

Abstract
Introduction
Children’s Interstitial Lung Diseases (cHILD) are a heterogeneous group of rare respiratory diseases. Their common characteristics are gas exchange abnormalities and diffuse pulmonary infiltrates on chest imaging. This group includes inherited surfactant protein deficiency (ISPD), a little-known etiology in Tunisia.
Case Presentation
A 22-month-old boy was referred to investigate recurrent respiratory infections. He had polypnea, cyanosis, finger clubbing, pectus carinatum, intercostal retraction, and bilateral crackles on pulmonary auscultation. The chest imaging revealed a diffuse ground-glass appearance consistent with cHILD. Lung biopsy was suggestive of ISPD. The infant was mainly treated with intravenous corticosteroids. At the age of nine, he was still dependent on oxygen but had better exercise tolerance.
Conclusion
This case showed that recurrent respiratory infections can hide cHILD which may be related to ISPD, particularly in infants. A better knowledge of this disease was necessary to start specific treatment. Early management would lead to better prognosis.
Keywords: Diffuse interstitial lung disease, Inherited surfactant protein deficiency, Recurrent respiratory infections, Infant
Résumé
Introduction
Les pneumopathies infiltrantes diffuses de l'enfant (PIDe) sont un groupe hétérogène de maladies respiratoires rares. Leurs caractéristiques communes sont des anomalies des échanges gazeux et des infiltrats pulmonaires diffus à l’imagerie thoracique. Ce groupe comprend le déficit héréditaire en protéines du surfactant (DHPS), une étiologie peu connue en Tunisie.
Observation
Un garçon de 22 mois était transféré pour explorer des broncho-pneumopathies récidivantes. Il avait une polypnée, une cyanose, un hippocratisme digital, un pectus carinatum, un tirage intercostal et des râles crépitants bilatéraux à l'auscultation pulmonaire. L'imagerie thoracique a révélé un aspect diffus en verre dépoli en faveur d’une PIDe. La biopsie pulmonaire était évocatrice d'un DHPS. Le nourrisson était principalement traité par des corticostéroïdes intraveineux. À l'âge de neuf ans, il était encore dépendant de l'oxygène mais avait une meilleure tolérance à l'effort.
Conclusion
Ce cas a montré que les broncho-pneumopathies récidivantes peuvent masquer une PIDe qui peut être liée à un DHPS, en particulier chez le nourrisson. Une meilleure connaissance de cette maladie était nécessaire pour débuter un traitement spécifique. Une prise en charge précoce conduirait à un meilleur pronostic.
Mots clés: Pneumopathies infiltrantes diffuses, Déficit héréditaire en protéines du surfactant, Broncho-pneumopathies récidivantes, Nourrisson
INTRODUCTION:
Children’s Interstitial Lung Diseases (cHILD) are a heterogeneous group of rare respiratory diseases responsible for high morbidity and mortality (1, 2). Their common characteristics are gas exchange abnormalities and diffuse pulmonary infiltrates on chest imaging (1). An annual cHILD incidence of 1.2 per million was reported in immunocompetent children (2). However, it remains largely underestimated (2). The clinical presentations are variable, nevertheless they are generally revealed by tachypnea associated with exercise intolerance (1, 2). We report a case of a rare cause of cHILD revealed by recurrent pneumonias.
CLINICAL CASE:
This observation was reported according to the CARE guideline (3 ).
A 22-month-old boy was referred to investigate recurrent respiratory infections reaching the stage of chronic respiratory failure. There was no family history other than first degree consanguineous parents. The patient was hospitalized at a regional hospital at the age of six, nine and 12 months for pneumonia, each episode treated with intravenous antibiotics for ten days. Since his last hospitalization, he maintained a chronic cough with worsening dyspnea on exertion.
On admission, the infant was hypotrophic (weight at 8.5 kg (-3 to -2 Standard Deviations (SD)), height at 84 cm (+1 SD)), and had polypnea at 42 cycles/min, oxygen saturation at 91% in room air and 98% under 1 L/min of oxygen, finger clubbing, pectus carinatum, intercostal retraction, and bilateral crackles on pulmonary auscultation. An increase in dyspnea with circumoral cyanosis was noted at the slightest effort.
