

Abstract
Frontotemporal dementia and amyotrophic lateral sclerosis (FTD-ALS) are associated with both a repeat expansion in the C9orf72 gene and mutations in the TANK-binding kinase 1 (TBK1) gene. We found that TBK1 is phosphorylated in response to C9orf72 poly(Gly-Ala) [poly(GA)] aggregation and sequestered into inclusions, which leads to a loss of TBK1 activity and contributes to neurodegeneration. When we reduced TBK1 activity using a TBK1-R228H (Arg228→His) mutation in mice, poly(GA)-induced phenotypes were exacerbated. These phenotypes included an increase in TAR DNA binding protein 43 (TDP-43) pathology and the accumulation of defective endosomes in poly(GA)-positive neurons. Inhibiting the endosomal pathway induced TDP-43 aggregation, which highlights the importance of this pathway and TBK1 activity in pathogenesis. This interplay between C9orf72, TBK1, and TDP-43 connects three different facets of FTD-ALS into one coherent pathway.
One-Sentence Summary:
Two FTD-ALS-associated mutations converge on the endosomal pathway, the disruption of which induces TDP-43 aggregation.
Frontotemporal dementia and amyotrophic lateral sclerosis (FTD-ALS) are characterized by the formation of protein inclusions, notably the aggregation of TAR DNA-binding protein 43 (TDP-43). Aggregates imply impaired protein clearance, and several FTD-ALS-associated genes influence proteostasis, including TANK-binding kinase 1 (TBK1) (1). Loss-of-function mutations in TBK1 are risk factors for FTD-ALS (2) and studies in SOD1 mouse models have suggested that they require a so-called second hit to become pathogenic (3–5). Accordingly, some TBK1 carriers also harbor a mutation in another disease-associated gene (6, 7). Rare patients who co-harbor a TBK1 mutation and a G4C2 repeat expansion in chromosome 9 open reading frame 72 (C9orf72) (8, 9) present with an earlier age of onset and a more rapid disease course (7). Thus, TBK1 could modulate C9orf72-associated FTD-ALS (c9FTD-ALS).
We used an adeno-associated virus (AAV)-based C9orf72-(G4C2)149 mouse model of c9FTD-ALS (10) to determine whether the repeat expansion influenced TBK1 phosphorylation at S172, a modification associated with its kinase-active form. Although total TBK1 levels were comparable in cortical brain lysates from C9orf72-(G4C2)149 mice and C9orf72-(G4C2)2 controls, mice expressing the expanded repeat showed an increase in the ratio of phosphorylated TBK1 (pTBK1) to total TBK1 (Fig. 1A, B). In the brains of C9orf72-(G4C2)149 mice, pTBK1 localized to puncta (Fig. 1C, D). We investigated whether these puncta corresponded to dipeptide repeat (DPR) protein aggregates. Five DPRs are produced from the repeat associated non-ATG translation of the G4C2 repeat; all five aggregate in the brains of c9FTD-ALS patients and mice (10–14). Three DPRs co-localized with pTBK1 in C9orf72-(G4C2)149 mice: poly(GA), poly(GR), and poly(PR) (Fig. 1E and Fig. S1A). These three DPRs are toxic in vivo (15–17).
Fig. 1. pTBK1 accumulates into DPR aggregates in c9FTD-ALS.
