

Abstract
Lung cancer is the primary cause of mortality in the United States and around the globe. Therapeutic options for lung cancer treatment include surgery, radiation therapy, chemotherapy, and targeted drug therapy. Medical management is often associated with the development of treatment resistance leading to relapse. Immunotherapy is profoundly altering the approach to cancer treatment owing to its tolerable safety profile, sustained therapeutic response due to immunological memory generation, and effectiveness across a broad patient population. Different tumor-specific vaccination strategies are gaining ground in the treatment of lung cancer. Recent advances in adoptive cell therapy (CAR T, TCR, TIL), the associated clinical trials on lung cancer, and associated hurdles are discussed in this review. Recent trials on lung cancer patients (without a targetable oncogenic driver alteration) reveal significant and sustained responses when treated with programmed death-1/programmed death-ligand 1 (PD-1/PD-L1) checkpoint blockade immunotherapies. Accumulating evidence indicates that a loss of effective anti-tumor immunity is associated with lung tumor evolution. Therapeutic cancer vaccines combined with immune checkpoint inhibitors (ICI) can achieve better therapeutic effects. To this end, the present article encompasses a detailed overview of the recent developments in the immunotherapeutic landscape in targeting small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). Additionally, the review also explores the implication of nanomedicine in lung cancer immunotherapy as well as the combinatorial application of traditional therapy along with immunotherapy regimens. Finally, ongoing clinical trials, significant obstacles, and the future outlook of this treatment strategy are also highlighted to boost further research in the field.
Keywords: Lung Cancer, SCLC, NSCLC, Immunotherapy, Nanomedicine, Cancer Vaccine, Antibody, Adaptive cell therapy, CAR T therapy, TCR T therapy, TIL therapy, Immunomodulators
Introduction
Globally, cancer incidence and death are rising, with lung cancer being the most commonly diagnosed form of cancer (11.6% of the total cases). In the United States, in 2022, there are expected to be ~ 236,740 new lung cancer cases, with ~ 130,180 human demise [1]. Lung cancer remains the leading cause of global cancer mortalities (18.4% of total cancer fatalities), causing significant societal burden and economic loss [1, 2]. Around 80% of lung cancer deaths are caused by smoking. Other risk factors for lung cancer include radon, asbestos, long-term and cumulative exposure to air pollution, especially polycyclic aromatic hydrocarbons (PAH) emissions, and personal or familial lung cancer history [3, 4]. Lung tumors are divided into two broad categories by the World Health Organization (WHO); non-small cell lung cancer (NSCLC), comprising 80–85% of all lung cancer cases, and small cell lung cancer (SCLC), constituting the other 15% incidences [5–7]. NSCLC can be further subcategorized into adenocarcinoma (LUAD), squamous cell carcinoma (LUSC), and large cell carcinoma (LCC). Each subcategory based on the molecular targetable genetic profile can be subcategorized into several types [8]. It turns out that the survival rates for metastatic lung cancer of both NSCLC and SCLC types are poor, with a 5-year survival of only about 4% [9, 10].
Although several anti-cancer strategies like surgery, chemotherapy, and irradiation are used to treat NSCLC and SCLC, there is an urgent need for effective strategies to cure or manage lung cancer, particularly late-stage cancers [11]. The prognosis of NSCLC is challenging due to the unavailability of a platform for early-stage diagnosis and the late appearance of symptoms in disease development, limiting treatment choices and survival [12]. Low-dose computed tomography (LDCT) is the gold standard for current lung cancer patient screening. So far, in the USA, only 5% of the 15 million high-risk individuals advised for screening have used LDCT. LDCT suffers from low early detection efficiency, false-positive detection, radiation hazard, and unavailability of resources for running an efficient CT-based screening program [13]. Though early detection increases the likelihood of tumor resection, treatment, and a successful outcome, the unavailability of an appropriate screening platform, metastatic nature, genetic heterogeneity, and minimal response to chemotherapy at late stages make lung cancer fatal [14]. However, chemotherapy and radiation are recommended (including neoadjuvant and/or adjuvant therapy) for locally advanced and metastatic cancers but have shown limited overall survival (OS) and toxic side effects. Targeted therapies along with chemotherapy have become standard therapies for NSCLC patients with actionable oncogenic alterations (driver mutations and fusions/rearrangements), resulting in increased progression-free survival (PFS) and the OS in several cases. Targeted therapies have differing side effect profiles compared to chemotherapy and may not necessarily have sustained treatment responses [15, 16].
SCLC is classified based on the extension of the disease into a limited disease SCLC (LD-SCLC) and an extensive disease SCLC (ED-SCLC). Although new chemotherapeutic agents are being continuously formulated, the prognosis remains poor due to aggressive progression, lack of early detection techniques, limited treatment options, and efficacy [16, 17]. For LD-SCLC, a standard strategy is chemotherapy (cisplatin or carboplatin with etoposide) combined with thoracic radiotherapy [18]. SCLC initially responds well to chemotherapy and radiation but often relapses, leading to poor survival. The median survival (MS) rate for this group of patients is approximately 7–12 months due to limited early detection modalities, dearth of tissue availability for clinical research, tumor genetic heterogeneity, and poor understanding of molecular mechanisms leading to rapid progression and therapeutic resistance [19, 20]. Clinical studies of new drugs and targeted molecular treatment for SCLC have shown limited, encouraging results [5, 21]. Hence, there is a pressing need for a new treatment modality with a persistent response.
Recent research has refined our understanding of the immune system's reaction to cancer and how to enhance it, leading to considerable improvements in cancer immunotherapy [22]. Immunotherapy possesses potential efficacy irrespective of the histology and driver mutational status, leading to sustained remission, especially for those patients who exhibit a response [23]. The goal of cancer immunotherapy is to elicit (or re-elicit) a cellular immune response, especially the T-cell-mediated tumor-specific antigen (TSA) and tumor-associated antigens (TAA)-directed cytotoxicity that can selectively destroy a tumor [24]. The immune-modulatory drugs can also counter cancer cells by increasing the concentration of tumor-specific antibodies, natural killer (NK) cells, dendritic cells (DCs), macrophages (MΦ), and cytokines in the blood plasma [25]. However, in the past few years, immunotherapy has been considered inapt for lung cancer due to minimal immune responses [26]. Lung cancer immunotherapy is challenging as the cells avoid immunosurveillance and reduce the overall immunological response by modulating the T-cell mediated cytotoxicity, secretion of immune-suppressive cytokines, and loss of major histocompatibility complex (MHC) expression [27]. Recent technical advances have helped determine the molecular granularity of lung cancer immunogenicity, and since then various types of immunotherapies have evolved for treating lung cancer. Immunotherapy treatment types include therapeutic vaccines, immune modulators, autologous cellular therapies, and monoclonal antibodies (mAbs) directed against checkpoint inhibitor signals associated with activated T-cells and/or with cancer cells. However, since each therapeutic approach has distinct advantages and disadvantages, combining multiple therapies or therapeutic strategies with immunotherapy is preferable [28]. The present article examines recent advances in lung cancer (NSCLC and SCLC) immunotherapy, continuing clinical studies of immunotherapeutic interventions, and future directions.
NSCLC and immunotherapy
LUAD is the most prevalent NSCLC, especially in the USA, accounts for around 40% of all lung cancer, and occurs in smokers and non-smokers regardless of their age and sex [29]. LUAD arises from the glandular cells of the alveoli (tiny air sacs) and tends to occur in the peripheral regions of the lung [30]. Due to its slow development rate than other types of lung cancer, it is more likely to be detected before it metastasizes beyond the lungs [31, 32]. LUSC is the second most common type of lung cancer, accounting for 25–30% of all lung cancer occurrences. LUSC is connected with smoking more than any other kind of NSCLC and is characterized by recurring somatically altered genes and pathways linked to smoking [33, 34]. Tracheobronchial squamous cells, particularly the basal cells, often give birth to squamous cell lung tumors, which are found mostly in the central part of the lung (the major airways) but may also occur peripherally [33, 34]. The third type, LCC accounts for approximately 5–10% of lung cancers and are also associated with smoking [35]. LCC generally shows no evidence of squamous or glandular maturation and remain undifferentiated, and as a result, it is often diagnosed through the exclusion of other possibilities. They habitually begin from the central part of the lungs, spread quickly, sometimes invading nearby lymph nodes, have chest wall involvement, and metastasize to distant organs [35, 36]. The lung cancer staging project by the International Association for the study of Lung Cancer (IASLC) revealed that patients were more likely to survive if diagnosed and treated in the early pathological stage with an MS of 95 months for stage IA, 75 months for stage IB, 44 months for stage IIA, 29 months for stage IIB and 19 months for stage IIIA. Also, a considerable influence factor on OS was the subtype of tumor cells [83 months for Bronchoalveolar carcinoma (uncommon type of LUAD), 45 months for LUAD, 44 months for LUSC, 34 months for LCC, and 26 months for Adenosquamous carcinoma] [37]. However, with the recent awareness about smoking cessation and improvements in early diagnosis and treatment, mortality rates of lung cancer have steadily dropped during the last two decades [38]. Immunotherapy is one such treatment advancement that has impacted patient survival in lung cancer, especially NSCLC. In this regard, understanding and accumulation of know-how about the immune mechanisms, driver mutations, neoantigens, and oncogenic pathways involved in NSCLC have brought about more clarity regarding the heterogeneity of tumor, mutational burden, and tumor microenvironment (TME), which has aided in designing new immunotherapeutic tools for targetable mutations [39].
The lung cancer genome is characterized by a unique mutational landscape. Specific oncogenic mutations confer a dominant gain of function and recessive loss of function mutations in tumor suppressor genes. Somatic mutations, homozygous gene deletions, gene amplifications, gene translocations, and epigenetic silencing may cause genomic changes and alterations in specific pathways leading to the transformation of normal cells to premalignant cells and finally into lung tumors [40]. Lung cancer genetic profiling indicated considerable patient heterogeneity. It has been possible to identify several oncogenes and tumor suppressor genes (Fig. 1). KRAS, ALK, c-MET, RET, BRAF V600E, ROS1, NTRK, TP53, and ERBB2 (HER2) [40] are among the actionable genetic changes found in NSCLC. Genomic alteration-associated generation of tumor-specific antigens or neoantigens expressed by the premalignant/tumor cells, following antigen-presenting cell (APC)-mediated antigen presentation, can activate the T-cell specific adaptive antitumor immune response. The three-signal activation dogma governs classical T-cell activation. APCs display antigenic peptides on MHC I molecules to naïve T cells via their cognate T-cell receptor (TCR) (signal 1). A positive costimulatory signal, termed signal 2 (interaction of DC-specific CD80/86 and T cells-specific CD28 receptor), is essential for T cell activation. DCs further secrete pro-inflammatory cytokines (signal 3) to induce T cells toward antigen-specific antitumor response (Fig. 2A) [40]. These primed and activated effector T cells can infiltrate lung TME and effects tumor cell killing. When placed in the context of the inflammatory milieu of the tumor, signal 3 may help clarify the connection between chronic inflammation and lung cancer. The expression of the immune checkpoints is linked to several of the genetic modifications, like TP53, KRAS, and STK11 gene mutations. The details of the genetic underpinnings of lung cancer are covered in other reviews and chapters [40, 41].
Fig. 1.

Genetic profiling of lung cancer (SCLC, LUAD, and LUSC) have shown changes in several oncogenes and tumor suppressor genes. Based on the total of somatic mutations, homozygous deletions, localized amplification, and substantial changes in gene expression, the values in each box represent the rates of genomic abnormalities. Other crucial proteins that mediate the pathways are also discussed. EGF, epidermal growth factor; FGF, fibroblast growth factor; GF, growth factor; DLL, deltalike; EGF, epidermal growth factor; FGF, fibroblast growth factor; GF, growth factor; HGF, hepatocyte growth factor; NRG, neuregulin; RTK, receptor tyrosine kinase. Figure reproduced with permission from Reference 39
Fig. 2.
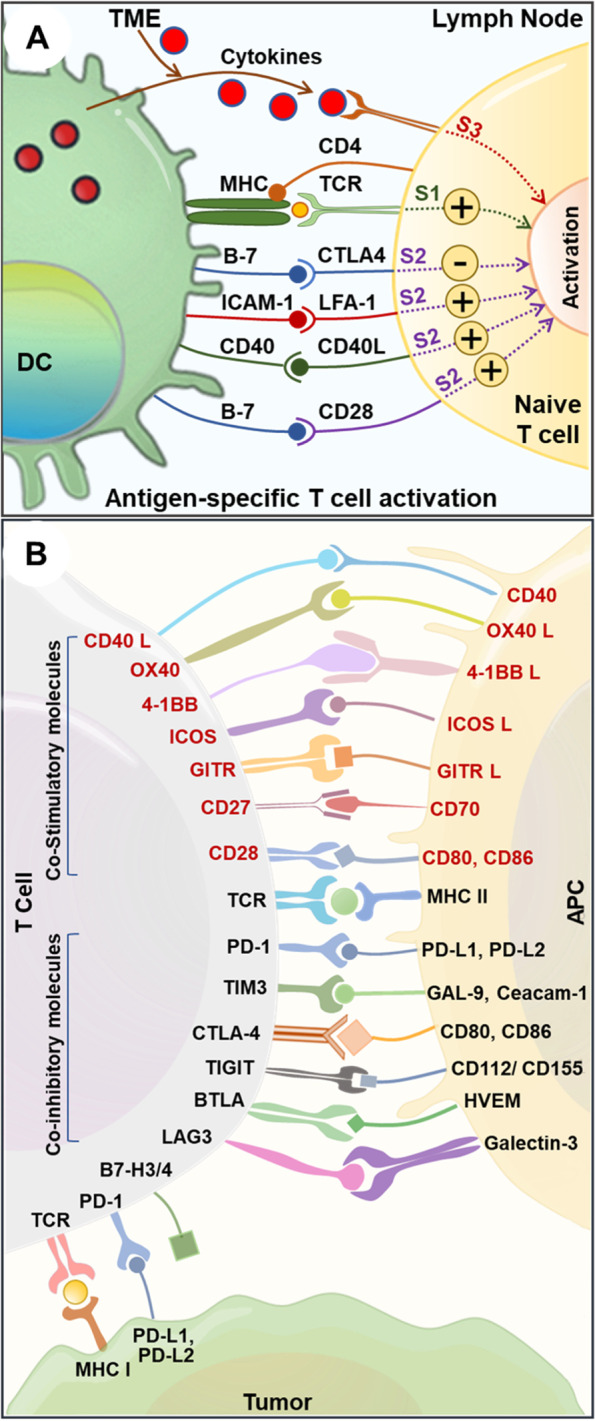
Immune interaction between T-cells, APCs, and cancer cells A. Schematic representing the mechanism of antigen-specific T cell activation. DCs play a crucial role in anti-tumor immunity due to their exceptional capacity to activate T cells following the central dogma of three signals. S1: Signal 1; S2: Signal 2; S3: Signal 3; TME: Tumor microenvironment; DC: Dendritic cell. B. Schematic showing immune interaction between T cell and APC; T cell with tumor cells. The T cell co-inhibitory and co-stimulatory molecules are shown in black and red fonts respectively. APC: Antigen presenting cell, L: Ligand
Lung cancer immunotherapy, which aids the immune system in identifying and eliminating cancer cells, has gotten much attention lately. The nature of the interaction of tumor cells with the immune cells in the TME defines the antitumor response. Recent studies exemplify a dichotomous role of the immune cells during lung tumor evolution and can either promote an anti-tumor response or modulate an immunosuppressive (pro-tumorigenic) TME [42, 43]. TME is a complex signal interaction space surrounding the tumor, constituted by the endothelial cells, stromal fibroblast, mesenchymal cells, adipocytes, immune cells, and the extracellular matrix. The composition and the pathological significance of the tumor immune microenvironment (TIME) have been a topic of intense investigation in the last decade. The discovery of immune checkpoints (ICP), which are proteins produced by some immune cells (like T cells) and cancer cells, is an unprecedented moment in the history of immunotherapy. Under normal physiological conditions, the ICPs bind with their complementary companion proteins (receptor-ligand interaction), activate inhibitory signals, turn off T cell response, and.
thereby preventing an indiscriminate attack on healthy cells. They are crucial for self-tolerance, normal regulation of the immune system, and immunostasis. Tumor cells use this crucial regulatory process to their advantage and express ICP proteins to evade immune cell-mediated tumor cell death. Targeting the immune checkpoint using checkpoint inhibitors (CKI) can lead to long-term clinical response and cancer cure. Since the discovery of CTLA-4, several ICPs have been discovered, including programmed death-1 (PD-1), T-cell immunoglobulin domain and mucin domain-containing molecule-3 (TIM-3), T-cell immunoglobulin and ITIM domain (TIGIT), B and T cell lymphocyte attenuator (BTLA), lymphocyte activation gene (LAG3), V-domain Ig suppressor of T cell activation (VISTA), and Cluster of Differentiation 200 (CD200) (Fig. 2B) [40].
Interaction of PD1 (expressed on effector T cells) with PD-L1 (expressed by tumor cells and TME-associated myeloid cells) acts as an inhibitory signal and causes effector T cell exhaustion. While CTLA-4 is upregulated in activated T cells and competes with the co-stimulatory CD80/86 expressed on APCs, thereby negatively affecting T cell activation and function. While PD-1 and CTLA-4 are the most studied ICPs, other ICPs may be effective. Tumor cell-expressed ligands (CD155, CD112) binds to TIGIT and impact T cell- and NK-cell-mediated tumor recognition. TIM3 and LAG3 inactivate T cell function and induce exhaustion (Fig. 2B) [15, 40]. A schematic showing immune interaction between T cell and APC; T cell with tumor cells is shown in Fig. 2B. Loss of CD4 + T cells and an increase in the expression of inhibitory receptors such as CD160, CD244, CTLA4, LAG-3, PD1, TIGIT, and TIM3, leads to a rapid decline in T cell effector activity. Advanced technologies have provided a comprehensive understanding of the complexity of the tumor-immune interactions. By parsing the distinct type of tumor-specific TIME, immunotherapeutic responsiveness may be predicted, and novel therapeutic targets can be identified for developing successful therapies.
CKIs as a therapy for advanced lung cancer have lately gained traction. U.S. Food and Drug Administration (FDA) in 2015 approved Nivolumab (blocks PD-1) for the treatment of LUSC (and subsequently for all NSCLC histological types) after the first-line treatment with platinum doublet chemotherapy had failed. Under normal conditions, the immune checkpoint receptor programmed cell death-1 (PD-1) is expressed on activated T cells. PD-1 inhibits immunological responses from being overstimulated, while its ligand, PD-L1, is expressed on immune cells and tumor cells. The PD-1/PD-L1 pathway interaction contributes significantly to tumor immune evasion. The anti-tumor immunity mediated by T cells is resurrected by inhibiting their connection, offering a survival advantage in various advanced, resistant cancers. For patients with PD-L1 positive cancers who had progressed after chemotherapy, Pembrolizumab was approved in 2015. In October 2016, the FDA authorized pembrolizumab as first-line therapy for patients with high (≥ 50%) PD-L1 expression. Atezolizumab was approved for use in patients with advanced NSCLC who had progressed after chemotherapy. Research on the role of the immune system in the treatment and prevention of cancer has been substantial. Immunotherapy is now a trendy topic, thanks to a flurry of FDA approvals. Immunotherapy encompasses cancer vaccines, MAbs, and adoptive cell transfer in addition to checkpoint inhibitors and will be discussed in further detail in the subsequent sections.
Tumor-specific vaccines
For long, vaccines have been old arsenals in medicine, primarily used to prevent the onset and spread of infectious disease, and to a smaller degree have been applied in oncology. The vaccines aim to promote antigen-specific immune responses in a patient by presenting TAAs to the individual’s immune system in the cancer environment [44]. Vaccine therapy aims to initiate or amplify adaptive anti-tumor immune responses by introducing tumor antigens to stimulate the host immune system to generate tumor antigen-specific effector and memory T-cell-based responses and not target non-malignant cells [45–47]. Vaccines targeting NSCLC have been investigated in several phase III trials throughout the last decade. Although they had a favorable toxicity profile and tolerability, almost all of them could not demonstrate survival advantages despite encouraging results in the preliminary phase II randomized trials. Tumor vaccination faces multiple challenges, and addressing them can lead to the path of therapeutic translation. Cancer vaccines suffer from limited penetrability in the tumor, wayning of the immuneresponse over time, and resistance. Multi-target vaccines generated against immunogenicity-optimized epitopes may address some of these challenges. Therefore, a greater knowledge of immune evasion mechanisms, designing effective formulations, and combination immunotherapy approaches (targeting TME and tumor cell-derived factors) can promote the development of the subsequent generation of cancer vaccines. The currently investigated vaccines, classified broadly into antigen-specific vaccines (peptide /protein vaccines, DNA vaccines, and vector-based vaccines) or whole-cell vaccines (allogeneic vaccines and autologous dendritic cell vaccines), are discussed in brief below (Fig. 3A).
Fig. 3.

Different aspects of lung cancer immunotherapy. A: Lung cancer immunotherapy by using a tumor-specific vaccine to combat cancer. B: Donor or patient T cells are collected in vitro, followed by the introduction of Chimeric Antigen Receptor (CAR) receptors and mass-produced in the lab to combat cancer. Following infusion back to the patient, the CAR T-cells attack the patient's tumor. C: Oncolytic virus and lung cancer cell oncolysis. D: Monoclonal antibodies (mAbs) may be effective against lung cancer by targeting a specific section of the cancer cell
Peptide/protein vaccines
The few protein-specific vaccines used in NSCLC are the CIMAvax epidermal growth factor (CIMAvax-EGF) vaccine, MAGE-A3, NY-ESO-1 and the BLP25 liposome vaccine (anti-MUC1). The CIMAvax-EGF, developed in Cuba, is a chemical conjugation of EGF with the P64 protein obtained from Meningitis B bacteria and the incomplete Freund's adjuvant Montanide ISA 51 [48]. The vaccine induces immune responses specifically against EGF, a molecular driver of cancer cells, aiming to block their proliferation. Its use is currently approved in the countries like Cuba, Peru, and Venezuela for treating stage IIIB and IV NSCLC patients who have progressed beyond the first line of chemotherapy. The CIMAvax-EGF was shown to be safe and immunogenic in patients with advanced NSCLC in a phase II randomized controlled study including 80 stage IIIB/IV NSCLC patients who had received a first-line chemotherapy [49]. Promising anti-EGF antibody response was documented in 51.3% of the vaccinated patients, and they survived significantly longer (11.7 months MS) than those that showed poor antibody response (3.6 months MS). Adverse events were recorded in fewer than a quarter of grades 1 and 2 patients. Subsequently, a phase III study was published in August 2016, showing the results of OS, safety, immunogenicity, and serum EGF concentration of 405 stage IIIB/IV NSCLC patients post-CIMAvax-EGF vaccination [50]. After completion of the first line of chemotherapy, patients were randomly assigned at a ratio of 2:1 for the vaccine with best supportive care (BSC) or to the control group. The survival was statistically higher (HR, 0.77; P = 0.036) in the treatment arm with an MS of 12.4 months for the vaccinated group contrasted with 9.4 months for the control patients. In January 2017, a new randomized phase I/II clinical trial against NSCLC with CIMAvax-EGF combined with the MAB Nivolumab (NCT02955290) began, for which the results are awaited.
Another type of protein targeting tumor vaccines is the ones that targets the cancer testis antigens (CTA) that include the New York oesophageal squamous cell cancer (NY-ESO-1) and the melanoma-associated antigen-A3 (MAGE-A3) antigens in case of NSCLC. The normal expression of CTA is primarily found in the male germ cells in the testis and rarely in the female ovary and trophoblast, while in some cases, due to genetic mutations, they become upregulated in a proportion of different malignant tumor types [51]. The MAGE was the first CTA to be identified, and its expression is evident in almost 30%—50% of NSCLC patients, especially in LUSC incidences [52, 53]. The phase III MAGRIT study evaluated the safety of a recombinant adjuvanted MAGE-A3 in patients with resected MAGE-A3 positive NSCLC. The trial included 2,312 patients with resected stage IB, IIA, or IIIA NSCLC. The vaccine comprises a recombinant protein comprising the MAGE-A3 and the fusion protein D of Haemophilus influenzae, along with the vaccine adjuvant AS02B. The trial's primary goal was to investigate disease-free survival (DFS). However, the study's final results did not show an improved DFS in the MAGE-A3 treated compared to the placebo control group (60.5 vs. 57.9 months, respectively) [54].
Similarly, NY-ESO-1, another CTA candidate, is expressed approximately in 30% of lung cancer specimens [55]. Many of its beneficial roles include a prognostic and a predictive factor for adjuvant and neoadjuvant chemotherapy treatment efficacy in NSCLC, and the capacity to induce specific antibodies in serum along with activation of the helper CD4 + and cytotoxic CD8 + T cells have already been demonstrated [56, 57]. Two concurrent phase I trials are at present recruiting patients to assess the safety and the immune response of ID-LV305 (immunotherapy targeting DCs in individuals with advanced cancer with the tumor cells expressing the NY-ESO-1 protein, NCT02122861) and of IDC-G305 (a new vaccine candidate containing recombinant NY-ESO-1 antigen and GLA-SE as an adjuvant, NCT02015416) in patients with NSCLC along with few other types of cancer patients. Another antigen expressed on NSCLC tumors is the mucin 1 (MUC-1) glycoprotein, which stimulates tumor cell proliferation pathologically via its cell surface receptor interaction [58]. It was chosen as a target for the development of the synthetic lipopeptide-based vaccine Tecemotide (L-BLP25), which was proved to be immunogenic and well-tolerated in a phase I study and was demonstrated as maintenance therapy for stage IIB-IV NSCLC patients through the achievement of stable disease or objective response, reported after the first-line chemotherapy in another phase IIB trial. In the phase III START trial, the treatment group showed no change in OS compared to the placebo-controlled groups (MS was 25.6 months vs. 22.3 months) [59–61]. Later, several studies, including the phase III START2 and INSPIRE trials were undertaken, but they were terminated owing to negative findings from the phase I/II Japanese EMR 63,325–009 study in unresectable stage III NSCLC patients. Use of pattern recognition receptor (PRR) activators and supramolecular peptide conjugates may enhance the potency of peptide vaccines. Hence further research is necessary to enhance the efficacy of peptide vaccines. Currently, personalized peptide-based vaccinations are being investigated for efficient therapeutic output.
