

Abstract
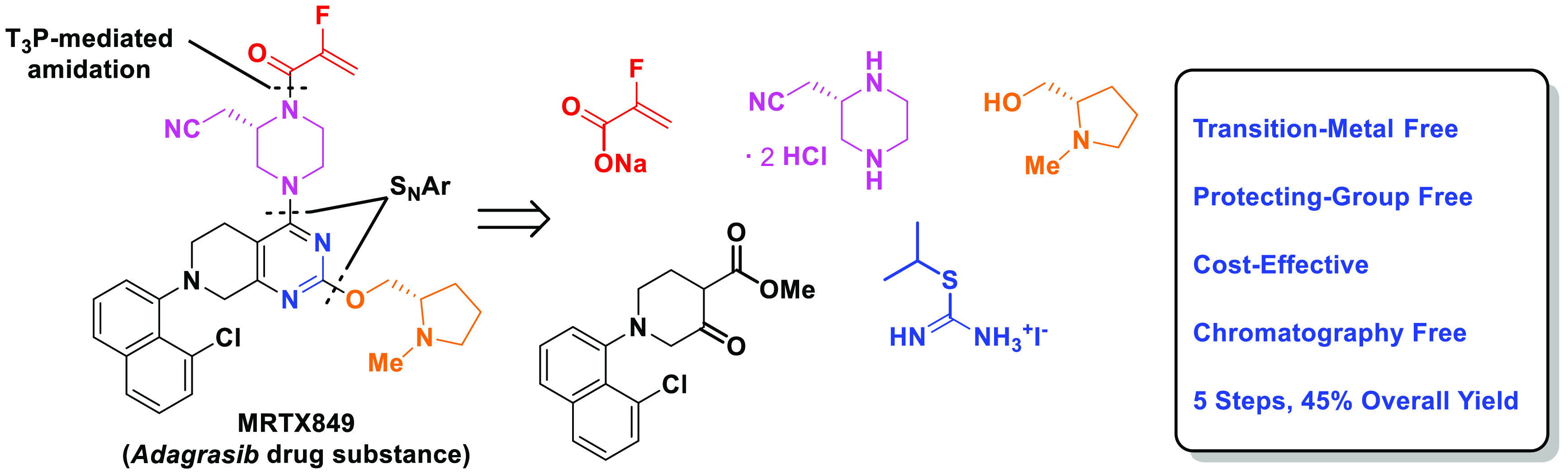
A concise, transition-metal and protection-free synthesis of adagrasib (MRTX849), a novel KRASG12C inhibitor drug recently approved by the FDA, is reported. Introduction of two chiral building blocks to the tetrahydropyridopyrimidine core was accomplished via two sequential SNAr reactions. Extensive reaction optimization led to a robust, transition-metal-free oxidation of the sulfide intermediate. A judicious choice of the leaving group with favorable steric and electronic characteristics at the 4-OH position of the tetrahydropyridopyrimidine core enabled a facile SNAr displacement to introduce the chiral piperazine. This new, five-step, chromatography-free synthesis of adagrasib from readily available starting materials obviated the palladium catalysis and protecting group manipulations in the current commercial route and significantly improved the efficiency of the process in 45% overall yield.
Kirsten rat sarcoma viral oncogene homologue (KRAS) is the notorious oncogene with the highest mutation rate among all cancers specifically associated with the top three most fatal cancers including pancreatic ductal adenocarcinoma (PDAC), non-small cell lung cancer (NSCLC), and colorectal cancer (CRC).1 Furthermore, KRAS has been considered as a challenging therapeutic target, famously designated as “undruggable”, and historically has had limited therapeutic options1,2 until recent discovery of sotorasib (AMG510)3 and adagrasib (MRTX849, 1 in Scheme 1)4 as drugs targeting the G12C variant. Adagrasib (1) is a highly selective and potent covalent KRASG12C inhibitor, which has recently been approved as a monotherapy drug and is being evaluated in a combination therapy in patients with advanced KRASG12C-mutated solid tumors.5−8Adagrasib (1) was reported to show only mild side effects when combined with the immunotherapy drugs.9 This positive clinical readout triggered the development of a more efficient process to this novel drug.
Scheme 1. Current Manufacturing Process to Adagrasib (1).
