

Abstract
Purpose:
To characterize MSH6/PMS2-associated mismatch repair deficient (MMR-D)/microsatellite instability-high (MSI-H) tumors given revised guidelines suggesting more modest phenotypes.
Methods:
Patients consented to IRB-approved protocols of tumor/germline sequencing or Lynch syndrome registry at a single institution from 2/2005–1/2021 with germline, heterozygous MSH6/PMS2 pathogenic/likely pathogenic (P/LP) variants were identified. Clinical data were abstracted and correlated with MMR/MSI status using non-parametric tests.
Results:
We identified 243 patients (133 sequencing, 110 registry) with germline MSH6/PMS2 P/LP variants; 186 (77%) had ≥1 cancer. Of 261 pooled tumors, colorectal (CRC) and endometrial cancers (EC) comprised 55% and 43% of cancers in MSH6 and PMS2 respectively; 192 tumors underwent molecular assessments, and 122 (64%) were MMR-D/MSI-H (77 in MSH6, 45 in PMS2). MMR-D/MSI-H cancers included CRC (n=56), EC (n=35), small bowel (n=6), ovarian (n=6), urothelial (n=5), pancreas/biliary (n=4), gastric/esophageal (n=3), non-melanoma skin tumors (n=3), prostate (n=2), breast (n=1), and CNS/brain (n=1). Among MMR-D/MSI-H CRC and EC, median age of diagnosis was 51.5 (range 27–80) and 55 (range 39–74) respectively; 9/56 (16%) of MMR-D/MSI-H CRCs were diagnosed at age <35.
Conclusions:
MSH6/PMS2 heterozygotes remain at risk for a broad-spectrum of cancers with 16% of MMR-D/MSI-H CRCs presenting before upper threshold of initiation of colonoscopy per guidelines.
Introduction
Lynch syndrome (LS) is an autosomal dominant cancer predisposition syndrome with up to an 80% lifetime risk of cancer of multiple types, including, but not limited to, colorectal (CRC), endometrial (EC), ovarian (OC), urothelial (UC), and small bowel (adenocarcinoma - SBA) with the most common cancers being CRC and EC.1–3 LS patients harbor germline pathogenic/likely pathogenic (P/LP) variants in the mismatch repair (MMR) genes (MLH1, MSH2, MSH6, PMS2, EPCAM promotor deletion).1,2 LS-associated cancers characteristically demonstrate MMR deficiency (MMR-D) and/or microsatellite instability-high (MSI-H) status, agnostic of tumor type.4
While it has been established that MLH1 and MSH2-associated LS carries higher lifetime cancer risks when compared to MSH6 and PMS2-associated LS,5–8 traditional clinical management guidelines per the National Comprehensive Cancer Network (NCCN) were universal, including recommendation for initiation of colonoscopic surveillance at age 20–25 and consideration of risk-reducing hysterectomy and bilateral salpingo-oophorectomy once childbearing is complete, irrespective of the causative MMR gene.9 However, recently updated NCCN guidelines10 for management of germline MSH6 and PMS2 P/LP heterozygotes have significantly changed due to accumulating evidence suggesting a more modest phenotype with later onset CRC and limited extra-colonic cancers compared to other MMR genes.5,8
A recent report from the Prospective Lynch Syndrome Database (PLSD) with 6350 participants and 51,646 follow-up years found decreased risk of cancers including CRC in MSH6 heterozygotes compared to MLH1 and MSH2 heterozygotes, with the exception of endometrial cancer where the risk was still elevated comparably. Notably, they found that PMS2 heterozygotes may not have increased risk of cancers in young to middle aged adults and that for older individuals the risk is still uncertain. They advocated that international management guidelines be updated to reflect these novel findings.5 Accordingly, many international guidelines have utilized these data and now provide gene-specific recommendations and delayed or reduced screening for MSH6/PMS2 heterozygotes.11–13 For example, the NCCN now recommend initiation of colonoscopy 10 years later (age 30–35) in MSH6 and PMS2-associated LS, and they also estimate the risk for OC among PMS2 heterozygotes to be similar to the general population.5,8,10 However, data are limited, and a comprehensive understanding of the risk spectrum and age of cancer onset is critical for cancer surveillance and risk-reduction.
We sought to characterize MSH6 and PMS2-associated cancers and age of diagnosis in those with MMR-D/MSI-H tumors, a hallmark of LS pan-cancer.4
Materials and Methods
Patient Selection
Patients were consented to an IRB-approved protocol of matched tumor/germline sequencing via MSK-IMPACT (ClinicalTrials.gov identifier, NCT01775072)14 or a prospective registry of LS patients at a single institution from 2/2005–1/2021. Query of the electronic medical record identified those with germline heterozygous MSH6 and PMS2 P/LP variants. Patients with variants of unknown significance (VUS) were excluded from this analysis as MSK-IMPACT does not report VUS findings. All germline genetic testing occurred in a commercial or in-house CLIA and New York State Department of Health (NYSDOH)-approved laboratory. Those confirmed to have constitutional mismatch repair deficiency (CMMR-D) were excluded.
Data Collection
Demographic and clinical information were abstracted from the medical record including sex, self-reported race/ethnicity, Ashkenazi Jewish (AJ) ancestry, and body mass index (BMI) at study enrollment. Smoking status was self-reported and defined as ever smoker, never smoker, or unknown. In cancer-affected patients, clinical data including age at cancer diagnosis, tumor type and histology were abstracted from the medical record (YL and AL).
MMR-D/MSI-H testing
Available tumors were evaluated for MMR-D/MSI-H status using either standard immunohistochemical (IHC) staining for MMR protein expression, PCR, or next-generation sequencing (NGS) based testing, with tumors considered positive if at least one method revealed an MMR-D or MSI-H phenotype. For tumors undergoing somatic testing via MSK-IMPACT, MSI assessment was conducted using MSIsensor, an NGS-based bioinformatics tool that incorporates data from more than 1,000 microsatellite regions, reporting the percentage of unstable loci as a cumulative score, with scores ≥10 considered MSI-H, scores <3 microsatellite stable (MSS), and scores 3 to <10 considered MSI-indeterminate (MSI-I). Any tumor found to be MSI-I was considered MMR-D if IHC analysis also demonstrated lack of MMR protein expression.15
Statistical Analysis
Demographic and clinical data were tabulated using summary statistics. In cancer-affected patients, the distribution of pooled tumor types, as several patients had multiple tumors, was assessed overall and for MMR-D/MSI-H tumors by gene (MSH6 or PMS2). Proportion of CRC, EC, and non-CRC/EC tumors were compared by gene (MSH6 vs. PMS2) and MMR-D/MSI-H status using non-parametric tests. Details of tumor histology were reported. Age at cancer diagnosis was analyzed by tumor type using summary statistics, overall, and in MMR-D/MSI-H tumors in the context of surveillance recommendations regarding age of initiation.