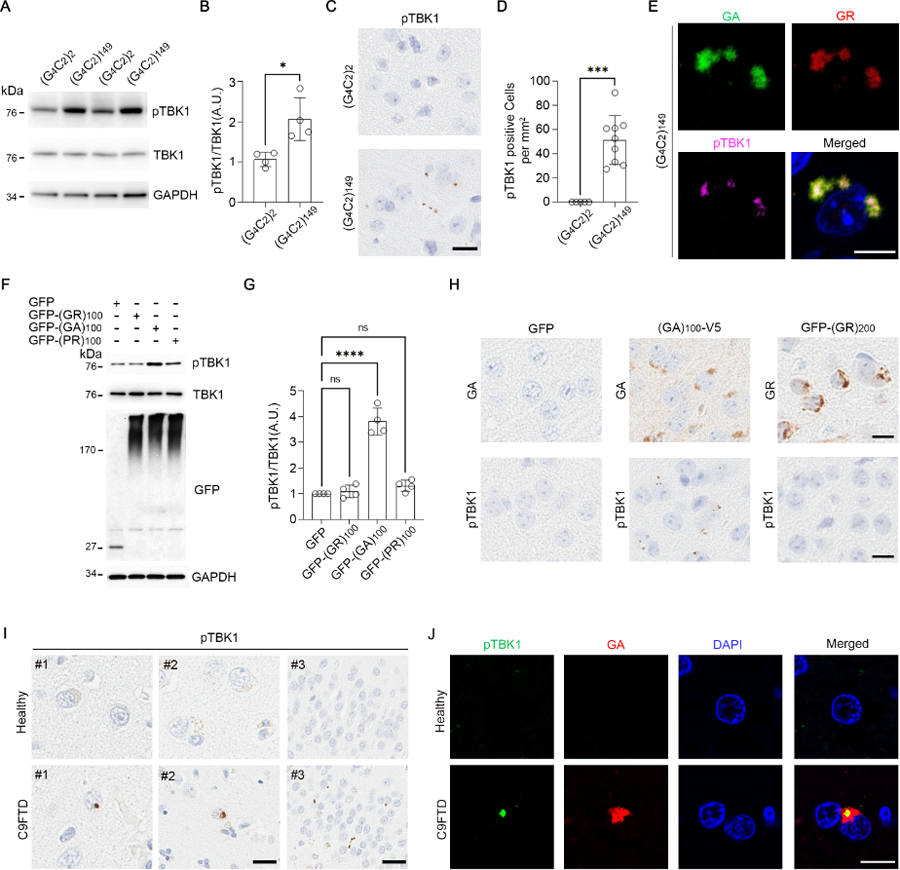
(A and B) Immunoblot (A) and quantification (B) of 6-month-old mouse cortical lysates (N = 4 mice). GAPDH, glyceraldehyde-3-phosphate dehydrogenase; A.U., arbitrary units. (C and D) Images (C) and quantification (D) of pTBK1 in 6-month-old mouse cortex (N = 9). Scale bar, 20 μm. (E) Immunofluorescence in 6-month-old mouse cortices. Scale bar, 10 μm. (F and G) Immunoblot (F) and quantification (G) of HEK293T lysates (N = 4 independent experiments). GFP monomer: ~27k Da; GFP-DPRs: high molecular weight (HMW) smears. (H) Immunostaining in the cortex of 3.5-month-old GFP and (GA)100-V5 mice and 2-week-old GFP-(GR)200 mice. Scale bars, 20 μm. (I) Immunostaining in healthy and C9-FTD frontal cortex (images labeled #1 and #2; scale bar, 10 μm) and hippocampus (images labeled #3; scale bar, 50 μm). (J) Immunofluorescence in healthy and C9-FTD frontal cortex. Scale bar, 10 μm. DAPI, 4′,6-diamidino-2-phenylindole. Data are means ± standard deviations (SDs). *P ≤ 0.05; ***P ≤ 0.001; ****P ≤ 0.0001; ns, nonsignificant. Statistics by Student’s t tests in (B) and (D) and one-way analysis of variance (ANOVA) in (G). Single-letter abbreviations for the amino acid residues are as follows: A, Ala; G, Gly; P, Pro; R, Arg.
