

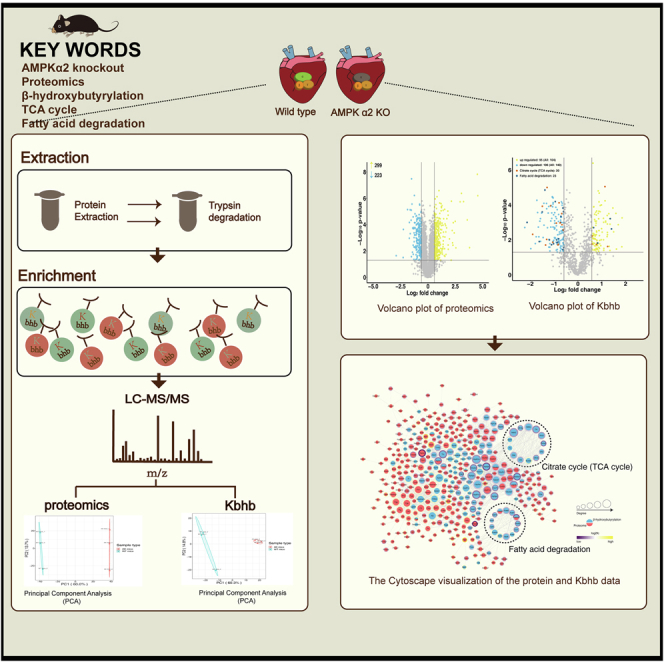
Abstract
AMP-activated protein kinase alpha 2 (AMPKα2) regulates energy metabolism, protein synthesis, and glucolipid metabolism myocardial cells. Ketone bodies produced by fatty acid β-oxidation, especially β-hydroxybutyrate, are fatty energy–supplying substances for the heart, brain, and other organs during fasting and long-term exercise. They also regulate metabolic signaling for multiple cellular functions. Lysine β-hydroxybutyrylation (Kbhb) is a β-hydroxybutyrate–mediated protein posttranslational modification. Histone Kbhb has been identified in yeast, mouse, and human cells. However, whether AMPK regulates protein Kbhb is yet unclear. Hence, the present study explored the changes in proteomics and Kbhb modification omics in the hearts of AMPKα2 knockout mice using a comprehensive quantitative proteomic analysis. Based on mass spectrometry (LC-MS/MS) analysis, the number of 1181 Kbhb modified sites in 455 proteins were quantified between AMPKα2 knockout mice and wildtype mice; 244 Kbhb sites in 142 proteins decreased or increased after AMPKα2 knockout (fold change >1.5 or <1/1.5, p < 0.05). The regulation of Kbhb sites in 26 key enzymes of fatty acid degradation and tricarboxylic acid cycle was noted in AMPKα2 knockout mouse cardiomyocytes. These findings, for the first time, identified proteomic features and Kbhb modification of cardiomyocytes after AMPKα2 knockout, suggesting that AMPKα2 regulates energy metabolism by modifying protein Kbhb.
Keywords: β-hydroxybutyrylation, proteomics, AMPKα2, heart, TCA cycle, fatty acid degradation
Abbreviations: β-OHB, β-hydroxybutyrate; AK mice, AMPKα2 knockout mice; AMPK, AMP-activated protein kinase; AMPKα2, AMP-activated protein kinase alpha 2; BP, biological process; BW, Body weight; CC, cellular component; CPT-1, carnitine O-palmitoyltransferase 1; E/A, the ratio of the early to late diastolic mitral inflow velocities; E'/A', the ratio of the early to late diastolic mitral annular velocities; FFA, free fatty acid; FS, fractional shortening; GO, Gene Ontology; HW, heart weight; KB, ketone body; Kbhb, Lysine β-hydroxybutyrylation; KEGG, Kyoto Encyclopedia of Genes and Genomes; KOG/COG, Clusters of Orthologous Groups of proteins; LFQ, Label-free quantification; LVEDD, Left ventricular end-diastolic diameter; LVEF, left ventricular ejection fraction; MF, molecular function; PCR, Polymerase Chain Reaction; PDH, pyruvate dehydrogenase; PTMs, posttranslational modifications; TCA cycle, tricarboxylic acid cycle; TG, triglyceride; TL, tibia length; WT mice, wildtype mice
Graphical Abstract
Highlights
-
•
Cardiac diastolic dysfunction of AMP-activated protein kinase alpha 2 knockout mice.
-
•
Proteomic and β-hydroxybutyrylation modification omics analysis of mice cardiac tissues.
-
•
Insights from β-hydroxybutyrylation modification omics on AMPK regulation.
In Brief
Quantitative β-hydroxybutyrylation modification omics was used to detect AMP-activated protein kinase alpha 2 (AMPKα2) knockout mice cardiac tissue. The β-hydroxybutyrylation sites of myocardial fatty acid degradation and TCA cycle–associated enzymes were dramatically downregulated after AMPKα2 knockout. Protein β-hydroxybutyrylation may have a role in AMPK-mediated energy metabolism regulation, providing a clue for future drug exploits for AMPK-related therapy.
The key enzyme in energy regulation, AMP-activated protein kinase (AMPK), plays a vital role in several metabolic processes (1, 2), such as energy production and utilization (3, 4, 5). AMPK consists of three subunits in various isoforms: the catalytic α (α1 and α2) subunits and the regulatory β (β1 and β2) and γ (γ1, γ2, and γ3) subunit. In mouse hearts, α subunit 2 (AMPKα2) is the dominant catalytic subtype that accounts for 70 to 80% of AMPK activity (6), regulating energy metabolism, cell growth, oxidative stress, cell growth, glucose, and fatty acid oxidation. AMPK is the main signaling molecule regulating myocardial energy. It acts on many metabolism-related transporters and enzymes; for example, hydroxymethylglutaryl-CoA reductase, glucose transporter 1 and GLUT4, glycogen synthase, and acetyl-CoA carboxylase (7, 8, 9). Previous studies have shown that through phosphorylation of key substrates, AMPK can promote the synthesis of acetyl-CoA and ketone bodies by suppressing the expression of carnitine palmitoyltransferase 1, which limits the rate-limiting enzymes in mitochondrial fatty acid β-oxidation (10, 11, 12).
Intermediates were formed from fatty acid β-oxidation: ketone bodies consisting of β-hydroxybutyrate (β-OHB), acetoacetate, and acetone. β-OHB and others were produced in the mitochondria of hepatocytes that are subsequently secreted into the blood and utilized by organs with ketolytic mechanisms, such as the heart (13). The classical role of β-OHB is an energy source; however, recent studies have shown that β-OHB induces lysine β-hydroxybutyrylation (Kbhb) in cells, and this modification may contribute to tumor progression and energy regulation (14, 15, 16). In 2021, Koronowski et al. (15) found that ketogenic diet leads to fatty acid oxidation to derive acetyl-CoA, produces excessive β-OHB, and increases S-adenosyl-L-homocysteine hydrolase Kbhb, which in turn decreases S-adenosyl-L-homocysteine hydrolase activity and alters the methionine cycle. To date, Kbhb is mostly described in histone proteins and barely described in nonhistone proteins, especially metabolism proteins. Thus, we conducted proteomic and posttranslational modification omics studies in AMPKα2 knockout (AK) mouse hearts to comprehensively demonstrate the regulatory mechanism of AMPKα2 in fatty acid oxidation, tricarboxylic acid cycle (TCA cycle), and other processes at the protein level. The current findings showed that AMPKα2-specific knockout did not alter weight, grip, heart weight (HW), and tibia length (TL) in mice but altered the myocardium mitochondrial ultrastructure and cardiac diastolic function. Finally, we identified 1582 Kbhb-modified sites in 585 proteins, and 244 Kbhb-modified sites in 142 proteins were significantly altered (fold change >1.5 or <1/1.5, p < 0.05) after AMPKα2 knockout. Functional enrichment analysis showed that dramatically downregulated Kbhb sites were related to fatty acid metabolism, amino acid metabolism, TCA cycle, cGMP-PKG signaling pathway, and congenital heart disease pathway. Upregulated Kbhb sites were involved in arginine and proline metabolism, antigen processing and presentation, and dilated cardiomyopathy.
