour cells in naive, age-matched, wild-type mice. The lung metastases for these mice were examined by luciferase imaging (Fig. 3b). All (100%) of the long-term survivors survived the rechallenge and completely eliminated the development of detectable tumours in the lungs (Fig. 3c). In contrast, all the control mice developed lung metastases and died by or before day 14 (Fig. 3d). These results show that the immunomodulatory scaffolds engender durable memory that can protect against the recurrence of cancer.
Fig. 3 |. Engineered scaffolds can suppress the growth of local and distant breast tumours and create anti-cancer memory.
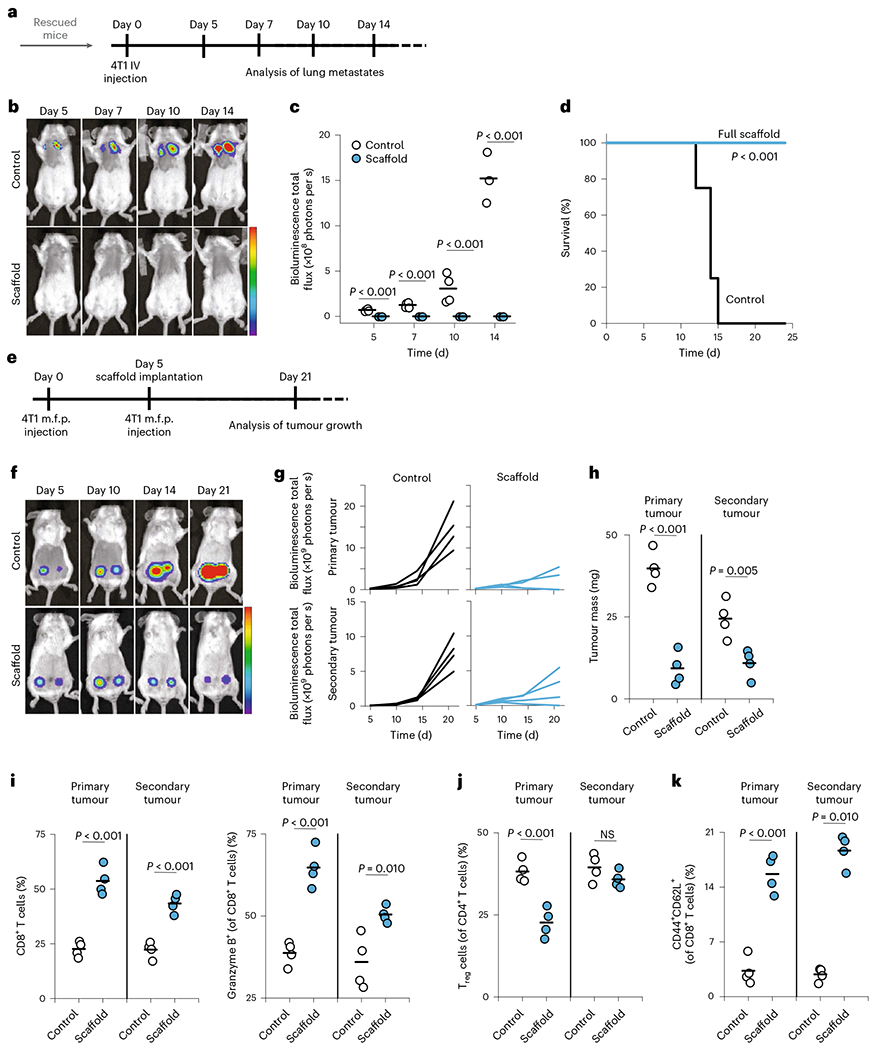
a, Timing of IV injection of 4T1 breast cancer cells at 100 days post injection of primary tumours in mice that eliminated primary tumours (n = 4). Naive mice (age matched) receiving IV injection of 4T1 breast cancer cells were used as control. b, Representative bioluminescence imaging of luciferase-producing cancer cells at different times post injection of 4T1-Luc in mice treated with immunoactive scaffolds. c, Quantification of bioluminescence intensity of mice at different times post injection of 4T1-Luc cancer cells. d, Survival of mice after IV injection of 4T1 breast cancer cells in treated and control groups. e, Timing of inoculation of primary and secondary tumours and follow-up surgical implantation of the cell-free scaffolds. Wild-type, immunocompetent syngeneic mice were implanted in the mammary fat pad with 5 × 105 4T1-Luc breast cancer cells. After 5 days, the scaffolds were implanted, and 3 × 105 tumour cells were injected into the contralateral mammary fat pad. f, Bioluminescence imaging of luciferase-producing cancer cells in mice treated with immunoactive scaffolds or (PBS) controls at various time points. g, Growth of primary and secondary 4T1 tumours in mice as quantified over time using bioluminescence imaging. h, Tumour masses were measured 15 days after inoculation of primary tumours in wild-type mice treated with immunoactive scaffolds. i, The frequency of CD8+ T cells and activated (granzyme B+) CD8+ T cells in primary and secondary tumours. j, The quantified frequency of Foxp3+CD25+CD4+ Treg cells in primary and secondary tumours. NS, not significant. k, The frequency of central memory (CD44+CD62L+CD8+) T cells in primary and secondary tumours (n = 4). Each point represents a mouse. Horizontal bars indicate means. The results were statistically analysed using one-way ANOVA.
Distant tumours
By the time many solid tumours are clinically detected, they have already spread regionally or distantly. We next assessed whether local depletion of intratumoural Treg cells in a primary tumour could alter the trajectory of the immune response in a distant secondary tumour. Wild-type mice received a mammary fat pad injection of 4T1-Luc cells, and as before immunoactive scaffolds were implanted subcutaneously after 5 days. On the same day, we injected tumour cells contralaterally to the primary one (Fig. 3e). We monitored tumour growth on both sides using bioluminescence imaging (Fig. 3f,g). The growth and the mass of the secondary tumour was significantly suppressed upon local treatment of the primary tumour with immunoactive scaffolds but not control scaffolds (Fig. 3h). We also noted an increase in the population of tumour-infiltrating, activated CD8+ T cells on both sides 15 days after the start of primary tumours (Fig. 3i). Activated T cells were also found in the draining lymph nodes, suggesting that local inhibition of Treg cells allows widespread trafficking of activated effector T cells at earlier time points, but the effect diminished at later times (Supplementary Fig. 13). We also observed the expansion of central memory (CD62L+CD44+CD8+) T cells in the tumour (Fig. 3k). This T-cell memory subset has been noted in mice and humans to be important for both clearance of tumours and prevention of their recurrence28. Notably, the proportion of Treg cells was markedly reduced only in the primary tumour (Fig. 3j) but not in the draining lymph nodes (Supplementary Fig. 13). These results show that localized suppression of intratumoural Treg cells resulted in both successful clearance of primary tumours and marked reduction of secondary tumours. The disproportionate increase in activated, cytotoxic CD8+ T cells in the distant lymph nodes and secondary tumour indicates that expansion of potent effector T cells in the primary tumour spreads to secondary sites. As further evidence of an immunological ‘abscopal’ effect arising from the scaffold acting on the primary tumour, we noted an elevated level of inflammatory serum cytokines in the circulation (Supplementary Fig. 12). Thus, while the release of TGFβi from the scaffold reduces Treg cells locally, there are global effects on effector T cells including clearing distant tumours and development of long-term protective memory.