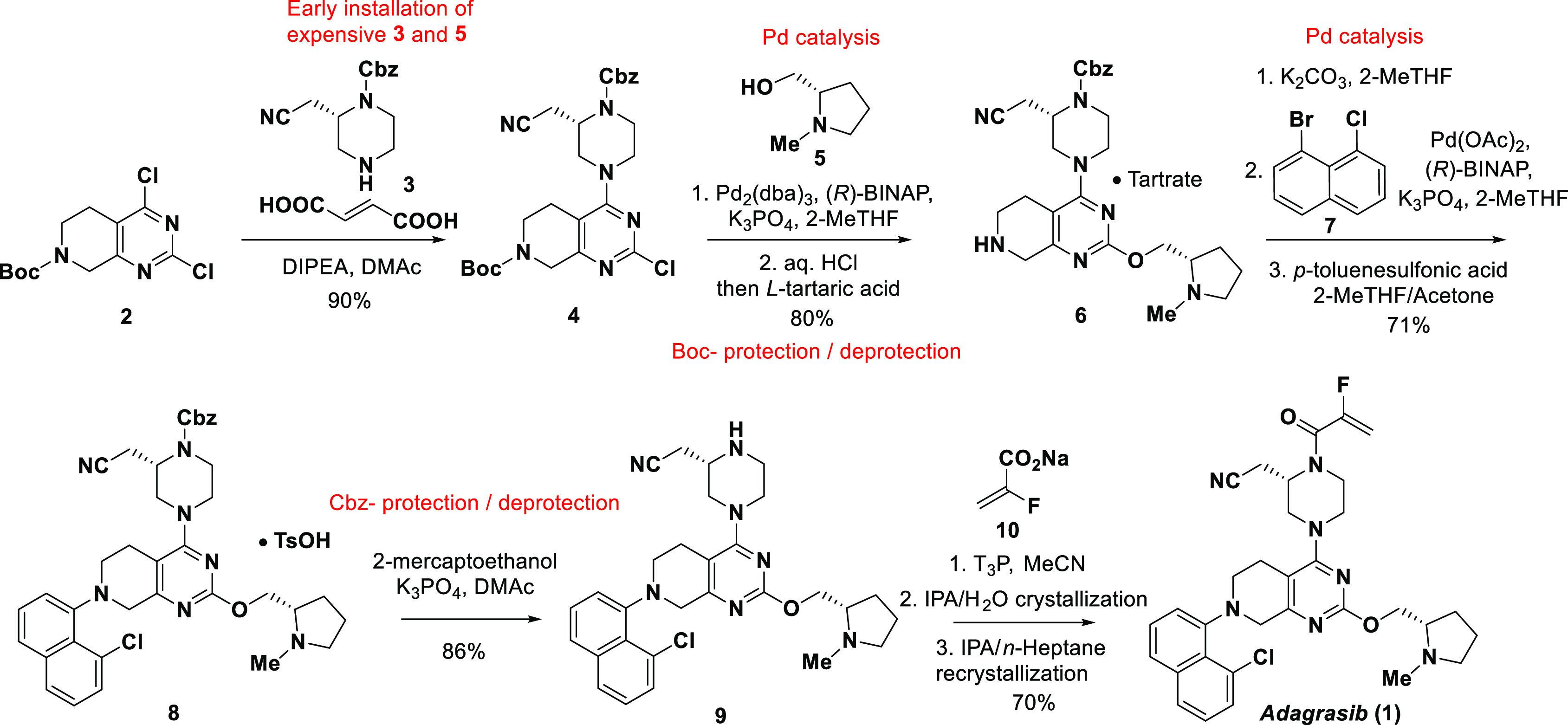
Adagrasib (MRTX849, 1) features three distinct subunits: N-methyl prolinol, chloronaphthyl, and substituted piperazine attached to the tetrahydropyridopyrimidine core. A unique 2-fluoroacrylamide warhead located at the distal side of the piperazine is responsible for covalent binding to the target protein. As shown in Scheme 1, the current synthetic route started with a Boc-protected tetrahydropyridopyrimidine 2.10 A regioselective SNAr reaction introduced the Cbz-masked piperazine moiety 3 at the more reactive 4-position. Installation of the prolinol subunit was achieved via palladium-catalyzed C–O bond formation between 4 at C-2 position and prolinol 5. Introduction of the chloronaphthyl subunit required Boc-deprotection followed by a Buchwald-Hartwig amination of the resulting 6 with chloronaphthyl bromide 7. Practical conditions employing 2-mercaptoethanol were developed to unmask the Cbz-protected piperazine to afford compound 9.11n-Propanephosphonic acid anhydride (T3P)-mediated coupling of 9 with the sodium 2-fluoroacrylate (10) completed the synthesis of adagrasib (1).