Furthermore, we observed that a subset of differential Kbhb sites were located on key proteins for fatty acid degradation and TCA cycle, suggesting that AMPKα2 probably affects the functions of the crucial proteins through Kbhb at these sites.
Experimental Procedures
Experimental Design and Statistical Rationale
Our study was based on AK mice and wildtype (WT) mice. Heart samples were analyzed from three mice per genotype (biological replicates). Based on three-dimensional assessments of reproducibility (Pearson's correlation coefficient, principal component analysis, relative standard deviation), statistical power was deemed to be sufficient (supplemental Fig. S1). Student’s t tests were used to calculate the p value, and p value < 0.05 was considered as the significance index. For physiological data, independent Student’s t test or one-way analysis of variance were used to assess differences between WT mice and AK mice.
Generation of AMPKα2 Knockout Mice
CRISPR/Cas9 gene-editing technology was used to cut the protein-coding region of the C57BL/6J mice target gene AMPKα2, and the mouse fertilized egg cells were repaired by nonhomologous end-joining, resulting in fragment deletion in the protein coding region, making the AMPKα2 protein ineffective, thereby achieving the gene knockout. The genotype of the AK was verified by PCR testing. Briefly stated, mouse tail tissue was used to extract DNA. Using the primers listed below: AMPKα2 wild type (Fig. 1A, a) (forward: 5′-TGACATCCTGTGGTGCTGAA-3′, reverse: 5′-CTGCCTAGTGCTGACTCTGA-3′) and AMPK α2 knockout (Fig. 1A, b) (forward: 5′-GCAGAGGCAGGCGAATTTC-3′, reverse:5′-GATTGTTCACTGGCTAATCTTAAGC-3′). For electrophoresis, the DNA products were placed on a 2% agarose gel, and pictures were acquired. Only DNA band near 582 bp indicates knockout homozygote, and both DNA band around 468 bp and 582 bp denotes heterozygote. WT genotype is indicated by both the 1500-bp and 468-bp DNA bands obtained using PCR (Fig. 1A).
Fig. 1.

Phenotype of AK and WT mice.A, genotyping PCR (a) AMPKα2 wildtype band. (b) AMPKα2 knockout band. Just one DNA band near 582 bp denotes knockout homozygote. B, Western blot was used to ascertain the expression of proteins such AMPKα2 and AMPKα1 in the heart. C, mice appearance and cardiac structure did not alter significantly after AMPKα2 knockout. D, histogram indicated that weight, grip, heart weight (HW), Tibia length (TL), HW/BW, HW/TL were not significant in WT mice and AK mice. (n = 6). AK, AMPKα2 knockout; AMPKα2, AMP-activated protein kinase alpha 2; BW, body weight.
Animals
Experimental mouse relative programs were approved to the Laboratory Animal Ethical and Welfare Committee of Shandong University Cheeloo College of Medicine (Approval No. 20157). WT C57BL/6J mice (WT mice) were purchased from Jinan Tengli Trade Co Ltd, and AK mice (C57BL/6J) were generated and identified by Beijing Viewsolid Biotechnology Co Ltd. A total of ten WT mice and ten AK mice were raised in the specific pathogen-free animal chamber of Shandong University Laboratory Animal Center. At the age of 3 months, the body weight (BW), grip, and echocardiographic of the animals were measured, and the changes in the activity, reaction, and fur were recorded. The room temperature was maintained at 20 to 25 °C, the humidity was 70%, and light was regulated (12 h of light/12 h of dark). The mice were allowed free access to the feed and potable water.
Grip Strength
A grip strength meter (Jiangsu Science Biological Technology Co Ltd) was used to measure the front paws grip strength of WT and AK mice. To test the mice's front paw grip, we took hold of the mouse at the base of the tail and lowered it vertically toward the bar. The mouse's tail pulled it slightly backward as its two forelimbs (paws) grabbed the bar. The peak force in newton (N) was recorded at the time the mouse released its paws from the bar in each measurement. With 30 s of rest in between each trial, mice received five trials. A final grip per mouse was determined by averaging the top three out of five trials.
Echocardiographic Imaging
After 3 months, 2% isoflurane anesthesia was used to perform transthoracic echocardiography on a heated platform utilizing the Vevo2100 imaging equipment (VisualSonics). M-mode echocardiography was used to evaluate the left ventricular ejection fraction, left ventricular end-diastolic diameter, and fractional shortening in the parasternal long-axis view. Pulsed Doppler was used to measure the early (E) and late (A) diastolic mitral flow velocities in the apical four-chamber view, and the ratio of E/A was calculated. The ratio of E'/A' was calculated by measuring the early (E') and late (A') diastolic mitral annulus velocities using tissue Doppler imaging in the apical four-chamber view.
Tissue Preparation
Mice were weighed and anesthetized by intraperitoneal injection with 10% chloral hydrate. After weighing the heart, a part of the myocardial tissue was stored at −80 °C, while the remaining was used for preparing light and electron microscopy sections.
Blood Parameter Determinations
Blood was drawn for testing. Serum concentrations of triglycerides and total cholesterol were determined using an automatized chemistry analyzer (Rayto). Free fatty acids (FFAs) test kit was used to measure the fatty acids in the serum of mice (Nanjing Jiancheng Bioengineering Institute). Serum β-OHB was assessed with Beta-Hydroxybutyric acid ELISA kits (USCN Life Science). All data were expressed as mean ± SD. p-value < 0.05 was considered statistically significant.
Evaluation of Myocardial Morphology
The mice cardiac tissue was fixed in 4% formaldehyde solution (3–5 days), dehydrated in ethanol (Kelong Chemical Reagent Factory), paraffin embedded, and paraffin sectionalized (LEICA, RM2235). Sections were cut into 4 μm thick and stained with hematoxylin-eosin (hematoxylin, Sigma, 041M0014V; eosin, Maikun Chemical Co Ltd, 20120831). Digital microscope (×400) (OLYMPUS, DX45) was used to capture images of the heart slice.
Electron Microscopy
Mice cardiac tissue was sliced into 1-mm3 organization blocks on ice and instantly immersed in 3% glutaraldehyde in cacodylate buffer at 4 °C for 2 h before postfixation in 1% osmium-tetroxide phosphate buffer about 2 h. Subsequently, the sections were embedded in epoxide resin after dehydration in a graded ethanol series with acetone. Using a Marada-G2 type CCD photo ultrastructure of pictures, the semithin slices were cut into ultrathin sections (about 70 nm). Finally, the sections were examined under a JEM-1200EX (JEOL, Japan) transmission electron microscope after dying with uranyl acetate and lead citrate.
