

Abstract
Mono- and bi-allelic variants in ALDH18A1 cause a spectrum of human disorders associated with cutaneous and neurological findings that overlap with both cutis laxa and spastic paraplegia. ALDH18A1 encodes the bifunctional enzyme pyrroline-5-carboxylate synthetase (P5CS) that plays a role in the de novo biosynthesis of proline and ornithine. Here we characterize a previously unreported homozygous ALDH18A1 variant (p.Thr331Pro) in four affected probands from two unrelated families, and demonstrate broad-based alterations in amino acid and antioxidant metabolism. These four patients exhibit variable developmental delay, neurological deficits and loose skin. Functional characterization of the p.Thr331Pro variant demonstrated a lack of any impact on the steady-state level of the P5CS monomer or mitochondrial localization of the enzyme, but reduced incorporation of the monomer into P5CS oligomers. Using an unlabeled NMR-based metabolomics approach in patient fibroblasts and ALDH18A1-null human embryonic kidney cells expressing the variant P5CS, we identified reduced abundance of glutamate and several metabolites derived from glutamate, including proline and glutathione. Biosynthesis of the polyamine putrescine, derived from ornithine, was also decreased in patient fibroblasts, highlighting the functional consequence on another metabolic pathway involved in antioxidant responses in the cell. RNA sequencing of patient fibroblasts revealed transcript abundance changes in several metabolic and extracellular matrix-related genes, adding further insight into pathogenic processes associated with impaired P5CS function. Together these findings shed new light on amino acid and antioxidant pathways associated with ALDH18A1-related disorders, and underscore the value of metabolomic and transcriptomic profiling to discover new pathways that impact disease pathogenesis.
Introduction
ALDH18A1 encodes delta-1-pyrroline-5-carboxylate synthase (P5CS), a bifunctional mitochondrial enzyme involved in the de novo biosynthesis of several amino acids and metabolites, including proline and ornithine (1,2). This enzyme catalyzes the conversion of glutamate to pyrroline-5-carboxylate, which is subsequently used by either ornithine aminotransferase (OAT) to make ornithine (3), or pyrroline-5-carboxylate reductase (PYCR1) to make proline (Fig. 1A). The metabolism of glutamate by P5CS connects this enzyme with both the urea cycle and tricarboxylic acid (TCA) cycle as well as the biosynthesis of several different amino acids and polyamines. In addition, several of the downstream reaction products of P5CS, including the polyamines and proline, are involved in cellular antioxidant responses (4–8). The P5CS substrate, glutamate, is also utilized by the cell for glutathione biosynthesis, further highlighting the importance of this metabolic pathway in establishing redox and antioxidant capacity.
Figure 1.

Overview of metabolic pathways connected to P5CS and the domain organization of the enzyme. (A) Schematic of the different metabolic pathways that are connected to the products of the P5CS-mediated reaction, and utilize the P5CS substrate, glutamate; a-KG, a-ketoglutarate; OAT, ornithine transaminase; PYCR1, pyrroline-5-carboxylate reductase; (B) pedigrees of the two families and four affected individuals included in this study; (C) domain organization of the P5CS enzyme and location of the homozygous p.Thr331Pro variant found in all four patients; MLS, mitochondrial localization signal; CFB, co-factor binding domain.
ALDH18A1 was first associated with human disease following the identification of two patients with a neurocutaneous disorder who were shown to bear biallelic mutations in the gene (9). ALDH18A1 has also been implicated in several human cancers including breast cancer and melanoma (10–12). The clinical complexity of ALDH18A1-related disorders expanded with the identification of variants in this gene associated with complicated forms of inherited spastic paraplegia (13,14). Based on the analysis of this broadening clinical spectrum and the impact of known ALDH18A1 mutations, ALDH18A1-related disorders in humans are thought to encompass at least two distinct syndromes—hereditary spastic paraplegia 9 (SPG9A and B) and cutis laxa 3 (ADCL3 and ARCL3A) (15). Dominant and recessive mutations have now been identified for both disorders. The cutaneous phenotypes associated with cutis laxa may arise in part from impaired proline biosynthesis, which may limit the production of collagen and elastin in the skin and other tissues. The mechanistic basis of the neurological manifestations is not fully understood. These impairments may relate to the altered availability of key metabolites for both the urea cycle and TCA cycle (reviewed in 15). The connection to the urea cycle is reinforced by similarities in motor neuron degeneration evident in patients with arginase and ornithine transporter defects (14).
Monitoring the fate of isotopically labeled glutamate in patient cells has been useful in establishing the pathogenicity of ALDH18A1 variants (16,17). This functional approach, however, does not provide insight regarding the complete landscape of metabolic changes that stem from impaired P5CS function. This limits our understanding of the factors and metabolites that contribute to disease pathogenesis. Here we describe a previously unreported missense homozygous variant of uncertain significance (p.Thr331Pro) in ALDH18A1 present in four affected probands from two unrelated families. These patients show many clinical features consistent with SPG9B, including global developmental delay and hypotonia, but share other symptoms with ARCL3A such as loose, hyperelastic skin. Studies were performed in patient cells to functionally resolve this variant of uncertain significance, and explore the metabolic and transcriptomic profiles that accompany impaired P5CS function. Nuclear magnetic resonance (NMR)-based metabolomic studies in two different cell systems and transcriptomic profiling on patient fibroblasts uncovered the involvement of numerous metabolites and pathways not identified in prior studies, including extensive mobilization of glutamate-derived antioxidant pathways, and abnormal abundance of extracellular matrix (ECM) proteins and ECM-modifying enzymes. The implications of these findings with regard to antioxidant responses, metabolic adaptations and cutaneous disease pathogenesis in these patients are discussed.
Results
Clinical summary of patients
The pedigrees of the two families are shown in Figure 1B. The domain organization of the P5CS enzyme and the position of the homozygous variant found in all four affected patients is depicted in Figure 1C. The clinical and diagnostic findings in the four patients from two unrelated families is described below, and the clinical features of these patients compared to those with ARCL3A and SPG9B are summarized in Table 1.
Table 1.