B16-F10 melanoma model
To determine whether local depletion of Treg cells could facilitate clearance of a second tumour type, we tested our scaffolds in the mouse melanoma model. Mice received a subcutaneous injection of B16-F10 cells to their right flank (Fig. 4a). We prepared scaffolds that were functionalized as above (‘full’) and control scaffolds that lacked functionalization. After 5 days, when the tumours first became palpable, we implanted the scaffolds adjacent to the tumour in the subcutaneous space (Fig. 4b,c); some mice remained entirely untreated. Unfunctionalized scaffolds served as controls for the surgery and wound healing. We ended the experiment for all mice on day 22, when the untreated mice required euthanasia for tumour size and morbidity. In all mice receiving the functionalized scaffold, tumour growth was suppressed (Fig. 4d) compared with untreated mice or those receiving a control scaffold. The tumour size for treated mice was roughly one-eighth the size of those receiving control scaffolds (Fig. 4d). For three (43%) of the treated mice, no residual tumour could be found at all (complete remission) (Fig. 4d). In a parallel set of experiments, we assessed long-term survival. Mice were euthanized as per institutional criteria when tumours were large or allowed to survive long term. We found that 80% of mice receiving the full immunoactive scaffold showed a complete response and survived long term, over a month beyond the mice receiving unfunctionalized scaffolds or no treatment (Fig. 4d, right). These results reveal that the immunomodulatory scaffold helps clear multiple types of solid tumours and promote long-term survival.
Fig. 4 |. Engineered scaffolds that curb melanoma.
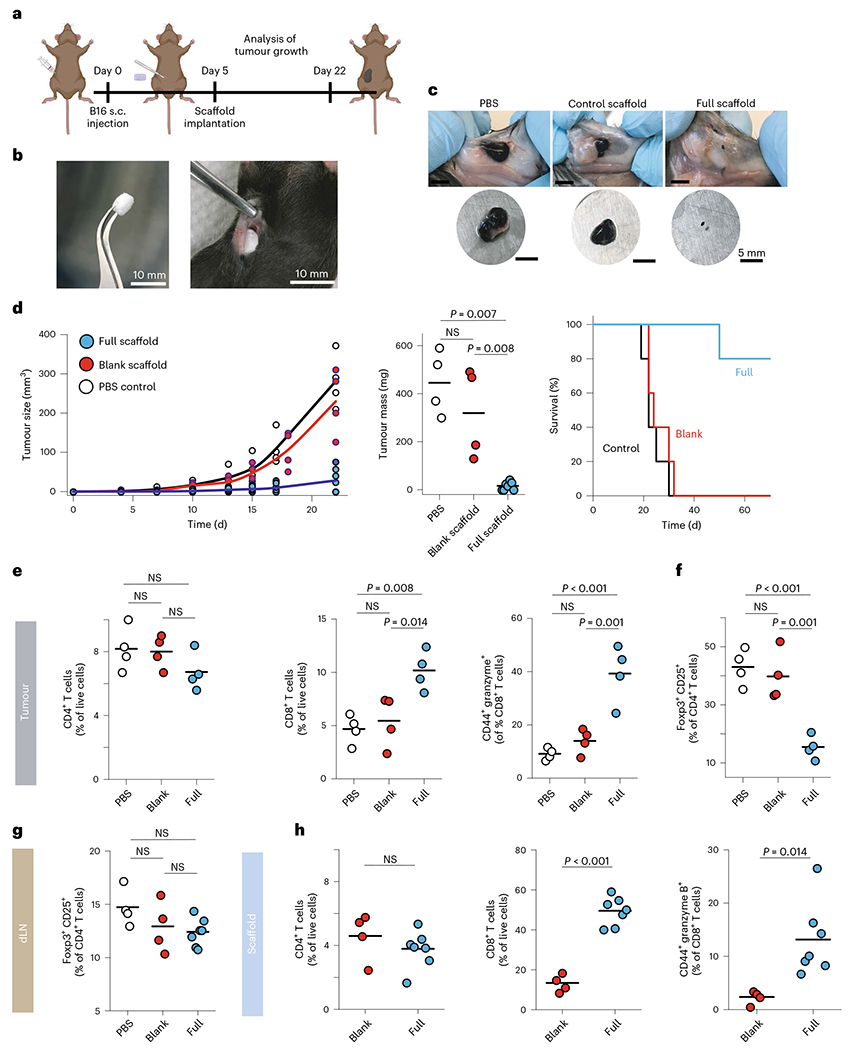
a, Schematic of B16 experiment. s.c., subcutaneous. b, A scaffold and its implantation into the subcutaneous space adjacent to the tumour. The engineered device is surgically implanted in a B16-F10-OVA bearing mice. c, Representative image of tumours extracted from wild-type mice 22 days after tumour inoculation. d, Melanoma (B16-F10-OVA) tumour growth and final tumour mass in wild-type mice implanted with either full (n = 7) or control scaffolds (n = 4) compared with PBS (n = 4). Each point represents one mouse. Survival of mice following treatment with either PBS, blank scaffold or immunoactive (full) scaffolds (n = 5). e, Status of recruited T cells in tumours. Flow cytometry analysis of CD4+ and CD8+ T cells recruited and expanded in the tumours 22 days after subcutaneous injection of B16-F10-OVA cells. Activation of recruited CD8+ T cells was monitored by measuring surface expression of CD44 as well as intracellular measurement of granzyme B expression. f,g, The frequency of Foxp3+CD25+CD4+ Treg cells in tumours (f) and tumour-draining lymph nodes (g). dLN, draining lymph nodes. h, Activated CD8+ T cells inside scaffolds. Flow cytometry analysis of the percentage of CD8+ T cells in explanted scaffolds 17 days after subcutaneous implantation of scaffolds. Horizontal bars indicate means.
Tumours, implanted scaffolds, tumour-draining lymph nodes and spleens were examined. The tumour-infiltrating T cells showed an increase in the proportion of CD8+ T cells, and they were more highly activated and cytotoxic (Fig. 4e) in the tumours adjacent to the functionalized scaffolds compared with those in the control scaffolds. These T cells were also significantly more activated and cytotoxic within the functionalized scaffolds compared with those within the control scaffolds (Fig. 4h and Supplementary Fig. 15). The proportions of total CD4+ T cells, on the other hand, were roughly consistent (Fig. 4e,h). The effect of recruitment and proliferation of the CD8+ T cells was local, as we saw no increase in the proportion, activation or cytotoxicity of CD8+ T cells in the draining lymph node at the late time point (22 days) (Supplementary Fig. 16a–c). These findings show that the functionalized scaffolds successfully recruited and locally activated T cells, promoted their entry into tumours and promoted the clearance of even aggressive tumours.
To better understand the impact of local TGFβ inhibition on the melanoma model, we examined various tissues for the presence of Treg cells. Treg cells were depleted by about 60% in the tumours adjacent to functionalized scaffolds compared with controls (Fig. 4f). As systemic administration of TGFβi can result in autoimmune disease29, we examined whether the local release of TGFβi adjacent to the tumour could suppress Treg cells in distant parts. We found no significant changes in Treg-cell proportions in the draining lymph node (Fig. 4g and Supplementary Fig. 16e). Exhaustion of tumour-infiltrating lymphocytes is commonly noted, and we saw no significant difference in PD-1hi T cells (Supplementary Figs. 16d and 17d).
To better understand the mechanism of these underlying findings, we examined the T cells recruited to the scaffolds. Previous work in melanomas overexpressing CCL21 showed recruitment of both CD4+ and CD8+ T cells, albeit with different tempos30. The scaffolds above used the chemokine CCL21 to recruit T cells into the milieu. We compared whether ligation of two different chemokine receptors on T cells, CCR7 and CXCR4, by two different chemokines, CCL21 and CXCL12 (also called stromal cell-derived factor 1), respectively, could explain the preferential recruitment of CD8+ T cells into the scaffold and tumour. Both chemokines successfully recruited T cells and helped clear tumours compared with control scaffolds (Supplementary Fig. 18a,b). We found that CCL21 favoured recruitment of CD8+ T cells and disfavoured CD4+ T cells compared with CXCL12 (Supplementary Fig. 18c). Despite the presence of identical activation antibodies CD3 and CD28 in the scaffold, co-ligating CCR7 with CCL21 also increased the cytotoxic state of the effector T cells (Supplementary Fig. 18d). These results show that CCL21 improves recruitment of cytotoxic effectors into the tumour compared with CXCL12. Moreover, ligation of CCR7 improves the killing potential of CD8+ T cells. These results show that activation of chemokine receptors on T cells improves the therapeutic capability of our scaffold.
