

ABSTRACT
Background
On approval of JYNARQUE (tolvaptan) for use in patients with autosomal dominant polycystic kidney disease (ADPKD) at risk for rapid progression, the US Food and Drug Administration required a Risk Evaluation and Mitigation Strategy (REMS) from the sponsor, which includes collection of post marketing liver safety data.
Methods
This is a retrospective interim analysis of the ongoing REMS. The period evaluated was from REMS implementation (14 May 2018) at tolvaptan commercialization to the analysis cutoff date (23 February 2021). Patients were previously tolvaptan-naïve and initiated tolvaptan in the post marketing setting. Reports of possible severe drug-induced liver injury (DILI) were evaluated for severity based on the evidence obtained (e.g. liver enzyme levels, symptoms, diagnostic tests and event outcomes). The incidence of DILI was compared between the REMS and tolvaptan clinical trials in ADPKD.
Results
Among 6711 REMS patients, 60 (0.9%) cases of possible severe DILI were reported, 4 of which were confirmed as serious and potentially fatal by the sponsor. One of these four patients met Hy's law criteria. In all four patients, liver enzymes normalized after tolvaptan discontinuation. The duration of tolvaptan exposure in the REMS is currently shorter than in completed clinical trials, but within this limitation, the incidence of possible severe DILI was lower in the REMS than in clinical trials (incidence rate ratio 0.587; P = .000411).
Conclusions
In interim data on >6000 tolvaptan REMS patients, <1% experienced possible severe DILI. Monthly monitoring, as described in the tolvaptan prescribing information, enables the prompt detection of liver enzyme abnormalities and appropriate drug discontinuation.
Keywords: autosomal dominant polycystic kidney disease (ADPKD), cystatin C, drug-induced liver injury (DILI), liver safety, post marketing surveillance, Risk Evaluation and Mitigation Strategy (REMS), tolvaptan
INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD) is a hereditary condition in which the development and growth of kidney cysts inexorably leads to a decline in the glomerular filtration rate (GFR) and eventually kidney failure [1]. In clinical trials, JYNARQUE (tolvaptan) demonstrated efficacy in slowing the loss of kidney function among subjects with ADPKD at risk of rapid progression [2–4]. Tolvaptan received US Food and Drug Administration (FDA) approval in April 2018 as the first pharmacotherapy for ADPKD in the USA, with an indication for slowing kidney function decline in adults at risk for rapidly progressing ADPKD [5].
During the clinical development program for tolvaptan as a treatment for ADPKD, liver safety signals were detected during routine safety monitoring by an independent data monitoring committee [6]. An imbalance was seen in the proportion of subjects with alanine aminotransferase (ALT) >3 × the upper limit of normal (ULN), 4.4% tolvaptan versus 1.0% placebo, in the Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes (TEMPO) 3:4 trial (NCT00428948). Additionally, two subjects in TEMPO 3:4 and one subject in the TEMPO 4:4 extension trial (NCT01214421) met Hy's law criteria, i.e. serum ALT or aspartate aminotransferase (AST) ≥3 × ULN and total bilirubin >2 × ULN, in the absence of cholestasis and without any other reason to explain the elevations [7]. The three cases were 0.26% of 1142 unique subjects who were treated with tolvaptan in TEMPO 3:4 (n = 961) and/or TEMPO 4:4 (n = 181). The transaminase elevations exhibited a characteristic pattern in which the window of susceptibility for clinical events was within the first 18 months of treatment, resolving within ∼4 months after tolvaptan discontinuation in all subjects [6]. After detection of these signals, liver chemistry testing—which had been conducted every 4 months throughout TEMPO 3:4 and initially every 6 months in TEMPO 4:4—was increased in frequency in TEMPO 4:4 to every 3 months, then to monthly. In the second pivotal trial [Replicating Evidence of Preserved Renal Function: An Investigation of Tolvaptan Safety and Efficacy in ADPKD (REPRISE); NCT02160145], liver function testing was conducted monthly for the entirety of the 1-year study [4, 6]. The frequency of ALT >3 × ULN (5.6% tolvaptan versus 1.2% placebo) in REPRISE was similar to that in the TEMPO program, but no additional cases meeting Hy's law criteria were reported, likely due to monthly monitoring and timely interruption of study medication in subjects experiencing liver enzyme elevations [4].
Given the clinical trial liver safety findings, the FDA required the implementation of a Risk Evaluation and Mitigation Strategy (REMS) on approval of tolvaptan for the treatment of ADPKD, which was initiated on 14 May 2018. Program requirements include educating healthcare providers and patients about the risk of serious and potentially fatal liver injury associated with the use of tolvaptan, conducting liver chemistry monitoring at specified times in patients prescribed tolvaptan [i.e. before treatment (baseline), 2 weeks and 4 weeks after tolvaptan initiation, then monthly for the first 18 months and every 3 months thereafter], and reporting of adverse events suggestive of serious and potentially fatal liver injury. Liver adverse event data were to be reported to the FDA 6 months after the initial approval of the REMS and annually thereafter [8].