The chest X-ray showed bilateral alveolar opacities and chest distension (Figure 1A). The chest computerized tomography (CT) scan revealed a diffuse ground-glass appearance (Figure 1B). The first investigations were normal (sweat test, echocardiography, immunity investigations, and chromatography of amino acids and organic acids). The flexible bronchoscopy showed moderate bilateral and diffuse bronchial inflammation. The analysis of the bronchoalveolar lavage fluid revealed a high cell count at 2,490,000/mm³, high neutrophil count (11.5%) and rare siderophages with low hemosiderin content at Perls staining. There was no eosinophilic deposit on Periodic Acid Shiff staining.
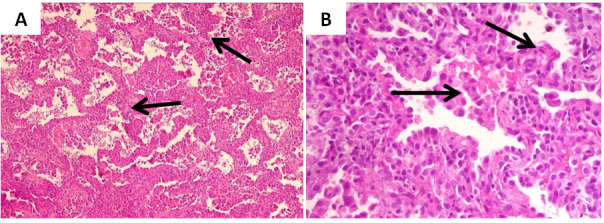
The histological examination of a lung biopsy fragment was evocative of a cHILD related to a possible primary pulmonary hemosiderosis. Both clinical and biological presentations of the patient were not consistent with this aetiology. Foreign experts’ review concluded that the patient’s histology showed a macrophagic alveolitis, type II cell pneumocyte hyperplasia, proteinaceous material Periodic Acid Shiff positive in some alveolar spaces and fibrosis of some interalveolar septa. This chronic infantile pneumonia was suggestive of an inherited deficiency in surfactant proteins, in particular SP-C or ABCA3 (ATP-binding cassette, sub-family A, member 3) (Figure 2). Genetics did not identify the mutation.
In adjunction to hypercaloric diet and long-term oxygen therapy at 1L/min, the infant was treated with series of intravenous pulse methylprednisolone at a dose of 300 mg/m2 once daily for three successive days. The series were spaced one month apart during the first six months, then three months given the clinical and chest imaging improvement. The infant also received anti-pneumococcal vaccination.
Two years later, hydroxychloroquine was added at a dose of 10 mg/kg/day in order to further space the pulses. Azithromycin was also introduced but not really administered until the age of seven due to lack of resources.
At the age of nine, the patient weighed 18 kg (-2.2 SD) and measured 125 cm (-1 SD). He was still dependent on oxygen but had better exercise tolerance. He was in 4th grade. His last chest CT scan, performed at the age of six, showed a decrease in the extent of ground-glass areas in the lower lobes (Figure 1C). Annual monitoring of echocardiography showed stable pulmonary arterial hypertension at 30 mmHg with moderate dilation of the right ventricle. Lung function tests were limited to a six-minute walk test due to poor patient’s cooperation. This test was satisfactory for age with continuous progress.
DISCUSSION:
To the best of the authors’ knowledge, our patient was the first case of inherited surfactant protein deficiency identified in Tunisia. Its diagnosis was difficult first because diffuse interstitial lung diseases are rare in infants, second this aetiology was unknown in Tunisia and third accessibility of its identification was limited.
Inherited disorders of surfactant metabolism are relatively a recent entity among cHILD causes and increasingly reported over the past two decades (4 ). They are the consequence of advances in molecular biology which have revealed mutations in the genes coding for proteins involved in the synthesis of surfactant, namely the SP-B and SP-C proteins, their transporter ABCA3, and the transcription factor NKX2-1 (NK2 Homeobox 1, also named Thyroid Transcription Factor -1).
Few epidemiological data about these conditions are available since their identification requires an expert team and a specialized technical platform. Thus, the reported cases were sporadic or grouped in small series. Nevertheless, ABCA3 deficiency would be the most frequent of them (5 ).
