

Summary
Antibodies are an important group of biological molecules that are used as therapeutics and diagnostic tools. Although millions of antibody sequences are available, identifying their structural and functional similarity and their antigen binding sites remains a challenge at large scale. Here, we present a fast, sequence-based computational method for antibody paratope prediction based on protein language models. The paratope information is then used to measure similarity among antibodies via protein language models. Our computational method enables binning of antibody discovery hits into groups as the function of epitope engagement. We further demonstrate the utility of the method by identifying antibodies targeting highly similar epitopes of the same antigens from a large pool of antibody sequences, using two case studies: SARS CoV2 Receptor Binding Domain (RBD) and Epidermal Growth Factor Receptor (EGFR). Our approach highlights the potential in accelerating antibody discovery by enhancing hit prioritization and diversity selection.
Subject areas: Computational chemistry, Protein, Bioinformatics, Artificial intelligence
Graphical abstract
Highlights
-
•
Protein language models were fine-tuned for paratope prediction
-
•
Model representations of antibody’s paratopes are used for similarity search
-
•
Antibodies with similar paratope representations show similar epitope engagement
Computational chemistry; Protein; Bioinformatics; Artificial intelligence
Introduction
Antibody (Ab) epitope knowledge is crucial for understanding B cell-mediated immunity. It is also essential information in developing therapeutic antibodies.1 To date, grouping a library of monoclonal antibodies (mAbs) by their epitopes against a common antigen (Ag), i.e., epitope binning, is recognized as one of the critical steps of a discovery campaign. Epitope binning early can reduce redundancies in Ab hit selection and accelerate our understanding of mechanism of action. The sooner such information is available, the greater the probability of maintaining epitope diversity in the selection process; thus, improving the identification of unique, functional, and effective antibodies with better immunogenicity, ADME, or developability properties is paramount.
Among the advancements in antibody technology platforms, Next Generation Sequencing (NGS) provides tens of thousands of antibody clones that can easily challenge the highest throughput experimental methodology currently employed. Consequently, an in silico approach is highly desired for its capacity, speed, and cost. Effective use of in silico binning can supply epitope relevant information much earlier in the process, on a greater diversity of molecules, to inform decision-making and focus labor/cost intensive experimental steps on molecules with increased probability of technical success. Moreover, reliable in silico binning is especially valuable for challenging targets, such as membrane antigens, unstable reagents, or targets with limited functional assay capacity. in silico Ab epitope prediction is a long-standing goal and challenged by the complexity of finding cognate pairs among the immense Ab repertoire and the multitude of Ag epitopes.2,3,4 As a result, there are no such in silico tools readily available. However, ever increasing computing power and emerging technologies,5,6 joined with accumulating experimental database and deep-learning algorithms,7,8,9,10 support the position that in silico prediction is possible. Toward the grand goal of Ab epitope prediction, we propose a divide-and-conquer approach by focusing on a specific task – in silico binning of thousands of Abs into functional groups of similar binding specificity by assessing their paratope properties, computationally. The establishment of an in silico binning tool, together with NGS, will further enhance the NGR discovery process. The ability to screen 100s–1000s Abs as a function of epitope engagement will speed up hit selection, ensure maintenance of maximal diversity, and contribute to intellectual property positioning versus competitors. There are several categories of computational methods that aim to find epitope similarity,11 including clonotyping, paratyping and structural profiling. Some of the methods rely on sequence similarity. But relying solely on sequence identity is not sufficient to identify Abs with similar epitopes, as antibodies can form similar interactions with notably low sequence identity.11 In addition, several works rely on structural complementary and docking methods, some in combination with machine learning, to predict epitope-paratope engagement.12,13,14,15,16,17
Abs have a conserved structural framework and their Ag specificity is determined by the surface of their binding site composed of six “hypervariable loops” referred to as Complementary Determining Regions (CDRs), which collectively comprise the paratope. Generally, the paratope of an Ab recognizes an epitope on the Ag with a high degree of specificity, which is achieved via unique molecular interactions. The specific interaction between paratope and epitope is driven by shape and physiochemical property complementarity, e.g., hydrophobicity, charge distribution, polarity, and shape index. Conceptionally, Abs with similar paratope features likely interact with similar epitopes on the Ag surface. Ab CDR sequences alone are not sufficient to distinguish epitope specificity; for example, necitumumab and cetuximab exhibit different CDR loop lengths and compositions, yet they bind to a very similar epitope on EGFR.18 Similarly, Ovalbumin (OVA) Abs with various H3 lengths can bind a similar epitope region evidenced by binding competition assays.19 This underpins the importance of analyzing the paratope features for binning, in addition to the sequence. Ab structure prediction by homology modeling is well-established, from which structural fingerprints of paratope can be retrieved. In the past, protein fingerprinting has demonstrated wide applications from searching for remotely homologous proteins in databases, screening ligand binding for targeted proteins. Along this line, works such as Ab-ligity,20 used structural similarities among modeled antibodies to present a structural comparison of paratopes, however, they rely on homology modeling, and inaccurate prediction of antibody’s loops may pose a challenge. In addition, generating homology for very large pools of antibodies can be computationally expensive.