In the tolvaptan REMS, the determination of adverse events suggestive of serious and potentially fatal liver injury was initially based solely on the clinical judgment of the prescriber or specialty pharmacist, with reports submitted by phone, or on a Liver Adverse Event Reporting Form or a Patient Status Form. In February 2020, more detailed and structured Liver Adverse Event Reporting and Patient Status forms and updated Prescriber Training content were introduced to better capture relevant patient data and provide precise criteria for healthcare providers to determine whether serious and potentially fatal liver injury had occurred. The criteria were based on FDA guidance for treatment discontinuation following the observation of specific liver enzyme elevations [7]. The revised Liver Adverse Event Reporting Form is provided in the Supplementary Appendix.
Hepatic injury does not occur in a predictable, dose-dependent manner. Rare, severe drug-induced liver injury (DILI) events may occur without a clear dose relationship and with a period of latency after drug initiation, suddenly manifesting after months of treatment without prior incident [9]. Such cases, known as idiosyncratic DILI, may or may not be identified during clinical trials due to their rarity. In general, the adequate characterization of drug liver safety for pharmaceuticals requires post marketing data collection from large numbers of patients [10]. For liver safety assessment, severe DILI is defined by the FDA as irreversible liver failure that is fatal or requires transplantation [7]. Less severe DILI is detected by elevated serum transaminase levels, as injured liver cells release abnormally high amounts of ALT and/or AST. Patients who meet Hy's law criteria have a high risk (∼10%) of experiencing a severe DILI event, and any identified case is therefore a major drug safety event [7]. Transaminase elevations beyond the ULN (≥3 × ULN) that do not meet Hy's law criteria are also cause for concern. An increased incidence of such elevations in a clinical trial population is a potential signal for risk of severe DILI [7].
In this report, interim liver safety data collected and analyzed in the tolvaptan REMS program are presented. The analysis period was from the start of the program (14 May 2018), which occurred immediately prior to tolvaptan commercialization (21 May 2018), to the data cutoff date of 23 February 2021.
MATERIALS AND METHODS
Analysis population
Eligibility criteria for inclusion in this analysis were patients with ADPKD who were enrolled in the REMS in the USA, were tolvaptan naïve, and who initiated treatment with tolvaptan in the post marketing setting.
Design
This is a retrospective analysis of adult patients with ADPKD who are enrolled in the tolvaptan REMS and initiated treatment with tolvaptan in the post marketing setting. A descriptive analysis was conducted using REMS data and the Otsuka Global Pharmacovigilance (GPV) database on patients with a possible diagnosis of severe DILI between 14 May 2018 (the REMS start date) and 23 February 2021 (data cutoff). The Otsuka GPV Department collects detailed data from healthcare providers on possible events of serious and potentially fatal liver injury using an Enhanced Pharmacovigilance Form. These data are entered into the Otsuka GPV safety database and relevant information required for this study is included in the analyses (Figure 1).
FIGURE 1:

Collection and assessment of data on possible events of serious and potentially fatal liver injury in the REMS program. *If no information/response is received from the HCP, Otsuka GPV does not determine event severity, and it remains classified as a serious and potentially fatal liver injury and included in the study analysis.
As with the liver adverse event reporting documents, the REMS Patient Enrollment Form was revised (in February 2019) to include more detailed information. The revised, FDA-approved form collects data on patient race/ethnicity and medical history (e.g. typical alcohol consumption), whereas this information may not have been provided prior to introduction of the new documents.
Identification of possible severe DILI
Adverse events of possible severe DILI reported to the REMS were identified for this analysis using broad criteria. A possible case was defined as any patient who met at least one of the following, regardless of causality assessment provided by the reporter:
Criterion 1: Diagnosis of acute hepatic failure, hepatic failure, hepatic necrosis, hepatic encephalopathy, ascites, hepatorenal failure, hepatorenal syndrome, hepatitis fulminant, liver transplant or death related to liver injury
Criterion 2: Development of any adverse event matching a lower-level Medical Dictionary for Regulatory Activities (MedDRA) term in one of the five hepatic Standardized MedDRA Queries (SMQs) (Table 1) and leading to liver transplantation, resulting in a fatal outcome or considered to be life-threatening
Criterion 3: Development of any adverse event matching a lower-level MedDRA term in one of the five hepatic SMQs and meeting any of the prespecified laboratory criteria for liver chemistry (Table 1).
Table 1.
Hepatic SMQs and laboratory criteria used in the definition of possible severe DILI (see accompanying text for full definition)
| Hepatic SMQs | Laboratory criteria |
|---|---|
| • Cholestasis and jaundice of hepatic origin • Hepatic failure, fibrosis and cirrhosis, and other liver damage-related conditions • Hepatitis, noninfectious • Liver-related investigations, signs, and symptoms • Liver-related coagulation and bleeding disturbances |
• ALT or AST >8 × ULN • ALT or AST >5 × ULN for >2 weeks • ALT or AST >3 × ULN and total bilirubin >2 × ULN or INR >1.5 (total bilirubin measurement can be within 30 days of the ALT elevation) • ALT or AST >3 × ULN with the appearance of fatigue, nausea, vomiting, right upper quadrant pain or tenderness, fever, rash, and/or eosinophilia (>5%) |
INR, international normalized ratio.
Cases of possible severe DILI captured in this manner were analyzed further to determine severity, timing and outcomes and to identify the following events:
Events that were confirmed as serious and potentially fatal liver injury by the Otsuka GPV safety team. Confirmation of an event as serious and potentially fatal, in accordance with FDA guidance [7], was based on the evidence presented in the case, including laboratory data and trends in those data over time, diagnostic tests, symptoms, medical history, the time course of the event, and information about potential confounding risk factors and concomitant medications
Hy's Law: ALT or AST ≥3 × ULN and total bilirubin >2 × ULN, in the absence of cholestasis and without any other reason to explain the elevations
Confirmed severe DILI by FDA criteria: irreversible liver failure that is fatal or requires transplantation [7].