An important detail in the therapeutic translation of this scaffold lies in its stability before use. Water-based hydrogels can have low stability because of spontaneous hydrolysis, which we sought to reduce by preparing our alginate-based scaffolds in a sterile, lyophilized and dry state. To test whether the scaffolds could retain their functional ability after 6 months of cold storage, we used functionalized scaffolds in the B16 melanoma mouse model and found no difference between freshly fabricated scaffolds and older ones (Supplementary Fig. 19). Also, we found sterilization through X-ray irradiation did not affect proteins within the scaffold (Supplementary Figs. 8 and 9).
These results showed that the immunoactive scaffold cleared tumours by recruiting and highly activating CD8+ T cells while depleting local Treg cells within the tumour.
Distant melanoma tumours
We next assessed whether local depletion of intratumoural Treg cells in a primary tumour could alter the trajectory of the immune response in a distant secondary tumour. Wild-type mice received a subcutaneous injection of B16-F10 cells in the flank, and as before functionalized or unfunctionalized scaffolds were implanted subcutaneously after 5 days, when the tumours first became palpable. On the same day, we injected tumour cells contralaterally to the primary one (Fig. 5a). We monitored tumour growth on both sides and measured tumour masses. The growth of the secondary tumour was suppressed by about 40% upon local treatment of the primary tumour with immunoactive scaffolds but not control scaffolds (Fig. 5b–d). The proportion of tumour-infiltrating CD8+ T cells was increased by more than twofold in the contralateral tumour of the mice that received functionalized scaffolds (Fig. 5e and Supplementary Fig. 21). Activated T cells were also found in the spleen and draining lymph nodes, suggesting that local inhibition of Treg cells allows widespread trafficking of activated effector T cells (Supplementary Figs. 23 and 24). Those T cells were more activated and expressed more granzyme B (Fig. 5f and Supplementary Fig. 21c,d). We noted the expansion at 22 days of an expanded population of CD62L+CD44+CD8+ T cells, suggesting the development of central memory T cells in the tumour. As before, the proportion of Treg cells was markedly reduced in the primary tumour. Importantly, Treg cells were also reduced in the secondary tumour, although to a lesser extent (Fig. 5h,i), but not in the lymph nodes (Supplementary Fig. 23e). These results show that localized suppression of intratumoural Treg cells resulted in both successful clearance of primary tumours and marked reduction of secondary tumours. The minor decrease in Treg cells in the distant tumour supports the model that antigen-specific Treg cells trained in the primary tumour may spread globally to facilitate distant tumours. By suppressing those Treg cells in the primary tumour, we suppressed Treg cells in the secondary tumours. However, the disproportionate increase in activated, cytotoxic CD8+ T cells in the secondary tumour despite the minor decrease in Treg cells there indicates that expansion of potent effector T cells in the primary tumour spreads through the body (abscopal effect) and affects distant sites arising from the scaffold acting on the primary tumour as seen also in our 4T1 breast cancer data.
Fig. 5 |. Engineered scaffolds can suppress the growth of local and distant tumours.
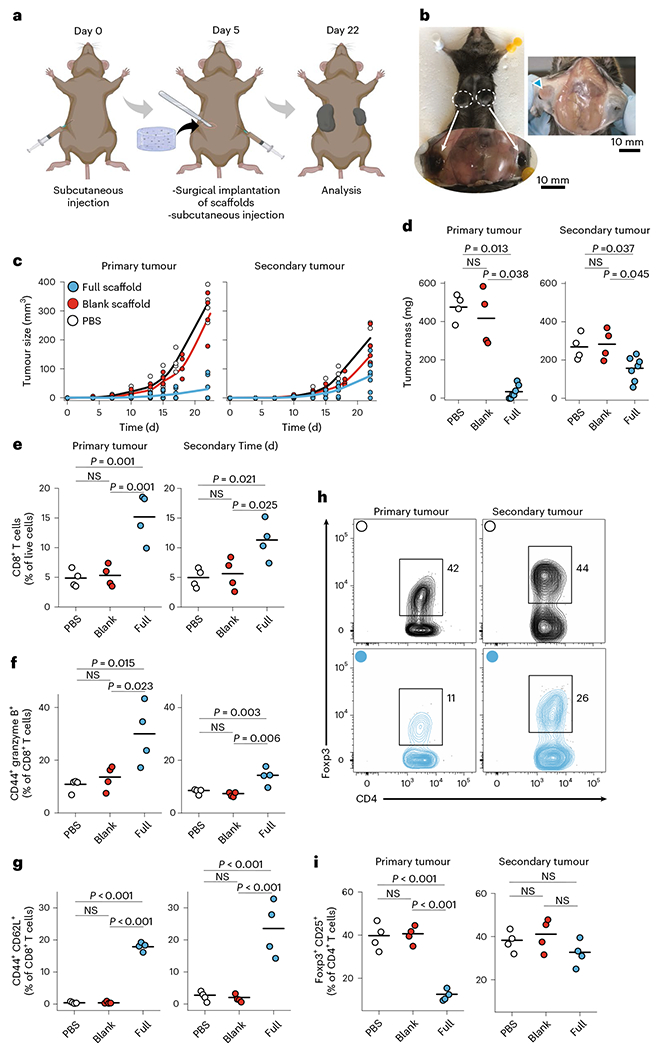
a, Timing of inoculation of primary and secondary tumours and follow-up surgical implantation of the cell-free scaffolds. b, Growth of primary and secondary tumours in the control mouse. c,d, Melanoma (B16-F10-OVA) tumour growth (c) and final tumour masses (d) were measured 22 days after inoculation of primary tumours in wild-type mice treated with full or control scaffolds (n = 4–7). Each point represents a mouse. e, The frequency of CD8+ T cells in primary and secondary tumours (n = 4). f,g, Flow cytometry study of the frequency of CD44+ granzyme B+CD8+ (activated) (f) and CD44+CD62L+CD8+ (central memory) (g) T cells in primary and secondary tumours (n = 4). h, Representative flow cytometry of Foxp3+CD25+CD4+ Treg cells in primary and secondary tumours for mice treated with full scaffolds (blue) and PBS (black). i, Frequency of Foxp3+CD25+CD4+ Treg cells in primary and secondary tumours. Horizontal bars indicate means.
Adoptive T-cell therapy
Delivery of engineered T cells such as chimeric antigen receptor (CAR) T cells is necessary for a variety of cancer therapies, but unlike in lymphomas, IV administration does not always allow for efficient delivery of tumour-specific T cells into solid tumours31. Alginate-based scaffolds that lack structure have been used to deliver T cells in mouse melanoma and breast cancer32,33, where the matrix served as a T-cell depot to support their local expansion. We sought to use the structural advantages of microporosity and mechanical rigidity of our scaffold to deliver T cells. Wild-type mice received a subcutaneous injection of B16-F10-OVA cells in the flank, and as before functionalized or unfunctionalized scaffolds were implanted after 5 days, when the tumours first became palpable (Fig. 6a–d). These scaffolds were loaded just before implantation with 5 × 106 in vitro activated OT-1 T cells, which bear a transgenic TCR that recognizes the cognate antigenic peptide from ovalbumin expressed by the tumors. Some mice instead received IV injection of either the same number (5 × 106) of OT-1 T cells or saline (Fig. 6d–f and Supplementary Fig. 25). Mice were euthanized at 22 days as before for further analysis.
Fig. 6 |. Engineered scaffolds deliver antigen-specific T cells, suppress Treg cells and control melanomas.