Western Blot
WT and AK mice heart lysates were separated on sodium dodecyl-polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred to nitrocellulose filter membrane (0.22 μm). Then, we blocked with 5% nonfat milk in TBST. The membrane was incubated with the primary antibody at 4 °C overnight and then with the secondary antibody at room temperature for 1 h. Chemiluminescence was used to detect proteins (apparatus: Chemiluminescent Imaging System). The following antibodies were used: AMPK alpha 1 polyclonal antibody (Proteintech), AMPK Alpha 2 Polyclonal antibody (Proteintech); anti-Kbhb antibody (PTMBioLab), anti-succinylated lysine (PTM Biolab), anti-malonylated lysine (PTM Biolab), anti-crotonylated lysine (PTM Biolab), anti-lactylated lysine (PTM Biolab), mouse IgG (H + L) secondary antibody (Pierce), rabbit IgG (H + L) secondary antibody (Pierce).
Protein Extraction and Trypsin Degradation
Briefly, AK and WT mice heart samples were ground into powder, followed by the addition of lysis buffer (8 M urea, 1% protease inhibitors, 3 μM Trichostatin A, and 50 mM nicotinamide) and centrifugation at 12,000g for 10 min at 4 °C. The supernatant was then gathered.
To analyze the proteomics of the heart tissue, the protein solution was alkylated with 11 mM iodoacetamide at room temperature in the dark after being reduced with 5 mM dithiothreitol at 56 °C. The protein sample was then diluted by adding 100 mM TEAB to urea concentration less than 2 M. Finally, trypsin was added for the first digestion (1:50 trypsin-to-protein mass ratio, overnight) and for a further digestion (1:100 trypsin-to-protein mass ratio, 4 h). The C18 SPE column desalted the peptides in the end. The peptides were separated using a reverse phase HPLC column (Agilent 300Extend C18, 5 m particle size, 4.6 mm id, 250 mm length). LC/MS analysis was performed after vacuum freeze-drying. For Kbhb, the supernatants of AK and WT mouse cardiac protein samples were mixed with 20% TCA (m/v) (Sigma-Aldrich) to deposit proteins and washed with precooled acetone. Subsequently, the protein pellets were resuspended in 200 mM triethylammonium bicarbonate buffer (Sigma-Aldrich), dispersed by sonication, and subjected to trypsinization. The samples were reduced with 5 mM dithiothreitol (Sigma-Aldrich) and alkylated with iodoacetamide (Sigma-Aldrich). Finally, the peptides were desalted by Strata X SPE column.
Kbhb-Modified Enrichment
To enrich Kbhb peptides, mouse cardiac samples were mixed with NETN buffer (100 mM NaCl, 1 mM EDTA, 50 mM Tris-HCl, 0.5% NP-40, pH 8.0), which was subsequently incubated with antibody beads (PTM Bio, Catalog # PTM1204) at 4 °C overnight. Then, the mixture was rinsed twice with H2O (ThermoFisher Scientific) and four times with NETN buffer solution. The attached peptides were washed out of the antibody beads with 0.1% trifluoroacetic acid (Sigma-Aldrich). Then, the eluted portions were pooled and vacuum dried. The cardiac samples were desalted for LC-MS/MS analysis using C18 ZipTips (Millipore), according to the manufacturer’s instructions.
LC-MS/MS Analysis
The tryptic peptides were added in solvent A (0.1% formic acid, 2% acetonitrile/in water) and injected on a home-made reverse-phase analytical column with integrated spray tip (100 μm i.d. × 25 cm) packed with 1.9 μm/120 Å ReproSil-PurC18 resins (Dr Maisch GmbH). The peptides were evaluated using a capillary source using Bruker Daltonics timsTOF Pro (Bruker Daltonics) mass spectrometry. The TOF detectors were used to examine the precursors and fragments, with a scan range of 100 to 1700 m/z on the MS/MS. In PASEF mode, the timsTOF Pro was employed (parallel accumulation serial fragmentation). Following the selection of precursors with charge states 0 to 5 for fragmentation, ten PASEF-MS/MS images were collected per cycle.
The final data of MS/MS were analyzed the MaxQuant search engine (v.1.6.15.0). Tandem mass spectra were retrieved against the Mus musculus SwissProt database (updated on 21/07/21, 17,089 entries) concatenated with the reverse decoy database. The cleavage enzyme, trypsin/P, was selected and allowed up to four missed cleavages (proteomics allowed up two missed cleavages). In the initial and primary searches, the mass tolerance for precursor ions was set to 20 ppm, while the fragment ion mass tolerance was set to 20 ppm. Carbamidomethyl on Cys was specified as fixed modification, but oxidation of Met, Kbhb of Lys, and acetylation of the protein’s N-terminal were variable (acetylation of the protein’s N-terminal and oxidation of Met were variable in proteomics). False discovery rate <1%.
Data Analysis
Proteins/peptides in the potential contaminant database and reverse decoy database were excluded. For the proteomic analysis, label-free quantification (LFQ) of MaxQuant was used to determine intensities and normalize protein quantities. LFQ intensity values were transformed to the relative quantitative value after centralization (the LFQ intensity of each sample divided by the mean of all samples). Proteins that were detected in at least two unique peptide samples were included for further analysis. For Kbhb proteomics, the cutoff of site with localization probability estimated by MaxQuant was required to be 0.75 or higher. Intensity values were transformed to the relative quantitative value after centralization (each sample's intensity of modified peptides divided by the average of all samples). The relative quantitative value of the modified peptide is typically divided by the relative quantitative value of the corresponding protein to remove the influence from protein expression of modifications. Proteins/sites with a p value < 0.05 were deemed significant using the Student t test. The fold change (mean values of AK mice/WT mice) >1.5 or 1/1.5, respectively, was used to define upregulated and downregulated proteins/sites.
Bioinformatics Methods
Gene Ontology (GO) annotated proteome from UniProt-GOA database (http://www.ebi.ac.uk/GOA/). Then, the differential proteins and differential Kbhb proteins were classified according to the three categories of biological process (BP), cellular component (CC), and molecular function (MF) using GO annotation. Kyoto Encyclopedia of Genes and Genomes (KEGG) connects known information on molecular interaction networks. In this study, we analyzed the metabolic pathways of differential proteins and differential Kbhb proteins to elucidate the effect of AK in mice heart based on KEGG enrichment. Differential Kbhb proteins and differential proteins were searched against the STRING database (v. 11.0) for protein–protein interactions between AK mice and WT mice. Followed by visualization using Cytoscape software (v. 3.7.2). In the outer circle, the ratio value following log2 treatment was displayed using the Omics Visualizer 1.3.0 plug-in. The color is more yellow the higher the value and more purple the lower the value. The protein's location with the biggest difference served as the ratio value for the modified group (ranked by the absolute value of ratio after log2 treatment). Confidence score ≥0.7.
Statistical Analysis
Heart samples were analyzed from three mice per genotype, and reproducibility was assessed using relative standard deviation, principal component analysis, and Pearson's Correlation Coefficient (supplemental Fig. S1). t test was used to analyze the differences in BW, grip, HW, TL, HW/BW and HW/TL between AK and WT mice, and graphs were drawn in GraphPad Prism 8 software. Quantitative values are presented as mean ± standard error of mean. The statistical significance was indicated by p < 0.05.
Results
General Condition of AK and WT Mice
Both WT and AK mice were sacrificed at 3 months of age. The genotype and the phenotype of the AK mice was verified by PCR testing and Western blot (WB) (Fig. 1, A and B). The appearance of AK mice was indistinguishable from that of the WT mice (Fig. 1C). Compared to WT mice, no conspicuous difference was detected in the BW, grip, HW, TL, HW/BW, and HW/TL after AMPKa2 knockout (Fig. 1D). AK mice were characterized by higher plasma FFA concentrations than were found in WT mice, while triglyceride, FFA, and β-OHB levels were no difference (Table 1)
Table 1.
