

Abstract
Glioblastoma is a highly aggressive brain tumor with limited treatment options. Several major challenges have limited the development of novel therapeutics, including the extensive heterogeneity of tumor cell states within each glioblastoma and the ability of glioma cells to diffusely infiltrate into neighboring healthy brain tissue, including the contralateral hemisphere. A T cell-mediated immune response could deal with these challenges based on the ability of polyclonal T cell populations to recognize diverse tumor antigens and perform surveillance throughout tissues. Here we will discuss the major pathways that inhibit T cell-mediated immunity against glioblastoma, with an emphasis on receptor–ligand systems by which glioma cells and recruited myeloid cells inhibit T cell function. A related challenge is that glioblastomas tend to be poorly infiltrated by T cells, which is not only caused by inhibitory molecular pathways but also currently utilized drugs, in particular high-dose corticosteroids that kill activated, proliferating T cells. We will discuss innovative approaches to induce glioblastoma-directed T cell responses, including neoantigen-based vaccines and sophisticated CAR T cell approaches that can target heterogeneous glioblastoma cell populations. Finally, we will propose a conceptual framework for the future development of T cell-based immunotherapies for glioblastoma.
Keywords: Glioblastoma, T cell-mediated immunity, Immunosuppressive pathways, Immunotherapy
What are the major challenges for the development of T cell-based immunotherapies for glioblastoma?
Heterogeneity and plasticity of glioblastoma cells
In many human solid tumors, heterogeneity is a significant challenge because a drug-resistant subpopulation can cause disease progression. Tumor cell heterogeneity is particularly striking in glioblastoma and very likely one of the major reasons why many therapeutic approaches have failed thus far. Recent single cell studies have highlighted the diversity of cellular states in glioblastoma: four major tumor cell states have been identified, three of which are reminiscent of neurodevelopmental programs, including neural progenitor-like cells (NPC), oligodendrocyte progenitor-like cells (OPC) and astrocyte-like cells (AC). A fourth cellular state, mesenchymal-like state (MES) is related to the interaction of glioma cells with immune cells, specifically macrophages and microglia [1, 2]. It is possible that additional cellular states in glioblastoma remain to be discovered. These four cellular states are found in each glioblastoma tumor, although at different cellular ratios, explaining previous classifications of glioblastomas into subtypes in which a particular state dominates, such as the mesenchymal state. It is important to note that these four states do not simply represent a linear cellular hierarchy, as frequently invoked in tumor stem cell models. Rather, these cellular states are continuous and proliferation is observed in all four states [1]. Most importantly, glioma cells are characterized by significant cellular plasticity: when cells of a particular state are isolated and implanted into the CNS of immunodeficient mice, resulting tumors re-emerge with the diversity of cellular states observed in the original tumor [1, 3, 4]. This diversity of cellular states and the inherent plasticity of glioma cells represent a major barrier for the development of small molecule drugs that target a particular oncogenic signaling pathway. Every therapeutic strategy will need to take the diversity and plasticity of glioma cells into account, including immunotherapies.
Diffuse infiltration of tumor cells into neighboring brain tissue
Surgery and radiation therapy cure many patients with solid tumors unless the tumor has spread to draining lymph nodes or distant organs. Although extracranial metastases occur very rarely in glioblastoma patients [5], glioma cells diffusely infiltrate into the neighboring healthy brain regions and can even cross through the corpus callosum and other structures into the contralateral hemisphere. Past surgical experience demonstrated that even removal of an entire brain hemisphere is not curative [6, 7]. Nevertheless, surgical removal of the main tumor mass remains a central aspect of the clinical management of patients with glioblastoma and the extent of resection is positively correlated with overall survival of patients [8–11]. Surgery also creates a time window for adjuvant therapies, including immunotherapy because overall survival is only 3 months without therapy [12, 13]. The diffuse infiltration of glioblastoma is also relevant for the application of immunotherapies: while the blood–brain barrier is frequently compromised in the main tumor mass (which can be identified by contrast enhanced regions on the T1 + contrast weighted preoperative MRI), allowing entry of therapeutic antibodies, the blood–brain barrier may be intact in distal brain areas (which can be identified by hyperintense signals in the T2 weighted or FLAIR sequence of the preoperative MRI) infiltrated by small numbers of glioma cells [14].
Defects in T cell priming, limited T cell infiltration and an immunosuppressive microenvironment
Most glioblastomas are poorly infiltrated by CD8 and CD4 T cells and instead highly infiltrated by immunosuppressive macrophages and microglia. Poor T cell infiltration into GBM may be caused by the immunosuppressive microenvironment and inefficient recruitment/activation of dendritic cells [15]. Inefficient T cell priming could be addressed therapeutically with cancer vaccines or CAR T cell therapies, as discussed in detail below. Also, lymphopenia due to T cell sequestration in the bone marrow has been observed in treatment naïve GBM patients and murine glioma models; such sequestration is observed following inhibition of the S1P1 receptor required for T cell recirculation from the bone marrow and lymphoid organs [16]. Nevertheless, a recent single cell RNA-seq analysis of glioblastoma-infiltrating T cells demonstrated a striking degree of clonal expansion by CD8 T cells which may be caused by T cell recognition of tumor antigens [17]. We will therefore discuss the major known immunosuppressive pathways in GBM and highlight potential opportunities for therapeutic intervention.
Depletion of T cells by currently used therapeutics, in particular high-dose corticosteroids
The diffusely infiltrative nature of glioblastomas can result in significant brain edema, and high-dose corticosteroids (such as dexamethasone) are the most widely utilized drug to deal with this serious issue [18]. From a clinical point of view, reducing brain edema with dexamethasone has significant advantages: 1) it reduces the intracranial pressure and facilitates surgical resection; 2) it reduces neurological deficits. However, high-dose corticosteroids efficiently induce apoptosis of activated and proliferating T cells [18]. A recent study analyzed T cell infiltration in glioblastomas from patients who did or did not require pre-surgical dexamethasone. This analysis demonstrated that pre-surgical dexamethasone resulted in a striking depletion of tumor-infiltrating T cells, affecting both CD8 and CD4 T cell populations (4.14-fold for all T cells, 7.72-fold for CD4 T cells) [17]. Continued treatment with corticosteroids can also be required after surgery or later at disease recurrence. Depletion of T cells by such drugs may be a major factor explaining why immunotherapies (such as PD-1 blocking mAbs) have not yet been successful in this challenging cancer type. Also, standard adjuvant therapy for glioblastoma, including radiation and concomitant chemotherapy with temozolomide, is known to induce lymphopenia and may also impair anti-tumor T cell function [19, 20]. It may be feasible to reduce the dose of corticosteroids by using bevacizumab, a VEGF blocking mAb, based on a number of studies which demonstrated that it can be successfully used to treat edema in patients with brain metastases [21, 22]. It has also been reported that bevacizumab can be used as a steroid-sparing agent in patients with brain metastases in an effort to improve the efficacy of immunotherapy [23].
