

ABSTRACT
Clostridia are a polyphyletic group of Gram-positive, spore-forming anaerobes in the Firmicutes phylum that significantly impact metabolism and functioning of the human gastrointestinal tract. Recently, Clostridia were divided into two separate classes, Clostridia and Erysipelotrichia, based on phenotypic and 16S rRNA gene-based differences. While Clostridia include many well-known pathogenic bacteria, Erysipelotrichia remain relatively uncharacterized, particularly regarding their role as a pathogen versus commensal. Despite wide recognition as a commensal, the erysipelotrichial species Clostridium innocuum has recently been associated with various disease states. To further understand the ecological and potential virulent role of C. innocuum, we conducted a genomic comparison across 38 C. innocuum isolates and 194 publicly available genomes. Based on colony morphology, we isolated multiple C. innocuum cultivars from the feces of healthy human volunteers (n = 5). Comparison of the 16S rRNA gene of our isolates against publicly available microbiota data sets in healthy individuals suggests a high prevalence of C. innocuum across the human population (>80%). Analysis of single nucleotide polymorphisms (SNPs) across core genes and average nucleotide identify (ANI) revealed the presence of four clades among all available genomes (n = 232 total). Investigation of carbohydrate and protein utilization pathways, including comparison against the carbohydrate-activating enzyme (CAZyme) database, demonstrated inter- and intraclade differences that were further substantiated in vitro. Collectively, these data indicate genetic variance within the C. innocuum species that may help clarify its role in human disease and health.
IMPORTANCE Clostridia are a group of medically important anaerobes as both commensals and pathogens. Recently, a new class of Erysipelotrichia containing a number of reassigned clostridial species has emerged, including Clostridium innocuum. Recent studies have implicated C. innocuum as a potential causative agent of diarrhea in patients from whom Clostridioides difficile could not be isolated. Using genomic and in vitro comparison, this study sought to characterize C. innocuum in the healthy human gut. Our analyses suggest that C. innocuum is a highly prevalent and diverse species, demonstrating clade-specific differences in metabolism and potential virulence. Collectively, this study is the first investigation into a broader description of C. innocuum as a human gut inhabitant.
KEYWORDS: Clostridium innocuum, genomics, metabolism, virulence, growth assay, gut microbiota, human microbiome
INTRODUCTION
Commensal bacteria, viruses, fungi, and protozoa, collectively termed microbiota, dominate all surfaces of multicellular hosts. The collective genes provided by individuals or groups of microbes maintain health of the host, providing functions such as colonization resistance against pathogens via multiple mechanisms, including nutrient niche exclusion (1–3), modulation of oxygen or pH gradients along the gut (4, 5), and production of metabolites that harm pathogens (6, 7). However, variability of the gut microbiota across individual hosts (8, 9) and lack of characterization of many common gut inhabitants (10) complicate discernible conclusions about many individual members. Recent genomic and phenotypic studies have highlighted strain-level diversity within prominent gut species that can extricate our understanding of the microbiota in health (11–13), which is not captured by 16S rRNA gene-based surveys.
The human gut microbiota is predominantly occupied by anaerobic bacteria, with the most abundant phyla being Firmicutes and Bacteroidetes (14). The diversity and function of several prevalent Bacteroidetes species have been extensively investigated (15), leading to their use as prominent model organisms to understand gut microbiota function (16). For example, species within the Bacteroides genus are known to harbor hundreds of polysaccharide utilization loci (PULs) that degrade different glycans and carbohydrates (16), ultimately supplying nutrients to both the host and surrounding microbes that contribute to protection from pathogens. Many prevalent taxa within the polyphyletic, Gram-positive Firmicutes phylum remain more nebulous. Within the gut, the Firmicutes phylum is comprised of three classes based on analyses of 16S rRNA nucleotide sequence, Bacilli, Clostridia, and Erysipelotrichia (17–19). Bacilli and Clostridia comprise well-studied members with major implications in industrial applications, health, and disease (20–23). Clostridia as a group have been demonstrated to induce beneficial immune responses, in part via their ability to produce short-chain fatty acids that can attenuate gut inflammation (24, 25). In comparison, the importance of Erysipelotrichia in the human gut microbiota remains relatively unexplored. Erysipelotrichia include species that share a genomic resemblance to Mollicutes, a class of parasitic bacteria that are characterized by their distinct lack of cell walls compared to the phylum Tenericutes (26). As a group, Erysipelotrichia in the gut have been associated with host lipid metabolism (27, 28) and disease in humans (29). In mice, expansion of Erysipelotrichia species has been observed following antibiotic treatment (30) or when fed a Western diet (31).
The role of the erysipelotrichial species Clostridium innocuum in human health remains especially ambiguous. C. innocuum was first isolated from an appendiceal abscess but was deemed innocuous due to a lack of virulence observed in mice and guinea pigs (32). Recently reclassified from its original clostridial designation (33), C. innocuum has been identified as part of the “normal” gut microbiota via its capability to biodegrade glucose ureide (34). Although initial phenotypic description of the organism suggests a nonmotile, nonvirulent nature of C. innocuum (32), current literature suggests otherwise. It has been implicated in extraintestinal infection and Clostridioides difficile-like antibiotic-associated diarrhea (34–36), as well as in case studies of bacteremia, endocarditis, osteomyelitis, and peritonitis (27, 37–39). Most recently, a study on Crohn’s disease (CD) conducted in mice identified C. innocuum in inflamed intestinal tissue of patients with CD (40). Despite these studies, a direct virulence mechanism has yet to be identified (36, 40–42).