Here, we propose a computational approach that predicts the paratope residues of antibodies first, then uses those to generate representations that can be utilized to measure similarity and used for clustering. There have been several works to predict antibody’s paratope residues from the sequence. Programs such as Parapred,21 ProABC22 and ProAbC-223 predict the paratopes using machine learning. In this work, we demonstrate a paratope prediction model based on protein language models and show the application of this method on two examples: binning of SARS CoV2 RBD antibodies and screening for novel antibodies which bind similar epitopes of commercial EGFR antibodies. Our proposed approach does not require a priori knowledge of antigen/epitope. Rather, we attempt to predict whether two or more Abs are likely to bind the same epitope via paratope similarity analysis; thus, computationally clustering Abs into functional bins. Our results show great promise for the method in acceleration and reducing the screening costs of novel antibody discovery.
Results
Paratope prediction
Table 1 shows the paratope prediction model’s performance measured by cross-validation according to several common metrics used for evaluating machine learning models. Several methods have been developed in the past for paratope prediction; however, one should take note of the factors that can affect performance, other than the model itself, such as dataset size, dataset preparation, train and test splitting method, CDR and paratope definition, etc. Needless to say, with available antibody structural data growing every year, newer methods have access to larger data compared to their predecessors. Also, different CDR and paratope definitions may cause the metrics values to be different even if the methods perform similarly. Such factors can make direct comparison among methods difficult.
Table 1.
Paratope prediction performance by different metrics of the fine-tuned protein language model on the Development (Dev.) and Test sets via 10-fold cross-validation
| Set | ROC AUC | F1 | MCC |
|---|---|---|---|
| Dev. | |||
| Test |
Average standard deviations among validation sets shown.
Nevertheless, here we report our method’s performance compared with another notable work, namely, Parapred21 on the same test set that we prepared for this comparison (Data S2), ensuring no sequences in the set are in either ours or Parapred’s training set. The set consists of 115 heavy and 94 light chains from 121 PDBs with redundant chains with higher than %95 sequence identity removed. Parapred uses the same criteria for CDR and paratope definition, and is freely available and therefore is suitable for this comparison.
Performance comparison with Parapred is shown in Table 2.
Table 2.
Paratope prediction performance comparison with Parapred by different metrics on the same test set
| Set | ROC AUC | F1 | MCC |
|---|---|---|---|
| Parapred | 0.876 | 0.663 | 0.533 |
| Ours | 0.660 | 0.541 |
Paratope similarity via neural representations
Measuring the similarity of words and sentences using neural representations is common in the field of natural language processing. Trained language models provide a convenient way of finding text and words with similar meanings by comparing the closeness of their neural embeddings. A language model learns similarity among words or sentences and generates similar representations for them during the pretraining phase. Works such as24 showed that such an approach is applicable for protein sequences as well by demonstrating that functional or structural similarities can be discovered using protein sequence embeddings generated by the model. The embeddings can directly be used as input to other models or be used for clustering and similarity measurements.
Although the paratope prediction by fine-tuning the language model is a supervised task, the generation and comparison of neural embeddings of the paratope residues does not require further training. This provides great advantage because novel antibodies for novel antigens can be compared immediately without requiring training data. Our method only measures paratope similarity: no information about the antigen has to be known, making this method antigen-agnostic.
Antibodies targeting SARS-CoV2 Receptor Binding Domain (RBD)
RBD epitope similarity analysis
Barnes et al. defined four major classes of epitopes for the RBD domain binders.25 A dataset of COVID’s spike protein’s binders with their corresponding epitope class labels was obtained from.26 The dataset contains 133 unique antibodies with the majority binding RBD. A collection of antibody pairs was generated by pairing each sequence with all other sequences. The pairs were labeled as “similar epitope” and “different epitope” depending on whether or not they belonged to the same epitope class. The embedding distance of each member of the pair with another was also calculated to see how our predicted paratope similarity relates to epitope similarity.
Low neural embedding distance indicates epitope similarity
The lower the Euclidean distance between two paratopes, the more likely they are to belong to the same class. This is revealed by the analysis shown in Figure 1. In this analysis, the pairwise embedding distances of all combinations of RBD binders collected from26 (Data S3), are measured. The analysis shows that distances lower than 0.9 are very likely to bind the same epitope.
Figure 1.

Fraction of same epitope pairs (blue) to the number of pairs within a certain distance
All possible pairs of the RBD binders in the dataset were generated and pairwise paratope embedding distances were measured. It is revealed that with distance under 0.9, it is highly likely for pairs to target the same epitope.