Why should the field continue to consider immunotherapies for glioblastoma despite the failure of PD-1 directed antibodies to prolong patient survival?
It is legitimate to ask why the field should continue to develop immunotherapy options for glioblastoma despite the failure of PD-1 blockade to induce a survival benefit in a phase 3 clinical trial [24]. Given the extensive heterogeneity of glioma cells at a single cell level and their inherent plasticity, it has proven challenging to develop small molecules inhibitors for glioblastoma because inhibition of a single (or possibly even two) oncogenic signaling pathways may not be sufficient to induce apoptosis of all tumor cells. Polyclonal T cell populations that target a diverse set of antigens, including neoantigens and shared tumor antigens, could at least in theory control a heterogeneous tumor cell population, and phase 1 clinical trials have been performed to test this concept, as will be discussed in detail below. Also, in a challenging cancer type in which the tumor cells continue to evolve, it could be advantageous for the therapeutic response to co-evolve. For example, induction of T cell-mediated immunity results in clonal expansion and recruitment of additional immune effector cell populations. Finally, T cells continuously migrate through tissues and could therefore target residual tumor cells, as will be discussed further in the context of novel CAR T cell therapies.
Which unique aspects of immune function within the brain are relevant for immunotherapy of glioblastoma?
Immune responses in the CNS are highly regulated, but the CNS is no longer considered to represent an ‘immune-privileged’ site. Interestingly, PD-1 blockade has shown significant efficacy for the treatment of brain metastases in patients with melanoma and non-small cell lung cancer (NSCLC), and such clinical responses can be durable [25, 26]. Thus, location of a malignancy within the CNS per se does not render it unresponsive to immune checkpoint blockade. Rather, major differences in the immune microenvironment have been identified between gliomas and brain metastases: gliomas are highly infiltrated by macrophages and microglia, while larger number of T cells and other leukocytes are present in brain metastases [15].
The absence of a lymphatic drainage for the brain parenchyma has been frequently cited as a cause for an ‘immune-privileged’ state of the CNS. Imaging studies have demonstrated the presence of lymphatic vessels within the meninges that provide a functional connection from the CNS to deep cervical lymph nodes. A significant population of dendritic cells is present in the meninges, including a population of migratory (CCR7 +) DCs. These vessels have the molecular hallmarks of lymphatic endothelial cells and carry immune cells (T cells, myeloid cells) as well as soluble factors from the CSF to these lymph nodes [28]. VEGF-C is a cytokine that promotes the formation of lymphatic vessels, and ectopic expression of this cytokine in murine glioma cells enhances priming of CD8 T cells in deep cervical lymph nodes [29]. Despite this anatomical connection between the meninges and cervical lymph nodes, it remains unknown how cell-associated tumor antigens are transported to cervical lymph nodes for priming of anti-tumor T cell responses. Also, T cell traffic into the CNS is highly regulated at the blood–brain barrier which restricts entry to activated T cells [30]. Nevertheless, activated CAR T cells injected systemically can migrate into glioblastomas both in humanized mouse models and in patients [31, 32].
A particularly prominent immunological feature of glioblastomas is their dense infiltration by recruited macrophages and brain-resident microglia. Osteopontin has been identified as one of the major soluble factors that induces macrophage infiltration in glioblastoma, and osteopontin expression negatively correlates with patient survival [33]. In glioblastoma, osteopontin is secreted both by tumor cells and recruited macrophages. Knockdown of the Opn gene in murine glioma cells results in a substantial survival benefit, demonstrating the importance of tumor cell-derived osteopontin. Interestingly, a striking survival benefit is also observed when wild-type glioma cells are implanted into the CNS of Opn knockout compared to wild-type mice. These findings are relevant for resistance to T cell-mediated immunity: knockdown of Opn expression by murine glioma cells substantially increases tumor infiltration by IL-2 producing CD8 and CD4 T cells; T cell infiltration is also enhanced when wild-type glioma cells are implanted into Opn knockout mice, again highlighting the dual source of osteopontin. Osteopontin binds to the integrin αvβ5 receptor on macrophages and also to the CD44 receptor expressed by glioma cells in a mesenchymal (MES) state. The recruitment of macrophages by osteopontin and other soluble factors plays a critical role in induction of the MES state of glioma cells, and tumor cells in the MES state express higher levels of osteopontin. These findings suggest a positive feedback loop that induces and maintains the MES state.
Gliomasphere cultures established from human glioblastomas frequently lose the MES state, but this state can be induced by treatment of gliomaspheres with TNFα, a cytokine produced by activated macrophages. The MES state is associated with impaired survival in glioblastoma, which may in part be explained by resistance of the MES state to radiation therapy [34]. Also, recent mechanistic studies have demonstrated that the interaction of murine glioma cells with macrophages results in epigenetic immunoediting of glioma cells that in turn results in more efficient recruitment of immunosuppressive myeloid cells and attenuated T cell-mediated immunity [35].
The unique localization of glioblastomas in the brain, surrounded by the bony skull, has major implications for monitoring of immunotherapy approaches for gliomas. Serial biopsies, which have provided important molecular insights into the action of immunotherapies in other solid tumor types, are not feasible for brain tumors. Thus, liquid biopsies (either CSF or blood) might become important tools for future clinical trials in glioblastoma [36–38].
Which receptor–ligand systems inhibit T cell-mediated immunity in glioblastoma?