The current synthetic route has been employed to produce sufficient drug substance to support all the drug development activities including clinical studies and commercial launch. However, considering the overall efficiency, cost and sustainability, we identified three areas for further improvement (Scheme 1): (i) reducing or eliminating usage of expensive palladium catalysts/ligands in both C–O and C–N bond formations;12 (ii) deferring the installation of the expensive chiral piperazine to a later stage in the synthesis; and (iii) avoiding protection/deprotection manipulations. Herein, we wish to report an efficient five-step synthesis of adagrasib (1) featuring late introduction of the costly piperazine and no need for the transition-metal catalysis or protecting groups.
The retrosynthesis of adagrasib featuring introduction of the costly chiral piperazine 12(8) at a late stage of the synthesis is shown in Scheme 2. We intended to keep the last step in the new synthesis the same as the existing commercial launch route (Scheme 1). We conceived that penultimate 9 could be prepared from compound 11 and chiral piperazine 12via a SNAr reaction. Compound 11, in turn, could be prepared from an appropriately activated pyrimidone (13a or 13b) via another SNAr reaction.13,14 Ketoester 17 could be obtained using conventional chemistry and would serve as starting material for the construction of the tetrahydropyridopyrimidine core.15
Scheme 2. Retrosynthesis of Adagrasib (1).

The synthesis began with the preparation of ketoester 17 via a three-step through process. As shown in Scheme 3, the starting material 19 underwent two sequential alkylations: first with bromoester 20 to afford intermediate 21 and then with Weinreb amide 22 to afford intermediate 23. Base-mediated intramolecular Dieckmann condensation of 23 afforded ketoester 17 in 60% overall yield. Notably, the use of Weinreb amide is essential to ensure the desired regioselectivity in the intramolecular Dieckmann condensation.16
Scheme 3. Synthesis of Ketoester 17.

With ketoester 17 in hand, a conventional two-step synthesis of sulfide 15a via thiourea condensation/alkylation was initially assessed. As shown in Scheme 4, the condensation of ketoester 17 with thiourea afforded thiol 18 and the ensuing alkylation provided sulfide 15a in good yield. However, this approach generated 2,4-bis-isopropylated side product 15c which proved to be difficult to purge in the downstream process. In addition, sulfide 15a contained residual iodide, which promoted severe H2O2 degradation in the subsequent oxidation reaction, raising safety concerns for a scale-up.17
Scheme 4. Synthesis of Sulfone 13a from Ketoester 17.

The formation of 2,4-bis-isopropylated side product 15c was circumvented by forming 15a via condensation of isopropyl isothiouronium iodide 16 with ketoester 17. The discovery of intermediacy of amide 24 was consistent with a one-pot two-step sequence (Scheme 5). A more in-depth study of the physical properties of sulfide 15 unearthed that it could be crystallized in its neutral form 15b. Indeed, simple acidic quench of the reaction afforded sulfide 15b in 76% isolated yield and importantly without any detectable residual iodide.18
Scheme 5. New Route for Sulfone 13a and 11 Synthesis.

