

Abstract
Deregulated de novo lipid synthesis (DNLS) is a potential druggable vulnerability in glioblastoma (GBM), a highly lethal and incurable cancer. Yet the molecular mechanisms that determine susceptibility to DNLS-targeted therapies remain unknown and the lack of brain-penetrant inhibitors of DNLS has prevented their clinical evaluation as GBM therapeutics. Here, we report that YTX-7739, a clinical-stage inhibitor of stearoyl CoA desaturase (SCD), triggers lipotoxicity in patient-derived GBM stem-like cells (GSCs) and inhibits fatty acid desaturation in GSCs orthotopically implanted in mice. When administered as a single agent, or in combination with Temozolomide (TMZ), YTX-7739 showed therapeutic efficacy in orthotopic GSC mouse models owing to its lipotoxicity and ability to impair DNA damage repair. Leveraging genetic, pharmacological, and physiological manipulation of key signaling nodes in gliomagenesis complemented with shotgun lipidomics, we show that aberrant MEK/ERK signaling and its repression of the energy sensor AMP-activated protein kinase (AMPK) primarily drive therapeutic vulnerability to SCD and other DNLS inhibitors. Conversely, AMPK activation mitigates lipotoxicity and renders GSCs resistant to the loss of DNLS, both in culture and in vivo, by decreasing the saturation state of phospholipids and diverting toxic lipids into lipid droplets. Altogether, our findings reveal mechanisms of metabolic plasticity in GSCs and provide a framework for the rational integration of DNLS-targeted GBM therapies.
One sentence summary
A clinical-grade inhibitor of SCD delays tumor growth and increases sensitivity to DNA-damaging agents in GBM preclinical mouse models
Introduction
Glioblastoma (GBM) is a highly aggressive and lethal form of primary brain tumor, enriched with cancer stem-like cells (GSCs), which are particularly resistant to conventional therapies (1). GSCs are endowed with metabolic plasticity and adaptability, allowing them to outcompete mature GBM cells with defined metabolic dependencies, survive in a harsh microenvironmental niche poor in nutrients, and overcome various therapeutic insults (2). These cells can metabolize multiple nutritional substrates and rely on oxidative phosphorylation as well as de novo lipid synthesis (DNLS) to support their energy demand, thus sustaining tumor growth and proliferation (2). The reprogramming of cellular lipid metabolism is intricately linked with malignant transformation, progression, metastasis, and therapeutic resistance (3). Oncogenic signaling in GBM, which regulates many such properties and allows GBM cells to cope with their energy demand, is primarily orchestrated by phosphatidylinositol 3-kinase (PI3K)/AKT and RAS/mitogen-activated protein kinase (MAPK) signaling (4). Although RAS mutations are rarely detected in malignant gliomas, the RAS/RAF/ extracellular signal-regulated kinase (ERK) kinase (MEK)/ERK pathway is aberrantly activated in these tumors (4). PI3K/AKT and RAS/MAPK can modulate the activity of various enzymes involved in lipid metabolism, thereby defining the tumor lipidome (3), and can regulate critical sensors of cellular metabolic demands such as AMP-activated protein kinase (AMPK), which plays a crucial role in restoring metabolic homeostasis (5).
Stearoyl CoA Desaturase 1 (SCD) is an enzyme responsible for the conversion of saturated fatty acids (SFA) to monounsaturated FA (MUFA). We have previously shown that GSCs depend on adaptive activation of SCD and that long-term depletion of MUFA in GSCs decreased their self-renewal properties and impaired tumor initiation in mice (6). On the other hand, pharmacological inhibition of SCD is primarily toxic due to the accumulation of SFAs (a process referred to as lipotoxicity), which induces ER stress and terminal unfolded protein response (UPR) signaling (6). Despite its poor ability to penetrate the blood-brain barrier (BBB), the SCD inhibitor CAY10566 showed therapeutic efficacy when delivered intranasally (6) or systemically (7) in mice bearing primary GBM tumors. An orally available SCD inhibitor, YTX-7739, was recently identified as a lead clinical candidate to treat synucleinopathies (8, 9). Based on its biochemical potency, safety, favorable pharmacokinetics, and the ability to cross the BBB in rats and cynomolgus monkeys (8), YTX-7739 is being evaluated in phase I clinical trials for the treatment of patients with Parkinson’s disease (https://www.trialregister.nl/trial/8258; https://www.trialregister.nl/trial/9172).
Translational efforts of SCD-targeted therapy for GBM have been hindered by the absence of clinically relevant, brain-penetrant inhibitors of this desaturase. Further, previous work has left unresolved whether precise genetic alterations could drive the dependence of GSCs on SCD or DNLS in general and whether defined molecular markers could predict vulnerability or resistance to DNLS-targeted therapies. Here, we explored the functional and therapeutic efficacy of YTX-7739 in GBM, and we undertook a discovery effort to understand how oncogenic signaling can impact lipid metabolism plasticity in GSCs.
RESULTS
A clinical-stage SCD inhibitor selectively inhibits fatty acid desaturation in patient-derived GSCs in vitro
To test whether YTX-7739 effectively reduces FA desaturation in GSCs by selectively inhibiting SCD, we performed a quantitative FA analysis of GSCs after genetic silencing of this desaturase with short-hairpin RNA (shRNA) or treatment with YTX-7739. Unsupervised hierarchical clustering of FA content revealed a comparable profile after drug treatment or SCD knockdown (Figure 1A). Consistent with prior studies (8), total FA desaturation, which was determined by calculating the desaturation index (DI = monounsaturated/saturated ratio) from measured C16 and C18 FA methyl ester, showed that treatment with YTX-7739 resulted in a 74% and 81% decrease in the DI of C16 and C18, respectively (Figure 1B). We then measured cell viability after treatment with YTX-7739 in normal human astrocytes (NHA) and 12 patient-derived GSCs lines with distinct genetic signatures isolated from primary and recurrent GBM (Table S1). NHA were the least sensitive to YTX-7739 with an EC50 of 81.41 μM (Figure 1C). Sensitivity to YTX-7739 varied among GSCs. 7/12 GSCs lines tested had an EC50 < 4μM and were therefore classified as sensitive to this inhibitor, whereas those with an EC50 > 20μM were classified as resistant for subsequent studies (Figure 1C). When plotting the normalized mRNA or protein expression of SCD in all 12 GSCs against the EC50 of YTX-7739, we did not observe any significant correlation (Figure S1A–B), thus indicating that SCD expression does not predict responsiveness to this inhibitor. Supplementing the cell culture medium with Oleate (C18:1n9), the main product of SCD, but not Palmitate (C16:0; PA), an SCD precursor, rescued GSCs from cell death caused by YTX-7739 (Figure 1D), thus confirming that the observed cell death is directly caused by SCD inhibition. Further, ectopic expression of SCD at supraphysiological concentrations diminished YTX-7739 toxicity in sensitive GSCs and had no effect in the resistant lines, further corroborating on-target activity (Figure 1E and S1C). Overall, YTX-7739 kills GSCs by specifically inhibiting SCD-mediated FA desaturation.
Figure 1: YTX-7739 is a selective inhibitor of FA desaturation in cultured GSCs.
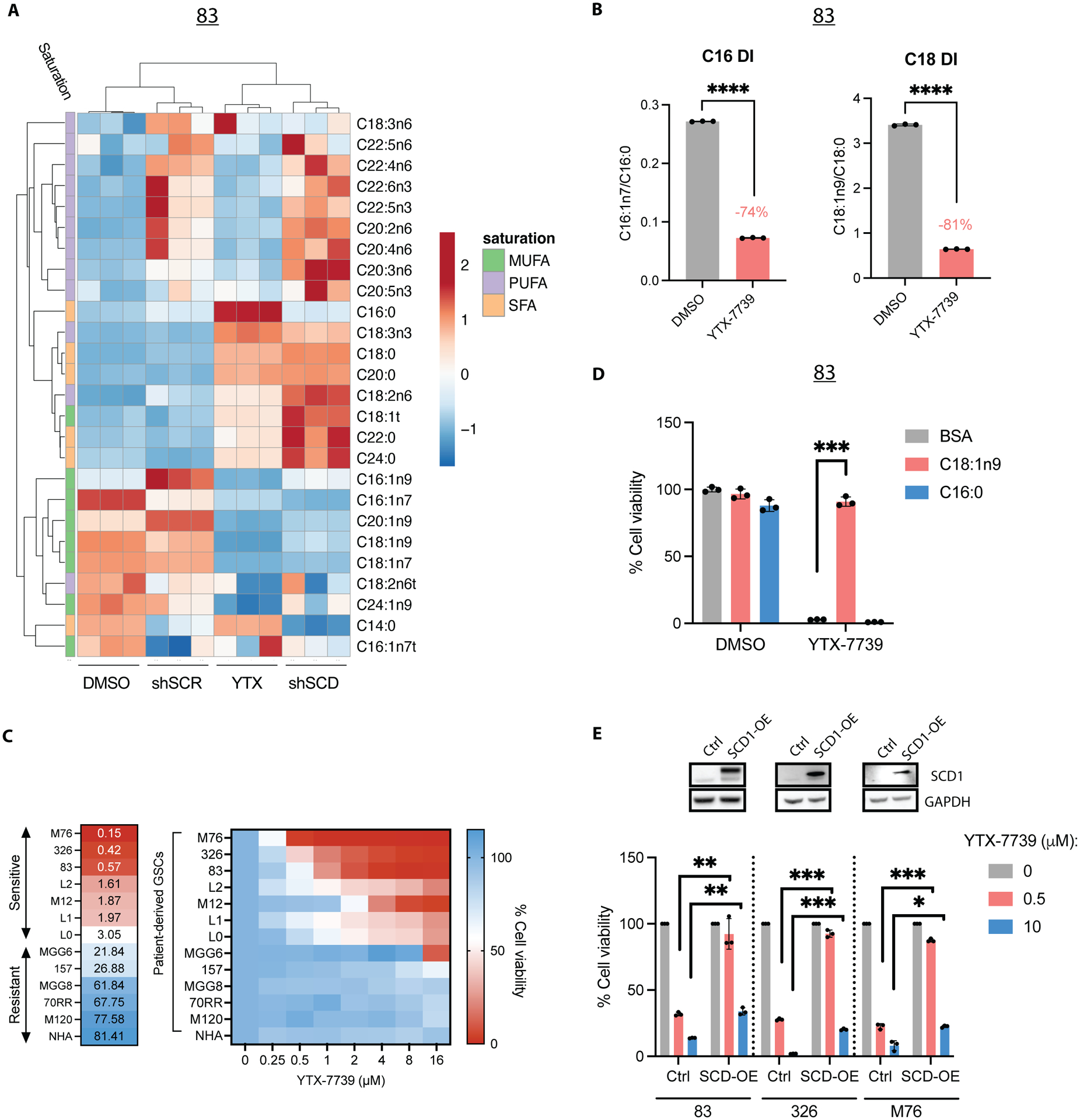
(A) Heat map representing unsupervised hierarchical clustering of FA content in 83 either transduced with a control shRNA (shSCR) or shSCD or treated with DMSO (control) or YTX-7739 (1μM) for 72h. (B) Desaturation index (DI) of C16 and C18 in 83 treated with YTX-7739 (1μM) for 72h. (C) Cell viability and EC50 of NHA and 12 GSC lines treated with YTX-7739 at the indicated doses for 96h, expressed as a percentage of control. (D) Cell viability in 83 treated with YTX-7739 (1μM) in the presence of BSA (control) or the indicated BSA-complexed FA for 96h. (E) Cell viability of GSCs ectopically expressing a control vector (Ctrl) or SCD1 (SCD1-OE) treated with YTX-7739 for 96h. The upper panel shows the immunoblots for SCD in Ctrl and SCD1-OE GSCs. ****P<0.0001; *** P<0.001; **P<0.01; *P<0.05. DI, desaturation index.