Results
Patient Characteristics
We identified 243 patients (147 females, 96 males) with germline P/LP MSH6/PMS2 variants (148 MSH6 and 95 PMS2), Table 1. Of the 243 patients, 133 underwent tumor-normal sequencing and were included in the MSK-IMPACT cohort. Although 132 patients were derived from the LS registry, 22 underwent tumor-normal sequencing and were included in the MSK-IMPACT cohort, so the LS registry cohort included the remaining 110 patients, Figure 1. Patients in the LS Registry cohort were more likely to be female compared to the MSK-IMPACT cohort (p=0.049), but there were no other significant differences between ascertainment groups, Supplementary Table 1. All patients in the MSK-IMPACT group were cancer-affected compared to 48% (53/110) of patients in the LS registry group. Among tumors that underwent MMR /MSI assessment, those from the LS registry cohort were more likely to be MMR-D/MSI-H compared to tumors from the MSK-IMPACT group, 94% (47/50) vs. 53% (75/142), p<0.001.
Table 1:
Patient Characteristics
| Variables | Overall (N=243) | MSH6 (N=148) | PMS2 (N=95) | p-valueb |
|---|---|---|---|---|
| Sex | ||||
| Male | 96 (40%) | 57 (39%) | 39 (41%) | 0.69 |
| Female | 147 (60%) | 91 (61%) | 56 (59%) | |
| Age at 1st cancer diagnosisa (median, range) | ||||
| Male | 53.5 (15–84) | 51.5 (15–84) | 54.5 (28–80) | 0.71 |
| Female | 53 (1–88) | 53 (27–88) | 51 (1–81) | 0.85 |
| Self-Reported Race/Ethnicity | ||||
| Non-Hispanic-White | 209 (86%) | 134 (90%) | 75 (79%) | 0.075c |
| Black | 14 (6%) | 5 (3%) | 9 (10%) | |
| Asian | 10 (4%) | 5 (3%) | 5 (5%) | |
| Other | 2 (1%) | 1 (1%) | 1 (1%) | |
| Hispanic | 5 (2%) | 1 (1%) | 4 (4%) | |
| Unknown | 3 (1%) | 2 (2%) | 1 (1%) | |
| Ashkenazi Jewish Ancestry | 42 (17%) | 38 (26%) | 4 (4%) | <0.001 |
| Body Mass Index (BMI) kg/m2 at study enrollment (median, range) | ||||
| Male | 26.7 (17.6–39.8) | 27.6 (17.6–39.8) | 26.0 (21.5–39.2) | 0.13 |
| Female | 25.3 (15–52.8) | 25.1 (18.4–52.8) | 25.7 (15–51.9) | 0.51 |
| Smoking Status | ||||
| Ever smoker | 67 (28%) | 41 (28%) | 26 (27%) | 0.502 |
| Never smoker | 142 (58%) | 80 (54%) | 62 (65%) | |
| Unknown/Missing | 34 (34%) | 27 (18%) | 7 (8%) | |
In cancer-affected patients only.
p-values are chi-squared/fisher’s exact or ranksum.
p=0.011 when comparing white to non-whites
Table depicts clinical characteristics for all patients overall and by gene (MSH6 and PMS2).
Figure 1: Patient Selection.
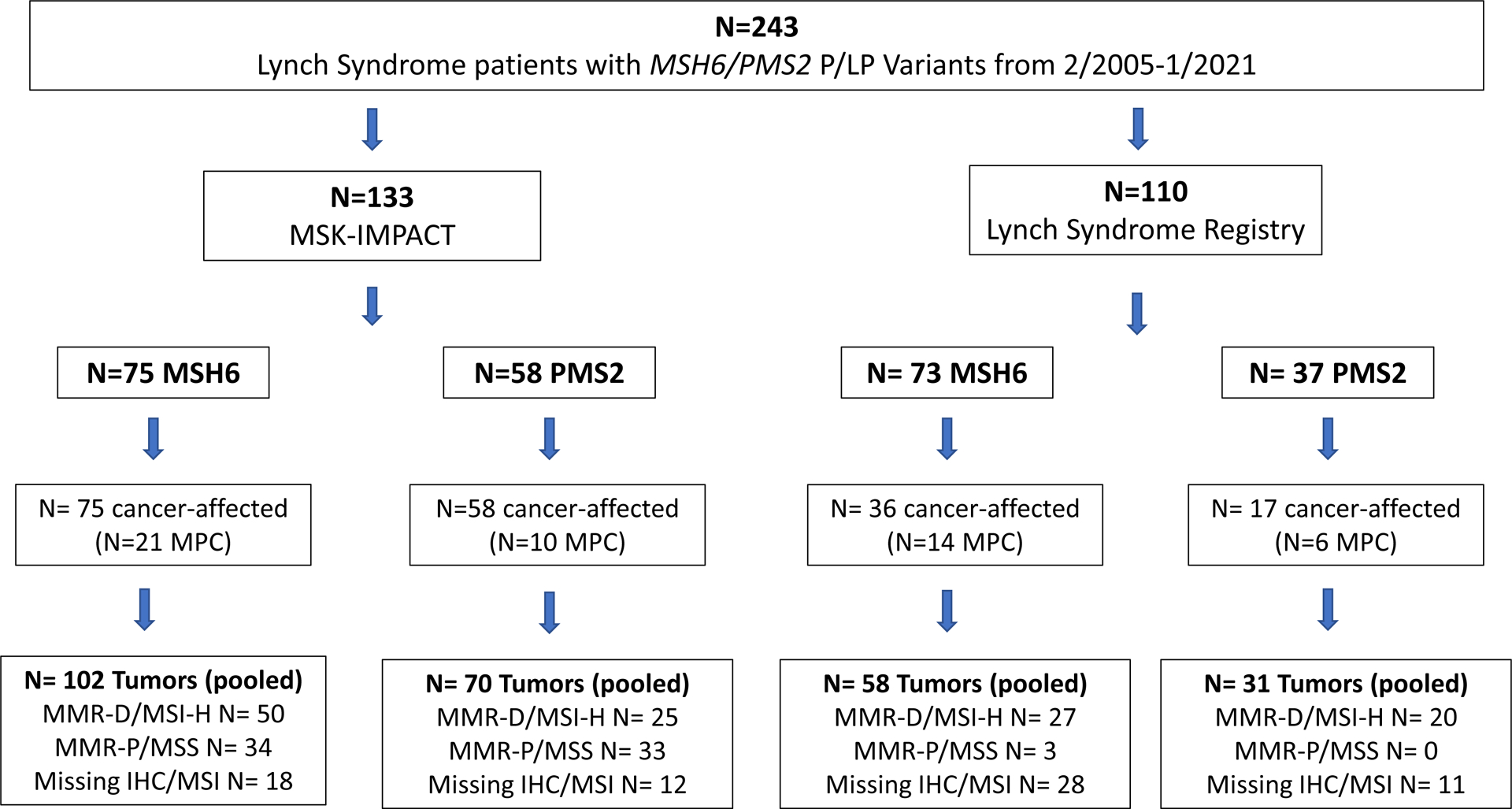
Figure depicts patients selection schema and demonstrates the number of cancer-affected and unaffected individuals as well as total pooled tumors and MMR-D/MSI status of tumors by ascertainment (MSK-IMPACT and LS registry cohort) and gene (MSH6 and PMS2)
Abbreviations: LS – Lynch Syndrome, MPC – Multiple primary cancer