PD-1–PD-L1 pathway
Monoclonal antibodies that block the PD-1–PD-L1 pathway have become major drugs in oncology based on their ability to enhance the function of tumor-infiltrating T cells [39, 40]. Generally speaking, these antibodies are more active against tumors with significant infiltration by PD-1 expressing CD8 T cells because they amplify a pre-existing T cell response [41]. Also, response rates are higher in tumors with PD-L1 expression by tumor cells or infiltrating myeloid cells. Finally, response rates are highest in tumors with a high mutational burden due to exposure to environmental carcinogens or mutations in key genes in DNA repair pathways [42]. It is important to consider that these three factors are interrelated: for example, tumors with a high mutational burden tend to be more densely infiltrated by CD8 T cells. In glioblastoma, infiltration by T cells is rather low, and tumor mutational burden is also not high [43]. These factors may explain why PD-1 blockade did not result in a significant survival benefit in a phase 3 clinical trial [24], but it also remains possible that use of corticosteroids and other drugs interfered with the therapeutic activity of these therapeutic agents. However, these results do not mean that PD-1–PD-L1 antibodies will not be useful for the treatment of glioblastoma (Fig. 1 A). Rather, it will be important to develop combination therapies that enhance T cell infiltration and expansion in glioblastomas. We will discuss such strategies later in the context of cancer vaccines.
Fig. 1. Inhibitory receptors on glioblastoma-infiltrating T cells.
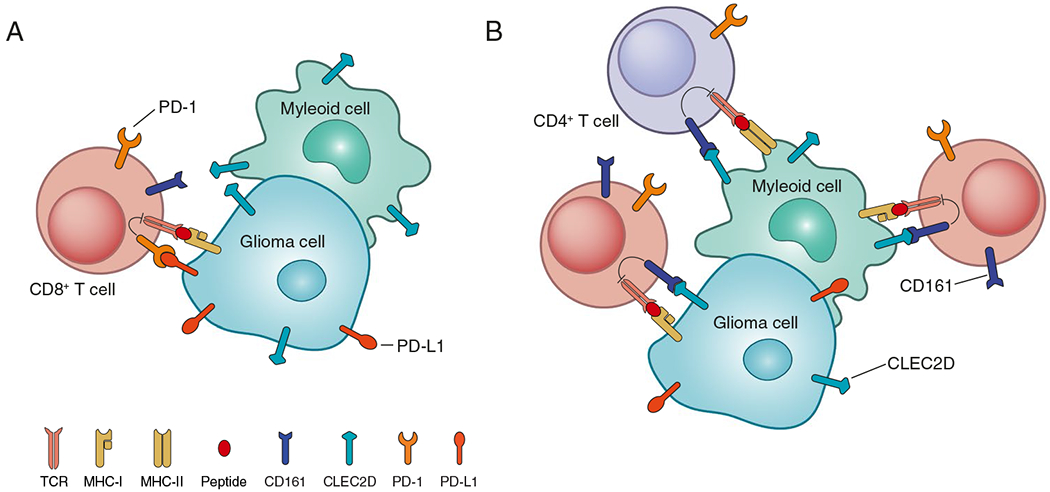
A. The PD-1 receptor inhibits early T cell signaling following binding to PD-L1 which can be expressed by glioma cells and myeloid cells. B. The CD161 receptor inhibits activation of CD8 and CD4 T cells following binding to its CLEC2D ligand expressed by glioma cells and myeloid cells
CD161–CLEC2D pathway
A recent single-cell RNA-seq study has highlighted the inhibitory CD161 receptor expressed by T cells infiltrating human diffuse gliomas (IDH wild-type glioblastoma and IDH mutant gliomas) [17]. CD161 (encoded by the KLRB1 gene) binds to the CLEC2D ligand expressed by malignant cells and myeloid cells in glioblastoma. This receptor had previously been identified in NK cells and shown to inhibit NK cell-mediated cytotoxicity of tumor cells [44]. When tumor-infiltrating T cells are activated following recognition of an MHC-bound peptide antigen, they undergo substantial clonal expansion. The scRNA-seq data demonstrate that KLRB1 is expressed at a higher level by clonally expanded versus non-expanded tumor-infiltrating CD8 T cells. Also, a cytotoxicity program in glioma-infiltrating CD8 T cells correlates with expression of multiple NK cell receptors, including KLRB1. At a protein level, CD161 is highly expressed by > 90% of infiltrating CD8 T cells and the majority of CD4 T cells in all examined gliomas. In contrast, expression of the PD-1 receptor is more variable between patients. Functional studies with short-term gliomaspheres co-cultured with human T cells demonstrate that inhibition of the CD161–CLEC2D pathway enhances T cell-mediated cytotoxicity against glioma cells as well as production of major cytokines required for antitumor immunity (Fig. 1 B). The relevance of this pathway has also been evaluated in a humanized mouse model. In this model, malignant cells from a gliomasphere culture of a recurrent glioblastoma are implanted into the brain of immunodeficient mice, followed by transfer of KLRB1 or control edited human T cells that express a tumor-specific TCR. The human glioma cells form highly aggressive, infiltrative tumors, yet transfer of KLRB1 versus control edited human T cells results in a significant survival benefit. Interestingly, editing of the KLRB1 gene also substantially reduces PD-1 expression by glioma-infiltrating human T cells, suggesting that this pathway may regulate T cell dysfunction in tumors. The CD161–CLEC2D pathway is also relevant in other human cancer types, including lung adenocarcinoma, colorectal cancer and hepatocellular carcinoma [17, 45]. CD161 is more broadly expressed by tumor-infiltrating lymphocytes than the PD-1 receptor, including CD8 T cells, CD4 T cells and NK cells. Also, it is expressed by tumor-infiltrating T cells with transcriptional programs of effector-memory and memory cells that may retain a higher degree of functionality than T cells with a terminal exhaustion program [17]. Targeting of this pathway is therefore of interest for the treatment of glioblastoma, potentially as part of a combination therapy strategy.
TIM-3, LAG3 and CTLA-4 inhibitory receptors
Monoclonal antibodies that target several additional inhibitory receptors expressed by T cells are in clinical development for a variety of cancer types. LAG-3 is an inhibitory receptor with homology to CD4 that binds to MHC class II proteins expressed by myeloid cells [46]. The combination of a LAG-3 mAb (relatlimab) and a PD-1 mAb has recently been shown to increase progression-free survival in patients with untreated metastatic or unresectable melanoma compared to monotherapy with a PD-1 mAb, and this combination therapy has been approved by the FDA for this indication [47]. In the murine GL-261 model, LAG-3 monotherapy shows moderate efficacy, which is increased when combined with a PD-1 mAb [48]. The TIM-3 inhibitory receptor is upregulated on exhausted T cells and binds to galectin-9 [46]. In the murine GL261 glioma model, a TIM-3 mAb does not show activity as a monotherapy, but a substantial survival benefit (~ 60% of mice) is observed in combination with stereotactic radiation therapy. Further addition of a PD-1 mAb results in survival of 100% of mice [49]. It should be noted that the GL261 model is immunogenic due to a high mutational burden, and shows monotherapy efficacy with PD-1 inhibition, in contrast to human GBM. The CTLA-4 inhibitory receptor binds to the CD80 and CD86 ligands on antigen presenting cells including dendritic cells [50]. CTLA-4 inhibition can thus enhance T cell priming. The CTLA-4 mAb ipilimumab has been approved by the FDA for the treatment of melanoma [51]. Combination therapy with PD-1 and CTLA-4 mAbs increases efficacy but is associated with a high frequency of severe (grade 3–4) adverse events [52]. The immunogenic GL261 glioma model is responsive to PD-1 monotherapy and PD-1 plus CTLA-4 combination therapy. However, in the SB28 glioma model this combination therapy does not show efficacy. Like human GBM, the SB28 model has a low mutation burden, low MHC class I expression and limited T cell infiltration [53].