We next explored which DPR or DPRs were responsible for these effects on TBK1 phosphorylation and localization. In HEK293T cells, green fluorescent protein (GFP)-(GA)100 increased the pTBK1-to-total TBK1 ratio and coimmunoprecipitated with TBK1, whereas GFP-(GR)100 and GFP-(PR)100 did not (Fig. 1F, G and Fig. S1B). Accordingly, pTBK1 formed puncta in the brains of mice expressing poly(GA)100-V5, but not in the brains of mice expressing GFP-(GR)200 or GFP alone (Fig. 1H). Thus, poly(GA) was responsible for triggering the phosphorylation and aggregation of TBK1. Additionally, we observed pTBK1 puncta in the frontal cortex and hippocampus of C9-FTD patients but not in healthy controls (Fig. 1I and Table S1); these puncta colocalized with poly(GA) (Fig. 1J and Fig. S1C). Puncta were also detected by a total TBK1 antibody in C9-FTD (Fig. S1D).
TBK1 is activated when the oligomerization of its interactors triggers its clustering and trans-autophosphorylation (18–20): The oligomerization of poly(GA) could induce TBK1 phosphorylation by a similar mechanism. In cells, the proline-interrupted poly(GA5P) – which is unable to form compact aggregates (15) – failed to induce the phosphorylation of TBK1 (Fig. 2A, B), did not colocalize with pTBK1 (Fig. 2C), and did not coimmunoprecipitate with TBK1 (Fig. 2F). Moreover, pTBK1 shifted to the insoluble fraction of cell lysates when poly(GA) was aggregated (Fig. 2D, E and Fig. S2A). The interaction of TBK1 with poly(GA) was independent of its phosphorylation, as coimmunoprecipitation still occurred when the S172 locus was mutated (Fig. S2B). Thus, TBK1 autophosphorylates after clustering into poly(GA) aggregates.
Fig. 2. Poly(GA) aggregation sequesters TBK1 into inclusions, which limits its activity.
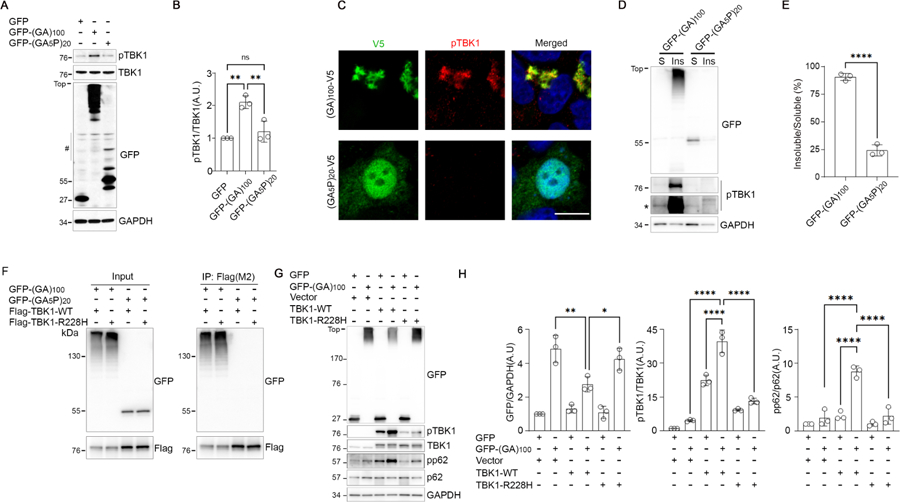
(A and B) Immunoblot (A) and quantification (B) of HEK293T lysates (N = 3 independent experiments). The number symbol in (A) indicates nonspecific bands. (C) Immunofluorescence of HEK293T cells. Scale bar, 10 μm. (D and E) Immunoblot (D) and quantification € of HEK293T lysate fractions (N = 3). S, soluble; Ins, insoluble. The asterisk in (D) indicates higher exposure. (F) Coimmunoprecipitation analysis of HEK293T lysates. P, immunoprecipitation. (G and H) Immunoblot (G) and quantification (H) of HEK293T lysates (N = 3). In all blots, GFP monomer: ~27 kDa; GFP-DPRs: ~50 kDa unaggregated, HMW oligomerized. Data are means ± SDs. *P ≤ 0.05; **P ≤ 0.01; ****P ≤ 0.0001; ns, nonsignificant. Statistics by one-way ANOVA.