DNA vaccines
DNA vaccines involve the insertion of a plasmid containing a particular DNA sequence encoding the target antigen to elicit specific immune responses in the presence of the antigen in situ. This approach is cost-effective and can be repeatedly administered. Another advantage is that the antigen post-expression can be presented by MHC class I and II, triggering CD4 and CD8 T cells and humoral immunity. While cytosolic sensors can recognize double-stranded plasmid DNA, which stimulates the innate immune response. Using a genetically engineered bi-transgenic KrasG12D inducible mouse (TetO-Kras4bG12D/Scgb1a1-rtTA) lung cancer model, Weng et al. used the Kras DNA vaccination. Vaccination yielded an efficient antitumor response and effectively targeted Kras-driven lung tumors [62]. MAGE-A3 protein (recMAGE-A3) vaccination has been used to target MAGE-A3, expressed in melanoma and NSCLC. Though effective in mouse melanoma models, when administered with or without adjuvant as a part of the large randomized MAGRIT MAGE-A3-positive NSCLC phase III trial demonstrated no advantage over the placebo [63]. DNA vaccines work in animal models but did not show promising results in clinical studies, necessitating the exploration of novel strategies. A comparison of xenogeneic antigens, neoantigens, and TAA in relation to therapeutic efficacy needs to be determined. Future research needs to investigate combination therapy approaches potentiate (targeted at activating antigen response and immunosuppression) to mediate synergistic and sustained immunogenic response in lung cancer. Another key area to investigate is the application of DNA vaccines in oncogenic virus-induced/activated cancers, including lung cancer. Advances in ex vivo DCs pulsing, nanotechnology, and surface functionalization approaches can help increase the efficacy of DNA vaccines. Considerations of immunodominance versus tolerance of immunogenic epitopes, poly-specific and poly-functional DNA vaccine, combination TAAs and neoantigens in a vaccine may boost vaccination-associated immunogenicity [64].
Vector vaccines
Vector-based vaccines are the constructs developed by manipulating specialized bacteria, viruses, yeast, or other structures to express any recombinant antigen. The TG4010 is a viral vector vaccine comprising a modified Vaccinia Virus Ankara (MVA) that encodes the human MUC1 and interleukin 2 [65]. Rochlitz et al. 2003, reported a good safety profile in a phase I clinical trial, where 13 patients having different solid tumors including lung cancer, were subjected to increasing doses of TG4010. Among them, one lung cancer patient showed a considerable reduction in the extent of metastasis over 14 months [66]. A phase II randomized clinical trial consisting of stages IIIB and IV NSCLC patients potentially pointed towards using TG4010 in combination with chemotherapy in first-line advanced or metastatic NSCLC for better chemotherapy results. [67]. The patients were administered the TG4010 in combination with the first-line chemotherapy (Cisplatin plus Vinorelbine doublet) or received the vaccine alone, and 29.5% of the patients who received treatment in the combination arm had a radiological response. A phase II trial (NCT00793208) that combines TG4010 with Nivolumab is ongoing [68]. Apart from this, other adenovirus vaccines expressing melanoma-associated antigen 3 (MAGE-A3) and MG1 maraba oncolytic virus (MG1-MAGEA3) were tested in phase I/II dose-escalation trial (NCT02879760) testing the combinatorial efficacy of the vaccine with Pembrolizumab, is presently recruiting NSCLC patients who have shown signs of radiological progression with at least one cycle of platinum-doublet chemotherapy [69]. The MAGE-A3, alone and in combination with MG1-MAGEA3 was tested in solid tumors, including lung cancer (NCT02285816) [69]. A better understanding of the molecular mechanism may enhance the efficacy of vector-based vaccines.
Dendritic cell vaccines
Cell-based immunotherapy helps immune cells identify tumor antigens and target cancer cells. This potential therapeutic immunotherapy technique is mainly explored in the context of dendritic cell-based vaccines, as DC therapy is safe and can elicit robust antigen-specific T cell responses owing to their antigen-presenting abilities [44]. Since the FDA authorization of Sipuleucel-T in April 2010 to treat metastatic prostate cancer, DC vaccines have progressed significantly, and several clinical trials are ongoing. A promising approach is the intra-tumoral delivery of autologous DC vaccine (CCL21 gene-modified DCs or AdCCL21-DC) targeting lung cancer. Lee et al. reported significant activation of CD8 + T cell tumor infiltration and antigen-specific immune response while using AdCCL21-DC in phase I clinical trial on stage IIIB, stage IV, or recurrent NSCLC (NCT00601094) [70]. Following the exciting results, another follow-up phase I trial is underway to evaluate the efficiency of pembrolizumab and AdCCL21-DC in combination on advanced-stage NSCLC patients (NCT03546361). The mechanistic effect of intratumoral CCL21-DC combined with anti-PD-1 therapy was further evaluated on murine NSCLC models [71]. Abascal et al. recently used murine CD103 + cDC1 (conventional DC type I) cells to produce soluble FLT3L (FLT3L cDC1) and conducted in situ vaccination experiments on anti-PD1 resistant murine NSCLC models and reported enhanced anti-tumor efficacy compared to non-modified cDC1 cells. Emerging research suggests DC vaccination may increase patient survival, calling for developing next-generation DC vaccines and testing new DC vaccine-immunotherapy combinations [71]. Nevertheless, the unique biology and classification of DCs, immune tolerance, weak and limited lifespan hamper their persistent and effective cancer immunity, and the production process are challenges that need to be addressed [72]. The role of different types of DCs (Mo-DC, cDC1, cDC2, pDC) and DC-derived exosomes in the context of the DC vaccine development may be further evaluated.
Allogeneic vaccines
Allogeneic vaccines contain non-self-cancer cells as the antigen source. Cancer cells of one patient are harvested and administered in another patient with the same tumor type, post necessary modifications and processing [73]. One such vaccine is the Belagenpumatucel-L. It is prepared by transfecting four radiated allogeneic NSCLC cell lines (H460, RH2, SKLU-1, H520, of which 2 are LUAD, one LUSC, and one LCC cell line) with a plasmid bearing the antisense of transforming growth factor β2 (TGF-β2) [74]. High levels of TGF-β have been correlated to immune suppression and worsening prognosis in NSCLC patients [75]. Inclusion of the antisense transgene in this vaccine inhibits TGF-β2 intending to increase immunogenicity. To assess its efficacy, a phase III randomized controlled trial (STOP), with 532 stage III/IV NSCLC patients who had no disease progression after a first line of platinum-based chemotherapy, was conducted that compared Belagenpumatucel-L with placebo. However, the study did not satisfy the primary endpoint as no difference in MS was observed between the vaccinated and the placebo arms (20.3 months vs. 17.8 months respectively, HR 0.94, P = 0.594). Similarly, another vaccine candidate comprising of autologous or allogeneic NSCLC cells plus GM.CD40L expressing K562 cells, when studied through phase I and II trials, could yield no affirmative results in terms of MS in NSCLC patients. Currently, two other allogeneic vaccines the Tergenpumatucel-L (NCT02460367, with 16 participants in a phase Ib/2 trial) and Viagenpumatucel-L (NCT02439450, with 121 participants in a phase Ib/2 DURGA trial), are being investigated in combination with ICIs. The major challenges of monotherapy include intratumoral heterogeneity, allogeneic vaccine-induced mutational divergence, and tumor escape, vaccines developed from cell lines/cellular components (non-self) may not reflect actual tumor antigens and tumor/ TME-induced immunosuppression. Hence better transcriptome analysis for antigen selection, including neoantigens, tackling immunosuppression, and combining T cell-based immunotherapies may become clinically translatable. Various clinical studies are underway on various solid tumors, and any success strategies may be expanded to lung cancer therapy.
Adoptive cell therapy
Adoptive cell therapy (ACT) utilizes tumor-reactive immune cells from patients, especially different types of T cells, that are grown and genetically engineered ex vivo before being re-administered to the patient as a therapy to identify and target cancer cells. In this regard, the most commonly used are Chimeric antigen receptor (CAR)-modified T cells (CAR T) therapy, Tumor-infiltrating lymphocyte (TIL) therapy, engineered T-cell receptor (TCR)-therapy, and Natural killer (NK) cell therapy [76]. Among these, the CAR-T cells and TCRs are both genetically modified synthetic biology approaches to target particular tumor antigens and exhibit prominent therapeutic effects [77, 78] (Fig. 3B). CAR-T cell immunotherapy has an 80–90% remission rate in hematological malignancies, and FDA has authorized CD19-targeting CAR-T for treating hematological cancers. This recent success with CAR-T therapy has changed the landscape of cancer therapy and spurred research efforts to translate these curative benefits to solid tumors like lung cancer [79].
CAR T cell therapy and lung cancer targeting
CAR-T cells are created genetically engineering autologous or allogeneic T cells in vitro by modifying T-cell receptors to identify and bind to antigens on cancer cells. Chimeric antigen receptor (CAR) are synthetic designed receptors, retrovirally transduced into T cells, and consists of three domains (Fig. 2, 3). CAR primarily consists of an extracellular antigen recognition domain (ectodomain), a transmembrane domain, and an intracellular signal transduction domain (endodomain). The ectodomain that determines the CAR’s affinity comprises an antibody-derived single-chain variable fragment (scFv) composed of the antigen-binding zone, including both the heavy and light chains of a monoclonal antibody (Fig. 4) [80]. On the one hand, the transmembrane domains connect the ectodomain via a hinge; on the other hand connect the endodomain, thereby anchoring the CAR to the cell.
Fig. 4.

Schematic representation of CAR architectural design. A: Evolution of CAR design through generations. B. Armored CAR T-cell design and functional mechanism. C. Schematic enumerates various building components that correlate to different CAR segments that may be exploited as CAR construction components. Gen: Generation; scFv: single-chain variable fragment; AD: activation domain; Co-S1: Co-stimulatory domain 1; Co-S2: Co-stimulatory domain 2; VL: variable light; VH: variable heavy; TRUCK: T-cells Redirected towards Universal Cytokine Killing. BiTEs: Bispecific T-cell engager
membrane. The CAR's intracellular domain has an activation domain (AD) and one or two co-stimulatory domains (Co-S1, 2) and can modulate the length, flexibility, surface density, downstream signal, and aggregation potential of CAR and thereby CAR-T functions (Fig. 4) [80, 81]. The activation domain is often associated with costimulatory molecules, which activate T cell activity and contribute to T cell proliferation and longevity. Phosphorylation of the immunoreceptor tyrosine-based activation motifs (ITAMs) of CAR endodomains activate and costimulate T lymphocytes. CAR structure evolution has seen several generations with hierarchical assimilation of functional modules (Fig. 4A) [82]. While the first-generation CAR lacked a costimulatory domain, the second and third-generation CAR adapted one and two costimulatory domains, respectively. Fourth-generation CAR-Ts combine the direct tumoricidal activity of CAR-Ts and the ability to overcome the immune-modulating ability of the tumor microenvironment without the systemic side effects (Fig. 4A).
CAR-T cells of this generation, called armored CAR T-cells, express proteins to minimize immunosuppression and increase anti-tumor activity (Fig. 4B) [83]. These CAR modifications can be summarized as follows, T-cells Redirected towards Universal Cytokine Killing) TRUCK, cytokine modulating CAR, and antibody modulating CAR. TRUCK CAR secretes cytokines to interfere with the immunosuppressive properties of solid tumors. By putting brakes on the immune-suppressive cytokine environment, it may be possible to enhance CAR T-cell and resident immune cell antitumor potential [80]. Cytokine modulation CARs depend on engineering CARs to express specific receptors, and ligands can help regulate CAR T-cells reaction to cytokines and may also affect cytokine function (Fig. 4B). To specifically target cancer antigens, CARs can be engineered to produce antibody-like proteins, called antibody modulating CARs. Another popular CAR design aspect is a nanobody or VHH antibody, composed of a single antibody's variable heavy chain or heavy chain-only antibodies (HcAbs) [84]. These armored CAR T-cells may boost solid tumor targeting efficacy due to their high affinity towards antigen, compactness, optimal stability, and manufacturability [85]. Bispecific T-cell engagers (BiTEs) are examples of innovative CAR design, where adaptive therapeutic benefit is achieved by a conjunction of two scFvs with differing antigenic specificities [86]. BiTEs are designed to bind to proteins on T cells and proteins on tumor cells, bringing them spatially closer, establishing an immune synapse, and unleashing immune cell killing (Fig. 4). Figure 4C lists several components that may be utilized as construction blocks corresponding to various CAR segments. CAR T treatment confronts various challenges when targeting solid tumors. However, increased attempts are being undertaken to target lung cancer in light of developments in synthetic biology, creative CAR design, and effectiveness against blood cancer [82].
Carcinoembryonic antigen (CEA) is strongly expressed in lung cancer compared to healthy cells, and high CEA expression is related to poor prognosis and metastasis. Therefore, anti-CEA CAR-T cell therapy and its safety efficacy are evaluated on CEA-positive lung cancer patients (NCT02349724, NCT04348643). Like CEA, Mucin 1 (MUC1) is also highly expressed in lung cancer, promotes metastasis, and is thus an exciting target for ACT against lung cancer [87, 88]. CAR-T cells targeting MUC1 successfully eliminate NSCLC tumor cells [88] and are evaluated in clinical trials for lung cancer (NCT03525782, NCT02587689, and NCT05239143). A third-generation anti-PSCA/MUC1/TGFβ/HER2/Mesothelin/Lewis-Y/GPC3/AXL/EGFR/B7-H3/Claudin18.2-CAR-T has been evaluated in an interventional Phase I trial (NCT03198052). Another interesting target is the cluster of differentiation 276 (CD276). CD276 is a new cancer diagnostic marker and an indicator of immunological status and prognosis that correlates favorably with the NSCLC tumor stage [89]. The silencing of CD276 modulates integrin signaling to ameliorate lung cancer metastatic potential [90]. Anti-PD-1 and anti-PD-L1 antibodies have radically improved NSCLC therapy with significantly increased ORR and PFS but suffers from multiple challenges. Recently PD-L1-targeting CAR T cells have been found effective in xenograft NSCLC tumors with high or low PD-L1 expression [91, 92]. MSLN-CAR T cells secreting PD-1 nanobody is being explored in an interventional phase I trial for NSCLC (NCT04489862). Immune infiltration in a tumor is a significant hurdle in effective ACT therapy. NSCLC tumors produce substantial amounts of the chemokine CXCL13, and an intelligent design would be to express its single receptor CXCR5 on the CAR T cells for enhanced CAR T cell tumor infiltration and heightened efficiency. An exploratory study of anti-EGFR CAR T cells modified by CXCR 5 is under phase I trials in NSCLC (NCT05060796) [93]. A third/fourth generation GPC3-CAR-T cell was engineered to target Glypican-3, a cell membrane proteoglycan and a potential marker specifically for LUSC and also for LUAD [94]. The GPC3-CAR was also engineered to express TGFβ targeting CAR (GPC3/TGF-CART), is effective in in vitro and in vivo experiments, and is under a phase I interventional investigation for LUSC (NCT03198546). TGFβ-CAR T cells can reduce the immunosuppressive tumor microenvironment making it more conducive for T cell activation [95]. A recent study examined PD-L1-targeting CAR T cells in PD-L1 high and low xenograft NSCLC tumors. PD-L1-CAR T cells exhibited robust effector T cell function and destroyed PD-L1 high and PD-L1 low tumors. Local irradiation improved PD-L1-CAR T cell efficacy and can be an innovative and effective strategy against PD-L1 low NSCLC patients [96]. Some exciting target antigens used for generating CAR against lung cancer include MUC-1, CEA, MSLN, HER2, GPC3, ROR1, and EGFR [97]. Although lung cancer CAR T immunotherapy is in its infancy, several challenges must be overcome before it can usher in widespread clinical implementation [98].
Developing an ACT for targeting lung cancer presents a big challenge. However, selecting the ideal TSA or TAA with minimal expression in normal tissue, eliminating off-tumor adverse effects and the immune-tolerant state posed by the tumor microenvironment, needs to be considered before developing CAR-T cells and TCRs for targeting NSCLC. Tackling CAR T -associated toxicity, antigen escape, heterogeneity of antigen, reduced CAR T proliferation in the tumor microenvironment, and CAR T tumor infiltration needs innovative synthetic biology approaches. Integrating appropriate modules to sense specific intra- and extracellular signals and actuator modules to coordinate precise transcriptional or translational control will effectively address the current lacunae. The use of multiple CARs on the same or different cell types (e.g., CD4/ CD8/ NK cells) and the ability to spatiotemporally controllable transient CAR activation using switchable CARs (inducible by ultrasound/ light/ drug/ adaptor) can strengthen the development of effective CAR T therapy [99]. Also, generating CAR-T cells with safety switches with inducible caspase-9 gene may be a successful approach [100–102]. To improve CAR T safety, novel self-driving and self-destruct CAR architecture are being engineered. CAR design, including multiple antigen-targeting abilities, can effectively address heterogeneous antigens in lung tumors [78, 98]. Another approach is to generate personalized.
CAR T for specific lung cancer genotypes. The rapid use of artificial intelligence (AI) in data analysis and synthetic biology design using CRISPR [103] may help scientists design synthetic receptors in order to correlate various chemical recognition events (e.g., SynNotch) [104]. Some researchers are trying to mass-produce CAR-T “off-the-shelf” cells, and this approach might make their CAR T therapy easier and cheaper. There is a need to identify neoantigens and develop enhanced high throughput screening tools to ensure that all structural components of the CAR T cells are optimized to target lung cancer.
T-cell receptor (TCR) engineering and lung cancer
TCR immunotherapy employs the innate mechanism of T cells to target tumor antigens by genetically modifying T cells ex vivo to express cancer-antigen-specific T cell receptors (TCRs) generated via TCR-engineering of patient-isolated T cells (TCR T) (Fig. 5) [105]. TCR therapy has distinct advantages over CAR T therapy targeting solid tumors like lung cancer [77]. TCRs identify only specific oncogenic peptides presented by human leukocyte antigen (HLA) class I on the surface of a tumor cell or an APC [106]. TCR T lymphocytes may target tumor mutation-derived neoantigens in a highly selective and non-toxic way, and a majority of clinical trials are focused on solid tumors.
Fig. 5.

Schematic showing TIL and TCR T cell therapy. TCR T and TILs are isolated from the patient and multiplied in the laboratory before being reintroduced to the patient
Multiple in vitro and in vivo investigations have shown the anti-cancer efficacy of TCR-T cells engineered to target lung cancer-specific antigens. Recently autologous TCR T-cell therapy against a lung tumor-specific protein called NY-ESO-1 is gaining traction. Letetresgene autoleucel (GSK3377794) is a first generation of TCR T-cells designed to target NY-ESO-1 and showed an objective response in Multiple Myeloma and myxoid/round cell liposarcoma trial (NCT03168438, NCT02992743). The safety and efficacy of Letetresgene autoleucel were evaluated after infusing back anti-NY-ESO-1 TCR to patients following lymphodepleting chemotherapy in a phase I trial that is now completed (NCT02588612). Other phase I trials using anti-NY-ESO-1 TCR transduced T cells alone or in combination with pembrolizumab on advanced NSCLC patient is underway (NCT03029273, NCT03709706). Another exciting target for TCR-T cell therapy is Kita-kyushu lung cancer antigen 1 (KK-LC-1), reported to be higher in LUAD than LUSC and exhibited a higher association with higher TMB tumors [107]. A clinical trial is currently investigating the safety and dosing of TCR Gene therapy targeting KK-LC-1 in KK-LC-1 + lung cancer patients (NCT05035407). TCR-T therapy has made a breakthrough in many tumors; however, there are few safe and effective targets due to possible off-target, dose-limiting cytotoxicity, autoimmune toxicity, and cytokine-related toxicity. Long-term safety evaluation studies with TCR-T cell therapy are also underway for lung and other cancers (NCT05292859, NCT05194735) and understanding their efficacy concerning somatic mutation and HLA typing (NCT05124743). A recent study used autologous TCR T therapy to target the hot spot KRAS G12D mutation in pancreatic cancer [108], with significant tumor regression and an overall partial response of 72%. Despite the survival of TCR-transduced T cells in the circulation, a second patient with the identical KRAS mutation and HLA allele did not benefit from T cell infusion, suggesting other potential TCR T failure mechanisms. The KRAS genetic variants are significant in lung cancer, especially in LUAD; the G12D mutation corresponds to ~ 3% of patients [109], and therefore a similar targeted TCR T therapy can bring a sea change in lung cancer treatment.
Though TCR T cell therapy is compelling, several challenges must be addressed for widespread clinical use [105]. Deficiencies in the antigen-processing and presentation machinery, downregulation of tumor HLA molecules and target mutations, augmentation of immune-suppressive cytokines, and complimentary activation of pathways (e.g., Wnt) inducing T cell tumor exclusion by the tumor/ TME can lead to attenuation of the TCR T therapy response [110, 111]. Moreover, HLA-coding genes are highly variable in the human genome, with about 20,000 alleles complicating TCR T HLA restriction. Unlike CAR T therapy, TCR T therapy TCRs are restricted to commonly found HLA alleles, such as HLA-A*02:01. TCR specificity is encoded by two distinct gene regions (TCR α and β transcripts) [112]. While doing TCR sequencing, intermixing can create ambiguity over which TCR α sequence corresponds with which TCR β sequence. Therefore, clonal T-cell isolation methods like limiting dilution, single Cell RT-PCR, and Single-Cell RNA Sequencing (scRNAseq) can be used [113]. Mispair heterodimerization of the TCR constant region of α and β chain of endogenous and engineered TCRs may lead to non-productive TCRs, unexpected autoantigen specificity, competition with engineered TCR, and may cause unwanted graft-versus-host-disease (GVHD). Techniques like murinization (integrating murine-derived TCR), the introduction of an additional disulfide bond (at residue 48 of Cα and residue 57 of Cβ), introducing stabilizing mutations (α-LVL) in α chain, domain swapping (Cα glycine and Cβ arginine), single-chain TCR, and combinations can improve TCR efficacy and safety profile [105]. Another issue is choosing and pre-clinically testing amongst the multiple antigen-specific TCR sequences discovered. An exciting tool is HLA restriction, where individually cloned HLA in COS7 cells are cocultured with TCR T cells to detect T cell activation. TCR affinity/avidity studies evaluate the association rate, dissociation rate, and binding constant using surface plasmon resonance. Cotransfected CD4 + T cells augment antitumor effects by boosting CD8 + T cell proliferation and survival [114]. Therefore, more studies with potential synergy with other immunotherapy, radiotherapy, and chemotherapy must be evaluated [106]. Novel genome engineering techniques, advances in single-cell genomics, and enhanced know-how through TCR T therapy trials may help design more effective TCR T cell therapy in lung cancer.
Tumor-infiltrating lymphocyte (TIL) and lung cancer
TILs can naturally recognize and target tumor cells, but the tumor cells usually evade this TIL-based immune response. TILs are often enriched with tumor-antigen specific T cell clones compared to T cells in peripheral blood and hence used for TIL therapy [115]. This novel immunotherapy approach uses TILs isolated from the patient's tumor and expanded ex vivo using recombinant IL-2 (rIL-2). IL-2 promotes TIL proliferation, activation, and tumor-killing activity [116]. Prior to TIL therapy, the patient is subjected to non-myeloablative lymphodepletion to eradicate the immune-suppressive TME. Billions of these TILs are then reintroduced in the patient, where they can proliferate, recognize tumor cells, and effectively ablate them (Fig. 5). TILs are primarily studied in solid tumors, including lung cancer [115].
In a similar context, ACT with TILs has been evaluated in NSCLC patients. One of the earlier studies examined the usefulness of TILs as a post-operative therapy for stage II–III NSCLC patients where tissue samples were surgically resected from the primary lung lesions of NSCLC patients [117]. Isolated lymphocytes and cancer cells were grown in a medium supplemented with rIL-2, and TILs were infused in stage stratified patients and subcutaneous injections of IL-2 daily until the maximum tolerable dosage was reached. The TIL arm had a favorable MS compared to the standard of care treatment arm (22.4 vs. 14.1 months). Recently, according to the results of a small phase I trial, TIL therapy (≥ 20 × 109 to 1010 CD3 + cells) along with IL-2 was found to be a feasible treatment option with a manageable toxicity profile [118]. It was observed that the TILs expanded in 95% of NSCLC patients with metastasis and had disease progression on Nivolumab (Opdivo). Another group investigated the effect of human double-negative T (DNT) cells (CD3 + CD4-CD8-) in targeting advanced lung cancer in vitro alone or in conjunction with Nivolumab (anti-PD-1 antibody). They observed that both patient- and healthy donor-derived DNT cells, expanded ex vivo, exhibited similar cytotoxicity against lung cancer cells [119]. It was also noted that DNT cells derived from healthy donors could considerably inhibit the growth of xenografts obtained from advanced-stage lung cancer patients, and the anti-cancer effect was further improved by the anti-PD-1 treatment that influenced augmented tumor infiltration of DNT cells. Autologous TIL therapy was also effective in a phase I trial on metastatic PD-1-resistant lung cancers (NCT03215810) [120]. However, the widespread application of TIL therapy has been mitigated by the demand for producing sufficient TILs in a stringent time period. Applications of rapid expansion protocols complying with good manufacturing practice conditions have improved the deficit [121]. Currently, a phase II study (NCT02133196, 85 participants) is recruiting patients to revise the utility of using autologous young TILs derived from NSCLC patients in combination with drugs like Aldesleukin, Fludarabine, and Cyclophosphamide. Further clinical trials are anticipated to predict the appositeness of TIL-based ACT in this new era of immunotherapy.