Identification of antibodies with similar epitopes on EGFR
During the process of antibody discovery, immunization against an antigen can yield a high number of antibodies that bind the antigen. However, not all epitopes are equally important: Blocking certain epitopes may cause a stronger inhibition of the antigen, while binding other epitopes may weakly interfere with the antigen’s normal functionality. Therefore, it is important to be able to identify those binders that bind the epitopes of interest; however, the experimental process can be costly for larger numbers of antibodies. Here, we demonstrate that how novel antibodies for specific epitopes can be identified if there are known antibodies that bind the same epitope. This can be done by finding nearest neighbors of the known antibody in the pool of antibodies with unknown epitopes. In other words, we find the antibodies with the most similar paratope representation to the known binder. The advantage of this approach is that antibodies with lower sequence similarities or different loop lengths to the known binder can be found, as long as the paratope representations are closely similar. We demonstrate the utility of this approach by finding 20 nearest neighbors of commercial antibodies (panitumumab and Necitumumab, 10 neighbors for each antibody) that target EGFR out of 1800 sequences. We identify the neighbors among antibody sequences obtained by single cell PCR (sequences not shown) from the human antibody repertoire and experimentally validate their epitope similarity.
High-throughput SPR experiments on the selected subset of 20 recombinantly-expressed antibodies (including benchmark controls) were performed to assess EGFR-ECD protein epitope coverage. These studies included epitope binning, isolated subdomain binding, and hEGFR-ECD-His binding. We employed two benchmark antibodies known to bind to the D3 domain and that share highly similar binding epitopes. The pairwise sequence similarity and paratope representation distances among the selected and benchmark antibodies are shown in Figures S1–S3. The antibodies (including benchmarks) were clustered into 3 bins using Carterra epitope analysis software based on the competition heatmap and were divided into bins among known subunits of the EGFR protein. Antibodies from each of the bins did not cross-block any of the antibodies belonging to the other bins (Figure 2). Affinity and competition data are shown in Table S1, and Figures S4 and S5.
Figure 2.

Epitope Binning Bins (Carterra LSA™) for 20 selected scPCR nearest neighbors of commercial antibodies
(A) The binning function of the Carterra Epitope software allows for clustering of antibodies that have identical blocking profiles. 20 selected antibodies are grouped into 3 bins.
(B) Antibodies are shown to bind the D3 domain cluster together (Red) with no overlap between D1 domain-binders (Yellow) and antibodies that bind D4 domain (Green).
Paratope and epitope similarity in sequence-dissimilar antibodies
It is reasonable to assume antibody sequences with high similarity possess similar paratopes and bind similar epitopes. Although measuring sequence similarity is trivial, an important question that may arise is whether the paratope similarity method can go beyond sequence similarity by only using sequence data. Previous works on protein language models showed the language model is able to derive evolutionary and structural patterns by performing the pretraining task, and the pretrained model was later used to make predictions regarding structure, such as secondary structure and protein contact prediction, in addition to learning the alignment of sequences.24 Furthermore, the comparison of neural embeddings instead of sequences themselves provides an alignment-free approach to searching for similarity, allowing similarities to be found in different residues and loop lengths.
Among the RBD’s antibodies, four crystal structures with PDB codes:7CH4, 6XC2, 7CH5 and 6XC4 present an interesting example, where the pair 7CH4 and 6XC2 and the pair 6XC2 and 7CH5 are derived from different patients and represent different clonotypes27; nevertheless, their paratope distances are in the range that imply similar epitopes: 0.81 and 0.79 respectively.
For the EGFR antibodies, the nearest neighbors picked were not necessarily the ones with highest sequence similarity. Particularly in the H3 loop region, all sequences differed by more than 3 residues, with some being of different loop length. The above observations indicate the applicability of the paratope similarity method regardless of sequence and lineage similarity. The pairwise sequence similarity and paratope representation distances among the selected and benchmark antibodies are shown in Figures S1–S3.
Discussion
In this work, we described a fast and effective method, based on the sequence only, to measure similarity among predicted antibody paratopes and showed its utility to estimate the epitope similarity regardless of the sequence similarity of the variable domain. We first fine-tuned a protein language model to predict the paratope residues of a given antibody sequence with high accuracy. Next, we used a pretrained language model to generate the neural representations of the predicted paratope residues and used them to measure the similarity between antibody pairs. Our study on two cases of RBD and EGFR binders showed promising results for using the similarity measurements for purposes such as computational epitope binning and diversity selection of antibody molecules in a discovery pipeline, making the process faster and reduce the costs. There are a number of potential applications for this work: Paratope predictions can be utilized to ranking in Ab:Ag docking, by finding the poses that agree with predicted paratopes. Furthermore, the nearest neighbor search enables identifying similar hits to the sequences of interest, which do not necessarily have high sequence similarity. In addition, antibodies can be clustered based on paratope similarities, providing an additional tool to experimental methods such as Carterra binning. Such clusterings can be also used for diversity selection – selecting a subset of sequences with high diversity that would increase the probability of finding antibodies with novel epitopes or unique functions. Given that language models can capture some structural information from the sequence,24 there are possible uses in B cell structural profiling,8 to investigate structural diversity among antibodies.