We hypothesize that as long as poly(GA) inclusions persist in diseased neurons, pTBK1 clusters cannot disassemble. Because TBK1 regulates the autophagic clearance of aggregates (18), its sequestration could increase poly(GA) inclusion levels. Replenishing TBK1 could help alleviate this burden. When we overexpressed wildtype TBK1 in cells expressing GFP-(GA)100, we saw a significant increase in the phosphorylation of the autophagy adaptor p62, a known target of TBK1 (18), and a decrease in poly(GA) aggregation in immunoblots (Fig. 2G, H). This decrease was not observed when we overexpressed TBK1-R228H (Arg228→His) (Fig. 2G, H), an ALS-associated partial loss-of-function mutant with reduced autophosphorylation activity (5). As expected, overexpression of wildtype TBK1 increased the ratio of pTBK1 to total TBK1 and the ratio of phosphorylated p62 to total p62, while TBK1-R228H only slightly increased these ratios (Fig. 2G, H). The TBK1-R228H mutation did not hinder its interaction with poly(GA) (Fig. 2F).
If TBK1 is sequestered by poly(GA), further impairing TBK1 activity would be detrimental to cells. Accordingly, mutations that reduce TBK1 activity would exacerbate defects in poly(GA)-positive neurons – explaining why patients with mutations in both C9orf72 and TBK1 show a more severe disease course (7). We modeled this double-mutant condition by using an established AAV-based protocol (10, 15) to express GFP or (GA)100-V5 in TBK1-R228H knockin (5) and wild-type mice. Mice expressing (GA)100-V5 developed a progressive hindlimb clasping defect (Fig. 3A) and showed impaired performance in the rotarod (Fig. S3A), hanging wire (Fig. 3B), and fear conditioning tests (Fig. S3B). These phenotypes were enhanced in TBK1-R228H mice (Fig. 3A, B and Fig. S3). Mice expressing GFP behaved normally (Fig. 3A, B and Fig. S3), consistent with the TBK1-R228H model being phenotypically normal (5).
Fig. 3. Reducing TBK1 activity exacerbates the behavioral, neurodegenerative, and pathological effects of poly(GA) in mice.
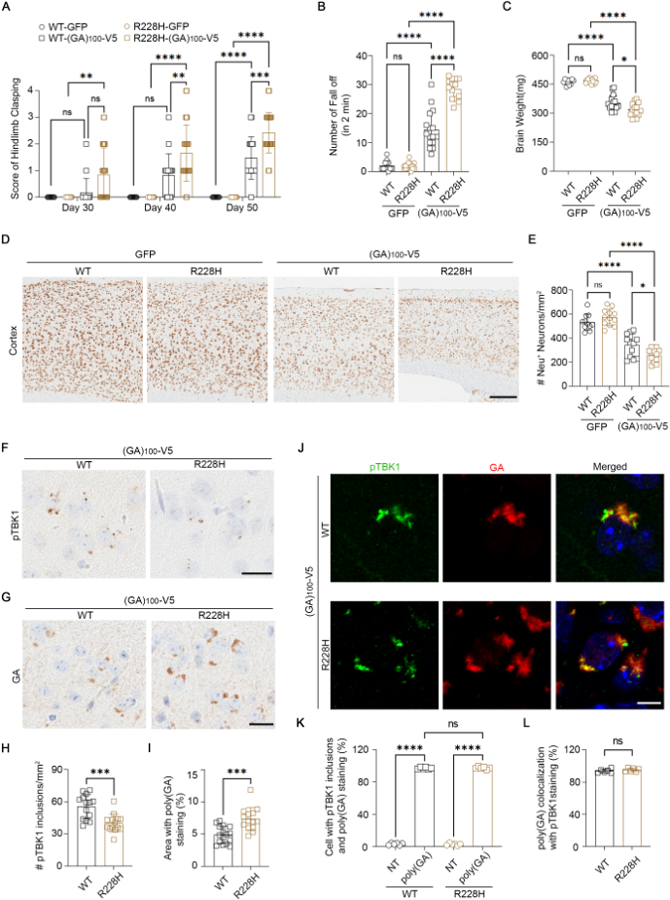
(A) Hindlimb clasping scores (N = 9, 10, 16, and 14 mice at each time point). WT, wild-type. (B) Hanging wire test in 3-month-old mice (N = 9, 10, 16, and 14). (C) Whole mouse brain weight (N = 12, 15, 16, and 14). (D and E) Images (D) and quantification (E) of NeuN in mouse cortex (N = 11, 12, 13, and 12). Scale bar, 200 µm. (F and H) Images (F) and quantification (H) of pTBK1 in mouse cortex (N = 14 each). Scale bar, 20 µm. (G and I) Images (G) and quantification (I) of poly(GA) in mouse cortex (N = 14 and 16). Scale bar, 20 µm. (J to L) Immunofluorescence (J) and quantification [(K) and (L)] in mouse cortex (N = 6 each). Scale bar, 10 μm. NT, non-transduced or poly(GA)-negative. Except in (A) and (B), mice were 3.5 months old. Data are means ± SDs. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001; ns, nonsignificant. Statistics by two-way ANOVA in (A), Student’s t test in (H) to (I), and one-way ANOVA in remaining panels.