Oncolytic viruses for lung cancer immunotherapy
Oncolytic Viruses (OVs) are genetically modified viruses that can identify, infect, and lyse diverse cell types in a tumor microenvironment, which can halt and often reduce tumor development [122]. They either possess a natural tropism to the cancer cells or can be genetically manipulated to recognize specific targets displayed by the cells. These targets often comprise of the nuclear transcription factors like human telomerase reverse transcriptase, osteocalcin, cyclooxygenase-2, prostate-specific antigen, or surface markers like folate receptor, prostate-specific membrane antigen, endothelial growth factor receptor, CD20, and HER2/Neu [123]. Moreover, it is well-known that different evasion mechanisms are in play in the tumor microenvironment that directly or indirectly downregulates the immune response, aiding the disease progression even in immunocompetent patients [124]. It has been observed that the OVs may stimulate the immune system against the tumor cells, hence affecting the establishment of an anticancer response [124–126]. Therefore, the clinical application of OVs emerges as a viable approach to induce an inflamed state in the tumor microenvironment whereby the immune system can detect and kill the abnormal cells [126, 127]. Additionally, the viruses exhibit a variety of pathways that direct the infected cells toward lysis, resulting in tumor cell death and enhancing immunotherapy effectiveness [128] (Fig.3C). Several such genetically modified OVs are currently undergoing investigation for lung cancer to determine their applicability and efficacy in the disease scenario. For example, the cytotoxic effect of oncolytic Herpes Simplex Virus-1 (HSV-1) regulated miRNA145 delivery was tested in vitro in human NSCLC cell lines (A549, H460, H838, and H197), showing therapeutic potential [129, 130]. Likewise, it has been proven that Coxsackievirus B3 (CVB3) holds precise oncolytic activities in nine human NSCLC cell lines. Also, it has been observed that intralesional injections of the virus in transplantable lung tumor models led to significant regression. The virus-infected NSCLC cells secreted ATP, abundantly expressed calreticulin on their surface, and translocated extranuclear HMGB-1, which are innate immune response markers that indicate immunogenic cell death (ICD) [131, 132]. Similarly, the application of oncolytic vaccinia viruses (OVVs) having three individual genetic backbones has been investigated in preclinical studies [133–135]. Moving onward, the applications of OVs are also under investigation through several completed and ongoing clinical trials. Lysogenic Adenovirus has an extensive tissue tropism, exploited in a two-intervention armed clinical trial (NCT01574729) including 58 patients, was conducted to evaluate an oncolytic Adenovirus (rAd-p53) mediated wild-type p53 gene transfer in stage III or IV NSCLC patients [136, 137]. 33% of patients in the experiment received a combination of rAd-p53 injection (through intratumoral or bronchial artery access) and chemotherapy instillation via the bronchial artery, while the rest (control group) received only the chemotherapy treatment. It was observed that the combinatorial treatment group showcased an extended disease progression than the control group (MS, 7.75 vs. 5.5 months; P = 0.018) of patients. Two patients with stage III NSCLC who received the combination treatment showed a complete response.
Similarly, much earlier in 2007, the potential of Seneca Valley Virus isolate 001 (SVV-001, now NTX-010) as an OV came to the forefront [138]. Additionally, it was reported that even the maximum viral dosage was well tolerated in SCLC and other malignancies, with predictable viral clearance kinetics and intra-tumoral viral replication [139]. However, data obtained from a recently published randomized placebo-controlled, double-blind, phase II clinical trial (NCT01017601) involving 50 patients with advanced-stage SCLC without advance of disease after platinum-based chemotherapy treatment suggests that the first-generation SVV-001 OV as a single agent may be incapable of generating desired clinical efficacy in the patients [140]. OV therapy faces multiple challenges, including ECM-based barrier to viral spread and tumor penetration leading to inadequate tumor trophism, passive targeting, previous immunization-associated anti-viral immune response, and tumor hypoxia inhibiting viral replication and functions. Different viral engineering approaches can help address some of the challenges, including using RGD-motifs, scFv fusion with capsid protein, bi-specific adaptors, capsid modification, stealthing, use of hypoxia-responsive promoters, novel theranostic modalities, and considering different serotypes. Other ongoing and completed clinical trials estimating the applicability of OVs in lung cancer are listed in Table 1.
Table 1.
Completed and current clinical trials evaluating the applicability of Oncolytic Viruses in lung cancer
| Clinical Trial ID | Oncolytic virus | Virus type | Transgene/ Target | Combination | Cancer | Status | Duration | Sponsor/ Agency |
|---|---|---|---|---|---|---|---|---|
| NCT 02,879,760 | Ad-MAGEA3 with MG1-MAGEA3 | Adenovirus vector Maraba virus | Melanoma associated antigen 3 | Pembrolizumab | NSCLC | Phase I / II | 2017–2020 | Turnstone Biologics, Corp |
| NCT 02,043,665 | CVA21 | Coxsackie virus | None (CAVATAK) | Pembrolizumab | NSCLC | Phase I | 2013–2020 | Viralytics |
| NCT 03,647,163 | VSV-IFNβ-NIS | Vesicular Stomatitis virus (VSV) | Interferon-beta (IFNβ) and the sodium iodide symporter (NIS) | Pembrolizumab | NSCLC | Phase I / II | 2019–2021 | Vyriad, Inc |
| NCT 00,861,627 | REOLYSIN | Reovirus Serotype 3—Dearing Strain | KRAS/EGFR | Carboplatin Paclitaxel | NSCLC | Phase II | 2009–2015 | Oncolytics Biotech |
| NCT 01,708,993 | REOLYSIN | Reovirus Serotype 3—Dearing Strain | KRAS/EGFR | Pemetrexed Docetaxel | NSCLC | Phase II | 2012–2016 | Canadian Cancer Trials Group |
| NCT 01,017,601 | (NTX-010) Seneca Valley virus-001 | Seneca virus | NA | NA | SCLC | Phase II | 2010–2013 | Alliance for Clinical Trials in Oncology |
| NCT 01,574,729 | rAd-p53 | Adenovirus | p53 | surgery | NSCLC | Phase II | 2012–2015 | Shenzhen SiBiono GeneTech Co.,Ltd |
| NCT 03,004,183 | ADV/HSV-tk | Herpes simplex virus | thymidine kinase | SBRT Pembrolizumab | NSCLC | Phase II | 2017–2022 | The Methodist Hospital Research Institute |
| NCT 02,831,933 | ADV/HSV-tk | Herpes simplex virus | thymidine kinase | SBRT Nivolumab | NSCLC | Phase II | 2017–2020 | Eric Bernicker, MD |
Targeted antibodies for lung cancer
Scientists have well exploited the capability of the antibodies to specifically target tumor antigens on cancer cells and created a plethora of targeted antibodies to impair tumor cell activities. They can be broadly classified into three categories: (i) mAbs, (ii) antibody–drug conjugates (ADCs) (iii) bispecific antibodies (Fig. 3D). A few mAbs that received FDA approval for the treatment of NSCLC in the preceding two decades are—Cetuximab, Bevacizumab, Nivolumab, and Pembrolizumab (Table 2). Cetuximab is an anti-EGFR mAb that shows specific binding to the extracellular domain of EGFR and disrupts its receptor tyrosine kinase (RTK)-associated downstream proliferative activity. It has furnished positive responses in various combination therapy. Other efficacious anti-EGFR mAbs under evaluation in NSCLC are Necitumumab, Nimotuzumab, and Ficlatuzumab [141–144]. An anti-VEGF mAb Bevacizumab that showed anti-angiogenic potential to inhibit tumor growth was the first to receive FDA approval and is discussed in other sections. Another approved anti-VEGF mAb is ramucirumab which showed significant promise in combination therapy for NSCLC [145].
Table 2.
Targeted antibodies for lung cancer therapy
| Targeted Antibodies | Lung cancer type | Related Molecule | Target / Bioactivity |
|---|---|---|---|
| Monoclonal Antibodies (MABs) | NSCLC | Cetuximab | Anti-EGFR |
| Necitumumab | Anti-EGFR | ||
| Nimotuzumab | Anti-EGFR | ||
| Ficlatuzumab | Anti-EGFR | ||
| Bevacizumab | Anti-VEGF | ||
| Ramucirumab | Anti-VEGF/VEGFR2 | ||
| Nivolumab | Anti-PD-1 | ||
| Pembrolizumab | Anti-PD-1 | ||
| Ipilimumab | Anti-CTLA-4 | ||
| Tremelimumab | Anti-CTLA-4 | ||
| Denosumab | Anti-RANKL | ||
| Figitumumab | Anti-IGF-1R | ||
| SCLC | Tarextumab | Anti-Notch2 / Notch3 | |
| Tucotuzumab | Anti-EpCAM | ||
| Bec2 | Anti-GD3 | ||
| Antibody–Drug Conjugate (ADC) | SCLC | Rovalpituzumab tesirine | Anti-DLL3 |
| Sacituzumab govitecan | Anti-Trop-2 | ||
| Lorvotuzumab mertansine | Anti-CD56 | ||
| NSCLC | Ado-Trastuzumab emtansine | Anti-HER2 | |
| Telisotuzumab vedotin | Anti-cMET | ||
| Enapotamab vedotin | Anti-AXL | ||
| Bispecific antibodies | NSCLC | Amivantamab | Anti-EGFR, Anti-MET |
Immune checkpoint inhibitor (ICI)-based targeting antibodies prevent tumor cells from being attacked by immune system components ready to combat them. Among them, Nivolumab and Pembrolizumab are anti-PD-1 mAbs currently used in clinics. Also, two other anti-CTLA-4 mAbs undergoing rigorous evaluation are Ipilimumab and Tremelimumab. Their anticancer activities are elaborately discussed in other sections. Denosumab, an anti-RANKL (receptor activator of nuclear factor-kappa B ligand) mAb, showed efficacy in metastatic lung cancer inhibition in a phase III study [146]. Phase I and II study with Figitumumab, a fully humanized anti-IGF-1R MAB, as first-line therapy combined with chemotherapy showed considerable promise, but a phase III trial was discontinued [147]. For SCLC, efforts have also been directed to develop mAbs such as Tarextumab (anti-Notch 2 / Notch 3), Tucotuzumab (anti-EpCAM), and Bec2 (anti-GD3), which furnished positive outcomes in various clinical trials [148–150]. Nonetheless, to further enhance the efficacies of the mAbs, tripartite ADC has been synthesized where a potent cytotoxin is conjugated to mAbs via a covalent linker. Few of them are already commercialized, and many are undergoing different phases of clinical trials [151]. For refractory and metastatic SCLC, ADCs such as Rovalpituzumab tesirine (anti-DLL3), Sacituzumab govitecan (anti-Trop-2), lorvotuzumab mertansine (anti-CD56) is undergoing phase I/II clinical trials and are showing encouraging results [152–154]. Recent studies did not find an apparent efficacy with Rovalpituzumab tesirine in SCLC patient trials [155]. For NSCLC, various ADCs such as Ado-trastuzumab emtansine (anti-HER2), Telisotuzumab vedotin (anti-cMET), Enapotamab vedotin (anti-AXL) are under development and showing promising outcomes [156–158]. The third category of antibody-based targeted cancer therapy, referred to as bispecific T cell engagers or bispecific antibodies (BiTEs), are developed by fusing two front-end regions of two antibodies. One of its categories, Amivantamab (anti-EGFR, anti-MET), is now approved for lung cancer treatment [159]. Considering the clinical evidence accumulated in recent times, we envisage that many of the targeted antibodies will be approved in the near future.
Immune checkpoint inhibitors and NSCLC
Inhibition of immune-checkpoint proteins by blocking the CTLA-4, PD-1, and PD-L1 has been the most successful immunotherapeutic strategy in NSCLC. Ipilimumab is a fully-humanized mAb capable of neutralizing the CTLA-4, thus enabling CTL activity and sustaining immune responses mostly by depletion of regulatory T cells (Tregs) that demonstrate high levels of CTLA-4 expression [160]. Lynch et al. demonstrated in a preliminary phase II study (CA184-041) that Ipilimumab, in combination with chemotherapy in the first-line treatment for metastatic stage IIIB/IV NSCLC showed an enhancement in immune-related progression-free survival (irPFS) compared to only chemotherapy, without significant added toxicities [161, 162]. Its combinatorial application with erlotinib, crizotinib, and nivolumab has also been studied in a phase Ib non-randomized clinical trial (NCT01998126) for EGFR and ALK translocation-positive stage IV NSCLC that was completed in 2018 [163]. MS was not reached, and an excessive toxicity profile led to the termination of the study. Few more studies that combine Ipilimumab with radiation (NCT02239900, phase I/II, randomized, 143 participants and NCT02221739, phase I/II, 39 participants) and PD-1 antibody (discussed underneath) are currently ongoing. Tremelimumab is a fully human mAb that explicitly targets human anti-CTLA4. After it recorded an initial failure in a phase II randomized trial (n = 87) when administered in patients with pre-treated advanced-stage NSCLC, recently it came forth from the phase III NEPTUNE trial (NCT02542293, phase III, randomized, 953 participants) that a combination of Tremelimumab plus Imfinzi (Durvalumab, anti-PD-L1 antibody) performed no better than standard chemotherapy at extending the survival of people with metastatic NSCLC [164].
On the contrary, antibodies targeting the PD-1 protein have shown greater therapeutic promise in NSCLC. While CTLA-4 pathway inhibitors increase the infiltration and repertoire of tumor-specific T cells, PD-L1/PD-1 inhibitors work by preventing the inhibition of T-cell functions. Nivolumab (brand name Opdivo) is a fully human anti-PD-1 IgG4 mAb that specifically targets the human PD-1 protein. Preliminary data obtained from a phase I clinical trial (NCT00730639, non-randomized, 395 participants) of Nivolumab was used in advanced or recurrent malignant patients, including NSCLC, spearheaded three key trials that presented their results in 2015 [165–167]. The phase II, single-arm CheckMate 063 trial (NCT01721759, 117 participants with advanced, refractory squamous NSCLC) demonstrated that intravenous administration of Nivolumab (3 mg/kg) every two weeks resulted in 14.5% (17 of 117) patients achieving an objective response (OR), the primary endpoint for the investigation while 26% of (30 of 117) patients showed stable disease [165]. 17% of patients were reported to have developed grade 3/4 adverse events (AE), the most frequent of which were: pneumonitis (3%), diarrhea (3%), and fatigue (4%). CheckMate 057 (NCT01673867, randomized, phase III study, 272 participants) evaluated Nivolumab's effectiveness and safety in patients with disease progression during or after first-line chemotherapy for patients with advanced squamous NSCLC and was compared to docetaxel [167]. Although the MS for Nivolumab was higher than with docetaxel (9.2 vs. 6.0 months), but PD-L1 expression was of neither predictive nor prognostic benefit. The phase III CheckMate 017 trial (NCT01642004, open-label, 352 participants) investigated the effect of Nivolumab (at 3 mg/kg every two weeks) as compared to docetaxel (at 75 mg/m2 every three weeks) in patients with IIIB/IV non-squamous NSCLC that advanced during or after first-line chemotherapy [166]. The MS in the Nivolumab group was 12.2 months, compared to 9.4 months in the docetaxel group. Nivolumab did not have the edge over docetaxel in terms of PFS; the study is ongoing. However, Nivolumab revealed a better efficacy than docetaxel across all categories determined by the degree of PD-L1 expression on the tumor cell membrane. Also, treatment-related severe AEs were observed in 10% of the patients treated with Nivolumab, against 54% with docetaxel. The FDA approved Nivolumab as the first anti-PD-1 drug to treat pre-treated advanced or metastatic NSCLC. The scheme of immunotherapy treatment in NSCLC patients is shown in Fig. 3.
Moreover, additional clinical trials like CheckMate 012 are currently underway to assess the efficacy of Nivolumab with or without Ipilimumab in first-line settings for advanced NSCLC. In the CheckMate 012 trial (NCT01454102, phase I, open-label, 472 participants to date), Nivolumab was initially tested as a monotherapy in first-line advanced stage IIIB/IV NSCLC, which resulted in a 23% (12 out of 52) ORR in newly diagnosed advanced NSCLC patients and the investigators found four patients with continuing complete responses [168]. The ORR was 28% (9 out of 32) in subjects with tumors expressing PD-L1 and 14% (2 of 14) in subjects with no detectable PD-L1 expression. Later, when tested in combination with Ipilimumab or another platinum-based chemotherapy cohort (n = 56), the trial showed a significant rate (45%) of AEs for which treatment discontinuation occurred in significant numbers [165]. ORR was achieved regardless of tumor PD-L1 expression and the respective ORRs were 33%, 47%, 47%, and 43% for Nivolumab (10 mg/kg) with gemcitabine/cisplatin, Nivolumab (10 mg/kg) along with pemetrexed/cisplatin, Nivolumab (10 mg/kg) in combination with paclitaxel/carboplatin, and Nivolumab (5 mg/kg) plus paclitaxel/carboplatin in this study. CheckMate 277 trial (NCT02477826, randomized, open-label, phase III, 2748 participants) evaluated Nivolumab or Nivolumab plus Ipilimumab, or Nivolumab in combination with platinum-doublet chemotherapy to platinum doublet chemotherapy in PD-L1-defined previously untreated NSCLC [169]. The results exhibited positive outcomes regarding OS with nivolumab plus ipilimumab compared to chemotherapy in patients irrespective of the expression of PD-L1 [170]. CheckMate 9LA presented an interesting improvement in the OS for advanced NSCLC patients with two cycles of chemotherapy in combination with Nivolumab and Ipilimumab [170, 171]. CheckMate 227 and CheckMate 9LA prompted a chemo-free doublet immunotherapy approach and improved the overall OS regardless of the patient’s PD-L1 profile.
Pembrolizumab (MK-3475) is another high-affinity humanized IgG4 mAb that targets the PD-1 protein. The drug's safety profile and therapeutic efficacy in NSCLC were initially assessed in the phase I clinical KEYNOTE-001 study (NCT01295827, phase I, randomized, open-label, 1260 participants), which demonstrated durable antitumor activity in advanced-stage NSCLC patients [172, 173]. Patients received Pembrolizumab at either 2 mg/kg (n = 55) or 10 mg/kg (n = 238) every 3 weeks or 10 mg/kg (n = 156) every 2 weeks and response was evaluated every 9 weeks. ORRs for the doses were 15% [95% CI, 7%-28%] at 2 mg/kg every three weeks, 25% (95% CI,18%-33%) at 10 mg/kg every three weeks, and 21% (95% CI,14%-30%) at 10 mg/kg every two weeks respectively, which suggest the use of a 2 mg/kg Pembrolizumab every three weeks as the optimum dosage in patients with previously treated, advanced NSCLC. Subsequently, KEYNOTE-010 (NCT01905657, randomized, phase II/III study, 1034 participants) compared the dosage gradient of Pembrolizumab with a fixed dose of docetaxel in patients who were pre-treated with advanced NSCLC (expressing PD-L1 ≥ 1%), keeping OS and PFS as the primary endpoints. Although the MS was considerably extended for Pembrolizumab 2 mg/kg as compared to docetaxel [HR 0.71, 95% confidence interval (CI) 0.58–0.88] and for Pembrolizumab 10 mg/kg when compared to docetaxel (HR 0.61, 95% CI 0.49–0.75), no statistically significant variance in the overall median PFS was observed. Notably, PFS was significantly longer with Pembrolizumab in patients whose tumor cells express at least 50% PD-L1. Following the results, the FDA approved Pembrolizumab to treat patients with advanced PD-L1 expressing NSCLC whose disease had worsened following chemotherapy in October 2015. Again, in the phase III KEYNOTE-024 trial (NCT02142738) that assessed the effectiveness of Pembrolizumab as first-line therapy compared to different chemotherapy regimens for metastatic treatment-naive NSCLC, the drug once again proved its advantage over only chemotherapy with significantly longer PFS (10.3 vs. 6.0 months; HR 0.50; P < 0.001) and OS (HR 0.60; 95% CI, 0.41 to 0.89; P = 0.005) in patients receiving the drug. Moreover, the Pembrolizumab group showed a response rate of 44.8% versus 27.8% in the chemotherapy-treated group, and severe AEs were reported to occur in ~ 26.6% of the Pembrolizumab group patients versus 56.6% of the patients in the chemotherapy-treated group. This led to the FDA approval of the drug for the first-line treatment of advanced metastatic NSCLC patients with high tumor PD-L1 expression (at least 50% tumor cells) [174].
The third group of check-point inhibitors target PD-L1 to inhibit the molecular interaction between PD-L1 and PD-1 or the molecular contact between PD-L1 and B7.1 (a T cell-specific inhibitory receptor). Durvalumab, Atezolizumab, and Avelumab are three fully-humanized anti-IgG1 mAbs that comprise this class of drugs. Durvalumab (MEDI-4736) was tested by a phase I/II trial in stage IIIB/IV NSCLC and other solid tumors. Durvalumab (10 mg/kg) was administered every two weeks for up to one year to treatment-naïve advanced NSCLC patients. The ORR was 25%, and the disease control rate was 56% with ≥ 12 weeks of follow-up, and grade ≥ 3 drug-related AEs (most frequent being diarrhea) were reported in 9% of patients [175]. Taking forward these encouraging outcomes, the efficacy of Durvalumab is being evaluated in trials for various aspects such as monotherapy (NCT02087423) after concurrent chemo-radiotherapy in stage III NSCLC (NCT02125461) also adjuvant therapy in patients with stage IB to IIIA NSCLC (BR31 trial; NCT02273375).
Again, a combinatorial study with Durvalumab and Tremelimumab was initiated to evaluate the postulate stating that co-inhibition of PD-1/PD-L1 and CTLA-4 may evoke synergy in immunotherapy in patients with advanced NSCLC [176]. The outcome demonstrated that 36% of patients developed AEs, and 23% of patients achieved ORR in a combined Tremelimumab 1 mg/kg cohort. Many phase II/III trials, including third-line ARCTIC (NCT02352948), the first-line MYSTIC (NCT02453282), and NEPTUNE (NCT02542293), have been commenced using a combination strategy of immuno-therapeutics. In the case of Atezolizumab (MPDL3280A), after a phase I study confirmed its efficacy for treatment in NSCLC (ORR of 23%, n = 53), especially in patients with tumor cell PD-L1 expression, a single-arm phase II study (BIRCH, NCT02031458, open-label, 667 participants) in PD-L1 selected (tumors or immune cells in TME) advanced NSCLC was initiated with ORR being the primary endpoint [177, 178]. The patients received Atezolizumab (1,200 mg) intravenously every three weeks and were distributed into three cohorts: first-line (cohort 1, with no prior chemotherapy; n = 139); second line (cohort 2, with one prior platinum chemotherapy; n = 268); and third-line or higher (cohort 3, with at least two prior chemotherapies of which one is platinum-based; n = 252). It was observed that BIRCH achieved its primary goal by exhibiting a significant increase in ORR (18% to 22% for the three cohorts) in Atezolizumab treated patients compared to historical controls, and most of the responses are ongoing. Also, the MS (minimum of 20 months follow-up) for cohort 1 was 23.5 months, cohort two was 15.5, and cohort 3 was 13.2 months. Thus, the trial showcased responses with good tolerability for Atezolizumab monotherapy in advanced-stage NSCLC patients with PD-L1 selected tumors.
Another phase II trial (POPLAR, NCT01903993, randomized, open-label, 287 participants) assessed the safety and efficacy of Atezolizumab-based immunotherapy compared to docetaxel therapy in NSCLC patients pre-treated with platinum-based chemotherapy [179]. Volunteers were randomly distributed (1:1) into two groups where one group received intravenous Atezolizumab (1,200 mg) and the other received docetaxel (75 mg/m2), both for three weeks intervals. As observed at the primary endpoint, the OS was improved for Atezolizumab (12.6 months) compared to Docetaxel (9.7 months, HR 0.73, P = 0.04). This can be attributed to the alteration in PD-L1 expression. Also, a diminution of treatment-induced grade 3/4 AEs in patients treated with Atezolizumab (11%) versus the docetaxel treated group (39%) strongly advocated for the possible benefits of Atezolizumab in NSCLC patients who received previous treatments [179]. Finally, the propitious outcomes obtained in the BIRCH and POPLAR trials led to the FDA approval of Atezolizumab for patients with advanced NSCLC. The NCT01846416 trial tested the drug as monotherapy for PD-L1 positive patients with advanced non-metastatic NSCLC. The third one, Avelumab, has demonstrated reasonable clinical efficacy and safety profile in NSCLC patients untreated previously and unselected for PD-L1 expression through NCT01772004 and NCT02395172 [180]. However, further investigations are required to prescribe the drug for practical applicability. NSCLC tumor PD-L1 expression has become an essential determinant of clinical pathology and frontline treatment. A summary of the treatment strategy in different crucial clinical trials is shown in Fig. 6. Though the studies help draw certain conclusions, more studies are needed to draw meaningful comparisons to improve NSCLC immunotherapy for both PD-L1 high and low populations.
Fig. 6.

Scheme of treatment with immunotherapy in NSCLC. The algorithm helps the physician decide on the available treatments for different types of lung cancers. Their consensus sequencing techniques. PD-L1 testing and histological subtype determination should be done by a multidisciplinary team for all patients. The role of driver mutation, as shown, is important in determining treatment modalities
Anti-T-cell immunoreceptor with Ig and ITIM domain (TIGIT) checkpoint inhibitor Tiragolumab suppresses innate and adaptive immunity in PD-L1-positive metastatic NSCLC and has received the Breakthrough Therapy Designation (BTD) by FDA [181]. LAG3 and FGL1 expression promote tumor development by suppressing the immune system and are amongst the most promising immune checkpoints. LAG3 accumulates on CD4 + and CD25-T cell surfaces in TILs and is also identified in the cytoplasm of NSCLC cells, while FGL1 is identified in NSCLC cell's cytoplasm [182]. LAG-3 antibody (liratrimab) may have therapeutic utility as a third ICI route after PD-1 and CTLA-4. A soluble LAG-3 protein eftilagimod alpha (Efti; IMP321) is also undergoing a phase II study (TACTI-002) (NCT03625323) in combination with pembrolizumab on metastatic NSCLC patients [183]. Nobel ICPs are emerging and need to be investigated as single agents and combinations. The current immunotherapy pipelines have concentrated on functional, cellular, and molecular readouts but lack mechanistic knowledge of immunotherapy targets.
Classification of lung cancer progression patterns and grading and updating the Common Terminology Criteria for Adverse Events (CTCAE) in relation to ICIs and associated diagnostic markers can help better design the clinical trial endpoints. Understanding the immune-related adverse events (irAEs) concerning different ICIs and therapeutic combinations can help plan better neutralizing mechanisms. Using appropriate pre-clinical models, including human-specific organoid models, humanized mice, and rigorous pharmacokinetic/ pharmacodynamic analysis, can help reduce clinical trial failures. Further investigation regarding the ICI treatment duration, rechallenge, and dose-ranging need attention. Advances in single-cell genomics, multiplex immunofluorescence, and multi-omic platforms encourage therapeutic discovery by finding and evaluating new immunotherapy targets. AI-based algorithms are currently being considered for radiological, pathological, and diagnostic data analysis and can help build successful immunotherapies and data-driven medication schedules and combinations. Recently Sun et al. developed a radiomics-based biomarker of tumor-infiltrating CD8 cells on patients in a phase I study with anti-PD-1 or PD-L1 monotherapy. They proposed imaging biomarkers may assess CD8 cell count, correlate with tumor immune profile, and predict immunotherapy patient outcomes in many solid tumors, including lung cancer [184]. The deployment of artificial intelligence in prospective clinical trials is constrained by various obstacles, such as the lack of a high-quality training data set, validation data set, code sharing, and transparency, despite the technology's innovation and significant promise [185].