Adjudication of possible causal relationship with tolvaptan
Any potential causal relationship of tolvaptan to liver adverse events reported to Otsuka is determined by an independent, blinded Hepatic Adjudication Committee (HAC). Composed of an external panel of gastroenterologists and hepatologists with therapeutic expertise in evaluating and assessing liver toxicity adverse events, the committee is charged with providing an independent, unbiased review of hepatic adverse event causality. The selection criteria for liver adverse events requiring adjudication by the committee were established by the HAC itself and can be found in the Supplementary Appendix. Causality assessment is conducted using the standardized scale of the DILI network, which classifies the likelihood of relationship to a drug as definite (>95% likelihood), highly likely (75%–95% likelihood), probable (50%–74% likelihood), possible (25%–49% likelihood), unlikely (<25% likelihood) or insufficient data [11].
Analyses
Available patient demographic and clinical characteristics from the REMS are presented as summary statistics. The incidence proportion and rates of possible events of severe DILI in the REMS were calculated as the percentage of patients affected and as events per 100 patient-years, respectively. The incidence rates from the REMS were compared with those from tolvaptan clinical trials, as required by the FDA [5]. The indirect comparison of the REMS and clinical trial incidence rates was performed by calculating an incidence rate ratio (IRR) using the Wald method. The clinical trials were TEMPO 3:4, the TEMPO 3:4 extension (TEMPO 4:4), REPRISE and long-term extension 156-13-211 (NCT02251275), which enrolled subjects from REPRISE, TEMPO 4:4 and previous tolvaptan trials.
RESULTS
Patient disposition
As of the cutoff date, 7329 patients with ADPKD were identified as receiving their first dose of commercial tolvaptan in the REMS program. Of these, 6711 patients were tolvaptan-naïve at the time of REMS enrollment and were included in this analysis. The remaining patients in the REMS program had been enrolled in clinical trials and transitioned from the investigational drug to commercial product; these patients were not included in the current analysis, as they had already been receiving tolvaptan for an extended period.
Demographic and clinical characteristics
The mean age at baseline was 45.9 years (range 15–95) and 51.7% of patients were female (Table 2). For the variables of race, ethnicity and alcohol classification, the majority of data were collected only after the implementation of the revised REMS Patient Enrollment Form, therefore, data on these variables are unavailable for ∼32% of patients. Of those with known race, 79.1% (3618/4573) were White, and of those with known ethnicity, 88.0% (4003/4551) were non-Hispanic/non-Latino.
Table 2.
Baseline demographic and clinical characteristics
| Variable | Tolvaptan-naïve safety population (N = 6711) | Subset with possible severe DILI (n = 60) | Subset without possible severe DILI (n = 6651) |
|---|---|---|---|
| Age (years) at enrollment | |||
| Mean (SD) | 45.9 (12.2) | 47.6 (11.3) | 45.9 (12.2) |
| Range | 15–95 | 21–68 | 15–95 |
| Gender, n (%) | |||
| Female | 3470 (51.7) | 36 (60.0) | 3434 (51.6) |
| Male | 3241 (48.3) | 24 (40.0) | 3217 (48.4) |
| Age (years), n (%) | |||
| ≤55 | 5203 (77.5) | 43 (71.7) | 5160 (77.6) |
| >55 | 1508 (22.5) | 17 (28.3) | 1491 (22.4) |
| Race, n (%) | |||
| Race unknown | 2138 (31.9) | 39 (65.0) | 2099 (31.6) |
| Race knowna | 4573 (68.1) | 21 (35.0) | 4552 (68.4) |
| White | 3618 (79.1) | 12 (57.1) | 3606 (79.2) |
| Black or African American | 435 (9.5) | 5 (23.8) | 430 (9.4) |
| American Indian or Alaska Native | 25 (0.5) | 1 (4.8) | 24 (0.5) |
| Asian | 220 (4.8) | 3 (14.3) | 217 (4.8) |
| Native Hawaiian or Pacific Islander | 16 (0.3) | 0 | 16 (0.4) |
| Other | 259 (5.7) | 0 | 259 (5.7) |
| Ethnicity | |||
| Ethnicity unknown | 2160 (32.2) | 39 (65.0) | 2121 (31.9) |
| Ethnicity knownb | 4551 (67.8) | 21 (35.0) | 4530 (68.1) |
| Hispanic or Latino | 548 (12.0) | 3 (14.3) | 545 (12.0) |
| Not Hispanic or Latino | 4003 (88.0) | 18 (85.7) | 3985 (88.0) |
| Alcohol classification, n (%) | |||
| Classification unknown | 2142 (31.9) | 39 (65.0) | 2103 (31.6) |
| Classification knownc | 4569 (68.1) | 21 (35.0) | 4548 (68.4) |
| Never drank | 1525 (33.4) | 8 (38.1) | 1517 (33.4) |
| Ex-drinker (stopped drinking ≥1 month ago) | 511 (11.2) | 4 (19.0) | 507 (11.1) |
| Current drinker | 2533 (55.4) | 9 (42.9) | 2524 (55.5) |
| Typical alcohol consumption (current drinkers only)d | |||
| Occasional (drink alcohol <1 time/week) | 1610 (63.6) | 6 (66.7) | 1604 (63.5) |
| Light (1–2 drinks/week) | 548 (21.6) | 1 (11.1) | 547 (21.7) |
| Moderate (3–7 drinks/week) | 313 (12.4) | 2 (22.2) | 311 (12.3) |
| Heavy (>7 drinks/week) | 62 (2.4) | 0 | 62 (2.5) |
The denominator for the percentage in each racial category shown is the number of patients with race known.