A search for favorable conditions to oxidize sulfide 15 to sulfone 13a included exploration of a range of oxidants such as oxone, m-chloroperbenzoic acid and H2O2 with catalytic Na2WO4, all known to be viable for this type of transformation.19−21 Employing H2O2 as the oxidant was particularly attractive due to its low cost, good atom economy and innocuous byproduct (water).22 However, neutral sulfide 15b (−OH form in Scheme 5) exhibited much poorer solubility than its salt 15a (−ONa form) in most organic solvents. For this reason, in situ salt formation of sulfide 15a for oxidation by pretreatment with NaOH was explored. Under these conditions, the sulfide was readily soluble in reasonable solvent volume and the oxidation of the in situ generated sodium salt 15a proceeded quickly to afford sulfone product 13a in 94% yield even in the absence of tungstate catalyst. However, significant H2O2 degradation in this strongly basic reaction medium (pH ≈ 14) posed safety concerns.23,24
It was reasoned that balancing sulfide solubility/reactivity versus H2O2 stability as a function of pH control could allow for efficient sulfide oxidation while minimizing unproductive H2O2 decomposition. Delightfully, we found that K3PO4 could resolve the issues described above (Scheme 5) as it readily converts the acidic pyrimidone sulfide 15b to its soluble potassium salt and the resulting PO43–/HPO42– buffer solution maintains the reaction pH at ∼12, effectively suppressing the undesired H2O2 degradation. Optimal conditions for this tungstate-free oxidation were as follows: 2.5 equiv. H2O2 with 2.5 equiv. K3PO4 in acetonitrile–water mixtures at 10 °C.25 At the end of reaction, acid quench of the reaction mixture enabled direct crystallization of the desired sulfone 13a (94% yield on multihundred-gram scale). Sulfone 13a sets the stage for prolinol installation via a SNAr reaction (Scheme 5). Extensive screening of bases showed that 2.5 equiv of NaOt-Am gave the best yield, albeit with around 3% hydrolyzed side product which could be easily rejected in the downstream reactions.
An alternative route to intermediate 11 from compound 18 was also demonstrated (Scheme 6). Chlorination of 18 with triphosgene selectively converted 2-SH group to 2-Cl with the 4-OH remaining intact and provided 13b in 81% yield.26 The chloropyrimidine performs equally well in the SNAr reaction using prolinol 14 to give compound 11 in 79% yield. This approach with better atom economy is complementary to the sulfone route (Scheme 5).
Scheme 6. Alternative Synthesis of Intermediate 11.

For the installation of the piperazine unit, the conversion of 11 to 9 could be accomplished in a two-step through process sequence as shown in Scheme 7. For example, activation of the OH group in 11 via either Tf2O or PhNTf2, followed by SNAr displacement afforded intermediate 9 in 70% and 78% yield, respectively (Scheme 7A, for details: see the SI). Preliminary screening of different leaving groups such as −OMs or −OTs showed significant amount of apparent hydrolysis to regenerate starting material (11) (Scheme 7B).
Scheme 7. SNAr Reaction Using Piperazine 12.

Based on these observations, we continued to explore alternatives to the −OTf leaving group to determine if the yield and purity profile could be improved and to better understand the mechanistic pathways involved in this SNAr process. Focusing on tosylate 25b, we initially speculated side-product 11 might come from hydrolysis of the intermediate 25. However, reaction in the presence of activated molecular sieves still gave the same reaction profile. The labeling experiment by adding H218O to the reaction solution did not form any 18O-labeled side product. In contrast, a tosylated piperazine 26 was identified as the sole byproduct. These results indicated that side product 11 did not result from the hydrolysis of intermediate 25. Rather, it was generated through a nucleophilic attack of the free piperazine nitrogen at the sulfur of the sulfonate (Pathway A in Scheme 7C). The major pathway with the attack at the carbon center of the pyrimidine ring affords the desired product 9 (Pathway B in Scheme 7C).
The formation of the side product 26 suggested that ortho-substituents on the benzene ring of the sulfonates might inhibit the Pathway A due to the steric effect of the ortho group, and consequently suppress the formation of 11 (Pathway B). To explore this hypothesis, we screened various substituted benzenesulfonates as the leaving groups for this SNAr reaction. As shown in Figure 1, 2-NO2, 2-Cl, 2,4-di-Cl, 2,6-di-Cl substituted benzenesulfonates with strong electron-withdrawing and steric effects exhibited a desirable reaction profile with much less side product 11 formed. When the readily available 2-nitrobenzenesulfonate was used (Scheme 8), the telescoped activation-SNAr process afforded product 9 in 90% yield and excellent purity.27 Finally, n-propanephosphonic acid anhydride (T3P)-mediated amidation of 9 with 10 afforded adagrasib (1) in 85% yield.10,28 It should be noted that using sodium acrylate 10 instead of the free acid minimized the decomposition of the acid and obviated the use of amine base in the T3P mediated coupling.
Figure 1.

Sulfonate screening for 2nd SNAr reaction.