Limitations of the study
The method described above was designed to be antigen-agnostic, because often detailed information about the antigen’s structure and the epitope is not available. Although being antigen-agnostic makes the method more general, only considering paratope similarity may pose a limitation for cases in which antibodies with different paratopes (sequentially and structurally) bind a similar epitope.
In addition, our method uses a CDR annotation to limit the paratope space, whereas it is known that in some instances, residues outside of the CDR may participate in binding28 and those are not predicted by the algorithm.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-hEGFR antibodies | This paper | N/A |
| Necitumumab | This paper | N/A |
| Cetuximab | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| hEGFR-ECD-His | This paper | N/A |
| hEGFR-D1-His | This paper | N/A |
| hEGFR-D1-D2-His | This paper | N/A |
| hEGFR-D3-D4-His | This paper | N/A |
| hEGFR-D4-His | This paper | N/A |
| Critical commercial assays | ||
| Carterra LSA | Carterra-Bio | https://carterra-bio.com/lsa/ |
| HC30M Chip | Carterra-Bio | 4279 |
| Software and algorithms | ||
| Carterra KITTM 1.7.2.3202 | Carterra-Bio | https://carterra-bio.com/resource-category/software/ |
| Carterra EpitopeTM 1.7.1.3055 | Carterra-Bio | https://carterra-bio.com/resource-category/software/ |
| Python 3.8.5 | Python Software Foundation | https://www.python.org/ |
| Pytorch 1.7.1 + cu101 | Paszke et al., 201929 | https://pytorch.org/ |
| Transformers 4.5.1 | Wolf et al., 201930 | https://huggingface.co/ |
| ProtBert | Elnaggar et al., 202031 | |
| Scikit-learn | Pedregosa et al., 201132 | https://scikit-learn.org/ |
| Matplotlib | Hunter, 200733 | https://matplotlib.org/ |
| Pandas | The pandas development team34 | https://pandas.pydata.org |
| Scipy | Virtanen et al., 202035 | https://scipy.org/ |
| Molecular Operating Environment | Chemical Computing Group, 202236 | https://www.chemcomp.com/ |
| Model Codes | Github | https://github.com/aghanbar-lilly/parasim |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Qing Chai (chai_qing_qc@lilly.com).
Materials availability
This study did not generate new unique reagents.
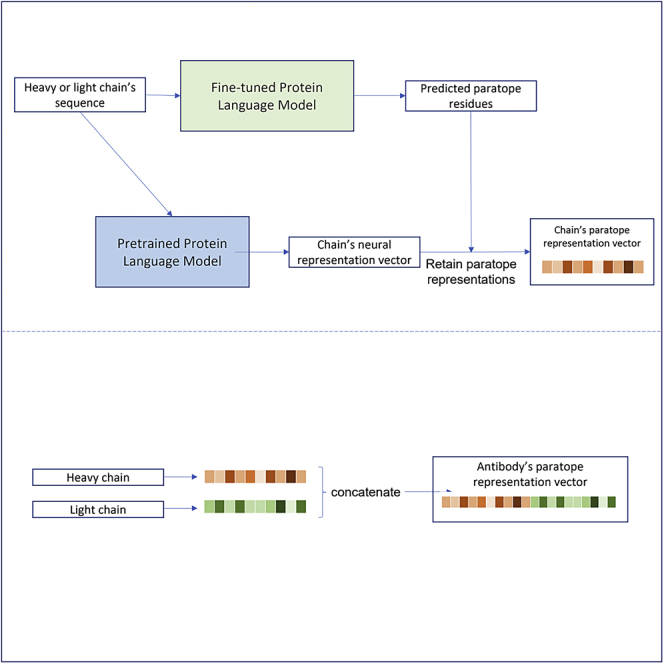
Paratope prediction using a protein language model
Dataset
We scanned the antigen-antibody complexes available in the PDB database by the MOE program36 to find structures with unique antibody sequences. 1479 antibody-antigen complexes (2958 heavy and light chains) were collected. The sequences were clustered using CD-HIT37 and redundant sequences with identity of %95 or higher were removed. The remaining sequences (1179 heavy and 955 light) were used as the dataset for training and validation of the paratope prediction model (Data S1).
CDR annotation and paratope definition
All antibodies were annotated using Chothia38 annotation scheme, while also adding two residues before and after the annotated region to also cover possible interactions outside of the annotated region, an approach that was also used by.21 Any residue in the annotated sequence with an atom within 4.5 Å of any atom of the antigen was labeled as paratope. The annotation and paratope detection were done using an SVL script39 run by MOE.36
Classification of paratope residues via a protein language model
The task of identifying paratope residues can be formulated as a token classification task, where each residue is a token and is labeled as being a paratope or not (binary classification). A pretrained language model40 is fine-tuned to carry out such task. Since antibody’s paratopes are expected to be on the CDR, the model’s loss is chosen so that it is optimized to predict the label of the residues only in the annotated region.