The denominator for the percentage in each ethnic category shown is the number of patients with ethnicity known.
The denominator for the percentage in each alcohol classification shown is the number of patients with classification known.
The denominator for the percentage in each typical alcohol consumption category shown is the number of patients who were current drinkers.
Exposure
Nearly two-thirds of patients (64.0%) had taken tolvaptan for >6 months, 44.9% had exposure >12 months, and 27.3% had exposure >18 months (Figure 2A). The most common dose was a daily split dose of 45/15 mg (Figure 2B).
FIGURE 2:

Duration of (A) tolvaptan exposure and (B) daily doses used in the safety population (N = 6711). A patient could change doses over the course of therapy; patients were counted at the dose they received most. If a patient received the same number of dispenses for more than one dose, the patient was counted at the higher dose.
Cases of possible severe DILI
A total of 60 (0.9%) of the 6711 patients on tolvaptan met the criteria for possible severe DILI, including 5 who met Criterion 1, 38 who met Criterion 2, and 27 who met Criterion 3 (some patients met multiple criteria). Evidence of a possible causal association with tolvaptan was not considered in the identification of possible severe DILI by any of the qualifying criteria. Levels of liver enzyme elevation in patients with possible severe DILI are shown in Figure 3. Laboratory data on ALT abnormalities were available for 34/60 patients and on AST abnormalities for 35/60 patients.
FIGURE 3:
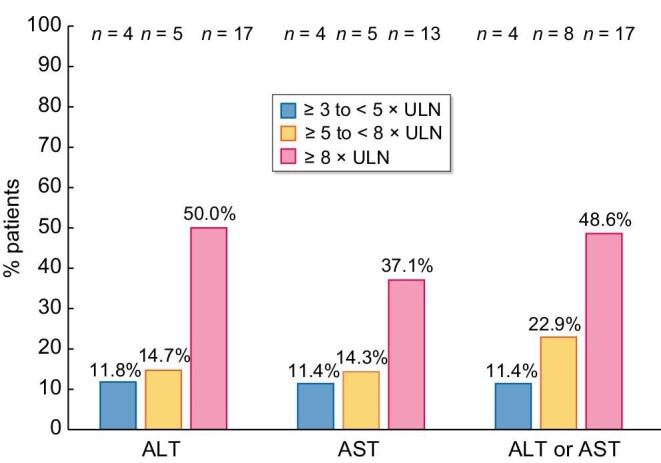
Transaminase elevations in patients with possible severe DILI for whom laboratory measurements were available (ALT, n = 34; AST, n = 35). Highest ALT/AST criteria met are shown. Patients with possible severe DILI for whom laboratory data were not provided are not shown.
The majority of patients with possible severe DILI [60.0% (n = 36)] were females; 71.7% of patients (n = 43) were ≤55 years of age, with a mean age at REMS enrollment of 47.6 11.32 years (Table 2). Based on the available patient information, no associations with possible severe DILI were seen for patient demographic and clinical characteristics, medical history, concurrent conditions or concurrent medications. The mean time to event in patients with possible severe DILI was 157.6 ± 146.49 days, with an interquartile range of 53–233 days. Analysis of the incidence of possible severe DILI events by time to onset indicated a pattern of greatest risk within the first 18 months of treatment and declining over time (Figure 4), consistent with the previously published clinical trial data [6]. However, conclusions regarding the time of susceptibility in the REMS are limited by the fact that only 27.3% of patients were exposed to tolvaptan for >18 months. Of the 60 patients with possible severe DILI, 48 (80%) are known to have discontinued tolvaptan. No data were available on the remaining 12 patients.
FIGURE 4:
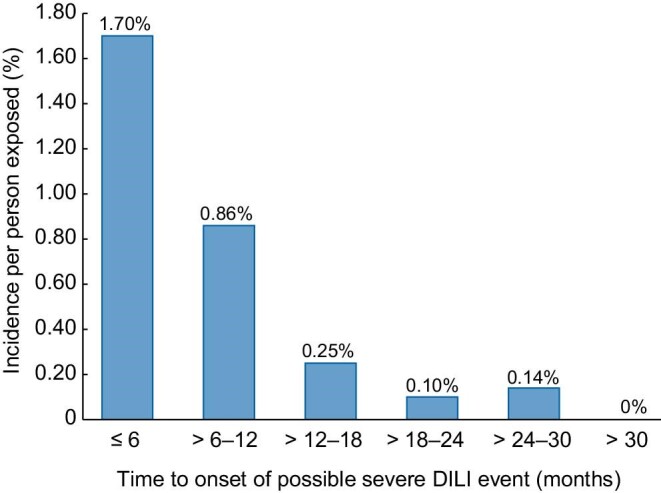
Incidence of possible severe DILI per person exposed by time (months) to onset. The figure excludes three patients for whom time to event could not be calculated.