YTX-7739 selectively kills GBM by exacerbating ER stress and promoting IRE1-JNK mediated cell death
Quantifying individual long-chain SFA revealed that SCD precursors, C16:0 and C18:0, were particularly abundant in GSCs and further enriched after SCD inhibition in YTX-7739 sensitive GSCs (Figure 1A and S2A), suggesting that this inhibitor kills GSCs by promoting SFA-mediated lipotoxicity. This increase in C16:0 or C18:0 amounts was not observed in three YTX-7739-resistant GSCs treated with this inhibitor (Figure S2B), despite an effective SCD inhibition evident through a decrease in the DI of C18 (but not C16 DI; Figure S2C). Following treatment with YTX-7739, we observed an upregulation of ER stress/UPR markers, including GRP78, spliced XBP1 (sXBP1), C/EBP-homologous protein (CHOP), and its direct transcriptional target GADD34. In addition, we observed increased phosphorylation of Ser51 on the α-subunit of eukaryotic translation initiation factor 2 (p-EIF2α), mainly in YTX-7739-sensitive GSCs lines (Figure 2A–B and S2D–E). We also observed increased phosphorylation of c-Jun N-terminal kinase (JNK), an essential regulator of the UPR-mediated apoptosis (10), and its downstream target c-Jun (Figure 2B). In accord with this, alleviating ER stress with Phenylbutyrate (PBA), Azoramide (AZO), or Salubrinal (SAL) protected against SCD inhibition (Figure 2C). Similarly, two inhibitors of the UPR sensor IRE1 (4μ8C and NSC95682) and two JNK inhibitors (SP600125 and DB07268) conferred protection against YTX-7739 (Figure 2C and S2F). Supplementing the cell culture medium with C16:0 to increase endogenous SFA pools strongly impaired cell viability in two GSCs lines resistant to YTX-7739 (Figure S2G). This data confirms that, although SCD is effectively inhibited in all GSCs, lipotoxicity and ensuing cell death only occurs when SFAs accumulate at cytotoxic concentrations. If this hypothesis is correct, then concomitant inhibition of SCD along with the upstream enzyme fatty acid synthase (FASN) or acetyl-CoA carboxylase (ACC) (to prevent SFA synthesis; Figure 2D) would be protective against SCD inhibition. Indeed, the combination of YTX-7739 with a relatively low dose of FASN or ACC inhibitor abrogated cytotoxicity (Figure 2E). Altogether, these results confirm that pharmacological inhibition of SCD promotes SFA-mediated lipotoxicity and ER stress activation of IRE1-JNK signaling in GSCs.
Figure 2: Treatment with YTX-7739 promotes a terminal UPR response and sensitizes to TMZ and radiation.
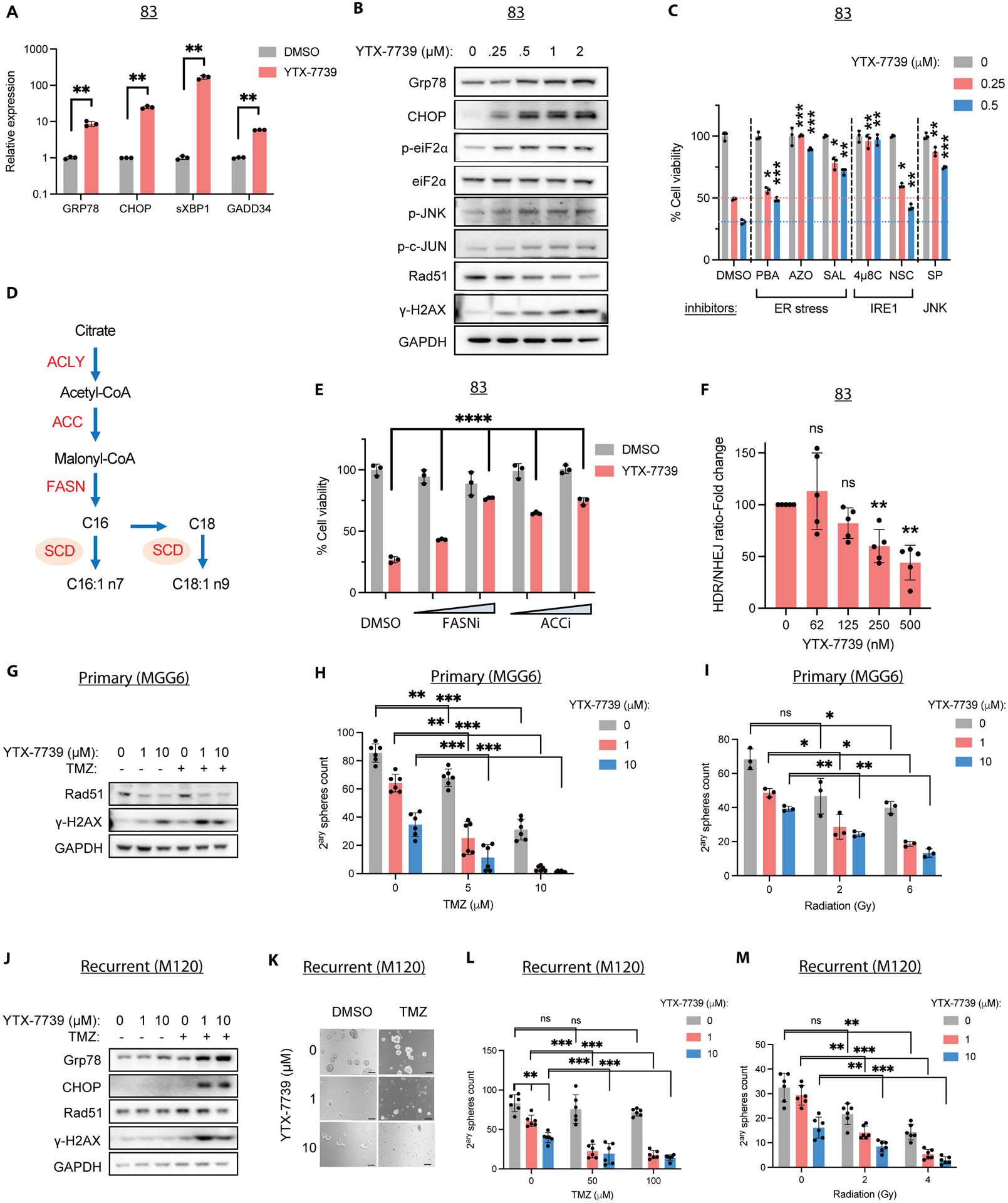
(A) Relative mRNA expression as compared to DMSO (control) and (B) immunoblot analysis of ER stress markers in 83 treated with YTX-7739 for 48h. (C) Cell viability of 83 treated with YTX-7739 in combination with inhibitors of ER stress (PBA: 4mM; Azoramide: 50μM; Salubrinal: 25μM), IRE1 (4μ8C: 25μM; NSC95682: 20μM) and JNK (SP600125: 20μM) for 96h. (D) Overview of de novo lipid synthesis (DNLS) pathway. (E) Cell viability of 83 treated with YTX-7739 (0.5μM) in combination with inhibitors of FASN (GSK2194069: 25–50nM) or ACC (CP-640186: 2.5–5μM) for 96h. (F) Fold-change in HDR/NHEJ normalized to cell viability in 83 treated with YTX-7739. (G-I) MGG6 were pretreated with YTX-7739 followed by TMZ or RT. (G) Immunoblot analysis of DNA repair and damage at 72h after treatment with YTX-7739 and TMZ (5μM). (H-I) Secondary sphere count 9 days after co-treatment with TMZ (H) or RT (I). (J-M) M120 were pretreated with YTX-7739, followed by TMZ or RT. (J) Immunoblot analysis after treatment with YTX-7739 and TMZ (100μM). (K) Representative micrographs of neurospheres. Scale bar, 100μm. (L-M) Secondary sphere count after co-treatment with TMZ (H) or RT (I). ****P<0.0001; *** P<0.001; **P<0.01; *P<0.05
Treatment with YTX-7739 increases sensitivity to radiation and Temozolomide in primary and recurrent GBM in culture
Double-strand breaks (DSB) at DNA-targeted sites are typically repaired through error-prone non-homologous end joining (NHEJ) or template-dependent homology-directed repair (HDR) that is mediated by RAD51 (11). We have previously reported that the upregulation of ER stress following pharmacological inhibition of SCD with CAY10566 decreased RAD51 expression, thus sensitizing GSCs to TMZ (6). Here we saw treatment with YTX-7739 also decreased RAD51 expression concomitant with an upregulation of the DNA damage marker γ-H2AX (Figure 2B). We used an HDR/NHEJ reporter system (12) and confirmed that treatment of GSCs with YTX-7739 decreased the ratio of HDR/NHEJ (Figure 2F), consistent with RAD51 downregulation. Next, we asked whether sensitization to TMZ or RT could occur in GSCs sensitive and resistant to the SCD inhibitor or those resistant to TMZ. We assessed cell viability, potentially altered by lipotoxicity or DNA damage, and stem cell properties, measured using a secondary sphere formation assay, in these experiments. The combined treatment of the GSC line 83 (YTX-7739 sensitive; TMZ semi-sensitive) with the SCD inhibitor and TMZ strongly depleted cell viability and secondary sphere formation (Figure S3A–B). Treatment of MGG6 (YTX-7739 resistant; TMZ sensitive) with YTX-7739 alone downregulated RAD51 and increased γ-H2AX, which was further increased when the SCD inhibitor was combined with TMZ (Figure 2G). Consequently, such a combination almost entirely abrogated long-term secondary sphere formation and decreased cell viability of MGG6 (Figures 2H and S3C). Further, combined treatment of YTX-7739 with RT also decreased secondary sphere counts (Figure 2I). In the recurrent GSC cell line M120 (YTX-7739- and TMZ-resistant), the combination of YTX-7739 with TMZ, but not the SCD inhibitor alone, decreased RAD51 protein expression along with an increase in ER stress and γ-H2AX (Figure 2J and S3D). A relatively high dose of TMZ (50–100μM) was ineffective (Figure 2K–L and S3E), and YTX-7739 (at 10 μM) did not change short-term cell viability but decreased sphere count (Figures 2K–M and S3E). However, YTX-7739 with TMZ strongly decreased both secondary sphere count and cell viability (Figures 2K–L and S3E). Treatment with YTX-7739 also sensitized these recurrent cells to RT (Figure 2M). Overall, our data suggests benefit from combining YTX-7739 and DNA damaging standard-of-care therapy for GBM to increase therapeutic efficacy.