Blood parameters of WT mice and AK mice
| Blood parameter | WT mice | AK mice |
|---|---|---|
| β- hydroxybutyrate(μg/ml) | 0.03 ± 0.01 | 0.04 ± 0.02 |
| FFA (mM) | 2.15 ± 0.29 | 2.72 ± 0.17a |
| TG (mM) | 0.65 ± 0.13 | 0.70 ± 0.12 |
| Total cholesterol (mM) | 2.01 ± 0.07 | 1.95 ± 0.04 |
All data are expressed as mean ± SEM (n = 6 per group).
Abbreviations: AK, AMPKα2 knockout; FFA, free fatty acids; TG, triglyceride.
p < 0.05.
Histological Examination
The histological myocardial alterations in WT mice and AK mice under a light microscope are depicted in Figure 2A, a and b. In the WT group, the color of myocardium was uniform, the morphology was consistent, and the boundaries were clear. A few cardiac cell nucleus in the AK mice had swollen, along with the loose cytoplasm. The ultrastructure of myocardial cells was displayed in Figure 2A, c and d. In the WT group, the myocardium mitochondria, round or oval shape, were arranged orderly, and the cristae in the whole mitochondrial cavity were dense and regularly distributed (Fig. 2A, c). However, compared to the WT group, the mitochondria of AK mice were distributed randomly, and the mitochondrial crest flattened (Fig. 2A, d).
Fig. 2.

Cardiac characteristics of AK mice and WT mice.A, AMPKα2 knockout destroyed the myocardial structure in mice. Representative images of HE staining of myocardium from WTY (a) and AKY (b) at ×400 magnification. Black scale bar represents 50 mm. Electron microscopic images of myocardium from WT mice (c) and AK mice (d). Black scale bar represents 1 mm. B, b1, left ventricular long-axis views on a two-dimensional echocardiography, with scale bar in millimeters. b2, M-mode echocardiograms displaying the size of the left ventricle, a scale bar in millimeters, and a time stamp in seconds on the right and bottom. b3, pulse-wave Doppler echocardiograms depicting mitral inflow velocities, time stamp in seconds is at the bottom, with the scale bar in mm/s on the right. b4, mitral annular velocities are shown on tissue Doppler echocardiograms with time stamp in seconds on the bottoms and a scale bar in mm/s on the right. AK, AMPKα2 knockout; AMPKα2, AMP-activated protein kinase alpha 2.
Echocardiography
To reveal functional changes induced by AK, we performed echocardiography in AK mice and WT mice (Fig. 2B and Table 2). To determine the left ventricular systolic function, fractional shortening and left ventricular ejection fraction were measured by M-mode measurements. All these parameters were not significantly decreased after AK. Worthwhile, after AK, an apparent decrease in the LV diastolic functional parameter, such as E/A was observed. Left ventricular end-diastolic diameter and E'/A' exhibited no differences after AK. Taken together, these results indicate that AK slightly impairs ventricular diastolic function.
Table 2.
Effects of AMPKα2 knockout on mouse left ventricular function and dimension in mice
| Parameter | WT mice | AK mice |
|---|---|---|
| LVEF (%) | 81.06 ± 6.99 | 69.93 ± 17.45 |
| FS (%) | 48.85 ± 8.26 | 40.08 ± 13.92 |
| LVEDD (mm) | 2.90 ± 0.09 | 3.18 ± 0.07 |
| E/A | 1.79 ± 0.25 | 0.68 ± 0.15a |
| E'/A' | 1.55 ± 0.84 | 1.24 ± 0.07 |
Abbreviations: AK, AMPKα2 knockout; E/A, the ratio of the early to late diastolic mitral inflow velocities; E'/A', the ratio of the early to late diastolic mitral annular velocities; FS, fractional shortening; LVEDD, Left ventricular end-diastolic diameter; LVEF, left ventricular ejection fraction.
p < 0.05, n = 5 per group.
Quantification and Identification of the Myocardial Proteome
A total of 3659 proteins were quantified in AK and WT mice. Finally, 522 differential proteins were obtained by defining the truncation value as 1.5-fold. Among, these, 57% (299/522) and 43% (223/522) were upregulated and downregulated differential proteins after AK (Fig. 3B). Supplemental data summarize the relevant information (name, molecular weight, subcellular localization, and main functions) of the proteins.
Fig. 3.

Whole proteome analysis of AK mice and WT mice.A, flowchart for the identification of omics in myocardial samples from WT and AK mice. B, volcano plot of differential protein quantify for proteomics. Yellow represents upregulated proteins and blue represents downregulated proteins. C, subcellular structure diagram of differential proteins. D, GO analysis results of differential proteins (yellow: biological process; green: cellular component; purple: molecular function). E, COG/KOG pathway of differential proteins (purple: cellular processes and signaling, green: information storage and processing, yellow: metabolism, blue: poorly characterized). F, top enriched items for KEGG pathway analysis in differential proteins. The vertical axis is the functional category or pathway, and the horizontal axis is the proportion of differentially expressed modified proteins in this functional type compared to the proportion of identified proteins. Gradation from yellow to purple indicates a decreasing p value. AK, AMPKα2 knockout; AMPKα2, AMP-activated protein kinase alpha 2; GO: Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; KOG/COG: clusters of orthologous groups of proteins.
In order to understand the effect of AMPKα2 on the mouse heart, differential proteins were divided into various subcellular locations, BPs, and MFs based on GO analysis, clusters of orthologous groups (COG/KOG) functional classification, and KEGG pathway analysis. The subcellular location of all the differentially expressed proteins is annotated in Figure 3C. The largest proportion of differential proteins was localized in the cytoplasm. Most of the differential proteins were in the cytoplasm (159, 30.46%), followed by nucleus (102, 19.54%) and extracellular differential proteins (99, 18.97%). In addition, only 14.56% of the differential proteins were distributed in mitochondria (Fig. 3C). GO analysis revealed that differential proteins were aggregated into multiple BPs, related to BPs (56%), CCs (26%), and MFs (18%) (Fig. 3D).
All differential proteins in the WT and AK mice were categorized into several cellular functions, associated with energy production and conversion, glycolipid metabolism, genetic material processing and modification, and regulation of cell cycle (Fig. 3E). Notably, after AMPK knockout, about 49% of the differential proteins were related to cellular processes and signal transduction (Fig. 3E). About 38% of proteins are involved in metabolism and information storage processing. In addition to classifying the functional sorts using GO analysis, proteins with prominent differential enrichment were noted based on KEGG analysis. The complement and coagulation cascades and Staphylococcus aureus infection were significant in the KEGG pathway analysis for differential proteins. Circadian rhythm and systemic lupus erythematosus were also related to the differential proteins (Fig. 3F).
Kbhb Modification Omics of WT and AK
Posttranslational modifications (PTMs) are crucial for protein function (activity and stability) and are closely related to various cardiovascular diseases (17, 18). Based on WB analysis, we showed five PTMs, including lysine succinylation, crotonylation, malonylation, β-hydroxybutyrylation, and lactylation (Fig. 4). Following AK, the levels of succinylation, crotonylation, malonylation, and β-hydroxybutyrylation were all noticeably reduced. Β-OHB-mediated Kbhb has attracted our attention. Ketone body (KB), considered efficient fuel, is meaningful in heart’s energy supply (19, 20). As an energy metabolism center, AMPK is also closely related to the metabolism of KB (21). At present, β-OHB–mediated Kbhb has not been reported in the heart. To further explore the effect of AMPK on the Kbhb of cardiac tissue, we conducted a proteomic analysis of Kbhb.