For residue (token) the model’s loss is calculated as follows:
| (Equation 1) |
where CDR is the set of residue indices annotated as CDR domains, and is the length of the chain, is the true label taking ether 1 or 0 which indicate the residue being a paratope or not, respectively, as defined by the distance criteria, and is the Softmax probability of the class.
Model’s was done using Huggingface’s transformers library.30
Fine-tuning the model
The model was fine-tuned for 8 epochs with the initial learning-rate set to and 3 as the batch size. 10-fold cross-validation was used to measure the prediction performance of the model. The validation set was divided into two sets, the development and test sets. During training, the best performing model on the development set was saved and the test set was used for final validation.
Neural representations of paratopes
In the next step, the predicted paratopes are used to generate neural representations of antibody’s paratopes. The details are described in the following subsections.
Embedding generation based on predicted paratopes
For an antibody chain’s sequence with length , given the final hidden representations retrieved from the pretrained version of the language model, the paratope embedding vector is represented as the weighted average of the representations of each residue where each weight is the probability of the residue belonging to the paratope class predicted by the fine-tuned model, with a value ranging from 0 to 1.
| (Equation 2) |
The final representation of the antibody using the two embedding vectors and for heavy and light chains, respectively, is obtained by concatenating the two vectors: .
Quantifying paratope similarity
Pairwise similarity of two antibodies in terms of their paratope representations are quantified by calculating the Euclidean (L2) distance of their representing vectors. Hence, antibodies with distance closer to zero, are assumed the most similar. Pairwise distance is used for visualization and search for nearest neighbors of antibodies as the ones with most similarity and likelihood to bind a similar epitope.
Experimental epitope binning of EGFR binders
Recombinant proteins
Recombinant human EGFR extracellular domain (ECD) was expressed in Chinese hamster ovary (CHO) cells as a His-tagged protein and purified by standard chromatography techniques. Domain specific proteins were designed by substituting D1, D1-D2, D3-D4, and D4 domains of EGFR into HER3 protein scaffold. Proteins were expressed as His-tagged fusions in 293 cells and purified by size exclusion chromatography technique. The purity percentages for D1, D1-D2, D3-D4, D4 domains and ECD were determined as 99.1, 98.9, 99.3, 100 and 100, respectively. Antibodies were expressed in CHO cells and supernatants were utilized for all kinetics and binning experiments. The antibodies were used as supernatants for the competition experiments and therefore their purity values were not determined.
Epitope binning
All epitope binning experiments were performed on a Carterra® LSATM instrument equipped with an HC-30M chip type (Carterra-bio), using a 384-ligand array format as previously described (Shu and McCauley, 2017). Assays were performed according to the manufacturer’s operational guidelines. The instrument used a multi-channel buffer of 25 mM 2-(N-morpholino) ethanesulfonic acid (MES), pH 5.5, and a single-channel buffer of 10 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), 150 nM NaCl, 3 mM EDTA, and 0.05% v/v surfactant P20 (HBS-EP+). The array preparation was performed as described above, using a HC30M chip and coupling unpurified antibody from CHO supernatant diluted to 3 μg/mL and 10 μg/mL in 10 mM acetate, pH 4.0, for 10 minutes, and deactivation for 7 minutes in 1 M ethanolamine, pH 8.5.