YTX-7739 inhibits fatty acid desaturation and extends survival in orthotopic GBM mouse models
We analyzed the in vivo pharmacokinetics (PK) of YTX-7739 in tumor free mice. We observed a dose-dependent increase in plasma concentrations of YTX-7739 following IP administration (Figure 3A). Importantly, IP administration of YTX-7739 (10 mg/kg) showed comparable brain and plasma concentrations, indicating brain permeability (detailed PK parameters are shown in Table S2). We then assessed the pharmacodynamics (PD) of YTX-7739 in plasma, brain, and tumor tissue in an orthotopic brain tumor mouse model. Animals bearing brain tumors generated by implanting GSC line 83 received a daily intraperitoneal (IP) dose of vehicle control or YTX-7739 (30mg/kg) for 11 days beginning on day 4 after implantation, before harvesting plasma and brain tissue from both hemispheres to measure the DI in plasma, brain, and brain tumor samples. We observed a decrease in C16 and C18 DI in all samples from the YTX-7739 group. Treatment with YTX-7739 resulted in a 50% and 30% decrease in C16 and C18 DI, respectively, in brain tumors (Figure 3B). In conjunction with a decreased DI, we observed upregulation of ER stress markers in brain tumor tissue after treatment with YTX-7739 (Figure 3C), along with increased apoptosis assessed by immunostaining for active caspase 3 (Figure 3D). To test the therapeutic efficacy of YTX-7739 in GBM mouse models, two GSC lines were selected based on a range of sensitivity to YTX-7739 and TMZ. In the first experiment, mice were implanted with the 83 GSC line, a highly aggressive; YTX-7739 sensitive, (EC50 = 0.57 μM, Figure 1C), TMZ semi-sensitive cell line, and allowed to establish tumors detected using bioluminescence imaging before initiating treatment. Mice received 16 daily IP doses of vehicle (Control group) or YTX-7739 at 10 and 30mg/kg. Albeit modest, treatment with this compound resulted in increased overall survival (Figure 3E). To compare the efficacy of this inhibitor to TMZ, we monitored tumor growth in 83-bearing mice treated with vehicle control, YTX-7739 (30mg/kg), TMZ (10mg/kg), or YTX-7739 (30mg/kg) + TMZ (10mg/kg). Consistent with a modest increase in survival, YTX-7739 as a monotherapy did not significantly decrease tumor growth. Whereas TMZ alone initially delayed tumor growth, it was quickly resumed 25 days after tumor implantation (Figure 3F). Conversely, combination of YTX-7739 and TMZ hindered tumor growth compared to TMZ alone (Figure 3F). Survival analysis demonstrated that the drug combination achieved a longer median survival (43.5 days) than TMZ alone (32 days; Figure 3G). Treatment with YTX-7739 or its combination with TMZ was well tolerated throughout the treatment, with no visible signs of discomfort, toxicity, or weight loss (Figure S4A). We repeated this experiment with mice implanted with MGG8, YTX-7739 resistant, (EC50 = 61.84 μM, Figure 1C), TMZ sensitive cell line. A decrease of C16 and C18 DI from the plasma of mice treated with YTX-7739 confirmed its effect on circulating fatty acids (Figure S4B). Longitudinal imaging of tumor growth showed a divergent response in the YTX-7739-treated group, whereby 4/8 mice showed a relatively stable tumor signal up to 10 weeks after tumor implantation, indicative of a strong therapeutic response (Figure 3H). In comparison, all mice in the control group (8/8) showed an increased tumor growth over time, and none were alive by week 10. This translated into an extended survival of 91 days in the treated group compared with 52 days in the control group (Figure 3I). Additionally, the combination of TMZ with YTX-7739 extended survival for 136.5 days compared to 106.5 days for TMZ alone. Despite these GSCs being highly sensitive to TMZ, there was no significant difference in overall survival between mice treated with either TMZ or YTX-7739 alone, suggesting a comparable therapeutic efficacy between YTX-7739 and the standard of care chemotherapeutic. The surviving mice showed no detectable tumors by bioluminescence imaging at 19 weeks post-implantation (Figure S4C). Further, the survival percentage at six months after tumor implantation, when this experiment was terminated, was 37.5% in mice treated with YTX-7739 alone compared to 25% in the TMZ-treated group. Finally, we did not observe any weight loss in the treated animals (Figure S4D), and long-term monitoring for up to 6 months did not reveal any visible signs of toxicity. These data demonstrate that YTX-7739 can cross the BBB, functionally inhibit SCD-mediated desaturation in brain tumors, and exert therapeutic efficacy in preclinical GBM models.
Figure 3: YTX-7739 inhibits fatty acid desaturation and delays tumor growth in mice.
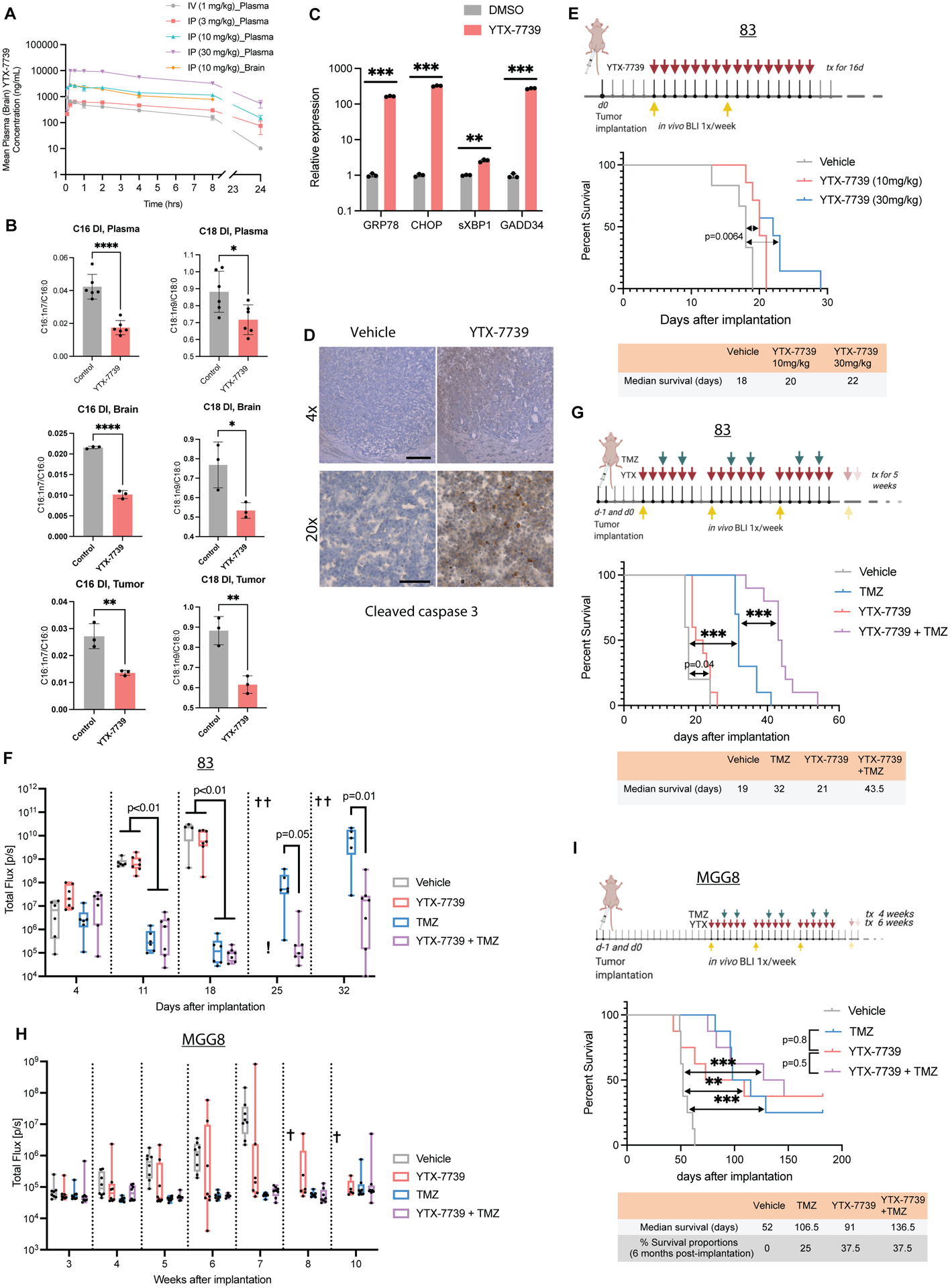
(A) Mean plasma and brain PK of YTX-7739 after IV or IP administration in mice (n=3/group). (B) DI of C16 and C18 in plasma, brain, and brain tumor samples from mice bearing 83 tumors and treated with YTX-7739 (30mg/kg) for 11 days beginning 4 days after inoculation (n=3/group). (C) Brain tumor tissue from that experiment was also used to measure mRNA expression of ER stress markers. (D) Representative immunostaining of cleaved caspase 3 in brain tumor tissue of DMSO and YTX-7739-treated mice. Scale bar, 4x: 250μm; 20x: 50μm. (E) Kaplan-Meier curve depicting median survival in mice bearing 83 tumors (n = 6/group) treated with a vehicle or YTX-7739 (10–30 mg/kg) for 16 days starting at day 5 post-implantation. (F) Overtime monitoring of tumor growth in mice bearing 83 tumors and treated with vehicle (n=6), YTX-7739 (30mg/kg; n=7), TMZ (25mg/kg; n=6) of YTX-7739+TMZ (n=7). Cross indicates that >50% of mice have expired due to tumor burden. (G) Median survival in mice bearing 83 and treated with vehicle (n=5), YTX-7739 (30mg/kg; n=10), TMZ (10mg/kg; n=10 or YTX-7739+TMZ (n=10). (H-I) Overtime monitoring of tumor growth (H) and median survival (I) in mice implanted with MGG8 (n=8/group) and treated with vehicle, YTX-7739 (30 mg/kg), TMZ (5mg/kg), or YTX-7739 + TMZ. P-value, two-sided log-rank test. ****P<0.0001; *** P<0.001; **P<0.01; *P<0.05
Signaling through RAS/MEK/ERK promotes vulnerability to SCD inhibitors
Given the disparate sensitivity to SCD inhibitors between NHA and GSCs (Figure 1C) and to define the precise genetic alterations driving vulnerability to such inhibitors, we used an astrocyte-based model of gliomagenesis initially described by Sonoda et al. (13). In this model, NHA are first engineered to express hTERT, combined with the loss of functional p53 and pRb pathways, to prevent senescence and increase cell proliferation. These immortalized non-transformed astrocytes are hereafter referred to as HA-NT. Ectopic expression of a mutant, constitutively active HRASG12V (HA-Ras) but not a truncated active form of EGFR (HA-EGFRvIII) in HA-NT promoted anchorage-independent growth (Figure S5A). Since RAS primarily signals through MEK/ERK, this anchorage-independent growth could be prevented by blocking this pathway with two specific MEK and ERK inhibitors (AZD8330 and AZD0364, respectively) (Figure S5B). HRAS activation in these cells also confers the ability to form tumors akin to a grade III anaplastic astrocytoma following intracranial implantation in mice (13). We confirmed increased cell proliferation in HA-NT compared to NHA (Figure S5C), increased phosphorylation of ERK1/2 and AKT in HA-Ras cells, and the activation of the truncated EGFR in HA-EGFRvIII (Figure 4A). SCD expression was moderately increased following the expression of HRASG12V or EGFRvIII (Figure 4A). Next, we examined sensitivity to two SCD inhibitors (YTX-7739 and CAY10566) in NHA and this isogenic astrocyte model. SCD inhibition led to a minor decrease in cell viability in NHA and HA-NT (Figure 4B), indicating that increased cell proliferation is insufficient to promote vulnerability to SCD inhibitors. Although EGFR signaling has been reported to sensitize GBM cell lines to inhibitors of fatty acid synthesis (14), HA-EGFRvIII showed a similar response to HA-NT. In contrast, HA-Ras cells were sensitive to SCD inhibitors with an EC50 of 209 nM and 18.6 nM for YTX-7739 and CAY10566, respectively (Figure 4B–C). This selective toxicity towards HA-Ras was further confirmed through increased apoptosis detected by caspase activation and Annexin V staining (Figure S5D–E). ER stress/UPR, DNA damage, and JNK activation were only observed in HA-Ras but not in HA-NT following SCD inhibition (Figure 4D–E and S5F). Cell death caused by SCD inhibitors could be reversed by supplementing HA-Ras with Oleate or inhibiting ER stress, IRE1, or JNK, similarly to what is observed in GSCs (Figure S5G–H). This implies that the molecular underpinnings of vulnerability and cell death triggered by SCD inhibition are similar in HA-Ras and GSCs. We also found that ectopic expression of HRAS, but not EGFRvIII, also increased sensitivity to FASN and ACC inhibitors (Figure S5I), thus indicating that RAS activation creates an overall dependence on DNLS. Further, blocking this pathway with MEK and ERK inhibitors prevented cell death caused by SCD inhibitors in HA-Ras, thus reverting this acquired sensitivity (Figure S5B and S5G). We found a significant negative correlation between ERK phosphorylation (p-ERK) and the EC50 of YTX-7739 in YTX-779-sensitive and resistant GSCs lines (r=−0.9; P=0.0046; Figure S5J). However, one of the inhibitor-resistant lines (157) showed a similar or greater p-ERK/ERK ratio than the three sensitive lines (83, 326, and L2). Importantly, inhibiting MEK/ERK in GSCs treated with YTX-7739 prevented ER stress and terminal UPR (Figure 4F–G) and the phosphorylation of IRE1, JNK, and c-Jun (Figure 4G). Consequently, inhibiting MEK/ERK averts PARP cleavage, an indicator of apoptosis, and protects against SCD inhibitors-induced cell death in three GSCs lines (Figure 4G–H and Figure S5K). Finally, ectopic expression of HRASG12V in three YTX-7739 resistant GSCs lines increased SCD expression in two of these lines (Figure 4I) and promoted vulnerability to this SCD inhibitor with a 20–45% increase in cell death after YTX-7739 treatment in all three lines (Figure 4J). Collectively, these results indicate that acquired or inherent sensitivity to SCD inhibitors is primarily mediated by MEK/ERK activity.