TGFβ and other inhibitory cytokines
TGFβ is a major immunosuppressive cytokine, and it is well-established that it potently inhibits CD8 T cell activation and proliferation. It also plays a major role in inducing the differentiation of CD4 T cells into FoxP3 + regulatory T cells [54]. It is an unusual cytokine because its activity is not regulated by the rate of secretion. Rather, a latent form of TGFβ is deposited on the extracellular matrix and later activated by integrin αv receptors, with integrin αvβ6 and αvβ8 being the most important receptors based on their high affinity for latent TGFβ [55]. Interestingly, TGFβ is not only relevant for inhibition of T cell-mediated immunity but also the aggressive infiltrative behavior of glioblastomas. A subpopulation of human glioma cells expresses high levels of the integrin αvβ8 receptor, and implantation of integrin αvβ8 expressing glioma cells into the brain of immunodeficient mice results in efficient formation of diffusely infiltrating gliomas. Also, genetic targeting of integrin β8 in low-passage glioma cells substantially reduces tumor initiation in vivo, even in immunodeficient mice that lack T cells, NK cells and B cells [56]. Thus, therapeutic targeting of integrin αvβ8 (or both integrin αvβ6 and αvβ8) may not only enhance T cell-mediated immunity but also inhibit a major tumor cell-intrinsic pathway.
Aryl hydrocarbon receptor–kynurenine pathway
The acryl hydrocarbon receptor (AHR) is an important ligand-regulated transcription factor in glioblastoma. Kynurenine, a byproduct of tryptophan catabolism, is an important oncometabolite produced by glioma cells. It is produced by the action of related enzymes, tryptophan 2,3-dioxygenase (TDO) and indoleamine 2,3-dioxygenase (IDO). Ligand binding by AHR results in nuclear translocation and association with ARNT. In glioblastoma, high-level expression of AHR is associated with reduced CD8 T cell infiltration. AHR has important immunosuppressive functions in myeloid cells. In macrophages, it promotes an immunosuppressive program, including expression of CCR2 (migration in response to CCL2 produced by glioma cells) and CD39 (a key enzyme in production of immunosuppressive adenosine from ATP released by dying tumor cells). In dendritic cells, it promotes an immunosuppressive program that favors differentiation of FoxP3 + regulatory T cells from naïve CD4 T cell precursors [57, 58].
Oncometabolite produced by IDH mutant glioma cells
IDH mutant gliomas have gain of function mutations in the IDH1 or IDH2 genes resulting in the production of the oncometabolite (R)-2-hydroxyglutarate (R-2-HG). Interestingly, IDH mutant gliomas have substantially lower levels of CD8 and CD4 T cell infiltration compared to IDH wild-type glioma [59]. R-2-HG is present at high concentrations in the millimolar range in the extracellular space in IDH mutant gliomas, and T cells take up R-2-HG through particular transporters, including the sodium-dependent dicarboxylate transporter SLC13A3. In functional in vitro studies, R-2-HG inhibits T cell proliferation and cytokine production in a concentration-dependent manner, and T cell proliferation is also lower in the tumor microenvironment of IDH mutant gliomas compared to IDH wild-type glioma. At a mechanistic level, R-2-HG interferes with the calcium-dependent transcriptional activity of NFAT, a key transcription factor in the T cell signaling cascade downstream of the TCR-CD3 complex. These findings are therapeutically relevant because a small molecule inhibitor of mutant IDH1 shows synergy with a PD-1 mAb in a murine IDH1 mutant glioma model. Of note, this difference between IDH-mutant versus IDH-wildtype gliomas has thus far only been shown in lower grade gliomas but not yet in IDH-mutant GBM versus IDH-wildtype GBM [59].
How can vaccines be used to enhance T cell-mediated immunity against glioblastoma?
Increasing the degree of T cell infiltration is one of the major goals in the development of immunotherapies for glioblastoma. Vaccines represent a potential strategy for induction of more vigorous T cell responses, but due to the heterogeneity of glioblastoma cell states and their functional plasticity, it will be important to target multiple antigens to enable T cell-mediated killing of glioma cells in all distinct cellular states (Fig. 2). Three recent studies highlight advances that have been made to achieve this goal.
Fig. 2. Targeting of a heterogeneous population of glioma cells by polyclonal T cells with multiple antigen specificities.