For epitope binning experiments, antibodies coupled to the chip surface were exposed to 400 nM hEGFR-ECD-His for 5 minutes followed injections of antibodies at 30 μg/mL both diluted in 1X HBSEP +0.1mg/mL BSA running buffer). Three regeneration cycles of 20 seconds were performed after each antibody sample by injecting 10 mM glycine pH 2.0 onto the chip surface. An antigen injection followed by a buffer-only injection was performed every 8 cycles to assess maximum binding to hEGFR-ECD-His protein and in-order to accurately determine the binning relationship.41
The data were analyzed using the Carterra Epitope analysis software for heatmap and competition network generation. Analyte binding signals were normalized to the antigen-only binding signal, such that the antigen-only signal average is equivalent to zero RU (response unit). A threshold window ranging from 0.2 RU to 0.25 RU above averaged signal obtained by antigen alone, was used to classify analytes into 3 categories: blockers (binding signal under the lower limit threshold), sandwiching (binding signal over the higher limit threshold) and ambiguous (binding signal between limit thresholds). Antibodies with low coupling to the chip, poor regeneration or with absence of self-blocking were excluded from the binning analysis. Like-behaved antibodies were automatically clustered to form a heatmap and competition plot where antibodies with identical blocking interactions within the sample set are clustered in a bin within the Carterra Epitope analysis software.42
Surface plasmon resonance (SPR) affinity measurements
A Carterra® LSATM instrument was used to measure binding kinetics of antibodies to hEGFR-ECD-His and domain-specific proteins. To measure binding kinetics and affinity, the mAb-coupled HC30M chip surface was exposed to injections of the proteins, with an association period of 5 minutes and dissociation period of 15 minutes. The tested concentrations of the full length hEGFR-ECD-His were 1000, 100, 33.3, 11.1, 3.70, 1.23, and 0.41 nM in HBS-EP+ containing 0.1 mg/mL BSA. Regeneration of the chip surface between the different concentrations was performed using 20 mM glycine, pH 2.0, for three 20 second cycles. Kinetic data was analyzed using Carterra KITTM software using a 1:1 Langmuir binding model. To determine binding to domain-specific proteins, the antibody-coupled HC30M chip surface was exposed to injections proteins, with an association period of 5 minutes and dissociation period of 15 minutes. The tested concentrations domain-specific proteins were 1000, 250, 111 and 62.5 nM in HBS-EP+ containing 0.1 mg/mL BSA. Regeneration of the chip surface between the different concentrations was performed using 20 mM glycine, pH 2.0, for three 20 second cycles. Kinetic data was analyzed using Carterra KITTM software using a 1:1 Langmuir binding model. Domain binding was simplified into a yes or no binding results for categorization only purpose.
Acknowledgments
We would like to acknowledge William Long from Chemical Computing Group for providing us with the SVL script to build our datasets. Also, we would like to thank William Smith at Eli Lilly and Company for his assistance in preparing the manuscript. We also thank Thomas Cujec, Julian Davies, and Elaine Conner and their group at Eli Lilly Biotechnology Center for generating data and experimental support. This work was funded by Eli Lilly and Company.
Author contributions
A.G. and Q.C. conceived the idea. Q.C. supervised the study. A.G. and M.J. developed the paratope prediction model including training and validation. A.G. analyzed the data and carried out the case studies. D.F. ran antibody competition assays and experiments, generated and analyzed the internal experimental data for EGFR antibodies. A.G. implemented the similarity search and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: January 25, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106036.
Supplemental information
Data and code availability
-
•
All original code and the paratope dataset generated from the PDB used to train and validate the paratope prediction model has been deposited at https://github.com/aghanbar-lilly/parasim and is publicly available.
-
•
EGFR antibody sequence data is confidential company data and therefore is not published.
-
•
Any additional inquiries regarding the data generated in this study should be directed to the lead contact.
References
- 1.Deng X., Ulrich S., Doranz B.J. Enhancing antibody patent protection using epitope mapping information. mAbs. 2018;10:204–209. doi: 10.1080/19420862.2017.1402998. Taylor & Francis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martin Closter Jespersen. Mahajan S., Peters B., Nielsen M., Marcatili P. Antibody specific b-cell epitope predictions: leveraging information from antibody-antigen protein complexes. Front. Immunol. 2019;10:298. doi: 10.3389/fimmu.2019.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hua C.K., Gacerez A.T., Sentman C.L., Ackerman M.E., Choi Y., Bailey-Kellogg C. Computationally-driven identification of antibody epitopes. Elife. 2017;6 doi: 10.7554/eLife.29023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sela-Culang I., Benhnia M.R.E.I., Matho M.H., Kaever T., Maybeno M., Schlossman A., Nimrod G., Li S., Xiang Y., Zajonc D., et al. Using a combined computational-experimental approach to predict antibody-specific b cell epitopes. Structure. 2014;22:646–657. doi: 10.1016/j.str.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 5.Pittala S., Bailey-Kellogg C. Mixture of experts for predicting antibody-antigen binding affinity from antigen sequence. bioRxiv. 2019 doi: 10.1101/511360. Preprint at. [DOI] [Google Scholar]
