

Summary
KRAS inhibitor AMG510 covalently modifies the G12C residue and inactivates the KRAS/G12C function. Because there are many reactive cysteines in the proteome, it is important to characterize AMG510 on-target modification and off-targets. Here, we presented a streamlined workflow to measure abundant AMG510 modified peptides including that of KRAS/G12C by direct profiling, and a pan-AMG510 antibody peptide IP workflow to profile less abundant AMG510 off-targets. We identified over 300 off-target sites with three distinct kinetic patterns, expanding the AMG510 modified proteome involved in the nucleocytoplasmic transport, response to oxidative stress, adaptive immune system, and glycolysis. We found that AMG510 covalently modified cys339 of ALDOA and inhibited its enzyme activity. Moreover, AMG510 modified KEAP1 cys288 and induced NRF2 accumulation in the nuclear of NSCLC cells independent of KRAS/G12C mutation. Our study provides a comprehensive resource of protein off-targets of AMG510 and elucidates potential toxicological sideeffects for this covalent KRASG12C inhibitor.
Subject areas: Biochemistry methods, Proteomics, Biochemical assay
Graphical abstract
Highlights
-
•
A direct streamlined workflow is developed to define the AMG510 modified proteome
-
•
AMG510 modifies proteins in response to oxidative stress, immune system, glycolysis, etc.
-
•
AMG510 covalently modifies cys339 of ALDOA and inhibits its enzyme activity
-
•
AMG510 covalently modifies cys288 of KEAP1 and induces nuclear accumulation of NRF2
Biochemistry methods; Proteomics; Biochemical assay
Introduction
KRAS is one of the most frequently mutated oncogenes.1,2,3,4 Efforts to develop drugs to inhibit KRAS have been largely unsuccessful because of the lack of a clear binding pocket.5 Recently, a class of drugs have been developed that covalently and irreversibly modify the KRAS/G12C at the cysteine 12 residue. These small molecules occupy a pocket in the switch II region (SIIP) when KRAS/G12C in its inactive GDP-bound conformation, and inhibit the KRAS oncogenic signaling and elicit anti-tumor response in tumor models.6,7,8 These new drugs include ARS853,9,10 1_AM,11 ARS1620,8 MRTX849 (adagrasib)7 and AMG510 (sotorasib).6,12 AMG510 and MRTX849 have been shown to potently suppress cell and xenograft tumor growth with half-maximal growth inhibitory concentration (IC50) in the low nanomolar range. An interaction with His95 groove in the helix 2 of the KRAS/G12C is reported to enhance their potency of inhibition.13 So far, both sotorasib and adagrasib showed promising anticancer activity in patients with KRAS/G12C mutated non–small-cell lung cancer (NSCLC), and sotorasib has been approved by FDA for the treatment of adult patients with KRAS/G12C-mutated locally advanced or metastatic NSCLC.14,15,16 However, despite the durable clinical benefits with KRAS/G12C mutant tumors, side effects such as diarrhea, anemia, liver enzyme abnormalities or QT prolongation have been reported for patients treated with sotorasib and adagrasib.12,15,16
Co-occurring mutations have the potential to alter the prognosis of KRASi treatment.16 Among the KRAS/G12C mutant NSCLC patients, co-occurring mutations were identified primarily with tumor suppressor drivers TP53 (35%), STK11 (31%) and KEAP1 (23%).17 Furthermore, advanced NSCLC patients with co-mutation of KRAS and KEAP1 have shorter duration on chemotherapy and overall survival in immunotherapy compared with only KRAS-mutant patients.18 Exploratory analyses for both sotorasib and adagrasib have analyzed the impact of co-occurring alterations on efficacy. For sotorasib, KEAP1 wild-type patients reported an ORR of 44% whereas those with KEAP1-mutated had an ORR of 20%.16 Adagrasib also exhibited a lower ORR in the KEAP1 mutant patients compared with the wild-type subgroup (36% versus 48%).15 All this suggested that KEAP1 mutation (loss of function) had negative impact on survival and immunotherapy efficacy in the KRAS/G12C mutant subgroup. The activity of KEAP1 was also switched off when the oxidative and electrophilic stresses occur.19 This KEAP1 loss was because of the direct covalent modification of electrophilic to the abundant cysteine residues of KEAP1.
Characterization of off-targets is always desirable and is particularly more urgent for covalent inhibitors targeting Cys residues because they often provide the crucial functionalities for proteins.20,21 Activity-based protein profiling (ABPP) is a frequently used chemical proteomics method for screening drug on/off targets,22,23 and has been applied to several covalent kinase inhibitors, including ibrutinib, PF-6274484, and osimertinib.24,25 A similar chemical proteomics approach has been used to characterize the target spectra of 243 clinical kinase inhibitors (kinobeads), which not only facilitated their repurposing, but also revealed the undesired side effects.26,27 Because the ABPP method typically requires the modification of the drug, which may alter the drug property, it may give rise to false-positive or false-negative identification of the original drug targets. Modification-free methods, e.g., thermal proteome profiling (TPP)28,29 and solvent-induced protein precipitation (SIP),30 which can avoid such complications, have been used for both covalent and noncovalent inhibitors such as TNP-470, staurosporine, ouabain, and dasatinib.31,32 However, these indirect characterization methods are often cumbersome and time-consuming, which requires substantial MS machine time. Off-targets of the KRAS inhibitor ARS-853, ARS-1620, and AMG510 have been studied previously using the desthiobiotin-IAA probe. This approach measured the decreased intensities of the desthiobiotin-IAA modified peptides after the inhibitor treatment, and has identified a few off-targets.6,8,10 This is an indirect measurement method that relies on the negative information (decreased IAA modifications) to infer modifications by the inhibitors.
Here we present a direct measurement workflow to characterize the AMG510 modifications and also develop a pan anti-AMG510 antibody to enrich the AMG510 modified peptides, allowing for global profiling of AMG510 off-targets. We identified a total of high confident 294 distinct AMG510 modified cysteines in H358 cells, and 62/174 AMG510 covalent bound cysteines in MIAPaca-2 CDX tumor/heart tissues. AMG510 can covalently modify protein targets to modulate the nuclear cytoplasmic transport, cell redox homeostasis, and glycolysis pathways. Specifically, we discovered that the AMG510 modification on cysteine 288 of KEAP1 might impair the KEAP1 activity and this might induce the nuclear accumulation of NRF2. Our study would provide a comprehensive resource for studying the anti-tumor function of AMG510 independent of KRAS signaling pathway and the pan anti-AMG510 antibody would serve as a potent tool for the drug development in future.