In the 186 (77%), cancer-affected patients [MSH6 111/148 (75%); PMS2 75/95 (79%)], the median age at initial cancer diagnosis was 53 (range 1–88) in females and 53.5 (15–84) in males. Median BMI at enrollment was 25.3 (range 15–52.8) in females and 26.7 (range 17.6–39.8) in males. Most patients were Non-Hispanic White (86%) and never smokers (58%). Ashkenazi Jewish (AJ) ancestry was self-reported in 42/243 (17%) patients with enrichment of AJ patients in the MSH6 compared to PMS2 group (26% vs. 4%, p<0.001), mostly due to the presence of two known AJ founder variants [MSH6 c.3959_3962delCAAG (p.Ala1320Glufs*6) and MSH6 c.3984_3987dupGTCA (p.Leu1330Valfs*12)]. Notably, there were more Non-Hispanic White compared to Non-White patients in the MSH6 group compared to PMS2 group (90% vs 79%, p=0.011), and 9/95 (10%) of patients in the PMS2 group self-identified as Black race. There were no other significant differences between the MSH6 and PMS2 groups, Table 1.
Characteristics of Pooled Tumors in Cancer-affected Patients
In our cohort, 51/243 (21%) patients had multiple primary cancers, defined using International Agency for Research on Cancer (IARC) criteria,16 of which 35 (24%) were in MSH6 and 16 (17%) were in PMS2 (p=0.20). This resulted in 261 total pooled tumors, 160 in MSH6 and 101 in PMS2, with the majority occurring in the MSK-IMPACT cohort (172/216, 66%). Of the 192 tumors with molecular assessment, 122 (64%) were MMR-D/MSI-H, 77 in the MSH6 and 45 in the PMS2 group, Figure 1. Synchronous tumors, defined as cancers with the same age at diagnosis, were found in 10 patients, including 7 in MSH6 and 3 in PMS2.
Overall, CRC and EC combined comprised 55% of all cancers in the MSH6 group and 43% of all cancers in the PMS2 group, Table 2, with PMS2 more likely than MSH6 to have non-CRC/EC tumors, p=0.021. CRC and EC were also more likely to be MMR-D/MSI-H (82% and 81% respectively) compared to other cancers (38%) (p<0.001) which was consistent for both groups (p<0.001 for MSH6, and p<0.001 for PMS2).
Table 2:
Cancer Spectrum in Lynch Syndrome Patients with P/LP Variants in MSH6 and PMS2
| MSH6 | PMS2 | |||
|---|---|---|---|---|
| Cancer | Overall (N=160) | MMR-D/MSI-H (N=77) | Overall (N=101) | MMR-D/MSI-H (N=45) |
| Colorectal (CRC) | 47 (29%) | 33 (43%) | 31 (31%) | 23 (51%) |
| Endometrial (EC) | 41 (26%) | 25 (32%) | 12 (12%) | 10 (23%) |
| Breast | 13 (8%) | 1 (1%) | 13 (13%) | 0 |
| Prostate | 11 (7%) | 1 (1%) | 3 (3%) | 1 (2%) |
| Urothelial (UC) | 5 (3%) | 4 (5%) | 2 (2%) | 1 (2%) |
| Pancreas/Biliary (PB) | 5 (3%) | 3 (4%) | 7 (7%) | 1 (2%) |
| Sarcoma | 2 (1%) | 0 | 0 | 0 |
| Gastric/Esophageal (GEJ) | 5 (3%) | 3 (4%) | 1 (1%) | 0 |
| Ovary (OC) | 5 (3%) | 2 (3%) | 12 (12%) | 4 (9%) |
| Lymphoma | 3 (2%) | 0 | 0 | 0 |
| Melanoma | 4 (3%) | 0 | 0 | 0 |
| CNS/Brain | 1 (1%) | 1 (1%) | 4 (4%) | 0 |
| Small Bowel (SB) | 1 (1%) | 1 (1%) | 5 (5%) | 5 (11%) |
| Skin Tumors, Non-melanoma | 6 (4%) | 3 (4%) | 5 (5%) | 0 |
| Kidney | 2 (1%) | 0 | 1 (1%) | 0 |
| Thyroid | 2 (1%) | 0 | 0 | 0 |
| Testicular/Germ Cell | 1 (1%) | 0 | 1 (1%) | 0 |
| Carcinoma of Unknown Primary | 0 | 0 | 1 (1%) | 0 |
| Lung | 2 (1%) | 0 | 0 | 0 |
| Other | 4 (3%) | 0 | 3 (3%) | 0 |
Table depicts cancer types and distribution for MSH6 and PMS2 heterozygotes, both overall and for MMR-D/MSI-H tumors.