Summary of clinical findings in the patients compared to ARCL3A and SPG9B
| Clinical features | Family 1 | Family 2 | |||||
|---|---|---|---|---|---|---|---|
| P1 | P2 | P3 | P4 | Total | ARCL3A | SPG9B | |
| Age of onset | 6 mo | 7 mo | 7 mo | 2 mo | 2–7 mo | 0–6 mo | ~5 yo |
| Cutis laxa | − | − | − | − | 0/4 | +++ | − |
| Skin hyperelasticity | − | + | + | + | 3/4 | − | − |
| Global developmental delay | + | + | + | + | 4/4 | +++ | +++ |
| Hypotonia | + | + | + | + | 4/4 | +++ | + |
| Joint hypermobility | − | + | + | + | 3/4 | +++ | + |
| Short stature | − | + | + | + | 3/4 | +++ | ++ |
| Microcephaly | + | + | + | − | 3/4 | +++ | ++ |
| Visible veins | + | + | + | + | 4/4 | ++ | − |
| Cataracts | − | + | − | − | 1/4 | ++ | + |
| Hypertonia/spasticity | + | − | − | − | 1/4 | + | +++ |
| Corpus callosum hypogenesis | + | + | ? | ? | 2/2 | +++ | + |
Family 1
Patient 1 (P1) is a 10-year-old female who initially presented for a genetics evaluation at 16 months old for developmental delay. She was first noted to have developmental delay at 6 months old. She sat at 16 months, started taking steps with assistance at 5 years, and walking at 7 years. She started saying mama when she was 2 years old. She now has a few signs but no other words. She has a history of pyloric stenosis, gastroesophageal reflux, constipation, gastric dysmotility, obstructive sleep apnea and bruxism. Her neurologic findings include a generalized resting tremor, hypotonia, periods of hyperventilation, head titubation and spastic diplegia. She wears splints on her arms to prevent self-injurious behavior. At her most recent physical exam, she had a weight of 28.1 kg (Z = −0.90), height of 128.6 cm (Z = −1.47), and head circumference of 48.2 cm (Z = −3.24). She was noted to have a broad face, deep-set eyes, synophrys, high nasal bridge, mild 2–3 toe syndactyly, central hypotonia and lower limb spasticity. Her skin exam was normal without loose skin. A brain magnetc resonance imaging (MRI) revealed prominent ventricles and subarachnoid spaces with thin corpus callosum and incomplete myelination. She has had normal urine organic acids, plasma amino acids and carnitine levels. Chromosomal microarray analysis (NCBI 36/hg18) did not identify any copy number variants but did reveal a 20-megabase block of homozygosity on chromosome 10q22.3q24.1. She has had normal methylation analysis for Angelman syndrome, sequencing and multiplex ligation-dependent probe amplification (MLPA) analysis of the MECP2 gene, myotonic dystrophy testing, high-resolution chromosome analysis and sequencing of the TCF4, UBE3A, SLC9A6, CDKL5 and FOXG1 genes. A syndromic autism panel revealed a variant in SHANK3, which was maternally inherited and not felt to be clinically significant. Whole exome sequencing revealed a variant in CACNA1G, which was maternally inherited and not felt to be clinically relevant. The (NM_002860.3:c.991G>T; p.Thr331Pro) variant in ALDH18A1 that was initially reported as a variant of uncertain significance but has subsequently been re-classified as likely pathogenic. This amino acid alteration is located in the C-lobe of the glutamate-5-kinase domain of the enzyme with no other known disease-causing variants in close proximity. With the new functional evidence provided below, the ALDH18A1 alteration has been reclassified as likely pathogenic based on American college of medical genetics (ACMG) criteria. The evidence used for this reclassification includes (1) it is present at a very low frequency in the public single nucleotide polymorphism (SNP) databases (PM2) (2) parental testing indicated all four parents are heterozygous carriers of this alteration which confirms homozygosity in the probands (PM3) and (3) functional analyses using patient fibroblasts and ALDH18A1 knockout HEK293 cells expressing the p.Thr331Pro variant enzyme demonstrated alterations in metabolites consistent with a pathogenic effect for this variant (PS3).
Family 2
Patient 2 (P2) is a 6-year-old female who initially presented for a genetics evaluation at 10 months old due to developmental delay. She has a history of poor weight gain, gastroesophageal reflux, cataract and hypotonia. She babbles but does not have any words. She can pull herself to a stand but does not walk. At her most recent physical exam, she had a weight of 12.1 kg (Z = −5.18), height of 96.2 cm (Z = −4.31) and head circumference of 45 cm (Z = −4.59). She has full cheeks, epicanthal folds, supraorbital fullness, deep-set eyes, full lips, open mouth, small jaw, mild tooth decay, prominent antihelices, decreased muscle mass, hypotonia, joint hypermobility, mild skin hyperelasticity, and visible veins on her face and trunk. Brain MRI at 7 months revealed thin corpus callosum and delayed myelination. She has had normal conventional karyotyping, whole genome chromosomal microarray analysis, plasma amino acids, acylcarnitine profile, carnitine levels, urine organic acids and myotonic dystrophy testing. Whole exome sequencing revealed a heterozygous variant in GJC2 but no evidence of an alteration of the other allele on deletion/duplication testing. She also was found to have a heterozygous secondary finding in PALB2 associated with increased cancer risk. Exome sequencing also revealed a homozygous NM_002860.3: c.991A>C (p.Thr331Pro) variant in ALDH18A1 that was initially reported as a variant of uncertain significance but has subsequently been re-classified as likely pathogenic.
Patient 3 (P3) is a 4-year-old male who initially presented for a genetics evaluation at 16 months old for developmental delay and poor growth. He has a history of hypotonia, gastroesophageal reflux and feeding difficulties. He is able to prop sit but is nonambulatory and nonverbal. At his most recent physical exam, he had a weight of 9.25 kg (Z = −7.06), height of 85.3 cm (Z = −4.62) and head circumference of 46.1 cm (Z = −3.48). He has full cheeks, epicanthal folds, supraorbital fullness, unfurled superior helices, full lips, open mouth, small jaw, tooth decay, decreased muscle mass, hypotonia, joint hypermobility, mild skin hyperelasticity and veins visible on his face and trunk (see Supplementary Material, File S1). He had normal plasma amino acids, acylcarnitine profile, carnitine levels and urine organic acids. Targeted gene testing revealed homozygosity for the p.Thr331Pro variant in ALDH18A1 previously identified in his older sister.
Patient 4 (P4) is a 16-month-old female who initially presented for a genetics evaluation at 4 months old due to the family history of two siblings with homozygous ALDH18A1 variants. She has a history of hypotonia, poor weight gain, constipation, gastroesophageal reflux and eczema. She can prop sit but is not walking. She has no words. At her most recent physical exam, she had a weight of 7.1 kg (Z = −2.80), height of 69.7 cm (Z = −3.29) and head circumference of 44.2 cm (Z = −1.43). She has full cheeks, epicanthal folds, supraorbital fullness, deep-set eyes, open mouth, protruding tongue, hypotonia, joint hypermobility, mild skin hyperelasticity and visible veins on her face and trunk. Her only genetic testing has been targeted testing which revealed homozygosity for the p.Thr331Pro variant in ALDH18A1.
Functional characterization of p.Thr331Pro variant in HEK293 and patient-derived cells
To explore the functional impact of this variant, we generated an ALDH18A1-knockout HEK293 cell line using CRISPR-Cas9 gene editing, and used this cell line to re-express either the WT or the p.Thr331Pro variant-bearing P5CS enzyme. As shown in Figure 2A, the ALDH18A1-KO cells show undetectable levels of the P5CS protein. Upon expression of either the WT or variant enzyme, no major differences in the abundance or electrophoretic mobility of the P5CS enzyme were noted when transfection efficiencies were equivalent, indicating that the variant protein is not inherently unstable or subject to proteolysis. This same set of cells were stained with an anti-P5CS antibody to examine whether the mitochondrial localization of the enzyme was altered (Fig. 2B). The P5CS enzyme localizes to structures consistent with mitochondria in WT HEK293 but is undetectable in the ALDH18A1 KO cells. The WT and p.Thr331Pro P5CS were shown to localize to the mitochondria when expressed in the KO HEK293 cells, as determined by co-staining with a TOM20 antibody. We did not note any substantial differences in the localization of the p.Thr331Pro P5CS in these immunostains. Skin fibroblasts from P2 were obtained and used to investigate the level and localization of endogenous P5CS. We showed that the steady-state level of the P5CS monomer in the patient cells is comparable to control fibroblasts (Fig. 2C). Prior studies on ALDH18A1 variants have demonstrated effects on the ability of the enzyme to form homo-oligomers as the basis for compromised function (16,18). This possibility was tested in the primary fibroblasts by resolving WT and patient cell lysates on a non-denaturing native gel. A reproducible decrease in a reactive band at 170 kDa, presumably the dimeric form of the enzyme, was detected in the patient fibroblasts, suggesting that the p.Thr331Pro variant may alter its ability to incorporate into P5CS oligomers (Fig. 2D). Quantification of the ratio of P5CS dimer/monomer, with the latter abundance determined following parallel resolution on a denaturing SDS-PAGE gel, showed a significant 40% reduction in this ratio in patient cells, supporting reduced formation of oligomers as the primary mechanism for impaired P5CS function.