Mice expressing (GA)100-V5 showed reduced total brain weight (Fig. 3C) and cortical neuronal loss (Fig. 3D, E). Both phenotypes were enhanced on the TBK1-R228H background (Fig. 3D, E), where the cortex was noticeably thinner (Fig. 3D). Neuroinflammation levels often correlate with neurodegeneration in mice, and as such, expression of (GA)100-V5 increased the expression of the microglial markers Iba1 (Fig. S4A) and CD68 (Fig. S4B) and the astroglial marker GFAP (Fig. S4C). This was enhanced on the TBK1-R228H background (Fig. S4A-C).
As in SOD1 models (3, 5), our “double mutant” study is consistent with the idea that reducing TBK1 activity in neurons cell-autonomously exacerbates FTD-ALS phenotypes. Loss of glial TBK1 may be beneficial (3, 5), however, because altered immune responses could modulate c9FTD-ALS (21). We investigated whether TBK1-R228H affected TBK1-regulated immune response pathways (18) in our mice. Poly(GA) increased the expression of two interferon-stimulated genes, IFIT1 (Fig. S4D) and MX1 (Fig. S4E), but their levels were comparable in wild-type and TBK1-R228H mice. Thus, the effects of TBK1-R228H on poly(GA)-induced defects may not involve the interferon pathway, but further experiments are required to confirm this hypothesis.
We observed pTBK1 puncta in the cortex of both wild-type and TBK1-R228H mice expressing (GA)100-V5, although pTBK1 puncta were less abundant in the TBK1-R228H mice due to the mutant’s reduced autophosphorylation activity (Fig. 3F, H). Poly(GA) inclusions were increased in TBK1-R228H mice compared to wild-type mice (Fig. 3G, I). These inclusions were intracellular and neuronal (Fig. S4F). There was also a corresponding increase in the number of puncta detected by a total TBK1 antibody in the TBK1-R228H mice (Fig. S4G, H). Total TBK1 and pTBK1 colocalized with poly(GA) aggregates in mice expressing (GA)100-V5 (Fig. S4I and Fig. 3J-L). Thus, the R228H mutation did not prevent TBK1 sequestration but did reduce its activity, impairing its ability to induce aggregate clearance and increasing poly(GA) pathology.
Loss of TBK1 impairs endosome maturation in human motor neurons (22). We asked whether TBK1 sequestration also impaired this pathway. Mice expressing (GA)100-V5 developed enlarged early endosomes in the cortex (Fig. 4A and Fig. S5A) that were more abundant on the TBK1-R228H background (Fig. 4A, B). We also observed aberrant Rab7-positive late endosomes in mice expressing (GA)100-V5 (Fig. S5B-D). These endosomal defects occurred in cells that contained poly(GA) aggregates (Fig. 4C, D), suggesting a causal link.