Results
Validation of a direct measurement assay for KRAS/G12C AMG510 covalent modification
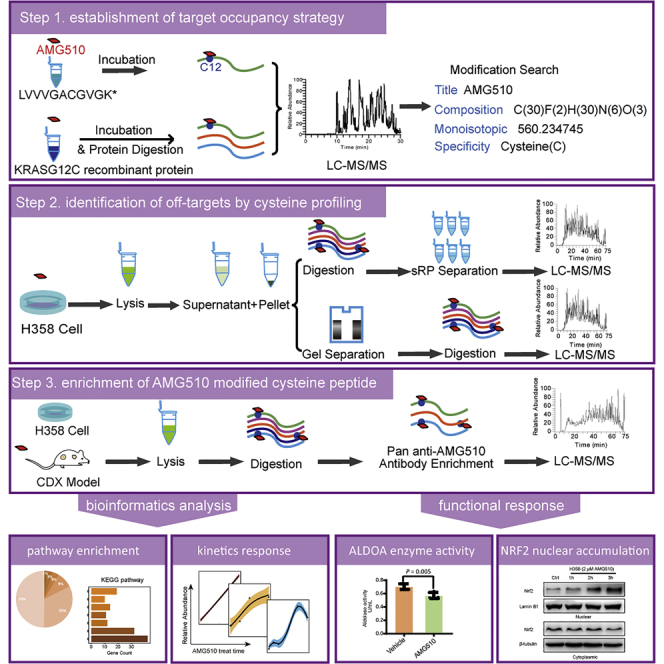
We first established a method to quantify the AMG510 modification of KRAS/G12C by using a synthetic tryptic peptide containing G12C (LVVVGACGVGK∗, ∗denote heavy isotope). AMG510 was incubated in vitro with the peptide and the resulting product was measured with LC-MS/MS. Both the free and AMG510 modified peptides were readily detected and distinguished by different retention times (RT = 10.79 min and 20.17 min, respectively). Furthermore, MS/MS demonstrated that all fragment ions containing the modified amino acid were mass-shifted according to the calculated mass of the AMG510 chemical formula (+560.2347 Da). These results confirmed that AMG510 modified the Cys residue in the peptide, and that the modification was stable (Figure 1A). Using the same procedure, we validated the Cys modification using a recombinant KRAS/G12C protein (Figure 1B). Based on these results, we set the AMG510 modification (+560.2347 Da) at cysteine as a dynamic modification in the subsequent MS analysis to search for AMG510 modified peptides.
Figure 1.

Mass spectrometric identification and verification of an AMG510 covalently modified peptide from KRAS/G12C protein
(A) MS/MS spectrum of the triply charged synthetic peptide (LVVVGACGVGK∗, ∗denote heavy isotope) covalently modified by AMG510 (left) and the extracted ion chromatograms (XICs) showing the retention times of free and modified peptides (right). Inset shows the precursor ion mass.
(B) MS/MS spectrum of the AMG510-modified LVVVGACGVGK from trypsin digested recombinant KRAS/G12C protein (left) and XICs of the free and modified peptides (right).
Direct profiling of AMG510 modified proteins in H358 cell line
We next set up an assay to directly measure AMG510 modification of KRAS/G12C and off-target modifications in cell lines. We treated the H358 cells with 2 μΜ AMG510 for 4 h and separated the soluble and insoluble protein fractions to simplify the proteome. Proteins were either in-solution digested followed by basic pH RP-LC separation into three fractions, or fractionated by SDS gel electrophoresis, cut into three bands, and extracted the in-gel digested peptides. LC/MS/MS was used to identify the proteins using a 75-min HPLC gradient for each fraction.
We filtered the data as described in the STAR Methods section. Briefly, the putative AMG510-modified peptides were first filtered with an ion-score≥10 and PEP≤0.01. Next, manual confirmation was performed to ensure that the isotopic peaks were in agreement with the charge state and the intensity distributions of the peptides. Finally, enough b/y fragment ions containing the AMG510-modified cysteine should be detected to insure the reliability of the candidate peptides.
As shown in Figure 2A, more than 20 AMG510 modified peptides were identified (Supplemental information, Table S1), with the KRAS/G12C peptide being the most abundant. In addition to the two off-targets that were identified in previous studies (RTN4 C1101 and HMOX2 C282), other off-targets were discovered for the first time. They included enzymes (PLOD3 C691, RPE C5, SPTLC2 C99, UBE2O C370, and TK1 C3), protein folding accessory protein (HSPD1 C237), differentiating epithelia cell markers (KRT4 C42), multifunctional membrane-binding protein (ANXA2 C9), tumor suppressor (PCBP1 C194) and transporters (RAE1 C106, S100A16C4, VDAC2 C91 and C225).33,34 The KRAS/G12C C12 containing peptide showed time and dose dependence on the AMG510 treatment (Figures 2B and 2C). A comparison of our results with previously published results is shown in Figure 2D and Supplemental information, Table S2.6,8,10,35 We conclude that direct proteomic profiling of three fractions with 75 min MS running time for each fraction was able to identify more than twenty AMG510 modified proteins.
Figure 2.

Summary of the AMG510 modified proteins by direct profiling
(A) AMG510 modified peptides with specific cysteine sites identified by direct profiling procedures: SDS-PAGE followed by in-gel digestion (In-Gel) or in-solution digestion followed by basic pH RP-LC (sRP). Two biological replicates are shown. Intensities are shown in log10 transformed AMG510 modified peptides areas.
(B) The time-dependence of the AMG510 modified KRAS/G12C peptide. The y axis on the left shows the intensity and the y axis on the right indicates the calculated target occupancy.
(C) The dose-dependence of the AMG510 modified KRAS/G12C peptide.
(D) A comparison of our method with previous studies on the number of off-targets modified by different covalent KRAS inhibitors.
Affinity profiling of AMG510 modified peptides in H358 cells using a pan anti-AMG510 antibody
To develop a more sensitive method that also requires less sample manipulation, we generated a polyclonal antibody targeting the AMG510 chemical moiety (pan anti-AMG510 antibody) and immobilized it on protein-A agarose beads to enrich AMG510 modified tryptic peptides. The pan anti-AMG510 antibody recognized the AMG510 modified peptide, but not MRTX849 modified and unmodified peptides although MRTX849 shares a similar covalent warhead with AMG510. (Figure 3A).
Figure 3.

Enrichment and quantification of AMG510 modified peptides by using a pan anti-AMG510 antibody
(A) Dot blot assay of the pan anti-AMG510 antibody on the unmodified LVVVGACGVGK peptide, or modified on the Cys12 by MRTX849 or AMG510. 100 ng of peptides or 10X serial diluted peptides were spotted on nitrocellulose membrane and detected with chemiluminescence.
(B) A Venn diagram showing the reproducibility of the peptide-IP profiling of AMG510 modification sites in three biological replicates.
(C) Scatterplots showing the Pearson correlation coefficients of modified peptide intensities between any two biological replicates on a log2 scale.
(D) Packcircles plot of peptide intensities of the off-targets identified in H358 cells treated with 2 μΜ AMG510 for 4h. The area of the circle and the color represents the relative intensities of each modified peptides.
(E) Bioinformatics analysis of the prominent 197 AMG510 covalent protein targets in terms of cellular location.
(F) The amino acid sequences flanking the AMG510 modified cysteine residue of the off-targets identified in (B) were analyzed by iceLogo.
We carried out peptide IPs using tryptic peptides obtained from H358 cells treated with AMG510 for 4 h, and analyzed the eluted peptides with a 75-min single shot LC-MS/MS runs. Using the stringent criteria described above, we identified on average 190 AMG510 modified peptides in a single peptide IP and a total of 274 AMG510-modified peptides on 233 proteins with >70% overlap in three replicates. (Supplemental information, Table S3). (Figure 3B). Moreover, an average square of Pearson correlation coefficient of 0.96 in peptide intensity was achieved between biological replicates, suggesting that the peptide-IP approach was highly reproducible (Figure 3C). The dynamic range of the modified peptide intensities was spanning approximately 3 orders of magnitude (6e6-1e9) with 4 h treatment (Figure 3D). Subcellular location of these off-targets proteins by Metascape revealed that the majority of them are cytoplasmic and nuclear proteins (Figure 3E). To investigate whether the pan-AMG510 antibody has sequence preference, we analyzed the amino acid sequences surrounding the modified cysteine sites to identify overrepresented sequence motifs using iceLogo.36 The analysis did not yield any enriched sequence motifs in the flanking sequences (Figure 3F), suggesting that the pan anti-AMG510 antibody is insensitive to the amino acid sequences in the vicinity of the modified cysteine and may be highly specific to the AMG510 chemical moiety. In conclusion, the workflow using a pan-AMG510 antibody peptide IP allowed the identification of low abundant off-targets.
