

Neurodegeneration with brain iron accumulation (NBIA) is a diverse collection of neurodegenerative illnesses defined by iron deposition in the basal ganglia. 1 This heterogeneous group of disorders is caused by mutations in 1 of the 13 identified NBIA genes (the different symptoms and magnetic resonance imaging [MRI] findings of these subtypes are described in Table 1). 2 NBIAs are characterized by gait abnormalities and clumsiness in childhood as well as spasticity, dystonia, and gradual cognitive and intellectual disability. 1 , 3 Diagnosis of these disorders is based on the patient's history and symptoms and evidence of iron deposition on brain MRI, 1 , 3 with the precise diagnosis verified by genetic study including whole exome sequencing (WES). 4 , 5 , 6 COASY protein associated neurodegeneration, or CoPAN, a new subtype of NBIA discovered in recent years, results from mutations in the Coenzyme A synthase gene (COASY). 6 , 7 In this article, we present the clinical manifestations and imaging of a known variant in the COASY gene, which is the first reported from Iran. The patient is a 24‐year‐old man who was born to healthy consanguineous parents. The prenatal and perinatal histories were found to be unremarkable, with normal physical and intellectual development during infancy. At the age of 2 years, his parents observed speech problems as well as delays in language development. He also had gait disturbance with imbalance and frequent falls. He was unable to attend regular schools owing to cognitive impairment, which was confirmed by standard neurologic examination despite being able to perform everyday tasks. There was no history of visual or hearing problems and no history of seizures. At the time of examination, speech appeared to be normal, in contrast with the history obtained from the parents. There were no abnormalities on examination of eye movements, and fundoscopy revealed no pathological finding. Further examination of the limbs showed mild hypokinesia, with mild dystonic posturing of the legs while walking (Video 1). Cerebellar testing, sensory exam, and muscle stretch reflexes were all normal, and plantar reflexes were bilaterally downward.
TABLE 1.
Summary of the differences between the various NBIA subtypes
| NBIA Subtype | Gene | Inheritance | Average Age of Onset | Symptoms | MRI Findings |
|---|---|---|---|---|---|
| PKAN | PANK2 | Autosomal recessive 5 |
3 years old (classic form) 18 years old (atypical form) 3 |
Classic form: ataxia and gait disturbances, cognitive impairment, and parkinsonism Atypical form: dystonia, dysarthria, and psychiatric symptoms 3 |
T2 hypointense GP with region of hyperintensity termed “eye of the tiger” 5 , 6 |
| PLAN | PLA2G6 | Autosomal recessive 5 |
6 months to 3 years (infantile onset) After 3 years old (childhood onset) 20 to 40 years old (adulthood onset) 3 |
Infantile: hypotonia, spastic tetraparesis, and seizures Childhood: dystonia, spasticity, ataxia, delayed speech, and autistic symptoms Adulthood: dystonia and mild cognitive impairment 3 |
Cerebellar atrophy, diffuse T2 hyperintensity in white matter, thinning of the optic chiasma and corpus callosum, hypointense GP late in disease progression 5 , 6 |
| MPAN | C19orf12 | Autosomal recessive 5 | Usually a childhood onset at 9 years old 3 | Dystonia, spasticity, gait abnormalities, and psychiatric symptoms 3 | T2 hypointense signaling in the GP and SN, cerebellar and cortical atrophy, T1 hyperintensity of caudate and putamen 5 , 6 |
| BPAN | WDR45 | X‐linked dominant 5 | Usually childhood onset 3 | Seizures, cognitive impairment, and ataxia 3 | T2 hypointense signaling in the GP and SN and cerebellar and cerebral atrophy 5 , 6 |
| FAHN | FA2H | Autosomal recessive 5 | Usually childhood onset at 4 years old 8 | Ataxic spastic gait, seizures, dystonia, and cognitive impairment 3 | T2 hypointense signaling of the GP and SN 5 , 6 |
| CoPAN | COASY | Autosomal recessive 5 | Usually childhood onset at 2.5 years old 8 | Gait disturbances and mild cognitive impairment with progressive dysarthria and dystonia 3 | T2 hypointense signaling of the GP and SN 5 , 6 |
| Aceruloplasminemia | CP | Autosomal recessive 5 | Usually mid‐ to late‐adulthood onset at 51 years old 3 , 8 | Ataxia, dystonia, and chorea 3 | T2 hypointense signaling of the GP, dentate, thalamus, and red nucleus 6 |
| Neuroferritinopathy | FTL | Autosomal dominant 5 | Usually adult onset at 40 years old 3 | Dystonia, ataxia, areflexia, dysarthria, parkinsonism, and cognitive impairment 3 | T2 hypointense signaling of the GP, SN, putamen, caudate, and thalamus 6 |
| Kufor‐Rakeb syndrome | ATP13A2 | Autosomal recessive 5 | Usually occurring before 20 years old 3 | Parkinsonism, spasticity, and cognitive impairment 3 , 5 | T2 hypointensity of the GP, caudate, and putamen 6 |
| Woodhouse‐Sakati syndrome | DCAF17 | Autosomal recessive 5 | Variable onset of juvenile to adulthood 8 | Dysarthria and sensorineural hearing loss 3 , 5 | T2 hypointensity in the GP 6 |
Abbreviations: PKAN; pantothenate kinase‐associated neurodegeneration, PLAN; PLA2G6‐associated neurodegeneration, MPAN; mitochondrial membrane protein‐associated neurodegeneration, BPAN; Beta‐propeller Protein Associated Neurodegeneration, FAHN; Fatty acid hydroxylase‐associated neurodegeneration, CoPAN, COASY protein associated neurodegeneration; GP, globus pallidus; MRI, magnetic resonance imaging; NBIA, neurodegeneration with brain iron accumulation; SN, substantia nigra;
Video 1.