Given the putative prevalence of C. innocuum in the human gut microbiota, we aimed to investigate the genomic and phenotypic diversity of C. innocuum. We compared genomic phylogeny, functional capacity, and virulence factors across single isolates and publicly available genomes. Using a custom 16S rRNA database, we identified C. innocuum as a highly prevalent human gut inhabitant. Single nucleotide polymorphisms in core genes suggest that the C. innocuum species splits into multiple clades, characterized by differences in metabolism. While comparison to known virulence factors did not identify a direct link to previously associated disease studies, we did identify clade-specific putative virulence factors. Together, these data support a role for C. innocuum and related erysipelotrichial species in modulating the gut nutrient landscape, as well as a strain-specific role for potential virulence.
RESULTS
Clostridium innocuum is a prevalent human gut microbe.
We screened five fresh fecal samples for the presence of C. innocuum strains as part of a larger study focused on cultivation of gut commensal bacteria (Fig. 1A). Sanger sequencing of the full-length 16S rRNA gene from morphologically distinct colonies confirmed the presence of 38 isolates that matched C. innocuum (80% similarity) (see Table S1 in the supplemental material), with each fecal sample yielding at least three distinct colonies. While metagenomic and 16S rRNA gene-based surveys suggest C. innocuum may be a common resident of the human gut microbiota, its prevalence across the human population is unknown. To broadly identify the presence of C. innocuum within the human gut microbiota, we compared multiple available 16S rRNA data sets from previous human gut microbiota studies to a curated database consisting of the 16S rRNA gene from our isolates (43, 44). Within these data sets, approximately 80% of samples (n = 420) contained C. innocuum sequences, suggesting a high prevalence of C. innocuum within the human gut microbiota (Fig. 1B). Although the relative abundance (RA) of C. innocuum in feces retrieved from 16S rRNA gene-based sequencing data was relatively low across all samples (mean RA = 0.22%), samples collected from patients on antibiotics (mean RA = 0.40%; n = 96) or with sepsis (mean RA = 0.46%; n = 24) were significantly increased compared to healthy controls (mean RA = 0.16%; n = 250).
FIG 1.
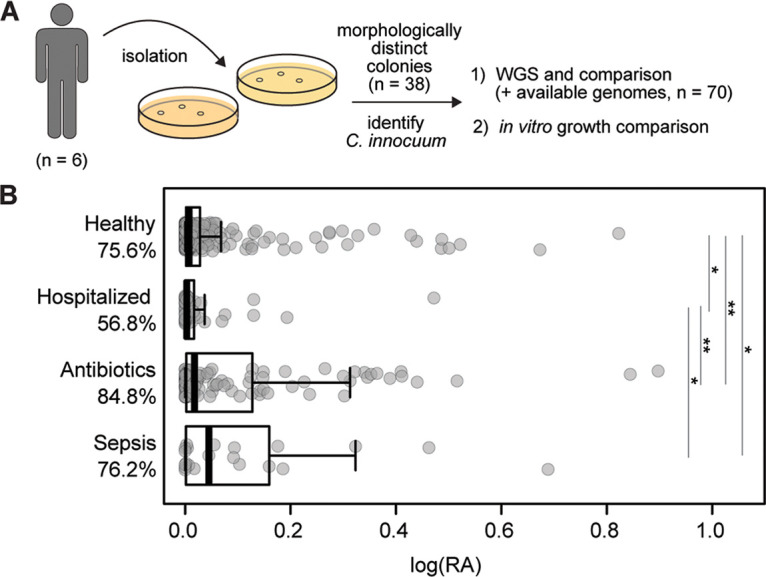
C. innocuum is a prevalent human gut bacterium. (A) Isolation pipeline design for C. innocuum. (B) Detection of C. innocuum in human feces across previously published 16S rRNA data sets using a custom classifier, categorized by published disease status. Log10 of relative abundance is displayed on the x axis, with prevalence (percent detected based on presence or absence). Pairwise Wilcoxon rank-sum test; *, P < 0.05; **, P < 0.005.
Excel sheet containing genome metadata. Download Table S1, XLSX file, 0.05 MB (48.8KB, xlsx) .
Copyright © 2023 Bhattacharjee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Whole-genome comparison reveals four C. innocuum clades.