Details on the outcomes of possible severe DILI events are limited to the available information. There were 30 (50.0%) patients with at least one event (patients could have more than one event) for which the outcome was not provided. Repeated follow-up attempts for these patients have been made, with attempts ongoing at the end of the reporting period. There were no fatal outcomes attributed to possible severe DILI events. At the time of data cutoff, 19 (31.7%) of 60 patients with possible severe DILI had recovered/resolved, 7 (11.7%) patients were recovering/resolving, 7 (11.7%) patients had not recovered, and for 27 (45.0%) patients the outcome data were not provided for any event(s) they had experienced. Two (3.3%) of 60 patients with possible severe DILI were reported to receive a treatment or intervention, 6 (10.0%) were hospitalized and 1 (1.7%) received an elective liver transplant. This liver transplant was not reported as a life-threatening event, no liver enzyme elevations were reported and the patient had underlying polycystic liver disease; the patient was on a liver transplant list and tolvaptan was stopped due to a request from the transplant team. It was not reported whether the patient was placed on the transplant list before or after initiating tolvaptan.
There was one known fatal event in a patient with possible severe DILI, with the death due to other causes that occurred in the setting of resolving transaminase elevations. A 61-year-old female experienced ALT/AST elevations ∼8 months after tolvaptan was initiated, with the elevations peaking at ALT >19 × ULN and AST >20 × ULN ∼2 months after onset. Alkaline phosphatase and total bilirubin levels were not provided. Events of acute renal and liver failure were also reported, but without any details or dates. Tolvaptan was withdrawn as a result of the transaminase elevations and the elevations were resolving 2.5 months after event onset, with ALT and AST >4 × ULN. Approximately 3 months from event onset, the patient underwent liver biopsy to rule out autoimmune hepatitis, which was complicated by post procedural hemorrhage and subsequent shock and hepatic encephalopathy. The patient died within 1 day of the procedure, with the causes of death listed as post procedural hemorrhage and shock. Information about the biopsy results or presence of coagulopathy was not reported. The independent HAC adjudicated the relationship of the liver enzyme elevations to tolvaptan as ‘possible’. The event could not be confirmed as DILI by the Otsuka GPV due to the lack of information in the case needed for complete and adequate medical assessment, as per the criteria for confirmation—such as data related to the time course of the event and possible confounding factors—as listed in the study Methods section. Based on the available information, the death of this patient was not a result of potential liver injury, but due to post-procedural complications.
The 60 events of possible severe DILI additionally included 4 events (6.7%) that were confirmed as serious and potentially fatal liver injury by the Otsuka GPV (Table 3). One patient, a 56-year-old female, exhibited ALT 1.25 × ULN and AST 1.05 × ULN 2 months after initiating tolvaptan. Three months later, she developed pruritus and experienced ALT >25 × ULN and AST >14 × ULN, and tolvaptan was withdrawn. The pruritus resolved and transaminases returned to the normal range. The HAC determined the relationship of the event with tolvaptan to be ‘possible’.
Table 3.
Confirmed cases of serious and potentially fatal liver injury in previously tolvaptan-naïve REMS participants (N = 6711)
| Patient sex, age (years) | Time to event after TLV initiation (months) | History of liver enzyme testing before eventa | Peak liver enzyme levelsb | Time to liver enzyme normalization after TLV was discontinued (months) | Additional information | Adjudicated causal relationship to TLV |
|---|---|---|---|---|---|---|
| F, 56 | 2 | Monthly monitoring of liver enzymes was performed | ALT >25 × ULN AST >14 × ULN Bilirubin < ULN |
5 | Patient experienced pruritus that also resolved TLV was discontinued 3 months after ALT/AST elevations were first noted |
Possible |
| F, 45 | 4 | Liver enzymes were within normal limits during monthly testing 1 month prior to event onset | ALT >13 × ULN AST >13 × ULN Bilirubin < ULN |
3 | Information was lacking on whether TLV was restarted after the event resolved After resolution of the event, ALT and AST increased to >4 × ULN; levels normalized again after TLV was presumably stopped a second time |
Possible |
| F, 68 | 5 | No testing for 49 days before the event | ALT >28 × ULN AST >41 × ULN Bilirubin >29 × ULN |
5 | Symptoms included fatigue, jaundice, abdominal distension and pain, bloating and nausea Losartan was also suspended, and the patient was hospitalized for observation during the event Not considered Hy's law because causes other than TLV could not be ruled out |
Insufficient data |
| F, 42 | 3 | Liver enzyme testing was not performed for 64 days prior to event onset. Baseline ALT/AST were within normal limits | ALT >55 × ULN AST >37 × ULN Bilirubin >9 × ULN |
10 | Negative hepatitis serology Event met Hy's law criteria |
Probable |
Testing before treatment and 2 and 4 weeks after TLV initiation, then monthly for the first 18 months of treatment is required.
The maximum elevation reached for each enzyme during the event is shown and the peaks for the different enzymes may have occurred at different times.
F, female; M, male; TLV, tolvaptan.
The second patient, a 45-year-old female, had ALT >13 × ULN and AST >12 × ULN 4 months after starting tolvaptan. Enzymes normalized 3 months after stopping tolvaptan. It was not stated whether tolvaptan was restarted, but 3 months after normalization, the patient had ALT and AST >4 × ULN. After tolvaptan was presumably stopped a second time, ALT and AST returned to normal. Although there were possibly two positive tolvaptan challenges and de-challenges, due to the lack of information provided, the HAC tentatively classified the relationship to tolvaptan as ‘possible’.