Charting a temporal profile of the off-targets on AMG510 treatment
To investigate the kinetics of AMG510 modification, we performed temporal AMG510 off-target profiling at various times (0, 30 min, 60 min, 120 min, 180 min and 240 min). We selected only off-targets that were detected in at least 3 out of 6 time points and appeared in at least 3 consecutive time points. We used the resulting 185 off-targets to carry out the Fuzzy C-Means (FCM) clustering analysis (Supplemental information, Table S4). Three distinct kinetic patterns were evident (Figure 4A). Cluster 1 contained the majority (38%) of the off-targets and showed a marked increase throughout the 240-min time course. This cluster contained nuclear exportins which transport protein (CSE1L) and RNA (RAE1 and XPO5) from nucleus into cytoplasm; subunits of nuclear pore complex (NUP93, NUP98, NUP153, and NUP155) which facilitate this translocation process. The oxidoreductases (CYP1B1 and HMOX2) and a substrate adaptor of E3 ligase (KEAP1) that degrades the master regulator of anti-oxidant and stress, NRF2, were also identified in this cluster, suggesting the AMG510 may impact the oxidative stress response (Figure 4B). Cluster 2 showed an early and pronounced increase throughout the first 180 min and then remained unchanged. Cluster 2 included the target protein KRAS/G12C and its downstream effectors, including ROCK2, PI3KCA, and RICTOR, and other components in receptor tyrosine kinases signaling (JUP, PXN) and glycolysis enzymes (PKM, TPI1). Moreover, this cluster also contained proteins in the adaptive immune systems, including the immunoproteasome PSME2 that is required for efficient antigen processing, purine metabolism regulator AMPD2, and UBA6 that not only activates ubiquitin but also the leukocyte antigen F-associated transcript 10 (FAT10).37,38,39 Furthermore, an immune checkpoint protein, ADAR, that is important for cancer immunotherapy was also found in cluster 2. These immune-related off-targets suggest possible complex effects on immunotherapy.40 Cluster 3 showed a rapid increase that reached maximum at 180 min, followed by a sharp decline. This cluster contained proteins involved in regulation of adaptive immune system, including MHC molecules HLA-A, proteasomal components PSME1, and E3 ubiquitin-protein ligase RNF213. Five glycolysis regulators, including ALDOA, GAPDH, PGK1, PKM, and LDHB were also classified to cluster 3. Another nuclear transporter KPNA4, which recognizes NLSs and docks NLS-containing proteins to the nuclear pore complex,41 was also identified in cluster 3. Taken together, these time course results led us to propose that AMG510 off-targets may impact biological processes of nucleocytoplasmic transport, oxidative stress response, immune response, and glycolysis.
Figure 4.

Temporal profiles of the off-targets identified in H358 cells
(A) The 185 AMG510 off-targets are classified into 3 clusters based on the distinct kinetics of its members.
(B) Heatmap of 45 differentially modified AMG510 off-targets identified in (A) and their associated biological pathways.
Identification of AMG510 off-targets in CDX tissues
We next investigated whether the approach could identify off-targets in the MIAPaCa-2 CDX mouse tissues. The mice were treated with AMG510 at an orally dose of 10 mg/kg, which produced partially blockade of tumor growth in the MIAPaCa-2 CDX. A total of 62 off-targets were identified in the CDX tumor tissue following the peptide IP (Figure 5A and Supplemental information, Table S5). Pathway enrichment analysis using Metascape showed that glycolysis and gluconeogenesis (Log(q-value) = −8.87), heterocycle catabolic process (Log(q-value) = −7.42), and neutrophil degranulation (Log(q-value) = −6) are the most significantly enriched groups, indicating a possible modulation of immune response and glycolysis by AMG510 (Figure 5B).
Figure 5.

AMG510 off-targets identified in tissues of MIAPaCa-2 derived tumor xenograft
Packcircles plot of intensities of off-targets from tumor (A) and heart tissue (C). The enriched pathways of the off-targets from tumor (B) and heart tissue (D) obtained from Metascape.
Two acrylamide-based drugs, osimertinib and MRTX849, have been reported to induce unanticipated cardiotoxicity which becomes evident only after long-term accumulation of the drugs.42,43 To investigate off-targets of AMG510 in the heart tissue, we performed peptide-IP and identified 186 modified peptides (Figure 5C and Supplemental information, Table S6). Pyruvate metabolism and citric acid (TCA) cycle (Log(q-value) = −7.72), cofactor metabolic process (Log(q-value) = −7.72), and muscle system process (Log(q-value) = −5.83) were the most significantly enriched groups (Figure 5D). These off-targets suggested that AMG510 may impact the TCA cycle and possibly the cardiac functions.
Potential pharmacological or toxicological effects of the off-targets
The above results showed that several key enzymes in the glycolysis pathway, including ALDOA, TPI1, GAPDH, PGK1, PGAM1, ENO1, PKM, and LDHB (Figure 6A), were modified by AMG510. ALDOA is located at the most upstream position in the glycolysis cascade, and plays an essential function. We first confirmed that Cys339 was also endogenously modified by AMG510 in H358 cells (Supplemental information, Figure S1). ALDOA Cys339 was previously found to be modified by itaconate that led to impaired glycolytic flux, suggesting that this cysteine is functionally important.44 Because of the proximity of the substrate binding site of ALDOA and the AMG510 modification site at cysteine 339 (Figure 6B), we hypothesized that AMG510 modification might affect the enzyme activity of ALDOA. The ALDOA enzyme activity assay showed that the activity was indeed inhibited by the AMG510 treatment (Figure 6C).
Figure 6.

AMG510 modifies ALDOA and RAE1 and inhibits aldolase activity
(A) Multiple cysteines in eight crucial glycolytic enzymes are modified by AMG510.
(B) Structural view of AMG510 modified Cys339 in ALDOA (PDB: 4ALD). The protein backbone is shown as slate cartoon. AMG510 is shown as cyan sticks and the fructose 1,6-bisphosphate is shown as deep salmon sticks. The distance between the two cysteines is shown.
(C) Aldolase activity in H358 cells treated with 2μΜ AMG510 or vehicle control for 120 min. p values was calculated in Student’s t-tests performed on aldolase activity. Data are represented as mean ± SD.
(D) Molecular docking modeling of the possible effect of AMG510 on the RAE1·NUP98 (PDB: 3MMY) complex. Cys106 and NUP98 are colored in cyan and magenta, respectively.