Among MMR-D/MSI-H tumors (n=122), 75% were comprised of CRC (n=56) and EC (n=35), with the remaining 25% comprised of 9 different cancer types: small bowel (adenocarcinoma-SBA, n=6), ovarian (OC, n=6), urothelial (n=5), pancreas/biliary (n=4), gastric/esophageal (n=3), non-melanoma skin tumors (n=3), prostate (n=2), breast (n=1), and CNS/brain (n=1). This distribution was similar between the MSH6 and PMS2 groups with slight differences in the MSH6 group being enriched in urothelial, pancreas/biliary, and gastric/esophageal cancers while the PMS2 group was enriched in small bowel and ovarian cancers, Table 2.
Among EC, the most common histology was endometrioid for both MSH6 and PMS2, overall, and for MMR-D/MSI-H tumors, Supplementary Table 2. Notably, there were 6 SBAs (5 in the PMS2 group, 1 in the MSH6 group), and all were MMR-D/MSI-H. There were 17 OCs (12 in PMS2, 5 in MSH6), and of the 15 that underwent molecular assessment, 6 (40%) were MMR-D/MSI-H (4 PMS2, 2 MSH6), Table 2. MMR-D/MSI-H OC histologies included clear cell (n=1) and carcinosarcoma (n=1) in the MSH6 group and endometrioid (n=2), poorly differentiated (n=1), and high grade serous (n=1) in the PMS2 group. The high-grade serous OC was reviewed by an expert gynecologic pathologist (RS) who concurred with the diagnosis and confirmed the loss of PMS2 on IHC and MSI-H status via MSIsensor. Of MMR-D/MSI-H non-melanoma skin tumors, all 3 were sebaceous adenomas, diagnostic of the Muir-Torre variant of LS17,18 Supplementary Table 2.
Age at Cancer Diagnosis for LS-associated Tumors
Among MMR-D/MSI-H CRC and EC, the median age at diagnosis was 51.5 (range 27–80) and 55 (range 39–74) respectively. Although less frequently observed, age at diagnosis of other LS-associated cancers with risk-reducing and/or surveillance measures (ovarian, gastric/esophageal, pancreas/biliary, urothelial and small bowel cancers) also exhibited a wide range, with the youngest patients with MMR-D/MSI-H SBA (PMS2 group) and EC (MSH6 group) being diagnosed at age <40, Figure 2/Supplementary Table 3. Among MMR-D/MSI-H CRC, 9/56 (16%) occurring in 7 patients (4 in MSH6, 3 in PMS2) were diagnosed under age 35, the suggested upper threshold for initiation of colonoscopy in MSH6 and PMS2-associated LS per revised guidelines, Figure 3. Notably, in 2 patients (1 MSH6 and 1 PMS2), the CRC was diagnosed at age <30, which is before the threshold for initiation of colonoscopy according to most guidelines.
Figure 2: Distribution of Age at Cancer Diagnosis: Overall and MMR-D/MSI-H.
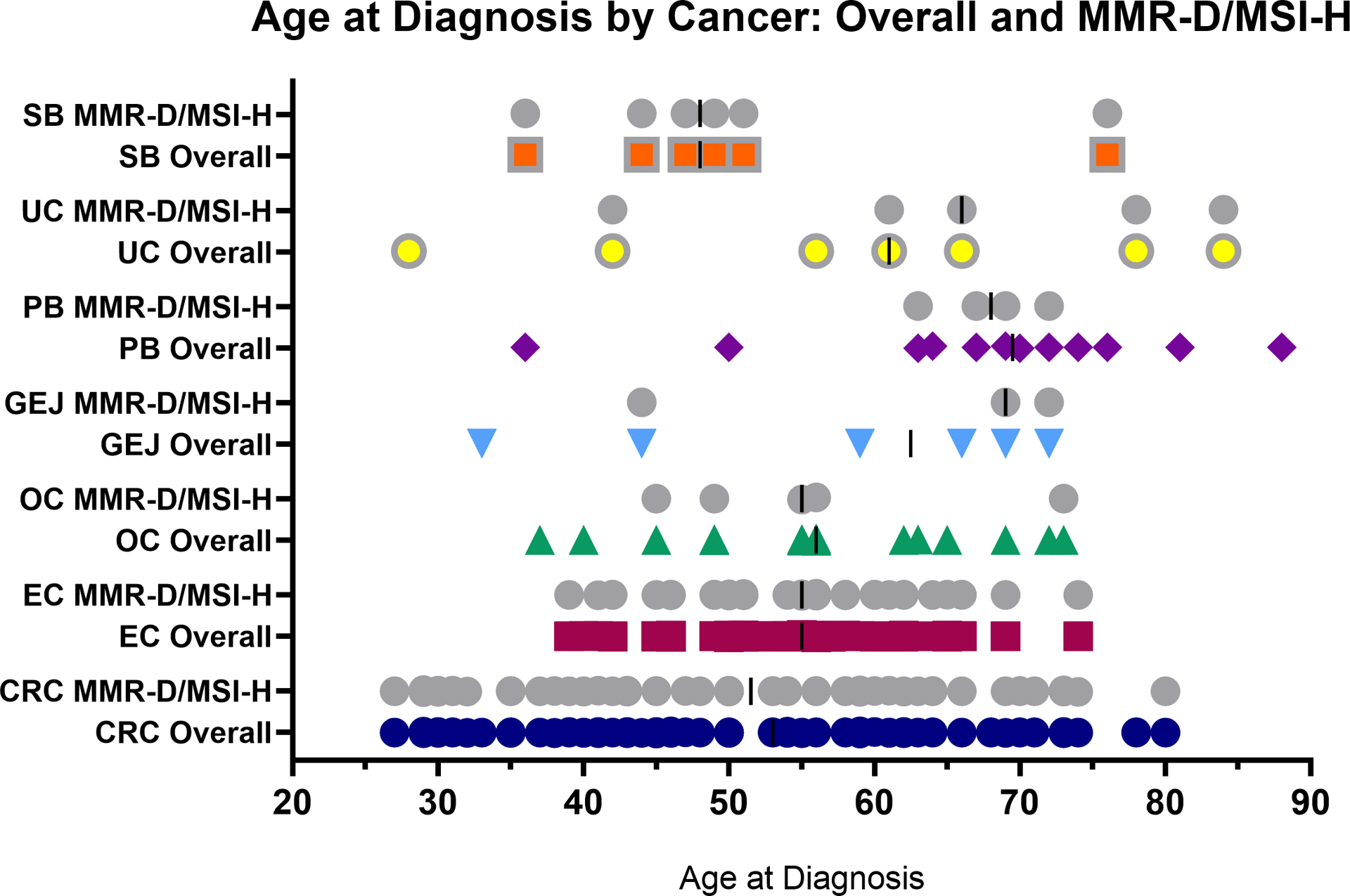
Figure depicts the distribution and median (dark bar) age at cancer diagnosis for individual tumors, overall (colors), which includes all tumors (MMR-D/MSI-H, MMR-P/MSS, and untested/unknown tumors), and for MMR-D/MSI-H tumors (gray) for all Lynch-associated cancers with screening protocols in patients with P/LP variants in MSH6 or PMS2.