Clinical symptoms of the inherited surfactant protein deficiencies are nonspecific. However, they vary according to the age of the disease revelation (6 ). In newborns, the situation is most often dramatic with a severe neonatal respiratory distress which does not improve despite exogenous surfactant administration and aggressive ventilation. Such a circumstance should lead to search for SP-B or ABCA3 mutation. The presence of extra-respiratory signs such as hypotonia and/or hypothyroidism suggests a NKX2-1 mutation. In infants, respiratory distress begins gradually from the first months of life. It is associated with a dry cough, polypnea, cyanosis, poor tolerance of effort (such as taking bottle) and failure to thrive. These non-specific signs may be responsible for a diagnostic delay as was our patient’s case. The disease can also be revealed by an unusual dependence on oxygen during a common respiratory infection. In children, the onset is most often insidious over several weeks or even months with non-specific respiratory symptoms as in infants. The disease can even be revealed at the stage of chronic respiratory failure with fingers clubbing, cyanosis, and denutrition. On another hand, a chest wall deformity such as pectus excavatum was described in six patients among a series of nine children with ABCA3 mutations (7 ). In our case, physical examination revealed pectus carinatum. The main surfactant protein deficiencies to evoke in infants and children are those of SP-C and ABCA3 (8-10 8, 9, 10 ).Besides, at any age, it is important to specify the family history. In fact, it can guide the diagnostic approach when histories of diffuse interstitial lung disease and of neonatal death from respiratory distress are present.
Chest CT scan is essential in inherited surfactant pathologies. It most often shows an interstitial syndrome with ground-glass images, reticular opacities and alveolar condensations (11 ).In patients with ABCA3 deficiency, parenchymal lung cysts were also reported (12 ).Later on, they, as well as the peripheral interstitial thickening, became more prominent. Among nine patients with ABCA3 mutations, five developed cysts that tended to increase in number and size over time (7 ).
The bronchoalveolar lavage analysis is also fundamental. It guides the diagnosis by showing a decrease in mature proteins and an increase in pro-proteins. However, this analysis requires a specialized laboratory as for the pulmonary histological study. The diagnosis of inherited disorders of surfactant metabolism is confirmed by molecular biology (13 ).Performing genetic testing for surfactant dysfunction is currently recommended prior to a lung biopsy in most cases. The biopsy should be considered when genetics does not identify the disease (14 ).In our case, histology was capital for the diagnosis as it showed suggestive lesions including type II cell pneumocyte hyperplasia, alveolar proteinaceous material, alveolar macrophages and fibrosis. Besides, the genetic confirmation of the disease is important for genetic counselling and for evaluating the risks of recurrence in families.
The treatment is based on intravenous pulse methylprednisolone at a dose of 10 mg/kg/day or 300 to 500 mg/m2/day for three successive days to be renewed monthly initially, then to space out according to the outcome. Hydroxychloroquine at a dose of 10 mg/kg/day, in combination with corticosteroid therapy, reinforces its immunosuppressive effect and prevents the accumulation of the structurally deficient protein by inhibiting the intracellular process of SP-C protein precursors’ synthesis (15 ).Finally, azithromycin seems to be beneficial due to its anti-inflammatory and immune-modulating effect (16 ).In adults, other agents such as pirfenidone, with anti-inflammatory and anti-fibrotic properties, and nintedanib, a tyrosine kinase inhibitor, revealed effective in idiopathic pulmonary fibrosis (17, 18).
On the other hand, nocturnal or continuous oxygen therapy may be necessary to correct hypoxia and prevent the onset of pulmonary arterial hypertension. Children should receive nutritional support with a high calorie diet. Attention must also be paid to the prevention of infections through hygiene measures and by carrying out anti-influenza and anti-pneumococcal vaccinations.
Patient monitoring is based on regular assessment of respiratory and growth parameters, oxygen requirement, respiratory functions, echocardiography, and chest CT scan. Such care ensured a prolonged survival for our patient while waiting for lung transplant, which is unavailable in our country.
Conclusion:
Recurrent respiratory infections may hide cHILD. Inherited disorders of surfactant metabolism are part of cHILD causes. A better knowledge of these causes is necessary in order to improve diagnostic means in our country. Early treatment would lead to better prognosis.
Declaration of interest
The authors declared no conflict of interest.
Aknowledgments
The authors would like to express their sincere gratitude to Professor Jacques de Blic, paediatric pulmonologist, and Doctor Jean Christophe Fournet, pathologist, at Necker-Enfants-Malades Hospital in Paris, for their help in making the diagnosis of inherited surfactant protein deficiency.