Because exportin RAE1 and a number of NUP subunits were significantly enriched in cytoplasmic nuclear transport pathway, we investigated its potential pharmacological or toxicological effects. We performed molecular docking of AMG510 to the covalent binding site of cysteine 106 on RAE1 by using AutoDock for Flexible Receptors.45 The modeling generated a total of 4 modes of low energy states. The detailed parameters and structural overviews of these possible models were depicted in the refining results (Supplemental information, Figure S2). As shown in Figure 6D, the surface groove of RAE1 was occupied by AMG510 at the Cys106 to form a covalent bond, which overlapped with the interaction groove within the structure of nucleoporin NUP98. Thus, covalent modification of AMG510 may disrupt the interaction of RAE1 and NUP98 and inactivate the functions of the RAE1/NUP98 complex.46
We also found AMG510 covalently modified the key KEAP1 cysteine residue 288, and this modification was validated by using AMG510 to modify synthetic KEAP1 peptide (CEILQSDSR) in vitro (Figure 7A). Cys288 serves as a primary sensor for some electrophiles (e.g., 15d-PGJ2, OA-NO2) with direct modification and is required for KEAP1-dependent ubiquitination of NRF2.47 Our hypothesis is that AMG510 might activate NRF2 by covalent modification of KEAP1 cysteine residue 288. WB analysis showed that AMG510 treatment induced the nuclear accumulation of NRF2 in a time-dependent manner but not for cytoplasmic extract (Figure 7B). The increasing nuclear NRF2 (western blot) and AMG510 modified KEAP1 peptide (peptide-IP) exhibited similar patterns (Figure 7C). Taken together, the data suggest that the newly translated NRF2 under AMG510 treatment circumvent KEAP1 binding and translocate to the nucleus, whereas the “old” NRF2 was still trapped in the cytosol.
Figure 7.

AMG510 modifies Cys288 of KEAP1 and induces the nuclear accumulation of NRF2 in vivo
(A) MS/MS spectrum of the AMG510-modified EILQSDSR from trypsin digested H358 cells after treatment with AMG510.
(B) Western blotting showed the effect of AMG510 on the nuclear and cytoplasmic fraction of NRF2 protein of H358 cells.
(C) Quantitative comparison of peptide-IP and western blot analysis on the activation of the covalent modification of KEAP1Cys288 and nuclear accumulation of NRF2.
(D) Western blotting showed the effect of AMG510 on the nuclear of NRF2 protein of two NSCLC cell lines, including HCC827 (KRAS WT) and A549 (KRAS G12S).
Discussion
We developed two approaches to identify the on- and off-targets of the KRAS inhibitor AMG510. The direct profiling approach allowed positive identification of moderate to high abundance off-targets with a few hours of MS measurement time; affinity profiling with a pan anti-AMG510 antibody enabled the identification of low to high abundance off-targets in 1 h of MS time. The deep coverage of the AMG510 modified proteome made it feasible to evaluate the biological and clinical impact of the AMG510 modification.
Pan-modification specific antibodies have been widely used in proteomics for efficient global analysis of specific modifications, as demonstrated in large-scale studies of protein lysine acetylation,48 arginine methylation,49 tyrosine phosphorylation,50,51 lysine succinylation,52 and lysine 2-hydroxyisobutyrylation.53 We now extended this method to analyze covalent chemical modifications. Using a relatively mature technique – peptide IP followed by a 75-min MS measurement, we identified more than 300 reliable AMG510 modification sites. This dataset represented the largest AMG510 modified targets described to date, which provides a rich resource for the community to explore the anti-tumor properties of AMG510. AMG510 was considered a “clean” drug, as only a few off-targets were reported. Our analysis revealed at least several hundred off-targets with varying abundance, mainly on proteins involved in nucleocytoplasmic transport, response to oxidative stress, adaptive immune system, and glycolysis processes.
Do these off-targets have functional relevance? Because no significant cell viability change was observed in cells without KRAS/G12C mutation, it suggests that AMG510 modifications on these off-targets are unlikely to exert strong toxicity under cell culture conditions in the absence of KRAS/G12C mutation. It remains unknown what the impact of these off-targets may have in vivo. Our data showed that the most significantly enriched pathways of the off-targets are nucleocytoplasmic transport, immune response, and glycolysis. It is intriguing that nucleocytoplasmic transport is a cancer vulnerability pathway, as a drug targeting XPO1 (selinexor) has been recently granted accelerated approval for adult patients with relapsed or refractory diffuse large B-cell lymphoma.54 Modification of RAE1 that may disrupt its interaction with NUP98 could impact on the nucleocytoplasmic transport function of the RAE1/NUP98 complex. AMG510 also modified ADAR, a protein that was demonstrated to be important in cancer immunotherapy. Recently, BALB/c mice treated with AMG510 in combination with the anti-PD1 antibody 29F.1A12 exhibited improved survival benefits.6 It would be interesting to investigate if AMG510 modifications on ADAR and other immune response proteins have synergistic effect on immunotherapy. Ank2 was modified by AMG510 at Cys3737 in the mouse heart tissue. ANK2 is involved in anchoring the Na/K-ATPase, Na/Ca exchanger, and InsP3 receptor to specialized microdomains in the cardiomyocyte transverse tubules.55,56 Its loss of function mutations could give rise to a spectrum of clinical phenotypes including long QT syndrome, atrial fibrillation, ventricular arrhythmia and so on.57,58,59 Recently, KRYSTAL-1 clinical trial showed that KRAS inhibitor MRTX849 induced several TRAE (treatment-related adverse events), including QT prolongation with 14% incidence in any grade TRAEs.42 The off-targets identified in the heart tissue may provide clues for future investigation and evaluation.
KEAP1 mutation (loss of function) was one of the most frequent co-mutations (15.8%) in non–small-cell lung cancer (NSCLC) patients bearing with KRAS G12C mutant. The co-occurring alterations of KEAP1 and KRAS were also associated with worse overall survival and PD-L1 treatment benefit compared with that of the mono-mutation of NSCLC patients. Moreover, the phase 2 trial of sotorasib also showed the KRASG12C NSCLC patients with mutated KEAP1 response higher than wild-type KEAP1.16 All these results suggested that KEAP1 function loss might serve as a poor predictive marker for the NSCLC patients with KRAS G12C mutation. In this study, we confirmed AMG510 covalently modified Cys 288 of KEAP1 and led to NRF2 accumulation in the nuclear of different NSCLC cell lines with distinct types of KRAS mutation (Figure 7D). So, we strongly suspect this AMG510 induced KEAP1 function loss might independent of KRAS mutation type and eventually exert bad clinical outcomes in KRASG12C NSCLC patients.
In summary, combining the anti-AMG510 antibody with peptide-IP strategy, we identified a total of 274 AMG510 modified cysteines. This dataset provides valuable resources for further exploring the functional roles of AMG510 in nucleocytoplasmic transport, response to oxidative stress, adaptive immune system, and glycolysis. Our in-depth studies on ALDOA Cys339 and KEAP1 Cys288 conclude that, by modifying their functional cysteines, AMG510 inhibits their enzymatic activities, which collectively contribute to its potential pharmacological or toxicological effects. We anticipate that the method described here should be broadly applicable to study other covalent small molecule inhibitors and the information is of great value to drug discovery and development. The pan anti-AMG510 antibody generated in our study might serves as a powerful tool for the drug development in future.