Figure 4: RAS/MEK/ERK promotes vulnerability to SCD inhibitors.
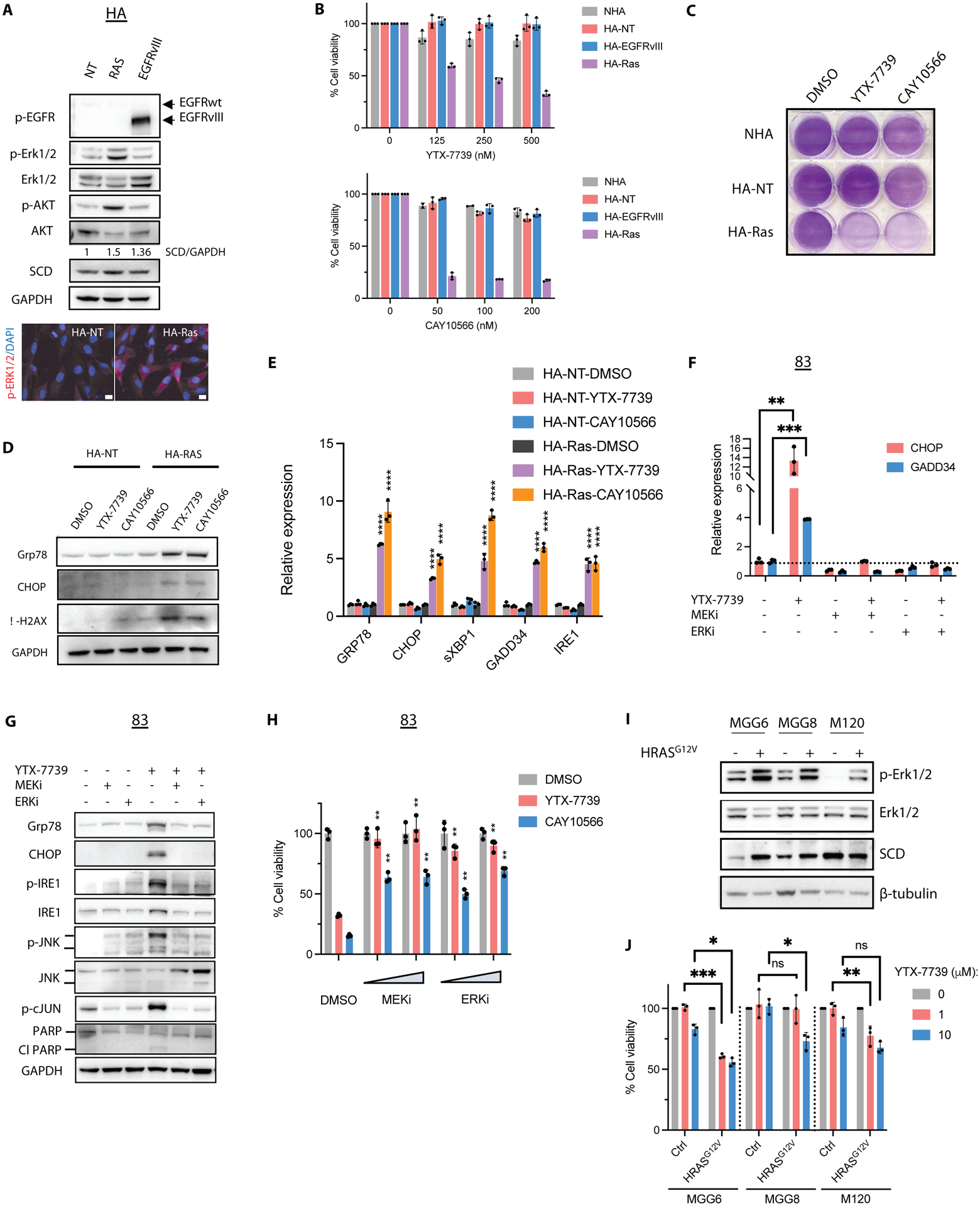
(A) Immunoblot analysis and immunostaining of HA-NT cells expressing a control vector, HRASG12V or EGFRvIII. Scale bar, 10μm. The ratio of SCD to GAPDH with values normalized to HA-NT is also shown. (B) Cell viability of primary (NHA) and transformed astrocytes (in 1% serum) treated with YTX-7739 (top) or CAY10566 (bottom) for 96h. (C) Cells were plated (in 1% serum) and treated with YTX-7739 (1μM) or CAY10566 (0.5μM) for five days before staining with crystal violet. (D-E) Immunoblot analysis (D) and relative mRNA expression (E) of ER stress, UPR, and DNA damage in HA-NT and HA-Ras treated with YTX-7739 (1μM) or CAY10566 (0.5μM) for 48 h. (F-G) Relative mRNA expression as compared to HA-NT DMSO (control) (F) and immunoblot analysis (G) of ER stress, UPR, JNK, and apoptosis in 83 treated with YTX-7739 (1μM) or its combination with AZD8330 (MEKi, 1μM), AZD0364 (ERKi, 1μM) for 72 hours. (H) Cell viability of 83 treated with YTX-7739 (1μM), CAY10566 (0.5 μM), or their combination with MEKi and ERKi (0.25–0.5μM) for 96 hours. (I) Immunoblot analysis of MGG6, MGG8, and M120 expressing a control vector or HRASG12V and (J) cell viability after treatment with YTX-7739 for 96h (J). ****P<0.0001; *** P<0.001; **P<0.01; *P<0.05; ns, not significant.
GSC subpopulations with low MEK-ERK signaling can escape DNLS-targeted therapy
Given the molecular heterogeneity and genetic plasticity of GSCs, we reasoned that cell subpopulations with low MEK-ERK activity could escape SCD-targeted therapy. To explore this possibility, we subjected two GSC lines sensitive to YTX-7739 (83 and M76) to several rounds of treatment with this inhibitor (YTX-7739 was added twice weekly at 1μm for 6 weeks). Whereas most cells cultured under such conditions failed to survive, few cells, referred to as 83R and M76R, could be propagated over an extended period. Rechallenging these cells with either YTX-7739 or CAY10566 resulted in lower cell death (Figure 5A and S6A) and an attenuated UPR (Figure 5B and S6B) as compared to their parental counterparts. In 83R, and to a lesser extent in M76R, we also observed a decreased sensitivity to FASN and ACC inhibitors (Figure S6C). Along with this increased resistance to DNLS inhibitors, we observed reduced phosphorylation of ERK1/2 and its downstream target RSK1 in resistant cells compared to their parental lines (Figure 5C). Finally, ectopic expression of HRASG12V in 83R restored sensitivity to YTX-7739 (Figure S6D). These results support an essential role of MEK-ERK signaling in defining sensitivity and resistance to SCD and DNLS inhibitors.
Figure 5: MEK-ERK signaling inhibits AMPK activity and defines the therapeutic response to DNLS inhibitors.
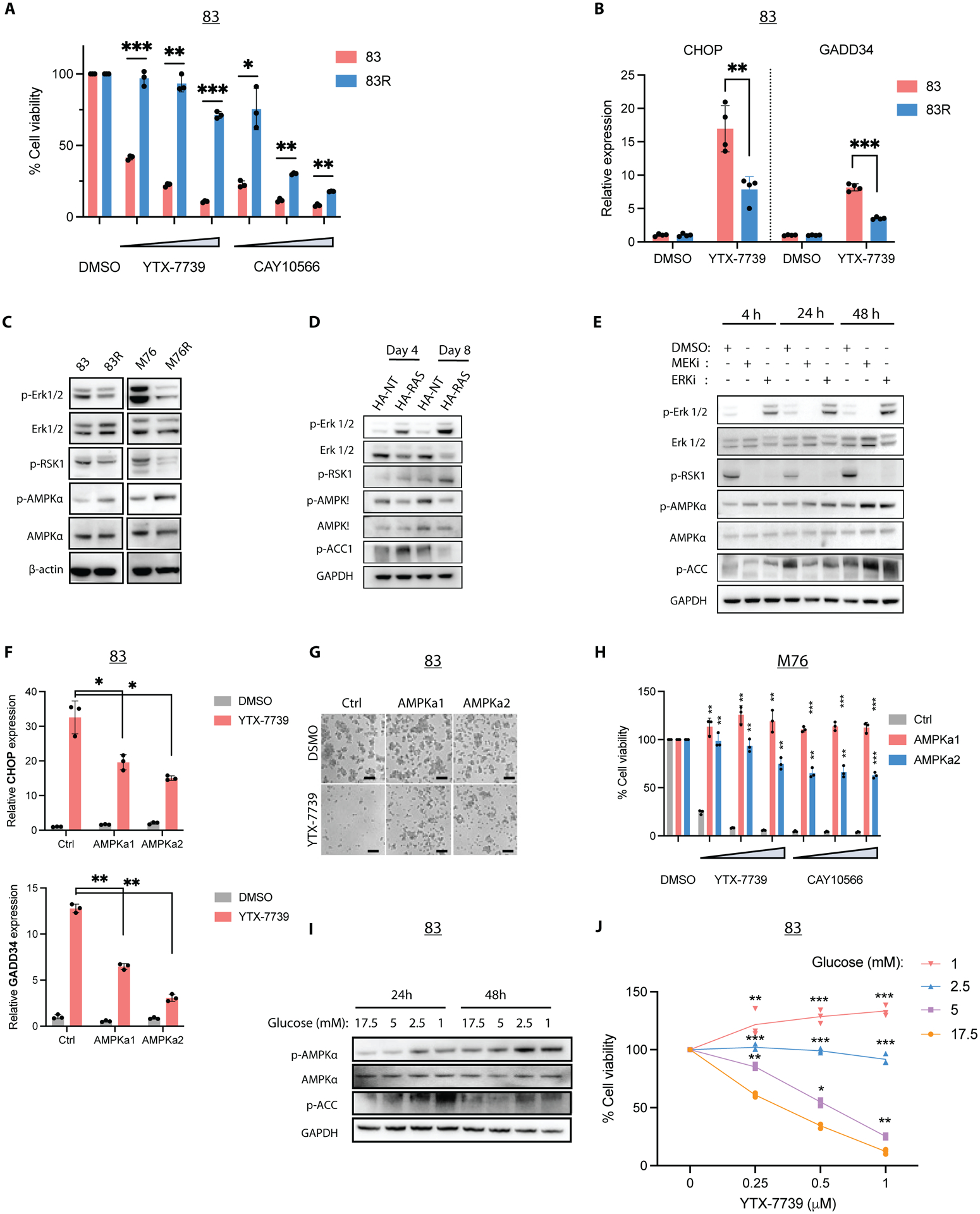
(A) Cell viability of 83/83R treated with YTX-7739 (0.25–1μM) and CAY10566 (0.125–0.5μM) for 96h. (B) Relative mRNA expression of UPR markers CHOP and GADD34 in 83/83R treated with DMSO (control) or YTX-7739 (1μM) for 72h. (C) Immunoblot analysis of ERK and AMPK signaling in 83/83R and M76/M76R. (D) Immunoblot analysis of ERK and AMPK signaling in transformed astrocytes on days 4 and 8 after expressing control (HA-NT) or HRASG12V (HA-Ras). (E) Immunoblot analysis of ERK and AMPK activity in 83 treated with MEK and ERK inhibitors. (F) Relative mRNA expression as compared to DMSO (control) of UPR markers CHOP and GADD34 in 83 Ctrl, AMPKa1, and AMPKa2 treated with or YTX-7739 (1μM) for 72h. (G) Representative micrographs of 83 expressing Ctrl, AMPKa1, or AMPKa2 after treatment with YTX-7739 (1μM) Scale bar: 100μm. (H) Cell viability of M76 after treatment with YTX-7739 (0.25–1μM) or CAY10566 (0.125–0.5μM) for 96h. (I) Immunoblot analysis of AMPK signaling in 83 cultured in decreasing concentrations of Glucose for 24–48h. (J) Cell viability of 83 cultured in decreasing concentrations of Glucose and concomitantly treated with YTX-7739 for 72h. ****P<0.0001; *** P<0.001; **P<0.01; *P<0.05.