Fig. 4. Poly(GA) induces endosomal defects that trigger TDP-43 proteinopathy, and a reduction in TBK1 activity intensifies this process.
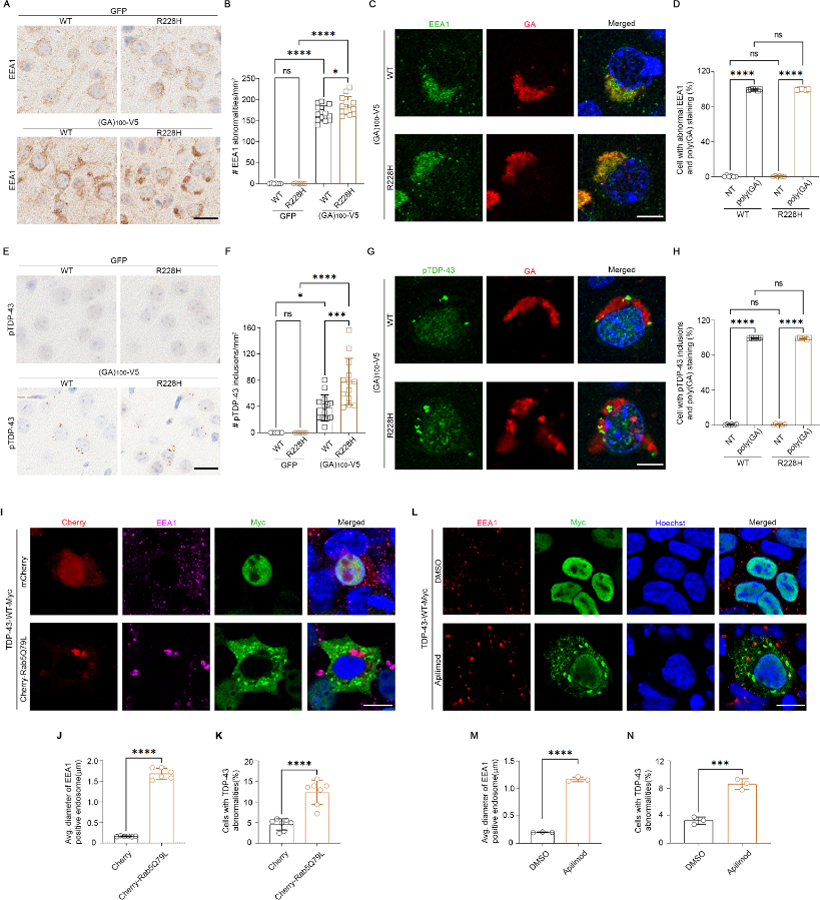
(A and B) Images (A) and quantification (B) of early endosome antigen 1 (EEA1) in mouse cortex (N = 6, 6, 13, and 13 mice). Scale bar, 20 µm. (C and D) Immunofluorescence (C) and quantification (D) in mouse cortex (N = 6 each). Scale bar, 10 μm. (E and F) Images (E) and quantification (F) of pTDP-43 in mouse cortex (N = 6, 6, 14, and 13). Scale bar, 20 µm. (G and H) Immunofluorescence (G) and quantification (H) in mouse cortex (N = 6 each). Scale bar, 10 μm. (I to K) Immunofluorescence (I) and quantification [(J) and (K)] in HEK293T cells. Scale bar, 10 μm. (L to M) Immunofluorescence (L) and quantification [(M) and (N)] in HEK293T cells ± apilimod treatment (N = 3 independent experiments). Scale bar, 10 μm. All mice were 3.5 months old. Data are means ± SDs. *P ≤ 0.05; ***P ≤ 0.001; ****P ≤ 0.0001; ns, nonsignificant. Statistics by Student’s t test in (J), (K), (M), and (N) and by one-way ANOVA in remaining panels.