Limitations of the study
Because of technical limitations, our method may fail to detect certain AMG510 modified peptides that are too long/short or unstable for LC-MS/MS detection, and the coverage should be improved by using alternative enzymes for digestion. Furthermore, because our study was only performed on preclinical cell line and CDX mouse models, clinical patients treated with AMG510 are necessary to further explore the AMG510 off-targets, providing comprehensive evaluation on the efficacy and safety of AMG510 therapy.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Recombinant Anti-Nrf2 antibody | Abcam | Cat# ab62352; RRID: AB_944418 |
| Recombinant Anti-Lamin B1 antibody | Abcam | Cat# ab133741; RRID: AB_2616597 |
| Anti-beta Tubulin antibody | Abcam | Cat# ab6046; RRID: AB_2210370 |
| Chemicals, peptides, and recombinant proteins | ||
| AMG510 | AbMole BioScience | M9356 |
| MRTX849 | AbMole BioScience | M9428 |
| LVVVGACGVGK∗ | Anhui Guoping Pharmaceutical Co., LTD | N/A |
| GDP-loaded, truncated (1-169) KRAS proteins | Reaction Biology | N/A |
| Critical commercial assays | ||
| Aldolase Activity Assay Kit | Abcam | Cat# ab196994 |
| Deposited data | ||
| Raw and analyzed data | This paper | PXD029449 |
| Experimental models: Cell lines | ||
| NCI-H358 cells | ATCC | Cat# CRL-5807; RRID: CVCL_1559 |
| MIA PaCa-2 cells | ATCC | Cat# CRL-1420; RRID: CVCL_0428 |
| A549 cells | ATCC | Cat# CCL-185; RRID: CVCL_0023 |
| HCC827 cells | ATCC | Cat# CRL-2868; RRID:CVCL_2063 |
| Experimental models: Organisms/strains | ||
| Mouse: BALB/c Nude Mouse | Gempharmatech Co., LTD | D000521 |
| Software and algorithms | ||
| R | R CRAN | https://cran.r-project.org/ |
| AutoDockFR | Sanner laboratory at Scripps Research | https://ccsb.scripps.edu/adfr/ |
Resource availability
Lead contact
Further information and requests for resources and data should be directed to and will be fulfilled by the lead contact, Jun Qin (jqin1965@126.com).
Materials availability
This study generates a new pan anti-AMG510 antibody, the antibody is available for anyone from the lead contact upon request.
Experimental model and subject details
Animals
Male BALB/c Nude Mouse (6-8 week) were purchased from the GemPharmatech. Co., LTD company and housed in a specific pathogen-free (SPF) environment with free access to food and water. All animal procedures and studies were conducted in accordance with the Medicine Animal Care and Use Committee at National Center for Protein Sciences (The PHOENIX Center, Beijing).
Method details
Cell lines and reagents
NCI-H358, MIA PaCa-2, A549 and HCC827 human cancer cell lines were purchased from AmericanType Culture Collection (ATCC) and maintained at 37°C in a humidified atmosphere at 5% CO2 and grown in RPMI-1640 growth media (H358 and HCC827) or DMEM (MIA PaCa-2 and A549) supplemented with 10% fetal bovine serum (Gibco), 50 units/mL penicillin, and 50 μg/mL streptomycin (Gibco). AMG510 (M9356) and MRTX849 (M9428) were purchased from AbMole BioScience.
KRAS biochemical modification assay
Heavy isotopic KRASG12C peptides covering residues 6−16 (LVVVGACGVGK∗) was used to react with 2 μM AMG510 or MRTX849 in 50 mM ammonium bicarbonate. GDP-loaded, truncated (1-169) KRAS proteins (G12C or wildtype) at 2 μM final concentration were incubated with 2 μM AMG510 in a buffer containing 20 mmol/L HEPES pH 7.5, 150 mmol/L NaCl, 1 mmol/L MgCl2, and 1 mmol/L DTT for 60minat 20°C. Reactions were quenched by adding formic acid to 0.2%. The resulting proteins were digested overnight at 37°C with trypsin in a 1:20 enzyme-to-protein ratio. Peptides were stored at -80°C until for MS analysis.
Direct profiling of AMG510 modified proteins in H358 cell line
NCI-H358 Cells (1 × 106) adhered overnight were treated with 2 μM AMG510 for 2 hours (Figure 2A) and 0.3 μM for the indicated incubation time (Figure 2B). After treatment, cells were washed twice with ice-cold PBS and then lysed in buffer containing 1% (w/v) sodium deoxycholate (DOC), 100 mM Tris (2-carboxyethyl) phosphine (TCEP), 400 mM 2-chloroacetamide (CAA) and 100 mM Tris-HCl (pH=8.8). The whole cell extracts were separated into soluble and insoluble fractions to simplify the proteome. The direct profiling included two peptide/protein separation methods. For small Reverse Phase (sRP) separation, the tryptic peptides were eluted from a C18 column sequentially with acetonitrile of different percentage as of acetonitrile (6%, 9%, 12%, 15%, 18%, 21%, 25%, 30% and 35%) and combined into three fractions. For gel separation, the supernatant and pellet fractions were fractionated by SDS gel electrophoresis, cut into 3 bands, followed by in-gel trypsin digestion. Peptides were stored at -80°C until redissolved for MS analysis. The KRAS/G12C occupancy fraction was calculated by dividing the decreased free peptides by the initial total free peptides.
Dot-blot of pan anti-AMG510 antibody
The pan anti-AMG510 antibody was co-developed with AbMax Biotechnology Co., LTD by immunizing the rabbit with AMG510 adducted KRAS/G12C peptide (LVVVGACGVGK) and isolated from the resulting serum with the highest ELISA titer. For dot blot assays, 100 ng of peptides (unmodified, MRTX849, and AMG510 modified peptide) or 10X serial diluted peptides were blotted onto the nitrocellulose membrane (Bio-Rad) and dried. The membrane was then blocked with freshly prepared PBS containing 5% non-fat milk for 1hat room temperature with constant agitation. The membrane was incubated with the pan anti-AMG510 antibody diluted in 1% freshly prepared PBS-milk solution for 1hat room temperature. After incubating the membrane with the secondary antibody (anti-rabbit HRP-conjugated IgG, Cell Signaling technology), chemiluminescent detection was performed using SuperSignal West Pico substrate (Thermo Scientific).
Affinity profiling of AMG510 modified peptides in H358 cell line
NCI-H358 cells (5 × 106) were treated with 2 μM AMG510 at the indicated concentration and incubation time (Figures 3 and 4). The temporal profiling in Figure 4 were performed in triplicates. The collected cells were lysed with DOC buffer, tryptic peptides were incubated with antibody-conjugated agarose beads at 4 °C for overnight. The peptides were then eluted with the extraction buffer (0.1M Glycine, pH 2.5), followed by desalting on a C18 column and dried in vacuum.
LC-MS/MS analysis using Q exactive HF
Dried peptide samples were re-dissolved in solvent A (0.1% formic acid in water). Liquid chromatographytandem mass spectrometry (LC-MS/MS) analysis was performed with Q-Exactive HF mass spectrometer (Thermo) equipped with an online Easy-nLC 1200 nano-HPLC system (Thermo). The injected peptides were separated on a reversed phase nano-HPLC C18 column (Pre-column: 3μm, 120 Å, 2 cm × 100 μm ID; analytical column: 1.9 μm, 120 Å, 15 cm × 150 μm ID) at a flow rate of 600 nL/min with different elution gradients: 8-40% solvent B (0.1% formic acid in 80% acetonitrile) in 30 min for synthetic peptide, 8-40% B in 30 min for recombinant KRAS protein, and 5-35% B in 75 min for direct and affinity profiling.
For MS analysis, the instrument was operated in the data-dependent acquisition mode. The full scan was processed in the Orbitrap from m/z 300–1400, at the resolution of 60 K, the AGC target was 3e6 and maximum injection time was 80 ms. The top 30 most intense ions in each scan cycle were selected for HCD fragmentation with normalized collision energy of 30%. For MS/MS scan, the fragment ions were detected in the Orbitrap with a resolution of 15 K, the AGC target was 5e4 and maximum injection time was 20 ms, dynamic exclusion was 15 s.