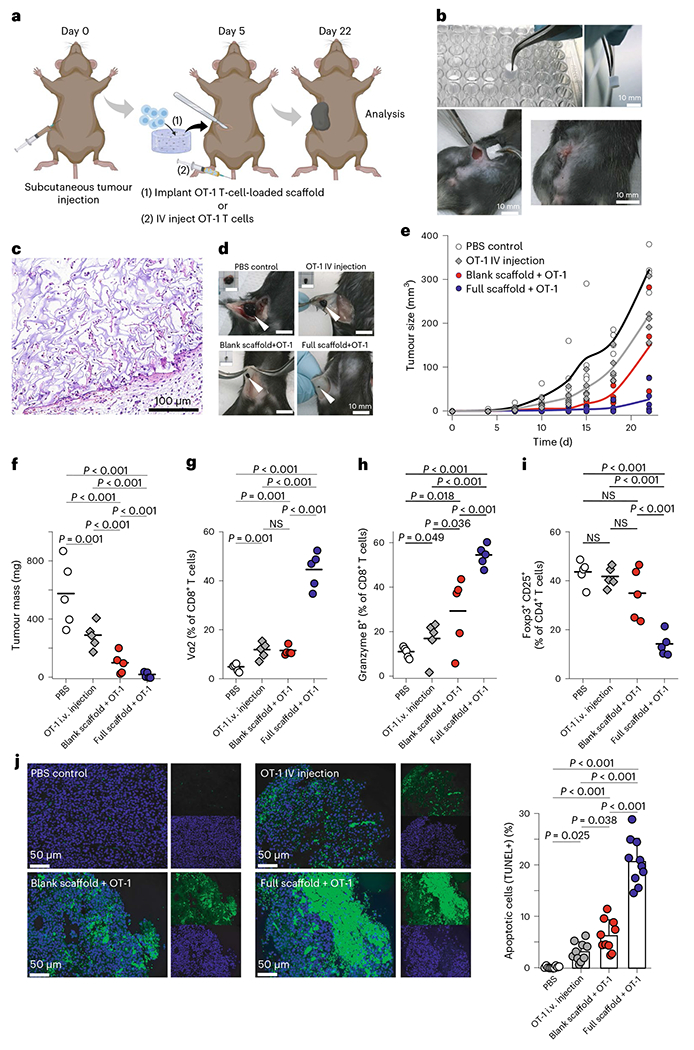
a, Timing of tumour inoculation and implantation of the scaffold loaded with activated OT-1 cells. b, The engineered scaffold is surgically implanted in a B16-F10-OVA bearing mouse. c, H&E staining showing connective tissue-like scaffolds with infiltrating cells. d, Representative images of subcutaneous tumours from wild-type mice 22 days after tumour inoculation. Alginate scaffolds carrying tumour-reactive T cells and T-cell-specific activator cues can eliminate melanoma tumours in mice. e,f, Melanoma (B16-F10-OVA) tumour growth (e) and final tumour mass (f) in wild-type mice with immunoactive or control scaffolds compared with PBS (n = 5). Each point represents one mouse. g, The presence of tumour-specific CD8+ T cells (OT-1) in tumours was studied 22 days after inoculation of tumour cells using flow cytometry. h, Frequency of CD8+ T cells with high expression of granzyme B. i, Frequency of Foxp3+CD25+CD4+ Treg cells in tumour was studied (n = 5). j, Tumour-cell apoptosis was detected using TUNEL staining after various treatments, examples of staining on the left and summarized data on the right. Horizontal bars indicate means.
H&E staining of the scaffolds adjacent to the tumour confirmed tissue engagement, successful delivery and proliferation of OT-1s plus recruitment of endogenous T cells (Fig. 6c). Our OT-1-loaded, functionalized scaffold significantly suppressed tumour growth compared with other treatments and controls, by ~16-fold compared with saline control and ~10-fold compared with IV injection of OT-1 T cells (Fig. 6e,f and Supplementary Fig. 26). We also observed significant improvement in survival of mice receiving the combination of full scaffolds and OT-1 T cells. All (100%) of the mice receiving OT-1-loaded immunoactive scaffolds showed long-term survival (tracked up to 70 days), even when they had been injected with more B16-F10-OVA (ovalbumin peptide) cancer cells (1 × 106 compared with 0.5 × 106) (Supplementary Fig. 29). These results show that scaffold-based delivery resulted in better tumour clearance than IV delivery of T cells.
Moreover, we found that the accumulation and activation of antigen-specific T cells was far more effective with the immunoactive scaffolds than with delivery by IV delivery or by an inert, control scaffold (Fig. 6g–i). Scaffold-based delivery of OT-1 T cells dramatically outperformed IV delivery when looking at the resulting number of OT-1 T cells in the tumour (Fig. 6g). Moreover, the scaffold-delivered T cells were more activated and showed greater cytotoxicity (Fig. 6h and Supplementary Fig. 26). Treg-cell populations in the tumour showed significant differences between treatment groups. Treg cells were suppressed by over 60% in the tumours adjacent to immunoactive scaffolds compared with the control scaffold, IV delivery of T cells or saline controls (Fig. 6i). Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling (TUNEL) staining showed a significant increase in apoptotic tumour cells in the functionalized scaffold that delivered OT-1 T cells compared with controls (Fig. 6j). These results confirm that the immunoactive scaffold delivers highly cytotoxic, antigen-specific T cells to the tumour environment.
Tumour-draining lymph nodes showed a comparable number of OT-1 T cells when they were delivered by the scaffold compared with IV injection (Supplementary Fig. 27). On the other hand, highly activated OT-1 cells were more present in the lymph node and spleens when mice received an IV injection compared with when scaffolds delivered the T cells (Supplementary Figs. 27 and 28). As before, the proportions of Treg cells in lymph nodes were comparable between groups (Supplementary Fig. 27c). These results explain why IV injection of engineered, tumour-specific T cells so often fails: these activated T cells end up in the lymph nodes and spleen rather than in the tumour. The direct access of effector T cells to the tumour microenvironment provided by the scaffold also bypasses the formidable barriers established by the tumour vasculature to the transport of tumour-specific T cells34.
We next assessed whether the improvements seen at the 3-week time point would reflect better long-term survival for mice receiving treatment of the immunoactive scaffold loaded with T cells for local delivery compared with those with the immunoactive scaffold without T-cell delivery (compare Figure 6e–i, blue versus red). We ran a longer experiment and compared head-to-head survival comparing the two conditions versus phosphate-buffered saline (PBS) control injections. We also compared injecting 1 × 106 B16-F10-OVA tumour cells, a higher tumour burden, versus 0.5 × 106 cells. We found significant improvement in survival of mice receiving the combination of immunoactive scaffolds plus local delivery of OT-1 T cells compared with mice with immunoactive scaffolds but no cell transfer. All (100%) of the mice receiving OT-1-loaded immunoactive scaffolds showed prolonged survival (up to 70 days), even with the higher tumour load (Supplementary Fig. 29). This experiment confirms that delivery of antigen-specific T cells by the immunoactive scaffolds can potently contribute to complete response and long-term survival.
We also assessed the toxicity of immunoactive scaffolds 1 week post-implantation in non-tumour-bearing mice. We found no significant changes in haematological counts (Supplementary Table 2). Major organs including the heart, liver, spleen and kidney were also examined for histological alterations. No detectable abnormalities or lesions in these organs were noticed (Supplementary Fig. 30).
Discussion
To facilitate the immune response against solid tumours, we introduced a new multifunctional biomaterial scaffold that is implanted adjacent to a tumour, which attracts cytotoxic T cells and enhances their function by suppressing tumour-resident Treg cells. Together these activities allow for the much sought-after effect of overcoming the immunosuppressive effects of the microenvironment of solid tumours without global adverse effects. This flexible platform offers localized immunomodulation and treatment of multiple cancer types. The approach is general and could be adapted for localized immunoregulation in autoimmune diseases and infections as well.