Immunotherapy in SCLC
Lung SCLC, mostly begotten by cigarette smoking, is an abjectly differentiated, fast-growing, high-grade, malignant epithelial cell carcinoma originating from neuroendocrine cells within the bronchial airways. SCLC cells are morphologically diverse, with poorly defined borders, tiny cytosol, granulated nuclear chromatin, and absence or unobtrusive nucleoli with a high mitotic count [186, 187]. 5% of SCLCs can be originated from extrapulmonary regions, including the nasopharynx, GI tract, and genitourinary tract [188–190]. However, SCLCs of pulmonary and extra-pulmonary origins have similar clinical and biological features characterized by rapid growth and early widespread metastasis [191–194]. About 70% of patients with ED-SCLC have apparent metastases at the time of diagnosis. The remaining 30% have LD-SCLC, defined by tumors confined to the hemithorax [195, 196]. This inevitably results in a poor prognosis for the patients, with MS being 15–20 months and 8–13 months for LD and ED-SCLC, respectively [195]. Similarly, SR on average over 5 years interval is 10%–13% and 1%–2% for LD and ED-SCLC patients, respectively [195, 197].
The aggressive nature of SCLC can be ascribed to its high TMB, which includes the bi-allelic deactivation of tumor suppressor genes, such as p53 and Rb1, in nearly all tumors incidences [198]. High TMB-associated generation of a higher number of neoantigens allows an enhanced presentation to the T cells leading to a heightened immunological response and may be exploited to create efficient immunotherapeutics. [199, 200]. SCLC has long been considered immunogenic due to its association with paraneoplastic disorders, such as Lambert-Eaton myasthenic syndrome (LEMS) in patients. This results from immune responses directed against specific antigenic targets (HuD, HuC, and Hel-N1) expressed on both SCLC and normal nerve cells [201–205]. Intriguingly, SCLC patients having LEMS are likely to obtain a better prognosis, which can be attributed to the phenomenon that the immune response directed against the nervous system can also target the tumor cells [203]. Also, better OS was observed in patients whose tumors were infiltrated with more CD45+ T cells, independent of stage and performance status. A correlation between higher effector-to-regulatory T-cell ratios and prolonged OS was established [202, 204, 205]. Recent developments have been remarkable in immunotherapy-based approaches for SCLC, such as ICIs, antigen-specific vaccines, and tumor vaccines, fostering hope for a general increase in the SR, OS, and patient quality of life.
Antigen-specific vaccines and SCLC
A vaccine-based approach has often been considered ideal for SCLC patients, especially those who have recently completed chemotherapy cycles, due to its minimal toxicity potential. Almost four decades ago, gangliosides and glycolipids were identified as therapeutic targets for the treatment of melanoma [206, 207]. Soon it was noted that most SCLC cell lines express GD3, a glycosphingolipid (ceramide and oligosaccharide) or oligoglycosylceramide containing one or multiple sialic acids (i.e., n-acetylneuraminic acid), which prompted the evaluation of BEC2, the anti-idiotypic mAbs mimicking GD3 ganglioside in SCLC patients [208]. In a pilot trial conducted on 15 patients who were administered BEC-2 adjuvanted with BCG after completion of initial chemotherapy, the OS was enhanced by 21 months [209]. Further large-scale trials, sponsored by the European Organization for the Research and Treatment of Cancer, detected a 40% increase in survival. The combination has currently been licensed by ImClone Systems, USA, and Merck Oncology in Europe, Australia, and New Zealand.
Upon further investigations and trials, four other antigens—GM2, Globo H, fucosyl GM1, and polysialic acid emerged as antigenic targets for immunotherapy in SCLC based on immune-histochemical analyses of tumor samples [210]. Although the immunogenicity for GM2 and Globo H was well-understood in other forms of malignant cancers, fucosyl GM1 and polysialic acid were selectively expressed in SCLC alone [211–215]. All the antigens were tested as conjugates with KLH and QS-21 as the adjuvant for administration in patients who completed initial chemotherapy or radiation therapy. Additionally, It was ensured that the patients were not taking systemic corticosteroids and were free from any underlying immune deficiencies or peripheral sensory neuropathy greater than Grade 1 [206]. Although some immune-specific responses had been noted in patients administered with the antigens, trials with a bigger cohort are still needed to validate their efficacy in the prognosis for SCLC patients.
Another target antigen recently surfaced in the treatment of SCLC is p53. An initial trial with 29 ED-SCLC patients given the p53 vaccine plus chemotherapy showed a high overall response rate (ORR). Similarly, induction of immune response (40–50%) and tolerability of a dendritic cell-based vaccine with modified p53 (INGN-225) was evaluated in a phase I/II study [216]. However, the optimal treatment in all of the above cases may not suffice with vaccination alone and ought to be coupled with chemotherapy scrupulously [217]. Thus, further trials are required to verify the role and extent of antigen-specific vaccines as a potential therapeutic approach in patients with SCLC. In another innovative approach by Sakamoto et al., the efficacy of a personalized peptide vaccination (PPV) was evaluated in a phase II trial where a maximum of 4 HLA-matched peptide sequences were selected from a previously established IgG response-specific peptide library [218]. The PPV was subcutaneously administered, in which 46 patients were enrolled. Each of the four groups of patients had a MS of 466 (0; n = 5), 397 (1; n = 15), 401 (2; n = 12), and 107 days (3; n = 14), respectively, in terms of prior treatment regimens. After one and two vaccination rounds, peptide-specific IgG responses increased in 70% and 95% of patients. OS was considerably longer in individuals with increased IgG responses after the second immunization cycle (1237 vs. 382 days; P = 0.010) than in patients with enhanced IgG responses. Despite some positive outcomes in OS prolongation and immune rejuvenation, further evaluation of its efficacy in eventual randomized trials is necessary.
Immune-checkpoint inhibitors for SCLC
Blocking the immune checkpoints with mAbs has gained significant attention as a promising therapeutic tool in oncology, including SCLC [5, 219]. While eliciting an antigen-specific T-cell response, costimulatory and co-inhibitory factors play a key role in immune regulation post-stimulation of the TCR [220]. After the TCR recognizes the antigenic peptides displayed by both the classes of MHC I & II molecules on the surface of antigen-presenting cells (APC), the entire T-cell activation process requires a second costimulatory signal generated by the costimulatory T-cell surface receptor CD28 that binds to B7 ligand subtypes CD80 and CD86 present on APCs surface [5, 221]. T-cell activation and subsequent immunological response are aided by the co-stimulation of CD28 with other related molecules, such as CD134 and CD137. Another fraction of molecules, viz., CTLA-4, PD-1, B7-H3, and B7x abate antigen-specific immune responses by restricting their magnitude and duration. These co-inhibition molecules are called “immune checkpoint proteins,” and inhibition of these protein pathways (immune-checkpoint inhibition) by blocking CTLA-4 and PD-L1 with mAbs, etc., have shown potential advances in cancer immunotherapy [219].
It has been postulated that PD-1 and its ligand on the SCLC cells may be involved in tumor cell growth inhibition. [222]. Pembrolizumab is an anti–PD-1 mAb designed to block the PD-1/PD-L1 pathway. To test the efficacy of this antibody, in the KEYNOTE-028 phase Ib trial, 24 patients with PD-L1+ ED-SCLC who had completed initial chemotherapy received Pembrolizumab. Although the study produced an ORR of 35%, with lasting responses over more than 16 weeks, the related AE rate was 53% [223]. In another study, using immunohistochemistry, Ishii et al. examined the expression of PD-L1 in 102 SCLC patients where 71.6% of volunteers expressed PD-L1, and its correlation with LD-SCLC was established. The results revealed that MS was 16.3 months in the PD-L1( +) subset and 7.3 months in the patients not expressing PD-L1 (P < 0.001) [224]. Few more prospective trials (NCT02359019 and NCT02403920) investigating Pembrolizumab with chemotherapy or Pembrolizumab with chemotherapy and radiotherapy for use in SCLC are ongoing. These clinical studies are trying to address the issue of limited treatment options for patients with metastatic SCLC who are on platinum-based chemotherapy, which may help SCLC patients.
Similarly, as previously mentioned, CTLA-4 is an immune checkpoint protein that is expressed on activated T cells, which is widely studied for its capacity to down-regulate T-cell activities [225]. With the development of fully human mAbs such as Ipilimumab, CTLA-4 has become an attractive therapeutic target for cancer. Of many recent clinical trials, a randomized phase II study has explored the combinatorial application of Ipilimumab with paclitaxel and carboplatin as the first-line treatment in ED-SCLC [226]. The outcome revealed that phased Ipilimumab improved the PFS compared to concurrent Ipilimumab. The OS of the two groups were 12.5 and 9.1 months respectively, with no significant difference (P = 0.13) [226]. Prompted by the outcomes, a phase III clinical trial (NCT01450761) of Ipilimumab along with chemotherapy compared to chemotherapy alone has commenced in ED-SCLC patients.
In addition, another phase I/II clinical trial (Checkmate 032, NCT01928394) evaluated the effectiveness of Nivolumab (an anti-PD-1 mAb) coupled with or without Ipilimumab in patients with limited-stage or extensive-stage SCLC relapse after at least one platinum-containing regimen [227]. The results presented that Nivolumab monotherapy and Nivolumab in combination with Ipilimumab exhibited anticancer activity with protracted responses and the adverse events were tolerable. More such trials are in-line. Thus, the constant efforts to improvise these strategies give us the hope of better future outcomes for SCLC prognosis and treatment.
Combination immunotherapy approaches
Because the immune response is dynamic, evidence suggests that combination therapy may improve cancer patient survival compared to monotherapy. Anti-angiogenic agents, chemotherapy, radiation therapy, and T-cell modulation are investigated in combination with immunotherapy. The arsenal of immunotherapeutics is rapidly gaining ground while unveiling new challenges. Finding the optimal therapy combinations, doses, and order of administration in specific cancer genotypes needs to be investigated. Other challenges are irAEs, finding suitable biomarkers for immunotherapy response, resistance to immunotherapy, and making the non-responders benefit from combination therapy. Understanding the tumor type, TME-mediated immunosuppression, immune profile of the tumor, and tumor genotypic profiling may help address some of the challenges. AI can analyze and categorize input data and produce models to anticipate molecular interactions, the efficiency of combination therapies, and predict poor prognosis associated genotypes and needs further attention. The forthcoming section summarizes the present state of combination therapy and clinical application.
Immunotherapy with Anti-angiogenic agents for targeting NSCLC
There is no dearth of preclinical evidence for the general validity of the proposition that angiogenesis is crucial for tumor growth, and it possesses a convoluted relation with tumor immunity, i.e., antiangiogenic compounds can invigorate the immune system while cancer immunotherapy turns to be antiangiogenic [228–230]. More importantly, a combination of these two can have a synergistic impact on inhibiting tumor growth. The immunosuppressive TME, comprising VEGF as a major modulator of immune response, endorses tumor cells towards evasion of immune surveillance [231]. VEGF (i) blocks lymphocyte trafficking across activated tumor endothelium by inducing clustering defects at the endothelial cell surface, (ii) inhibits tumor infiltration of the T cells via upregulation of the Fas ligand, (iii) induces proliferation in immune suppressors, viz., Tregs and myeloid-derived suppressor cells (MDSCs), etc. [232–234]. Antiangiogenic therapy normalizes the tumor blood vessels decreasing interstitial fluid pressure, thereby enhancing drug penetration within the tumor and synergizing with chemotherapy and immunotherapy [235, 236]. A study comprising 125 advanced NSCLC patients showed that anti-VEGF therapy, bevacizumab, mediated metabolic changes of the tumor through the LKB1/AMPK pathway, which correlates with increased survival [237]. Many preclinical and clinical data also hint at the possibility of synergy between immunotherapy and anti-angiogenic therapy in NSCLC, enhancing the potential of both [238]. For instance, using an in vivo NSCLC model, Tao et al. showed that immunotherapy in combination with bevacizumab inhibits tumor growth synergistically, and the approach holds promise for clinical translation [239]. Multiple clinical trials are ongoing with this combinatorial approach and, hopefully, will move from bench side to bedside shortly [240, 241].
Chemo-immunotherapy in lung cancer
As discussed in the earlier sections, inhibition of the PD-1 receptor or PD-L1 is the mainstay of action of current immunotherapy agents in NSCLC and SCLC. Nonetheless, the combinatorial applications of immunotherapeutic agents with Chemotherapy drugs have emerged as a much-practiced method for managing progressive disease. Below we discuss the same elaborately. Few more trials like the NCT02486718 (IMpower010), NCT02657434 (IMpower132), NCT02409342 (IMpower110), NCT02367781 (IMpower130), and NCT02366143 (IMpower150) are ongoing to test the efficacy of Atezolizumab with chemotherapy. Furthermore, several combination treatments of the drug with other intervention approaches such as the MEK inhibitor cobimetinib (NCT01988896), the tyrosine kinase inhibitor drug cabozantinib (NCT03170960), the anti-VEGF-A humanized monoclonal IgG Bevacizumab (NCT03836066, NCT03616691), etc. are also being evaluated.
Chemo-immunotherapy in NSCLC
The primary immunotherapy agents employed are Pembrolizumab, Atezolizumab, Durvalumab, Nivolumab, and Ipilimumab. Figure 6 briefly depicts the strategy taken for the treatment with immunotherapy in NSCLC.
Pembrolizumab
It is an immune checkpoint inhibitor for the PD-1 receptor, which improves anti-tumor immunity. As per treatment guidelines, every patient with metastatic NSCLC should undergo IHC testing for PD-L1 expression and other driver mutations like EGFR, ALK, ROS-1, and BRAF before starting treatment [242].
As first-line therapy
In eligible patients (Performance status 0–2 and no contraindication for immunotherapy) with metastatic NSCLC irrespective of histology with PD-L1 level ≥ 50% and with none of the driver mutations (EGFR, ALK, ROS-1, BRAF), Pembrolizumab is recommended as first-line monotherapy. Patients with PD-L1 expression 1–49% and all driver mutations remaining negative are recommended Pembrolizumab plus chemotherapy as the first-line therapy. For Non-Squamous/Adenocarcinoma, combination chemotherapy of Carboplatin or Cisplatin with Pemetrexed is preferred with Pembrolizumab. In patients with squamous cell histology, combination chemotherapy of Carboplatin and Paclitaxel/Albumin bound paclitaxel is preferred with Pembrolizumab [243]. KEYNOTE-024 trial aimed at comparing Pembrolizumab monotherapy vs. platinum-based chemotherapy as first-line therapy for NSCLC patients with PD-L1 expression level ≥ 50% and driver mutations negative. It showed an improved response rate and MS with Pembrolizumab monotherapy (30.0 vs. 14.2 months) [174]. KEYNOTE-189, a randomized phase III trial with metastatic non-squamous NSCLC patients, compared Pembrolizumab and Carboplatin or Cisplatin and Pemetrexed vs. chemotherapy alone. It showed better OS at 1 year with Pembrolizumab plus chemotherapy (69.2% vs. 49.4%) [244]. KEYNOTE-407, a randomized phase III trial performed with metastatic squamous cell NSCLC patients, also evaluated the efficacy of Pembrolizumab together with Carboplatin and Paclitaxelor Albumin bound Paclitaxel. It resulted with improved MS in case of Pembrolizumab plus chemotherapy (15.9 vs. 11.3 months) [245].
As subsequent therapy
KEYNOTE 010, a phase III randomized trial carried out with advanced NSCLC patients who were previously treated and PD-L1 positive ≥ 1% and driver mutation-negative compared Pembrolizumab with chemotherapy (Docetaxel). It showed more prolonged overall survival for Pembrolizumab [246].
Atezolizumab
As first-line therapy
It is recommended as first-line therapy with ABCP regimen (Atezolizumab + Bevacizumab + Carboplatin + Paclitaxel) in patients with metastatic non-squamous NSCLC [247]. IMpower 150, a phase III randomized trial, evaluated the efficacy of first-line therapy with the ABCP regimen in patients with metastatic non-squamous NSCLC vs. Bevacizumab and chemotherapy, resulting in better MS (19.2 vs. 14.7 months) [248].
As subsequent therapy
Patients with metastatic NSCLC, during or after systemic therapy, are recommended with Atezolizumab as subsequent therapy. In a phase III randomized trial, OAK, Atezolizumab, and Docetaxel were compared as subsequent therapy in metastatic NSCLC patients, where Atezolizumab exhibited a better OS. [249, 250].
Durvalumab
PACIFIC, In a phase III randomized trial, PACIFIC, the efficacy of adjuvant treatment with Durvalumab was estimated as consolidation immunotherapy compared to placebo with unresectable stage III NSCLC after receiving treatment with concurrent treatment chemoradiation. The result showed increased OS and PFS (17.2 vs. 5.6 months) after Durvalumab consolidation therapy [251].
Nivolumab with or without ipilimumab
Nivolumab and Ipilimumab are immune checkpoint inhibitors with a complementary mechanism of action on T-cells where Nivolumab inhibits PD-1 receptors, and Ipilimumab is a human CTLA-4 blocking antibody.
As first-line therapy
In a phase III randomized trial, CheckMate 227, first-line Nivolumab/Ipilimumab was evaluated in comparison to Nivolumab monotherapy and chemotherapy in patients with metastatic NSCLC with high TMB (TMB ≥ 10 mutations per megabase) and negative for any driver mutations. It reports a better median PFS for Nivolumab/Ipilimumab (7.2 vs. 5.5 months) [169].
As subsequent therapy
In CheckMate 057, a phase III randomized trial, the efficacy of Nivolumab was compared with Docetaxel as subsequent therapy for metastatic non-squamous NSCLC patients during or after first-line chemotherapy. The trial reports better MS in the Nivolumab arm (12.2 vs. 9.4 months) [166]. In another phase III randomized trial, CheckMate 017, the efficacy of Nivolumab was also compared with Docetaxel as subsequent therapy for metastatic squamous cell NSCLC patients who had disease progression after chemotherapy. The report showed better MS with Nivolumab (9.2 vs. 6 months) [167].
Chemo-immunotherapy in SCLC
Immunotherapy is only indicated in patients with extensive-stage SCLC
As first-line therapy
IMpower 133, a phase III randomized trial with ED-SCLC patients, adding Atezolizumab to Platinum and etoposide demonstrates improved MS (12.3 vs. 10.3 months) than platinum plus etoposide, which was standard of care for many years [252]. The observed survival advantage in IMpower 133 trial is independent of the PD-L1 expression and the TMB levels. Atezolizumab, in combination with the etoposide-platinum, is now recommended as first-line therapy by the NCCN panel for patients with ED-SCLC [243, 253]. CASPIAN trial is another randomized phase III trial that evaluated Durvalumab with etoposide-platinum in comparison to etoposide-platinum only as first-line therapy for ED-SCLC patients. Durvalumab, in combination with chemotherapy, significantly improved the OS [254].
As subsequent therapy
Pembrolizumab is recommended as subsequent therapy in patients with relapsed SCLC regardless of PD-L1 level following the recent analysis of two studies, KEYNOTE 028 and KEYNOTE 158 [255]. Nivolumab alone or with Ipilimumab is recommended as new subsequent therapy for patients who have relapsed within 6 months after first-line therapy. Checkmate 331, a randomized phase III trial, suggested using Nivolumab monotherapy. Also, CheckMate 032, a phase II trial, reported using Nivolumab in combination with Ipilimumab in relapsed patients [227]. Patients who progress after receiving first-line Atezolizumab should not be treated with any other immunotherapy.
Radiation and immunotherapy
Though few trials are ongoing to evaluate the efficacy of any immune CKIs for use as concurrent therapy with radiation in cases of early or locally advanced lung cancer, there is no definite recommendation to date [256]. After the PACIFIC trial, Durvalumab has been recently recommended as consolidation immunotherapy in patients of unresectable stage III NSCLC post-treatment with concurrent chemoradiation [166].
Immunomodulatory nanomedicine for use in lung cancer
Our prior discussion shows that immune stimulation is required for cancer treatment to detect and annihilate the non-self-antigens and create a memory effect as a future remedy. While a myriad of immunotherapeutics has obtained commendable results in the treatment of various cancers, they also faced some daunting challenges such as low water solubility, poor pharmacokinetic profiles viz., less absorption, less accumulation in the tumor region, thereby less bioactivity after prolonged circulation, and enhanced immune-mediated off-target toxicity [257, 258]. To our intrigue, nanotechnology, with all its propitious facets, is capable of addressing the existing and ensuing issues, thereby accomplishing the expected achievement level in terms of therapeutic benefits [259–262]. With a better understanding of the tumor microenvironment, smart stimuli-responsive nanocarriers are being developed to take advantage of acidic pH, hypoxia, increased ATP synthesis, changed redox state of cancer cells, and other factors [263]. It turns out that nanoparticles enhance the benefits of cancer immunotherapy via (i) providing protection to antigens and adjuvants, (ii) simultaneous delivery to the APC (iii) TME reprogramming to recommence immune surveillance.
As of now, a multitude of nanomaterials have demonstrated their immunomodulatory potential in pre-clinical, and few of them have undergone various phases of clinical trials [264, 265]. For instance, a liposomal cancer vaccine (L-BLP25) was developed by Oncothyreon Canada Inc., where the antigen tecemotide (carcinoma-associated human MUC-1) and an adjuvant 3-O-Deacyl-4’-monophosphoryl lipid A (MPL) were inserted into the lipid bilayer made up of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dimyristoyl-sn-glycero-3-phospho-(1'-rac-glycerol) (DMPG), and cholesterol. Of many clinical trials performed with it worldwide, a phase IIb trial (NCT00157209) with patients with stage IIIB or IV NSCLC demonstrated an increment of 4.2 months in MS in the group receiving L-BLP25 compared to the cohort receiving BSC only. In a subset comprising patients with stage IIIB loco-regional NSCLC, an enhancement of 17.3 months in MS was observed, where the treatment group showed 49% 3-year survival compared to the control group with BSC showing 27% [266]. More light should be shed on the tailoring of patient-oriented cancer immunotherapy in concordance with the eventful and changeful dynamics of TME, which will help determine the timing and dosing of the therapeutic schedule. For instance, another phase III trial revealed that the liposomal tecemotide vaccine with concurrent chemoradiotherapy (NCT00409188) improved the OS to 9 months while sequential administration of chemoradiotherapy could not extend the OS, which hints at the importance of the timing of combinatorial therapy [267]. Table 3 summarizes a few clinical trials where the safety and efficacy of nanomaterials were evaluated for use in lung cancer immunotherapy.
Table 3.
Clinical trials where the safety and efficacy of nanomaterials were evaluated for use in lung cancer immunotherapy
| Clinical Trial ID | Drug | Delivery System | Cancer | Status | Duration | Sponsor/ Agency |
|---|---|---|---|---|---|---|
| NCT 02,049,151 | Tecemotide Following Concurrent Chemo-radiotherapy | Liposome | Unresectable Stage III NSCLC | Phase III (USA) | 2014–2015 | EMD Serono/Oncothyreon Canada Inc |
| NCT 00,960,115 | Tecemotide Following Chemotherapy | Liposome | Unresectable Stage III NSCLC | phase I/II (Japan) | 2009–2015 | Merck KGaA, Darmstadt, Germany |
| NCT 01,015,443 | Tecemotide Following primary Chemo-radiotherapy | Liposome | Unresectable Stage III NSCLC | Phase III (Asian population) | 2009–2015 | Merck KGaA, Darmstadt, Germany |
| NCT 00,828,009 | Tecemotide with bevacizumab after chemotherapy and radiation therapy | Liposome | Unresectable Stage IIIA/IIIB NSCLC | Phase II (USA) | 2010–2019 | ECOG-ACRIN Cancer Research Group |
| NCT 00,157,196 | Tecemotide with single-dose low CPA | Liposome | Stage IIIA NSCLC | Phase II | 2005–2012 | Merck KGaA, Darmstadt, Germany |
| NCT 00,157,209 | Tecemotide with single-dose low CPA | Liposome | Stage IIIB/Stage IV NSCLC | Phase IIB (Germany) | 2000–2012 | Merck KGaA, Darmstadt, Germany |
| NCT 00,409,188 | Tecemotide with single-dose low CPA | Liposome | Unresectable Stage III NSCLC | Phase III (23 countries) | 2007–2012 | EMD Serono |
| NCT 03,836,352 | DPX-Survivac with low dose Cyclophosphamide & Pembrolizumab | Liposome | NSCLC and other carcinomas | Phase II (USA/Canada) | 2018–2023 | ImmunoVaccine Technologies, Inc. (IMV Inc.) |
| NCT 00,291,473 | Mixed cancer vaccines, CHP-HER2 and CHP-NY-ESO-1 with OK-432 (Picibanil) | Cholesterol-Bearing Hydrophobized Pullulan | Lung cancer and other carcinomas | Phase I (Japan) | 2005–2008 | Ludwig Institute for Cancer Research |
| NCT 01,853,878 | Recombinant PRAME protein combined with the AS15 Adjuvant System GSK2302032A | Liposome | NSCLC after removal of the tumor | Phase II (9 countries) | 2013–2016 | GlaxoSmithKline |
| NCT 01,258,868 | Tumor Cell Vaccines with ISCOMATRIX Adjuvant and Celecoxib | Liposome | Lung cancer and other carcinomas | Phase I (USA) | 2010–2016 | National Cancer Institute (NCI) |
Few preclinical studies also elicited remarkable potential of nano-platforms for cancer immunotherapy. Interestingly, Moon and colleagues in 2017 synthesized a stable and homogeneous lipoprotein nanodisc, comprising phospholipids and 22 amino acid apolipoprotein mimetic peptides, to deliver neoantigen vaccines to draining lymph nodes. The nanodiscs evoked a strong anti-tumor T-cell response, eradicating established tumors and inhibiting metastatic tumor growth on murine lungs [268]. Alongside tissue and cellular targeting, molecular targeting has also shown great potential in cancer treatment, and their combinations with conventional chemotherapy have improved PFS in a phase III clinical trial with NSCLC patients having EGFR mutations [269]. It has also been proven now that the molecular targeting drugs can initiate immune responses via various mechanisms such as (i) aiding in antigen presentation by APC, (ii) instigating ICD in tumor cells, (iii) promoting T cell infiltration in TME, (iv) triggering NK cells, (v) attenuating the number of MDSCs, Treg, TAMs in TME. Nanotechnology and nanomaterials were used to improve molecularly targeted immunomodulation [270]. In 2020, Norvaline/Sunitinib encapsulating CuS photo-thermal nanoparticles were developed by Domvri et al. to target and exhaust MDSC subsets in lung TME. A549 tumor xenograft experiment revealed a marked anti-tumor effect simultaneously evoking strong innate and adaptive immune responses. It was shown that tumor infiltration of both CD8 + and CD4 + T cells was enhanced, and NK cells were activated with the diminution of MDSCs and Foxp3 + Treg cells (immune tolerance) [271]. A significant number of studies are ongoing to explore further the role of immunomodulatory nanomedicine in lung cancer immunotherapy, and we envisage an upsurge in their implications in the near future.