Figure 2.

Functional analysis of the p.Thr331Pro variant in HEK293 cells and patient fibroblasts demonstrate normal steady-state levels and mitochondrial localization but impaired incorporation of the P5CS monomer into homo-oligomeric structures. (A) Representative Western blot of the P5CS enzyme in WT, ALDH18A1-null cells, and ALDH18A1-null cells transfected with either WT or p.Thr331Pro ALDH18A1 DNA; (B) immunostaining of P5CS and TOM20 in the KO HEK293 cells transfected with WT and p.Thr331Pro ALDH18A1 DNA; overall transfection efficiencies ranged from 50% to 60% across the different runs and were roughly equivalent with WT or p.Thr331Pro ALDH18A1 DNA in individual experiments; (C) representative western blot of the P5CS monomer in WT and patient fibroblasts and quantification of relative abundance from four independent experiments; (D) native gel electrophoresis and western blot analysis of P5CS in WT and patient fibroblast lysates. The ratio of P5CS oligomer to monomer was quantified from four independent experiments, and average values plotted. A one-way ANOVA was performed to determine statistical significance. P-value smaller than 0.05 is considered statistically significant; *P < 0.05; **P < 0.01; ***P < 0.001; ns = not significant.
NMR metabolomics reveals broad-based adaptations in amino acid metabolism and antioxidant biosynthesis
We next sought to understand global metabolic alterations that arise from impaired P5CS function. Using both the HEK293 cell system and WT and patient fibroblasts, 1H NMR-based metabolomics were performed on cells grown under standard culture conditions. Features in the resulting spectra were annotated and quantified, culminating in 26 quantified and annotated metabolites. We observed broad and significant alterations in the abundance of several different metabolites when comparing the WT and ALDH18A1 KO HEK293 cells, highlighting the adaptations these cells undergo in order to maintain viability and essential components of P5CS-related metabolic pathways. To what extent such differences also arise due to karyotype differences in the WT and KO cells, as opposed to the underlying metabolic defect, is not known. Nonetheless, we were able to compare the relative abundance of several key metabolites in the ALDH18A1 KO cells expressing either the WT or p.Thr331Pro P5CS enzyme. These findings show that while the profiles of both enzyme expressing cells are more similar to each other than the KO, the p.Thr331Pro enzyme restored metabolite levels less effectively in comparison to the WT enzyme (Fig. 3; Supplementary Material, File S2). This is evident for metabolites such as proline, glycine, succinate, UDP-GlcNAc and glutamine. There were several examples where the opposite trend was noted, including glutamate and aspartate. In total, there were six metabolites that showed significant changes in abundance between p.Thr331Pro and WT enzyme expressing cells, including proline, glycine, succinate, NAD+, phosphorylcholine and creatine (Fig. 3).
Figure 3.

Expression of p.Thr331Pro variant enzyme partially restores metabolic profile of ALDH18A1 knockout compared to expression of WT enzyme. (A) Average 1H NMR spectra of HEK 293 cell extracts. KO − CRISPR knockout of ALDH18A1, KO + p.Thr331Pro (labeled T331P in the figure)—knockout cells exogenously expressing T331P variant of ALDH18A1 enzyme. KO + WT—knockout cells exogenously expressing wild type ALDH18A1 enzyme; overall transfection efficiencies ranged from 50% to 60% across the different runs and were largely equivalent with WT or p.Thr331Pro ALDH18A1 DNA; (B) principal component analysis scores plot of KO, KO + T331P, and KO + WT cells. Integrated spectral features used for analysis; (C) box and whisker plots of metabolites annotated from NMR spectra found significantly different by one-way ANOVA (FDR adjusted P-value < 0.05). All comparisons significant between KO and KO + T331P, KO and KO + WT unless otherwise noted. † indicates additional significant comparison between KO + T331P and KO + WT. * indicates significant comparisons between KO and KO + WT, and KO + T331P and KO + WT only.
A parallel analysis was undertaken using WT and patient fibroblasts. Reduction in the abundance of both proline and glutathione were observed in the patient cells (Fig. 4A). The decrease in proline levels was consistent with the analysis in HEK293 cell system but diminished glutathione levels has not been previously shown in ALDH18A1 patient cells. Another striking observation was the overall reduction in the level of several amino acids in the patient cells, likely indicating the need for these cells to consume other amino acids for cataplerosis and/or conversion to glutamate in order to replenish the TCA cycle and glutamate-derived metabolites. Pathway enrichment analysis of metabolites significantly altered between WT and p.Thr331Pro enzyme-expressing cells in both the HEK293 system and primary fibroblasts showed consistency in the pathways being impacted. These include arginine and proline metabolism, glycine and serine metabolism, and glutamate metabolism (Fig. 4B). Metabolite changes that were consistent between the two cell systems suggest that these trends stem directly from impaired P5CS function. For metabolites where the changes varied between the two cell systems, we believe the patient fibroblasts may more accurately reflect the biochemistry of the underlying disease, as these are untransfected primary cells with residual P5CS function that have not undergone clonal proliferation or selection.
Figure 4.
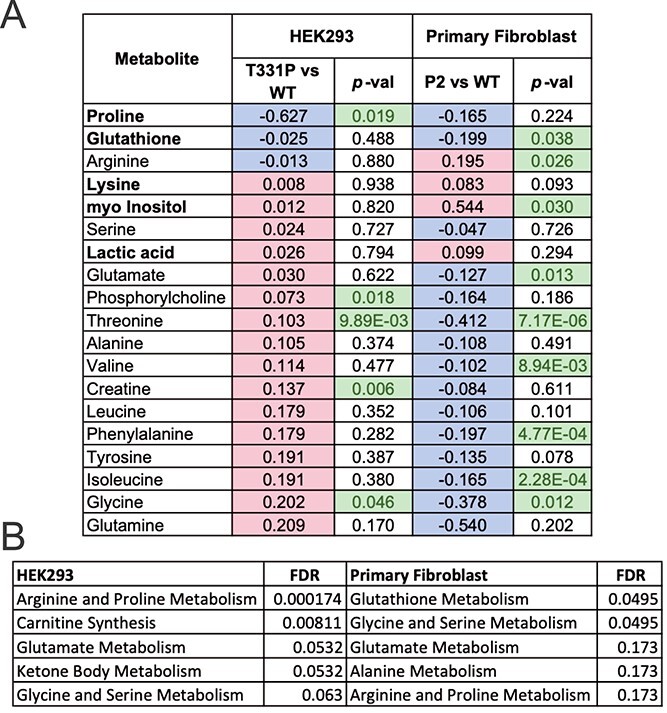
Patient fibroblasts show unique metabolite changes but consistent alterations of proline and glutamyl metabolism. (A) Table of fold change and P-values for all annotated metabolites common to both HEK 293 and primary fibroblast cell extracts. Red indicates relative increase in T331P variant enzyme expressing cells, blue a decrease. Bold metabolite names indicate those with consistent trend between cell systems. Green values indicate significant change (P-value < 0.05 by two-tailed T-test) for that comparison. These significant metabolites were used (but not exclusively) as input for metabolite set over representation analysis in (B); (B) metabolite set enrichment analysis results of significant metabolites for each cell system. Top 5 significant metabolite sets from small molecule pathway database (SMPDB) shown for each cell system, along with FDR P-values. List of all annotated metabolites with raw P-value < 0.05 between p.Thr331Pro and WT enzyme expressing cells was used for over representation analysis in each cell system.