Abbreviations: SB – small bowel, UC – urothelial, PB – pancreas/biliary, GEJ – Gastric/esophageal, OC – ovarian cancer, EC – endometrial cancer, CRC – colorectal cancer
Figure 3: Distribution of Age at Cancer Diagnosis in Colorectal Cancer: Overall and MMR-D/MSI-H.
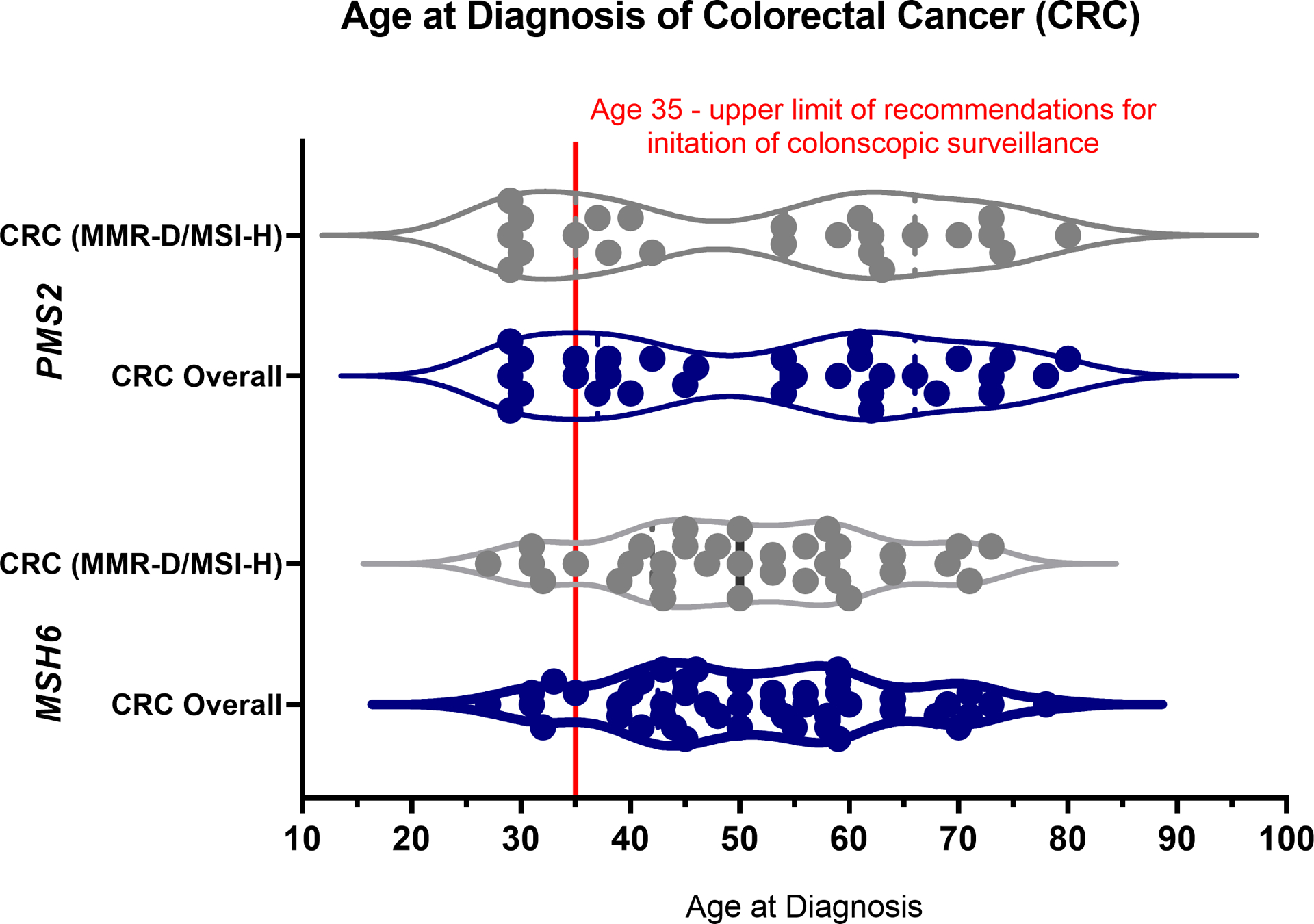
Figure depicts the age at diagnosis for individual colorectal cancers specifically, stratified by gene group (MSH6 vs. PMS2), overall (blue), which includes all tumors (MMR-D/MSI-H, MMR-P/MSS, and untested/unknown tumors), and for MMR-D/MSI-H tumors (gray) in patients with P/LP variants in MSH6 or PMS2. The red line depicts age = 35, the upper limit of guideline recommendations for initiation of surveillance colonoscopies.