Immunomodulatory nutraceuticals in lung cancer
In recent years, the use of nutraceuticals in the prevention and supportive care of cancer patients has drawn significant attention from researchers across the globe, and enormous efforts have been directed toward deciphering their therapeutic role and immunomodulatory mechanism of action in different cancers. For instance, the impact of curcumin had already been demonstrated by various groups in many tumor growth inhibition studies, which revealed its anti-cancer, anti-angiogenesis, and anti-metastatic properties. Recently, Zou et al. conducted a study with lung cancer patients, which showed that a two-week treatment with curcumin arrested the Treg cells and enhanced peripheral Th1 cells. Importantly, a conversion of Tregs to Th1 cells was observed via downregulating transcription of Foxp3 and upregulating the expression of IFN-γ, which can be attributed to curcumin treatment [272]. Even more recently, in a mechanistic investigation, dose-dependent regulation of PD-L1 expression by resveratrol was observed. At a lower concentration (< 5 μM), it induced PD-L1 expression in various lung cancer cells (H1299, A549, H460) via activating the Wnt pathway, but it inhibited PD-L1 expression at a higher dose (> 40 μM) [273]. Other nutraceuticals include apigenin, luteolin, phloretin, saponin, capsaicin, gallic acid, caffeic acid phenethyl ester, zerumbone, quercetin, etc. have also demonstrated their immunomodulatory potential against lung cancer. We provide a collection of evidence accumulated in recent years for immunomodulatory nutraceutical intervention in Table 4 [274–283]. It has been demonstrated that tumor-related inflammatory responses had the unanticipated effect of tumorigenesis and progression, assisting incipient abysmally grown tumors. Inflammation promotes neoplastic progression by generating an increasing number of signaling molecules, including EGF.
Table 4.
Anti-proliferative, anti-inflammatory, and antioxidant potential of nutraceuticals against lung cancer
| Target | Compound | Source | Bioactivity |
|---|---|---|---|
| Lung Cancer | Curcumin | Curcuma longa | Converted Foxp3 + regulatory T cells to Th1 cells in patients with lung cancer |
| Resveratrol | grapes, wine, peanuts | Enhanced binding of β-catenin/TCF to PD-L1 promoter and increased PD-L1 expression. A higher dose (> 40 μM) resulted in a progressive reduction of PD-L1 | |
| Apigenin | Parsley | Suppressed the translocation of NF-κB from the cytoplasm to the nucleus, which further inhibited target genes that block apoptosis | |
| Phloretin | Apples | Exerted anti-inflammatory activities on A549 cancer cell lines by reducing intercellular adhesion molecule 1 (ICAM-1) and cyclooxygenase (COX)-2 expression and suppressing monocyte adhesion through inhibition of MAPK, PI3K/Akt, and NF-κB signaling pathways | |
| Edible macro-fungi | beta-glucan | Significantly increased IFN-γ mRNA expression, increased M1 phenotype, and attenuated M2 phenotype of TAM | |
| Saponin | P. polyphylla Smith var. chinensis (Franch.) Hara | Decreased serum levels of TNF-α, IL-8, and IL-10, decreased expressions of proinflammatory cytokines MCP-1, IL-6, and TGF-β1 | |
| Capsaicin | Chili | Inhibited the upregulation of TNF-α, IL-6, COX-2, and NF-κB expression | |
| Luteolin | Broccoli, Apple skin | Inhibited monocyte recruitment and cancer cell migration via suppression of the TAM-secreted CCL2 | |
| Gallic acid | Grape seeds, rose flowers, sumac, oak, and witch hazel | Suppressed LPS-induced NF-κB activation in A549 lung cancer cells, inhibited IFN-γ, IL-6, IL-1β & NF-κB regulated anti-apoptotic genes expression | |
| Caffeic acid phenethyl ester | Propolis | Inhibited NF-kB, caffeic acid phenethyl ester significantly diminished the induction of inflammatory gene | |
| Zerumbone | Zingiber officinalis | Inhibited lung carcinogenesis by modulating the expression of NF-κB and heme oxygenase-1 | |
| Quercetin | Grapes, Berries | Suppressed the nuclear translocation of NF-κB and reduced levels of inflammatory cytokine TNF-α, IL-1, and IL-6, significantly increasing the NK-cell-mediated cytotoxic activity against lung cancer cells | |
| EPA, DHA, and Se | Fish oil and Selenium yeast | Synergistically decreased the population of splenic Tregs and MDSCs and thus augmented host anti-tumor immunity against lung carcinoma |
It has been elicited that tumor-related inflammatory responses had the unanticipated effect of exacerbating tumorigenesis and progression, helping incipient abysmally grown tumors acquire cancer-specific salient characteristics and promote their impending development into full-blown cancers. Inflammation contributes to neoplastic growth by providing a growing list of signaling molecules: EGF, VEGF, FGF2, chemokines, cytokines that promote the inflammatory state, and matrix-degrading MMPs, cysteine cathepsin proteases, heparanase, and inductive signals that lead to activation of epithelial-mesenchymal transition (EMT), etc. Moreover, inflammatory cells secrete chemicals, particularly reactive oxygen species (ROS), which are proactively mutagenic for neighboring tumor cells, facilitating their genetic evolution.
oxidant properties have extensively been used in research to explore their anti-cancer, anti-angiogenesis, and anti-metastatic properties [284]. Anti-inflammatory agents have increasingly been used as effective adjuvants for conventional therapies, and three mechanisms have been proposed—(i) chemo-protection, (ii) alterations in pharmacokinetics or metabolism, and (iii) chemo-sensitization [285, 286]. Notably, a study by Menendez and colleagues in 2016 revealed that oral intake of silibinin significantly inhibited brain metastasis of NSCLC patients. Marked tumor growth inhibition was also shown in NSCLC xenograft by oral gavage of silibinin [287]. Taken together, therapeutic immunomodulatory nutraceutical intervention has conferred several benefits in lung cancer in both preclinical and clinical settings.
Ongoing clinical trials for immunotherapeutics
Different clinical trials are ongoing to assess various existing and new immunotherapeutic agents developed worldwide to assess their application in the management of lung cancer. A few recent trials conducted on NSCLC and SCLC patients are mentioned in Tables 5 and 6 [242, 243]. It is to be noted that the important phase II/III trials whose results have been analyzed have already been discussed in the previous sections. A few important ongoing trials are discussed below.
Table 5.
Ongoing randomized phase II and phase III Trials in Early and Locally Advanced NSCLC
| Neoadjuvant Strategy | Trial Identifier (NCT reference) | Phase | Drug/Treatment |
|---|---|---|---|
| IB-IIIA | CheckMate 816 (NCT 02,998,528) | III | Nivo + Ipi vs. chemotherapy |
| IIIA | (NCT03081689) | II | Nivo + chemotherapy |
| IB (> 3 cm)-IIIA | TOP 1501 (NCT02818920) | II | Pembro |
| IB (> 4 cm)-IIIA | PRINCEPS (NCT02994576) | II | Atezo |
| IB-IIIA | MAC (NCT02716038) | II | Atezo + chemotherapy |
| I (> 2 cm)-IIIA | (NCT02904954) | II | Durva or Durva + SRBT |
| IB(> 4 cm)-IIIA | ANVIL (NCT 02,595,944) | III | Nivo vs Obs |
| IB(> 4 cm)-IIIA | PEARLS(NCT02504372) | III | Pembro vs. placebo |
| IB(> 4 cm)-IIIA | IMpower010(NCT02486718) | III | Atezo vs. BSC |
| IB(> 4 cm)-IIIA | BR-31(NCT02273375) | III | Durva vs. placebo |
| Unresectable III A/B | RTOG3505(NCT02768558) | III | CRT Nivo vs. placebo |
| Early stage unresected NSCLC | NCT03833154 | III | Durvalumab + SBRT vs placebo + SBRT vs Osimertinib + SBRT |
| IIA- IIIB NSCLC | ALCHEMIST (NCT04267848) | III | Pembrolizumab + chemotherapy |
| Early stage NSCLC | CANOPY-N(NCT03968419) | II | Canakinumab ± Pembro |
| III-IV NSCLC | NCT03793179 | III | Pembro ± chemotherapy |
| III-IV NSCLC | IGNYTE(NCT03767348) | II | RP 1 ± Nivolumab |
| PD-L1 expressing NSCLC | NCT04432207 | I | IMU-201 (PD1-Vaxx) |
| III-IV NSCLC | NCT04007744 | I | Pembro + Sonidegib |
Abbreviations: Nivo Nivolumab, Ipi ipilimumab, Pembro Pembrolizumab, Atezo Atezolizumab, Durva Durvalumab, SBRT Stereotactic body radiotherapy, Obs Observation, BSC Best supportive care, CRT chemoradiotherapy
Table 6.
Ongoing Clinical Trials Investigating Immunotherapy in SCLC
| Trial Identifier (NCT reference) | Drug/Target | Phase | Treatment Regimen |
|---|---|---|---|
| Limited stage | |||
| NCT02402920 | Pembro/PD-1 | I | MK-3475 + PE + Thoracic RT |
| NCT02934503 | Pembro/PD-1 | II | Pembro + PE + Thoracic RT |
| Extensive stage | |||
| NCT03066778 | Pembro/PD-1 | III | Pembro + PE vs Placebo + PE |
| NCT02763579 | Atezo/PD-L1 | I/II | Atezo + Carboplatin + Etoposide Vs. Placebo + Carboplatin + Etoposide |
| Extensive stage-Maintenance | |||
| NCT03043599 | Ipi Nivo/CTLA-4 PD-1 | I/II | Consolidative Ipi + Nivo and maintenance Nivo with Thoracic RT after platinum-based chemotherapy |
| RAPTOR (NCT04402788) | Atezo | II/III | Atezo vs Atezo + RT |
| Extensive stage-Second-line and beyond | |||
| NCT03026166 | Ipi Nivo/CTLA-4 PD-1 | I/II | Rova-T and Nivo or Rova-T and Nivo plus Ipi 1 mg/kg or Rova-T and Nivo plus Ipi 3 mg/kg |
Abbreviations: Pembro Pembrolizumab, PE platinum/etoposide, Rt Radiotherapy, Atezo Atezolizumab, PD-1 Programmed cell death-1, Ipi ipilimumab, Nivo Nivolumab, CTLA-4 cytotoxic T-lymphocyte antigen, Rova-T Rovalpituzumab tesirine, Durva Durvalumab
CheckMate 816 is a phase III trial among 326 stage IB-IIIA NSCLC patients for comparing treatment outcomes with Nivolumab and Ipilimumab versus standard chemotherapy. PEARLS is a phase III trial in stage IB-IIIA of 1380 NSCLC patients comparing DFS between Pembrolizumab versus placebo. IMpower010, a phase III trial in stage IB-III A of 1127 NSCLC patients, is directed to assess the difference in DFS between Atezolizumab and best supportive care. Another crucial ongoing phase III trial of unresectable III A/B, 660 NSCLC patients is RTOG3505 and aims to compare OS and PFS after chemotherapy followed by Nivolumab versus placebo. Another phase III ALCHEMIST clinical trial (ANVIL) examines the effect of adjuvant Nivolumab in OS and/or DFS over standard observation after surgery and standard adjuvant therapy in 714 stage IB-IIIA NSCLC patients. The other notable phase III trial is NCT03066778, in 430 extensive-stage SCLC patients expressing PD-1 compares PFS with Pembrolizumab and Platinum/Etoposide versus placebo and Platinum/Etoposide. In another ongoing phase II/III trial (RAPTOR) with extensive-stage SCLC patients in maintenance therapy,138 patients in phase II, and 186 patients in phase III are compared for PFS with Atezolizumab alone versus Atezolizumab and radiotherapy. While NCT02934503: This is a phase II trial on 60 extensive stages SCLC patients assessing progression-free survival with Pembrolizumab, Platinum/Etoposide, and Radiotherapy.
Major obstacle and future perspective for lung cancer treatment
Immunotherapy has shown tremendous promise in lung cancer therapy but is still in its infancy. A significant hurdle is the absence of optimal appropriately standardized in vitro and in vivo laboratory-based, preclinical, and clinical model systems in evaluating the efficacy, mechanism, kinetics, and toxicity of immunotherapy and immune modulators [288]. Immunocompetent mouse-in-mouse models are often used, including the genetically engineered mouse models (GEMMs), chemically induced models, and syngeneic tumor graft models [289, 290]. Though existing models address some aspects, they do not provide the complete coverage needed to understand the basic mechanisms of immune biology and to evaluate new immunotherapies. Humanized mice, or human immune system (HIS) mice, which have both the human immune system and human tumors, are increasingly employed in preclinical immunotherapy investigations but are extremely costly [291]. Organoids or tumoroid with immune cell 3D co-culture models and microfluidic-based organoids-on-a-chip models are developed to use patient-derived tumor cells. These technologies have great potential but are still in the early stage of development. Thus, there is an urgent need to develop trustworthy models for understanding the dominant drivers of cancer immunity, immune mechanism, therapeutics, primary and secondary immune escape, synthetic immunity, and toxicity studies for translational cancer immunotherapy.
Further research is essential to understand better different immune aspects of lung cancer, including immune escape, immunosuppression, immune editing, and tumor-intrinsic adaptive response to immunotherapeutic stress, to reactivate and reliably channel the patient's immunity against cancer. Combination immunotherapy is effective, but there is no consensus on selecting the treatment strategy. Future NSCLC therapies will possibly comprise a combination of chemotherapy, neoantigen vaccinations, and several ICIs to target the rewired tumor signaling pathways. In combination with ipilimumab, nivolumab is the most potential immunotherapy cocktail for advanced NSCLC patients. The prospect of different classes of novel immune modulators in combination immunotherapy is almost unexplored. The safety, tolerability, and effectiveness of monotherapy and combination immunotherapeutics are now being studied in clinical trials. Epigenetic alterations in a complex interaction with genetic alterations lead to lung tumorigenesis. Changes in the epigenetic landscape can cause dysregulation of oncogenes and tumor suppressor genes, leading to heightened proliferation, faster cell cycles, apoptosis resistance, and immune modulation [292]. Epigenetic therapy is mainly focused on DNA methyltransferase inhibitors (DNMTi), histone deacetylase inhibitors (HDACi), Janus kinase 2 inhibitors and RNA-based therapeutics in combination with immunotherapeutics can boost the effectiveness of current lung cancer therapies for long-term patient survival [293]. Co-targeting tumor-intrinsic and tumor ecosystem-associated adaptive stress pathways, including metabolic, oxidative, endoplasmic reticulum, DNA damage, and replication stress response, can help design translatable combination therapies. Immunotherapeutics can be combined with epigenetic inhibitors (e.g., DNMTi, HDACi, EZH2i) to target epigenetics-mediated tumorigenesis and immunosuppression.
The advent of bulk and single-cell multi-omics studies has paved the way to evaluate and customize immunotherapy at a personalized level. Each patient's tumor is unique, and the collection of all somatic cancer mutations found in a single tumor is termed ‘mutanome’. Lung cancer and smoking are interlinked and may exhibit smoking-associated neoepitope signatures. Personalized and unique lung cancer-specific neoantigens can be detected and.
targeted by ACT therapy. The ACT arsenal for lung cancer treatment is expanding. CAR T, TCR, and TIL therapy have made significant clinical inroads, but several challenges exist and are discussed in the respective section. Chemokines influence T cell recruitment and infiltration into lung tumors, and a higher CD4 + and CD8 + T lymphocyte infiltration is a favorable prognostic indicator [294]. Jin et al. recently used CCR6-expressing CAR T cells to target lung cancer using a xenograft mouse model and showed promising T cell infiltration and tumor killing [295]. Earlier Adachi et al. engineered CAR-T cells to express IL-7 and CCL19 (crucial for maintaining T-cell zones in lymphoid organs) and showed promising results with Lewis Lung carcinoma in a mouse model [296]. Additionally, improvements in multi-omic platforms at a single cell level and access to publicly available data sets can help better apprehend the immune landscape for educated therapeutic selection.
A personalized immunotherapy approach needs to be investigated in lung cancer. Tumor heterogeneity can be a significant bottleneck in designing personalized immunotherapies. The immune microenvironment is dynamic, and spatiotemporal analysis of different types of lung cancer can help understand the immune repertoire, antigen-presentation modes, and immune editing. Recent developments in multi-spectral analysis techniques, like multiplex immunofluorescence (MIF), Imaging mass cytometry (IMC), Chipcytometry, Multiplexed Ion Beam Imaging (MIBI), DNA barcoding-based mIHC/IF, and insitu-plex can help in deciphering the tumor immune repertoire [297]. Whole exome sequencing (WES), RNA-seq, single-cell sequencing, and TCR sequencing can reveal the tumor mutanome and cell-specific TCRs that are neoantigen-specific and aid in the cancer vaccination approach [298]. Several factors like TCR diversity, TCR degeneracy, neoantigen clonality, neoantigen subtype, and differential agretopicity index offer challenges. Each patient's cancer cells have a unique cocktail of neoantigen–MHC complexes (termed the neoantigenome). Multiple AI-based multi-component computational algorithms can examine the binding complementarity of the mutant peptide and the patient's HLA alleles and evaluate the potential to develop an anti-tumor T cell response. AI approaches like TSNAD, pVAC-Seq, INTEGRATE-neo, NetMHCpan, MARIA, EDGE, and DeepHLApan employ multi-layer architecture to find patterns and predict MHC-I/II binding and neoantigen binding efficacy and immunogenicity [299–301]. Though in silico binding prediction can yield helpful information, LC–MS/MS analysis of MHC molecules immunoprecipitation and peptide identification will produce an accurate and robust database, e.g., The Immune Epitope Database (IEDB) [301]. Personalized neoantigen identification can help develop next-generation immunotherapeutics. For preclinical and clinical applications, the binding affinity of the neoantigen to the corresponding MHC is predicted, and affinities greater than 500 nM are considered immunogenic neoepitopes and subsequently selected for the development of customized cancer vaccines [302]. There is an urgent need to discover lung cancer-specific composite biomarkers for categorizing tumor immunogenicity, patient stratification, pharmacodynamic prediction, and finalizing regulatory endpoint to improve lung cancer immunotherapy. Recent advances in AI-based algorithms and models will soon be able to predict immunotherapy and combination therapy responses at a personalized level to further the efficacious use of immunotherapy at a clinical level [303].
Though immunotherapy incites immunememory and is the most promising way to treat cancer and is better tolerated with minimum side effects as per clinical data, some patients suffer from immune-related adverse effects (irAEs) [304]. The side effects include flu-like symptoms, skin rash, pain, edema, heart palpitations, diarrhea, an overly activated immune status, and damaging organ systems. Cytokine release syndrome and onset of diabetes are also observed in CAR-T and immunotherapy patients. Interstitial and alveolar infiltrates followed by pneumonitis is the most common immune-related adverse event in the lung [305]. Though immunosuppressive corticosteroids are the choice for treating irAEs but are also reported to reduce the efficacy of the immunotherapy. Studies regarding immunomodulatory nanomaterials and nutraceuticals may open up new avenues in addressing irAEs and autoimmune toxicities. Additional research is required to comprehend the process of irAE better to manage the adverse effects of lung cancer immunotherapy.
Conclusion
With the discovery of immunotherapy, the therapeutic paradigm for patients with advanced lung cancer has fundamentally transformed lung cancer treatment and is still evolving. ICIs have improved patient OS while causing fewer adverse effects than traditional chemotherapeutic medicines and have become an integral part of treatment algorithms. Several possible therapeutic options are available for advanced-stage lung cancer patients, from single-agent immunotherapy to quadruple therapy, which combines immunotherapy with chemotherapy and anti-vascular endothelial growth factor medications. In order to treat advanced lung cancer patients, the U.S. FDA has approved immunotherapy medications alone or in combination with other immunotherapies and chemotherapy, as reviewed in this article. Generation of resistance to ICIs, whether intrinsic or acquired, is a significant issue for the oncology community. Cellular therapy is a potential and practical addition to the arsenal of lung cancer immunotherapies. Due to the absence of tumor-specific antigens, a hostile TME, and toxicity, cellular treatment is an attractive but unquestionably challenging prospect. Clinical studies evaluate innovative therapeutic methods, including the combination and sequencing of PD-1/PD-L1 inhibitors with different ICIs and DNA repair targeting medicines. Overall, the therapeutic advantages of ICIs and ACT have exhibited promising trends for efficacious lung cancer therapy in the future.
Acknowledgements
A.M. takes this opportunity to acknowledge Raktim Chattopadhyay of Esperer Onco Nutrition Pvt. Ltd. for his constant support. M.K.P. acknowledges S. Dubinett and B. Gomperts from UCLA for providing constant support and mentoring.
Abbreviations
- PAH
Polycyclic aromatic hydrocarbons
- WHO
World Health Organization
- NSCLC
Non-small cell lung cancer
- SCLC
Small cell lung cancer
- LUAD
Lung adenocarcinoma
- LUSC
Lung squamous cell carcinoma
- LCC
Large cell carcinoma
- LDCT
Low-dose computed tomography
- LD-SCLC
Limited disease SCLC
- ED-SCLC
Extensive disease SCLC
- MS
Median survival
- TSA
Tumor-specific antigen
- TAA
Tumor-associated antigens
- NK
Natural killer cells
- DC
Dendritic cells
- MΦ
Macrophages
- APC
Antigen presenting cell
- mAb
Monoclonal antibody
- MHC
Major histocompatibility complex
- IASLC
International Association for the study of Lung Cancer
- CKI
Checkpoint inhibitor
- PD-1
Programmed cell death-1
- PD-L1
Programmed death-ligand 1
- FDA
U.S. Food and Drug Administration
- EGF
Epidermal growth factor
- BSC
Best supportive care
- CTA
Cancer testis antigen
- NY-ESO-1
New York esophageal squamous cell carcinoma 1
- MAGE-A3
Melanoma-associated antigen-A3
- DFS
Disease-free survival
- MUC-1
Mucin 1
- MVA
Modified Vaccinia Virus Ankara
- TGF-β2
Transforming growth factor β2
- CTL
Cytotoxic T lymphocytes
- ACT
Adoptive cell therapy
- TCR
T-cell receptor
- TIL
Tumor-infiltrating lymphocytes
- CAR-T
Chimeric antigen receptor-modified T cells
- CEA
Carcinoembryonic antigen
- MSLN
Mesothelin
- HER2
Human epidermal growth factor receptor 2
- GPC3
Glypican-3
- CAR T
Chimeric antigen receptor (CAR)-modified T cells
- TIL
Tumor-infiltrating lymphocyte
- TCR
T-cell receptor
- HLA
Human leukocyte antigen
- TMB
Tumor mutational burden
- BiTEs
Bispecific T-cell engagers
- ROR1
Receptor tyrosine kinase-like orphan receptor
- DNT
Double-negative T cells
- OV
Oncolytic Virus
- HSV-1
Herpes Simplex Virus-1
- CVB3
Coxsackievirus B3
- ICD
Immunogenic cell death
- OVV
Oncolytic vaccinia viruses
- SVV-001
Seneca Valley Virus isolate 001
- ADC
Antibody–drug conjugate
- BiTE
Bispecific antibodies
- irPFS
Immune-related progression-free survival
- OR
Objective response
- AE
Adverse event
- TIGIT
T cell immunoreceptor with Ig and ITIM domains
- BTD
Breakthrough therapy designation
- ORR
Overall response rate
- PPV
Personalized peptide vaccination
- TMB
Tumor mutational burden
- LEMS
Lambert-Eaton myasthenic syndrome
- EMT
Epithelial-mesenchymal transition
- ROS
Reactive oxygen species
- GEMM
Genetically engineered mouse models
- HIS-mice
Human immune system mice
- MPL
3-O-Deacyl-4’-monophosphoryl lipid A
- DPPC
1,2-Dipalmitoyl-sn-glycero-3-phosphocholine
- DMPG
1,2-Dimyristoyl-sn-glycero-3-phospho-(1'-rac-glycerol)
- MDSC
Myeloid-derived suppressor cells
- Tregs
Regulatory T cells
Authors’ contributions
AL, AM, AnM, MP, wrote the paper; PP, NS, PuP, BB, AnM, MP, edited the paper; MP made the figures; MP, final edits and submission. The author(s) read and approved the final manuscript.
Funding
No Funding available.
Availability of data and materials
Not applicable.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
On behalf of the author(s), the corresponding author certifies the accuracy of the content given to the journal. The corresponding author ensures that all the co-authors have agreed to all of the contents and will notify all the authors when the manuscript is accepted. The corresponding author is answerable to all the inquiries on behalf of all the co-authors. The corresponding author ensures that all authors have seen and approved the final version of the paper and that all are aware of the submission of the paper. The corresponding author is solely responsible for maintaining a proper communication with the journal and between co-authors before and after publication.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Anubhab Mukherjee, Email: anubhabrsv@gmail.com.
Manash K. Paul, Email: manashp@ucla.edu
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics 2022. CA Cancer J Clin. 2022;72(1):7–33. doi: 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- 2.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 3.American Cancer Society. Cancer Facts and Figures 2015. 2015. https://www.cancer.org/cancer-facts-and-figures-2015.pdf.