Analysis of polyamine levels demonstrates a reduction in putrescine in patient fibroblasts
The apparent increased utilization of glutathione in the patient cells suggests a possible response to oxidative stress caused by mitochondrial dysfunction or metabolic toxicity. To look at other antioxidant molecules derived from glutamate metabolism, polyamines including putrescine, spermidine and spermine were analyzed in WT and patient fibroblasts (Fig. 5). None of these molecules were detected or identified in the NMR-based metabolomics but can be quantified using high-performance liquid chromatography (19). De novo production of these polyamines is derived almost exclusively from ornithine in cells. These results showed a robust reduction in putrescine levels (and total polyamine levels), with spermidine and spermine not significantly impacted. It is possible the patient fibroblasts prioritize the synthesis of spermidine and spermine as part of a broader antioxidant response, leading to overutilization of their precursor putrescine.
Figure 5.
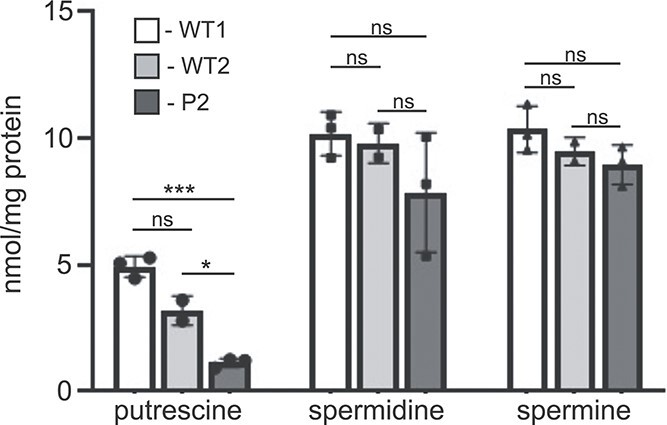
Reduced levels of the polyamine putrescine in patient fibroblasts. The abundance of putrescine, spermidine and spermine in the two WT and patient fibroblast lines was determined using mass spectrometry in three independent experiments. A one-way ANOVA was performed to determine statistical significance; *P < 0.05; **P < 0.01; ***P < 0.001; ns = not significant.
Altered transcript abundance of several ECM and metabolic genes in ALDH18A1 patient fibroblasts
RNA sequencing was performed on WT and patient fibroblasts to examine alterations in gene expression in response to impaired P5CS activity. The volcano plot in Figure 6A shows upregulation of 2391 genes and downregulation of 456 genes in the patient fibroblasts (full dataset is shown in Supplementary Material, File S3). Among the dysregulated genes in the patient fibroblasts, several genes involved in glutathione metabolism, including GPX1, GSTT2B and GSS, were noted. Transcript abundance of the glutamate transporter SLC7A11 was decreased by 1.7-log fold in the patient cells. If this reduction corresponds to lower amounts of the SLC7A11 transporter, these cells may have a limited capacity to resupply glutamate for the necessary metabolic reactions, including proline and glutathione biosynthesis. In addition to differential expression of several ECM-related genes, numerous genes involved in cell signaling were also altered in patient cells. This is highlighted by the Reactome and Panther pathway analysis, which shows overrepresentation of several genes in the MAPK, SEMA3 and RUNX2 pathways, as well as genes involved in cholesterol metabolism (Fig. 6B and Supplementary Material, File S3). Among the most significantly altered transcripts in the patient cells were the metalloproteinase MMP3 (up 4.9-log fold) and the matrix protein, COMP (cartilage oligomeric matrix protein; down 6.6-log fold) (Fig. 6C). Elevation in the transcript and protein abundance of several MMPs has been observed in cutis laxa patient fibroblasts (20,21). Western blot analysis was performed on WT and patient fibroblast lysates revealing that levels for both proteins correspond to their relative transcript abundance (Fig. 6D and E). The reduction in COMP levels in the patient cells is accompanied by altered transcript abundance of other ECM genes, as well as several ECM-related growth factors (TGFB3R) and enzyme inhibitors (TIMP3) (Fig. 6B). Together, these findings point to altered transcript abundance in response to impaired P5CS function, and alterations in ECM-associated proteins that appear to be consistent across human disorders with cutis laxa.
Figure 6.

RNA sequencing of WT and patient fibroblasts reveals altered transcript abundance of multiple genes. (A) Volcano plot of the differentially expressed genes (2391 upregulated and 456 downregulated compared to WT control fibroblasts); data represent analysis of three independent biological replicates for each cell line; (B) overrepresented genes/pathways from the Reactome and Panther GO analyses; (C) table of the most significant transcript abundance changes in patient cells compared to WT controls; (D) western blot analysis of COMP protein and quantification of relative levels (n = 4; statistical significance determined using a Student t-test, ****P < 0.0001); (E) western blot analysis of MMP3 protein and quantification of relative levels (n = 4; statistical significance determined using a student t-test, ****P < 0.0001). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is shown as a loading control.
Discussion
We describe a previously unreported homozygous missense variant in the ALDH18A1 gene that is present in four affected patients from two unrelated families. The amino acid change resides in the glutamate-5-kinase domain of the P5CS enzyme, in a region where few other variants have been identified. The clinical phenotype of these patients appears to be intermediate between ARCL3 and SPG9B with regard to severity and specificity. They share many neurological features associated with SPG9B, including hypotonia, but also had loose skin characteristic of ARCL3A. Interestingly, P1 has hypertonia and spasticity consistent with SPG9B but the other patients do not. P1 also did not have any evidence of loose skin whereas all three of the affected patients in Family 2 have this cutaneous phenotype. The intermediate nature of the clinical manifestations highlights the complexity of ALDH18A1-related disease, and the competition between different metabolic pathways for common substrates. The current functional evidence strongly supports pathogenicity of the p.Thr331Pro variant, despite no reduction in the steady-state level of the monomeric P5CS enzyme nor impaired mitochondrial localization. A defect in homo-oligomer formation was observed using native gel electrophoresis, suggesting the p.Thr331Pro variant may affect complex formation like other reported variants (16,18). The pathogenicity of this variant and evidence for impaired P5CS function is most clearly supported by the metabolomics data showing reduction in products downstream of P5CS including proline and the ornithine-derived polyamine, putrescine. The new insights into the pathogenic mechanisms and the global alterations in metabolism associated with ALDH18A1-related disorders are summarized in Figure 7 and discussed below.
Figure 7.

Summary of metabolic and transcriptomic findings in the context of P5CS-mediated metabolism. Metabolomic findings point to the possible involvement of glutathione and polyamines in the context of ALDH18A1-related disorders. As both are relevant to antioxidant responses, their reduction (e.g. total polyamines) and/or overutilization (e.g. glutathione) may indicate the presence of oxidative stress. The reduced levels of several amino acids, and increased transcript abundance of TCA cycle genes, in the patient cells may indicate the need to replenish glutamate pools depleted by the increased metabolic demand for this amino acid. In addition to the lower levels of proline (which can directly impact the production of ECM proteins), altered transcript and protein abundance of the matrix metalloproteinase, MMP3, and the matrix component, COMP, were noted in patient fibroblasts, highlighting broader alterations in ECM homeostasis associated with impaired P5CS function. Lastly, the increase in arginine levels indicates that this amino acid may not be efficiently converted to ornithine by arginase, thus exacerbating the reduction in ornithine-derived polyamines.