In a third case, a 68-year-old female with stage 3 chronic kidney disease, hypertension, and cystic liver exhibited ALT 26 × ULN, AST 39 × ULN, bilirubin 7.2 × ULN and alkaline phosphate 1.99 × ULN while experiencing symptoms of fatigue, jaundice, abdominal distension and pain, bloating and nausea. The event started after 5 months on tolvaptan and 49 days since the patient's previous liver enzyme assessment. Concomitant medications included losartan, loratadine and vitamin supplementation. Tolvaptan was discontinued, losartan was suspended and the patient was hospitalized for evaluation of liver enzymes. The patient's laboratory values returned to normal within 5 months and the HAC adjudicated the causality of the event as ‘insufficient data’. The limited information available for the event did not enable potential causes other than tolvaptan to be ruled out; accordingly, the event could not be confirmed as meeting the definition of Hy's law.
The fourth patient, who met Hy's law criteria, was a 42-year-old female with ADPKD and an autoimmune condition. She had transaminase levels in the normal range at baseline. Three months after starting tolvaptan, while taking a dose of 90 mg/day, she experienced ALT >11 × ULN and AST >6 × ULN and discontinued tolvaptan. The abnormality was observed after 2 months without liver function testing (monthly testing is required during the first 18 months of tolvaptan therapy for ADPKD). Transaminase levels peaked after tolvaptan discontinuation (ALT >55 × ULN, AST >37 × ULN), with concomitant total bilirubin >4 × ULN. Hepatitis serology for hepatitis A, B and C virus were negative. Ten months after tolvaptan was stopped, ALT, AST and total bilirubin had returned to the normal range without further medical procedure, indicating recovery. The HAC adjudicated the relationship of the event with tolvaptan to be ‘probable’.
No liver transplants or fatalities due to tolvaptan were reported in the REMS. As described above, an elective liver transplant was performed in a patient without elevated liver enzymes and a fatality occurred in another patient with possible severe DILI following a liver biopsy to rule out autoimmune hepatitis. Both events were included in the analyses because the criteria to identify possible severe DILI were applied regardless of suspected causality. However, neither event appeared to be attributable to tolvaptan. Thus no confirmed cases of severe DILI by FDA criteria were identified in the REMS dataset.
Comparison with clinical trial program in ADPKD
The incidence proportion of possible severe DILI (by Criteria 1, 2 and 3) on a population basis in the REMS was 0.9% (60/6711 patients) and in the ADPKD clinical trial program (TEMPO 3:4, REPRISE and their extension trials) was 5.5% (151/2743 patients). The incidence rate per 100 patient-years was 0.93 (60 events in 6486 patient-years) in the REMS, which was lower than the rate of 1.57 (154 events in 9786 patient-years) in the clinical trial program {IRR 0.587 [95% confidence interval (CI) 0.436–0.792]; P = .000411 by Wald method}. The number of events (n = 154) in the clinical trial program differed from the number of patients with possible severe DILI (n = 151) because some patients experienced more than one event.
It should be noted that this is an interim analysis and the comparison of incidence rates is still at an early stage. In clinical trials, susceptibility for liver toxicity was greatest within the first 18 months of tolvaptan treatment [6]. Subjects in the ADPKD clinical trial program on average had an exposure of 3.57 years, which was well beyond the 18-month susceptible period, whereas in the REMS population, exposure was greater than the 18-month period of susceptibility in only 27.3% of patients and >12 months in only 44.9% at the data used for this report. Although the present analysis of the REMS data suggests that the greatest risk for DILI is in the initial 18 months, the comparison of incidence should be repeated in the future when the follow-up duration in the REMS program achieves more maturity. This comparison will be performed on an annual basis and submitted to the FDA under the REMS program.
The incidence rate of possible severe DILI by Criterion 1 was 0.08 per 100 patient-years in the REMS population and 0.11 per 100 patient-years in the clinical trial population [IRR 0.686 (95% CI 0.238–1.973); P = .48]. The incidence rate by Criterion 2 was 0.59 per 100 patient-years in the REMS; no comparison could be made because there were no cases that met Criterion 2 in the clinical trial sample. The incidence rate by Criterion 3 was lower in the REMS than in the clinical trial population [0.42 versus 1.46 per 100 patient-years; IRR 0.285 (95% CI 0.189–0.430); P < .001].
DISCUSSION
This article provides real-world safety data on tolvaptan in a large population of patients. No case of severe DILI (an irreversible liver failure that is fatal or requires liver transplantation) related to the use of tolvaptan has been reported to date in the REMS program. One patient, with normal liver enzymes, underwent elective liver transplantation in a case that was not considered related to tolvaptan. Another patient died from post-procedural complications following a liver biopsy performed to rule out autoimmune hepatitis after experiencing possible severe DILI. One case meeting Hy's law criteria was identified, in which liver enzyme levels recovered to normal following discontinuation of tolvaptan. In this patient, abnormal transaminase levels were first observed 3 months after initiating tolvaptan and after 2 months without liver enzyme testing, indicating the importance of adherence to the required monthly testing schedule during the first 18 months of therapy. In addition to the case meeting Hy's law criteria, three other patients experienced events confirmed as serious and potentially fatal liver injury, one of whom had a 49-day gap since her previous liver function test. In all four patients, the event resolved after event detection and tolvaptan discontinuation.