Scheme 8. 2nd SNAr Reaction and Amidation.

In summary, a new, transition-metal and protection-free synthesis of adagrasib (MRTX849, 1) has been developed. This chromatography-free synthesis proceeds in five linear steps to produce adagrasib (MRTX849, 1) in 45% overall yield and with excellent quality. This approach takes advantage of a late-stage installation of the costly chiral subunits onto the tetrahydropyridopyrimidine core by two sequential SNAr reactions. The new synthesis obviates palladium catalysis and protecting group manipulations, which were necessary in the existing commercial route. A robust, catalyst-free oxidation of sulfide also alleviated safety concerns caused by the H2O2 degradation. The study on the leaving groups shows both steric and electronic effect enhanced the efficiency of the SNAr reaction. We believe this newly developed route offers a preferred means to produce the important anticancer therapeutic adagrasib (1). Furthermore, the methodology developed here could be applied to modular syntheses of analogues of the KRASG12C inhibitors.29
Acknowledgments
The authors are grateful to WuXi STA, PharmaBlock China and US for the general support in chemistry; to Stanley Yu (Mirati Therapeutics) and Todd Baumgartner (Mirati Therapeutics) for the analytical support; and to Dr. Michał Achmatowicz (Mirati Therapeutics) for the helpful discussion during the preparation of the manuscript.
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.2c04266.
Experimental details, NMR data, and characterization for all new compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Huang L.; Guo Z.; Wang F.; Fu L. KRAS Mutation: from Undruggable to Druggable in Cancer. Signal Transduct. Target. Ther. 2021, 6, 386–405. 10.1038/s41392-021-00780-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Désage A.-L.; Léonce C.; Swalduz A.; Ortiz-Cuaran S. Targeting KRAS Mutant in Non-Small Cell Lung Cancer: Novel Insights Into Therapeutic Strategies. Front. Oncol. 2022, 12, 1–19. 10.3389/fonc.2022.796832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Griffin D. J.; Beaver M. G.; Blue L. E.; Borths C. J.; Brown D. B.; Caille S.; Chen Y.; Cherney A. H.; Cochran B. M.; Colyer J. T.; Corbett M. T.; Correll T. L.; Crockett R. D.; Dai X.-J.; Dornan P. K.; Farrell R. P.; Hedley S. J.; Hsieh H.-W.; Huang L.; Huggins S.; Liu M.; Lovette M. A.; Quasdorf K.; Powazinik W. I. V.; Reifman J.; Robinson J. A.; Sangodkar R. P.; Sharma S.; Kumar S. S.; Smith A. G.; St-Pierre G.; Tedrow J. S.; Thiel O. R.; Truong J. V.; Walker S. D.; Wei C. S.; Wilsily A.; Xie Y.; Yang N.; Parsons A. T. Development of a Commercial Manufacturing Process for Sotorasib, a First-in-Class KRASG12C Inhibitor. Org. Process Res. Dev. 2022, 26, 3115–3125. 10.1021/acs.oprd.2c00249. [DOI] [Google Scholar]
- Adagrasib is marketed under the brand name Krazati, see: https://www.krazati.com.