Western blot analysis
H358 cells were treated with or without 2 μM AMG510 for indicated time periods and harvested. Proteins were extracted from the cells, separated in 10% SDS polyacrylamide gel, and then transferred to PVDF membranes. The membranes were blocked in TBST buffer (0.125 M NaCl, 25 mMTris pH 8.0) containing 5% defatted milk for 1 h and then incubated with specific first antibody overnight at 4 °C. Protein bands were detected using the SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, USA) after hybridization with horseradish peroxidase-conjugated second antibody. NRF2, Lamin B1, and β-tubulin antibodies (#ab62352, #ab133741, #ab6046 separately) were the first antibodies for NRF2 detection.
Preparation of cytosolic and nuclear extracts
Cells were pelleted by centrifugation after washing with cold PBS and suspended in ice-cold hypotonic buffer. Following incubation in an ice bath for 15 min, cells were centrifuged again, and the supernatant was collected as a cytosolic fraction. The remaining cell pellets were resuspended in ice-cold buffer C containing 20 mM HEPES (pH 7.9), 20% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, and 0.2 mM PMSF and were incubated at 0 °C for 2 h. After vortex mixing, the resulting suspension was centrifuged (12,000 rpm, 15 min), and the supernatant was collected as a nuclear extract and stored at -80°C.
ALDOA enzymatic activity assay
The aldolase activity was measured using the Aldolase Activity Assay Kit (Colorimetric) (ab196994, Abcam). In brief, cells were treated with 2 μM AMG510 and vehicle control for 120 min. After treatment, the cells were incubated with reaction mix and the aldolase activity was determined by measuring the colorimetric product by a Benchmark microplate reader (Molecular Devices Corporation, USA).
Cell derived xenografts in nude mice
BALB/c nude mice (4–6 weeks old) obtained from Gempharmatech Co., LTD (Beijing China) were housed under a standard SPF (specific pathogen-free) laboratory environment. For xenograft production, MIA PaCa-2 human pancreatic cancer cells (2 × 106 cells in 200 μl PBS) were subcutaneously injected into the right flanks of each mouse to establish tumor. After 3 weeks, mice were then received oral doses of either vehicle control (5% DMSO +10% Solutol HS 15 + 85% (6%HP-β-CD in water)) or AMG510 (10 mg/kg/day). Treatment continued for a total of 3 days. The tumor size was measured using digital caliper and tumor volume was calculated with the following formula: volume = 0.5 × width2 × length. After mice were euthanized by isoflurane anesthesia, the tumors and heart tissues were excised and collected for peptide-IP analyses.
Quantification and statistical analysis
Database search and data processing
The acquired raw data were searched by Thermo Proteome Discoverer 2.0 equipped with Mascot 2.3 (Matrix Science Inc) against the human National Center for Biotechnology Information (NCBI) RefSeq protein databases (updated on 10-11-2019, added KRASG12C mutation, 32020 protein entries, https://www.ncbi.nlm.nih.gov/refseq/). The parameter settings were: two missed cleavages were allowed (trypsin); the mass tolerances were 20 ppm for precursor and 50 mmu for product ions; dynamic modifications were set as oxidation (M), carbamidomethyl (C), acetyl (protein N-term), AMG510 (C) (Monoisotopic: 560.2347). Assigned peptides were filtered with 1% false discovery rate (FDR) using Percolator validation based on q-value. The filters for modified peptides were Ion score≥10 and PEP≤0.01. Further manual verification was performed to exclude false identification including two steps, step 1 consists of checking isotope peaks of parent ions to ensure validity of charge and m/z of monoisotopic peaks; step 2 is a rigorous procedure which compared practical MS/MS spectra with theoretical spectra to evaluate credibility of every filtered PSMs.
Statistical analysis
R (version 3.6.1) statistical programming language was used for statistical analyses (Student’s t-tests for Figure 6) and visualization. Pearson correlation coefficient was used to measure the degree of correlation between independent biological replicates (Figure 3). Fuzzy C-means (FCM) clustering analysis was performed to partition off-targets proteome into k groups based on their temporal response kinetics (Figure 4). The FCM function from basic R stats package was executed with optimization of k and other parameters by default. The corresponding original expression profile was required to transform into Z-score as input before plotting, the expression pattern generated by the average of intensity in each cluster depicted its typical characteristic. Non-redundant pathway enrichment analysis for these off-targets (Figures 4 and 5) were enriched using Metascape (q-value <0.05).
Acknowledgments
This research work was supported by the National Key Research and Development Program of China (2022YFA1303200, 2018YFA0507503, 2022YFC0867400), National Natural Science Foundation of China (32088101, 81874237).
Author contributions
YiniW., B.Z., and J.Q. designed research; YiniW., B.Z., C.X., X.L., D.C., Q.D., B.Z., J.L., and Q.L., performed research; S.Z., D.Z., and M.L., contributed new reagents/analytic tools; YiniW. and B.Z. analyzed data; YiniW., B.Z., YiW., and J.Q. wrote the paper.
Declaration of interests
H.W. was an employee of Beijing Pineal Diagnostics at the time of data collection. X.L., D.C., Q.D., B.Z., J.L., and Q.L. were employees of GenFleet Therapeutics at the time of data collection.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: January 28, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106080.
Supplemental information
Data and code availability
-
•
The MS raw data generated in this study have been submitted to ProteomeXchange database via the iProX partner repository under accession number PXD029449.
-
•
This paper does not report original code. Any additional information required to reanalyze the reported data is available from the lead contact upon request.