The NMR-based metabolomics in two different cell systems uncovered the involvement of several pathways and metabolites that had not be appreciated in other studies. Most striking, and consistent between the two cell systems, was the reduction in glutathione levels in cells with impaired P5CS function. Glutathione is synthesized in a multistep pathway beginning with the conversion of glutamate to γ-glutamyl-cysteine by the enzyme, glutamate-cysteine ligase. The reduction in glutathione levels may be interpreted as an overutilization of this pathway, further stressed by the need for glutamate in the biosynthesis of both ornithine and proline. Levels of several amino acids were significantly reduced in the patient fibroblasts, possibly reflecting their conversion to glutamate in response to increased need for this key substrate. It will be of interest to explore whether the depletion of these amino acids might trigger other consequences such as mTOR inhibition or an imbalance in proteostasis. We are currently exploring whether the depletion of these amino acids, such as the nutrient sensor leucine, may result in the upregulation of autophagic pathways (22–24). The inhibition of mTOR-related pathways and increased autophagy is intriguing as it could also increase mitophagy and the degradation of mitochondria or other organelles (25,26). Reduction in the level of other amino acids such as arginine could indirectly impact the urea cycle leading to additional insults to sensitive tissues such as neurons. The upregulation of MMP3 and other matrix-degrading enzymes may be part of the same response to amino acid deprivation and increased need for glutamate to support key metabolic processes.
In the HEK293 expression system, significant changes in metabolites such as lactate, succinate, NAD+, and creatine suggest broad differences in the energy and/or redox status of the cells based on which enzyme is expressed. Similarly, transcriptome analysis of patient fibroblasts shows an enrichment of TCA cycle genes that are upregulated, and significant downregulation of several biosynthetic off-ramp transcripts such as PCK and GOT1. These observations imply that the metabolic adaptations required to compensate for P5CS dysfunction are energy intensive, and require increased TCA cycle activity to maintain energy requirements within the cell. Alternatively, increased expression of TCA cycle genes could be generating intermediates and cofactors to be used for glutamate/glutathione biosynthesis, such as 2-oxoglutarate and NADPH. In addition, phospholipid precursors myo-inositol and phosphorylcholine were observed to be significantly altered in patient cells and HEK cells, respectively. These, in addition to the enrichment of differentially expressed genes in several lipid pathways, suggest broad changes in lipid metabolism. These again could be indicative of metabolic adaptations to balance the energy needs of the cell, or reflective of altered membrane lipid composition. We believe some caution is warranted in the interpretation of trends from the metabolomics results from the HEK293 cells in light of the large differences in some metabolites in the WT versus KO cells.
The reduction and/or overutilization of glutathione may indicate the presence of oxidative stress. Preliminary experiments performed to identify oxidative stress in the patient cells were inconclusive, although it is possible that the antioxidant responses, including the production of glutathione, is sufficient to prevent detectable oxidative stress in fibroblasts. Whether such antioxidant responses are inadequate in other cell types such as neurons remains to be determined. We speculate that the increased need for glutamate in the brain as a precursor for neurotransmitters such as GABA may put further stress on P5CS-related metabolism, creating mitochondrial stress and production of reactive oxygen species that impact survival and function of neurons and other sensitive cell types. Aside from the requirement for proline in the biosynthesis of many ECM proteins, this amino acid is also an antioxidant (4). Thus, under conditions where oxidative stress is abundant, competition for proline may create an imbalance that can contribute to pathogenesis.
Polyamines are critical cellular components required for a multitude of functions, including the regulation of receptor ion channels in the central nervous system (CNS) (27,28). As such, their concentrations are strictly controlled to maintain homeostasis (29). The altered polyamine levels detected in the ALDH18A1-variant patient fibroblasts may therefore implicate a role for polyamines in the patient phenotype. The recent identification of a growing number of patients with gene variants affecting polyamine metabolic enzymes has created a family of polyamine-related disorders. Although yet to be fully characterized, these rare, neurodevelopmental syndromes, including Snyder-Robinson syndrome (30,31) and Bachmann-Bupp syndrome (32), share certain components of the clinical phenotype, including developmental delay and hypotonia (33,34). As each of these syndromes, including the ALDH18A1 variant, causes a different perturbance in the individual polyamine pools, it will be important to decipher the contributions of each to the associated phenotypes. Additionally, the combined total amount of polyamines may be important, particularly regarding antioxidant capacity, as polyamines can protect against oxidative damage (35). To our knowledge, this is the first report of a genetic variant causing an overall reduction in polyamine concentration.
The transcriptome analysis of patient fibroblasts revealed altered abundance of numerous ECM-related transcripts. Of note, transcript abundance of the cartilage oligomeric matrix protein (COMP) was reduced nearly 7-log fold in the patient cells, while the matrix-degrading enzyme, MMP-3, had a 6-log fold elevation in transcript abundance. Analysis of enriched pathways among the differentially expressed genes also uncovered several metabolic pathways, including cholesterol metabolism, that had not been previously noted in the context of ALDH18A1-related disease. We confirmed that altered transcript abundance of both COMP and MMP3 correlate to the same changes in protein level by western blot. It is possible that impaired P5CS function and de novo proline biosynthesis cause a shift in the types of ECM proteins made by the patient fibroblasts. Collectively, the combination of transcriptomics and metabolomics in patient cells has uncovered multiple new pathways and processes that are sensitive to impaired P5CS, setting the stage for a deeper investigation of how these altered pathways relate to the tissue pathogenesis and phenotypic specificity in patients with ALDH18A1 variants.
Materials and Methods
Exome sequencing
DNA libraries were prepared from genomic DNA isolated from the proband fibroblasts, using the Agilent SureSelectXT Clinical Research Exome v2 capture kit (Agilent Technologies, Santa Clara, CA). Briefly, DNA was fragmented using the Covaris ME220 system (Covaris, Woburn, MA), and fragments of 150–200 bp were selected using AMPure XP beads (Beckman Coulter, Brea, CA). Fragments were subsequently end-repaired, adenylated at the 3′ end, ligated to sequencing adaptors, and then PCR-amplified using the SureSelectXT Library Preparation kit (Agilent Technologies) with DNA being purified using AMPure XP beads after each of these steps. 750 ng of each DNA library was used for hybridization and capture with the SureSelectXT Clinical Research Exome v2 probes (Agilent Technologies). Captured fragments were amplified by PCR and purified. The quality of the enriched libraries was evaluated using a D1000 Tape on the TapeStation 4200 (Agilent Technologies). Libraries were quantified on a Victor Nivo Fluorometer (PerkinElmer, Waltham, MA) using a Quant-IT Broad Range kit (Life Technologies, Carlsbad, CA), and separate libraries were pooled and sequenced using an Illumina NovaSeq 6000™ Sequencing System at 157× coverage (Illumina Inc., San Diego, CA) per the manufacturer’s protocol.
The Agilent SureSelectXT Clinical Research Exome V2 kit was used to target known disease-associated exonic regions of the genome (coding sequences and splice junctions of known protein-coding genes associated with disease, as well as an exomic backbone) using genomic DNA isolated from peripheral blood samples. The targeted regions were sequenced using the Illumina NovaSeq™ 6000 System with 150 bp paired-end reads. Using Illumina DRAGEN Bio-IT Platform® software, the DNA sequence was aligned and compared to the human genome build 19 (hg19/NCBI build 37). The emedgene® software was used to filter and analyze sequence variants identified in the patient and compare them to the sequences of affected and unaffected family members. Sanger sequencing was subsequently performed to confirm the variant of interest.
Antibodies
The rabbit anti-P5CS polyclonal antibody was purchased from Novus Biologicals (cat# NBP1-83324; Centennial, CO, USA), the mouse anti-TOM20 monoclonal antibody was obtained from Santa Cruz (cat# sc-17764; Dallas, TX, USA) the goat anti-COMP polyclonal antibody was from R&D Systems (cat# AF3134; Minneapolis, MN, USA), the mouse anti-MMP3 monoclonal antibody was obtained from R&D Systems (cat# MAB513; Minneapolis, MN, USA), and the HRP-conjugated, rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) monoclonal antibody was purchased from Cell Signaling Technology (cat# 3683; Danvers, MA, USA).