For such patients, we considered the possibility of autosomal recessive constitutional mismatch repair deficiency (CMMR-D) syndrome, and manually reviewed patient clinical and pathology records for additional details and relevant diagnostic work-up, Supplementary Table 4. None of the patients met clinical diagnostic criteria for CMMR-D. Among these seven patients, four underwent tumor molecular profiling in addition to IHC analysis. While all tumors demonstrated MSI-H status and high tumor mutational burden, the majority of tumors did not demonstrate the ultrahypermutated (>150 somatic variants) phenotype that is classically associated with CMMR-D,19,20 with the one ultrahypermutated tumor being a CRC diagnosed in a 31-year-old MSH6 P/LP variant cancer. In addition to somatic MMR gene variants, the tumor molecular profile identified three variants in POLD1 and four variants in POLE, which have been well-described as somatic causes of the ultrahypermutated phenotype.21 This patient underwent both commercial germline genetic testing of 80 cancer susceptibility genes and MSK-IMPACT research-based testing assessing 88 genes, with no additional variant of either clinical or uncertain significance identified in MSH6, PMS2, MLH1, MSH2, EPCAM, MSH3, POLD1, or POLE.
Notably, there was one patient in the PMS2 group with three primary synchronous CRC’s diagnosed at age 29. All CRC’s demonstrated PMS2 absence on IHC, with retained expression of MLH1, MSH2, and MSH6. This patient had extensive clinical-grade genetic testing and was found to be negative for any additional variants of either clinical or uncertain significance in PMS2, MLH1, MSH2, MSH6, TP53, APC, or MUTYH. While the patient also had history of a high-grade urothelial carcinoma and myxopapillary ependymoma at age 28, he did not meet proposed clinical diagnostic criteria for CMMR-D, scoring a zero on the C4CMMRD algorithm for clinical diagnostic assessment of CMMR-D.22,23
Discussion
Despite being lower penetrance LS-associated genes, patients with MSH6/PMS2 P/LP variants remained at risk for a broad-spectrum of cancers and early-onset CRC. While the majority of MMR-D/MSI-H cancers were CRC and EC, 25% of MMR-D/MSI-H tumors were comprised of non-CRC/EC tumors, notably ovarian and small bowel cancers. While the age at initial cancer diagnosis varied for different cancer types, many cancers, particularly CRC, were diagnosed prior to guideline recommended ages to initiate surveillance protocols in MSH6/PMS2-associated LS.
Although there was a wide range in age at diagnosis (27–80), 16% of MMR-D/MSI-H CRC’s presented prior to the upper threshold of initiation of colonoscopic surveillance per revised guidelines.10,12,13 Interestingly, this early-onset CRC group consisted of an equal distribution between the MSH6 and PMS2 groups. This is important to highlight, as colonoscopy is the only proven effective surveillance modality for LS that may also be preventive, given the removal of precursor lesions (e.g., adenomatous polyps) at time of procedure.24,25 As our study only assessed patients affected with CRC rather than precancerous polyps, presumably these patients would have benefited from earlier colonoscopic surveillance. Future studies assessing colonoscopy surveillance and pathologic findings in cancer-unaffected patients will better inform age of initiation of colonoscopy among MSH6 and PMS2 heterozygotes.
In contrast, no EC’s were diagnosed prior to age 35, the upper limit of NCCN guideline considerations regarding screening endometrial biopsies.10 This also further emphasizes that risk-reducing hysterectomy, with or without bilateral salpingo-oophorectomy (BSO), should not be performed before age 35 based on P/LP variants alone. Most MMR-D/MSI-H EC’s were of endometrioid or clear cell histology as previously reported;26 however, we also observed one leiomyosarcoma.
Notably, we identified 17 ovarian cancers (5 in MSH6, 12 in PMS2) among 147 female patients. Unfortunately, only 15 had MMR/MSI analysis completed, resulting in the identification of 6 (2 in MSH6, 4 in PMS2) MMR-D/MSI-H OC’s. Of the 12/17 OC cases where tumor-normal sequencing data were available, 5 tumors exhibited LOH or a 2nd hit (3 PMS2 group, 2 MSH6 group). Of these, 3 were MMR-D/MSI-H (2 PMS2, 1 MSH6). These data are limited by sample size and tumor purity. Unfortunately, family history data were also limited, but of the 16/17 patients with some data available, 5/16 (31%) had a family history of a GYN cancer. Of note, one patient in the PMS2 group had two primary ovarian cancers, the first presenting as an early stage MMR-D endometrioid ovarian cancer that was synchronous with her endometrial cancer (tumor on biopsy specimen only) in the setting of endometriosis, and the second presented as a pelvic mass 6 years later as a poorly differentiated carcinoma of GYN origin that was PMS2 absent on IHC, emphasizing the distinct nature of endometriosis-associated ovarian/peritoneal cancers in LS.27 The remaining two patients were without any additional tumor tissue available for further MMR/MSI testing. As such, LS-associated OC in this cohort may, in fact, be an underestimation. Although the median age of OC diagnosis overall was 56, 6 OCs (2 MSH6 and 4 PMS2) were diagnosed in pre-menopausal females (age <50). Further studies are needed to understand the underlying OC incidence in these patients, particularly those with PMS2 heterozygotes given the recent changes to NCCN guidelines pertaining to risk-reducing BSO no longer being recommended universally.28 Although most MMR-D/MSI-H OC’s were of endometrioid or clear cell histology, there was one high-grade serous OC and one carcinosarcoma, findings that should be verified in larger studies.
Although gastric cancers were rare and mostly diagnosed at age >40 (NCCN recommended age for initiation of endoscopic surveillance), one gastric cancer in a PMS2 heterozygote was diagnosed at age 33. Similarly, although most urothelial cancers were diagnosed at age >30 (NCCN recommended age to initiate urinalysis surveillance), one MMR-D/MSI-H urothelial cancer in a PMS2 heterozygote was diagnosed at age 28. Interestingly, we found 6 small bowel cancers, all MMR-D/MSI-H, with 83% (5/6) being in the PMS2 group. While it has been reported that small bowel cancer is a rare tumor type within the LS-spectrum,10,29,30 data are limited, with higher prevalence seemingly in MLH1 and MSH2-associated LS.29 As such, this may be a phenotype that needs more awareness and study. Skin tumors that exhibited an MMR-D/MSI-H phenotype were sebaceous adenomas, diagnostic of the Muir-Torre syndrome variant of LS.