Fig. 4.

The level oflysine succinylation (K-succ), crotonylation (K-Cr), malonylation (K-Mal), and β-hydroxybutyrylation (K-Bhb) were all noticeably reduced after AK. AK, AMPKα2 knockout; AMPKα2, AMP-activated protein kinase alpha 2.
A total of 1582 Kbhb sites were identified from 585 proteins. Among these, 313 (53.5%) had only one Kbhb site, and 272 (46.5%) had >2 Kbhb sites (Fig. 5A), while, some proteins had >10 Kbhb sites and were heavily modified (Fig. 5B). Furthermore, Kbhb substantially modified some proteins. Titin (Ttn) protein is a key component of vertebrate rhabdoid muscle assembly and function, consisting of 180 Kbhb sites. Myosin 6 (Myh 6), involved in muscle contraction, contains 48 Kbhb sites, while isocitrate dehydrogenase consisting of 16 Kbhb site is involved in the TCA cycle and acts in the intermediary metabolism and energy production (Fig. 5B). Interestingly, several Kbhb sites in these proteins might have an influence on their activity and functions.
Fig. 5.

Quantitative Kbhb-modified proteomics in AK and WT mice.A, distribution of protein Kbhb sites. 313 (53.5%) had only one Kbhb site. B, statistical map of proteins with >10 Kbhb sites. C, volcano plot of Kbhb-modified differential sites. D, subcellular distribution of Kbhb protein. E, functional enrichment of Kbhb-modified proteins by the Gene Ontology database (yellow: biological process; green: cellular component; purple: molecular function). F, COG/KOG functional classification chart of differential Kbhb proteins. G, top enriched items for KEGG pathway analysis. (Orange: up KEGG pathway, purple: down KEGG pathway). AK, AMPKα2 knockout; AMPKα2, AMP-activated protein kinase alpha 2; Kbhb, Lysine β-hydroxybutyrylation; KEGG, Kyoto Encyclopedia of Genes and Genomes; KOG/COG, Clusters of Orthologous Groups of proteins.
WB analysis confirmed that AK significantly reduces the overall Kbhb level in the heart. A 1.5-fold change resulted in 244 differential Kbhb sites of 142 proteins. Subsequently, 140 differential Kbhb sites of 84 proteins were downregulated, while 104 differential Kbhb sites of 70 proteins were upregulated after AK (Fig. 5C). The subcellular localization of all differentially Kbhb proteins is annotated in Figure 5D. Most of the differential Kbhb proteins were located in the mitochondria (50/35.46%) and cytoplasm (49/34.75%), suggesting that Kbhb mainly acts through the mitochondria (Fig. 5D).
The differential Kbhb-modified proteins were sorted according to BPs, CCs, and MFs by GO annotation (Fig. 5E). The top two BPs were cellular process (134, 20%) and metabolic processes (107, 16%). Further functional analysis based on COG/KOG category revealed that several Kbhb proteins were associated with metabolism, especially lipid metabolism (19, 25%) (Fig. 5F). KEGG pathway analysis showed that prominent downregulated Kbhb-modified sites were associated with amino acid and fatty acid metabolism, cGMP-PKG signaling, and TCA cycle, and congenital heart disease pathways. The upregulated Kbhb sites were involved in arginine and proline metabolism, antigen processing and presentation, and dilated cardiomyopathy (Fig. 5G). Next, the flanking region of the site was analyzed, and we found that glycine (G), tryptophan (W), leucine (L), phenylalanine (F), and tyrosine (Y) were overexpressed at −1 and +1 positions around the Lys Kbhb sites (Fig. 6A).
Fig. 6.

Characterizing Kbhb in mouse myocardium.A, the map shows the frequency change of amino acids close to the Kbhb site. Glycine (G), tryptophan (W), leucine (L), phenylalanine (F), and tyrosine (Y) were overexpressed, −1 and +1 positions around the Lys Kbhb sites. B and C, all Kbhb were equally divided into four groups according to their fold difference (Q1:< 0.5, Q2: 0.5–1/1.5, Q3: 1.5–2.0, Q4: >2.0). B, statistical chart of quantity of each group. C, KEGG pathway of the Kbhb proteome. Fat digestion and absorption, TCA cycle, cholesterol metabolism, and glyoxylate and dicarboxylate metabolism pathway were enriched with low AK/WT ratio. D, interaction network of differential Kbhb and proteome between TCA cycle and fatty acid degradation pathway. The Kbhb group is blue, while the protein group is red. The number of interacting nodes determines the size of the circle. In the outer circle, the ratio value following log2 treatment was displayed using the Omics Visualizer 1.3.0 plug-in. The color is more yellow the higher the value and more purple the lower the value. The protein’s location with the biggest difference served as the ratio value for the modified group (ranked by the absolute value of ratio after log2 treatment). Kbhb, Lysine β-hydroxybutyrylation; KEGG, Kyoto Encyclopedia of Genes and Genomes.
To further study the effect of Kbhb after AMPKα2 gene knockout, Kbhb sites were divided into four groups based on differential expression (Q1: <0.5, Q2: 0.5–1/1.5, Q3: 1.5–2.0, Q4: >2.0), followed by KEGG pathway analysis for each group (Fig. 6B). In the KEGG category, pathways related to RNA degradation, HIF-1 signaling pathway, and glycolysis/gluconeogenesis were enriched with high AK/WT ratio. Conversely, fat digestion and absorption, TCA cycle, cholesterol metabolism, and glyoxylate and dicarboxylate metabolism pathways were significantly downregulated with low AK/WT ratio (Fig. 6C).
In this study, KEGG category showed that the part of significantly downregulated Kbhb sites in AK mice were located in proteins interrelated to mitochondrial fatty acid degradation and TCA cycle (Tables 3 and 4). Therefore, the interaction network of differential Kbhb proteins and differential proteins was analyzed based on the interaction gene/protein retrieval tool (STRING v.11.0) database, including 26 proteins related to lipid degradation and TCA cycle (Fig. 6D and supplemental Fig. S2). Notably, ten of the key enzymes had >2 different Kbhb sites. Citrate synthase had five differential Kbhb sites, and carnitine O-palmitoyltransferase 1 (CPT-1) had two differential Kbhb sites. Multiple differential Kbhb sites in these fatty acid oxidation- and TCA cycle-related proteins might have significant effects on energy metabolism (Supplemental Data 2).
Table 3.