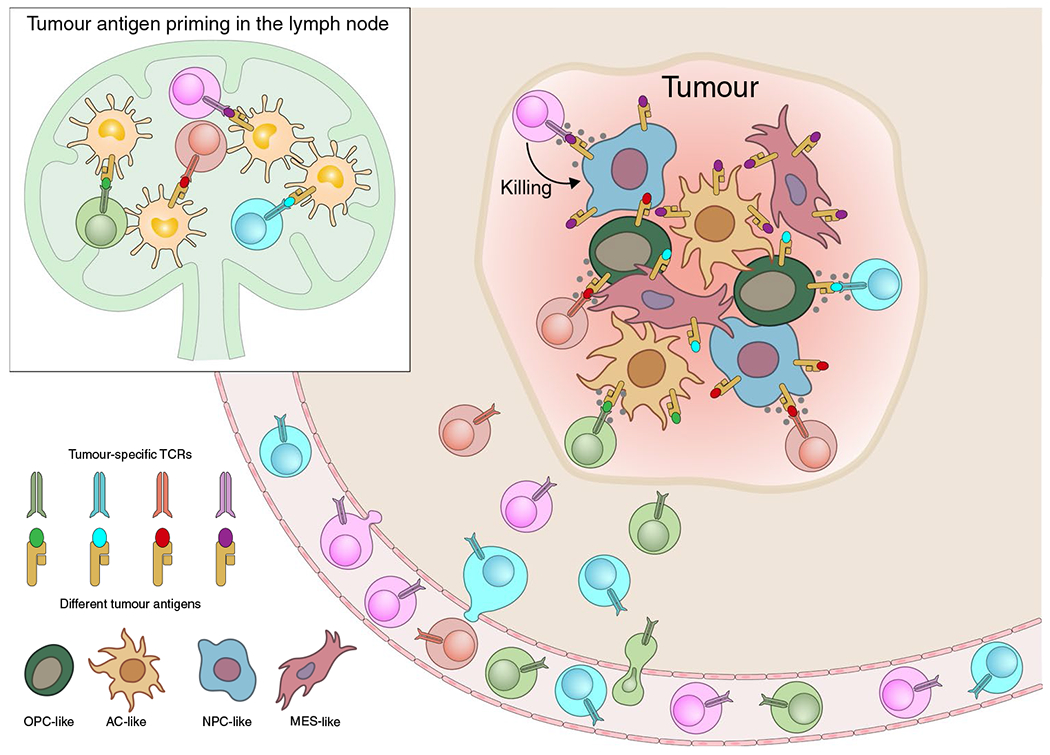
Vaccines that target multiple antigens, including neoantigens and/or shared tumor antigens, can induce polyclonal T cell populations in lymph nodes. Such polyclonal T cell populations could deal with heterogeneous glioma cell populations by inhibiting the outgrowth of tumor cells that have down-regulated or lost expression of a single antigen
The first study pursued the hypothesis that it could be advantageous to target a full repertoire of tumor antigens in glioblastoma, including non-mutated shared tumor antigens and neoantigens [60]. The team emphasized the precise identification of peptides presented by HLA class I proteins using mass spectrometry and then established a ‘warehouse’ of pre-manufactured synthetic peptides for two HLA class I molecules, HLA-A*02:01 (33 peptides) and HLA-A*24:02 (26 peptides). Patients received two sets of peptide-based vaccines. The first vaccine was based on shared tumor antigens, and for each patient the highest-ranking peptides were identified by HLA immunopeptidome and transcriptome analyses. Ex vivo HLA class I tetramer staining demonstrated that 45 of 87 selected peptides (51.7%) were immunogenic. The second vaccine used up to two patient-specific neoantigens, when feasible. These neoantigen peptides induced a CD4 T cell response (84.7% of peptides) or CD4 plus CD8 T cell responses (38.5% of peptides). The median overall survival was 29.0 months (n = 15 patients), and 6 patients were alive at the time of reporting. These survival data compare favorably to historical datasets, but a survival benefit needs to be formally established in future clinical trials. This study demonstrates the value of targeting non-mutated antigens by careful definition of the HLA class I immunopeptidome.
The second study tested the hypothesis that neoantigen-based vaccines could be developed for glioblastoma patients by careful identification of neoantigen peptides predicted to bind to the HLA class I proteins of individual patients [61]. Newly diagnosed glioblastoma patients were vaccinated following surgical resection and conventional radiotherapy. A median of 64.5 HLA binders (range of 30–163) could be predicted per tumor, and a median number of 12 peptides (7–20) were included in the vaccine. All three patients who required dexamethasone treatment during vaccine priming failed to induce a T cell response, again highlighting the detrimental effects of high-dose corticosteroids on the induction of therapeutic T cell responses. In contrast, patients who did not receive dexamethasone during vaccine priming generated robust de novo responses against multiple predicted neoantigens. Multiplex immunofluorescence analysis of patients who required surgery post-vaccination demonstrated increased tumor infiltration by CD8 T cells. In one of these patients, scRNA-seq analysis of tumor-infiltrating T cells was used to define their TCR sequences to determine which of these cells were specific for neoantigen peptides used in the vaccine. Neoantigen-specific T cells were isolated from post-vaccination blood specimens and their TCR sequences were compared to those from tumor-infiltrating T cells. Interestingly, six neoantigen-specific T cell clones could be identified in both tumor and blood samples (2 CD8 T cell clones, 4 CD4 T cell clones), demonstrating that neoantigen-specific T cells had infiltrated the patient’s tumor. Furthermore, the neoantigen specificity of both CD8 and CD4 T cell clones was confirmed by transfection of their TCRs [61].
In IDH mutant tumors, the most common IDH1 mutation affects codon 132 (R132H), and this neoantigen is presented by MHC class II proteins. A recent clinical trial demonstrated that vaccination with this IDH1 neoantigen is safe and that activated neoantigen-specific CD4 T cells can be identified in the tumor tissue [62].
Taken together, these three recent studies demonstrate that personalized vaccines can be developed for both shared tumor antigens and neoantigens. It will be important to further optimize these vaccination platforms to enhance the magnitude of T cell expansion and to endow induced T cells with optimal functional programs. Also, it will be important to compare peptide-based approaches to mRNA-based technologies because this platform may enable more rapid formulation of personalized vaccines [63], an aspect that is relevant in aggressive tumor types such as glioblastoma.
How can CAR T cells be designed to deal with the high level of tumor heterogeneity in glioblastoma?
CAR T cells represent another exciting avenue for the induction of T cell-mediated immunity against glioblastoma. While CAR T cells have shown striking activity in hematological malignancies, it has been more challenging to develop CAR T cell therapies for solid tumor indications. In B cell malignancies, CD19 is homogeneously expressed at a high level by malignant cells, while a more heterogeneous expression pattern is observed for most antigens considered for CAR T cell therapy of solid tumors [64]. The central question therefore is how CAR T cells could target a highly heterogeneous tumor such as glioblastoma. Two creative approaches point to potential strategies for overcoming this important barrier for T cell-based therapies.