- 4.Kanwal M, Ding XJ, Cao Y. Familial risk for lung cancer. Oncol Lett. 2017;13(2):535–42. 10.3892/ol.2016.5518. Epub 2016 Dec 20. [DOI] [PMC free article] [PubMed]
- 5.Li Q, Yuan D, Ma C, Liu Y, Ma L, Lv T, et al. A new hope: the immunotherapy in small cell lung cancer. Neoplasma. 2016;63(3):342–350. doi: 10.4149/302_151001N511. [DOI] [PubMed] [Google Scholar]
- 6.Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83(5):584–594. doi: 10.1016/S0025-6196(11)60735-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JHM, Beasley MB, et al. The 2015 world health organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thor Oncol Publication Int Assoc Study Lung Cancer. 2015;10(9):1243–1260. doi: 10.1097/JTO.0000000000000630. [DOI] [PubMed] [Google Scholar]
- 8.Inamura K. Lung cancer: understanding its molecular pathology and the 2015 WHO classification. Front Oncol. 2017;7:193. doi: 10.3389/fonc.2017.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boloker G, Wang C, Zhang J. Updated statistics of lung and bronchus cancer in United States (2018) J Thorac Dis. 2018;10(3):1158–1161. doi: 10.21037/jtd.2018.03.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siegel RL, Miller KD, Jemal A. Cancer statistics 2018. CA Cancer J Clin. 2018;68(1):7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 11.Yasumoto K, Hanagiri T, Takenoyama M. Lung cancer-associated tumor antigens and the present status of immunotherapy against non-small-cell lung cancer. Gen Thorac Cardiovasc Surg. 2009;57(9):449–457. doi: 10.1007/s11748-008-0433-6. [DOI] [PubMed] [Google Scholar]
- 12.Gulley JL, Spigel D, Kelly K, Chandler JC, Rajan A, Hassan R, et al. Avelumab (MSB0010718C), an anti-PD-L1 antibody, in advanced NSCLC patients: A phase 1b, open-label expansion trial in patients progressing after platinum-based chemotherapy. J Clin Oncol. 2015;33(15_suppl):8034. doi: 10.1200/jco.2015.33.15_suppl.8034. [DOI] [Google Scholar]
- 13.Dyer O. US task force recommends extending lung cancer screenings to over 50s. BMJ. 2021;372:n698. doi: 10.1136/bmj.n698. [DOI] [PubMed] [Google Scholar]
- 14.Khanna P, Blais N, Gaudreau PO, Corrales-Rodriguez L. Immunotherapy comes of age in lung cancer. Clin Lung Cancer. 2017;18(1):13–22. doi: 10.1016/j.cllc.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Rolfo C, Caglevic C, Santarpia M, Araujo A, Giovannetti E, Gallardo CD, et al. Immunotherapy in NSCLC: a promising and revolutionary weapon. Adv Exp Med Biol. 2017;995:97–125. doi: 10.1007/978-3-319-53156-4_5. [DOI] [PubMed] [Google Scholar]
- 16.Arcaro A. Targeted therapies for small cell lung cancer: Where do we stand? Crit Rev Oncol Hematol. 2015;95(2):154–164. doi: 10.1016/j.critrevonc.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 17.Jotte R, Conkling P, Reynolds C, Galsky MD, Klein L, Fitzgibbons JF, et al. Randomized phase II trial of single-agent amrubicin or topotecan as second-line treatment in patients with small-cell lung cancer sensitive to first-line platinum-based chemotherapy. J Clin Oncol. 2011;29(3):287–293. doi: 10.1200/JCO.2010.29.8851. [DOI] [PubMed] [Google Scholar]
- 18.Slotman BJ, van Tinteren H. Which patients with extensive stage small-cell lung cancer should and should not receive thoracic radiotherapy? Transl Lung Cancer Res. 2015;4(3):292–294. doi: 10.3978/j.issn.2218-6751.2015.04.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalemkerian GP, Akerley W, Bogner P, Borghaei H, Chow LQ, Downey RJ, et al. Small cell lung cancer. J Natl Compr Cancer Netw JNCCN. 2013;11(1):78–98. doi: 10.6004/jnccn.2013.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kalemkerian GP, Gadgeel SM. Modern staging of small cell lung cancer. J Natl Compr Cancer Netw JNCCN. 2013;11(1):99–104. doi: 10.6004/jnccn.2013.0012. [DOI] [PubMed] [Google Scholar]
- 21.Spigel DR, Greco FA, Rubin MS, Shipley D, Thompson DS, Lubiner ET, et al. Phase II study of maintenance sunitinib following irinotecan and carboplatin as first-line treatment for patients with extensive-stage small-cell lung cancer. Lung Cancer (Amsterdam, Netherlands) 2012;77(2):359–364. doi: 10.1016/j.lungcan.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 22.Seegobin K, Majeed U, Wiest N, Manochakian R, Lou Y, Zhao Y. Immunotherapy in non-small cell lung cancer with actionable mutations other than EGFR. Front Oncol. 2021;11:750657. doi: 10.3389/fonc.2021.750657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forde PM, Kelly RJ, Brahmer JR. New strategies in lung cancer: translating immunotherapy into clinical practice. Clin Cancer Res. 2014;20:1067. doi: 10.1158/1078-0432.CCR-13-0731. [DOI] [PubMed] [Google Scholar]
- 24.Pardoll D. Does the immune system see tumors as foreign or self? Annu Rev Immunol. 2003;21:807–839. doi: 10.1146/annurev.immunol.21.120601.141135. [DOI] [PubMed] [Google Scholar]
- 25.Malhotra J, Jabbour SK, Aisner J. Current state of immunotherapy for non-small cell lung cancer. Transl Lung Cancer Res. 2017;6(2):196–211. doi: 10.21037/tlcr.2017.03.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raez LE, Cassileth PA, Schlesselman JJ, Sridhar K, Padmanabhan S, Fisher EZ, et al. Allogeneic vaccination with a B7.1 HLA-A gene-modified adenocarcinoma cell line in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2004;22(14):2800–7. doi: 10.1200/JCO.2004.10.197. [DOI] [PubMed] [Google Scholar]
- 27.Massarelli E, Papadimitrakopoulou V, Welsh J, Tang C, Tsao AS. Immunotherapy in lung cancer. Transl Lung Cancer Res. 2014;3(1):53–63. doi: 10.3978/j.issn.2218-6751.2014.01.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Zou ZH, Xia HL, He JX, Zhong NS, Tao AL. Strengths and weaknesses of immunotherapy for advanced non-small-cell lung cancer: a meta-analysis of 12 randomized controlled trials. PLoS ONE. 2012;7(3):e32695. doi: 10.1371/journal.pone.0032695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Couraud S, Zalcman G, Milleron B, Morin F, Souquet PJ. Lung cancer in never smokers--a review. Eur J Cancer (Oxford, England 1990) 2012;48(9):1299–311. doi: 10.1016/j.ejca.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 30.Noguchi M, Morikawa A, Kawasaki M, Matsuno Y, Yamada T, Hirohashi S, et al. Small adenocarcinoma of the lung. Histol Characteristics Prognosis Cancer. 1995;75(12):2844–2852. doi: 10.1002/1097-0142(19950615)75:12<2844::aid-cncr2820751209>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 31.Travis WD, Travis LB, Devesa SS. Lung cancer. Cancer. 1995;75(1 Suppl):191–202. doi: 10.1002/1097-0142(19950101)75:1+<191::AID-CNCR2820751307>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 32.Stellman SD, Muscat JE, Hoffmann D, Wynder EL. Impact of filter cigarette smoking on lung cancer histology. Prev Med. 1997;26(4):451–456. doi: 10.1006/pmed.1997.0212. [DOI] [PubMed] [Google Scholar]
- 33.Aros CJ, Paul MK, Pantoja CJ, Bisht B, Meneses LK, Vijayaraj P, et al. High-throughput drug screening identifies a potent wnt inhibitor that promotes airway basal stem cell homeostasis. Cell Rep. 2020;30(7):2055–64.e5. doi: 10.1016/j.celrep.2020.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kenfield SA, Wei EK, Stampfer MJ, Rosner BA, Colditz GA. Comparison of aspects of smoking among the four histological types of lung cancer. Tob Control. 2008;17(3):198–204. doi: 10.1136/tc.2007.022582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muscat JE, Stellman SD, Zhang ZF, Neugut AI, Wynder EL. Cigarette smoking and large cell carcinoma of the lung. Cancer Epidemiol Biomarkers Prev. 1997;6(7):477–480. [PubMed] [Google Scholar]
- 36.Brambilla EPB, Geisinger K, et al. et al. Large cell carcinoma. In: Travis WBE, Müller-Hermelink H, et al.et al., editors. World Health Organization Classification of Tumours Pathology and Genetics of Tumours of the Lung. Pleura, Thymus and Heart Geneva: WHO Press; 2004. pp. 45–50. [Google Scholar]
- 37.Chansky K, Sculier JP, Crowley JJ, Giroux D, Van Meerbeeck J, Goldstraw P. The international association for the study of lung cancer staging project: prognostic factors and pathologic TNM stage in surgically managed non-small cell lung cancer. J Thorac Oncol. 2009;4(7):792–801. doi: 10.1097/JTO.0b013e3181a7716e. [DOI] [PubMed] [Google Scholar]
- 38.Siegel RL, Miller KD, Jemal A. Cancer Statistics,2017. CA Cancer J Clin. 2017;67(1):7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 39.Thomas A, Rajan A, Giaccone G. Tyrosine kinase inhibitors in lung cancer. Hematol/Oncol Clinics North Am. 2012;26(3):589–605. doi: 10.1016/j.hoc.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salehi-Rad R, Li R, Paul MK, Dubinett SM, Liu B. The biology of lung cancer. Clin Chest Med. 2020;41(1):25–38. doi: 10.1016/j.ccm.2019.10.003. [DOI] [PubMed] [Google Scholar]
- 41.Ruwali M, Moharir K, Singh S, Aggarwal PK, Paul M. Updates in pharmacogenetics of non-small cell lung cancer. Pharmacogenetics. 2021:1–20. https://www.intechopen.com/books/10578.
- 42.S. Chauhan D, Mudaliar P, Basu S, Aich J, K. Paul M. Tumor-derived exosome and immune modulation. Extracellular vesicles - role in diseases pathogenesis and therapy [Working Title]. Physiology 2022
- 43.Genova C, Dellepiane C, Carrega P, Sommariva S, Ferlazzo G, Pronzato P, et al. Therapeutic implications of tumor microenvironment in lung cancer: focus on immune checkpoint blockade. Front Immunol. 2022;12:799455. doi: 10.3389/fimmu.2021.799455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sellars MC, Wu CJ, Fritsch EF. Cancer vaccines: Building a bridge over troubled waters. Cell. 2022;185:2770. doi: 10.1016/j.cell.2022.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelly RJ, Giaccone G. Lung cancer vaccines. Cancer J (Sudbury, Mass) 2011;17(5):302–308. doi: 10.1097/PPO.0b013e318233e6b4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Finn OJ. Cancer vaccines: between the idea and the reality. Nat Rev Immunol. 2003;3(8):630–641. doi: 10.1038/nri1150. [DOI] [PubMed] [Google Scholar]
- 47.Cuppens K, Vansteenkiste J. Vaccination therapy for non-small-cell lung cancer. Curr Opin Oncol. 2014;26(2):165–170. doi: 10.1097/CCO.0000000000000052. [DOI] [PubMed] [Google Scholar]
- 48.Saavedra D, Crombet T. CIMAvax-EGF: a new therapeutic vaccine for advanced non-small cell lung cancer patients. Front Immunol. 2017;8:269. doi: 10.3389/fimmu.2017.00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neninger Vinageras E, de la Torre A, Osorio Rodríguez M, Catalá Ferrer M, Bravo I, Mendoza del Pino M, et al. Phase II randomized controlled trial of an epidermal growth factor vaccine in advanced non-small-cell lung cancer. J Clin Oncol . 2008;26(9):1452–8. doi: 10.1200/JCO.2007.11.5980. [DOI] [PubMed] [Google Scholar]
- 50.Rodriguez PC, Popa X, Martínez O, Mendoza S, Santiesteban E, Crespo T, et al. A phase III clinical trial of the epidermal growth factor vaccine CIMAvax-EGF as switch maintenance therapy in advanced non-small cell lung cancer patients. Clin Cancer Res. 2016;22(15):3782–3790. doi: 10.1158/1078-0432.CCR-15-0855. [DOI] [PubMed] [Google Scholar]
- 51.Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev. 2002;188:22–32. doi: 10.1034/j.1600-065X.2002.18803.x. [DOI] [PubMed] [Google Scholar]
- 52.Gjerstorff MF, Andersen MH, Ditzel HJ. Oncogenic cancer/testis antigens: prime candidates for immunotherapy. Oncotarget. 2015;6(18):15772–15787. doi: 10.18632/oncotarget.4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grah JJ, Katalinic D, Juretic A, Santek F, Samarzija M. Clinical significance of immunohistochemical expression of cancer/testis tumor-associated antigens (MAGE-A1, MAGE-A3/4, NY-ESO-1) in patients with non-small cell lung cancer. Tumori. 2014;100(1):60–68. doi: 10.1177/1430.15817. [DOI] [PubMed] [Google Scholar]
- 54.Vansteenkiste JF, Cho BC, Vanakesa T, De Pas T, Zielinski M, Kim MS, et al. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17(6):822–835. doi: 10.1016/S1470-2045(16)00099-1. [DOI] [PubMed] [Google Scholar]
- 55.Gure AO, Chua R, Williamson B, Gonen M, Ferrera CA, Gnjatic S, et al. Cancer-testis genes are coordinately expressed and are markers of poor outcome in non-small cell lung cancer. Clin Cancer Res. 2005;11(22):8055–8062. doi: 10.1158/1078-0432.CCR-05-1203. [DOI] [PubMed] [Google Scholar]
- 56.John T, Starmans MH, Chen YT, Russell PA, Barnett SA, White SC, et al. The role of Cancer-Testis antigens as predictive and prognostic markers in non-small cell lung cancer. PLoS ONE. 2013;8(7):e67876. doi: 10.1371/journal.pone.0067876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murphy R, Green S, Ritter G, Cohen L, Ryan D, Woods W, et al. Recombinant NY-ESO-1 cancer antigen: production and purification under cGMP conditions. Prep Biochem Biotechnol. 2005;35(2):119–134. doi: 10.1081/PB-200054732. [DOI] [PubMed] [Google Scholar]
- 58.Raina D, Kosugi M, Ahmad R, Panchamoorthy G, Rajabi H, Alam M, et al. Dependence on the MUC1-C oncoprotein in non-small cell lung cancer cells. Mol Cancer Ther. 2011;10(5):806–816. doi: 10.1158/1535-7163.MCT-10-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Butts C, Murray N, Maksymiuk A, Goss G, Marshall E, Soulières D, et al. Randomized phase IIB trial of BLP25 liposome vaccine in stage IIIB and IV non-small-cell lung cancer. J Clin Oncol. 2005;23(27):6674–6681. doi: 10.1200/JCO.2005.13.011. [DOI] [PubMed] [Google Scholar]
- 60.Butts C, Maksymiuk A, Goss G, Soulières D, Marshall E, Cormier Y, et al. Updated survival analysis in patients with stage IIIB or IV non-small-cell lung cancer receiving BLP25 liposome vaccine (L-BLP25): phase IIB randomized, multicenter, open-label trial. J Cancer Res Clin Oncol. 2011;137(9):1337–1342. doi: 10.1007/s00432-011-1003-3. [DOI] [PubMed] [Google Scholar]
- 61.Butts C, Murray RN, Smith CJ, Ellis PM, Jasas K, Maksymiuk A, et al. A multicenter open-label study to assess the safety of a new formulation of BLP25 liposome vaccine in patients with unresectable stage III non-small-cell lung cancer. Clin Lung Cancer. 2010;11(6):391–395. doi: 10.3816/CLC.2010.n.101. [DOI] [PubMed] [Google Scholar]
- 62.Weng TY, Yen MC, Huang CT, Hung JJ, Chen YL, Chen WC, et al. DNA vaccine elicits an efficient antitumor response by targeting the mutant Kras in a transgenic mouse lung cancer model. Gene Ther. 2014;21(10):888–896. doi: 10.1038/gt.2014.67. [DOI] [PubMed] [Google Scholar]
- 63.Vansteenkiste JF, Cho BC, Vanakesa T, De Pas T, Zielinski M, Kim MS, et al. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17(6):822–835. doi: 10.1016/S1470-2045(16)00099-1. [DOI] [PubMed] [Google Scholar]
- 64.Lopes A, Vandermeulen G, Préat V. Cancer DNA vaccines: current preclinical and clinical developments and future perspectives. J Exp Clin Cancer Res. 2019;38(1):146. doi: 10.1186/s13046-019-1154-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Limacher JM, Quoix E. TG4010: A therapeutic vaccine against MUC1 expressing tumors. Oncoimmunology. 2012;1(5):791–792. doi: 10.4161/onci.19863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rochlitz C, Figlin R, Squiban P, Salzberg M, Pless M, Herrmann R, et al. Phase I immunotherapy with a modified vaccinia virus (MVA) expressing human MUC1 as antigen-specific immunotherapy in patients with MUC1-positive advanced cancer. J Gene Med. 2003;5(8):690–699. doi: 10.1002/jgm.397. [DOI] [PubMed] [Google Scholar]
- 67.Ramlau R, Quoix E, Rolski J, Pless M, Lena H, Lévy E, et al. A phase II study of Tg4010 (Mva-Muc1-Il2) in association with chemotherapy in patients with stage III/IV Non-small cell lung cancer. J Thorac Oncol. 2008;3(7):735–744. doi: 10.1097/JTO.0b013e31817c6b4f. [DOI] [PubMed] [Google Scholar]
- 68.Ramlau R, Quoix E, Rolski J, Pless M, Lena H, Lévy E, et al. A Phase II Study of Tg4010 (Mva-Muc1-Il2) in association with chemotherapy in patients with stage III/IV non-small cell lung cancer. J Thorac Oncol. 2008;3(7):735–744. doi: 10.1097/JTO.0b013e31817c6b4f. [DOI] [PubMed] [Google Scholar]
- 69.Zhao Y, Liu Z, Li L, Wu J, Zhang H, Zhang H, et al. Oncolytic adenovirus: prospects for cancer immunotherapy. Front Microbiol. 2021;12:707290. doi: 10.3389/fmicb.2021.707290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee JM, Lee M-H, Garon E, Goldman JW, Salehi-Rad R, Baratelli FE, et al. Phase I trial of intratumoral injection of CCL21 gene-modified dendritic cells in lung cancer elicits tumor-specific immune responses and CD8+ T-cell infiltration. Clin Cancer Res. 2017;23(16):4556–4568. doi: 10.1158/1078-0432.CCR-16-2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Salehi-Rad R, Li R, Liu B, Lim R, Tran L, Ong S, et al. Intratumoral CCL21-Gene Modified Dendritic Cells (CCL21-DC) Combined with Checkpoint Blockade in Murine Models of Non-Small Cell Lung Cancer (NSCLC) with Varying Mutational Load. B98 Targeting the Immune System in Lung Cancer Progression. 2019;199:A4034A. https://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2019.199.1_MeetingAbstracts.A4034.
- 72.Fu C, Zhou L, Mi Q-S, Jiang A. DC-based vaccines for cancer immunotherapy. Vaccines. 2020;8(4):706. doi: 10.3390/vaccines8040706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Srivatsan S, Patel JM, Bozeman EN, Imasuen IE, He S, Daniels D, et al. Allogeneic tumor cell vaccines: the promise and limitations in clinical trials. Hum Vaccin Immunother. 2014;10(1):52–63. doi: 10.4161/hv.26568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Suzuki H, Owada Y, Watanabe Y, Inoue T, Fukuharav M, Yamaura T, et al. Recent advances in immunotherapy for non-small-cell lung cancer. Hum Vaccin Immunother. 2014;10(2):352–357. doi: 10.4161/hv.26919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Miyazono K. Positive and negative regulation of TGF-beta signaling. J Cell Sci. 2000;113(Pt 7):1101–1109. doi: 10.1242/jcs.113.7.1101. [DOI] [PubMed] [Google Scholar]
- 76.Houot R, Schultz LM, Marabelle A, Kohrt H. T-cell–based Immunotherapy: adoptive cell transfer and checkpoint inhibition. Cancer Immunol Res. 2015;3(10):1115–1122. doi: 10.1158/2326-6066.CIR-15-0190. [DOI] [PubMed] [Google Scholar]
- 77.Tan E, Gakhar N, Kirtane K. TCR gene-engineered cell therapy for solid tumors. Best Pract Res Clin Haematol. 2021;34(3):101285. doi: 10.1016/j.beha.2021.101285. [DOI] [PubMed] [Google Scholar]
- 78.Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11(4):69. doi: 10.1038/s41408-021-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhong S, Cui Y, Liu Q, Chen S. CAR-T cell therapy for lung cancer: a promising but challenging future. J Thorac Dis. 2020;12(8):4516–4521. doi: 10.21037/jtd.2020.03.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Caliendo F, Dukhinova M, Siciliano V. Engineered cell-based therapeutics: synthetic biology meets immunology. Front Bioeng Biotechnol. 2019;7:43. doi: 10.3389/fbioe.2019.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xiao B-F, Zhang J-T, Zhu Y-G, Cui X-R, Lu Z-M, Yu B-T, et al. Chimeric antigen receptor T-cell therapy in lung cancer: potential and challenges. Front Immunol. 2021;12:782775. doi: 10.3389/fimmu.2021.782775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chocarro L, Arasanz H, Fernández-Rubio L, Blanco E, Echaide M, Bocanegra A, et al. CAR-T cells for the treatment of lung cancer. Life. 2022;12(4):561. doi: 10.3390/life12040561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Qu J, Mei Q, Chen L, Zhou J. Chimeric antigen receptor (CAR)-T-cell therapy in non-small-cell lung cancer (NSCLC): current status and future perspectives. Cancer Immunol Immunother. 2020;70(3):619–631. doi: 10.1007/s00262-020-02735-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bao C, Gao Q, Li L-L, Han L, Zhang B, Ding Y, et al. The application of nanobody in CAR-T therapy. Biomolecules. 2021;11(2):238. doi: 10.3390/biom11020238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hawkins ER, D’Souza RR, Klampatsa A. Armored CAR T-cells: the next chapter in T-cell cancer immunotherapy. Biologics Targets Ther. 2021;15:95–105. doi: 10.2147/BTT.S291768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhou S, Liu M, Ren F, Meng X, Yu J. The landscape of bispecific T cell engager in cancer treatment. Biomarker Res. 2021;9(1):38. doi: 10.1186/s40364-021-00294-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Saltos A, Khalil F, Smith M, Li J, Schell M, Antonia SJ, et al. Clinical associations of mucin 1 in human lung cancer and precancerous lesions. Oncotarget. 2018;9(86):35666–35675. doi: 10.18632/oncotarget.26278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wei X, Lai Y, Li J, Qin L, Xu Y, Zhao R, et al. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. Oncoimmunology. 2017;6(3):e1284722. doi: 10.1080/2162402X.2017.1284722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang C, Hao X. Prognostic significance of CD276 in non-small cell lung cancer. Open Med. 2019;14(1):805–812. doi: 10.1515/med-2019-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li F, Chen H, Wang D. Silencing of CD276 suppresses lung cancer progression by regulating integrin signaling. J Thorac Dis. 2020;12(5):2137–2145. doi: 10.21037/jtd.2020.04.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McGowan E, Lin Q, Ma G, Yin H, Chen S, Lin Y. PD-1 disrupted CAR-T cells in the treatment of solid tumors: promises and challenges. Biomed Pharmacother. 2020;121:109625. doi: 10.1016/j.biopha.2019.109625. [DOI] [PubMed] [Google Scholar]
- 92.Qu J, Mei Q, Liu L, Cheng T, Wang P, Chen L, et al. The progress and challenge of anti-PD-1/PD-L1 immunotherapy in treating non-small cell lung cancer. Therapeutic advances in medical oncology. 2021;13. [DOI] [PMC free article] [PubMed]
- 93.Li G, Guo J, Zheng Y, Ding W, Han Z, Qin L, et al. CXCR5 guides migration and tumor eradication of anti-EGFR chimeric antigen receptor T cells. Molecular Therapy - Oncolytics. 2021;22:507–517. doi: 10.1016/j.omto.2021.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ning J, Jiang S, Li X, Wang Y, Deng X, Zhang Z, et al. GPC3 affects the prognosis of lung adenocarcinoma and lung squamous cell carcinoma. BMC Pulm Med. 2021;21(1):199. doi: 10.1186/s12890-021-01549-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hou AJ, Chang ZL, Lorenzini MH, Zah E, Chen YY. TGF-β-responsive CAR-T cells promote anti-tumor immune function. Bioeng Transl Med. 2018;3(2):75–86. doi: 10.1002/btm2.10097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu M, Wang X, Li W, Yu X, Flores-Villanueva P, Xu-Monette ZY, et al. Targeting PD-L1 in non-small cell lung cancer using CAR T cells. Oncogenesis. 2020;9(8):72. doi: 10.1038/s41389-020-00257-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Villanueva N, Bazhenova L. New strategies in immunotherapy for lung cancer: beyond PD-1/PD-L1. Ther Adv Respir Dis. 2018;12:1753466618794133. doi: 10.1177/1753466618794133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zeltsman M, Dozier J, McGee E, Ngai D, Adusumilli PS. CAR T-cell therapy for lung cancer and malignant pleural mesothelioma. Transl Res. 2017;187:1–10. doi: 10.1016/j.trsl.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang C, Zhuang Q, Liu J, Liu X. Synthetic Biology in Chimeric Antigen Receptor T (CAR T) Cell engineering. ACS Synth Biol. 2022;11(1):1–15. doi: 10.1021/acssynbio.1c00256. [DOI] [PubMed] [Google Scholar]