PMS2 heterozygotes had a higher proportion of non-CRC/EC cancers compared to MSH6 heterozygotes overall, although rates were similar for MMR-D/MSI-H cancers, likely suggesting incidental, sporadic cancers not associated with LS in the PMS2 group among mismatch repair proficient (MMR-P)/MSS cases. Of note, there were no differences in BMI or smoking status, traditional exposures related to cancer, between the MSH6 and PMS2 groups, although other environmental factors not captured in this study may still modify risk and should be studied in larger, epidemiological studies. MMR-D/MSI-H OC’s and small bowel cancers were also enriched in PMS2, a finding that should be verified in larger studies, given potential implications for surveillance and risk-reduction.
Among the 16 LS patients with two or more P/LP germline variants (Supplementary Table 5), there were 3 patients in which the 2nd germline finding more likely contributed to cancer development. One patient had an MSS pancreatic cancer and a P/LP BRCA2 germline variant in addition to their PMS2 germline variant. Another patient had an MMR-P/MSS ovarian cancer with a P/LP RAD51D germline variant in addition to their PMS2 germline variant. Finally, one other patient had an MMR-P/MSS CRC and a P/LP CHEK2 germline variant in addition to their MSH6 germline variant. This suggests that some cancers may not be related to underlying LS but rather to other inherited cancer predisposition syndromes in a minority of LS patients. Of note, the FH c.1431_1433dupAAA (p.Lys477dup) variant identified in 2 of our patients has not been associated with renal cell carcinoma.31
There were several limitations to our study. First, this was a retrospective study from a single tertiary cancer center, limiting its generalizability. The two ascertainment cohorts, one a LS registry from a genetics clinic and the other a tumor-normal sequencing cohort of cancer-affected patients, may have also influenced the cancer spectrum and ages at initial cancer diagnosis. Additionally, assessments of family history, which may modify cancer risk, were limited and not standardized. Although inconclusive cases on IHC underwent strict pathology review, data on MMR/MSI status were not available for all tumors, which limited our ability to define causality in our LS patients. As such, our LS-associated cancers may be underestimations. However, our study adds important insights in light of recent MSH6 and PMS2-associated LS clinical management guidelines, which were revised based on evidence supporting the reduced penetrance compared to MLH1 and MSH2-associated LS.10 While we agree and acknowledge the importance of weighing the risks and benefits for invasive procedures, we also note that the vast majority of such data was extrapolated from overlapping studies, mostly European,5,8,32 which may not reflect the same risk estimate in the United States (US) population.
Notably, 14% of our overall cohort self-identified as Non-White, reflecting a diversity not seen in previous, mostly European cohorts, and there may be differences in rates of LS and tumor phenotype amongst ancestry groups33,34 that could potentially contribute to health disparities. In particular, disparities in EC outcomes by self-reported race have been well described, and Black women have worse survival compared to White women,35 a trend that is projected to worsen over time.36,37 Although we saw differences in self-reported race/ethnicity by gene group with the MSH6 cohort being enriched in Non-Hispanic White patients compared to the PMS2 group, this is likely confounded by the higher prevalence of those with AJ ancestry in the MSH6 group. Additionally, the International Mismatch Repair Consortium (IMRC) recently reported variations in CRC risk for patients with LS by geographical location (Europe, North America and Australia),38 and future studies should further investigate differences in gene penetrance and cancer risks by geography and ancestry with consideration of environmental factors such as obesity, smoking, and other lifestyle characteristics.
While our study is primarily descriptive in nature, it provides important information on the breadth of cancers observed in MSH6/PMS2-associated LS and describes tumor phenotype, including MMR-D/MSI-H status, and age of diagnosis. This forms the foundation for additional collaborative research in this area, which may better inform future updates to clinical management guidelines. Larger, prospective studies are needed to validate these findings, and individualized risk-assessment may be needed for surveillance recommendations.
Supplementary Material
Acknowledgements:
We would like to thank all the patients and their families who participated in these studies. This work was supported by the Romeo Milio Lynch Syndrome Foundation, Fieldstone Family Fund, and the Robert and Kate Niehaus Center for Inherited Cancer Genomics. MSKCC is supported by the NCI Core grant P30 CA008748.
Footnotes
Ethics Declaration: Patients were consented to an IRB-approved protocol of matched tumor/germline sequencing via MSK-IMPACT (ClinicalTrials.gov identifier, NCT01775072) or a prospective registry of LS patients at a single institution from 2/2005–1/2021. This work was approved by MSK IRB under protocol numbers 12–245 and 04–144.
Data Availability:
The full deidentified clinical dataset is available upon request to the corresponding author.
References:
- 1.de la Chapelle A The incidence of Lynch syndrome. Fam Cancer. 2005;4(3):233–237. [DOI] [PubMed] [Google Scholar]
- 2.Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med. 2005;352(18):1851–1860. [DOI] [PubMed] [Google Scholar]
- 3.Hampel H, Frankel W, Panescu J, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66(15):7810–7817. [DOI] [PubMed] [Google Scholar]
- 4.Latham A, Srinivasan P, Kemel Y, et al. Microsatellite Instability Is Associated With the Presence of Lynch Syndrome Pan-Cancer. J Clin Oncol. 2019;37(4):286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dominguez-Valentin M, Sampson JR, Seppala TT, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genetics in medicine : official journal of the American College of Medical Genetics. 2020;22(1):15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Møller P, Seppälä T, Bernstein I, et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut. 2017;66(3):464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Møller P, Seppälä TT, Bernstein I, et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the Prospective Lynch Syndrome Database. Gut. 2018;67(7):1306–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.ten Broeke SW, Brohet RM, Tops CM, et al. Lynch syndrome caused by germline PMS2 mutations: delineating the cancer risk. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33(4):319–325. [DOI] [PubMed] [Google Scholar]
- 9.Gupta S, Provenzale D, Llor X, et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Colorectal, Version 2.2019. J Natl Compr Canc Netw. 2019;17(9):1032–1041. [DOI] [PubMed] [Google Scholar]
- 10.(NCCN) NCCN, https://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf. Accessed 10/24/2020.