An innovative CAR T cell design addresses this issue of tumor heterogeneity. Approximately 30% of glioblastomas express mutant EGFRvIII, but expression of this antigen is heterogeneous in such tumors. In a phase 1 clinical trial, treatment with EGFRvIII CAR T cells was shown to result in the outgrowth of EGFRvIII negative tumors [65]. CAR T cell-based selection of antigen loss variants has also been observed in hematological malignancies: outgrowth of leukemia cells with loss of CD19 expression is one of the major causes of disease relapse following treatment with CD19 CAR T cells [66]. Secretion of a bispecific antibody (BiTE) by CAR T cells that targets CD3 (T cell activation) and EGFR (tumor surface antigen) enables targeting of EGFR by CAR T cells, an antigen that is frequently overexpressed in glioblastoma but also expressed in healthy epithelial tissues [67]. Local BiTE secretion also enables killing of EGFR + glioma cells by other T cells in the tumor microenvironment and could thereby enhance endogenous antitumor immunity directed against a diverse set of antigens (Fig. 3). A number of other glioblastoma antigens have been reported as potential targets for CAR T cells that could be used to target EGFRvIII negative tumors, including EphA2, IL13Rα2 and GD2 [68–70].
Fig. 3. CAR T cell approaches that enable targeting of highly heterogeneous glioma cell populations.
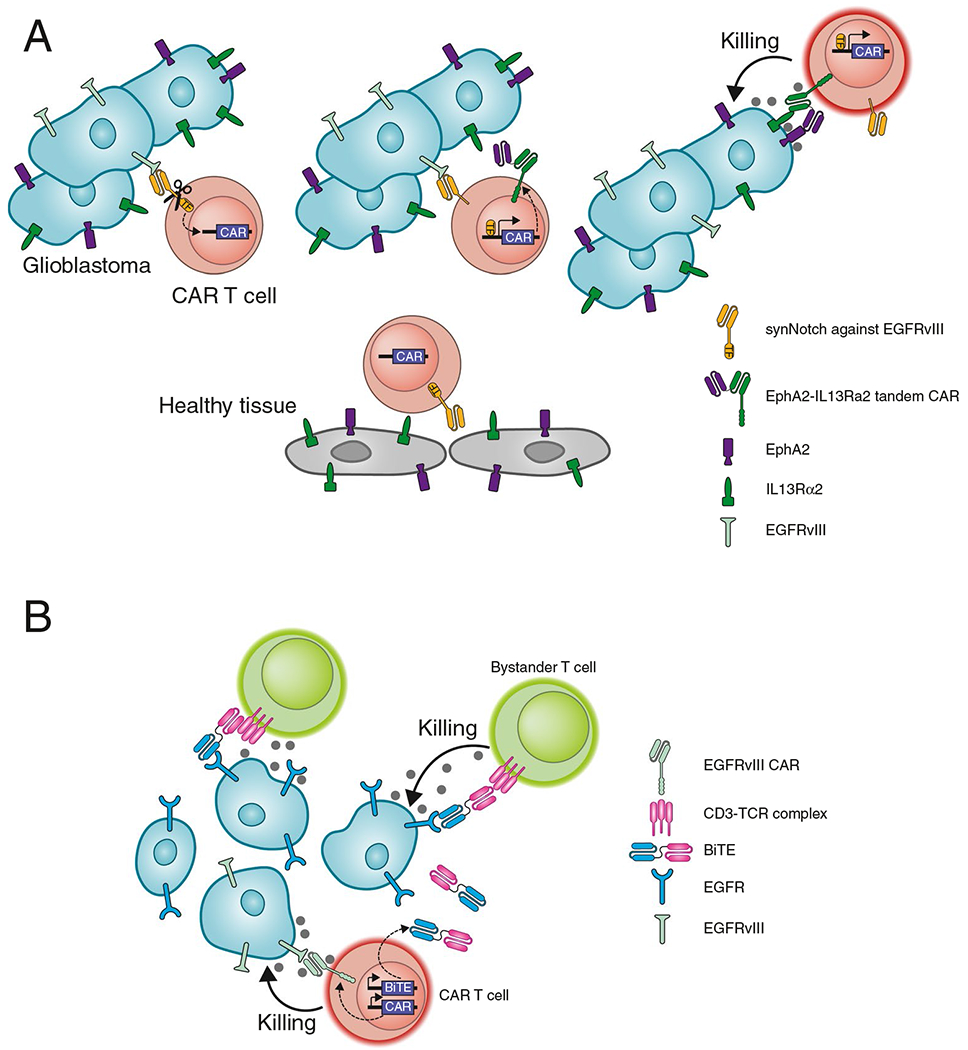
A. SynNotch receptors enable induction of a transcriptional program in T cells. EGFRvIII is a highly specific mutant protein in glioblastoma but is only expressed by a subpopulation of glioma cells. Binding of EGFRvIII by a Syn-Notch receptor results in cleavage of the cytoplasmic domain which then acts as a transcription factor that induces expression of a killing CAR. This CAR is directed against EphA2 and IL13Rα2 antigens on glioma cells. These antigens are broadly expressed by glioma cells but not entirely tumor specific. However, expression of the killing CAR is only induced in the tumor, thus avoiding toxicity against healthy tissues. B. Secretion of a bispecific T cell engager (BiTE) by CAR T cells enables targeting of a broadly expressed tumor antigen. The CAR recognizes EGFRvIII, conferring tumor specificity. Secretion of a BiTE that binds to EGFR on tumor cells and CD3 on T cells enables killing of tumor cells by bystander T cells
SynNotch receptors enable customized design of a transcriptional circuit in CAR T cells. This design is based on the biological principles of the Notch receptor which is activated by mechanical stress, resulting in proteolytic release of the cytoplasmic domain that then acts as a transcription factor. Incorporation of the juxtamembrane and transmembrane domains of Notch can be used to create a SynNotch receptor in which ligand binding by the extracellular domain results in release of a transcription factor that induces the expression of a killing CAR [71]. This design enables expression of the killing CAR only in the relevant tissue microenvironment and can be considered to represent an IF–THEN circuit [72]. The killing CAR targets two antigens—EphA2 and IL13Rα2—that are broadly expressed by glioma cells but are not entirely tumor specific (Fig. 3). The first SynNotch design targets the EGFRvIII mutant protein that is highly tumor specific but is expressed in a heterogeneous pattern by glioma cells. This CAR circuit enables long-term control of a glioblastoma patient-derived xenograft (PDX) model with heterogeneous expression of EGFRvIII. The second SynNotch design targets a brain-specific antigen (MOG) expressed on the surface of oligodendrocytes and myelin. This circuit induces expression of the killing CAR only in the CNS, again enabling T cell-mediated control of a glioblastoma PDX model in immunodeficient mice. This conceptual approach thus addresses two major challenges: 1. Specificity: The SynNotch receptor induces expression of the killing CAR only within the tumor (EGFRvIII design) or the relevant organ (MOG design), 2. Heterogeneity: Tumor/organ specific expression of the killing CAR enables targeting of antigens that are broadly expressed by tumor cells but are not entirely tumor specific. The MOG SynNotch design is also of particular interest for targeting of glioma cells that have infiltrated deeply into neighboring brain regions, including the contralateral hemisphere [72].