- Jänne P. A.; Riely G. J.; Gadgeel S. M.; Heist R. S.; Ou S.-H. I.; Pacheco J. M.; Johnson M. L.; Sabari J. K.; Leventakos K.; Yau E.; Bazhenova L.; Negrao M. V.; Pennell N. A.; Zhang J.; Anderes K.; Der-Torossian H.; Kheoh T.; Velastegui K.; Yan X.; Christensen J. G.; Chao R. C.; Spira A. I. Adagrasib in Non-Small-Cell Lung Cancer Harboring a KRASG12C Mutation. N. Engl. J. Med. 2022, 387, 120–131. 10.1056/NEJMoa2204619. [DOI] [PubMed] [Google Scholar]
- Ou S.-H. I.; Jänne P. A.; Leal T. A.; Rybkin I. I.; Sabari J. K.; Barve M. A.; Bazhenova L.; Johnson M. L.; Velastegui K. L.; Cilliers C.; Christensen J. G.; Yan X.; Chao R. C.; Papadopoulos K. P. First-in-Human Phase I/IB Dose-Finding Study of Adagrasib (MRTX849) in Patients With Advanced KRASG12C Solid Tumors (KRYSTAL-1). J. Clin. Oncol. 2022, 40, 2530–2538. 10.1200/JCO.21.02752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallin J.; Engstrom L. D.; Hargis L.; Calinisan A.; Aranda R.; Briere D. M.; Sudhakar N.; Bowcut V.; Baer B. R.; Ballard J. A.; Burkard M. R.; Fell J. B.; Fischer J. P.; Vigers G. P.; Xue Y.; Gatto S.; Fernandez-Banet J.; Pavlicek A.; Velastagui K.; Chao R. C.; Barton J.; Pierobon M.; Baldelli E.; Patricoin E. F. III; Cassidy D. P.; Marx M. A.; Rybkin I. I.; Johnson M. L.; Ou S.-H. I.; Lito P.; Papadopoulos K. P.; Jänne P. A.; Olson P.; Christensen J. G. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discovery 2020, 10, 54–71. 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell J. B.; Fischer J. P.; Baer B. R.; Blake J. F.; Bouhana K.; Briere D. M.; Brown K. D.; Burgess L. E.; Burns A. C.; Burkard M. R.; Chiang H.; Chicarelli M. J.; Cook A. W.; Gaudino J. J.; Hallin J.; Hanson L.; Hartley D. P.; Hicken E. J.; Hingorani G. P.; Hinklin R. J.; Mejia M. J.; Olson P.; Otten J. N.; Rhodes S. P.; Rodriguez M. E.; Savechenkov P.; Smith D. J.; Sudhakar N.; Sullivan F. X.; Tang T. P.; Vigers G. P.; Wollenberg L.; Christensen J. G.; Marx M. A. Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 6679–6693. 10.1021/acs.jmedchem.9b02052. [DOI] [PubMed] [Google Scholar]
- Ledford H. Cancer drugs are closing in on some of the deadliest mutations. Nature 2022, 610, 620–622. 10.1038/d41586-022-03392-2. [DOI] [PubMed] [Google Scholar]
- Snead D.; Gan Y.; Scattolin T.; Paymode D.; Achmatowicz M.; Rudisill D., et al. Development of Adagrasib’s Commercial Manufacturing Route. ChemRxiv.; Cambridge Open Engage: Cambridge, 2022. 10.26434/chemrxiv-2022-9b3s1. [DOI]
- Scattolin T.; Gharbaoui T.; Chen C.-y. A Nucleophilic Deprotection of Carbamate Mediated by 2-Mercaptoethanol. Org. Lett. 2022, 24, 3736–3740. 10.1021/acs.orglett.2c01410. [DOI] [PubMed] [Google Scholar]
- Palladium price has risen dramatically (≈ $100,000/kg) over the past decade and remains highly volatile: Williams G.Palladium Price Update: H1 2022, Investing news Network, 2022. https://investingnews.com/daily/resource-investing/precious-metals-investing/palladium-investing/palladium-price-update/.
- Hoffmann-Emery F.; Niedermann K.; Rege P. D.; Konrath M.; Lautz C.; Kraft A. K.; Steiner C.; Bliss F.; Hell A.; Fischer R.; Carrera D. E.; Beaudry D.; Angelaud R.; Malhotra S.; Gosselin F. Development of a Practical and Greener Process for the Dual Leucine Zipper Kinase Inhibitor GDC-0134 Comprising Two SNAr Reactions, Oxidation and Suzuki Coupling. Org. Process Res. Dev. 2022, 26, 313–322. 10.1021/acs.oprd.1c00389. [DOI] [Google Scholar]
- Li C.; Haeffner F.; Wang S.; Yuan C.; Shang D.; Shi X.; Ma B.; Hopkins B. T.; O’Brien E. M. Sulfone Displacement Approach for Large-Scale Synthesis of 4-Chloro-N-(1-methyl-1H-pyrazol-4-yl) pyrimidin-2-amine. Org. Process Res. Dev. 2022, 26, 137–143. 10.1021/acs.oprd.1c00314. [DOI] [Google Scholar]
- Brown T. H.; Blakemore R. C.; Durant G. J.; Emmett J. C.; Ganellin C. R.; Parsons M. E.; Rawlings D. A.; Walker T. F. Isocytosine H2-receptor Histamine Antagonists I. Oxmetidine and Related Compounds. Eur. J. Med. Chem. 1988, 23, 53–62. 10.1016/0223-5234(88)90167-5. [DOI] [Google Scholar]
- Intramolecular Dieckmann condensation of bis-ester was explored and the reaction is not regioselective.