Figure 1. Chest imaging: (A) Bilateral alveolar opacities and thoracic distension on chest X-ray, (B) Diffuse ground-glass appearance on chestcomputerized tomography (CT) scan, (C) Decrease in areas of ground-glass in the lower lobes on the control chest CT scan.
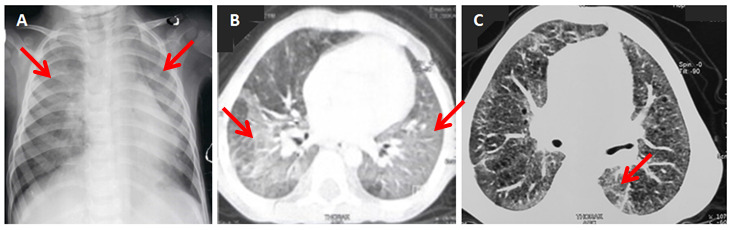
Figure 2. Histological examination of the lung biopsy: (A) alveolar septa thickened by fibrosis, (B) slight inflammatory infiltrate with macrophages along thesepta and hyperplasia of pneumocytes II (Hematoxylin and eosin staining, low magnification on (A) and high magnification on (B)).
References
- Cunningham S, Jaffe A, Young L R. Children’s interstitial and diffuse lung disease. Lancet Child Adolesc Health. 2019;3(8):30117–30118. doi: 10.1016/S2352-4642(19)30117-8. [DOI] [PubMed] [Google Scholar]
- Clement Annick, Nathan Nadia, Epaud Ralph, Fauroux Brigitte, Corvol Harriet. Orphanet Journal of Rare Diseases. 1. Vol. 5. Springer Science and Business Media LLC; 2010. Interstitial lung diseases in children; pp. 22–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JJ Gagnier, G Kienle, DG Altman, D Moher, H Sox, D Riley. The CARE Guidelines: Consensus-based Clinical Case Reporting Guideline Development. G. Glob Adv Health Med. 2013;2(5):38–43. doi: 10.7453/gahmj.2013.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa Hiroshi, Kure Shigeo. Clinical Medicine Insights: Circulatory, Respiratory and Pulmonary Medicine. 1. 9s1. SAGE Publications; 2015. Interstitial Lung Disease in Childhood: Clinical and Genetic Aspects; pp. CCRPM.S23282–CCRPM.S23282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alanazi Alnashmi, Epaud Ralph, Heena Humariya, Becdelievre Alix De, Miqdad Abeermohammad, Fanen Pascale M. Annals of Thoracic Medicine. 3. Vol. 12. Medknow; 2017. The most frequent ABCA3 nonsense mutation -p.Tyr1515* (Y1515X) causing lethal neonatal respiratory failure in a term neonate; pp. 213–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole F Sessions, Nogee Lawrence M, Hamvas Aaron. Pediatric Clinics of North America. 5. Vol. 53. Elsevier BV; 2006. Defects in Surfactant Synthesis: Clinical Implications; pp. 911–927. [DOI] [PubMed] [Google Scholar]
- Doan M L, Guillerman R P, Dishop M K, Nogee L M, Langston C, Mallory G B, Sockrider M M, Fan L L. Thorax. 4. Vol. 63. BMJ; 2008. Clinical, radiological and pathological features of ABCA3 mutations in children; pp. 366–373. [DOI] [PubMed] [Google Scholar]
- Flamein Florence, Riffault Laure, Muselet-Charlier Céline, Pernelle Julie, Feldmann Delphine, Jonard Laurence, Durand-Schneider Anne-Marie, Coulomb Aurore, Maurice Michèle, Nogee Lawrence M, Inagaki Nobuya, Amselem Serge, Dubus Jean Christophe, Rigourd Virginie, Brémont François, Marguet Christophe, Brouard Jacques, De Blic Jacques, Clement Annick, Epaud Ralph, Guillot Loïc. Human Molecular Genetics. 4. Vol. 21. Oxford University Press (OUP); 2012. Molecular and cellular characteristics of ABCA3 mutations associated with diffuse parenchymal lung diseases in children; pp. 765–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thouvenin G, Taam R A, Flamein F, Guillot L, Le Bourgeois M, Reix P, Fayon M, Counil F, Depontbriand U, Feldmann D, Pointe H D L, De Blic J, Clement A, Epaud R. Archives of Disease in Childhood. 6. Vol. 95. BMJ; 2010. Characteristics of disorders associated with genetic mutations of surfactant protein C; pp. 449–454. [DOI] [PubMed] [Google Scholar]