This video shows mild hypokinesia and mild dystonic posture of the feet on walking.
Brain MRI revealed hypointensity of both the globi pallidi and substantia nigra on T2, fluid‐attenuated inversion recovery, and susceptibility weighted imaging sequences (Fig. 1). WES was performed on the DNA of the patient, and a known homozygous variant c.1495C>T: p.Arg499Cys in the COASY gene (NM_025233; rs140709867) was revealed. The variant was only found in the heterozygous state in GnomAD (https://gnomad.broadinstitute.org/) with a minor allele frequency 0.000014. The variant was not detected in other genome databases, including the Iranome (www.iranome.com) with WES data of 800 healthy Iranian individuals. The variant was predicted as a disease causing/damaging variant by several in silico prediction tools, such as Sorting Intolerant From Tolerant (SIFT) (https://sift.bii.a-star.edu.sg/), Polyphen2 (http://genetics.bwh.harvard.edu/pph2/), Protein Variation Effect Analyzer (PROVEAN) (https://bio.tools/provean), Mutation Taster (https://www.mutationtaster.org/), and Mutation Assessor (http://mutationassessor.org/r3/). The variant was cosegregated with the disease status in the family members and predicted as a likely pathogenic variant based on the American College of Medical Genetics and Genomics criteria. The detected variant causes CoPAN as a rare subtype of NBIAs. No other mutations or NBIA disease‐causing genes, including ATP13A2, C19orf12, CP, DCAF17, FA2H, FTL, PANK2, PLA2G6, and WDR45, were detected using WES, so we concluded the variant is the cause of disease and results in CoPAN. As treatment, we prescribed levodopa carbidopa 100/10 mg three times a day and amantadine 100 mg two times a day, which were partially effective in reducing the dystonia and hypokinesia.
FIG. 1.
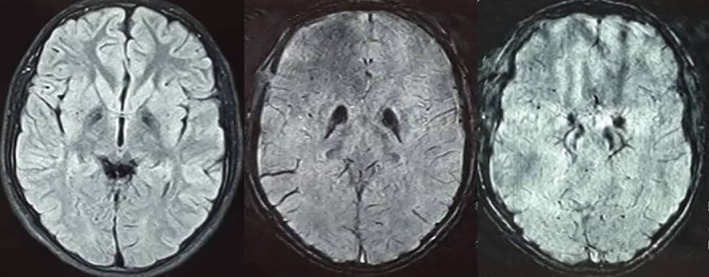
Brain magnetic resonance imaging. Fluid‐attenuated inversion recovery sequence (right) and susceptibility weighted imaging images (middle and left) showing iron deposition in the globus pallidus and substantia nigra.