TBK1-mediated endosomal defects induce TDP-43 pathology in human motor neurons (22). Rare pTDP-43 inclusions are also observed in mice expressing poly(GA) (15). We asked whether our double-mutant mice would show more pronounced pTDP-43 pathology. In fact, pTDP-43 inclusions were more abundant in mice expressing (GA)100-V5 on the TBK1-R228H background (Fig. 4E, F). These inclusions occurred in poly(GA)-positive cells (Fig. 4G, H), which suggests that poly(GA) aggregates, endosomal defects, and TDP-43 pathology are linked.
To determine whether endosomal defects are sufficient to induce TDP-43 pathology, we used a constitutively active Rab5-Q79L mutant to induce enlarged early endosomes in cells (Fig. 4I, J and Fig. S6A-C). In a subset of these cells, exogenously expressed TDP-43, but not GFP, showed cytoplasmic aggregation and reduced nuclear localization (Fig. 4I, K and Fig. S6A). We saw similar results when we used the phosphatidylinositol-3-phosphate 5-kinase inhibitor apilimod to disrupt endosome maturation (Fig. 4L-N). Rab5-Q79L also induced endogenous TDP-43 aggregation but not its nuclear depletion (Fig. S6B, D). Therefore, endosomal defects induce TDP-43 aggregation, but TDP-43 nuclear depletion may be downstream of initial cytoplasmic seeding. Defects in nucleocytoplasmic transport predate TDP-43 mislocalization in c9FTD-ALS (23), and TDP-43 aggregates sequester nuclear pore components and related factors (24).
We suggest a model in which poly(GA) inclusions sequester TBK1, leading to a reduction in TBK1 function that disrupts endosome maturation and induces TDP-43 aggregation. Although our work focuses on C9orf72 and TBK1, mutations in the multiple FTD-ALS genes affet endosomes or lysosomes in cells (25–28). Enlarged endosomes are also seen in an ALS-like mouse models and sporadic ALS tissue (27, 29). Furthermore, filaments of lysosomal transmembrane protein 106B (TMEM106B) are a pathological hallmark in multiple neurodegenerative diseases, including FTD-ALS (30–32), and variants in TMEM106B are associated with FTD (33). Several disease processes therefore converge on the endolysosomal pathway, the disruption of which could sensitize cells to pathogenic insults, like protein aggregation, and act as a primary driver of TDP-43 proteinopathy and neurodegeneration.
Supplementary Material
Acknowledgments:
We are grateful to all individuals and their families who agreed to donate post-mortem tissue to the Mayo Clinic Brain Bank. We also acknowledge V. R. Phillips and A. L. Librero for their help in tissue sectioning and counter staining and L. J. Lewis-Tuffin for her help with confocal microscopy and the scanning of tissue slides.
Funding:
National Institutes of Health/National Institute of Neurological Disorders and Stroke grant R35NS097273 (L.P.)
National Institutes of Health/National Institute of Neurological Disorders and Stroke grant U54NS123743 (M.P., L.P.)
National Institutes of Health/National Institute of Neurological Disorders and Stroke grant P01NS084974 (D.W.D., T.F.G., B.O., Y.-J.Z., L.P.)
National Institutes of Health/National Institute of Aging grant RF1AG062077 (J.D.F., L.P.)
National Institutes of Health/National Institute of Aging grant RF1AG062171 (L.P.)
National Institutes of Health/National Institute of Neurological Disorders and Stroke grant R01NS117461 (T.F.G., Y.-J.Z)
National Institutes of Health/ National Institute of Neurological Disorders and Stroke grant RF1NS120992 (M.P.)
The Mayo Clinic Foundation (L.P.)
The Biogen, Medicinova, Cytokinetics, Mitsubishi grants (B.O.)
Footnotes
Competing interests: L.P. and Mayo Clinic have licensed technology involving C9orf72 Repeat Expansion Constructs and Virus and AAV-C9orf72–149 Repeat Expansion Mouse Model. L.P. is a consultant for Expansion Therapeutics. M.P. is a consultant for Target ALS. B.O. has consulted for Biogen, Medicinova, Mitsubishi, AnnJi and Tsumura. The other authors declare that they have no competing interests.