What are the key principles for future development of immunotherapies for glioblastoma?
These recent advances can be used to distill several principles to guide the future development of T cell-based immunotherapies for glioblastoma.
Develop approaches for avoiding or mitigating drug-induced T cell depletion. T cells are the key effector cells of all FDA approved immunotherapies (including immune checkpoint blockade and CAR T cells) and also essential for the efficacy of cancer vaccines. Experiments in experimental glioma models and clinical trials demonstrate that dexamethasone interferes with therapeutic activity, such as the failure to induce neoantigen-specific T cell responses in patients who received corticosteroids during vaccine priming [61].
Induce T cell responses to multiple antigens in order to deal with the extensive heterogeneity and plasticity of glioma cells. Alternatively, CAR T cells can be designed in which recognition of a tumor or tissue specific antigen induces an effector program that enables targeting of antigens that are not entirely tumor specific. It will be also important to study in clinical trials how effectively tumor heterogeneity can be controlled, and which tumor cell states are a major cause for disease relapse.
Develop immunotherapies that target multiple inhibitory pathways, including pathways related to T cell–tumor cell interactions (such as PD-1–PD-L1 or CD161–CLEC2D) and pathways responsible for the detrimental crosstalk between tumor cells and myeloid cells (such as osteopontin or AHR). Further mechanistic studies on human glioblastoma and murine glioma models will be required to identify additional pathways for intervention.
Consider early intervention with immunotherapies, including a pre-surgical (neoadjuvant) setting. Neoadjuvant-based approaches have shown promising activity in a number of other cancer types, including melanoma, lung cancer, colon cancer and head and neck cancer. These studies have demonstrated that neoadjuvant immunotherapy enhances both local and systemic tumor immunity [73].
Develop better approaches to monitor the efficacy of immunotherapy regimens to optimize the development of combination therapies, including liquid biopsies of cerebrospinal fluid (CSF) to monitor tumor evolution (circulating tumor DNA, ctDNA) and quantification of immune cell derived cytokines.
Acknowledgements
This work was supported by a grant from NIH (P01 CA236749 to K.W.W.) and The Jennifer Oppenheimer Cancer Research Initiative at Dana-Farber Cancer Institute (to K.W.W.). S.M. is supported by a fellowship from the Deutsche Forschungsgemeinschaft (DFG, grant MA 8489/1-1).
Conflict of interest statement
K.W.W. serves on the scientific advisory board of T-Scan Therapeutics, SQZ Biotech and Nextechinvest and receives sponsored research funding from Novartis. He is a scientific co-founder of Immunitas Therapeutics.
References
- 1.Neftel C et al. (2019) An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 178(4):835–849 e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel AP et al. (2014) Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344(6190):1396–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tirosh I, Suva ML (2020) Tackling the Many Facets of Glioblastoma Heterogeneity. Cell Stem Cell 26(3):303–304 [DOI] [PubMed] [Google Scholar]
- 4.Chaligne R et al. (2021) Epigenetic encoding, heritability and plasticity of glioma transcriptional cell states. Nat Genet 53(10):1469–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosen J et al. (2018) Extracranial Metastases of a Cerebral Glioblastoma: A Case Report and Review of the Literature. Case Rep Oncol 11(2):591–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jusue-Torres I, Prabhu VC, Jones GA 2021. Dandy’s hemispherectomies: historical vignette. J Neurosurg, 1–7 [DOI] [PubMed] [Google Scholar]
- 7.Dandy WE (1928) Removal of right cerebral hemisphere for certain tumors with hemiplegia: preliminary report. J Am Med Assoc 90:823–825 [Google Scholar]
- 8.Brown TJ et al. (2016) Association of the Extent of Resection With Survival in Glioblastoma: A Systematic Review and Meta-analysis. JAMA Oncol 2(11):1460–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duffau H (2013) A new philosophy in surgery for diffuse low-grade glioma (DLGG): oncological and functional outcomes. Neurochirurgie 59(1):2–8 [DOI] [PubMed] [Google Scholar]
- 10.Sanai N, Berger MS (2008) Glioma extent of resection and its impact on patient outcome. Neurosurgery 6(4):753–64 [DOI] [PubMed] [Google Scholar]
- 11.Sanai N et al. (2011) An extent of resection threshold for newly diagnosed glioblastomas. J Neurosurg 115(1):3–8 [DOI] [PubMed] [Google Scholar]
- 12.Nieder C et al. (2005) Treatment of unresectable glioblastoma multiforme. Anticancer Res 25(6C):4605–4610 [PubMed] [Google Scholar]
- 13.Simpson JR et al. (1993) Influence of location and extent of surgical resection on survival of patients with glioblastoma multiforme: results of three consecutive Radiation Therapy Oncology Group (RTOG) clinical trials. Int J Radiat Oncol Biol Phys 26(2):239–244 [DOI] [PubMed] [Google Scholar]
- 14.van Tellingen O et al. (2015) Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat 19:1–12 [DOI] [PubMed] [Google Scholar]
- 15.Friebel E et al. (2020) Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 181(7):1626–164220 [DOI] [PubMed] [Google Scholar]
- 16.Chongsathidkiet P et al. (2018) Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med 24(9):1459–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathewson ND et al. (2021) Inhibitory CD161 receptor identified in glioma-infiltrating T cells by single-cell analysis. Cell 184(5):1281–1298 e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iorgulescu JB et al. (2021) Concurrent Dexamethasone Limits the Clinical Benefit of Immune Checkpoint Blockade in Glioblastoma. Clin Cancer Res 27(1):276–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sengupta S et al. (2012) Impact of temozolomide on immune response during malignant glioma chemotherapy. Clin Dev Immunol 2012:831090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song AJ et al. (2021) Impact of lymphopenia on survival for elderly patients with glioblastoma: A secondary analysis of the CCTG CE6 (EORTC 26062–22061 TROG0301) randomized clinical trial. Neurooncol Adv 3(1):vdab153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bai X et al. (2021) Efficacy of bevacizumab in the treatment of refractory brain edema of metastatic tumors from different sources. Neurol Res 43(12):955–960 [DOI] [PubMed] [Google Scholar]
- 22.Kurkjian C, Kim ES (2012) Risks and benefits with bevacizumab: evidence and clinical implications. Ther Adv Drug Saf 3(2):59–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banks PD et al. (2019) Bevacizumab as a steroid-sparing agent during immunotherapy for melanoma brain metastases: A case series. Health Sci Rep 2(3):e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reardon DA et al. (2020) Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol 6(7):1003–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bander ED et al. (2021) Melanoma brain metastasis presentation, treatment, and outcomes in the age of targeted and immunotherapies. Cancer 127(12):2062–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li W, et al. 2021. Efficacy of PD-1/L1 inhibitors in brain metastases of non-small-cell lung cancer: pooled analysis from seven randomized controlled trials. Future Oncol [DOI] [PubMed] [Google Scholar]