- 11.Crosbie EJ, Ryan NAJ, Arends MJ, et al. The Manchester International Consensus Group recommendations for the management of gynecological cancers in Lynch syndrome. Genetics in medicine : official journal of the American College of Medical Genetics. 2019;21(10):2390–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monahan KJ, Bradshaw N, Dolwani S, et al. Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut. 2020;69(3):411–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seppälä TT, Latchford A, Negoi I, et al. European guidelines from the EHTG and ESCP for Lynch syndrome: an updated third edition of the Mallorca guidelines based on gene and gender. Br J Surg. 2021;108(5):484–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng DT, Prasad M, Chekaluk Y, et al. Comprehensive detection of germline variants by MSK-IMPACT, a clinical diagnostic platform for solid tumor molecular oncology and concurrent cancer predisposition testing. BMC medical genomics. 2017;10(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niu B, Ye K, Zhang Q, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics. 2014;30(7):1015–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.International Agency for Reserach on Cancer W. International Rules for for Multiple Primary Cancers. 2004.
- 17.Kruse R, Rütten A, Lamberti C, et al. Muir-Torre phenotype has a frequency of DNA mismatch-repair-gene mutations similar to that in hereditary nonpolyposis colorectal cancer families defined by the Amsterdam criteria. Am J Hum Genet. 1998;63(1):63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suspiro A, Fidalgo P, Cravo M, et al. The Muir-Torre syndrome: a rare variant of hereditary nonpolyposis colorectal cancer associated with hMSH2 mutation. Am J Gastroenterol. 1998;93(9):1572–1574. [DOI] [PubMed] [Google Scholar]
- 19.Shlien A, Campbell BB, de Borja R, et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat Genet. 2015;47(3):257–262. [DOI] [PubMed] [Google Scholar]
- 20.Tabori U, Hansford JR, Achatz MI, et al. Clinical Management and Tumor Surveillance Recommendations of Inherited Mismatch Repair Deficiency in Childhood. Clin Cancer Res. 2017;23(11):e32–e37. [DOI] [PubMed] [Google Scholar]
- 21.Muzny DM, Bainbridge MN, Chang K, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wimmer K, Kratz CP, Vasen HFA, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium ‘Care for CMMRD’ (C4CMMRD). Journal of Medical Genetics. 2014;51(6):355–365. [DOI] [PubMed] [Google Scholar]
- 23.Aronson M, Colas C, Shuen A, et al. Diagnostic criteria for constitutional mismatch repair deficiency (CMMRD): recommendations from the international consensus working group. Journal of Medical Genetics. 2021:jmedgenet-2020–107627. [DOI] [PubMed] [Google Scholar]
- 24.Rubenstein JH, Enns R, Heidelbaugh J, Barkun A. American Gastroenterological Association Institute Guideline on the Diagnosis and Management of Lynch Syndrome. Gastroenterology. 2015;149(3):777–782; quiz e716–777. [DOI] [PubMed] [Google Scholar]
- 25.Vasen HF, Abdirahman M, Brohet R, et al. One to 2-year surveillance intervals reduce risk of colorectal cancer in families with Lynch syndrome. Gastroenterology. 2010;138(7):2300–2306. [DOI] [PubMed] [Google Scholar]
- 26.Gordhandas S, Kahn RM, Gamble C, et al. Clinicopathologic features of endometrial cancer with mismatch repair deficiency. Ecancermedicalscience. 2020;14:1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hallas-Potts A, Dawson JC, Herrington CS. Ovarian cancer cell lines derived from non-serous carcinomas migrate and invade more aggressively than those derived from high-grade serous carcinomas. Scientific reports. 2019;9(1):5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu YL, Breen K, Catchings A, et al. Risk-Reducing Bilateral Salpingo-Oophorectomy for Ovarian Cancer: A Review and Clinical Guide for Hereditary Predisposition Genes. JCO Oncol Pract. 2021:Op2100382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Latham A, Shia J, Patel Z, et al. Characterization and Clinical Outcomes of DNA Mismatch Repair-deficient Small Bowel Adenocarcinoma. Clin Cancer Res. 2021;27(5):1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benson AB, Venook AP, Al-Hawary MM, et al. Small Bowel Adenocarcinoma, Version 1.2020, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2019;17(9):1109–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang L, Walsh MF, Jairam S, et al. Fumarate hydratase FH c.1431_1433dupAAA (p.Lys477dup) variant is not associated with cancer including renal cell carcinoma. Human mutation. 2020;41(1):103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Møller P, Seppälä T, Bernstein I, et al. Incidence of and survival after subsequent cancers in carriers of pathogenic MMR variants with previous cancer: a report from the prospective Lynch syndrome database. Gut. 2017;66(9):1657–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenblum RE, Ang C, Suckiel SA, et al. Lynch Syndrome-Associated Variants and Cancer Rates in an Ancestrally Diverse Biobank. JCO Precis Oncol. 2020;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu YL, Maio A, Kemel Y, et al. Disparities in pan-cancer patients undergoing germline cancer risk assessment by self-reported race/ethnicity and ancestry. Journal of Clinical Oncology. 2021;39(15_suppl):10508–10508. [Google Scholar]
- 35.Cote ML, Ruterbusch JJ, Olson SH, Lu K, Ali-Fehmi R. The Growing Burden of Endometrial Cancer: A Major Racial Disparity Affecting Black Women. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2015;24(9):1407–1415. [DOI] [PubMed] [Google Scholar]
- 36.Gaber C, Meza R, Ruterbusch JJ, Cote ML. Endometrial Cancer Trends by Race and Histology in the USA: Projecting the Number of New Cases from 2015 to 2040. J Racial Ethn Health Disparities. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giaquinto AN, Broaddus RR, Jemal A, Siegel RL. The Changing Landscape of Gynecologic Cancer Mortality in the United States. Obstetrics & Gynecology. 9900: 10.1097/AOG.0000000000004676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Variation in the risk of colorectal cancer in families with Lynch syndrome: a retrospective cohort study. The Lancet Oncology. 2021;22(7):1014–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The full deidentified clinical dataset is available upon request to the corresponding author.