Downregulated Kbhb sites on mitochondrial fatty acid degradation
| Protein description | Gene name | Position | Amino acid | AK/WT ratio | AK/WT p value | Regulated type |
|---|---|---|---|---|---|---|
| Long-chain-fatty-acid--CoA ligase 1 | Acsl1 | 641 | K | 0.61 | 0.000 | Down |
| Long-chain-fatty-acid--CoA ligase 1 | Acsl1 | 217 | K | 0.545 | 0.014 | Down |
| Long-chain-fatty-acid--CoA ligase 1 | Acsl1 | 396 | K | 0.663 | 0.027 | Down |
| Enoyl-CoA delta isomerase 1 | Eci1 | 229 | K | 0.383 | 0.000 | Down |
| Medium-chain specific acyl-CoA dehydrogenase | Acadm | 212 | K | 0.571 | 0.009 | Down |
| Very long-chain specific acyl-CoA dehydrogenase | Acadvl | 373 | K | 0.658 | 0.000 | Down |
| Carnitine O-palmitoyltransferase 2 | Cpt2 | 433 | K | 0.493 | 0.017 | Down |
| Hydroxyacyl-coenzyme A dehydrogenase | Hadh | 87 | K | 0.559 | 0.013 | Down |
| Trifunctional enzyme subunit alpha | Hadha | 644 | K | 0.647 | 0.003 | Down |
| Trifunctional enzyme subunit alpha | Hadha | 60 | K | 0.603 | 0.001 | Down |
| Trifunctional enzyme subunit alpha | Hadha | 214 | K | 0.325 | 0.012 | Down |
| Trifunctional enzyme subunit alpha | Hadha | 519 | K | 0.643 | 0.036 | Down |
| 3-ketoacyl-CoA thiolase | Acaa2 | 214 | K | 0.518 | 0.000 | Down |
| Acetyl-CoA acetyltransferase | Acat1 | 265 | K | 0.437 | 0.010 | Down |
| Carnitine O-palmitoyltransferase 1 | Cpt1b | 584 | K | 0.354 | 0.000 | Down |
| Carnitine O-palmitoyltransferase 1 | Cpt1b | 150 | K | 0.465 | 0.019 | Down |
| Trifunctional enzyme subunit beta | Hadhb | 202 | K | 0.645 | 0.000 | Down |
| Trifunctional enzyme subunit beta | Hadhb | 189 | K | 0.228 | 0.001 | Down |
| Trifunctional enzyme subunit beta | Hadhb | 53 | K | 0.538 | 0.010 | Down |
Abbreviations: AK, AMPKα2 knockout; Kbhb, Lysine β-hydroxybutyrylation.
Table 4.
Downregulated Kbhb sites on TCA cycle
| Protein description | Gene name | Position | Amino acid | AK/WT ratio | AK/WT p value | Regulated type |
|---|---|---|---|---|---|---|
| Dihydrolipoyl dehydrogenase | Dld | 430 | K | 0.481 | 0.014 | Down |
| Dihydrolipoyl dehydrogenase | Dld | 143 | K | 0.626 | 0.000 | Down |
| Malate dehydrogenase | Mdh2 | 239 | K | 0.408 | 0.000 | Down |
| Pyruvate dehydrogenase E1 component subunit alpha | Pdha1 | 313 | K | 0.268 | 0.005 | Down |
| Isocitrate dehydrogenase [NADP] | Idh2 | 180 | K | 0.578 | 0.000 | Down |
| Isocitrate dehydrogenase [NAD] subunit gamma 1 | Idh3g | 226 | K | 0.403 | 0.009 | Down |
| Aconitate hydratase | Aco2 | 517 | K | 0.451 | 0.000 | Down |
| Succinate dehydrogenase [ubiquinone] iron-sulfur subunit | Sdhb | 235 | K | 0.629 | 0.001 | Down |
| Citrate synthase | Cs | 375 | K | 0.565 | 0.018 | Down |
| Citrate synthase | Cs | 321 | K | 0.654 | 0.003 | Down |
| Citrate synthase | Cs | 393 | K | 0.56 | 0.019 | Down |
| Citrate synthase | Cs | 459 | K | 0.596 | 0.001 | Down |
| Citrate synthase | Cs | 52 | K | 0.355 | 0.000 | Down |
| Dihydrolipoyllysine-residue succinyltransferase component of 2-oxoglutarate dehydrogenase complex | Dlst | 268 | K | 0.533 | 0.001 | Down |
| Isocitrate dehydrogenase [NAD] subunit alpha | Idh3a | 350 | K | 0.391 | 0.020 | Down |
Abbreviations: AK, AMPKα2 knockout; Kbhb, Lysine β-hydroxybutyrylation.
Discussion
ATP production in the mouse heart is mainly dependent on mitochondrial fatty acid β-oxidation, with KB and others supplying only fraction of the precursor (21). AMPK is an energy regulator involved in various physiological processes, such as restoring energy balance, protein synthesis, and glucose and fatty acid metabolism (15, 16). β-OHB along with acetoacetate and acetone are the constituents of ketone bodies. Β-OHB is an intermediate in mitochondrial fatty acid β-oxidation and is defined as a substrate that maintains metabolic homeostasis and a metabolic signal-regulating lipolysis, oxidative stress, and cellular function (13). Some studies have shown that β-OHB catalyzes lysine Kbhb of histones (14). Herein, we showed that the AK disrupts the normal ultrastructure of the cardiomyocytes with ventricular diastolic function impaired as detected by echocardiography and reported a proteomic analysis of Kbhb in WT mice and AK mice. Based on HPLC-MS/MS, we identified 522 upregulated or downregulated differential proteins in cardiac tissues, while 244 differential Kbhb–modified sites were detected after AK. This study, for the first time, observed that the Kbhb sites of key enzymes of energy metabolism, for instance, mitochondrial fatty acid β-oxidation and TCA cycle, were significantly downregulated after AK.
The current data indicated that AK did not elicit significant changes in appearance, BW, grip and HW. (Fig. 1, C and D), indicating that specific knock out of AMPKα2 did not affect the general condition in young mice. We also observed that AK provokes unfavorable changes in the myocardium, along with the ventricular diastolic function impaired, in line with ultrastructural disarray and mitochondria distributed randomly (Fig. 2 and Table 2). Notably, AMPK knockout induced flattened and disappeared mitochondrial crest in mouse cardiac tissue, suggesting that the knockout affects energy metabolism, which is the primary event in the mitochondria.
PTMs are crucial for protein function (activity and stability) and are closely related to various cardiovascular diseases (17, 18). To date, >400 PTMs, containing methylation, ubiquitination, phosphorylation, acetylation, and glycosylation, have been reported (22, 23, 24, 25). Kbhb modification was first reported by Xie et al. in 2016 (14). In recent years, Kbhb modification has been shown to participate in energy metabolism, tumor metabolism, DNA damage repair, and other processes (15, 16, 26). Hence, we performed proteomics and Kbhb proteomics analyses to elucidate the potential molecular mechanisms of AMPK energy regulators. Quantitative Kbhb proteomics analysis discovered 244 Kbhb significantly altered protein sites, of which 42.6% (104/244) were upregulated and 57.4% (144/244) were downregulated after AK (Fig. 5C). The Kbhb modified proteins were mainly distributed in the mitochondria (35.45%). Further analysis showed that pathway were enriched for Kbhb, including macronutrient, energy metabolism, and congenital heart disease. Our data investigated the functions and roles in AK progression by GO and KEGG pathway enrichment analyses. Several nutrient pathways were enriched for Kbhb-containing glycolysis, amino acid metabolism, fatty acid β-oxidation, and the TCA cycle.