- 27.Sun BL et al. (2018) Lymphatic drainage system of the brain: A novel target for intervention of neurological diseases. Prog Neurobiol 163–164:118–143 [DOI] [PubMed] [Google Scholar]
- 28.Louveau A et al. (2015) Structural and functional features of central nervous system lymphatic vessels. Nature 523(7560):337–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song E et al. (2020) VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature 577(7792):689–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engelhardt B (2006) Molecular mechanisms involved in T cell migration across the blood-brain barrier. J Neural Transm (Vienna) 113(4):477–485 [DOI] [PubMed] [Google Scholar]
- 31.Johnson LA et al. (2015) Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med 7(275):275ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Rourke DM et al. (2017) A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 9(399):eaaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei J et al. (2019) Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J Clin Invest 129(1):137–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhat KPL et al. (2013) Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 24(3):331–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gangoso E et al. (2021) Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell 184(9):2454–2470 e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramkissoon LA et al. (2020) Genomic Profiling of Circulating Tumor DNA From Cerebrospinal Fluid to Guide Clinical Decision Making for Patients With Primary and Metastatic Brain Tumors. Front Neurol 11:544680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shankar GM et al. (2017) Liquid biopsy for brain tumors. Expert Rev Mol Diagn 17(10):943–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siravegna G et al. (2017) Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol 14(9):531–548 [DOI] [PubMed] [Google Scholar]
- 39.Chen DS, Mellman I (2017) Elements of cancer immunity and the cancer-immune set point. Nature 541(7637):321–330 [DOI] [PubMed] [Google Scholar]
- 40.Sharpe AH, Pauken KE (2018) The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol 18(3):153–167 [DOI] [PubMed] [Google Scholar]
- 41.Tumeh PC et al. (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515(7528):568–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klempner SJ et al. (2020) Tumor Mutational Burden as a Predictive Biomarker for Response to Immune Checkpoint Inhibitors: A Review of Current Evidence. Oncologist 25(1):e147–e159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gromeier M et al. (2021) Very low mutation burden is a feature of inflamed recurrent glioblastomas responsive to cancer immunotherapy. Nat Commun 12(1):352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosen DB et al. (2005) Cutting edge: lectin-like transcript-1 is a ligand for the inhibitory human NKR-P1A receptor. J Immunol 175(12):7796–7799 [DOI] [PubMed] [Google Scholar]
- 45.Sun Y et al. (2021) Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell 184(2):404–421 e16 [DOI] [PubMed] [Google Scholar]
- 46.Anderson AC, Joller N, Kuchroo VK (2016) Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 44(5):989–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tawbi HA et al. (2022) Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N Engl J Med 386(1):24–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harris-Bookman S et al. (2018) Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int J Cancer 143(12):3201–3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim JE et al. (2017) Combination Therapy with Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin Cancer Res 23(1):124–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baumeister SH et al. (2016) Coinhibitory Pathways in Immunotherapy for Cancer. Annu Rev Immunol 34:539–573 [DOI] [PubMed] [Google Scholar]
- 51.Hodi FS et al. (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363(8):711–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Larkin J et al. (2015) Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 373(1):23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Genoud V et al. (2018) Responsiveness to anti-PD-1 and anti-CTLA-4 immune checkpoint blockade in SB28 and GL261 mouse glioma models. Oncoimmunology 7(12):e1501137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanjabi S, Oh SA, Li MO (2017) Regulation of the Immune Response by TGF-beta: From Conception to Autoimmunity and Infection. Cold Spring Harb Perspect Biol 9(6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Munger JS et al. (1999) The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96(3):319–328 [DOI] [PubMed] [Google Scholar]
- 56.Guerrero PA et al. (2017) Glioblastoma stem cells exploit the alphavbeta8 integrin-TGFbeta1 signaling axis to drive tumor initiation and progression. Oncogene 36(47):6568–6580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gabriely G, Quintana FJ (2020) Role of AHR in the control of GBM-associated myeloid cells. Semin Cancer Biol 64:13–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takenaka MC et al. (2019) Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci 22(5):729–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bunse L et al. (2018) Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med 24(8):1192–1203 [DOI] [PubMed] [Google Scholar]
- 60.Hilf N et al. (2019) Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 565(7738):240–245 [DOI] [PubMed] [Google Scholar]
- 61.Keskin DB et al. (2019) Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565(7738):234–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Platten M et al. (2021) A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 592(7854):463–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kreiter S et al. (2015) Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 520(7549):692–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maude SL et al. (2014) Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371(16):1507–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.O’Rourke DM et al. (2017) A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 9(399) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Park JH et al. (2018) Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med 378(5):449–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Choi BD et al. (2019) CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol 37(9):1049–1058 [DOI] [PubMed] [Google Scholar]
- 68.Krebs S et al. (2014) T cells redirected to interleukin-13Ralpha2 with interleukin-13 mutein–chimeric antigen receptors have antiglioma activity but also recognize interleukin-13Ralpha1. Cytotherapy 16(8):1121–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin Q et al. (2021) First-in-Human Trial of EphA2-Redirected CAR T-Cells in Patients With Recurrent Glioblastoma: A Preliminary Report of Three Cases at the Starting Dose. Front Oncol 11:694941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mount CW et al. (2018) Potent antitumor efficacy of anti-GD2 CAR T cells in H3–K27M(+) diffuse midline gliomas. Nat Med 24(5):572–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morsut L et al. (2016) Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 164(4):780–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Choe JH, et al. (2021) SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med 13(591). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Menzies AM et al. (2021) Pathological response and survival with neoadjuvant therapy in melanoma: a pooled analysis from the International Neoadjuvant Melanoma Consortium (INMC). Nat Med 27(2):301–309 [DOI] [PubMed] [Google Scholar]