- 6.Di Rienzo L., Milanetti E., Lepore R., Olimpieri P.P., Tramontano A. Superposition-free comparison and clustering of antibody binding sites: implications for the prediction of the nature of their antigen. Sci. Rep. 2017;7:45053. doi: 10.1038/srep45053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martini S., Nielsen M., Peters B., Sette A. The immune epitope database and analysis resource program 2003–2018: reflections and outlook. Immunogenetics. 2020;72:57–76. doi: 10.1007/s00251-019-01137-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kovaltsuk A., Raybould M.I.J., Wong W.K., Marks C., Kelm S., Snowden J., Trück J., Deane C.M. Structural diversity of b-cell receptor repertoires along the b-cell differentiation axis in humans and mice. PLoS Comput. Biol. 2020;16 doi: 10.1371/journal.pcbi.1007636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gainza P., Sverrisson F., Monti F., Rodolà E., Boscaini D., Bronstein M.M., Correia B.E. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat. Methods. 2020;17:184–192. doi: 10.1038/s41592-019-0666-6. [DOI] [PubMed] [Google Scholar]
- 10.Mahajan S., Yan Z., Jespersen M.C., Jensen K.K., Marcatili P., Nielsen M., Sette A., Peters B. Benchmark datasets of immune receptor-epitope structural complexes. BMC Bioinf. 2019;20:490–497. doi: 10.1186/s12859-019-3109-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raybould M.I.J., Rees A.R., M Deane C. Current strategies for detecting functional convergence across b-cell receptor repertoires. mAbs. 2021;13 doi: 10.1080/19420862.2021.1996732. Taylor & Francis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahita J., Kim D.-G., Son S., Choi Y., Kim H.-S., Bailey-Kellogg C. Computational epitope binning reveals functional equivalence of sequence-divergent paratopes. Comput. Struct. Biotechnol. J. 2022;20:2169–2180. doi: 10.1016/j.csbj.2022.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sunny S., Pebbeti Bhanu Prakash, Gopakumar G., Jayaraj P.B. Deepbindppi: epitope-paratope prediction using attention based graph convolutional network. Research Square. 2022 doi: 10.21203/rs.3.rs-1975678/v1. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pittala S., Bailey-Kellogg C. Learning context-aware structural representations to predict antigen and antibody binding interfaces. Bioinformatics. 2020;36:3996–4003. doi: 10.1093/bioinformatics/btaa263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vecchio A.D., Deac A., Liò P., Veličković P. Neural message passing for joint paratope-epitope prediction. arXiv. 2021 doi: 10.48550/arXiv.2106.00757. Preprint at. [DOI] [Google Scholar]
- 16.Davila A., Xu Z., Li S., Rozewicki J., Wilamowski J., Kotelnikov S., Kozakov D., Teraguchi S., Standley D.M. Abadapt: an adaptive approach to predicting antibody–antigen complex structures from sequence. Bioinformatics Advances. 2022;2 doi: 10.1093/bioadv/vbac015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernández-Quintero M.L., Vangone A., Loeffler J.R., Seidler C.A., Georges G., Liedl K.R. Paratope states in solution improve structure prediction and docking. Structure. 2022;30:430–440.e3. doi: 10.1016/j.str.2021.11.001. [DOI] [PubMed] [Google Scholar]
- 18.Bagchi A., Haidar J.N., Eastman S.W., Vieth M., Topper M., Iacolina M.D., Walker J.M., Forest A., Shen Y., Novosiadly R.D., Ferguson K.M. Molecular basis for necitumumab inhibition of egfr variants associated with acquired cetuximab resistance. Mol. Cancer Therapeut. 2018;17:521–531. doi: 10.1158/1535-7163.MCT-17-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsiao Y.-C., Chen Y.-J.J., Goldstein L.D., Wu J. Zhonghua Lin, Kellen Schneider, Subhra Chaudhuri, Aju Antony, Kanika Bajaj Pahuja, Zora Modrusan, et al. Restricted epitope specificity determined by variable region germline segment pairing in rodent antibody repertoires. mAbs. 2020;12 doi: 10.1080/19420862.2020.1722541. Taylor & Francis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong W.K., Robinson S.A., Alexander B., Georges G., Lewis A.P., Shi J., James S., Taddese B., M Deane C. Ab-ligity: identifying sequence-dissimilar antibodies that bind to the same epitope. mAbs. 2021;13 doi: 10.1080/19420862.2021.1873478. Taylor & Francis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liberis E., Veličković P., Sormanni P., Vendruscolo M., Liò P. Parapred: antibody paratope prediction using convolutional and recurrent neural networks. Bioinformatics. 2018;34:2944–2950. doi: 10.1093/bioinformatics/bty305. [DOI] [PubMed] [Google Scholar]