The richest KB, β-OHB, has diverse bioactive properties, not only as an energy source but also as an inhibitor of histone deacetylases. Shimadzu et al. showed that ketogenic diet or fasting and other conditions connected with increasing β-OHB abundance, and corresponding increase in overall histone acetylation in mouse tissues is similar to the Kbhb-induced conditions (23). KBs, especially β-OHB, represent a transported form of acetyl-CoA. In mouse myocardial mitochondria, acetyl-CoA could be formed from pyruvate by pyruvate dehydrogenase (PDH) and fatty acids through β-oxidation (27). Acetyl-CoA levels, together with acetyltransferase and deacetylase, adjust protein in acetylation through mitochondrial fatty acid β-oxidation and TCA cycle pathways. As a regulatory center of energy metabolism, AMPK regulates a variety of metabolic enzymes. Previous studies have shown that the activation of the AMPK/acetyl-CoA carboxylase/CPT-1 pathway promotes fatty acid β-oxidation and increases the yield of β-OHB (KB), which in turn increases the overall histone acetylation in mouse tissues (27, 28, 29). According to Koronowski et al., as the concentration of β-OHB increased, its activated CoA formed β-OHB-CoA, which acted as the substrate of Kbhb, alter the overall Kbhb level (15). AMPK facilitates the transition from pyruvate metabolism to the TCA cycle by tight regulation of PDH activity (30). In our findings, the Kbhb sites of key enzymes in β-oxidation and TCA cycle (for instance CPT-1 and PDHA1) were significantly downregulated after AK, which appears to overlap significantly with the acetylation target pathways. Rardin et al. (31) showed that the proteins and sites of kac and Kbhb overlapped 75% when mitochondrial analysis was limited. This opens the possibility that protein Kbhb might regulate AMPK-mediated energy metabolism by cooperating with or competing with acetylation.
AMPK maintains energy metabolic homeostasis by coordinating various cellular processes, such as glycolysis, fatty acid degradation, and TCA cycle (28). Since AMPK has been linked to abnormal metabolic pathway activation and epigenetic adjustment, the characteristics of carcinoma, affecting AMPK could provide novel targets for carcinoma therapy (32). AMPK is implicated in suppressing tumor growth in early stage, and at the same time, it may also act as a cancer gene to promote tumorigenesis (33, 34, 35).
Previously, it was thought that tumor tissue could bypass the TCA cycle and exploit glycolysis (Warburg effect); however, recent evidence suggested that a large number of cancer cells depend on the TCA cycle for supply energy (36). Zhen et al. demonstrated that AMPK can convert the metabolic processes to the TCA cycle by regulating PDH activity, thereby preventing oxidative stress and metabolism-induced cell death, which ultimately leads to cancer cell metastasis (30, 37). The present study showed that after AK, the site of the key enzyme PDH in the TCA cycle was downregulated by 3.73-fold. Recent studies have reported that AMPK sustains the activity of the PDH complex by phosphorylating PDHA (37). Whether AMPK can directly affect the activity of PDH complex by mediating the Kbhb PDHA, thereby regulating the TCA cycle, requires further investigation.
Taken together, our study is the first analysis of Kbhb substrates post-AK and provides a dataset of cardiac Kbhb in mammals. Further studies on the function of protein Kbhb modifications in energy metabolism will provide an in-depth insight into protein PTMs, discuss the distinctive characteristic of AMPK in regulating energy metabolism, and provide a clue for future drug exploits for AMPK-targeted cancer therapy.
For the first time, Kbhb modification was reported in the cardiac tissue of AK mice; nevertheless, the present study had some limitations. Although a considerable number of Kbhb peptides have been identified in this study, the exact modification site function remains to be elucidated.
Data Availability
Annotated spectra for identified β-hydroxybutyrylation modification have been deposited on MS-viewer (https://msviewer.ucsf.edu/cgi-bin/msform.cgi?form=msviewer) with search key “vau9y04iwv”. Mass spectrometry proteomics data have been deposited on the ProteomeXchange Consortium via the iProX partner repository. Project name: The role of β-hydroxybutyrylation in cardiac energy metabolism using AK mouse model. Dataset identifier: PXD034269.
Supplemental data
This article contains supplemental data.
Conflict of interest
The authors declare no competing interests.
Acknowledgments
Funding and additional information
This work was supported by the Key Technology Research and Development Program of Shandong [2022CXGC010510]; the Natural Science Foundation of Shandong Province [ZR2020MH313]; the Key Technology Research and Development Program of Shandong [2017GSF218101]; the National Natural Science Foundation of China [81700725]; the National Major Science and Technology projects of China [2012ZX09303016-003]; the Natural Science Foundation of Shandong Province [ZR2017BH003].
Author contributions
W.-j. D., X.-h. L., Y. S., Y.-p. S., R. Y., and H.-q. G. data curation; W.-j. D., X.-h. L., C.-m. T., X.-c. Y., R. Y., N. Y, Z. Z., and Y.-q. X. software; W.-j. D. and X.-h. L. validation; W.-j. D. writing-original draft; X.-h. L. writing-review and editing; C.-m. T., Y.-p. S., M.-y. L., and J.-c. F. visualization; X.-c. Y., Y. S., and M.-y. L. investigation; W.-h. Z. resources; Z. Z. and Y.-q. X. methodology; Y.-q. X. supervision; Y.-q. X. conceptualization.
Contributor Information
Zhen Zhang, Email: 18560088221@163.com.
Yan-qiu Xing, Email: xingyanqiu@sina.com.
Supplemental Data
Supplemental Figure S1.

Biological replicates test: A, Repeatability test of proteomics. a, Principal Component Analysis (PCA); b, Pearson's Correlation Coefficient (PCC); c, relative standard deviation (RSD); B, Repeatability test of Kbhb omics. a, Principal Component Analysis (PCA); b, Pearson's Correlation Coefficient (PCC); c, relative standard deviation (RSD).
Supplemental Figure S2.

The proteome and modification group's different proteins were integrated and compared to the STRING (v.11.0) protein interaction network database, and the protein interaction connection was retrieved based on the confidence score > 0.7 (high confidence). The modification group is blue, while the protein group is red. The number of interacting nodes determines the size of the circle. In the outer circle, the ratio value following log2 treatment was displayed using the Omics Visualizer 1.3.0 plug-in. The color is more yellow the higher the value and more purple the lower the value. The protein's location with the biggest difference served as the ratio value for the modified group (ranked by the absolute value of ratio after log2 treatment).