Generation of ALDH18A1-KO HEK293T cells and cell culture
ALDH18A1-knockout HEK293 cells were generated by CRISPR-Cas9 editing using a parental HEK293T cell line (Canopy Biosciences, Saint Louis, MO). The clonal knockout line was sequenced and shown to bear a 1 bp insertion in the first allele and a 1 bp deletion in the second allele. Both the parental and KO HEK293 cell lines were shown to have abnormal numbers of certain chromosomes, and a translocation of chromosomes 4 and 10, following conventional karyotyping.
Allele 1: 1 bp deletion of exon 2
CGCAGCATGTTGAGTCAAGTTTACCGCTG -GGGTTCCAGCCCTTCAACCAACATCTTCTG
CGCAGCATGTTGAGTCAAGTTTACCGCTGTGGGTTCCAGCCCTTCAACCAACATCTTCTG
Allele 2: 1 bp insertion of exon 2
CGCAGCATGTTGAGTCAAGTTTACCGCTGGTGGGTTCCAGCCCTTCAACCAACATCTTCT
CGCAGCATGTTGAGTCAAGTTTACCGCTG TGGGTTCCAGCCCTTCAACCAACATCTTCT
Patient fibroblasts were obtained from P2 following consent. All cell lines were maintained in DMEM with 10% fetal bovine serum and penicillin/streptomycin in a humidified incubator with 5% CO2. Monolayers were subcultured every 2–3 days using trypsin/EDTA.
Transfection, gel electrophoresis and western blotting
Transfections in the HEK293 cell system were performed using Lipofectamine Plus. Transfection efficiency was determined for each experiment by immunostaining for the P5CS enzyme and calculating the percentage of cells with detectable P5CS enzyme. For denaturing western blot analysis, 20 μg of the cell lysates prepared in RIPA buffer were separated on a 10% SDS-PAGE gel. For the native gel analysis, cell lysates were prepared using buffer containing 1% NP-40, passed five times through a 21-gauge needle syringe and separated on a 6% PAGE gel using running buffer without SDS. Resolved protein was transferred to 0.45 μm pore nitrocellulose at 110 volts for 2 h at 4°C. The membrane was rinsed and blocked membranes were blocked with 5% milk/TBST for 1 h at room temperature. Blots were incubated overnight with antibodies 1:1000 at 4°C, washed and then incubated with HRP conjugated anti-rabbit (1:2000) for 1 h before washing and developing with SuperSignal West Pico PLUS ECL reagent (ThermoFisher #34577). All images were captured on the Bio-Rad ChemiDoc MP Imaging System (Bio-Rad #12003154). Analysis was done with Image Lab Software (Bio-Rad #1709690, ver5.2.1). For the native gel, NativeMark unstained protein standards (Fisher; cat# LC0725) were used.
NMR-based metabolomics studies
Each plate of adherent HEK293 cells was washed with PBS, scraped and pellets flash-frozen in liquid nitrogen. After transport on dry-ice and just prior to extraction, ice-cold 80% methanol/water extraction solvent was added to pellets. Primary fibroblast culture plates were flash-frozen and transported on dry ice. Due to lower cell density, fibroblasts were scraped from three 10 cm culture plates in ice-cold extraction solvent and combined to create each experimental replicate prior to extraction. Aqueous metabolites were extracted by vortexing/lysing cell pellets in the extraction solvent, pelleting cell debris by centrifugation and collecting the supernatant. 10% of the supernatant was taken from each sample to form an internal pooled sample. The solvent was then evaporated to produce dried extracts using a CentriVap Benchtop Vacuum Concentrator (Labconco, Kansas City, MO, USA). Extracts were reconstituted in a deuterium oxide phosphate buffer (pH 7.4) and kept at 4°C before data acquisition. 1H NMR spectra were acquired on all samples using noesypr1d pulse sequence on a 600 MHz Bruker Avance III HD spectrometer using a 5 mm TCI cryoprobe. Additional 1H-13C heteronuclear single quantum correlation (1H–13C HSQC) and 1H–1H total correlation spectroscopy (1H–1H TOCSY) spectra were acquired on the internal pooled sample and used for metabolite annotation with COLMARm (36). One dimensional 1H NMR spectra were referenced, solvent/water regions removed and normalized with probabilistic quotient normalization (PQN) algorithm (37) using an in-house MATLAB toolbox (github.com/artedison/Edison_Lab_Shared_Metabolomics_UGA). Relative quantification of all spectral features was performed using a semi-automatic workflow within the toolbox. Statistical analysis of these features including PCA, fold-change calculations and ANOVA was performed using Metaboanalyst (38). One-way ANOVA was performed using these features to identify significant features, using Tukey post-hoc test to determine significant comparisons. Non-overlapped features were chosen to represent relative abundance of annotated metabolites, and assigned a confidence score as previously described (39). Pathway enrichment analysis was also performed in Metaboanalyst using hypergeometric test for overrepresentation, mapping to pathways described in small molecule pathway database (SMPD) (40). For pairwise comparisons of p.Thr331Pro versus WT HEK293 and WT versus P2 primary fibroblast cells, a student’s T-test was performed. Metabolites with a raw P-value < 0.05 were included in the list of compounds for pathway enrichment. For integrated transcriptome and metabolome pathway analysis of primary fibroblast cells, fold changes for all annotated metabolites and transcripts between WT and P2 cells, along with lists of metabolites and transcripts with P-value < 0.05 were submitted to Paintomics 3.0 to establish pathway enrichment using Fisher exact test (41).
Polyamine measurements
Intracellular polyamine concentrations were measure by the reverse-phase HPLC methods of Kabra et al. (19) using 1,7-diaminoheptane as an internal standard.
RNA sequencing
To prepare libraries for RNA sequencing, triplicate fibroblast cultures at 80–90% confluence in 10 cm culture dishes were collected directly in 1.5 ml Trizol. Total RNA was extracted using the Direct-Zol miniprep kit RNA extraction kit with a DNA digestion step according to the manufacturer’s instructions (Zymo Research, Irvine, CA). RNA was eluted with 30 μl water. We depleted ribosomal RNA using the Universal Plus Total RNA Seq kit with Human AnyDeplete (Tecan, Männedorf, Switzerland) and prepared bar-coded cDNA libraries for sequencing on an SP flow cell on the NovaSeq 6000 platform (Illumina, San Diego, CA) similar to previously described work (42,43). We performed the initial steps of raw read processing and normalization as previously described except that we used the human reference genome (GRCh38) for alignment (42,43). We used the edgeR package for differential expression analysis and performed Gene Ontology analysis by statistical overrepresentation tests using PantherDB (44–46). The code for all the analyses is available on Github and the data are deposited in GEO (https://github.com/snehamokashi/ALDH_RNAseq and GEO accession number: GSE202424).
Supplementary Material
Acknowledgements
We are grateful to the patients and families for their participation in this study. We thank Bonne Lethco and Dr Barb Dupont at the GGC for assistance with the conventional karyotyping of the WT parental and ALDH18A1 KO cells.
Contributor Information
Maxwell B Colonna, Complex Carbohydrate Research Center, University of Georgia, Athens, GA 30602, USA.
Tonya Moss, Greenwood Genetic Center, Greenwood, SC 29646, USA.
Sneha Mokashi, Greenwood Genetic Center, Greenwood, SC 29646, USA.
Sujata Srikanth, Greenwood Genetic Center, Greenwood, SC 29646, USA.
Julie R Jones, Greenwood Genetic Center, Greenwood, SC 29646, USA.
Jackson R Foley, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins School of Medicine; Baltimore, MD 21287, USA.
Cindy Skinner, Greenwood Genetic Center, Greenwood, SC 29646, USA.