Recent data from the Canadian Hepatic Safety Monitoring and Distribution Program (HSMDP) indicated that 8/1556 patients (0.5%) treated with tolvaptan for ADPKD in the real-world setting met criteria for permanent discontinuation (i.e. ALT or AST >8 × ULN, ALT or AST >5 × ULN for >2 weeks, ALT or AST >3 × ULN and total bilirubin >2 × ULN or international normalized ratio >1.5, ALT or AST >3 × ULN with persistent manifestations of hepatic injury). No cases of severe DILI were reported [12]. The laboratory criteria for permanent discontinuation in the HSMDP analysis were the same as those used in Criterion 3 of the REMS, which captured 27/6711 patients (0.4%). The frequency of enzyme elevations meeting criteria for permanent discontinuation in the REMS was thus comparable with that reported by the HSMDP.
The imbalance in cases of possible severe DILI by Criterion 2 between the REMS population (38 cases) and the clinical trial population (0 cases) may have been due to the need for assessment by the reporter on whether the event was life-threatening. Healthcare providers in clinical practice settings might be more conservative in their judgment of event severity (i.e. be more likely to consider an event life-threatening) than clinical trial investigators (who may be more familiar with criteria for serious liver injury). The difference highlights the need for follow-up information collection for REMS adverse event reports to characterize the reported incidents as fully as possible and enable precise assessment of event severity. Revisions made to REMS data collection forms to better define the criteria for serious liver injury have reduced the potential for missing information in the initial data collection.
Regarding limitations of this analysis, information for 30 patients has not been obtained to determine if the reported events met criteria for severe liver injury. Follow-up attempts are ongoing, including multiple phone calls and mailings, and severity classification of these possible severe DILI events is pending. Post marketing safety surveillance faces inherent limitations in the follow-up of patients treated in routine practice relative to the carefully controlled setting of a clinical trial. Patients’ monthly laboratory values are not captured within the REMS program, given that such reporting places an additional burden on healthcare providers. However, prescribing physicians are required to attest every 3 months for the first 18 months of treatment, and every 6 months thereafter, that they are conducting periodic liver monitoring per REMS guidance for all patients enrolled in the REMS, and the absence of such data for a patient should not be construed as evidence that the tests are not being performed as required. Once a potential DILI event is reported to Otsuka, laboratory values are requested from the prescriber as a part of routine pharmacovigilance activities.
There are also limitations resulting from the shorter duration of exposure to tolvaptan in the REMS program relative to the tolvaptan clinical trials in ADPKD. A majority of previously tolvaptan-naïve patients in the REMS have been on tolvaptan therapy for <12 months, whereas the window of susceptibility for risk of liver injury with tolvaptan administration in ADPKD is up to 18 months [6]. Given that patients sometimes discontinue tolvaptan after a brief period due to aquaretic side effects, the duration of treatment for some individuals with <12 months of exposure may have been short, potentially biasing the analysis by the inclusion of patients unlikely to have experienced liver-related adverse events.
The REMS population presents a lower incidence rate of possible severe DILI when compared with the clinical trial population; however, caution should be applied in interpreting these data at this time due to the limited duration of exposure to the drug in the REMS population. Based on the findings of this interim analysis and assuming these observations are confirmed in continuous longitudinal analyses of these data, it is likely that a major contributor to the lower rate in the REMS population is the monthly monitoring of liver enzyme levels for the first 18 months of tolvaptan treatment in ADPKD and every 3 months thereafter, which enables prompt detection of abnormalities and, if necessary, drug discontinuation. Thus the REMS program is facilitating management of risk in the real world and limiting harm to patients appropriately. In contrast, in TEMPO 3:4, the monitoring of liver enzymes was being conducted every 4 months prior to the change in frequency of monitoring in the clinical trials, which may have resulted in more subjects experiencing higher elevations of liver enzymes in the trial population, as any elevations in transaminases were not being detected early enough to monitor closely or allow therapy interruption when necessary. Recent surveillance data from the real-world use of tolvaptan in Japanese patients with ADPKD support the value of monthly liver enzyme monitoring in mitigating the risk of DILI events [13].
Within the context of the study limitations, the preliminary incidence of possible severe DILI appears to be lower or no worse than that reported in clinical trials; however, this assessment needs to be repeated in the future when more data are collected in this ongoing REMS program. Following each annual liver adverse event report that will be submitted to the FDA under the terms of the REMS program, additional analyses will be conducted to further characterize the liver safety profile of this drug. Otsuka is committed to reporting these data as they become available to optimize patient benefit and care.
Supplementary Material
ACKNOWLEDGEMENTS
This study was funded by Otsuka, which participated in the design of the study; the collection, analysis and interpretation of data; writing of the report and the decision to submit the report for publication. Writing and editorial services in preparation of the manuscript were provided by BioScience Communications (New York, NY), activities that were also funded by Otsuka.
Contributor Information
Alvin Estilo, Otsuka Pharmaceutical Development & Commercialization, Inc., Rockville, MD, USA.
LaRee Tracy, Otsuka Pharmaceutical Development & Commercialization, Inc., Rockville, MD, USA.
Carol Matthews, UBC, Blue Bell, PA, USA.
Michele Riggen, UBC, Blue Bell, PA, USA.
Annette Stemhagen, UBC, Blue Bell, PA, USA.