AMPK signaling is repressed by MEK-ERK activation
Our effort to identify regulators downstream of MEK-ERK prompted us to consider AMPK, given its previously reported roles in phosphorylating and inactivating ACC (15), promoting a catabolic switch (16), and suppressing JNK activity (17). We observed increased AMPK phosphorylation in 83R and M76R compared to their parental cells (Figure 5C). AMPK signaling is reportedly downregulated after constitutive activation of Ras, Raf, or MEK/ERK pathway (18, 19). This was confirmed in our astrocytes model, where we saw a decreased phosphorylation of AMPK and ACC, at day 8 post-transduction with HRASG12V-expressing lentivirus (Figure 5D). Conversely, in parental 83 treated with MEK or ERK inhibitors, we saw overall time-dependent phosphorylation of AMPK and ACC (Figure 5E), although p-ACC was reduced at 48h following MEK/ERK inhibition. Treatment with the ERK inhibitor AZD0364 increased ERK phosphorylation, as previously reported for several ERK inhibitors and attributed to feedback activation (20). However, a complete blockade of MEK/ERK signaling is reflected through the downregulation of RSK1 phosphorylation (Figure 5E). Overall, these results confirm that MEK/ERK signaling inhibits AMPK activity.
Therapeutic response to DNLS inhibitors is dictated by AMPK
To test whether vulnerability to DNLS inhibitors is directly mediated by AMPK, we explored genetic, pharmacological, and physiological modulation of AMPK activity. First, we expressed two constitutively active mutants of AMPK (AMPKa1 (21) and AMPKa2 (22)) in two YTX-7739 sensitive GSCs and HA-Ras. AMPK activation dampened or suppressed terminal UPR induced by YTX-7739 (Figure 5F and S7A–B) and rendered GSCs and HA-Ras cells insensitive to SCD, FASN, and ACC inhibitors (Figures 5G–H and S7C–D). Similarly, pharmacological activation of AMPK with the synthetic ligand A-769662 provided a dose-dependent protection against YTX-7739 in GSCs and HA-Ras cells (Figure S7E–F). A second AMPK activator (AICAR) also conferred protection, whereas an AMPK inhibitor (dorsomorphin) increased the cytotoxicity of YTX-7739 in two GSC lines (Figure S7G). Further, treatment of 83R with dorsomorphin restored sensitivity to YTX-7739 (Figure S7H). Dorsomorphin is a non-specific inhibitor of AMPK (23), prompting us to explore shRNA-mediated silencing of AMPKA1 and AMPKA2 as an alternative. However, in agreement with the essential role of AMPK activity in GSCs (24), cell viability was decreased following AMPK silencing (Figure S7I), which precluded further testing. Since AMPK is activated by glucose deprivation (25), we also tested whether activating endogenous AMPK by lowering glucose concentrations in the GSC culture medium (which contains 17.5mM of Glucose) would also protect against SCD inhibition. Along with increased phosphorylation of AMPK and ACC at the lowest glucose concentrations (Figure 5I), we observed protection from SCD inhibitors in 83 and M76 (Figure 5J and Figure S7J). Our results show that AMPK activation drives resistance to SCD and DNLS inhibitors whereas targeting AMPK increases the therapeutic response.
GSCs adapt to lipotoxicity and impaired DNLS by activating AMPK
We reasoned that AMPK activation could protect against SFA-triggered lipotoxicity. Indeed, constitutive activation of AMPK strongly repressed terminal UPR caused by C16:0 (Figure S8A) and protected against its lipotoxicity in two GSC lines (Figure S8B). Despite the reported inactivating phosphorylation of ACC by AMPK, there was no significant decrease in cell proliferation or the sphere-forming ability of GSCs expressing AMPKa1 or AMPKa2. Further, as mentioned earlier, AMPK activation was protective against FASN and ACC inhibition (Figure S7D), which, unlike SCD inhibition, does not promote lipotoxicity. This suggested that, in addition to being protected against lipotoxicity, GSCs with activated AMPK could become refractory to DNLS. Indeed, long-term silencing of SCD over 30 days impaired sphere formation in the control group but only partially impaired sphere formation in AMPKa1 or AMPKa2 expressing GSCs (Figure 6A). Silencing of SCD also prevented tumor growth following intracranial implantation of GSCs in mice (6), whereas AMPKa1 expression did not significantly alter tumor growth (Figure 6B). Despite delayed tumor growth, mice in the AMPKa1/shSCD group developed tumors that gradually grew over time (Figure 6B).
Figure 6: AMPK reverts lipotoxicity by altering lipid composition.
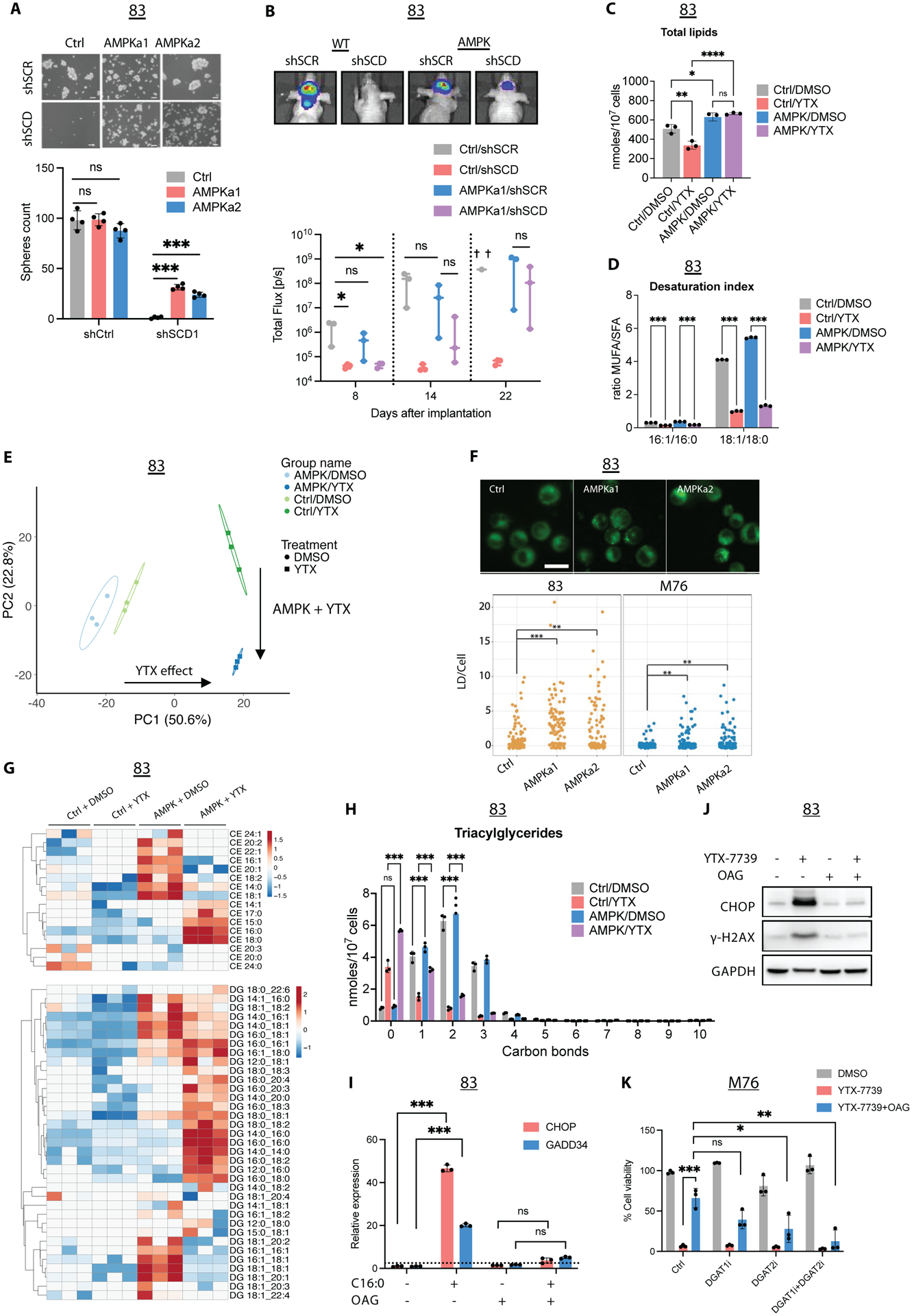
(A) Micrographs and spheres count in 83 Ctrl, AMPKa1, and AMPKa2 thirty days following two rounds of transduction with shCtrl or shSCD lentivirus. Scale bar, 50μm. (B) 83-Fluc Ctrl or AMPKa1 GSCs were transduced with shCtrl or shSCD1 and intracranially implanted in mice (n=3). Representative images and longitudinal Fluc imaging of mice in each group. (C) Total lipids in untreated (DMSO) and treated (YTX) 83 Ctrl/AMPK for 72h. (D) Desaturation index (DI) of 16:0 and 18:0 in 83 Ctrl/AMPK treated with YTX-7739 for 72h. (E) PCA of individual lipid species from untreated and treated 83 Ctrl/AMPK. Percentage of total variance explained by individual principal components (PC1 and PC2). (F) Fluorescence imaging and quantification of lipid droplets (LD) in GSC expressing Ctrl, AMPKa1, and AMPKa2. Scale bar, 25 μm (G) Heatmap of CE and DG after YTX-7739 treatment for 72h. (H) The amount of TG with 0–10 carbon bonds in 83 Ctrl/AMPK treated with YTX-7739 for 72h. (I) Relative expression of UPR markers in 83 supplemented with BSA or C16:0 (300μM) ± OAG (60μM). (J) Immunoblot analysis of 83 treated with YTX-7739 (1μM) ± OAG. (K) Cell viability of M76 co-treated with YTX-7739 (0.5μM) and OAG (60μM) in addition to DGAT1i and GAT2i for 96h. ****P<0.0001; *** P<0.001; **P<0.01; *P<0.05; ns, not significant.
AMPK signaling protects from SCD inhibition by altering lipid composition
To understand how AMPK mitigates lipotoxicity and impacts lipogenesis, we performed shotgun lipidomics on GSCs expressing a control vector (Ctrl) or AMPKa1 (AMPK) and treated with vehicle (DMSO) or YTX-7739 for 72 hrs. We quantified approximately 900 lipid species across 17 lipid subclasses. We found that enforced AMPK signaling increased the total amount of lipids per cell (Figure 6C and Figure S8C). Treatment of Ctrl GSCs with YTX-7739 decreased cellular lipids and AMPK signaling attenuated the YTX-7739-mediated decrease in lipid amounts (Figure 6C). Analysis of acyl tail composition in the lipidome confirmed that YTX-7739 treatment decreased MUFA incorporation into complex lipids and the DI (16:1/16:0 and 18:1/18:0), irrespective of AMPK status (Figure 6D). Thus, these data show that SCD inhibition alters both lipid desaturation and total lipid accumulation in GSCs.