References
- 1.AACR Project GENIE Consortium AACR Project GENIE: powering precision medicine through an international consortium. Cancer Discov. 2017;7:818–831. doi: 10.1158/2159-8290.CD-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manchado E., Weissmueller S., Morris J.P., Chen C.-C., Wullenkord R., Lujambio A., de Stanchina E., Poirier J.T., Gainor J.F., Corcoran R.B., et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature. 2016;534:647–651. doi: 10.1038/nature18600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Misale S., Yaeger R., Hobor S., Scala E., Janakiraman M., Liska D., Valtorta E., Schiavo R., Buscarino M., Siravegna G., et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ratner E., Lu L., Boeke M., Barnett R., Nallur S., Chin L.J., Pelletier C., Blitzblau R., Tassi R., Paranjape T., et al. A KRAS-variant in ovarian cancer acts as a genetic marker of cancer risk. Cancer Res. 2010;70:6509–6515. doi: 10.1158/0008-5472.CAN-10-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uprety D., Adjei A.A. KRAS: from undruggable to a druggable Cancer Target. Cancer Treat Rev. 2020;89:102070. doi: 10.1016/j.ctrv.2020.102070. [DOI] [PubMed] [Google Scholar]
- 6.Canon J., Rex K., Saiki A.Y., Mohr C., Cooke K., Bagal D., Gaida K., Holt T., Knutson C.G., Koppada N., et al. The clinical KRAS (G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–223. doi: 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 7.Hallin J., Engstrom L.D., Hargis L., Calinisan A., Aranda R., Briere D.M., Sudhakar N., Bowcut V., Baer B.R., Ballard J.A., et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020;10:54–71. doi: 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Janes M.R., Zhang J., Li L.-S., Hansen R., Peters U., Guo X., Chen Y., Babbar A., Firdaus S.J., Darjania L., et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell. 2018;172:578–589.e17. doi: 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 9.Lito P., Solomon M., Li L.-S., Hansen R., Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 2016;351:604–608. doi: 10.1126/science.aad6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patricelli M.P., Janes M.R., Li L.-S., Hansen R., Peters U., Kessler L.V., Chen Y., Kucharski J.M., Feng J., Ely T., et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016;6:316–329. doi: 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- 11.Zeng M., Lu J., Li L., Feru F., Quan C., Gero T.W., Ficarro S.B., Xiong Y., Ambrogio C., Paranal R.M., et al. Potent and selective covalent quinazoline inhibitors of KRAS G12C. Cell Chem. Biol. 2017;24:1005–1016.e3. doi: 10.1016/j.chembiol.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 12.Hong D.S., Fakih M.G., Strickler J.H., Desai J., Durm G.A., Shapiro G.I., Falchook G.S., Price T.J., Sacher A., Denlinger C.S., et al. KRASG12C inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med. 2020;383:1207–1217. doi: 10.1056/NEJMoa1917239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim D., Xue J.Y., Lito P. Targeting KRAS(G12C): from inhibitory mechanism to modulation of antitumor effects in patients. Cell. 2020;183:850–859. doi: 10.1016/j.cell.2020.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakajima E.C., Drezner N., Li X., Mishra-Kalyani P.S., Liu Y., Zhao H., Bi Y., Liu J., Rahman A., Wearne E., et al. FDA approval summary: sotorasib for KRAS G12C-mutated metastatic NSCLC. Clin. Cancer Res. 2022;28:1482–1486. doi: 10.1158/1078-0432.CCR-21-3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jänne P.A., Riely G.J., Gadgeel S.M., Heist R.S., Ou S.-H.I., Pacheco J.M., Johnson M.L., Sabari J.K., Leventakos K., Yau E., et al. Adagrasib in non–small-cell lung cancer harboring a KRASG12C mutation. N. Engl. J. Med. 2022;387:120–131. doi: 10.1056/NEJMoa2204619. [DOI] [PubMed] [Google Scholar]
- 16.Skoulidis F., Li B.T., Dy G.K., Price T.J., Falchook G.S., Wolf J., Italiano A., Schuler M., Borghaei H., Barlesi F., et al. Sotorasib for lung cancers with KRAS p. G12C mutation. N. Engl. J. Med. 2021;384:2371–2381. doi: 10.1056/NEJMoa2103695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palma G., Khurshid F., Lu K., Woodward B., Husain H. Selective KRAS G12C inhibitors in non-small cell lung cancer: chemistry, concurrent pathway alterations, and clinical outcomes. NPJ Precis. Oncol. 2021;5:98. doi: 10.1038/s41698-021-00237-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arbour K.C., Jordan E., Kim H.R., Dienstag J., Yu H.A., Sanchez-Vega F., Lito P., Berger M., Solit D.B., Hellmann M., et al. Effects of Co-occurring genomic alterations on outcomes in patients with KRAS-mutant non–small cell lung CancerCo-occurring genomic alterations in KRAS-mutant NSCLC. Clin. Cancer Res. 2018;24:334–340. doi: 10.1158/1078-0432.CCR-17-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi A., Kang M.-I., Watai Y., Tong K.I., Shibata T., Uchida K., Yamamoto M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell Biol. 2006;26:221–229. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McConnell E.W., Smythers A.L., Hicks L.M. Maleimide-based chemical proteomics for quantitative analysis of cysteine reactivity. J. Am. Soc. Mass Spectrom. 2020;31:1697–1705. doi: 10.1021/jasms.0c00116. [DOI] [PubMed] [Google Scholar]
- 21.Weerapana E., Wang C., Simon G.M., Richter F., Khare S., Dillon M.B.D., Bachovchin D.A., Mowen K., Baker D., Cravatt B.F. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pichler C.M., Krysiak J., Breinbauer R. Target identification of covalently binding drugs by activity-based protein profiling (ABPP) Bioorg. Med. Chem. 2016;24:3291–3303. doi: 10.1016/j.bmc.2016.03.050. [DOI] [PubMed] [Google Scholar]
- 23.van Rooden E.J., Florea B.I., Deng H., Baggelaar M.P., van Esbroeck A.C.M., Zhou J., Overkleeft H.S., van der Stelt M. Mapping in vivo target interaction profiles of covalent inhibitors using chemical proteomics with label-free quantification. Nat. Protoc. 2018;13:752–767. doi: 10.1038/nprot.2017.159. [DOI] [PubMed] [Google Scholar]
- 24.Lanning B.R., Whitby L.R., Dix M.M., Douhan J., Gilbert A.M., Hett E.C., Johnson T.O., Joslyn C., Kath J.C., Niessen S., et al. A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat. Chem. Biol. 2014;10:760–767. doi: 10.1038/nchembio.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niessen S., Dix M.M., Barbas S., Potter Z.E., Lu S., Brodsky O., Planken S., Behenna D., Almaden C., Gajiwala K.S., et al. Proteome-wide map of targets of T790M-EGFR-directed covalent inhibitors. Cell Chem. Biol. 2017;24:1388–1400.e7. doi: 10.1016/j.chembiol.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bantscheff M., Eberhard D., Abraham Y., Bastuck S., Boesche M., Hobson S., Mathieson T., Perrin J., Raida M., Rau C., et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 2007;25:1035–1044. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- 27.Klaeger S., Heinzlmeir S., Wilhelm M., Polzer H., Vick B., Koenig P.-A., Reinecke M., Ruprecht B., Petzoldt S., Meng C., et al. The target landscape of clinical kinase drugs. Science. 2017;358:eaan4368. doi: 10.1126/science.aan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franken H., Mathieson T., Childs D., Sweetman G.M.A., Werner T., Tögel I., Doce C., Gade S., Bantscheff M., Drewes G., et al. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 2015;10:1567–1593. doi: 10.1038/nprot.2015.101. [DOI] [PubMed] [Google Scholar]
- 29.Reinhard F.B.M., Eberhard D., Werner T., Franken H., Childs D., Doce C., Savitski M.F., Huber W., Bantscheff M., Savitski M.M., Drewes G. Thermal proteome profiling monitors ligand interactions with cellular membrane proteins. Nat. Methods. 2015;12:1129–1131. doi: 10.1038/nmeth.3652. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X., Wang Q., Li Y., Ruan C., Wang S., Hu L., Ye M. Solvent-induced protein precipitation for drug target discovery on the proteomic scale. Anal. Chem. 2020;92:1363–1371. doi: 10.1021/acs.analchem.9b04531. [DOI] [PubMed] [Google Scholar]