- 22.Olimpieri P.P., Chailyan A., Tramontano A., Marcatili P. Prediction of site-specific interactions in antibody-antigen complexes: the proabc method and server. Bioinformatics. 2013;29:2285–2291. doi: 10.1093/bioinformatics/btt369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ambrosetti F., Olsen T.H., Olimpieri P.P., Jiménez-García B., Milanetti E., Marcatilli P., Bonvin A.M.J.J. proabc-2: prediction of antibody contacts v2 and its application to information-driven docking. Bioinformatics. 2020;36:5107–5108. doi: 10.1093/bioinformatics/btaa644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rives A., Meier J., Sercu T., Goyal S., Lin Z., Liu J., Guo D., Ott M., Zitnick C.L., Ma J., Fergus R. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2016239118. e2016239118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barnes C.O., Jette C.A., Abernathy M.E., Dam K.M.A., Esswein S.R., Gristick H.B., Malyutin A.G., Sharaf N.G., Huey-Tubman K.E., Lee Y.E., et al. Sars-cov-2 neutralizing antibody structures inform therapeutic strategies. Nature. 2020;588:682–687. doi: 10.1038/s41586-020-2852-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gowthaman R., Guest J.D., Yin R., Adolf-Bryfogle J., Schief W.R., Pierce B.G. Cov3d: a database of high resolution coronavirus protein structures. Nucleic Acids Res. 2021;49:D282–D287. doi: 10.1093/nar/gkaa731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan T.J.C., Yuan M., Kuzelka K., Padron G.C., Beal J.R., Chen X., Wang Y., Rivera-Cardona J., Zhu X., Stadtmueller B.M., et al. Sequence signatures of two public antibody clonotypes that bind sars-cov-2 receptor binding domain. Nat. Commun. 2021;12:3815. doi: 10.1038/s41467-021-24123-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sela-Culang I., Kunik V., Ofran Y. The structural basis of antibody-antigen recognition. Front. Immunol. 2013;4:302. doi: 10.3389/fimmu.2013.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adam P., Gross S., Massa F., Adam L., James B., Gregory Chanan, Killeen T., Lin Z., Gimelshein N., Antiga L., et al. Pytorch: an imperative style, high-performance deep learning library. Adv. Neural Inf. Process. Syst. 2019;32 [Google Scholar]
- 30.Wolf T., Debut L., Sanh V., Chaumond J., Clement D., Anthony M., Cistac P., Tim Rault, Louf R., Morgan F., et al. Huggingface’s transformers: state-of-the-art natural language processing. arXiv. 2019 doi: 10.48550/arXiv.1910.03771. Preprint at. [DOI] [Google Scholar]
- 31.Ahmed E., Heinzinger M., Dallago C., Rihawi G., Wang Y., Jones L., Gibbs T., Feher T., Angerer C., Martin S., et al. Prottrans: towards cracking the language of life’s code through self-supervised deep learning and high performance computing. arXiv. 2020 doi: 10.48550/arXiv.2007.06225. Preprint at. [DOI] [Google Scholar]
- 32.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Bertrand T., Grisel O., Blondel M., Peter P., Weiss R., Vincent D., et al. Scikit-learn: machine learning in python. J. Mach. Learn. Res. 2011;12:2825–2830. [Google Scholar]
- 33.Hunter J.D. Matplotlib: a 2d graphics environment. Comput. Sci. Eng. 2007;9:90–95. [Google Scholar]
- 34.The Pandas Development Team. Pandas-Dev/pandas: Pandas, February 2020.
- 35.Virtanen P., Gommers R., Oliphant T.E., Haberland M., Reddy T., Cournapeau D., Burovski E., Peterson P., Weckesser W., Bright J., et al. Scipy 1.0: fundamental algorithms for scientific computing in python. Nat. Methods. 2020;17:261–272. doi: 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Molecular operating environment (moe), 2020. 09 chemical computing group ulc, 1010 sherbooke st. west, suite 910, montreal, qc, canada, h3a, 2r7, 2022. https://www.chemcomp.com/Research-Citing_MOE.htm.
- 37.Li W., Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 38.Al-Lazikani B., Lesk A.M., Chothia C. Standard conformations for the canonical structures of immunoglobulins. J. Mol. Biol. 1997;273:927–948. doi: 10.1006/jmbi.1997.1354. [DOI] [PubMed] [Google Scholar]
- 39.antibody_contacts.svl, Scientific Vector Language (Svl) Source Code provided by Chemical Computing Group Ulc, 1010 Sherbooke St. West, suite 910, montreal, qc, canada, h3a 2r7, 2022. https://www.chemcomp.com/Research-Citing_MOE.htm.
- 40.Ahmed E., Heinzinger M., Dallago C., Rehawi G., Wang Y., Jones L., Gibbs T., Feher T., Angerer C., Martin S., et al. Prottrans: towards cracking the language of life’s code through self-supervised deep learning and high performance computing. bioRxiv. 2020 doi: 10.48550/arXiv.2007.06225. Preprint at. [DOI] [Google Scholar]
- 41.Abdiche Y.N., Miles A., Eckman J., Foletti D., Van Blarcom T.J., Yeung Y.A., Pons J., Rajpal A. High-throughput epitope binning assays on label-free array-based biosensors can yield exquisite epitope discrimination that facilitates the selection of monoclonal antibodies with functional activity. PLoS One. 2014;9 doi: 10.1371/journal.pone.0092451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brooks B.D., Closmore A., Yang J., Holland M., Cairns T., Cohen G.H., Bailey-Kellogg C. Characterizing epitope binding regions of entire antibody panels by combining experimental and computational analysis of antibody: antigen binding competition. Molecules. 2020;25:3659. doi: 10.3390/molecules25163659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All original code and the paratope dataset generated from the PDB used to train and validate the paratope prediction model has been deposited at https://github.com/aghanbar-lilly/parasim and is publicly available.
-
•
EGFR antibody sequence data is confidential company data and therefore is not published.
-
•
Any additional inquiries regarding the data generated in this study should be directed to the lead contact.