Next, we applied Principal Component Analysis (PCA) to the lipidomics data to better discern the lipids most contributing to the observed variance between the treatments. PCA revealed minimal differences between untreated Ctrl and AMPK GSCs (Figure 6E). Principal component 1 (PC1) captured approximately 50% of the total variance and primarily reflected changes to the lipidome in response to YTX-7739 treatment. Principal component 2 (PC2) delineated approximately 23% of the total variance and reflected changes induced by SCD inhibition combined with AMPK signaling. As expected, the top 50 lipids positively contributing to PC1 were enriched for phospholipids containing two saturated acyl tails, such as PG 16:0_18:0, PC 16:0_18:0, PE 16:0_16:0, and phospholipids containing one saturated acyl tail and one polyunsaturated acyl tail, such asPG 16:0_18:2, PE 16:0_18:3, and PC 18:0_18:2 (Figure S8D). Similarly, triacylglycerides (TG) enriched in saturated acyl tails, including TG 50:0, TG 52:0, and TG 54:0, were observed in PC1. These data demonstrate an underrepresentation of MUFA acyl tails in the lipidome and support the idea that YTX-7739 treatment of cells decreases the incorporation of MUFAs into complex lipids irrespective of AMPK signaling.
To better understand the changes to the lipidome induced by SCD inhibition in the presence of constitutive AMPK signaling, we examined the lipids positively and negatively contributing to PC2 (Figures S8E–F). Positive contributors to PC2 were largely phospholipids containing two saturated acyl tails, such as PE 18:0_16:0, PE 16:0_16:0, and PC 18:0_18:0. In contrast, we observed that negative contributors to PC2, reflective of AMPK signaling and SCD inhibition, were enriched for phospholipids containing MUFAs, including PG 16:1_18:1, PC 12:0_18:1, and PE 18:0_18:1, or PUFAs, including PE 18:0_20:4, PC 18:2_18:2. These data suggest that AMPK signaling lowers the saturation state of phospholipids in the membrane of SCD-inhibited cells, thereby decreasing engagement of ER stress pathways, lipotoxicity, and cell death.
We also observed several TG and cholesteryl esters (CE) species containing SFAs negatively contributing to PC2, such as TG 42:0, CE 18:0, and CE 16:0, which suggests that AMPK signaling was shuttling SFAs into neutral lipid storage. BODIPY staining of Ctrl and AMPK cells confirmed that AMPK signaling increased lipid droplets (LD) in GSCs (Figure 6F). Cluster analysis of neutral lipids species, including CEs and diglycerides (DGs), showed that AMPK signaling increased the amounts of individual CEs and DGs containing MUFA or PUFA acyl tails (Figure 6G). SCD inhibition drove the accumulation of SFA-containing neutral lipid species only in AMPK GSCs. Likewise, analysis of TGs desaturation revealed a similar pattern where SCD inhibition resulted in the accumulation of saturated or monounsaturated TGs (Figure 6H), thus suggesting that GSC-AMPK cells treated with YTX-7739 shuttle lipotoxic SFAs, mainly 16:0 and 18:0, into LD to preserve cellular viability. To mimic the intracellular increase of DG, we used 1-oleoyl-2-acetyl-glycerol (OAG), a membrane-permeable analog of DG. Supplementing GSCs lines with OAG prevented UPR, DNA damage, and lipotoxicity induced by excess C16:0, YTX-7739, or SCD silencing (Figure 6I–K, S9A–D). Diacylglycerol O-acyltransferase (DGAT) catalyzes the terminal step converting DG and fatty acyl-CoA into TG, which subsequently form lipid droplets in the cytosol (26). Pharmacological inhibition of DGAT1 (with A922500) and DGAT2 (with PF-06424439), which prevents TG and LD formation in GBM (27), reversed OAG-mediated protection against YTX-7739 and C16:0 (Figure 6K and S9E–F). Taken together, these results reveal that AMPK activation can overcome DNLS inhibition and resultant lipotoxicity by altering membrane lipid composition and channeling SFAs into neutral lipids, such as CEs and TGs.
Discussion
This current study explored the therapeutic efficacy of a clinical-stage inhibitor of SCD and the genetic drivers conferring vulnerability to lipogenesis inhibitors in GBM. From a clinical perspective, several of our findings are noteworthy. First, we show that YTX-7739, the first clinical-stage inhibitor of SCD, triggers lipotoxicity in GSCs and provide evidence supporting the clinical evaluation of this inhibitor in patients with GBM. Second, we demonstrate that MEK/ERK signaling renders GSCs particularly vulnerable to pharmacological inhibitors of lipogenesis. At the same time, the dependence on DNLS is dictated by AMPK activity, thus illustrating a protective role of AMPK and its contribution to metabolic plasticity in GBM.
Our previous PK studies in mice and preclinical studies in rats and cynomolgus monkeys revealed favorable properties of YTX-7739, including bioavailability, low clearance, and a favorable brain-to-plasma ratio of greater than 1 (8). Our PD studies in mice bearing GBM tumors showed that YTX-7739 could achieve a comparable reduction in FA desaturation in normal brain and brain tumors, demonstrating its ability to penetrate the blood-brain barrier effectively. In our preclinical orthotopic GSCs mouse models, treatment with YTX-7739 improved overall survival, particularly in the MGG8 model, whereas its efficacy in the 83 model was less robust. Mice implanted with 83 presented a short survival of 17–19 days on average, which narrows the therapeutic window and limits the overall treatment regimen. In rats for instance, the maximal reduction of C16 DI was obtained after dosing with YTX-7739 for 7 days (8). For these fast-growing tumors, a higher dose of YTX-7739 or twice-daily (BID) dosing might deplete endogenous pools of SCD products more rapidly, thereby increasing therapeutic efficacy.
Overall, the efficacy of this inhibitor in our mouse models was transient and, in most cases, failed to eradicate GBM tumors. Therefore, from a clinical perspective, combination therapies with TMZ, radiation, or other agents are more likely to provide a better therapeutic outcome. This is supported by our observation that inhibiting SCD impairs DNA damage repair through HDR by downregulating RAD51 (6). In the highly aggressive 83 model, YTX-7739 with TMZ extended overall survival compared to either monotherapy. In the MGG8 model, which is highly sensitive to TMZ, YTX-7739 as monotherapy was equally effective as the standard of care adjuvant chemotherapeutic currently used for GBM treatment, TMZ.
Identifying genetic drivers of vulnerability to SCD and other DNLS inhibitors in GBM is of major clinical importance. We show that constitutive activation of HRAS/MEK/ERK promotes susceptibility to DNLS inhibitors, whereas impaired MEK/ERK activity renders GSCs insensitive to the SCD inhibitor. We identified AMPK as a downstream target for MEK/ERK and the primary regulator of sensitivity to DNLS inhibitors. Importantly, our findings reveal an adaptive metabolic reprogramming orchestrated by AMPK, allowing GSCs to overcome DNLS inhibition and maintain proliferation and tumor initiation properties. We show that GSCs that survive long-term pharmacological inhibition of SCD have decreased MEK/ERK signaling and increased AMPK activity. AMPK activation confers protection against metabolic stress (24, 28). By employing shotgun lipidomics, we could show that AMPK activation allows GSCs to overcome lipotoxicity, sustain proliferation, and retain their in vivo tumor initiation ability when DNLS is impaired by altering membrane lipid composition and sequestering toxic SFAs into neutral lipids.
Our findings show that the therapeutic response to DNLS-targeted therapy is contingent on a dynamic shift in molecular signaling, particularly MEK/ERK and AMPK, which, as a nutrient and energy sensor, could be regulated in response to the cellular energetic needs or the biochemical microenvironment of the tumor. This implies bidirectional plasticity where sensitive GSCs could become more resistant to the inhibitor (as seen with 83R and M76R). In contrast, other GSC lines (such as MGG8) failed to respond to YTX-7739 mediated lipotoxicity in culture yet displayed an excellent therapeutic response following intracranial implantation in mice. Neither the metabolic demands nor the biochemical environment of GBM is faithfully recapitulated under in vitro cell culture conditions. Additionally, various intrinsic or exogenous stimuli pertinent to the GBM microenvironment can impact MAPK/ERK and AMPK signaling. For instance, the rapid growth of GBM tumors creates hypoxic niches enriched in GSCs (29), whereas ERK1/2 activation is triggered by hypoxia (30). Therefore, GSCs residing in these hypoxic niches may be particularly vulnerable to DNLS inhibition. On the other hand, glucose concentrations that potently impact AMPK activity and the response to SCD inhibitors, as we have shown, are typically high in the GBM culture medium relative to restricted glucose in the brain tumor environment (31). Therefore, it is likely that AMPK is activated in glucose-deprived tumors. Other physiological and pathological conditions relevant to GBM, including aging and inflammation, suppress AMPK activity (32). Therefore, we propose that MEK/ERK and AMPK activities, detected by immunohistochemistry of tumor biopsies, could be predictive biomarkers to guide patient selection and stratification. Our findings should also help tailor treatment paradigms to maximize therapeutic efficacy. For instance, some widely used drugs, such as the anti-inflammatory agent salicylate or anti-diabetic compound Metformin, are potent activators of AMPK (33, 34) and could be detrimental to the efficacy of YTX-7739 or other DNLS-targeting therapies.
There are limitations to this study. First, in vivo therapeutic efficacy of YTX-7739 was only tested using two GSCs lines. Second, we used immunocompromised mice, which precludes any assessment of the immune response following pharmacological inhibition of SCD. Inhibiting lipid synthesis in regulatory T cells (Treg) reportedly elicits an anti-tumor immune response (35). Given the role of Tregs in suppressing the anti-tumor immune response in GBM (36), drugging the tumor immune microenvironment with SCD inhibitors might enhance the anti-tumor response. Third, in light of the role of AMPK in promoting a switch from anabolic to catabolic processes (16), including the inactivation of ACC (15), a defined mechanism for a seemingly contradictory upregulation of MUFA and LDs in GSCs with a constitutively active AMPK remains to be elucidated. AMPK increases FA uptake by enhancing the translocation of the FA transporter CD36 to the plasma membrane (37). Therefore, AMPK may enhance CD36-mediated uptake of exogenous FA in GSCs. From that perspective, impeding AMPK activity, particularly in vivo, could maximize the efficacy of DNLS-targeting agents. However, this remains a challenge in the absence of highly specific, brain-penetrant inhibitors of AMPK.
In conclusion, this work highlights metabolic adaptability in GSCs and provides an integrative framework for targeting DNLS in GBM. Our findings may inform patient stratification strategies, drug combinations, and clinical trial design to maximize therapeutic efficacy for this morbid and incurable brain cancer.
MATERIALS AND METHODS
Study design
This study sought to evaluate the therapeutic efficacy of a clinical grade brain-penetrant inhibitor of SCD in patient-derived GSCs and preclinical mouse models of GBM. We also used an astrocyte-based model of gliomagenesis to define oncogenic signaling that renders cells susceptible to DNLS-targeted therapies. Our cell culture experiments were designed to determine the specificity, mode of action, and therapeutic efficacy of YTX-7739 in GSCs. Cell culture experiments were complemented with in vivo validation of YTX-7739 PK and PD properties and its antitumor efficacy in orthotopic GSC mouse models either as a monotherapy or in combination with TMZ. Cell culture experiments consisted of at least three independent biological replicates and were repeated at least three times. For shotgun lipidomics analysis, we used three biological replicates for each group; these experiments were performed only once. The investigators were blinded to group allocations or subsequent data analysis for all experiments involving fatty acid analysis, including shotgun lipidomics, tumor sectioning, staining, and microscopy. Blinding was not applied to other experiments. Statistical methods were not used to predetermine the sample size. However, sample sizes were determined based on previous experience with our GSCs models or estimates from pilot experiments. For all cell culture and in vivo experiments, we used a minimum of three samples per group unless stated otherwise. Animals were first imaged to determine tumor volume, and randomization was performed based on tumor size. Animals were euthanized if they lost 20% of their weight or exhibited signs of morbidity, according to the approved animal protocol. The n values of mice used for each experiment and the corresponding statistical methods are indicated in the figure legend.