CoPAN is a rare form of NBIA caused by an autosomal recessive inherited mutation of the CoA‐synthase enzyme located on 17q21, with 5 confirmed mutations documented. 1 , 3 , 4 , 7 , 8 This bifunctional protein synthesizes CoA from pantothenic acid by catalyzing the last 2 steps of its pathway and is located downstream of pantothenate kinase‐associated neurodegeneration 2 (PKAN2), which performs the first step in this pathway. 1 , 4 , 7 , 8 Previous studies showed that mutations in this gene result in a significant reduction of the COASY protein and subsequent CoA and acetyl–CoA biosynthesis. 3 , 7 As the pathogenic pathway for CoPAN is similar to that of PKAN, the possible etiologies might also be similar. For PKAN, it is hypothesized that the accumulation of cysteine as the precursor for CoA might be responsible for neurodegeneration through rapid oxidization and the release of free radicals in the presence of iron. 6 , 7 Reports have shown that symptoms of CoPAN start from the first decade, with progressive early childhood‐onset dystonia, spasticity, and gait abnormalities along with mild cognitive impairment. 1 , 4 , 6 , 7 Psychiatric symptoms can also be seen in CoPAN manifesting as obsessive‐compulsive disorder. 1 , 4 In the history taken from this patient, cognitive impairment and frequent falling were noticed in early childhood, and although at the time of examination no speech defects or dysarthria was noticed, the parents revealed a history of speech problems in his early childhood. Similar to other NBIAs, the globus pallidus and substantia nigra are affected in CoPAN, with brain MRI showing hypointense signaling on T2‐weighted images, 1 , 3 , 4 , 5 , 6 , 9 as was seen in this patient. Genetic testing using WES revealed a homozygous variant c.1495C>T:p.R499C in the COASY gene that has been reported by Dusi et al. 7 Due to the nonspecific symptoms and MRI findings, the diagnosis is based on genetic study. Aside from the typical NBIA findings in a brain MRI, CoPAN can also be seen associated with T2‐weighted hyperintensities of the caudate and thalamus, which were not seen in this case. It is important to note that these hyperintensities and swelling of the striatum and thalamus may regress over time, subsequently being replaced with the more identifiable pallidal iron deposits. These changes typically occur between the ages of 7 and 9 years, which could explain why signal changes were not detected in the striatum and thalamus in our older patient. Bilateral pallidal changes with calcifications can also mimic the imaging pattern seen in PKAN referred to as “eye of the tiger.” 10 In summary, this is the first case of CoPAN from Iran with a known mutation with typical symptoms and brain MRI.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Manuscript: A. Writing of the First Draft, B. Review and Critique.
N.H.: 1C, 2A
R.N.S.A.: 2A
A.A.: 2C
A.R.T.: 1B, 2B
M.R.: 1A, 1B, 2B
Disclosures
Ethical Compliance Statement: The authors confirm that the approval of an institutional review board was not required for this work. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines. The patient's father signed the consent for publishing the video. Informed consent was obtained from the patient's family.
Funding Sources and Conflicts of Interest: The authors declare that there are no funding sources or conflicts of interest relevant to this work.
Financial Disclosures for the Previous 12 Months: The authors declare that there are no additional disclosures to report.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Tonekaboni SH, Mollamohammadi M. Neurodegeneration with brain iron accumulation: An overview. Iran J child Neurol 2014;8(4):1–8. [PMC free article] [PubMed] [Google Scholar]
- 2. Kolarova H, Tan J, Strom TM, Meitinger T, Wagner M, Klopstock T. Lifetime risk of autosomal recessive neurodegeneration with brain iron accumulation (NBIA) disorders calculated from genetic databases. EBioMedicine 2022;77:103869. 10.1016/j.ebiom.2022.103869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hajati R, Emamikhah M, Danaee Fard F, Rohani M, Alavi A. Neurodegeneration with brain iron accumulation and a brief report of the disease in Iran. Can J Neurol Sci 2021;49:338–351. 10.1017/cjn.2021.124. [DOI] [PubMed] [Google Scholar]
- 4. Hayflick SJ, Kurian MA, Hogarth P. Neurodegeneration with brain iron accumulation. Handb Clin Neurol 2018;147:293–305. 10.1016/B978-0-444-63233-3.00019-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hogarth P. Neurodegeneration with brain iron accumulation: Diagnosis and management. J Mov Disord 2015;8(1):1–13. 10.14802/jmd.14034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Evers C, Seitz A, Assmann B, et al. Diagnosis of CoPAN by whole exome sequencing: Waking up a sleeping tiger's eye. Am J Med Genet A 2017;173(7):1878–1886. 10.1002/ajmg.a.38252. [DOI] [PubMed] [Google Scholar]
- 7. Dusi S, Valletta L, Haack TB, et al. Exome sequence reveals mutations in CoA synthase as a cause of neurodegeneration with brain iron accumulation. Am J Hum Genet 2014;94(1):11–22. 10.1016/j.ajhg.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arber CE, Li A, Houlden H, Wray S. Review: Insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: Unifying theories. Neuropathol Appl Neurobiol 2016;42(3):220–241. 10.1111/nan.12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ma K, Mcneill A, Lin J‐P, Er M. Childhood disorders of neurodegeneration with brain iron accumulation (NBIA). Dev Med Child Neurol 2011;53(5):394–404. 10.1111/j.1469-8749.2011.03955.x. [DOI] [PubMed] [Google Scholar]
- 10. Lehéricy S, Roze E, Goizet C, Mochel F. MRI of neurodegeneration with brain iron accumulation. Curr Opin Neurol 2020;33(4):462–473. [DOI] [PubMed] [Google Scholar]