- 31.Martinez Molina D., Jafari R., Ignatushchenko M., Seki T., Larsson E.A., Dan C., Sreekumar L., Cao Y., Nordlund P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 32.Savitski M.M., Reinhard F.B.M., Franken H., Werner T., Savitski M.F., Eberhard D., Martinez Molina D., Jafari R., Dovega R.B., Klaeger S., et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 2014;346:1255784. doi: 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- 33.DeWard A.D., Cramer J., Lagasse E. Cellular heterogeneity in the mouse esophagus implicates the presence of a nonquiescent epithelial stem cell population. Cell Rep. 2014;9:701–711. doi: 10.1016/j.celrep.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo J., Jia R. Splicing factor poly(rC)-binding protein 1 is a novel and distinctive tumor suppressor. J. Cell. Physiol. 2018;234:33–41. doi: 10.1002/jcp.26873. [DOI] [PubMed] [Google Scholar]
- 35.Wijeratne A., Xiao J., Reutter C., Furness K.W., Leon R., Zia-Ebrahimi M., Cavitt R.N., Strelow J.M., Van Horn R.D., Peng S.-B., et al. Chemical proteomic characterization of a covalent KRASG12C inhibitor. ACS Med. Chem. Lett. 2018;9:557–562. doi: 10.1021/acsmedchemlett.8b00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colaert N., Helsens K., Martens L., Vandekerckhove J., Gevaert K. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods. 2009;6:786–787. doi: 10.1038/nmeth1109-786. [DOI] [PubMed] [Google Scholar]
- 37.Hipp M.S., Kalveram B., Raasi S., Groettrup M., Schmidtke G. FAT10, a ubiquitin-independent signal for proteasomal degradation. Mol. Cell Biol. 2005;25:3483–3491. doi: 10.1128/MCB.25.9.3483-3491.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kloetzel P.-M. The proteasome and MHC class I antigen processing. Biochim. Biophys. Acta. 2004;1695:225–233. doi: 10.1016/j.bbamcr.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 39.Aichem A., Pelzer C., Lukasiak S., Kalveram B., Sheppard P.W., Rani N., Schmidtke G., Groettrup M. USE1 is a bispecific conjugating enzyme for ubiquitin and FAT10, which FAT10ylates itself in cis. Nat. Commun. 2010;1:13. doi: 10.1038/ncomms1012. [DOI] [PubMed] [Google Scholar]
- 40.Bhate A., Sun T., Li J.B. ADAR1: a new target for immuno-oncology therapy. Mol. Cell. 2019;73:866–868. doi: 10.1016/j.molcel.2019.02.021. [DOI] [PubMed] [Google Scholar]
- 41.Beck M., Schirmacher P., Singer S. Alterations of the nuclear transport system in hepatocellular carcinoma–New basis for therapeutic strategies. J. Hepatol. 2017;67:1051–1061. doi: 10.1016/j.jhep.2017.06.021. [DOI] [PubMed] [Google Scholar]
- 42.Jänne P., Rybkin I.I., Spira A., Riely G., Papadopoulos K., Sabari J., Johnson M., Heist R., Bazhenova L., Barve M., et al. KRYSTAL-1: activity and safety of adagrasib (mrtx849) in advanced/metastatic non–small-cell lung cancer (NSCLC) harboring KRAS G12C mutation. Eur. J. Cancer. 2020;138:S1–S2. [Google Scholar]
- 43.Anand K., Ensor J., Trachtenberg B., Bernicker E.H. Osimertinib-induced cardiotoxicity: a retrospective review of the FDA adverse events reporting system (FAERS) JACC. CardioOncol. 2019;1:172–178. doi: 10.1016/j.jaccao.2019.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qin W., Qin K., Zhang Y., Jia W., Chen Y., Cheng B., Peng L., Chen N., Liu Y., Zhou W., et al. S-glycosylation-based cysteine profiling reveals regulation of glycolysis by itaconate. Nat. Chem. Biol. 2019;15:983–991. doi: 10.1038/s41589-019-0323-5. [DOI] [PubMed] [Google Scholar]
- 45.Ravindranath P.A., Forli S., Goodsell D.S., Olson A.J., Sanner M.F. AutoDockFR: advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Comput. Biol. 2015;11:e1004586. doi: 10.1371/journal.pcbi.1004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ren Y., Seo H.-S., Blobel G., Hoelz A. Structural and functional analysis of the interaction between the nucleoporin Nup98 and the mRNA export factor Rae1. Proc. Natl. Acad. Sci. USA. 2010;107:10406–10411. doi: 10.1073/pnas.1005389107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamamoto M., Kensler T.W., Motohashi H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018;98:1169–1203. doi: 10.1152/physrev.00023.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Svinkina T., Gu H., Silva J.C., Mertins P., Qiao J., Fereshetian S., Jaffe J.D., Kuhn E., Udeshi N.D., Carr S.A. Deep, quantitative coverage of the lysine acetylome using novel anti-acetyl-lysine antibodies and an optimized proteomic workflow. Mol. Cell. Proteomics. 2015;14:2429–2440. doi: 10.1074/mcp.O114.047555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo A., Gu H., Zhou J., Mulhern D., Wang Y., Lee K.A., Yang V., Aguiar M., Kornhauser J., Jia X., et al. Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell. Proteomics. 2014;13:372–387. doi: 10.1074/mcp.O113.027870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bergström Lind S., Molin M., Savitski M.M., Emilsson L., Aström J., Hedberg L., Adams C., Nielsen M.L., Engström A., Elfineh L., et al. Immunoaffinity enrichments followed by mass spectrometric detection for studying global protein tyrosine phosphorylation. J. Proteome Res. 2008;7:2897–2910. doi: 10.1021/pr8000546. [DOI] [PubMed] [Google Scholar]
- 51.Grønborg M., Kristiansen T.Z., Stensballe A., Andersen J.S., Ohara O., Mann M., Jensen O.N., Pandey A. A mass spectrometry-based proteomic approach for identification of serine/threonine-phosphorylated proteins by enrichment with phospho-specific antibodies: identification of a novel protein, Frigg, as a protein kinase A substrate. Mol. Cell. Proteomics. 2002;1:517–527. doi: 10.1074/mcp.m200010-mcp200. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Z., Tan M., Xie Z., Dai L., Chen Y., Zhao Y. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 2011;7:58–63. doi: 10.1038/nchembio.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dai L., Peng C., Montellier E., Lu Z., Chen Y., Ishii H., Debernardi A., Buchou T., Rousseaux S., Jin F., et al. Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat. Chem. Biol. 2014;10:365–370. doi: 10.1038/nchembio.1497. [DOI] [PubMed] [Google Scholar]
- 54.Ben-Barouch S., Kuruvilla J. Selinexor (KTP-330)-a selective inhibitor of nuclear export (SINE): anti-tumor activity in diffuse large B-cell lymphoma (DLBCL) Expert Opin. Investig. Drugs. 2020;29:15–21. doi: 10.1080/13543784.2020.1706087. [DOI] [PubMed] [Google Scholar]
- 55.Mohler P.J., Davis J.Q., Bennett V. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP 3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. 2005;3:e423. doi: 10.1371/journal.pbio.0030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mohler P.J., Davis J.Q., Davis L.H., Hoffman J.A., Michaely P., Bennett V. Inositol 1, 4, 5-trisphosphate receptor localization and stability in neonatal cardiomyocytes requires interaction with ankyrin-B. J. Biol. Chem. 2004;279:12980–12987. doi: 10.1074/jbc.M313979200. [DOI] [PubMed] [Google Scholar]
- 57.Mohler P.J., Splawski I., Napolitano C., Bottelli G., Sharpe L., Timothy K., Priori S.G., Keating M.T., Bennett V. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc. Natl. Acad. Sci. USA. 2004;101:9137–9142. doi: 10.1073/pnas.0402546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mohler P.J., Le Scouarnec S., Denjoy I., Lowe J.S., Guicheney P., Caron L., Driskell I.M., Schott J.J., Norris K., Leenhardt A., et al. Defining the cellular phenotype of “ankyrin-B syndrome” variants. Circulation. 2007;115:432–441. doi: 10.1161/CIRCULATIONAHA.106.656512. [DOI] [PubMed] [Google Scholar]
- 59.Tester D.J., Ackerman M.J. Genetics of long QT syndrome. Methodist Debakey Cardiovasc. J. 2014;10:29–33. doi: 10.14797/mdcj-10-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The MS raw data generated in this study have been submitted to ProteomeXchange database via the iProX partner repository under accession number PXD029449.
-
•
This paper does not report original code. Any additional information required to reanalyze the reported data is available from the lead contact upon request.