- 100.Barrett DM, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum Gene Ther. 2013;24(8):717–727. doi: 10.1089/hum.2013.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365(18):1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2(2):112–120. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dimitri A, Herbst F, Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer. 2022;21(1):78. doi: 10.1186/s12943-022-01559-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Saffern M, Samstein R. Taking CAR T cells up a synthetic Notch. Nat Rev Immunol. 2021;21(3):135. doi: 10.1038/s41577-021-00514-1. [DOI] [PubMed] [Google Scholar]
- 105.Shafer P, Kelly LM, Hoyos V. Cancer therapy with TCR-Engineered T cells: current strategies, challenges, and prospects. Front Immunol. 2022;13:835762. doi: 10.3389/fimmu.2022.835762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chandran SS, Klebanoff CA. T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol Rev. 2019;290(1):127–147. doi: 10.1111/imr.12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hsu R, Baca Y, Xiu J, Wang R, Bodor JN, Kim C, et al. Molecular characterization of Kita-Kyushu lung cancer antigen (KK-LC-1) expressing carcinomas. Oncotarget. 2021;12(25):2449–2458. doi: 10.18632/oncotarget.28132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Leidner R, Sanjuan Silva N, Huang H, Sprott D, Zheng C, Shih Y-P, et al. Neoantigen T-cell receptor gene therapy in pancreatic cancer. N Engl J Med. 2022;386(22):2112–2119. doi: 10.1056/NEJMoa2119662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gao G, Liao W, Ma Q, Zhang B, Chen Y, Wang Y. KRAS G12D mutation predicts lower TMB and drives immune suppression in lung adenocarcinoma. Lung Cancer (Amsterdam, Netherlands) 2020;149:41–45. doi: 10.1016/j.lungcan.2020.09.004. [DOI] [PubMed] [Google Scholar]
- 110.Kankeu Fonkoua LA, Sirpilla O, Sakemura R, Siegler EL, Kenderian SS. CAR T cell therapy and the tumor microenvironment: Current challenges and opportunities. Mol Ther - Oncolytics. 2022;25:69–77. doi: 10.1016/j.omto.2022.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rath JA, Arber C. Engineering strategies to enhance TCR-based adoptive T cell therapy. Cells. 2020;9(6):1485. doi: 10.3390/cells9061485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhao Q, Jiang Y, Xiang S, Kaboli PJ, Shen J, Zhao Y, et al. Engineered TCR-T cell immunotherapy in anticancer precision medicine: pros and cons. Front Immunol. 2021;12:658753. doi: 10.3389/fimmu.2021.658753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.He Q, Jiang X, Zhou X, Weng J. Targeting cancers through TCR-peptide/MHC interactions. J Hematol Oncol. 2019;12(1):139. doi: 10.1186/s13045-019-0812-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ben Khelil M, Godet Y, Abdeljaoued S, Borg C, Adotévi O, Loyon R. Harnessing antitumor CD4+ T cells for cancer immunotherapy. Cancers. 2022;14(1):260. doi: 10.3390/cancers14010260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Veatch JR, Simon S, Riddell SR. Tumor-infiltrating lymphocytes make inroads in non–small-cell lung cancer. Nat Med. 2021;27(8):1339–1341. doi: 10.1038/s41591-021-01445-z. [DOI] [PubMed] [Google Scholar]
- 116.Wang S, Sun J, Chen K, Ma P, Lei Q, Xing S, et al. Perspectives of tumor-infiltrating lymphocyte treatment in solid tumors. BMC Med. 2021;19(1):140. doi: 10.1186/s12916-021-02006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ratto GB, Zino P, Mirabelli S, Minuti P, Aquilina R, Fantino G, et al. A randomized trial of adoptive immunotherapy with tumor-infiltrating lymphocytes and interleukin-2 versus standard therapy in the postoperative treatment of resected nonsmall cell lung carcinoma. Cancer. 1996;78(2):244–251. doi: 10.1002/(SICI)1097-0142(19960715)78:2<244::AID-CNCR9>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 118.Creelan B WC, Teer J, et al., editor Durable complete responses to adoptive cell transfer using tumor infiltrating lymphocytes (TIL) in non-small cell lung cancer (NSCLC): a phase I trial. AACR Virtual Annual Meeting I; 2020 April 27-28, 2020. https://www.cancer.org/cancer-facts-and-figures-2015.pdf
- 119.Fang L, Ly D, Wang S-s, Lee JB, Kang H, Xu H, et al. Targeting late-stage non-small cell lung cancer with a combination of DNT cellular therapy and PD-1 checkpoint blockade. J Exp Clin Cancer Res. 2019;38(1):123. doi: 10.1186/s13046-019-1126-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Creelan BC, Wang C, Teer JK, Toloza EM, Yao J, Kim S, et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: a phase 1 trial. Nat Med. 2021;27(8):1410–1418. doi: 10.1038/s41591-021-01462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ben-Avi R, Farhi R, Ben-Nun A, Gorodner M, Greenberg E, Markel G, et al. Establishment of adoptive cell therapy with tumor infiltrating lymphocytes for non-small cell lung cancer patients. Cancer Immunol Immunother : CII. 2018;67(8):1221–1230. doi: 10.1007/s00262-018-2174-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Santos Apolonio J, Lima de Souza Gonçalves V, Cordeiro Santos ML, Silva Luz M, Silva Souza JV, Rocha Pinheiro SL, et al. Oncolytic virus therapy in cancer: a current review. World J Virol. 2021;10(5):229–55. doi: 10.5501/wjv.v10.i5.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Russell SJ, Peng K-W, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30(7):658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35:S185–S198. doi: 10.1016/j.semcancer.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 125.Hemminki O, dos Santos JM, Hemminki A. Oncolytic viruses for cancer immunotherapy. J Hematol Oncol. 2020;13(1):84. doi: 10.1186/s13045-020-00922-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang Y, Li Y, Chen K, Qian L, Wang P. Oncolytic virotherapy reverses the immunosuppressive tumor microenvironment and its potential in combination with immunotherapy. Cancer Cell Int. 2021;21(1):262. doi: 10.1186/s12935-021-01972-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rosewell Shaw A, Suzuki M. Oncolytic viruses partner with T-cell therapy for solid tumor treatment. Front Immunol. 2018;9:2103. doi: 10.3389/fimmu.2018.02103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lawler SE, Speranza M-C, Cho C-F, Chiocca EA. Oncolytic viruses in cancer treatment. JAMA Oncol. 2017;3(6):841. doi: 10.1001/jamaoncol.2016.2064. [DOI] [PubMed] [Google Scholar]
- 129.Hernandez FP, Sandri-Goldin RM. Herpes simplex virus 1 regulatory protein ICP27 undergoes a head-to-tail intramolecular interaction. J Virol. 2010;84(9):4124–4135. doi: 10.1128/JVI.02319-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li J-M, Kao K-C, Li L-F, Yang T-M, Wu C-P, Horng Y-M, et al. MicroRNA-145 regulates oncolytic herpes simplex virus-1 for selective killing of human non-small cell lung cancer cells. Virology J. 2013;10(1):241. doi: 10.1186/1743-422X-10-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez AB, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J ImmunoTher Cancer. 2020;8(1):e000337. doi: 10.1136/jitc-2019-000337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Miyamoto S, Inoue H, Nakamura T, Yamada M, Sakamoto C, Urata Y, et al. Coxsackievirus B3 Is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Can Res. 2012;72(10):2609–2621. doi: 10.1158/0008-5472.CAN-11-3185. [DOI] [PubMed] [Google Scholar]
- 133.McCart JA, Ward JM, Lee J, Hu Y, Alexander HR, Libutti SK, et al. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Can Res. 2001;61(24):8751–8757. [PubMed] [Google Scholar]
- 134.Guo ZS, Thorne SH, Bartlett DL. Oncolytic virotherapy: Molecular targets in tumor-selective replication and carrier cell-mediated delivery of oncolytic viruses. Biochimica et Biophysica Acta (BBA) Rev Cancer. 2008;1785(2):217–31. doi: 10.1016/j.bbcan.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Guo ZS, Lu B, Guo Z, Giehl E, Feist M, Dai E, et al. Vaccinia virus-mediated cancer immunotherapy: cancer vaccines and oncolytics. J ImmunoTher Cancer. 2019;7(1):6. doi: 10.1186/s40425-018-0495-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Niemann J, Kühnel F. Oncolytic viruses: adenoviruses. Virus Genes. 2017;53(5):700–706. doi: 10.1007/s11262-017-1488-1. [DOI] [PubMed] [Google Scholar]
- 137.Guan Y-s, Liu Y, Zou Q, He Q, La Z, Yang L, et al. Adenovirus-mediated wild-type p53 gene transfer in combination with bronchial arterial infusion for treatment of advanced non-small-cell lung cancer, one year follow-up. J Zhejiang Univ SCIENCE B. 2009;10(5):331–40. doi: 10.1631/jzus.B0820248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Reddy PS, Burroughs KD, Hales LM, Ganesh S, Jones BH, Idamakanti N, et al. Seneca Valley virus, a systemically deliverable oncolytic picornavirus, and the treatment of neuroendocrine cancers. JNCI J Natl Cancer Inst. 2007;99(21):1623–33. doi: 10.1093/jnci/djm198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rudin CM, Poirier JT, Senzer NN, Stephenson J, Loesch D, Burroughs KD, et al. Phase I Clinical Study of Seneca Valley Virus (SVV-001), a replication-competent picornavirus, in advanced solid tumors with neuroendocrine features. Clin Cancer Res. 2011;17(4):888–895. doi: 10.1158/1078-0432.CCR-10-1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schenk EL, Mandrekar SJ, Dy GK, Aubry MC, Tan AD, Dakhil SR, et al. A Randomized double-blind phase II study of the seneca valley virus (NTX-010) versus placebo for patients with extensive-stage SCLC (ES SCLC) who were stable or responding after at least four cycles of platinum-based chemotherapy: north central cancer treatment group (Alliance) N0923 study. J Thorac Oncol. 2020;15(1):110–119. doi: 10.1016/j.jtho.2019.09.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Santini D, Hilbe W, Pall G, Kocher F, Pircher A, Zabernigg A, et al. Multicenter phase II study evaluating two cycles of docetaxel cisplatin and cetuximab as induction regimen prior to surgery in chemotherapy-naive patients with NSCLC stage IB-IIIA (INN06-Study. PloS one. 2015;10(5):0125364. doi: 10.1371/journal.pone.0125364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Díaz-Serrano A, Sánchez-Torre A, Paz-Ares L. Necitumumab for the treatment of advanced non-small-cell lung cancer. Future Oncol. 2019;15(7):705–716. doi: 10.2217/fon-2018-0594. [DOI] [PubMed] [Google Scholar]
- 143.Yamamoto N, Harada H, Okamoto I, Masuda N, Hayakawa K, Satouchi M, et al. Phase 2 study of nimotuzumab in combination with concurrent chemoradiotherapy in patients with locally advanced non–small-cell lung cancer. Clin Lung Cancer. 2021;22(2):134–141. doi: 10.1016/j.cllc.2020.12.012. [DOI] [PubMed] [Google Scholar]
- 144.Arcangelo M, Cappuzzo F. Focus on the potential role of ficlatuzumab in the treatment of non-small cell lung cancer Biologics. Targets Ther. 2013;7:61. doi: 10.2147/BTT.S28908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Garon EB, Ciuleanu T-E, Arrieta O, Prabhash K, Syrigos KN, Goksel T, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. The Lancet. 2014;384(9944):665–673. doi: 10.1016/S0140-6736(14)60845-X. [DOI] [PubMed] [Google Scholar]
- 146.Scagliotti GV, Hirsh V, Siena S, Henry DH, Woll PJ, Manegold C, et al. Overall survival improvement in patients with lung cancer and bone metastases treated with denosumab versus zoledronic acid: subgroup analysis from a randomized phase 3 study. J Thorac Oncol. 2012;7(12):1823–1829. doi: 10.1097/JTO.0b013e31826aec2b. [DOI] [PubMed] [Google Scholar]
- 147.Langer CJ, Novello S, Park K, Krzakowski M, Karp DD, Mok T, et al. Randomized, phase III trial of first-line Figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin alone in patients with advanced non–small-cell lung cancer. J Clin Oncol. 2014;32(19):2059–2066. doi: 10.1200/JCO.2013.54.4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yen W-C, Fischer MM, Axelrod F, Bond C, Cain J, Cancilla B, et al. Targeting notch signaling with a Notch2/Notch3 antagonist (Tarextumab) inhibits tumor growth and decreases tumor-initiating cell frequency. Clin Cancer Res. 2015;21(9):2084–2095. doi: 10.1158/1078-0432.CCR-14-2808. [DOI] [PubMed] [Google Scholar]
- 149.Gladkov O, Ramlau R, Serwatowski P, Milanowski J, Tomeczko J, Komarnitsky PB, et al. Cyclophosphamide and tucotuzumab (huKS-IL2) following first-line chemotherapy in responding patients with extensive-disease small-cell lung cancer. Anticancer Drugs. 2015;26(10):1061–1068. doi: 10.1097/CAD.0000000000000281. [DOI] [PubMed] [Google Scholar]
- 150.Bottomley A, Debruyne C, Felip E, Millward M, Thiberville L, Addario GD, et al. Symptom and quality of life results of an international randomised phase III study of adjuvant vaccination with Bec2/BCG in responding patients with limited disease small-cell lung cancer. Eur J Cancer. 2008;44(15):2178–2184. doi: 10.1016/j.ejca.2008.06.036. [DOI] [PubMed] [Google Scholar]
- 151.Mukherjee A, Waters AK, Babic I, Nurmemmedov E, Glassy MC, Kesari S, et al. Antibody drug conjugates: progress, pitfalls, and promises. Hum Antibodies. 2018;27(1):53–62. doi: 10.3233/HAB-180348. [DOI] [PubMed] [Google Scholar]
- 152.Morgensztern D, Besse B, Greillier L, Santana-Davila R, Ready N, Hann CL, et al. Efficacy and safety of rovalpituzumab tesirine in third-line and beyond patients with DLL3-Expressing, relapsed/refractory small-cell lung cancer: results from the phase II trinity study. Clin Cancer Res. 2019;25(23):6958–6966. doi: 10.1158/1078-0432.CCR-19-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gray JE, Heist RS, Starodub AN, Camidge DR, Kio EA, Masters GA, et al. Therapy of Small Cell Lung Cancer (SCLC) with a topoisomerase-I–inhibiting Antibody-Drug Conjugate (ADC) targeting trop-2. Sacituzumab Govitecan Clin Cancer Res. 2017;23(19):5711–5719. doi: 10.1158/1078-0432.CCR-17-0933. [DOI] [PubMed] [Google Scholar]
- 154.Shah MH, Lorigan P, O’Brien MER, Fossella FV, Moore KN, Bhatia S, et al. Phase I study of IMGN901, a CD56-targeting antibody-drug conjugate, in patients with CD56-positive solid tumors. Invest New Drugs. 2016;34(3):290–299. doi: 10.1007/s10637-016-0336-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hann CL, Burns TF, Dowlati A, Morgensztern D, Ward PJ, Koch MM, et al. A phase 1 study evaluating rovalpituzumab tesirine in frontline treatment of patients with extensive-stage SCLC. J Thorac Oncol. 2021;16(9):1582–1588. doi: 10.1016/j.jtho.2021.06.022. [DOI] [PubMed] [Google Scholar]
- 156.Hotta K, Aoe K, Kozuki T, Ohashi K, Ninomiya K, Ichihara E, et al. A phase II study of trastuzumab emtansine in HER2-positive non-small cell lung cancer. J Thorac Oncol. 2018;13(2):273–279. doi: 10.1016/j.jtho.2017.10.032. [DOI] [PubMed] [Google Scholar]
- 157.Waqar SN, Redman MW, Arnold SM, Hirsch FR, Mack PC, Schwartz LH, et al. A phase II study of Telisotuzumab Vedotin in patients with c–MET-positive stage IV or recurrent squamous cell lung cancer (LUNG-MAP sub-study S1400K, NCT03574753) Clin Lung Cancer. 2021;22(3):170–177. doi: 10.1016/j.cllc.2020.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Koopman LA, Terp MG, Zom GG, Janmaat ML, Jacobsen K, van den Heuvel Gresnigt E, et al. Enapotamab vedotin, an AXL-specific antibody-drug conjugate, shows preclinical antitumor activity in non-small cell lung cancer. JCI Insight. 2019;4(21):e128199. doi: 10.1172/jci.insight.128199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Park K, Haura EB, Leighl NB, Mitchell P, Shu CA, Girard N, et al. Amivantamab in EGFR Exon 20 insertion-mutated non–small-cell lung cancer progressing on platinum chemotherapy: initial results from the CHRYSALIS phase I study. J Clin Oncol. 2021;39(30):3391–3402. doi: 10.1200/JCO.21.00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009;206(8):1717–1725. doi: 10.1084/jem.20082492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Tomasini P, Khobta N, Greillier L, Barlesi F. Ipilimumab: its potential in non-small cell lung cancer. Ther Adv Med Oncol. 2012;4(2):43–50. doi: 10.1177/1758834011431718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lynch TJ, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol. 2012;30(17):2046–2054. doi: 10.1200/JCO.2011.38.4032. [DOI] [PubMed] [Google Scholar]
- 163.Chalmers AW, Patel S, Boucher K, Cannon L, Esplin M, Luckart J, et al. Phase I trial of targeted EGFR or ALK therapy with ipilimumab in metastatic NSCLC with long-term follow-up. Target Oncol. 2019;14(4):417–421. doi: 10.1007/s11523-019-00658-0. [DOI] [PubMed] [Google Scholar]
- 164.Zatloukal P, Heo DS, Park K, Kang J, Butts C, Bradford D, et al. Randomized phase II clinical trial comparing tremelimumab (CP-675,206) with best supportive care (BSC) following first-line platinum-based therapy in patients (pts) with advanced non-small cell lung cancer (NSCLC) J Clin Oncol. 2009;27(15_suppl):8071. doi: 10.1200/jco.2009.27.15_suppl.8071. [DOI] [Google Scholar]
- 165.Rizvi NA, Mazières J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol. 2015;16(3):257–265. doi: 10.1016/S1470-2045(15)70054-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in advanced Nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus Docetaxel in advanced squamous-cell non–small-cell lung cancer. N Engl J Med. 2015;373(2):123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gettinger S, Rizvi NA, Chow LQ, Borghaei H, Brahmer J, Ready N, et al. Nivolumab monotherapy for first-line treatment of advanced non-small-cell lung cancer. J Clin Oncol. 2016;34(25):2980–2987. doi: 10.1200/JCO.2016.66.9929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, et al. Nivolumab plus Ipilimumab in advanced non-small-cell lung cancer. N Engl J Med. 2019;381(21):2020–2031. doi: 10.1056/NEJMoa1910231. [DOI] [PubMed] [Google Scholar]
- 170.Deng H, Zhou C. From CheckMate 227 to CheckMate 9LA: rethinking the status of chemotherapy in the immunotherapy era—chemo-free or chemo-reform? Transl Lung Cancer Res. 2021;10(4):1924–1927. doi: 10.21037/tlcr-21-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Paz-Ares L, Ciuleanu T-E, Cobo M, Schenker M, Zurawski B, Menezes J, et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): an international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22(2):198–211. doi: 10.1016/S1470-2045(20)30641-0. [DOI] [PubMed] [Google Scholar]
- 172.Chatterjee M, Turner DC, Felip E, Lena H, Cappuzzo F, Horn L, et al. Systematic evaluation of pembrolizumab dosing in patients with advanced non-small-cell lung cancer. Ann Oncol. 2016;27(7):1291–1298. doi: 10.1093/annonc/mdw174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non–small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 174.Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive non–small-cell lung cancer. N Engl J Med. 2016;375(19):1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 175.Antonia SJ, Kim S-W, Spira AI, Ahn MJ, Ou S-HI, Stjepanovic N, et al. Safety and clinical activity of durvalumab (MEDI4736), an anti-PD-L1 antibody, in treatment-naïve patients with advanced non-small-cell lung cancer. J Clin Oncol. 2016;34(15_suppl):9029. doi: 10.1200/JCO.2016.34.15_suppl.9029. [DOI] [Google Scholar]
- 176.Antonia SJ, Goldberg SB, Balmanoukian A, Chaft JE, Sanborn RE, Gupta A, et al. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: a multicentre, phase 1b study. Lancet Oncol. 2016;17(3):299–308. doi: 10.1016/S1470-2045(15)00544-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Peters S, Gettinger S, Johnson ML, Jänne PA, Garassino MC, Christoph D, et al. Phase II trial of Atezolizumab as first-line or subsequent therapy for patients with programmed death-ligand 1-selected advanced non-small-cell lung cancer (BIRCH) J Clin Oncol. 2017;35(24):2781–2789. doi: 10.1200/JCO.2016.71.9476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Herbst RS, Soria J-C, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet (London, England) 2016;387(10030):1837–1846. doi: 10.1016/S0140-6736(16)00587-0. [DOI] [PubMed] [Google Scholar]
- 180.Verschraegen CF, Chen F, Spigel DR, Iannotti N, McClay EF, Redfern CH, et al. Avelumab (MSB0010718C; anti-PD-L1) as a first-line treatment for patients with advanced NSCLC from the JAVELIN Solid Tumor phase 1b trial: Safety, clinical activity, and PD-L1 expression. J Clin Oncol. 2016;34(15_suppl):9036. doi: 10.1200/JCO.2016.34.15_suppl.9036. [DOI] [Google Scholar]
- 181.Rotte A, Sahasranaman S, Budha N. Targeting TIGIT for immunotherapy of cancer: update on clinical development. Biomedicines. 2021;9(9):1277. doi: 10.3390/biomedicines9091277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Shan C, Li X, Zhang J. Progress of immune checkpoint LAG-3 in immunotherapy (Review) Oncol Lett. 2020;20(5):1. doi: 10.3892/ol.2020.12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Felip E, Doger B, Majem M, Carcereny E, Krebs M, Peguero JA, et al. Initial results from a phase II study (TACTI-002) in metastatic non-small cell lung or head and neck carcinoma patients receiving eftilagimod alpha (soluble LAG-3 protein) and pembrolizumab. J Clin Oncol. 2020;38(15):3100. doi: 10.1200/JCO.2020.38.15_suppl.3100. [DOI] [Google Scholar]
- 184.Sun R, Limkin EJ, Vakalopoulou M, Dercle L, Champiat S, Han SR, et al. A radiomics approach to assess tumour-infiltrating CD8 cells and response to anti-PD-1 or anti-PD-L1 immunotherapy: an imaging biomarker, retrospective multicohort study. Lancet Oncol. 2018;19(9):1180–1191. doi: 10.1016/S1470-2045(18)30413-3. [DOI] [PubMed] [Google Scholar]
- 185.Xu Z, Wang X, Zeng S, Ren X, Yan Y, Gong Z. Applying artificial intelligence for cancer immunotherapy. Acta Pharmaceutica Sinica B. 2021;11(11):3393–3405. doi: 10.1016/j.apsb.2021.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Travis WD. Update on small cell carcinoma and its differentiation from squamous cell carcinoma and other non-small cell carcinomas. Mod Pathol. 2012;25 Suppl 1:S18–30. 10.1038/modpathol.2011.150. [DOI] [PubMed]
- 187.Travis WDB, E; Müller-Hermelink, H.K.; et al. Pathology and Genetics: Tumours of the Lung, Pleura, Thymus and Heart. IARC: Lyon. 2004;1.