Angie Lichty, Greenwood Genetic Center, Greenwood, SC 29646, USA.
Anthony Kocur, Greenwood Genetic Center, Greenwood, SC 29646, USA.
Tim Wood, Department of Pathology and Laboratory Medicine, Children’s Hospital Colorado, Aurora, CO 80045, USA.
Tracy Murray Stewart, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins School of Medicine; Baltimore, MD 21287, USA.
Robert A Casero Jr., Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins School of Medicine; Baltimore, MD 21287, USA.
Heather Flanagan-Steet, Greenwood Genetic Center, Greenwood, SC 29646, USA.
Arthur S Edison, Complex Carbohydrate Research Center, University of Georgia, Athens, GA 30602, USA.
Michael J Lyons, Greenwood Genetic Center, Greenwood, SC 29646, USA.
Richard Steet, Greenwood Genetic Center, Greenwood, SC 29646, USA.
Conflict of Interest statement
The Greenwood Genetic Center receives revenue from diagnostic testing performed in the GGC Molecular Diagnostic Laboratory.
Funding
Greenwood Genetic Center, the National Science Foundation (NSF 1946970 and 1648035 to A.S.E. and M.C.); and the University of Pennsylvania Orphan Disease Center Million Dollar Bike Ride (MDBR-20-135-SRS) and the Chan Zuckerberg Initiative (to R.A.C./T.M.S.); and the National Cancer Institute (R01 CA235863 to R.A.C.).
Ethics Statement
Informed consent was obtained from the families of the affected individuals involved in this study (Self Regional Healthcare; IRB Number: Pro00085001). This consent included permission to use patient photos in published manuscripts.
References
- 1. Hu, C.A., Khalil, S., Zhaorigetu, S., Liu, Z., Tyler, M., Wan, G. and Valle, D. (2008) Human Delta1-pyrroline-5-carboxylate synthase: function and regulation. Amino Acids, 35, 665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hu, C.A., Delauney, A.J. and Verma, D.P. (1992) A bifunctional enzyme (delta 1-pyrroline-5-carboxylate synthetase) catalyzes the first two steps in proline biosynthesis in plants. Proc. Natl. Acad. Sci. U. S. A., 89, 9354–9358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ginguay, A., Cynober, L., Curis, E. and Nicolis, I. (2017) Ornithine aminotransferase, an important glutamate-metabolizing enzyme at the crossroads of multiple metabolic pathways. Biology (Basel), 6, 18–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Krishnan, N., Dickman, M.B. and Becker, D.F. (2008) Proline modulates the intracellular redox environment and protects mammalian cells against oxidative stress. Free Radic. Biol. Med., 44, 671–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang, Z., Zhao, X., Shang, W., Liu, Y., Ji, J.F., Liu, J.P. and Tong, C. (2021) Pyrroline-5-carboxylate synthase senses cellular stress and modulates metabolism by regulating mitochondrial respiration. Cell Death Differ., 28, 303–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ha, H.C., Sirisoma, N.S., Kuppusamy, P., Zweier, J.L., Woster, P.M. and Casero, R.A., Jr. (1998) The natural polyamine spermine functions directly as a free radical scavenger. Proc. Natl. Acad. Sci. U. S. A., 95, 11140–11145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ha, H.C., Yager, J.D., Woster, P.A. and Casero, R.A., Jr. (1998) Structural specificity of polyamines and polyamine analogues in the protection of DNA from strand breaks induced by reactive oxygen species. Biochem. Biophys. Res. Commun., 244, 298–303. [DOI] [PubMed] [Google Scholar]
- 8. Rider, J.E., Hacker, A., Mackintosh, C.A., Pegg, A.E., Woster, P.M. and Casero, R.A., Jr. (2007) Spermine and spermidine mediate protection against oxidative damage caused by hydrogen peroxide. Amino Acids, 33, 231–240. [DOI] [PubMed] [Google Scholar]
- 9. Kamoun, P., Aral, B. and Saudubray, J.M. (1998) A new inherited metabolic disease: delta1-pyrroline 5-carboxylate synthetase deficiency. Bull. Acad. Natl Med., 182, 131–137 discussion 138-139. [PubMed] [Google Scholar]
- 10. Craze, M.L., Cheung, H., Jewa, N., Coimbra, N.D.M., Soria, D., El-Ansari, R., Aleskandarany, M.A., Wai Cheng, K., Diez-Rodriguez, M., Nolan, C.C. et al. (2018) MYC regulation of glutamine-proline regulatory axis is key in luminal B breast cancer. Br. J. Cancer, 118, 258–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu, W., Le, A., Hancock, C., Lane, A.N., Dang, C.V., Fan, T.W. and Phang, J.M. (2012) Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl. Acad. Sci. U. S. A., 109, 8983–8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Phang, J.M., Liu, W., Hancock, C.N. and Fischer, J.W. (2015) Proline metabolism and cancer: emerging links to glutamine and collagen. Curr. Opin. Clin. Nutr. Metab. Care, 18, 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Coutelier, M., Goizet, C., Durr, A., Habarou, F., Morais, S., Dionne-Laporte, A., Tao, F., Konop, J., Stoll, M. and Charles, P. (2015) Alteration of ornithine metabolism leads to dominant and recessive hereditary spastic paraplegia. Brain, 138, 2191–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Panza, E., Martinelli, D., Magini, P., Dionisi Vici, C. and Seri, M. (2019) Hereditary spastic paraplegia is a common phenotypic finding in ARG1 deficiency, P5CS deficiency and HHH syndrome: Three inborn errors of metabolism caused by alteration of an interconnected pathway of glutamate and urea cycle metabolism. Front. Neurol., 10, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marco-Marin, C., Escamilla-Honrubia, J.M., Llacer, J.L., Seri, M., Panza, E. and Rubio, V. (2020) Delta(1)-pyrroline-5-carboxylate synthetase deficiency: an emergent multifaceted urea cycle-related disorder. J. Inherit. Metab. Dis., 43, 657–670. [DOI] [PubMed] [Google Scholar]
- 16. Fischer-Zirnsak, B., Escande-Beillard, N., Ganesh, J., Tan, Y.X., Al Bughaili, M., Lin, A.E., Sahai, I., Bahena, P., Reichert, S.L., Loh, A. et al. (2015) Recurrent de novo mutations affecting residue Arg138 of pyrroline-5-carboxylate synthase cause a progeroid form of autosomal-dominant cutis laxa. Am. J. Hum. Genet., 97, 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bicknell, L.S., Pitt, J., Aftimos, S., Ramadas, R., Maw, M.A. and Robertson, S.P. (2008) A missense mutation in ALDH18A1, encoding Delta1-pyrroline-5-carboxylate synthase (P5CS), causes an autosomal recessive neurocutaneous syndrome. Eur. J. Hum. Genet., 16, 1176–1186. [DOI] [PubMed] [Google Scholar]
- 18. Panza, E., Escamilla-Honrubia, J.M., Marco-Marin, C., Gougeard, N., De Michele, G., Morra, V.B., Liguori, R., Salviati, L., Donati, M.A., Cusano, R. et al. (2016) ALDH18A1 gene mutations cause dominant spastic paraplegia SPG9: loss of function effect and plausibility of a dominant negative mechanism. Brain, 139, e3. [DOI] [PubMed] [Google Scholar]
- 19. Kabra, P.M., Lee, H.K., Lubich, W.P. and Marton, L.J. (1986) Solid-phase extraction and determination of dansyl derivatives of unconjugated and acetylated polyamines by reversed-phase liquid chromatography: improved separation systems for polyamines in cerebrospinal fluid, urine and tissue. J. Chromatogr., 380, 19–32. [DOI] [PubMed] [Google Scholar]