- Hansen J. C. The Iodide-Catalyzed Decomposition of Hydrogen Peroxide: A Simple Computer-Interfaced Kinetics Experiment for General Chemistry. J. Chem. Educ. 1996, 73, 728–732. 10.1021/ed073p728. [DOI] [Google Scholar]
- Upon screening of various alkyl groups in the condensation reaction, the i-Pr thiourea showed superior reaction performance and highest isolated yield. For experimental details, see the SI.
- Liu N.-W.; Liang S.; Manolikakes G. Recent Advances in the Synthesis of Sulfones. Synthesis 2016, 48, 1939–1973. 10.1055/s-0035-1560444. [DOI] [Google Scholar]
- Matavos-Aramyan S.; Soukhakian S.; Jazebizadeh M. H. Selected Methods for the Synthesis of Sulfoxides and Sulfones with Emphasis on Oxidative Protocols. Phosphorus, Sulfur Silicon Relat. Elem. 2020, 195, 181–193. 10.1080/10426507.2019.1672691. [DOI] [Google Scholar]
- a Laudadio G.; Straathof N. J. W.; Lanting M. D.; Knoops B.; Hessel V.; Noël T. An Environmentally Benign and Selective Electrochemical Oxidation of Sulfides and Thiols in a Continuous-flow Microreactor. Green Chem. 2017, 19, 4061–4066. Electrochemical oxidation was also reported recently: 10.1039/C7GC01973D. [DOI] [Google Scholar]; b Bottecchia C.; Lehnherr D.; Lévesque F.; Reibarkh M.; Ji Y.; Rodrigues V. L.; Wang H.; Lam Y.-h.; Vickery T. P.; Armstrong B. M.; Mattern K. A.; Stone K.; Wismer M. K.; Singh A. N.; Regalado E. L.; Maloney K. M.; Strotman N. A. Kilo-Scale Electrochemical Oxidation of a Thioether to a Sulfone: A Workflow for Scaling up Electrosynthesis. Org. Process Res. Dev. 2022, 26, 2423–2437. 10.1021/acs.oprd.2c00111. [DOI] [Google Scholar]
- Shilcrat S. Process Safety Evaluation of a Tungsten-Catalyzed Hydrogen Peroxide Epoxidation Resulting In a Runaway Laboratory Reaction. Org. Process Res. Dev. 2011, 15, 1464–1469. 10.1021/op200133q. [DOI] [Google Scholar]
- Galbács Z. M.; Csányi L. J. Alkali-induced Decomposition of Hydrogen Peroxide. J. Chem. Soc., Dalton Trans. 1983, 2353–2357. 10.1039/DT9830002353. [DOI] [Google Scholar]
- Koubek E.; Haggett M. L.; Battaglia C. J.; Ibne-Rasa K. M.; Pyun H. Y.; Edwards J. O. Kinetics and Mechanism of the Spontaneous Decompositions of Some Peroxoacids, Hydrogen Peroxide and t-Butyl Hydroperoxide. J. Am. Chem. Soc. 1963, 85, 2263–2268. 10.1021/ja00898a016. [DOI] [Google Scholar]
- Jereb M. Highly Atom-Economic, Catalyst- and Solvent-Free Oxidation of Sulfides into Sulfones Using 30% Aqueous H2O2. Green Chem. 2012, 14, 3047–3052. Under these conditions, sulfone was formed through sulfoxide intermediate. For an example of catalyst-free oxidation of sulfide with H2O2, see: 10.1039/c2gc36073j. [DOI] [Google Scholar]
- Callingham M.; Blum F.; Pavé G. One-Step Synthesis of 2-Chloropyrimidin-4-ol Derivatives: An Unusual Reactivity of Thiophosgene. Org. Lett. 2015, 17, 4930–4932. 10.1021/acs.orglett.5b02375. [DOI] [PubMed] [Google Scholar]
- The SNAr reaction of 12 is regioselective as shown in reference (8).
- The current process with no impurity > 0.08% detected is under evaluation for commercialization of the drug.
- We are actively exploring this area and will report it in due course.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.