Primary cells and cell lines
Primary GBM cells were derived from surgical specimens obtained from patients with GBM at the Massachusetts General Hospital (provided by Dr. H. Wakimoto) under the appropriate Institutional Review Board approval (MGG6, MGG8, 70RR) or provided by Dr. I. Nakano (157, 83, 326), Dr. B.Reynolds (L0, L1, L2) and Dr. J.Sarkaria at The Brain Tumor PDX National Resource at Mayo Clinic (M76, M12, M120). All GBM cells used in this study have been previously characterized (38–42). Cells were expanded as neurospheres and maintained in DMEM/F12 (Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12) medium supplemented with B27 without vitamin A (1:50; Life Technologies), heparin (2 μg/mL; Sigma Aldrich), human recombinant Epidermal Growth Factor (EGF; 20 ng/mL; ABM), and human recombinant basic fibroblast growth factor (bFGF-2; 10ng/mL; ABM). DMEM-F12 without L-Glutamine without Hepes without Glucose (Biowest) was used to control glucose concentrations in the culture medium. NHAs were obtained from Sciencell Research Laboratories and grown in Astrocyte Medium (Sciencell). Human astrocytes transformed with E6/E7/hTERT were provided by Dr. A. Krichevsky and Dr. E. Uhlmann and generated as previously described (13). Ionizing radiation of GSCs was performed using a 137Cs irradiator.
Mouse Orthotopic Brain Tumor Models
All animal experiments were approved by the Massachusetts General Hospital Subcommittee on Research Animal Care and conformed to the guidelines of the NIH Guide for the Care and Use of Laboratory Animals. GSCs (1–5 × 104) expressing Firefly luciferase (Fluc) were implanted into the left forebrain of male and female athymic nude mice (1.0 mm anterior and 2.0 mm laterally to bregma at a depth of 2.5 mm from the skull surface) using a stereotactic frame. Brain tumors were detected and monitored over time using Fluc bioluminescence imaging using a Xenogen IVIS 200 imaging system (PerkinElmer) following intraperitoneal (IP) injections of D-luciferin (150 mg/kg body weight) (Gold Biotechnology). Signal intensity was quantified with Living Image software 4.3.1 (PerkinElmer). For all in vivo studies, YTX-7739 was administered by IP injections. Control groups were administered with 100 μl of vehicle (0.5% methylcellulose + 0.2% Tween80 in sterile saline solution) by IP injections. TMZ was dissolved in DMSO (100 mM stock solution) and freshly resuspended in 5% dextrose before IP administration in mice.
Shotgun Lipidomics Analysis
83 cells were seeded at a density of 1×105 cells/well in a 6-well plate and treated with DMSO (Control) or YTX-7739 (1 μM) in four replicates. At 72h after treatment, cells were counted and collected for lipid extraction. Shotgun lipidomics analysis was performed as previously described (43). Briefly, after the addition of an internal standard mixture consisting of 70 lipid standards across 17 subclasses (AB Sciex, 5040156, Avanti 330827, Avanti 330830, Avanti 330828, Avanti 791642), and two successive extractions, organic layers were vacuum dried in a Thermo SpeedVac SPD300DDA machine, using ramp setting 4 at 35°C for 45 minutes with a total run time of 90 minutes. Samples were then resuspended in methanol/dichloromethane (1:1 ratio) with 10 mM Ammonium Acetate and transferred to robovials (Thermo 10800107) for subsequent lipidomics analysis. Samples were analyzed by direct infusion using the Sciex 5500 with Differential Mobility Device (DMS) (comparable to Sciex Lipidyzer platform) for quantitative measurement of 1450 lipid species belonging to 17 subclasses. The DMS was tuned with EquiSPLASH LIPIDOMIX (Avanti 330731). Quantitative values of lipid species were normalized to cell counts. Heatmaps and PCA plots were generated using Clustvis (https://biit.cs.ut.ee/clustvis/). The color scale on heatmaps refers to the row Z score. Nipals PCA was utilized to calculate principal components (PC) on PCA. X and Y axis show PC1 and PC2, which refer to the percentage of the total variance, respectively.
Mouse Pharmacokinetics and Pharmacodynamics studies
Mouse pharmacokinetic (PK) studies were performed by ChemPartner in an IACUC-accredited facility, following the guidelines of their ethics committee. Male CD1 mice were used in these studies with n=3mice/group. YTX-7739 was administered at 1 mg/kg intravenously (IV) in the foot dorsal vein (formulated in 10% DMSO/10% Solutol HS 15/80% HP-β-CD in saline) or IP at 3, 10, and 30 mg/kg (formulated in 0.5% methylcellulose in water). Approximately 150 μL blood/time point was collected at each time point into K2EDTA tubes by the tail vein. Blood samples were prepared and processed as previously described (8). Samples were analyzed using standard LCMS/MS methods (API6500, Qtriple) and compared to a reference standard. Brain concentrations of YTX-7739 were determined from homogenized tissue prior to LC-MS/MS analysis. YTX-7739 concentrations were compared to a standard curve of YTX-7739 spiked into naïve brain homogenate. Pharmacokinetic (PK) parameters were determined using WinNonlin. To measure the pharmacodynamics (PD) of YTX-7739 in plasma, brain, and tumor tissue, female athymic nude mice bearing 83 GSCs-Fluc brain tumors (confirmed using Fluc imaging) were treated for 11 days with solvent control or YTX-7739 (30mg/kg). Mice were sacrificed 4h after the last YTX-7739 injection. Plasma and brain tissue samples which include the brain tumor as well as the contralateral hemisphere (brain), were collected for fatty acid profiling.
Statistical Analysis
The results are presented as individual values or mean ± standard deviation (SD) unless stated otherwise. Statistical significance was determined using GraphPad Prism 9 (GraphPad Software). For two groups comparison, statistical significance was calculated using the two-tailed Student’s t-test when data were normally distributed and the Mann-Whitney test when data were not normally distributed. For multiple groups comparison, p values were generated using one-way ANOVA followed by Dunnett’s multiple comparisons test when data were normally distributed and the Kruskal-Wallis test followed by Dunn’s multiple comparison test when data were not normally distributed to compare different groups to controls. Normality was tested using the Shapiro-Wilk test. Animal survival curves were analyzed for statistical significance using a log-rank test (Mantel-Cox), and survival curves were plotted as Kaplan-Meier. P values of 0.05 or less were considered significant. Correlation studies were performed using Spearman nonparametric correlation testing. The group size was determined based on preliminary experiments, and no statistical method was used to determine the sample size. Graphs were generated using GraphPad Prism 9 or R package version 0.4.0. https://CRAN.R-project.org/package=ggpubr. Data analysis for lipidomics studies was conducted using R. Schematic figures were created with BioRender.com.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Drs. H. Wakimoto (MGH), the Brain Tumor PDX National Resource at Mayo Clinic, I. Nakano and B. Reynolds for providing primary GBM cells; Dr. A. Krichevsky and Dr. E. Uhlmann for providing HA-NT cells; Dr. S. Plotkin and Dr. I. Arrillaga-Romany for their valuable feedback on this project. We thank Dana-Farber/Harvard Cancer Center for the use of the Specialized Histopathology Core, which provided immunohistochemistry service.
FUNDING
This work is supported by NIH/NINDS R01 NS113822 (C.E.B), DoD Peer Reviewed Cancer Research CA191075 to C.E.B, NIH/NCI P50 CA165962 SPORE in Brain Tumor Research subaward to C.E.B, American Brain Tumor Association (ABTA) Discovery Grant supported by the Uncle Kory Foundation to C.E.B, Concern Foundation Conquer Cancer Now Award to C.E.B., NIH R01 NS121319 to S.J.B., German Academic Exchange Service (DAAD) scholarship to K.M.E., and American-Italian Cancer Foundation Postdoctoral Research Fellowship to A.S..
COMPETING INTERESTS:
D.F.T. and C.Y.C. are past employees of Yumanity Therapeutics. C.E.B. received in-kind support from Yumanity Therapeutics. K.J.W. serves as a consultant for Verso Biosciences Inc.. D.F.T. is an inventor on patent application PCT/US2020/023913 submitted by Yumanity Therapeutics, Inc. for compounds and uses thereof, including SCD inhibitors and their uses, and PCT/US2019/023737 submitted by Yumanity Therapeutics, Inc. for compounds and uses thereof, including SCD inhibitors and their uses.
Footnotes
This manuscript has been accepted for publication in Science Translational Medicine. This version has not undergone final editing. Please refer to the complete version of record at www.sciencetranslationalmedicine.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior written permission of AAAS.
List of Supplementary Materials
DATA AND MATERIALS AVAILABILITY:
All data associated with this study are present in the paper or supplementary materials. When applicable, materials can be made available under a material transfer agreement (MTA) with the Mass General Brigham Innovation office. YTX-7739 was provided by Yumanity under an MTA. pLenti-BLRR and pLenti-trGluc are available through Addgene (plasmids #158958 and #158959).