References
- 1.Viollet B., Andreelli F., Jørgensen S.B., Perrin C., Geloen A., Flamez D., et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J. Clin. Invest. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hardie D.G., Hawley S.A. AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays. 2001;23:1112–1119. doi: 10.1002/bies.10009. [DOI] [PubMed] [Google Scholar]
- 3.Carvajal K., Zarrinpashneh E., Szarszoi O., Joubert F., Athea Y., Mateo P., et al. Dual cardiac contractile effects of the alpha2-AMPK deletion in low-flow ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2007;292:H3136–H3147. doi: 10.1152/ajpheart.00683.2006. [DOI] [PubMed] [Google Scholar]
- 4.Young L.H., Li J., Baron S.J., Russell R.R. AMP-activated protein kinase: a key stress signaling pathway in the heart. Trends Cardiovasc. Med. 2005;15:110–118. doi: 10.1016/j.tcm.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 5.Sambandam N., Lopaschuk G.D. AMP-activated protein kinase (AMPK) control of fatty acid and glucose metabolism in the ischemic heart. Prog. Lipid Res. 2003;42:238–256. doi: 10.1016/s0163-7827(02)00065-6. [DOI] [PubMed] [Google Scholar]
- 6.Zaha V.G., Young L.H. AMP-activated protein kinase regulation and biological actions in the heart. Circ. Res. 2012;111:800–814. doi: 10.1161/CIRCRESAHA.111.255505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carling D., Zammit V.A., Hardie D.G. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987;223:217–222. doi: 10.1016/0014-5793(87)80292-2. [DOI] [PubMed] [Google Scholar]
- 8.Munday M.R., Campbell D.G., Carling D., Hardie D.G. Identification by amino acid sequencing of three major regulatory phosphorylation sites on rat acetyl-CoA carboxylase. Eur. J. Biochem. 1988;175:331–338. doi: 10.1111/j.1432-1033.1988.tb14201.x. [DOI] [PubMed] [Google Scholar]
- 9.Wu N., Zheng B., Shaywitz A., Dagon Y., Tower C., Bellinger G., et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell. 2013;49:1167–1175. doi: 10.1016/j.molcel.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X., Liu C., Liu C., Wang Y., Zhang W., Xing Y., et al. Trimetazidine and lcarnitine prevent heart aging and cardiac metabolic impairment in rats via regulating cardiac metabolic substrates. Exp. Gerontol. 2019;119:120–127. doi: 10.1016/j.exger.2018.12.019. [DOI] [PubMed] [Google Scholar]
- 11.Hardie D.G., Pan D.A. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem. Soc. Trans. 2002;30:1064–1070. doi: 10.1042/bst0301064. [DOI] [PubMed] [Google Scholar]
- 12.Folmes C.D., Lopaschuk G.D. Role of malonyl-CoA in heart disease and the hypothalamic control of obesity. Cardiovasc. Res. 2007;73:278–287. doi: 10.1016/j.cardiores.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 13.Abdul Kadir A., Clarke K., Evans R.D. Cardiac ketone body metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2020;1866:165739. doi: 10.1016/j.bbadis.2020.165739. [DOI] [PubMed] [Google Scholar]
- 14.Xie Z., Zhang D., Chung D., Tang Z., Huang H., Dai L., et al. Metabolic regulation of gene expression by histone lysine beta-hydroxybutyrylation. Mol. Cell. 2016;62:194–206. doi: 10.1016/j.molcel.2016.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koronowski K.B., Greco C.M., Huang H., Kim J.K., Fribourgh J.L., Crosby P., et al. Ketogenesis impact on liver metabolism revealed by proteomics of lysine beta-hydroxybutyrylation. Cell Rep. 2021;36:109487. doi: 10.1016/j.celrep.2021.109487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu K., Li F., Sun Q., Lin N., Han H., You K., et al. p53 beta-hydroxybutyrylation attenuates p53 activity. Cell Death Dis. 2019;10:243. doi: 10.1038/s41419-019-1463-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pejaver V., Hsu W.L., Xin F., Dunker A.K., Uversky V.N., Radivojac P., et al. The structural and functional signatures of proteins that undergo multiple events of post-translational modification. Protein Sci. 2014;23:1077–1093. doi: 10.1002/pro.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao J., Shao K., Chen X., Li Z., Liu Z., Yu Z., et al. The involvement of post-translational modifications in cardiovascular pathologies: focus on SUMOylation, neddylation, succinylation, and prenylation. J. Mol. Cell. Cardiol. 2020;138:49–58. doi: 10.1016/j.yjmcc.2019.11.146. [DOI] [PubMed] [Google Scholar]
- 19.Aubert G., Martin O.J., Horton J.L., Lai L., Vega R.B., Leone T.C., et al. The failing heart relies on ketone bodies as a fuel. Circulation. 2016;133:698–705. doi: 10.1161/CIRCULATIONAHA.115.017355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bedi K.C., Jr., Snyder N.W., Brandimarto J., Aziz M., Mesaros C., Worth A.J., et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation. 2016;133:706–716. doi: 10.1161/CIRCULATIONAHA.115.017545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H., Ma Z., Zhai Y., Lv C., Yuan P., Zhu F., et al. Trimetazidine ameliorates myocardial metabolic remodeling in isoproterenol-induced rats through regulating ketone body metabolism via activating AMPK and PPAR alpha. Front. Pharmacol. 2020;11:1255. doi: 10.3389/fphar.2020.01255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ke M., Shen H., Wang L., Luo S., Lin L., Yang J., et al. Identification, quantification, and site localization of protein posttranslational modifications via mass spectrometry-based proteomics. Adv. Exp. Med. Biol. 2016;919:345–382. doi: 10.1007/978-3-319-41448-5_17. [DOI] [PubMed] [Google Scholar]
- 23.Scott J.D., Pawson T. Cell signaling in space and time: where proteins come together and when they’re apart. Science. 2009;326:1220–1224. doi: 10.1126/science.1175668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia B.A., Shabanowitz J., Hunt D.F. Characterization of histones and their post-translational modifications by mass spectrometry. Curr. Opin. Chem. Biol. 2007;11:66–73. doi: 10.1016/j.cbpa.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 25.Xie Z., Dai J., Dai L., Tan M., Cheng Z., Wu Y., et al. Lysine succinylation and lysine malonylation in histones. Mol. Cell. Proteomics. 2012;11:100–107. doi: 10.1074/mcp.M111.015875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang H., Chang Z., Qin L.N., Liang B., Han J.X., Qiao K.L., et al. MTA2 triggered R-loop trans-regulates BDH1-mediated beta-hydroxybutyrylation and potentiates propagation of hepatocellular carcinoma stem cells. Signal Transduct. Target. Ther. 2021;6:135. doi: 10.1038/s41392-021-00464-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimazu T., Hirschey M.D., Newman J., He W., Shirakawa K., Le Moan N., et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–214. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ke R., Xu Q., Li C., Luo L., Huang D. Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism. Cell Biol. Int. 2018;42:384–392. doi: 10.1002/cbin.10915. [DOI] [PubMed] [Google Scholar]
- 29.Kolb H., Kempf K., Röhling M., Lenzen-Schulte M., Schloot N.C., Martin S. Ketone bodies: from enemy to friend and guardian angel. BMC Med. 2021;19:313. doi: 10.1186/s12916-021-02185-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cai Z., Peng D., Lin H.K. AMPK maintains TCA cycle through sequential phosphorylation of PDHA to promote tumor metastasis. Cell Stress. 2020;4:273–277. doi: 10.15698/cst2020.12.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rardin M.J., He W., Nishida Y., Newman J.C., Carrico C., Danielson S.R., et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013;18:920–933. doi: 10.1016/j.cmet.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu C.C., Peng D., Cai Z., Lin H.K. AMPK signaling and its targeting in cancer progression and treatment. Semin. Cancer Biol. 2021;85:52–68. doi: 10.1016/j.semcancer.2021.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vila I.K., Yao Y., Kim G., Xia W., Kim H., Kim S.J., et al. A UBE2O-AMPKalpha2 axis that promotes tumor initiation and progression offers opportunities for therapy. Cancer Cell. 2017;31:208–224. doi: 10.1016/j.ccell.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Faubert B., Boily G., Izreig S., Griss T., Samborska B., Dong Z., et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17:113–124. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eichner L.J., Brun S.N., Herzig S., Young N.P., Curtis S.D., Shackelford D.B., et al. Genetic analysis reveals AMPK is required to support tumor growth in murine Kras-dependent lung cancer models. Cell Metab. 2019;29:285–302.e7. doi: 10.1016/j.cmet.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson N.M., Mucka P., Kern J.G., Feng H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell. 2018;9:216–237. doi: 10.1007/s13238-017-0451-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cai Z., Li C.F., Han F., Liu C., Zhang A., Hsu C.C., et al. Phosphorylation of PDHA by AMPK drives TCA cycle to promote cancer metastasis. Mol. Cell. 2020;80:263–278.e7. doi: 10.1016/j.molcel.2020.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Annotated spectra for identified β-hydroxybutyrylation modification have been deposited on MS-viewer (https://msviewer.ucsf.edu/cgi-bin/msform.cgi?form=msviewer) with search key “vau9y04iwv”. Mass spectrometry proteomics data have been deposited on the ProteomeXchange Consortium via the iProX partner repository. Project name: The role of β-hydroxybutyrylation in cardiac energy metabolism using AK mouse model. Dataset identifier: PXD034269.