- Nogee L M, Garnier G, Dietz H C, Singer L, Murphy A M, Demello D E, Colten H R. Journal of Clinical Investigation. 4. Vol. 93. American Society for Clinical Investigation; 1994. A mutation in the surfactant protein B gene responsible for fatal neonatal respiratory disease in multiple kindreds. pp. 1860–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechri M, Epaud R, Emond S, Coulomb A, Jaubert F, Tarrant A, Feldmann D, Flamein F, Clement A, De Blic J, Taam R Abou, Brunelle F, Le Pointe H Ducou. Pediatric Pulmonology. 10. Vol. 45. Wiley; 2010. Surfactant protein C gene (<i>SFTPC</i> ) mutation-associated lung disease: High-resolution computed tomography (HRCT) findings and its relation to histological analysis; pp. 1021–1029. [DOI] [PubMed] [Google Scholar]
- Kröner Carolin, Wittmann Thomas, Reu Simone, Teusch Veronika, Klemme Mathias, Rauch Daniela, Hengst Meike, Kappler Matthias, Cobanoglu Nazan, Sismanlar Tugba. Thorax. 3. Vol. 72. BMJ Publishing Group Ltd; 2017. Lung disease caused by ABCA3 mutations; pp. 213–220. [DOI] [PubMed] [Google Scholar]
- Gupta Atul, Zheng Sean Lee. Archives of Disease in Childhood. 1. Vol. 102. BMJ; 2017. Genetic disorders of surfactant protein dysfunction: when to consider and how to investigate; pp. 84–90. [DOI] [PubMed] [Google Scholar]
- Vece Timothy J, Young Lisa R. Chest. 3. Vol. 149. Elsevier BV; 2016. Update on Diffuse Lung Disease in Children; pp. 836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush Andrew, Cunningham Steve, De Blic Jacques, Barbato Angelo, Clement Annick, Epaud Ralph, Hengst Meike, Kiper Nural, Nicholson Andrew G, Wetzke Martin, Snijders Deborah, Schwerk Nicolaus, Griese Matthias. Thorax. 11. Vol. 70. BMJ; 2015. European protocols for the diagnosis and initial treatment of interstitial lung disease in children; pp. 1078–1084. [DOI] [PubMed] [Google Scholar]
- Clement A, Tamalet A, Leroux E, Ravilly S, Fauroux B, Jais J-P. Thorax. 10. Vol. 61. BMJ; 2006. Long term effects of azithromycin in patients with cystic fibrosis: a double blind, placebo controlled trial; pp. 895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghu Ganesh, Craig Johnson W, Lockhart Diane, Mageto Yolanda. American Journal of Respiratory and Critical Care Medicine. 4. Vol. 159. American Thoracic Society; 1999. Treatment of Idiopathic Pulmonary Fibrosis with a New Antifibrotic Agent, Pirfenidone; pp. 1061–1069. [DOI] [PubMed] [Google Scholar]
- Richeldi Luca, Du Bois Roland M, Raghu Ganesh, Azuma Arata, Brown Kevin K, Costabel Ulrich, Cottin Vincent, Flaherty Kevin R, Hansell David M, Inoue Yoshikazu, Kim Dong Soon, Kolb Martin, Nicholson Andrew G, Noble Paul W, Selman Moisés, Taniguchi Hiroyuki, Brun Michèle, Le Maulf Florence, Girard Mannaïg, Stowasser Susanne, Schlenker-Herceg Rozsa, Disse Bernd, Collard Harold R. New England Journal of Medicine. 22. Vol. 370. Massachusetts Medical Society; 2014. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis; pp. 2071–2082. [DOI] [PubMed] [Google Scholar]