REFERENCES
- 1.Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN, Cancer stem cells in glioblastoma. Genes & development 29, 1203–1217 (2015); published online EpubJun 15 ( 10.1101/gad.261982.115). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badr CE, Silver DJ, Siebzehnrubl FA, Deleyrolle LP, Metabolic heterogeneity and adaptability in brain tumors. Cellular and molecular life sciences : CMLS 77, 5101–5119 (2020); published online EpubDec ( 10.1007/s00018-020-03569-w). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koundouros N, Poulogiannis G, Reprogramming of fatty acid metabolism in cancer. British journal of cancer 122, 4–22 (2020); published online EpubJan ( 10.1038/s41416-019-0650-z). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pearson JRD, Regad T, Targeting cellular pathways in glioblastoma multiforme. Signal Transduct Target Ther 2, 17040 (2017) 10.1038/sigtrans.2017.40). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia D, Shaw RJ, AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol Cell 66, 789–800 (2017); published online EpubJun 15 ( 10.1016/j.molcel.2017.05.032). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinkham K, Park DJ, Hashemiaghdam A, Kirov AB, Adam I, Rosiak K, da Hora CC, Teng J, Cheah PS, Carvalho L, Ganguli-Indra G, Kelly A, Indra AK, Badr CE, Stearoyl CoA Desaturase Is Essential for Regulation of Endoplasmic Reticulum Homeostasis and Tumor Growth in Glioblastoma Cancer Stem Cells. Stem Cell Reports 12, 712–727 (2019); published online EpubApr 9 ( 10.1016/j.stemcr.2019.02.012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oatman N, Dasgupta N, Arora P, Choi K, Gawali MV, Gupta N, Parameswaran S, Salomone J, Reisz JA, Lawler S, Furnari F, Brennan C, Wu J, Sallans L, Gudelsky G, Desai P, Gebelein B, Weirauch MT, D’Alessandro A, Komurov K, Dasgupta B, Mechanisms of stearoyl CoA desaturase inhibitor sensitivity and acquired resistance in cancer. Sci Adv 7, (2021); published online EpubFeb ( 10.1126/sciadv.abd7459). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tardiff DF, Lucas M, Wrona I, Chang B, Chung CY, Le Bourdonnec B, Rhodes KJ, Scannevin RH, Non-clinical Pharmacology of YTX-7739: a Clinical Stage Stearoyl-CoA Desaturase Inhibitor Being Developed for Parkinson’s Disease. Mol Neurobiol, (2022); published online EpubJan 20 ( 10.1007/s12035-021-02695-1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nuber S, Chung CY, Tardiff DF, Bechade PA, McCaffery TD, Shimanaka K, Choi J, Chang B, Raja W, Neve E, Burke C, Jiang X, Xu P, Khurana V, Dettmer U, Fanning S, Rhodes KJ, Selkoe DJ, Scannevin RH, A Brain-Penetrant Stearoyl-CoA Desaturase Inhibitor Reverses alpha-Synuclein Toxicity. Neurotherapeutics, (2022); published online EpubApr 20 ( 10.1007/s13311-022-01199-7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szegezdi E, Logue SE, Gorman AM, Samali A, Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO reports 7, 880–885 (2006); published online EpubSep ( 10.1038/sj.embor.7400779). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang HHY, Pannunzio NR, Adachi N, Lieber MR, Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 18, 495–506 (2017); published online EpubAug ( 10.1038/nrm.2017.48). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chien JC, Tabet E, Pinkham K, da Hora CC, Chang JC, Lin S, Badr CE, Lai CP, A multiplexed bioluminescent reporter for sensitive and non-invasive tracking of DNA double strand break repair dynamics in vitro and in vivo. Nucleic acids research 48, e100 (2020); published online EpubSep 25 ( 10.1093/nar/gkaa669). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sonoda Y, Ozawa T, Hirose Y, Aldape KD, McMahon M, Berger MS, Pieper RO, Formation of intracranial tumors by genetically modified human astrocytes defines four pathways critical in the development of human anaplastic astrocytoma. Cancer research 61, 4956–4960 (2001); published online EpubJul 1 ( [PubMed] [Google Scholar]
- 14.Guo D, Prins RM, Dang J, Kuga D, Iwanami A, Soto H, Lin KY, Huang TT, Akhavan D, Hock MB, Zhu S, Kofman AA, Bensinger SJ, Yong WH, Vinters HV, Horvath S, Watson AD, Kuhn JG, Robins HI, Mehta MP, Wen PY, DeAngelis LM, Prados MD, Mellinghoff IK, Cloughesy TF, Mischel PS, EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal 2, ra82 (2009); published online EpubDec 15 ( 10.1126/scisignal.2000446). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeh LA, Lee KH, Kim KH, Regulation of rat liver acetyl-CoA carboxylase. Regulation of phosphorylation and inactivation of acetyl-CoA carboxylase by the adenylate energy charge. The Journal of biological chemistry 255, 2308–2314 (1980); published online EpubMar 25 ( [PubMed] [Google Scholar]
- 16.Trefts E, Shaw RJ, AMPK: restoring metabolic homeostasis over space and time. Mol Cell 81, 3677–3690 (2021); published online EpubSep 16 ( 10.1016/j.molcel.2021.08.015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma Y, Wang J, Gao J, Yang H, Wang Y, Manithody C, Li J, Rezaie AR, Antithrombin up-regulates AMP-activated protein kinase signalling during myocardial ischaemia/reperfusion injury. Thromb Haemost 113, 338–349 (2015); published online EpubFeb ( 10.1160/TH14-04-0360). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hardie DG, AMPK--sensing energy while talking to other signaling pathways. Cell Metab 20, 939–952 (2014); published online EpubDec 2 ( 10.1016/j.cmet.2014.09.013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanduja S, Feng Y, Mathis RA, Sokol ES, Reinhardt F, Halaban R, Gupta PB, AMPK promotes tolerance to Ras pathway inhibition by activating autophagy. Oncogene 35, 5295–5303 (2016); published online EpubOct 6 ( 10.1038/onc.2016.70). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pratilas CA, Solit DB, Targeting the mitogen-activated protein kinase pathway: physiological feedback and drug response. Clinical cancer research : an official journal of the American Association for Cancer Research 16, 3329–3334 (2010); published online EpubJul 1 ( 10.1158/1078-0432.CCR-09-3064). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X, Xiao ZD, Han L, Zhang J, Lee SW, Wang W, Lee H, Zhuang L, Chen J, Lin HK, Wang J, Liang H, Gan B, LncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress. Nature cell biology 18, 431–442 (2016); published online EpubApr ( 10.1038/ncb3328). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang JR, Lingeman E, Luong T, Ahmed S, Muhar M, Nguyen T, Olzmann JA, Corn JE, A Genome-wide ER-phagy Screen Highlights Key Roles of Mitochondrial Metabolism and ER-Resident UFMylation. Cell 180, 1160–1177 e1120 (2020); published online EpubMar 19 ( 10.1016/j.cell.2020.02.017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dasgupta B, Seibel W, Compound C/Dorsomorphin: Its Use and Misuse as an AMPK Inhibitor. Methods Mol Biol 1732, 195–202 (2018) 10.1007/978-1-4939-7598-3_12). [DOI] [PubMed] [Google Scholar]
- 24.Chhipa RR, Fan Q, Anderson J, Muraleedharan R, Huang Y, Ciraolo G, Chen X, Waclaw R, Chow LM, Khuchua Z, Kofron M, Weirauch MT, Kendler A, McPherson C, Ratner N, Nakano I, Dasgupta N, Komurov K, Dasgupta B, AMP kinase promotes glioblastoma bioenergetics and tumour growth. Nature cell biology 20, 823–835 (2018); published online EpubJul ( 10.1038/s41556-018-0126-z). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, Foretz M, Viollet B, 5’-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Molecular and cellular biology 26, 5336–5347 (2006); published online EpubJul ( 10.1128/MCB.00166-06). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greenberg AS, Coleman RA, Kraemer FB, McManaman JL, Obin MS, Puri V, Yan QW, Miyoshi H, Mashek DG, The role of lipid droplets in metabolic disease in rodents and humans. The Journal of clinical investigation 121, 2102–2110 (2011); published online EpubJun ( 10.1172/JCI46069). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng X, Geng F, Pan M, Wu X, Zhong Y, Wang C, Tian Z, Cheng C, Zhang R, Puduvalli V, Horbinski C, Mo X, Han X, Chakravarti A, Guo D, Targeting DGAT1 Ameliorates Glioblastoma by Increasing Fat Catabolism and Oxidative Stress. Cell Metab 32, 229–242 e228 (2020); published online EpubAug 4 ( 10.1016/j.cmet.2020.06.002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chae YC, Caino MC, Lisanti S, Ghosh JC, Dohi T, Danial NN, Villanueva J, Ferrero S, Vaira V, Santambrogio L, Bosari S, Languino LR, Herlyn M, Altieri DC, Control of tumor bioenergetics and survival stress signaling by mitochondrial HSP90s. Cancer cell 22, 331–344 (2012); published online EpubSep 11 ( 10.1016/j.ccr.2012.07.015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, Hjelmeland AB, Rich JN, Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer cell 15, 501–513 (2009); published online EpubJun 2 ( 10.1016/j.ccr.2009.03.018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minet E, Arnould T, Michel G, Roland I, Mottet D, Raes M, Remacle J, Michiels C, ERK activation upon hypoxia: involvement in HIF-1 activation. FEBS letters 468, 53–58 (2000); published online EpubFeb 18 ( 10.1016/s0014-5793(00)01181-9). [DOI] [PubMed] [Google Scholar]
- 31.Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, Weil RJ, Nakano I, Sarkaria JN, Stringer BW, Day BW, Li M, Lathia JD, Rich JN, Hjelmeland AB, Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci 16, 1373–1382 (2013); published online EpubOct ( 10.1038/nn.3510). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeon SM, Regulation and function of AMPK in physiology and diseases. Exp Mol Med 48, e245 (2016); published online EpubJul 15 ( 10.1038/emm.2016.81). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green KA, Mustard KJ, Kemp BE, Sakamoto K, Steinberg GR, Hardie DG, The ancient drug salicylate directly activates AMP-activated protein kinase. Science 336, 918–922 (2012); published online EpubMay 18 ( 10.1126/science.1215327). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE, Role of AMP-activated protein kinase in mechanism of metformin action. The Journal of clinical investigation 108, 1167–1174 (2001); published online EpubOct ( 10.1172/JCI13505). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lim SA, Wei J, Nguyen TM, Shi H, Su W, Palacios G, Dhungana Y, Chapman NM, Long L, Saravia J, Vogel P, Chi H, Lipid signalling enforces functional specialization of Treg cells in tumours. Nature 591, 306–311 (2021); published online EpubMar ( 10.1038/s41586-021-03235-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pombo Antunes AR, Scheyltjens I, Duerinck J, Neyns B, Movahedi K, Van Ginderachter JA, Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. Elife 9, (2020); published online EpubFeb 4 ( 10.7554/eLife.52176). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Habets DD, Coumans WA, El Hasnaoui M, Zarrinpashneh E, Bertrand L, Viollet B, Kiens B, Jensen TE, Richter EA, Bonen A, Glatz JF, Luiken JJ, Crucial role for LKB1 to AMPKalpha2 axis in the regulation of CD36-mediated long-chain fatty acid uptake into cardiomyocytes. Biochimica et biophysica acta 1791, 212–219 (2009); published online EpubMar ( 10.1016/j.bbalip.2008.12.009). [DOI] [PubMed] [Google Scholar]
- 38.Wakimoto H, Mohapatra G, Kanai R, Curry WT Jr., Yip S, Nitta M, Patel AP, Barnard ZR, Stemmer-Rachamimov AO, Louis DN, Martuza RL, Rabkin SD, Maintenance of primary tumor phenotype and genotype in glioblastoma stem cells. Neuro-oncology 14, 132–144 (2012); published online EpubFeb ( 10.1093/neuonc/nor195). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanaka S, Luk S, Kiyokawa J, Onozato ML, Iafrate AJ, Shah K, Martuza RL, Rabkin SD, Batchelor TT, Cahill DP, Chi AS, Wakimoto H, Genetically distinct glioma stem-like cell xenografts established from paired glioblastoma samples harvested before and after molecularly targeted therapy. Scientific reports 9, 139 (2019); published online EpubJan 15 ( 10.1038/s41598-018-37437-2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mao P, Joshi K, Li J, Kim SH, Li P, Santana-Santos L, Luthra S, Chandran UR, Benos PV, Smith L, Wang M, Hu B, Cheng SY, Sobol RW, Nakano I, Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proceedings of the National Academy of Sciences of the United States of America 110, 8644–8649 (2013); published online EpubMay 21 ( 10.1073/pnas.1221478110). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siebzehnrubl FA, Silver DJ, Tugertimur B, Deleyrolle LP, Siebzehnrubl D, Sarkisian MR, Devers KG, Yachnis AT, Kupper MD, Neal D, Nabilsi NH, Kladde MP, Suslov O, Brabletz S, Brabletz T, Reynolds BA, Steindler DA, The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol Med 5, 1196–1212 (2013); published online EpubAug ( 10.1002/emmm.201302827). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vaubel RA, Tian S, Remonde D, Schroeder MA, Mladek AC, Kitange GJ, Caron A, Kollmeyer TM, Grove R, Peng S, Carlson BL, Ma DJ, Sarkar G, Evers L, Decker PA, Yan H, Dhruv HD, Berens ME, Wang Q, Marin BM, Klee EW, Califano A, LaChance DH, Eckel-Passow JE, Verhaak RG, Sulman EP, Burns TC, Meyer FB, O’Neill BP, Tran NL, Giannini C, Jenkins RB, Parney IF, Sarkaria JN, Genomic and Phenotypic Characterization of a Broad Panel of Patient-Derived Xenografts Reflects the Diversity of Glioblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research 26, 1094–1104 (2020); published online EpubMar 1 ( 10.1158/1078-0432.CCR-19-0909). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsieh WY, Williams KJ, Su B, Bensinger SJ, Profiling of mouse macrophage lipidome using direct infusion shotgun mass spectrometry. STAR Protoc 2, 100235 (2021); published online EpubMar 19 ( 10.1016/j.xpro.2020.100235). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chien JC, Badr CE, Lai CP, Multiplexed bioluminescence-mediated tracking of DNA double-strand break repairs in vitro and in vivo. Nature protocols 16, 3933–3953 (2021); published online EpubAug ( 10.1038/s41596-021-00564-8). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or supplementary materials. When applicable, materials can be made available under a material transfer agreement (MTA) with the Mass General Brigham Innovation office. YTX-7739 was provided by Yumanity under an MTA. pLenti-BLRR and pLenti-trGluc are available through Addgene (plasmids #158958 and #158959).