- 20. Hatamochi, A., Kuroda, K., Shinkai, H., Kohma, H., Oishi, Y. and Inoue, S. (1998) Regulation of matrix metalloproteinase (MMP) expression in cutis laxa fibroblasts: upregulation of MMP-1, MMP-3 and MMP-9 genes but not of the MMP-2 gene. Br. J. Dermatol., 138, 757–762. [DOI] [PubMed] [Google Scholar]
- 21. Hatamochi, A., Mori, K., Arakawa, M., Ueki, H. and Kondo, M. (1996) Collagenase gene expression in cutis laxa fibroblasts is upregulated by transcriptional activation of the promoter gene through a 12-0-tetradecanoyl-phorbol-13-acetate (TPA)-responsive element. J. Invest. Dermatol., 106, 631–636. [DOI] [PubMed] [Google Scholar]
- 22. Tsien, C., Davuluri, G., Singh, D., Allawy, A., Ten Have, G.A., Thapaliya, S., Schulze, J.M., Barnes, D., McCullough, A.J., Engelen, M.P. et al. (2015) Metabolic and molecular responses to leucine-enriched branched chain amino acid supplementation in the skeletal muscle of alcoholic cirrhosis. Hepatology, 61, 2018–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wyant, G.A., Abu-Remaileh, M., Wolfson, R.L., Chen, W.W., Freinkman, E., Danai, L.V., Vander Heiden, M.G. and Sabatini, D.M. (2017) mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell, 171, 642–654.e612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zheng, L., Zhang, W., Zhou, Y., Li, F., Wei, H. and Peng, J. (2016) Recent advances in understanding amino acid sensing mechanisms that regulate mTORC1. Int. J. Mol. Sci., 17, 1636–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Markaki, M., Tsagkari, D. and Tavernarakis, N. (2021) Mitophagy mechanisms in neuronal physiology and pathology during ageing. Biophys. Rev., 13, 955–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sukhorukov, V., Voronkov, D., Baranich, T., Mudzhiri, N., Magnaeva, A. and Illarioshkin, S. (2021) Impaired mitophagy in neurons and glial cells during aging and age-related disorders. Int. J. Mol. Sci., 22, 10251–10264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cheriyan, J., Balsara, R.D., Hansen, K.B. and Castellino, F.J. (2016) Pharmacology of triheteromeric N-methyl-D-aspartate receptors. Neurosci. Lett., 617, 240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hirose, T., Saiki, R., Yoshizawa, Y., Imamura, M., Higashi, K., Ishii, I., Toida, T., Williams, K., Kashiwagi, K. and Igarashi, K. (2015) Spermidine and Ca(2+), but not Na(+), can permeate NMDA receptors consisting of GluN1 and GluN2A or GluN2B in the presence of Mg(2+). Biochem. Biophys. Res. Commun., 463, 1190–1195. [DOI] [PubMed] [Google Scholar]
- 29. Pegg, A.E. (2016) Functions of polyamines in mammals. J. Biol. Chem., 291, 14904–14912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arena, J.F., Schwartz, C., Ouzts, L., Stevenson, R., Miller, M., Garza, J., Nance, M. and Lubs, H. (1996) X-linked mental retardation with thin habitus, osteoporosis, and kyphoscoliosis: linkage to Xp21.3-p22.12. Am. J. Med. Genet., 64, 50–58. [DOI] [PubMed] [Google Scholar]
- 31. Cason, A.L., Ikeguchi, Y., Skinner, C., Wood, T.C., Holden, K.R., Lubs, H.A., Martinez, F., Simensen, R.J., Stevenson, R.E., Pegg, A.E. and Schwartz, C.E. (2003) X-linked spermine synthase gene (SMS) defect: the first polyamine deficiency syndrome. Eur. J. Hum. Genet., 11, 937–944. [DOI] [PubMed] [Google Scholar]
- 32. Bupp, C.P., Schultz, C.R., Uhl, K.L., Rajasekaran, S. and Bachmann, A.S. (2018) Novel de novo pathogenic variant in the ODC1 gene in a girl with developmental delay, alopecia, and dysmorphic features. Am. J. Med. Genet. A, 176, 2548–2553. [DOI] [PubMed] [Google Scholar]
- 33. Peron, A., Spaccini, L., Norris, J., Bova, S.M., Selicorni, A., Weber, G., Wood, T., Schwartz, C.E. and Mastrangelo, M. (2013) Snyder-Robinson syndrome: a novel nonsense mutation in spermine synthase and expansion of the phenotype. Am. J. Med. Genet. A, 161A, 2316–2320. [DOI] [PubMed] [Google Scholar]
- 34. VanSickle, E.A., Michael, J., Bachmann, A.S., Rajasekaran, S., Prokop, J.W., Kuzniecky, R., Hofstede, F.C., Steindl, K., Rauch, A., Lipson, M.H. and Bupp, C.P. (2021) Expanding the phenotype: four new cases and hope for treatment in Bachmann-Bupp syndrome. Am. J. Med. Genet. A, 185, 3485–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Murray Stewart, T., Dunston, T.T., Woster, P.M. and Casero, R.A., Jr. (2018) Polyamine catabolism and oxidative damage. J. Biol. Chem., 293, 18736–18745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bingol, K., Li, D.W., Zhang, B. and Bruschweiler, R. (2016) Comprehensive metabolite identification strategy using multiple two-dimensional NMR spectra of a complex mixture implemented in the COLMARm web server. Anal. Chem., 88, 12411–12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dieterle, F., Ross, A., Schlotterbeck, G. and Senn, H. (2006) Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal. Chem., 78, 4281–4290. [DOI] [PubMed] [Google Scholar]
- 38. Chong, J., Wishart, D.S. and Xia, J. (2019) Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinformatics, 68, e86. [DOI] [PubMed] [Google Scholar]
- 39. Walejko, J.M., Chelliah, A., Keller-Wood, M., Gregg, A. and Edison, A.S. (2018) Global metabolomics of the placenta reveals distinct metabolic profiles between maternal and fetal placental tissues following delivery in non-labored women. Meta, 8, 10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Frolkis, A., Knox, C., Lim, E., Jewison, T., Law, V., Hau, D.D., Liu, P., Gautam, B., Ly, S. and Guo, A.C. (2010) SMPDB: the small molecule pathway database. Nucleic Acids Res., 38, D480–D487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hernandez-de-Diego, R., Tarazona, S., Martinez-Mira, C., Balzano-Nogueira, L., Furio-Tari, P., Pappas, G.J., Jr. and Conesa, A. (2018) PaintOmics 3: a web resource for the pathway analysis and visualization of multi-omics data. Nucleic Acids Res., 46, W503–W509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Johnstun, J.A., Shankar, V., Mokashi, S.S., Sunkara, L.T., Ihearahu, U.E., Lyman, R.L., Mackay, T.F.C. and Anholt, R.R.H. (2021) Functional diversification, redundancy, and epistasis among paralogs of the Drosophila melanogaster Obp50a-d gene cluster. Mol. Biol. Evol., 38, 2030–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mokashi, S.S., Shankar, V., Johnstun, J.A., Huang, W., Mackay, T.F.C. and Anholt, R.R.H. (2021) Systems genetics of single nucleotide polymorphisms at he drosophila Opb56H locus. BioRxiv, 06.28.450219. https://doi.org.10.1101/2021.06.28.450219. [Google Scholar]
- 44. Chen, Y., Lun, A.T. and Smyth, G.K. (2016) From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Res, 5, 1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McCarthy, D.J., Chen, Y. and Smyth, G.K. (2012) Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res., 40, 4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Robinson, M.D., McCarthy, D.J. and Smyth, G.K. (2010) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.