Existing clinical approaches to circumvent the immunosuppressive tumour microenvironment have serious downsides. For example, global inhibition of Treg cells can lead to autoimmune side effects, whereas systemic activation of T cells can lead to cytokine storms. Immunomodulatory solutions that act locally, thereby avoiding global side effects, would be attractive and may indeed be necessary. At the same time, intratumoural or intralymphatic injections of Treg-cell-depleting therapies have shown limited promise because of the high oncotic pressure that rapidly dissipates the therapeutic effect. Another approach entails the release of cytokines to T cells via nanogel ‘backpacks’, which has been shown to enhance tumour clearance35. However, the continual and uncontrolled release of cytokines risks systemic effects. IV administration of collagen-binding domains fused to the cytokine IL-12 prolonged the release of cytokine into the tumour stroma36; however, this approach increases risks of cardiovascular disease. We have previously developed NPs that deliver cytokines to CD8+ T cells at carefully controlled rates and showed that prolonged exposure to IL-2 impacts their differentiation and effector function37. Besides cytokines, antibodies that stimulate CD3 and CD28 are known to augment T-cell activation in situ. Intratumoural injection of stimulatory antibodies has been attempted, but rapid transport away from the tumour environment has prevented potential clinical application38,39. As we demonstrated in this work, sustained release of cytokines and other immunomodulatory factors from a biomaterial into the local tumour microenvironment, while at the same time stably anchoring stimulatory antibodies near the tumour, addresses these shortcomings.
In addition to biochemical cues, we and others have shown in recent work that T cells sense the mechanical stiffness of 3D microenvironments and undergo augmented activation when the environment reaches an elastic modulus of ~40 kPa (ref. 24). Mechanistically, we have shown that the stiffness of the local environment affects T-cell activation by modulating their metabolic programme23. Others have used biomechanically soft hydrogels in vivo to activate T cells33, but we now know that mechanical rigidity is important to maximize activation. The mechanical stiffness of our biomaterial was optimized to mimic that of activated lymph nodes33, hence its key role in our approach.
Adoptive cell therapy of antigen-specific T cells, in particular CAR T cells, have been successful in the treatment of liquid cancers because intravenously delivered cells are preferentially trafficked to the lymph nodes and spleen, where these cancers reside. However, in the context of solid tumours, the IV delivery of antigen-specific T cells has shown major inadequacies. Regional or local injection of CAR T cells has been superior to IV delivery for a variety of cancers40–42. Delivery of CAR T cells into the tumour microenvironment from a biopolymer device has been shown in mouse models of cancer, suggesting the promise of this approach32,33. These strategies required the activation of T cells ex vivo before delivery, which works well for CAR T cells but does not work for enabling endogenous T-cell responses.
In summary, we have shown that an approach orchestrating multiple immunological mechanisms simultaneously can overcome barriers imposed by solid tumours to allow endogenous or engineered T cells to infiltrate and eradicate otherwise aggressive cancers. Moreover, these T cells clear secondary tumours and metastases, retain long-term memory and protect from cancer recurrence.
Methods
Chemicals and biologicals
Unless noted otherwise, all chemicals were purchased from Sigma-Aldrich. All glassware was cleaned overnight using concentrated sulfuric acid and then thoroughly rinsed with Milli-Q water. All the other cell culture reagents, solutions and dishes were obtained from ThermoFisher Scientific, except as indicated otherwise.
Preparation and characterization of cytokine-producing microparticles
Monodisperse mesoporous silica microparticles (5–20 μm) were formed using a microfluidic jet spray-drying process, using cetyl-trimethylammonium bromide and/or pluronic F127 as templating agents, and tetraethylorthosilicate for silica as reported before43,44. To conjugate heparin to the particles, we first added amine groups to the particles. We suspended the mesoporous silica microparticles (800 mg) in dehydrated methanol (50 ml) then added (3-aminopropyl) triethoxysilane (3 ml) and stirred at room temperature overnight. The final product was centrifuged (1,500 rpm, 3 min) and washed with methanol five times, followed by drying under high vacuum. To functionalize the aminated-silica particles with heparin, heparin sodium salt (216 mg) was dissolved in deionized water (8 ml) and activated via successive addition of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (63 mg) and N-hydroxysulfosuccinimide (sulfo-NHS) (71.4 mg). After stirring for 5 min, the ethanolic solution of amino-functionalized silica (20 mg in 1.12 ml) was added to the reaction mixture and stirred for 12 h at room temperature. Afterwards, the particles were separated by centrifugation and washed several times with deionized water and ethanol to remove unreacted reagents.
To incorporate IL-2 (BRB Preclinical Repository, National Cancer Institute, National Institutes of Health (NIH)) into these heparin-coated microparticles, the microparticles were incubated with different amounts of cytokine in PBS buffer containing bovine serum albumin (BSA; 0.1% w/v) and were gently shaken overnight at 4 °C. The microparticles were then centrifuged and washed several times to remove unabsorbed cytokines. BSA offers the potential for immunogenicity and cross-reactivity with ovalbumin45, an immunogen for the tumours where we used OT-1 T cells. To test for immunogenicity of BSA in our ovalbumin experiments, we started by assessing whether BSA could activate naive OT-1 CD8+ primary mouse T cells co-cultured with 1 μg ml−1 (~1 μM) ovalbumin antigen (SIINFEKL) as a positive control or BSA (1 and 10 mg ml−1, which are 22 and 222 times the molar concentration of the OVA) in the presence of lipopolysaccharide (100 ng ml−1). We saw no response of OT-1 T cells to large amounts of BSA (Supplementary Fig. 6). To assess whether endogenous T cells (not OT-1) could be primed by BSA and have heightened responses to ovalbumin-bearing tumours, we compared tumour trajectories for melanoma tumours expressing ovalbumin and melanoma tumours lacking ovalbumin (Supplementary Fig. 20), both after exposure to BSA in the scaffold. We saw no differences, whereas we should have instead seen an improvement in outcomes in the melanomas containing ovalbumin if BSA was able to prime those responses. Taken together, these results suggest that the amount of BSA in our scaffold was so small that it was not able to cross-react or enhance T-cell responses to ovalbumin.
To estimate the in vitro release of IL-2 from microparticles, we incubated 20 × 106 microparticles in 2 ml PBS (pH 7.4; supplemented with 1 mM CaCl2) at 37 °C. At different time intervals, 500 μl of the supernatant was collected and replaced with an equivalent volume of PBS. The concentration of released IL-2 was determined using a human IL-2 enzyme-linked immunosorbent assay (ELISA) kit (ThermoFisher). The final particles used for formulating scaffolds (below) used 50 μg IL-2 per mg of particles.
In some experiments, the silica particles were also covalently conjugated with T-cell activating antibodies: anti-CD3 (clone 2C11; Bio-X-Cell) and anti-CD28 (clone 37.51; Bio-X-Cell). We used 1 μg anti-CD3 and 0.25 μg anti-CD28 per 106 silica particles. After activation of antibodies’ carboxylic groups for 10 min with EDC/NHS, microparticles were added and incubated under gentle stirring at 4 °C overnight. The functionalized microparticles were then separated from the solution and washed several times. Unreacted functional groups were quenched by washing samples in Tris buffer (100 mM, pH 8) for 30 min. Micro-BCA assay was used to quantify total amount of surface conjugated antibodies according to the manufacturer’s protocol.
TGFβi particles
We prepared PLGA NPs to release the TGFβi LY2157299 (also called galunisertib, Cayman Chemical). We prepared the PLGA using a nanoprecipitation method as previously reported46: briefly, Resomer RG 503 PLGA (50:50; molecular weight: 28 kg mol−1) was used in this study. LY2157299 and PLGA were dissolved in 5 ml dichloromethane and sonicated into 1% poly vinyl alcohol (PVA) solution (50 ml) by probe sonicator (12 W) for 2 min. The resulting emulsification was then added to 100 ml 0.5% PVA solution. The solution was agitated, and the dichloromethane was allowed to evaporate for 4 h. The solution was then centrifuged at 3,000 × g for 5 min to pellet out any non-nano size material. The supernatant was removed and ultracentrifuged and washed three times at 21,000 × g for 20 min to wash away the PVA. The resulting NP solution was flash frozen in liquid nitrogen and lyophilized for 2 days before characterization and use. The hydrodynamic diameter and surface charge of the formed PLGA NPs were studied using dynamic light scattering and zeta potential measurements (Zetasizer Nano).