- 188.Azzopardi JG. Oat-cell carcinoma of the bronchus. J Pathol Bacteriol. 1959;78:513–519. doi: 10.1002/path.1700780218. [DOI] [PubMed] [Google Scholar]
- 189.Nicholson SA, Beasley MB, Brambilla E, Hasleton PS, Colby TV, Sheppard MN, et al. Small cell lung carcinoma (SCLC): a clinicopathologic study of 100 cases with surgical specimens. Am J Surg Pathol. 2002;26(9):1184–1197. doi: 10.1097/00000478-200209000-00009. [DOI] [PubMed] [Google Scholar]
- 190.Travis WD. Advances in neuroendocrine lung tumors. Ann Oncol . 2010;21(Suppl-7):vii65–71. doi: 10.1093/annonc/mdq380. [DOI] [PubMed] [Google Scholar]
- 191.Fruh M, De Ruysscher D, Popat S, Crino L, Peters S, Felip E. Small-cell lung cancer (SCLC) ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol J Eur Soc Med Oncol. 2013;24(6):99–105. doi: 10.1093/annonc/mdt178. [DOI] [PubMed] [Google Scholar]
- 192.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359(13):1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Johnson BE, Crawford J, Downey RJ, Ettinger DS, Fossella F, Grecula JC, et al. Small cell lung cancer clinical practice guidelines in oncology. J Natl Compr Cancer Network. 2006;4(6):602–622. doi: 10.6004/jnccn.2006.0050. [DOI] [PubMed] [Google Scholar]
- 194.Rudin CM, Ismaila N, Hann CL, Malhotra N, Movsas B, Norris K, et al. Treatment of small-cell lung cancer: American society of clinical oncology endorsement of the american college of chest physicians guideline. J Clin Oncol. 2015;33(34):4106–4111. doi: 10.1200/JCO.2015.63.7918. [DOI] [PubMed] [Google Scholar]
- 195.Chan BA, Coward JI. Chemotherapy advances in small-cell lung cancer. J Thorac Dis. 2013;5(Suppl 5):S565–S578. doi: 10.3978/j.issn.2072-1439.2013.07.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Simon GR, Turrisi A. Management of small cell lung cancer: ACCP evidence-based clinical practice guidelines (2nd edition) Chest. 2007;132(3 Suppl):324s–39s. doi: 10.1378/chest.07-1385. [DOI] [PubMed] [Google Scholar]
- 197.Lally BE, Urbanic JJ, Blackstock AW, Miller AA, Perry MC. Small cell lung cancer: have we made any progress over the last 25 years? Oncologist. 2007;12(9):1096–1104. doi: 10.1634/theoncologist.12-9-1096. [DOI] [PubMed] [Google Scholar]
- 198.George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524(7563):47–53. doi: 10.1038/nature14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Horn L, Reck M, Spigel DR. The future of immunotherapy in the treatment of small cell lung cancer. Oncologist. 2016;21(8):910–921. doi: 10.1634/theoncologist.2015-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (New York, NY) 2015;348(6230):124–8. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Darnell RB. Onconeural antigens and the paraneoplastic neurologic disorders: at the intersection of cancer, immunity, and the brain. Proc Natl Acad Sci USA. 1996;93(10):4529–4536. doi: 10.1073/pnas.93.10.4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Koyama K, Kagamu H, Miura S, Hiura T, Miyabayashi T, Itoh R, et al. Reciprocal CD4+ T-cell balance of effector CD62Llow CD4+ and CD62LhighCD25+ CD4+ regulatory T cells in small cell lung cancer reflects disease stage. Clin Cancer Res. 2008;14(21):6770–6779. doi: 10.1158/1078-0432.CCR-08-1156. [DOI] [PubMed] [Google Scholar]
- 203.Maddison P, Newsom-Davis J, Mills KR, Souhami RL. Favourable prognosis in Lambert-Eaton myasthenic syndrome and small-cell lung carcinoma. Lancet (London, England) 1999;353(9147):117–118. doi: 10.1016/S0140-6736(05)76153-5. [DOI] [PubMed] [Google Scholar]
- 204.Tani T, Tanaka K, Idezuka J, Nishizawa M. Regulatory T cells in paraneoplastic neurological syndromes. J Neuroimmunol. 2008;196(1–2):166–169. doi: 10.1016/j.jneuroim.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 205.Wang W, Hodkinson P, McLaren F, Mackean MJ, Williams L, Howie SEM, et al. Histologic assessment of tumor-associated CD45(+) cell numbers is an independent predictor of prognosis in small cell lung cancer. Chest. 2013;143(1):146–151. doi: 10.1378/chest.12-0681. [DOI] [PubMed] [Google Scholar]
- 206.Krug LM. Vaccine therapy for small cell lung cancer. Semin Oncol. 2004;31(1 Suppl 1):112–116. doi: 10.1053/j.seminoncol.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 207.Livingston PO, Calves MJ, Jr. Natoli EJ. Approaches to augmenting the immunogenicity of the ganglioside GM2 in mice purified GM2 is superior to whole cells. J Immunol (Baltimore Md :1950) 1987;138(5):1524–9. doi: 10.4049/jimmunol.138.5.1524. [DOI] [PubMed] [Google Scholar]
- 208.Fuentes R, Allman R, Mason MD. Ganglioside expression in lung cancer cell lines. Lung Cancer (Amsterdam, Netherlands) 1997;18(1):21–33. doi: 10.1016/S0169-5002(97)00049-4. [DOI] [PubMed] [Google Scholar]
- 209.McCaffery M, Yao TJ, Williams L, Livingston PO, Houghton AN, Chapman PB. Immunization of melanoma patients with BEC2 anti-idiotypic monoclonal antibody that mimics GD3 ganglioside: enhanced immunogenicity when combined with adjuvant. Clin Cancer Res. 1996;2(4):679–686. [PubMed] [Google Scholar]
- 210.Zhang S, Cordon-Cardo C, Zhang HS, Reuter VE, Adluri S, Hamilton WB, et al. Selection of tumor antigens as targets for immune attack using immunohistochemistry: I. Focus on gangliosides. Int J Cancer. 1997;73(1):42–9. doi: 10.1002/(SICI)1097-0215(19970926)73:1<42::AID-IJC8>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 211.Dickler MN, Ragupathi G, Liu NX, Musselli C, Martino DJ, Miller VA, et al. Immunogenicity of a fucosyl-GM1-keyhole limpet hemocyanin conjugate vaccine in patients with small cell lung cancer. Clin Cancer Res. 1999;5(10):2773–2779. [PubMed] [Google Scholar]
- 212.Gilewski T, Ragupathi G, Bhuta S, Williams LJ, Musselli C, Zhang XF, et al. Immunization of metastatic breast cancer patients with a fully synthetic globo H conjugate: a phase I trial. Proc Natl Acad Sci USA. 2001;98(6):3270–3275. doi: 10.1073/pnas.051626298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Helling F, Zhang S, Shang A, Adluri S, Calves M, Koganty R, et al. GM2-KLH conjugate vaccine: increased immunogenicity in melanoma patients after administration with immunological adjuvant QS-21. Can Res. 1995;55(13):2783–2788. [PubMed] [Google Scholar]
- 214.Krug LM, Ragupathi G, Ng KK, Hood C, Jennings HJ, Guo Z, et al. Vaccination of small cell lung cancer patients with polysialic acid or N-propionylated polysialic acid conjugated to keyhole limpet hemocyanin. Clin Cancer Res. 2004;10(3):916–923. doi: 10.1158/1078-0432.CCR-03-0101. [DOI] [PubMed] [Google Scholar]
- 215.Slovin SF, Ragupathi G, Adluri S, Ungers G, Terry K, Kim S, et al. Carbohydrate vaccines in cancer: immunogenicity of a fully synthetic globo H hexasaccharide conjugate in man. Proc Natl Acad Sci USA. 1999;96(10):5710–5715. doi: 10.1073/pnas.96.10.5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Chiappori AA, Soliman H, Janssen WE, Antonia SJ, Gabrilovich DI. INGN-225: a dendritic cell-based p53 vaccine (Ad.p53-DC) in small cell lung cancer: observed association between immune response and enhanced chemotherapy effect. Expert Opin Biol Ther. 2010;10(6):983–91. doi: 10.1517/14712598.2010.484801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Antonia SJ, Mirza N, Fricke I, Chiappori A, Thompson P, Williams N, et al. Combination of p53 cancer vaccine with chemotherapy in patients with extensive stage small cell lung cancer. Clin Cancer Res. 2006;12(3 Pt 1):878–887. doi: 10.1158/1078-0432.CCR-05-2013. [DOI] [PubMed] [Google Scholar]
- 218.Sakamoto S, Yamada T, Terazaki Y, Yoshiyama K, Sugawara S, Takamori S, et al. Feasibility study of personalized peptide vaccination for advanced small cell lung cancer. Clin Lung Cancer. 2017;18(6):e385–e394. doi: 10.1016/j.cllc.2017.03.011. [DOI] [PubMed] [Google Scholar]
- 219.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Asmar R, Rizvi NA. Immunotherapy for advanced lung cancer. The Cancer J. 2015;21(5):383–391. doi: 10.1097/PPO.0000000000000151. [DOI] [PubMed] [Google Scholar]
- 222.Yamane H, Isozaki H, Takeyama M, Ochi N, Kudo K, Honda Y, et al. Programmed cell death protein 1 and programmed death-ligand 1 are expressed on the surface of some small-cell lung cancer lines. Am J Cancer Res. 2015;5(4):1553–1557. [PMC free article] [PubMed] [Google Scholar]
- 223.Ott PA, Elez E, Hiret S, Kim DW, Morosky A, Saraf S, et al. Pembrolizumab in patients with extensive-stage small-cell lung cancer: results from the phase Ib KEYNOTE-028 Study. J Clin Oncol. 2017;35(34):3823–3829. doi: 10.1200/JCO.2017.72.5069. [DOI] [PubMed] [Google Scholar]
- 224.Ishii H, Azuma K, Kawahara A, Yamada K, Imamura Y, Tokito T, et al. Significance of programmed cell death-ligand 1 expression and its association with survival in patients with small cell lung cancer. J Thorac Oncol. 2015;10(3):426–430. doi: 10.1097/JTO.0000000000000414. [DOI] [PubMed] [Google Scholar]
- 225.Salama AK, Hodi FS. Cytotoxic T-lymphocyte-associated antigen-4. Clin Cancer Res. 2011;17(14):4622–4628. doi: 10.1158/1078-0432.CCR-10-2232. [DOI] [PubMed] [Google Scholar]
- 226.Reck M, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol. 2013;24(1):75–83. doi: 10.1093/annonc/mds213. [DOI] [PubMed] [Google Scholar]
- 227.Antonia SJ, López-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016;17(7):883–895. doi: 10.1016/S1470-2045(16)30098-5. [DOI] [PubMed] [Google Scholar]
- 228.De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. 2017;17(8):457–474. doi: 10.1038/nrc.2017.51. [DOI] [PubMed] [Google Scholar]
- 229.Hall RD, Le TM, Haggstrom DE, Gentzler RD. Angiogenesis inhibition as a therapeutic strategy in non-small cell lung cancer (NSCLC) Transl Lung Cancer Res. 2015;4(5):515–523. doi: 10.3978/j.issn.2218-6751.2015.06.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Ohm JE, Gabrilovich DI, Sempowski GD, Kisseleva E, Parman KS, Nadaf S, et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood. 2003;101(12):4878–4886. doi: 10.1182/blood-2002-07-1956. [DOI] [PubMed] [Google Scholar]
- 231.Ohm JE, Carbone DP. VEGF as a mediator of tumor-associated immunodeficiency. Immunol Res. 2001;23(2–3):263–272. doi: 10.1385/IR:23:2-3:263. [DOI] [PubMed] [Google Scholar]
- 232.Bouzin C, Brouet A, De Vriese J, Dewever J, Feron O. Effects of vascular endothelial growth factor on the lymphocyte-endothelium interactions: identification of caveolin-1 and nitric oxide as control points of endothelial cell anergy. J Immunol (Baltimore, Md : 1950) 2007;178(3):1505–11. doi: 10.4049/jimmunol.178.3.1505. [DOI] [PubMed] [Google Scholar]
- 233.Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20(6):607–615. doi: 10.1038/nm.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, et al. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Can Res. 2013;73(2):539–549. doi: 10.1158/0008-5472.CAN-12-2325. [DOI] [PubMed] [Google Scholar]
- 235.Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Can Res. 2004;64(11):3731–3736. doi: 10.1158/0008-5472.CAN-04-0074. [DOI] [PubMed] [Google Scholar]
- 236.Wu JB, Tang YL, Liang XH. Targeting VEGF pathway to normalize the vasculature: an emerging insight in cancer therapy. Onco Targets Ther. 2018;11:6901–6909. doi: 10.2147/OTT.S172042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Bonanno L, De Paoli A, Zulato E, Esposito G, Calabrese F, Favaretto A, et al. LKB1 expression correlates with increased survival in patients with advanced non-small cell lung cancer treated with chemotherapy and Bevacizumab. Clin Cancer Res. 2017;23(13):3316. doi: 10.1158/1078-0432.CCR-16-2410. [DOI] [PubMed] [Google Scholar]
- 238.Manegold C, Dingemans AC, Gray JE, Nakagawa K, Nicolson M, Peters S, et al. The Potential of Combined Immunotherapy and Antiangiogenesis for the Synergistic Treatment of Advanced NSCLC. J Thorac Oncol. 2017;12(2):194–207. doi: 10.1016/j.jtho.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 239.Tao L, Huang G, Shi S, Chen L. Bevacizumab improves the antitumor efficacy of adoptive cytokine-induced killer cells therapy in non-small cell lung cancer models. Med Oncol (Northwood, London, England) 2014;31(1):777. doi: 10.1007/s12032-013-0777-3. [DOI] [PubMed] [Google Scholar]
- 240.Liang H, Wang M. Prospect of immunotherapy combined with anti-angiogenic agents in patients with advanced non-small cell lung cancer. Cancer Manag Res. 2019;11:7707–7719. doi: 10.2147/CMAR.S212238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Yi M, Jiao D, Qin S, Chu Q, Wu K, Li A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol Cancer. 2019;18(1):60. doi: 10.1186/s12943-019-0974-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Gadgeel SM, Stevenson J, Langer CJ, Gandhi L, Borghaei H, Patnaik A, et al. Pembrolizumab (pembro) plus chemotherapy as front-line therapy for advanced NSCLC: KEYNOTE-021 cohorts A-C. J Clin Oncol. 2016;34(15_suppl):9016. doi: 10.1200/JCO.2016.34.15_suppl.9016. [DOI] [Google Scholar]
- 243.NCCN Clinical practice guideline in oncology (NCCN Guidelines). Non Small cell Lung Cancer. 2020. https://www.cancer.org/cancer-facts-and-figures-2015.pdf
- 244.Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378(22):2078–2092. doi: 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 245.Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gümüş M, Mazières J, et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med. 2018;379(21):2040–2051. doi: 10.1056/NEJMoa1810865. [DOI] [PubMed] [Google Scholar]
- 246.Herbst RS, Baas P, Kim DW, Felip E, Pérez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet (London, England) 2016;387(10027):1540–1550. doi: 10.1016/S0140-6736(15)01281-7. [DOI] [PubMed] [Google Scholar]
- 247.Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378(24):2288–2301. doi: 10.1056/NEJMoa1716948. [DOI] [PubMed] [Google Scholar]
- 248.Reck M, Mok TSK, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir Med. 2019;7(5):387–401. doi: 10.1016/S2213-2600(19)30084-0. [DOI] [PubMed] [Google Scholar]
- 249.Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet (London, England) 2017;389(10066):255–265. doi: 10.1016/S0140-6736(16)32517-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.F. Barlesi KP, F. Ciardiello, J. von Pawel, S. Gadgeel, T. Hida, D. Kowalski, M. Cobo Dols, D. Cortinovis, J. Leach, J. Polikoff, D.R. Gandara, C. Barrios, D.S. Chen, P. He, M. Kowanetz, M. Ballinger, D. Waterkamp, A. Sandler, A. Rittmeyer, editor PRIMARY ANALYSIS FROM OAK, A RANDOMIZED PHASE III STUDY COMPARING ATEZOLIZUMAB WITH DOCETAXEL IN 2L/3L NSCLC. Annals of Oncology; 2016.
- 251.Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N Engl J Med. 2018;379(24):2342–2350. doi: 10.1056/NEJMoa1809697. [DOI] [PubMed] [Google Scholar]
- 252.Horn L, Mansfield AS, Szczęsna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med. 2018;379(23):2220–2229. doi: 10.1056/NEJMoa1809064. [DOI] [PubMed] [Google Scholar]
- 253.Liu SV, Reck M, Mansfield AS, Mok T, Scherpereel A, Reinmuth N, et al. Updated overall survival and PD-L1 subgroup analysis of patients with extensive-stage small-cell lung cancer treated with atezolizumab, carboplatin, and etoposide (IMpower133) J Clin Oncol. 2021;39(6):619–630. doi: 10.1200/JCO.20.01055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Paz-Ares L, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, et al. Durvalumab plus platinum–etoposide versus platinum–etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. The Lancet. 2019;394(10212):1929–1939. doi: 10.1016/S0140-6736(19)32222-6. [DOI] [PubMed] [Google Scholar]
- 255.Chung HC, Piha-Paul SA, Lopez-Martin J, Schellens JHM, Kao S, Miller WH, Jr, et al. Pembrolizumab after two or more lines of previous therapy in patients with recurrent or metastatic SCLC: results from the KEYNOTE-028 and KEYNOTE-158 studies. J Thorac Oncol. 2020;15(4):618–627. doi: 10.1016/j.jtho.2019.12.109. [DOI] [PubMed] [Google Scholar]
- 256.Wirsdörfer F, de Leve S, Jendrossek V. Combining radiotherapy and immunotherapy in lung cancer: can we expect limitations due to altered normal tissue toxicity? Int J Mol Sci. 2018;20(1):24. doi: 10.3390/ijms20010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Gordon JR, Ma Y, Churchman L, Gordon SA, Dawicki W. Regulatory dendritic cells for immunotherapy in immunologic diseases. Front Immunol. 2014;5:7. doi: 10.3389/fimmu.2014.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Shen Y, Hao T, Ou S, Hu C, Chen L. Applications and perspectives of nanomaterials in novel vaccine development. MedChemComm. 2018;9(2):226–238. doi: 10.1039/C7MD00158D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Mukherjee A, Madamsetty VS, Mukherjee S. Emerging trends in immunomodulatory nanomaterials toward cancer therapy. Synth Lect Biomed Eng. 2021;16(1):i–84. doi: 10.1007/978-3-031-01669-1. [DOI] [Google Scholar]
- 260.Mukherjee A, Paul M, Mukherjee S. Recent progress in the theranostics application of nanomedicine in lung cancer. Cancers. 2019;11(5):597. doi: 10.3390/cancers11050597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Madamsetty VS, Paul MK, Mukherjee A, Mukherjee S. Functionalization of nanomaterials and their application in melanoma cancer theranostics. ACS Biomater Sci Eng. 2019;6(1):167–181. doi: 10.1021/acsbiomaterials.9b01426. [DOI] [PubMed] [Google Scholar]
- 262.Mukherjee S, Madamsetty VS, Bhattacharya D, Roy Chowdhury S, Paul MK, Mukherjee A. Recent advancements of nanomedicine in neurodegenerative disorders theranostics. Adv Funct Mater. 2020;30(35):2003054. doi: 10.1002/adfm.202003054. [DOI] [Google Scholar]
- 263.Ovais M, Mukherjee S, Pramanik A, Das D, Mukherjee A, Raza A, et al. Designing stimuli-responsive upconversion nanoparticles that exploit the tumor microenvironment. Adv Mater. 2020;32(22):e2000055. doi: 10.1002/adma.202000055. [DOI] [PubMed] [Google Scholar]
- 264.Chiang C-S, Lin Y-J, Lee R, Lai Y-H, Cheng H-W, Hsieh C-H, et al. Combination of fucoidan-based magnetic nanoparticles and immunomodulators enhances tumour-localized immunotherapy. Nat Nanotechnol. 2018;13(8):746–754. doi: 10.1038/s41565-018-0146-7. [DOI] [PubMed] [Google Scholar]
- 265.Zhang Q, Wei W, Wang P, Zuo L, Li F, Xu J, et al. Biomimetic magnetosomes as versatile artificial antigen-presenting cells to potentiate t-cell-based anticancer therapy. ACS Nano. 2017;11(11):10724–10732. doi: 10.1021/acsnano.7b04955. [DOI] [PubMed] [Google Scholar]
- 266.Butts C, Maksymiuk A, Goss G, Soulières D, Marshall E, Cormier Y, et al. Updated survival analysis in patients with stage IIIB or IV non-small-cell lung cancer receiving BLP25 liposome vaccine (L-BLP25): phase IIB randomized, multicenter, open-label trial. J Cancer Res Clin Oncol. 2011;137(9):1337–1342. doi: 10.1007/s00432-011-1003-3. [DOI] [PubMed] [Google Scholar]
- 267.Mitchell P, Thatcher N, Socinski MA, Wasilewska-Tesluk E, Horwood K, Szczesna A, et al. Tecemotide in unresectable stage III non-small-cell lung cancer in the phase III START study: updated overall survival and biomarker analyses. Ann Oncol. 2015;26(6):1134–1142. doi: 10.1093/annonc/mdv104. [DOI] [PubMed] [Google Scholar]
- 268.Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater. 2016;16(4):489–496. doi: 10.1038/nmat4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Hosomi Y, Morita S, Sugawara S, Kato T, Fukuhara T, Gemma A, et al. Gefitinib alone versus gefitinib plus chemotherapy for non–small-cell lung cancer with mutated epidermal growth factor receptor: NEJ009 study. J Clin Oncol. 2020;38(2):115–123. doi: 10.1200/JCO.19.01488. [DOI] [PubMed] [Google Scholar]
- 270.Li Z, Liu Y, Fang X, Shu Z. Nanomaterials enhance the immunomodulatory effect of molecular targeted therapy. Int J Nanomed. 2021;16:1631–1661. doi: 10.2147/IJN.S290346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Domvri K, Petanidis S, Anestakis D, Porpodis K, Bai C, Zarogoulidis P, et al. Dual photothermal MDSCs-targeted immunotherapy inhibits lung immunosuppressive metastasis by enhancing T-cell recruitment. Nanoscale. 2020;12(13):7051–7062. doi: 10.1039/D0NR00080A. [DOI] [PubMed] [Google Scholar]
- 272.Zou JY, Su CH, Luo HH, Lei YY, Zeng B, Zhu HS, et al. Curcumin converts Foxp3+ regulatory T cells to T helper 1 cells in patients with lung cancer. J Cell Biochem. 2017;119(2):1420–1428. doi: 10.1002/jcb.26302. [DOI] [PubMed] [Google Scholar]
- 273.Yang M, Li Z, Tao J, Hu H, Li Z, Zhang Z, et al. Resveratrol induces PD-L1 expression through snail-driven activation of Wnt pathway in lung cancer cells. J Cancer Res Clin Oncol. 2021;147(4):1101–1113. doi: 10.1007/s00432-021-03510-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Cardenas H, Arango D, Nicholas C, Duarte S, Nuovo G, He W, et al. Dietary apigenin exerts immune-regulatory activity in vivo by reducing NF-κB activity, halting leukocyte infiltration and restoring normal metabolic function. Int J Mol Sci. 2016;17(3):323. doi: 10.3390/ijms17030323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Chen M, Wang X, Zha D, Cai F, Zhang W, He Y, et al. Apigenin potentiates TRAIL therapy of non-small cell lung cancer via upregulating DR4/DR5 expression in a p53-dependent manner. Sci Rep. 2016;6(1):35468. doi: 10.1038/srep35468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Sul O-J, Ra SW. Quercetin prevents LPS-induced oxidative stress and inflammation by modulating NOX2/ROS/NF-kB in lung epithelial cells. Molecules. 2021;26(22):6949. doi: 10.3390/molecules26226949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Oo AM, Mohd Adnan LH, Nor NM, Simbak N, Ahmad NZ, Lwin OM. Immunomodulatory effects of flavonoids: An experimental study on natural-killer-cell-mediated cytotoxicity against lung cancer and cytotoxic granule secretion profile. Proc Singapore Healthc. 2020;30(4):279–285. doi: 10.1177/2010105820979006. [DOI] [Google Scholar]
- 278.Choi K-C, Lee Y-H, Jung MG, Kwon SH, Kim M-J, Jun WJ, et al. Gallic acid suppresses lipopolysaccharide-induced nuclear factor-κB signaling by preventing RelA acetylation in A549 lung cancer cells. Mol Cancer Res. 2009;7(12):2011–2021. doi: 10.1158/1541-7786.MCR-09-0239. [DOI] [PubMed] [Google Scholar]
- 279.Joo JH, Jetten AM. NF-κB-dependent transcriptional activation in lung carcinoma cells by farnesol involves p65/RelA(Ser276) phosphorylation via the MEK-MSK1 signaling pathway. J Biol Chem. 2008;283(24):16391–16399. doi: 10.1074/jbc.M800945200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Cao S-Y, Li Y, Meng X, Zhao C-N, Li S, Gan R-Y, et al. Dietary natural products and lung cancer: effects and mechanisms of action. J Funct Foods. 2019;52:316–331. doi: 10.1016/j.jff.2018.11.004. [DOI] [Google Scholar]
- 281.Wang W-J, Wu Y-S, Chen S, Liu C-F, Chen S-N. Mushroomβ-Glucan may immunomodulate the tumor-associated macrophages in the lewis lung carcinoma. Biomed Res Int. 2015;2015:1–15. doi: 10.1155/2015/604385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Li Y, Gu JF, Zou X, Wu J, Zhang MH, Jiang J, et al. The anti-lung cancer activities of steroidal saponins of P. polyphylla Smith var. chinensis (Franch) Hara through enhanced immunostimulation in experimental Lewis tumor-bearing C57BL/6 mice and induction of Apoptosis in the A549 cell line. Molecules. 2013;18(10):12916–36. doi: 10.3390/molecules181012916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Shiku H, Wang H, Chan Y-L, Li T-L, Bauer BA, Hsia S, et al. Reduction of splenic immunosuppressive cells and enhancement of anti-tumor immunity by synergy of fish oil and selenium yeast. PloS one. 2013;8(1):e52912. doi: 10.1371/journal.pone.0052912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Maiuolo J, Gliozzi M, Carresi C, Musolino V, Oppedisano F, Scarano F, et al. Nutraceuticals and cancer: potential for natural polyphenols. Nutrients. 2021;13(11):3834. doi: 10.3390/nu13113834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Niedzwiecki A, Roomi M, Kalinovsky T, Rath M. Anticancer efficacy of polyphenols and their combinations. Nutrients. 2016;8(9):552. doi: 10.3390/nu8090552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Zhou Y, Zheng J, Li Y, Xu D-P, Li S, Chen Y-M, et al. Natural polyphenols for prevention and treatment of cancer. Nutrients. 2016;8(8):515. doi: 10.3390/nu8080515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Bosch-Barrera J, Sais E, Cañete N, Marruecos J, Cuyàs E, Izquierdo A, et al. Response of brain metastasis from lung cancer patients to an oral nutraceutical product containing silibinin. Oncotarget. 2016;7(22):32006–32014. doi: 10.18632/oncotarget.7900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Sanmamed MF, Chester C, Melero I, Kohrt H. Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies. Ann Oncol. 2016;27(7):1190–1198. doi: 10.1093/annonc/mdw041. [DOI] [PubMed] [Google Scholar]
- 289.Chulpanova DS, Kitaeva KV, Rutland CS, Rizvanov AA, Solovyeva VV. Mouse tumor models for advanced cancer immunotherapy. Int J Mol Sci. 2020;21(11):4118. doi: 10.3390/ijms21114118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Salehi-Rad R, Li R, Tran LM, Lim RJ, Abascal J, Momcilovic M, et al. Novel Kras-mutant murine models of non-small cell lung cancer possessing co-occurring oncogenic mutations and increased tumor mutational burden. Cancer Immunol Immunother. 2021;70(8):2389–2400. doi: 10.1007/s00262-020-02837-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Zloza A, Karolina Palucka A, Coussens LM, Gotwals PJ, Headley MB, Jaffee EM, et al. Workshop on challenges, insights, and future directions for mouse and humanized models in cancer immunology and immunotherapy: a report from the associated programs of the 2016 annual meeting for the Society for Immunotherapy of cancer. J ImmunoTher Cancer. 2017;5(1):77. doi: 10.1186/s40425-017-0278-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Shi Y-X, Sheng D-Q, Cheng L, Song X-Y. Current landscape of epigenetics in lung cancer: focus on the mechanism and application. J Oncol. 2019;2019:1–11. doi: 10.1155/2019/8107318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Hirsch FR, Scagliotti GV, Mulshine JL, Kwon R, Curran WJ, Wu Y-L, et al. Lung cancer: current therapies and new targeted treatments. The Lancet. 2017;389(10066):299–311. doi: 10.1016/S0140-6736(16)30958-8. [DOI] [PubMed] [Google Scholar]
- 294.Liu F, Wu H. CC Chemokine receptors in lung adenocarcinoma: the inflammation-related prognostic biomarkers and immunotherapeutic targets. J Inflamm Res. 2021;14:267–285. doi: 10.2147/JIR.S278395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Jin L, Cao L, Zhu Y, Cao J, Li X, Zhou J, et al. Enhance anti-lung tumor efficacy of chimeric antigen receptor-T cells by ectopic expression of C-C motif chemokine receptor 6. Sci Bull. 2021;66(8):803–812. doi: 10.1016/j.scib.2020.12.027. [DOI] [PubMed] [Google Scholar]
- 296.Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. 2018;36(4):346–351. doi: 10.1038/nbt.4086. [DOI] [PubMed] [Google Scholar]
- 297.Tan WCC, Nerurkar SN, Cai HY, Ng HHM, Wu D, Wee YTF, et al. Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun. 2020;40(4):135–153. doi: 10.1002/cac2.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Lu Y-C, Zheng Z, Lowery FJ, Gartner JJ, Prickett TD, Robbins PF, et al. Direct identification of neoantigen-specific TCRs from tumor specimens by high-throughput single-cell sequencing. J ImmunoTher Cancer. 2021;9(7):e002595. doi: 10.1136/jitc-2021-002595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Chen B, Khodadoust MS, Olsson N, Wagar LE, Fast E, Liu CL, et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat Biotechnol. 2019;37(11):1332–1343. doi: 10.1038/s41587-019-0280-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Wu J, Wang W, Zhang J, Zhou B, Zhao W, Su Z, et al. DeepHLApan: a deep learning approach for neoantigen prediction considering both HLA-peptide binding and immunogenicity. Front Immunol. 2019;10:2559. doi: 10.3389/fimmu.2019.02559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Bulik-Sullivan B, Busby J, Palmer CD, Davis MJ, Murphy T, Clark A, et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat Biotechnol. 2018;37(1):55–63. doi: 10.1038/nbt.4313. [DOI] [PubMed] [Google Scholar]
- 302.Zaidi N, Soban M, Chen F, Kinkead H, Mathew J, Yarchoan M, et al. Role of in silico structural modeling in predicting immunogenic neoepitopes for cancer vaccine development. JCI Insight. 2020;5(17):e136991. doi: 10.1172/jci.insight.136991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Lapuente-Santana Ó, van Genderen M, Hilbers PAJ, Finotello F, Eduati F. Interpretable systems biomarkers predict response to immune-checkpoint inhibitors. Patterns. 2021;2(8):100293. doi: 10.1016/j.patter.2021.100293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Haanen JBAG, Carbonnel F, Robert C, Kerr KM, Peters S, Larkin J, et al. Corrections to “Management of toxicities from immunotherapy: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29:iv264–iv6. doi: 10.1093/annonc/mdy162. [DOI] [PubMed] [Google Scholar]
- 305.Porcu M, De Silva P, Solinas C, Battaglia A, Schena M, Scartozzi M, et al. Immunotherapy associated pulmonary toxicity: biology behind clinical and radiological features. Cancers. 2019;11(3):305. doi: 10.3390/cancers11030305. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.