Data and materials availability: All data are available in the main text or the supplementary materials.
References and Notes
- 1.Shahheydari H et al. , Protein Quality Control and the Amyotrophic Lateral Sclerosis/Frontotemporal Dementia Continuum. Front Mol Neurosci 10, 119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cui R, Tuo M, Li P, Zhou C, Association between TBK1 mutations and risk of amyotrophic lateral sclerosis/frontotemporal dementia spectrum: a meta-analysis. Neurol Sci 39, 811–820 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Brenner D et al. , Heterozygous Tbk1 loss has opposing effects in early and late stages of ALS in mice. J Exp Med 216, 267–278 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu D et al. , TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell 174, 1477–1491 e1419 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerbino V et al. , The Loss of TBK1 Kinase Activity in Motor Neurons or in All Cell Types Differentially Impacts ALS Disease Progression in SOD1 Mice. Neuron 106, 789–805 e785 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Lattante S et al. , Coexistence of variants in TBK1 and in other ALS-related genes elucidates an oligogenic model of pathogenesis in sporadic ALS. Neurobiol Aging 84, 239 e239–239 e214 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Van Mossevelde S et al. , Clinical features of TBK1 carriers compared with C9orf72, GRN and non-mutation carriers in a Belgian cohort. Brain 139, 452–467 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeJesus-Hernandez M et al. , Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Renton AE et al. , A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chew J et al. , Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol Neurodegener 14, 9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ash PE et al. , Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gendron TF et al. , Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol 126, 829–844 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mori K et al. , The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Mori K et al. , Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 126, 881–893 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Zhang YJ et al. , C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci 19, 668–677 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cook CN et al. , C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci Transl Med 12, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang YJ et al. , Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 363, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oakes JA, Davies MC, Collins MO, TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain 10, 5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang C et al. , Structural basis of STING binding with and phosphorylation by TBK1. Nature 567, 394–398 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu S et al. , Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Burberry A et al. , C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature 582, 89–94 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hao J et al. , Loss of TBK1 activity leads to TDP-43 proteinopathy through lysosomal dysfunction in human motor neurons. bioRxiv, 2021.2010.2011.464011 (2021). [Google Scholar]
- 23.Coyne AN et al. , G4C2 Repeat RNA Initiates a POM121-Mediated Reduction in Specific Nucleoporins in C9orf72 ALS/FTD. Neuron 107, 1124–1140 e1111 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chou CC et al. , TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci 21, 228–239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varga RE et al. , In Vivo Evidence for Lysosome Depletion and Impaired Autophagic Clearance in Hereditary Spastic Paraplegia Type SPG11. PLoS Genet 11, e1005454 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kunita R et al. , Homo-oligomerization of ALS2 through its unique carboxyl-terminal regions is essential for the ALS2-associated Rab5 guanine nucleotide exchange activity and its regulatory function on endosome trafficking. J Biol Chem 279, 38626–38635 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Chow CY et al. , Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 448, 68–72 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skibinski G et al. , Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 37, 806–808 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Palmisano R et al. , Endosomal accumulation of APP in wobbler motor neurons reflects impaired vesicle trafficking: implications for human motor neuron disease. BMC Neurosci 12, 24 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang A et al. , Homotypic fibrillization of TMEM106B across diverse neurodegenerative diseases. Cell 185, 1346–1355 e1315 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang YX et al. , Amyloid fibrils in disease FTLD-TDP are composed of TMEM106B not TDP-43. Nature, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schweighauser M et al. , Age-dependent formation of TMEM106B amyloid filaments in human brains. Nature, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Deerlin VM et al. , Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 42, 234–239 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zolotukhin S et al. , Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther 6, 973–985 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Chew J et al. , Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 348, 1151–1154 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Janocko NJ et al. , Neuropathologically defined subtypes of Alzheimer’s disease differ significantly from neurofibrillary tangle-predominant dementia. Acta Neuropathol 124, 681–692 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.