Preparation and characterization of scaffolds
To form the scaffolds, we first oxidized the alginate (molecular weight ≈ 250 kDa, high G blocks; Novamatrix UP MVG, FMC Biopolymer) with sodium periodate (1.5%) overnight at room temperature, then quenched the reaction by drop-wise addition of ethylene glycol for 45 min. We then dialysed the solution (molecular weight cut off 3.5 kDa) against deionized water for 3 days followed by lyophilization. Afterwards, the alginate was dissolved in 2-(4-morpholinyl)ethanesulfonic acid (150 mM, NaCl 250 mM, pH 6.5) and covalently conjugated to arginine–glycine–aspartic acid (RGD)-containing peptide (GGGGRGDY; GenScript) using carbodiimide chemistry (NHS/EDC). The reaction was continued for 24 h followed by dialysis (MWCO 20 kDa) and lyophilization. This alginate–RGD complex in PBS was mixed with CCL21 protein (Peprotech Recombinant Mouse Exodus-2, 250 ng per scaffold). To prepare scaffolds, 20 × 106 IL-2-loaded heparin-functionalized silica microparticles were mixed with 1 ml of alginate. The alginate–chemokine–cytokine–partide–TGFβi-NP mixture was cross-linked using calcium sulphate solution. Each resulting scaffold bore 400 IU of IL-2. The gels were casted in desired 24- or 96-well plates followed by two overnight washes to remove extra calcium ions. These scaffolds were frozen at −80 °C, lyophilized for 3 days and stored at 4 °C before use.
To load these NPs into alginate-based scaffolds, LY2157299-loaded PLGA NPs were mixed with alginate before crosslinking via calcium. The concentration of released LY2157299 from NPs before and after loading into alginate scaffolds was determined by measuring the ultraviolet absorption of LY2157299.
We prepared an array of different alginate formulations by varying either the polymer content or the amount of crosslinker (here CaSO4). To measure the mechanical stiffness of our gels, we used an Instron 5542 mechanical tester, and all the samples were tested at a rate of 1 mm min−1. Young’s modulus was then calculated from the slope of the linear region that corresponds with 0–10% strain. The final alginate formulation used for in vivo studies comprised alginate 2.5% with 40 mM CaSO4.
Calcium measurement in the scaffold
Some calcium is sequestered into the alginate matrix for crosslinking, and some is extra. We perform 3-day wash (two overnights with change) by immersing in PBS (PBS + 0.42 mM CaCl2). We measured the calcium content of the scaffolds before and after washing. We also immersed the scaffolds after washing into in vitro culture media (RPMI +10% FBS + antibiotics and amino acids) for 3 days and 7 days. Then we measured the calcium content of the scaffolds. For all these measurements, scaffolds were lysed using EDTA (50 mM) and alginate lyase (porcine, Sigma-Aldrich, 3.4 mg ml−1). Then we triggered the dissociation of the EDTA-Ca2+ complex and released calcium ions for measurement by addition of acetic acid (0.05 vol%). Finally, the calcium content was measured using a Calcium Colorimetric Assay Kit (BioVision).
To immobilize anti-CD3 and anti-CD28 to the scaffolds, the freeze-dried scaffolds were activated with EDC/NHS for 15 min. Then the scaffolds were washed twice with PBS (supplemented with 0.42 mM CaCl2) before addition of anti-CD3 and anti-CD28. Then they were incubated at 4 °C overnight. Unreacted functional groups were quenched by washing the scaffolds with Tris buffer (100 mM, pH 8) for 30 min. For T-cell activation studies, 5 × 106 primary naive T cells were added to the scaffolds and cultured for 3–5 days to study their effector functions.
CCL21 release from the scaffolds was measured by mouse CCL21 ELISA kits as a function of time.
To sterilize the fabricated scaffolds before in vitro or in vivo functional assays, we used X-ray irradiation (Gulmay Medical RS320 X-ray unit) as per ISO 11137-2:2013 recommended protocols47. A dose of 25 kGy (2.5 Mrad) was used for sterilization. We showed that this sterilization approach did not alter the physical properties, including changes in morphology and mechanical stiffness of the scaffolds, or T-cell activation properties (Supplementary Fig. 8).
We also measured the endotoxin level of digested scaffolds using ToxinSensor Chromogenic LAL Endotoxin Assay Kit (Genscript). Each scaffold was dissolved in 1 ml of digestion buffer, and the endotoxin level was calculated to be around 0.14 ± 0.02 endotoxin units per ml. The FDA Centre for Devices and Radiological Health has adopted the USP Endotoxin Reference Standard, which limits and regulates the acceptable level of endotoxin contamination for medical devices to 0.5 endotoxin units per ml. Thus, our scaffolds contained a very low level of endotoxin at a level that is FDA-allowable.
Scanning electron microscopy (SEM) images of the gels were taken to see the cross-sectional microstructure and porosity of the alginate-based scaffolds. The lyophilized scaffolds were freeze-fractured (using liquid nitrogen) for cross-sectional images. The scaffolds were sputtered with iridium (South Bay Technology Ion Beam Sputtering) before imaging with a ZEISS Supra 40VP scanning electron microscope (Carl Zeiss Microscopy). The sizes of pores from different parts of the SEM images were then measured and analysed using ImageJ 1.53h software (NIH). For SEM imaging of cell-loaded scaffolds, the cell-laden hydrogels were fixed with 2.5% glutaraldehyde, followed by post-fixation in osmium tetroxide before serial dehydration in increasing concentrations of ethanol (25%, 50%, 75%, 90% and 100%, for 15 min each) and iridium sputtering.
We also used confocal microscopy to study the association of T cells with the scaffolds. For these experiments, alginate was stained with amine-fluorescein before hydrogel formation. T cells were incubated with the scaffold in eight well-plate Labtek II chambers for 30 min before fixation and staining. The supernatant was removed from Labtek chambers, and then scaffolds were fixed with 4% PFA. Then to permeabilize cells for intracellular staining, 0.1% Triton X-100 in PBS was incubated with cells for 5 min. To stain the cells, we used Alexa Fluor 568-conjugated phalloidin (Life Technologies). After several washes, samples were covered with Fluoromount-G with DAPI (eBioscience) and stored at 4 °C before imaging with the Leica Confocal SP8-STED microscope. Images were analysed using Leica software, Imaris and ImageJ/Fiji.
Ethics
All mouse experiments were conducted in accordance with University of California, Los Angeles’s (UCLA’s) institutional policy on humane and ethical treatment of animals following protocols approved by the UCLA Animal Research Committee.
T-cell isolation and activation
For adoptive transfer of antigen-specific T cells intravenously or for peritumoural delivery by scaffolds, we used 5-to 8-week-old wild-type or OT-1 TCR transgenic mice (Jackson Labs). T cells were purified from spleens using the EasySep immunomagnetic negative selection enrichment kit (Stem Cell Technologies). T cells were cultured in media comprising RPMI-1640 supplemented with 10% heat-inactivated FBS, 1% penicillin/streptomycin, 1% sodium pyruvate, 1% HEPES buffer and 0.1% μM 2-mercaptoethanol.
In vitro activation of T cells was performed by culturing 1 × 106 cells per ml in tissue culture-treated 24-well plates that were pre-coated with anti-CD3 (clone 2C11; Bio X Cell) at a concentration of 10 μg ml−1 plus addition of 2 μg ml−1 soluble anti-CD28 (clone 37.51; Bio X Cell). T cells were then collected from wells and allowed to proliferate in IL-2 (BRB Preclinical Repository, National Cancer Institute, NIH)-containing medium (50 U ml−1), before being used for experiments.