Timothy Wilt, Otsuka Pharmaceutical Development & Commercialization, Inc., Rockville, MD, USA.
Anatoliy Krakovich, Otsuka Pharmaceutical Development & Commercialization, Inc., Rockville, MD, USA.
Charlotte Jones-Burton, Otsuka Pharmaceutical Development & Commercialization, Inc., Rockville, MD, USA.
Vinu George, Otsuka Pharmaceutical Development & Commercialization, Inc., Rockville, MD, USA.
Robert McQuade, Otsuka Pharmaceutical Development & Commercialization, Inc., Rockville, MD, USA.
Mirza Rahman, Otsuka Pharmaceutical Development & Commercialization, Inc., Rockville, MD, USA.
FUNDING
This study was funded by Otsuka Pharmaceutical Development & Commercialization (Rockville, MD).
AUTHORS’ CONTRIBUTIONS
A.E., T.W., R.M.Q. and M. Rahman conceptualized the research idea and study design. A.E., T.W., A.K. and M. Rahman were responsible for data acquisition. A.E., C.M., M. Riggen, T.W., A.K., C.J.B., V.G., R.M.Q. and M. Rahman performed data analysis/interpretation. M. Rahman and L.T. performed statistical analysis. A.S., C.J.B., V.G. and M. Rahman were responsible for supervision or mentorship. Each author contributed important intellectual content during manuscript drafting or revision, had full access to the data and approved the submitted version.
DATA AVAILABILITY STATEMENT
To submit inquiries related to Otsuka clinical research or to request access to individual participant data (IPD) associated with any Otsuka clinical trial, please visit https://clinical-trials.otsuka.com/. For all approved IPD access requests, Otsuka will share anonymized IPD on a remotely accessible data-sharing platform.
CONFLICT OF INTEREST STATEMENT
A.E., L.T., T.W., A.K., C.J.B., V.G., R.M.Q., and M. Rahman are employees of Otsuka Pharmaceutical Development & Commercialization (Rockville, MD). C.M., M. Riggen and A.S. are employees of UBC, which provides REMS administration and analytic services for Otsuka.
REFERENCES
- 1. Chebib FT, Torres VE. Autosomal dominant polycystic kidney disease: core curriculum 2016. Am J Kidney Dis 2016; 67: 792–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Torres VE, Chapman AB, Devuyst O et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2012; 367: 2407–2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Torres VE, Chapman AB, Devuyst O et al. Multicenter, open-label, extension trial to evaluate the long-term efficacy and safety of early versus delayed treatment with tolvaptan in autosomal dominant polycystic kidney disease: the TEMPO 4:4 Trial. Nephrol Dial Transplant 2018; 33: 477–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Torres VE, Chapman AB, Devuyst O et al. Tolvaptan in later-stage autosomal dominant polycystic kidney disease. N Engl J Med 2017; 377: 1930–1942 [DOI] [PubMed] [Google Scholar]
- 5. US Food and Drug Administration . Jynarque Approval Letter. 23 April 2018. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/204441Orig1s000Approv.pdf (7 November 2021, date last accessed) [Google Scholar]
- 6. Watkins PB, Lewis JH, Kaplowitz N et al. Clinical pattern of tolvaptan-associated liver injury in subjects with autosomal dominant polycystic kidney disease: analysis of clinical trials database. Drug Saf 2015; 38: 1103–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. US Food and Drug Administration . Guidance for Industry, Drug-Induced Liver Injury: Premarketing Clinical Evaluation. 2009. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM174090.pdf. (5 November 2021, date last accessed) [Google Scholar]
- 8. US Food and Drug Administration . Risk Evaluation and Mitigation Strategy (REMS) Document. JYNARQUE (tolvaptan) REMS Program. December 2019. https://www.accessdata.fda.gov/drugsatfda_docs/rems/Jynarque_2019_12_03_REMS_Full.pdf. (7 November 2021, date last accessed) [Google Scholar]
- 9. Mosedale M, Watkins PB. Drug-induced liver injury: advances in mechanistic understanding that will inform risk management. Clin Pharmacol Ther 2017; 101: 469–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. European Association for the Study of the Liver . EASL clinical practice guidelines: drug-induced liver injury. J Hepatol 2019; 70: 1222–1261 [DOI] [PubMed] [Google Scholar]
- 11. Rockey DC, Seeff LB, Rochon J et al. Causality assessment in drug-induced liver injury using a structured expert opinion process: comparison to the Roussel-Uclaf causality assessment method. Hepatology 2010: 51: 2117–2126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McFarlane P, Parfrey P, Bichet DG et al. Canadian real-world assessment of tolvaptan in autosomal polycystic kidney disease (ADPKD): C-MAJOR study and safety monitoring and distribution program. Poster presented at American Society of Nephrology Kidney Week 2020 Reimagined, 22–25 October 2020, virtual [Google Scholar]
- 13. Mochizuki T, Muto S, Miyake M et al. Safety and efficacy of tolvaptan in real-world patients with autosomal dominant polycystic kidney disease—interim results of SLOW-PKD surveillance. Clin Exp Nephrol 2021; 25: 1231–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
To submit inquiries related to Otsuka clinical research or to request access to individual participant data (IPD) associated with any Otsuka clinical trial, please visit https://clinical-trials.otsuka.com/. For all approved IPD access requests, Otsuka will share anonymized IPD on a remotely accessible data-sharing platform.