To study T-cell proliferation and function, T cells were recovered from scaffolds at selected time points by digesting the scaffolds using EDTA (50 mM) and alginate lyase (porcine, SigmaAldrich, 3.4 mg ml−1) for 20 min. The number of viable T cells recovered from the scaffolds were determined by trypan blue exclusion.
For Treg-cell formation experiments CD4+ T cells were purified from mouse spleen as mentioned above. Cells were then either activated on scaffolds or on anti-CD3e antibody (8 μg ml−1) coated plates with the anti-CD28 antibody (2 μg ml−1) supplemented medium. At the same time TGFβ (15 ng ml−1) was added to the media. After 4 days Treg cells were removed, fixed, permeabilized and stained with antibodies for flow cytometric analysis of Foxp3 expression.
Flow cytometry
For flow cytometry analysis, antibodies to mouse antibodies were purchased from eBioscience, BioLegend or BD Biosciences. To study proliferation behaviour of T-cell responses during various treatments, their expansion was measured by 5-(and-6)-carboxyfluorescein diacetate, succinimidyl ester (CFSE) dilution. For CFSE dilution experiments, 5 ×105 naive CD4+/CD8+ T cells were labelled with 2 μM CFSE for 13 min, followed by two washes and then incubation with splenocytes. Splenocytes were extracted from the spleens of wild-type C57BL/6 mice. Then the cells were incubated in ACK lysis buffer (Gibco) for 5 min at room temperature to remove red blood cells. The remaining cells were then treated with ova peptide as above to present to T cells. Cells were analysed on a Cytek DxP10 flow cytometer using FlowJo software (version 10.8.1, Treestar/BD).
For intracellular staining of granzyme B and Foxp3, we followed the manufacturer’s protocol (Foxp3/Transcription Factor Staining Buffer Set, eBioscience). The following antibodies were used for intracellular staining from Biolegend: Foxp3 (clone MF-14, AF647, catalogue number 126408); granzyme B (clone GB11, AF647, catalogue number 515406), Mouse IgG1, κ Isotype Ctrl (clone MOPC-21, AF647, catalogue number 400130).
Migration assay
The migration assay to evaluate the role of chemokines on recruitment of T cells and melanoma cells in the presence and absence of chemokine-functionalized scaffolds was performed using regular Transwell migration as we reported37. The number of migrated cells was evaluated after 4 h using an automatic cell counter.
Breast cancer model
About 5 × 105 4T1-Luc or 4T1 cells were injected orthotopically into the mammary fat pad of female wild-type BALB/c mice (6–8 weeks old, Jackson Lab). The growth of tumours was monitored by measuring the bioluminescence signal with the IVIS Imaging System (Perkin Elmer) and/or using a digital calibre. Stable signals were determined by monitoring the photon count up to 30 min after intraperitoneal injection of luciferase substrate, D-luciferin (15 mg ml−1 diluted in PBS), into animals anaesthetized with inhaled 2.5% isoflurane. Upper body metastases were also assessed in the same way on day 30 after covering the primary tumour sites. The Living Image software was used to analyse bioluminescence flux.
At 100 days post injection of the primary tumour, surviving mice were rechallenged with retro-orbital (IV) injection of 2 × 105 4T1-Luc tumour cells in 100 μl of HBSS. Development of metastases in lung was monitored by bioluminescence imaging.
We also tested the immunological abscopal effects for the breast cancer model. Immunoactive scaffolds and PBS control were prepared. Wild-type, immunocompetent syngeneic mice were implanted in the mammary fat pad with 5 × 105 4T1-Luc breast cancer cells. After 5 days, the scaffolds were implanted, and 3 × 105 tumour cells were injected into the contralateral mammary fat pad. We then tracked tumour growth using bioluminescence imaging.
Melanoma model
About 2 × 105 to 5 × 105 B16-F10-OVA or B16-F10 tumour cells were subcutaneously injected into the right or both (in the contralateral tumour model) right and left flanks of wild-type C57BL/6 J WT mice (6–8 weeks old, Jackson Lab). These melanoma-derived cells are transfected to express chicken OVA48. Five days after tumour cell injection, scaffolds were surgically implanted subcutaneously into the same approximate region of the tumours in both flanks. For cell-loaded studies, ex vivo activated OT-1 T cells were transferred either intravenously using retro-orbital injections (100 μl per animal) or with implantable scaffolds on the same day. About 5 × 106 OT-1 T cells were given in all adoptive transfer experiments. Tumour size was assessed over time using a digital caliper until day 22 at which animals were killed and the tumour, draining lymph nodes and spleen were extracted.
We also tested the immunological abscopal effects for the melanoma model. Wild-type mice received a subcutaneous injection of 5 × 105 B16-F10-OVA cancer cells in the flank, and as before functionalized or unfunctionalized scaffolds were implanted subcutaneously after 5 days, when the tumours first became palpable. On the same day, we injected 3 × 105 tumour cells contralaterally to the primary one.
Tumour assays
Tumour masses were measured using a digital balance before digesting the tumour tissue for flow cytometry or fixing it for tissue sectioning. Tumours were digested by incubating in collagenase and DNase I (50 μg ml−1) at 37 °C for 15 min. These enzymes were inactivated with EDTA (20 μl per ml of solution). Tissues were then mechanically disaggregated and passed through a 70 μm cell strainer to obtain a single-cell suspension. Cells were then stained with the fluorochrome-conjugated antibodies on ice. For intracellular staining, cells were permeabilized with granzyme B Fix/Perm buffer according to the manufacturer’s instructions (BioLegend) before staining. Detection of apoptotic cells in the tumour tissue was achieved using TUNEL staining following the manufacturer’s directions. TUNEL-positive cells were indicated as apoptotic melanoma cells. Tissue sections were imaged by a fluorescence microscope (Keyence BZ-X800).
Blood was collected 7 days post implantation of the scaffolds for cytokines analysis. Concentrations of interferon gamma and tumour necrosis factor alpha in serum was determined by ELISA (eBioscience).
Haematology, biochemistry and tissue collection
After 7 days of treatment, all mice were killed, and blood and major organs were collected for blood analysis and organ studies. Using a standard blood-collection technique, we drew 1,000 μl blood into LTT-Lavender top EDTA anticoagulant collection tubes for haematology analysis. The blood serums were also isolated using SST-serum Separator Tube containing clot activator with gel for blood chemistry analyses. Samples were analysed by IDEXX BioAnalytics. Major organs as well as the scaffolds from these mice were also extracted, fixed and processed, and stained with H&E. Slides were examined using a digital microscope.
Statistical analysis
The Kruskal–Wallis rank sum test, one-way analysis of variance (ANOVA) and two-tailed Student’s t-test were utilized as appropriate to analyse the data at a significance of α or P < 0.05. Quantitative data were expressed as mean ± s.d. To determine the number of specimens for the proposed experiments, a power analysis was conducted based on pilot experiments.
Supplementary Material
Acknowledgements
We thank L. Bentolila and S. Xu for their technical assistance with confocal imaging and animal imaging studies, respectively, and S. Haile for her assistance with flow cytometry. M.J.B. discloses support in part for the research described in this study from the University of California, Los Angeles (UCLA) Jonsson Comprehensive Cancer Center (JCCC). L.-S.B. discloses support in part for the research described in this study from the UCLA CTSI (UL1TR001881). S.L. discloses support in part for the research described in this study from the National Institutes of Health (NIH) (R01 CA234343). F.S.M. discloses support in part for the research described in this study from the R&D fund by Symphony Bioscience. Confocal laser scanning microscopy was performed at the Advanced Light Microscopy/Spectroscopy Laboratory and the Leica Microsystems Centre of Excellence at the California NanoSystems Institute at UCLA, which is supported by funding from the NIH Shared Instrumentation Grant (S10OD025017) and NSF Major Research Instrumentation grant (CHE-0722519). Scanning electron microscopy was performed at California NanoSystems Institute Electron Imaging Centre for NanoMachines Shared Resource Facility at UCLA. Flow cytometry was partially performed at the UCLA JCCC and Centre for AIDS Research Flow Cytometry Core Facility, which are partially supported by NIH (P30 CA016042 and 5P30 AI028697), and by the JCCC, the UCLA AIDS Institute, the David Geffen School of Medicine at UCLA, the UCLA Chancellor’s Office and the UCLA Vice Chancellor’s Office of Research. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIH (United States) or/and other agencies of the State of California.
Footnotes
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41551-022-00977-0.
Competing interests
The Regents of the University of California filed patent applications (PCT/US2020/051363 and WO2021055658A1) related to this study, with F.S.M, M.M.H.-S. and M.J.B. as inventors. F.S.M, M.M.H.-S. and M.J.B. are founders and equity holders in Symphony Biosciences. The other authors declare no competing interests.
Peer review information Nature Biomedical Engineering thanks the anonymous reviewers for their contribution to the peer review of this work.
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. All data generated in this study, including source data for the figures, are available from figshare with the identifier https://doi.org/10.6084/m9.figshare.21406713.
References
- 1.Dahan R et al. FcγRs modulate the anti-tumor activity of antibodies targeting the PD-1/PD-L1 axis. Cancer Cell 28, 285–295 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Simpson TR et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med 210, 1695–1710 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haanen JBAG et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol 28, iv119–iv142 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Champiat S et al. Management of immune checkpoint blockade dysimmune toxicities: a collaborative position paper. Ann. Oncol 27, 559–574 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Naidoo J et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann. Oncol 27, 1362 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumagai S. et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol 21, 1346–1358 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Sasada T, Kimura M, Yoshida Y, Kanai M & Takabayashi A CD4+CD25+ regulatory T cells in patients with gastrointestinal malignancies: possible involvement of regulatory T cells in disease progression. Cancer 98, 1089–1099 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Curiel TJ et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med 10, 942–949 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Sato E. et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl Acad. Sci. USA 102, 18538–18543 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shang B, Liu Y, Jiang SJ & Liu Y Prognostic value of tumor-infiltrating Foxp3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci. Rep 5, 15179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xydia M. et al. Common clonal origin of conventional T cells and induced regulatory T cells in breast cancer patients. Nat. Commun 12, 1119 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas DA & Massagué J TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8, 369–380 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Mariathasan S. et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Batlle E & Massagué J Transforming growth factor-β signaling in immunity and cancer. Immunity 50, 924–940 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stauber AJ, Credille KM, Truex LL, Ehlhardt WJ & Young JK Nonclinical safety evaluation of a transforming growth factor β receptor I kinase inhibitor in Fischer 344 rats and beagle dogs. J. Clin. Toxicol 4, 1–10 (2014). [Google Scholar]
- 16.Herbertz S et al. Clinical development of galunisertib (Ly2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Devel. Ther 9, 4479–4499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park J. et al. Combination delivery of TGF-β inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat. Mater 11, 895–905 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colombo MP & Piconese S Regulatory T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat. Rev. Cancer 7, 880–887 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Zhou X et al. Precise spatiotemporal interruption of regulatory T-cell-mediated CD8+ T-cell suppression leads to tumor immunity. Cancer Res. 79, 585–597 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Onizuka S et al. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor α) monoclonal antibody. Cancer Res. 59, 3128–3133 (1999). [PubMed] [Google Scholar]
- 21.Tanaka A & Sakaguchi S Targeting Treg cells in cancer immunotherapy. Eur. J. Immunol 49, 1140–1146 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Sato K et al. Spatially selective depletion of tumor-associated regulatory T cells with near-infrared photoimmunotherapy. Sci. Transl. Med 8, 352ra110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meng KP, Majedi FS, Thauland TJ & Butte MJ Mechanosensing through YAP controls T cell activation and metabolism. J. Exp. Med 217, e20200053 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Majedi FS et al. T-cell activation is modulated by the 3D mechanical microenvironment. Biomaterials 252, 120058 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khazaie K & von Boehmer H The impact of CD4+CD25+ Treg on tumor specific CD8+ T cell cytotoxicity and cancer. Semin. Cancer Biol 16, 124–136 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Budhu S et al. Blockade of surface-bound TGF-β on regulatory T cells abrogates suppression of effector T cell function in the tumor microenvironment. Sci. Signal 10, eaak9702 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pulaski BA & Ostrand-Rosenberg S Mouse 4T1 breast tumor model. Curr. Protoc. Immunol 10.1002/0471142735.im2002s39 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Han J, Khatwani N, Searles TG, Turk MJ & Angeles CV Memory CD8+ T cell responses to cancer. Semin. Immunol 49, 101435 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wrzesinski SH, Wan YY & Flavell RA Transforming growth factor-β and the immune response: implications for anticancer therapy. Clin. Cancer Res 13, 5262–5270 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Novak L, Igoucheva O, Cho S & Alexeev V Characterization of the CCL21-mediated melanoma-specific immune responses and in situ melanoma eradication. Mol. Cancer Ther 6, 1755–1764 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Riley RS, June CH, Langer R & Mitchell MJ Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov 18, 175–196 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith TT et al. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J. Clin. Invest 127, 2176–2191 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stephan SB et al. Biopolymer implants enhance the efficacy of adoptive T-cell therapy. Nat. Biotechnol 33, 97–101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tahmasebi S, Elahi R & Esmaeilzadeh A Solid tumors challenges and new insights of CAR T cell engineering. Stem Cell Rev. Rep 15, 619–636 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Tang L et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat. Biotechnol 10.1038/nbt.4181 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mansurov A et al. Collagen-binding IL-12 enhances tumour inflammation and drives the complete remission of established immunologically cold mouse tumours. Nat. Biomed. Eng 10.1038/s41551-020-0549-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Majedi FS et al. Cytokine secreting microparticles engineer the fate and the effector functions of T-cells. Adv. Mater 30, 1703178 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stohrer M, Boucher Y, Stangassinger M & Jain RK Oncotic pressure in solid tumors is elevated. Cancer Res. 60, 4251–4255 (2000). [PubMed] [Google Scholar]
- 39.Jain RK Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumors. Cancer Res. 50, 814–819 (1990). [PubMed] [Google Scholar]
- 40.Adusumilli PS et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med 6, 261ra151 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katz SC et al. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther. 23, 142–148 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nellan A et al. Durable regression of medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J. Immunother. Cancer 6, 1–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waldron K et al. Formation of monodisperse mesoporous silica microparticles via spray-drying. J. Colloid Interface Sci 10.1016/j.jcis.2013.12.027 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Liu W, Chen XD & Selomulya C On the spray drying of uniform functional microparticles. Particuology 22, 1–12 (2015). [Google Scholar]
- 45.Dearman RJ, Caddick H, Basketter DA & Kimber I Divergent antibody isotype responses induced in mice by systemic exposure to proteins: a comparison of ovalbumin with bovine serum albumin. Food Chem. Toxicol 38, 351–360 (2000). [DOI] [PubMed] [Google Scholar]
- 46.Hasani-Sadrabadi MM et al. On-chip synthesis of fine-tuned bone-seeking hybrid nanoparticles. Nanomedicine 10, 3431–3449 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Sterilization of Health Care Products—Radiation—Part 2: Establishing the Sterilization Dose—Technical Corrigendum 1 (ISO, 2009). [Google Scholar]
- 48.Bellone M et al. Relevance of the tumor antigen in the validation of three vaccination strategies for melanoma. J. Immunol 165, 2651–2656 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main data supporting the results in this study are available within the paper and its Supplementary Information. All data generated in this study, including source data for the figures, are available from figshare with the identifier https://doi.org/10.6084/m9.figshare.21406713.