We next sought to identify genomic heterogeneity among all available C. innocuum genomes, including isolates within the current study (n = 38), genomes available on the Genome Taxonomy Database (GTDB) (n = 40) (40), genomes sequenced in a previously published study associated with Crohn’s disease (n = 31) (45), a newly published data set from clinical samples (n = 119) (46), and four fully annotated genomes (C. innocuum strains 14501, I46, LC-LUMC, and 2959) (47–49) (Table S1). Isolates were sequenced using Illumina technology, assembled (average depth of coverage, 90×), and annotated using Prokka (50). The newly assembled full-length 16S rRNA gene from all 232 C. innocuum genomes was used for taxonomic identification using both NCBI BLAST and EZBioCloud databases. These comparisons, as well as the full genomic assembly compared against the GTDB database, confirmed all genomes as Erysipelotrichaceae species. An initial maximum-likelihood tree of the full-length 16S rRNA gene obtained from the whole-genome assemblies revealed that most species clustered under one branch (Fig. S1A), suggesting that the 16S rRNA gene may not be an appropriate proxy for distinguishing C. innocuum strains.
Maximum-likelihood trees of C. innocuum based on full-length 16S rRNA gene- and PhyloPhlAn-selected markers. (A) Maximum-likelihood tree of full-length 16S rRNA generated by RAxML, aligned in Clustal Omega (99). (B) Maximum-likelihood tree of C. innocuum genomes based on 400 selected markers generated using PhyloPhlAn (500 bootstraps). The C. difficile genomes (CD630 and R20291) were used as an outgroup for both. Download FIG S1, EPS file, 1.4 MB (1.4MB, eps) .
Copyright © 2023 Bhattacharjee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We next analyzed the pangenome from de novo-assembled whole genomes of all putative C. innocuum genomes available (n = 232) using Roary (95% blast percentage identity) (Table S1) (51). Assemblies of 232 unique genomes averaged 4.6 Mbp, close to the type strain C. innocuum ATCC 14501 at 4.7 Mbp (48). We observed an average of 4,400 protein-encoding genes by coding sequences (CDS) obtained from Prokka (50). The gene accumulation curve followed Heap’s law with γ = 0.3322 ± 0.06 (R2 = 94.68%) (Fig. S2A). Heap’s decay parameter, alpha, totaled less than one (alpha = 0.794), suggesting an open pangenome for C. innocuum. This was further supported by the distribution of gene abundance across the number of genomes (Fig. S2B), which demonstrated that the number of unique genes (524 genes) common to all 232 genomes was less than those observed in a single genome, indicating extensive gene transference within and outside the species.
The pangenome of C. innocuum and related species is open. (A) Total number of genes as a function of number of all included genomes within the four clades (n = 232). Yellow-colored solid line represents the average number of total genes from five subsamplings, with transparent yellow as error bars. The function follows Heap’s law (top left corner). The alpha is from Heap’s law estimate, run over 500 iterations using micropan in R (P < 2e-16). (B) Total number of unique genes as a function of number of genomes (P < 2e-16). Download FIG S2, EPS file, 1.4 MB (1.5MB, eps) .
Copyright © 2023 Bhattacharjee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The average nucleotide identity (ANI) (52) computed across all genomes revealed four distinct clades. Clades III and IV were 90% or less similar to clades I and II, less than an expected cutoff for a genus (90%)- or species-level (95%) comparison (53, 54) (Fig. 2). Clades I and II were more closely related with >95% ANI, and they included all four available reference genomes. We then used Anvi’o to assign core (present in all), soft-shell (present in 95 to 99% of genomes), and accessory genes (present in less than 95% of the genomes), which totaled 524 core and 28,915 accessory genes across all 232 genomes (Fig. 2). When excluding clade III and IV genomes, these totaled 2,058 core and 18,256 accessory genes, with the pangenome still open as indicated by the alpha and gamma parameters (Fig. S3).
FIG 2.

C. innocuum exhibits an open pangenome. C. innocuum pangenome displaying presence or absence of core (orange; present in 100% of genomes), soft-shell core (green; present in 95 to 99% of genomes), and accessory (blue; present in <95% genomes) genes, with hierarchical clustering based on average nucleotide identity (ANI) in the right-hand corner (coloring based on 40 to 100% similarity). Clade and cluster designated in the legend. Analyses incorporate all available unique C. innocuum genomes (n = 232).
The pangenome for canonical C. innocuum strains is open. (A) Total number of genes as a function of number of genomes within clades I and II (n = 193). Yellow-colored boxplots represent the average number of total genes from five subsamplings. The function follows Heap’s law (top left corner). The alpha is from Heap’s law estimate, run over 500 iterations using micropan in R (P < 2e-16). (B) Total number of unique genes as a function of number of genomes (P < 2e-16). (C) Core, soft-shell core, and accessory/singleton gene representation of the pangenome in clade I (green) or clade II (yellow) C. innocuum strains. Download FIG S3, TIF file, 2.7 MB (2.8MB, tif) .
Copyright © 2023 Bhattacharjee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
A maximum-likelihood tree using the core genome of all strains reiterated clustering of C. innocuum genomes into four clades as observed by ANI (Fig. 3A), demonstrating widespread distribution of the isolates from this study across clades I, II, and IV. Additionally, there was not an observable overrepresentation of C. innocuum strains isolated from clinical cases within any clade. A maximum-likelihood tree based on a set of 400 selected protein markers present across all bacteria and archaebacteria constructed using PhyloPhlAn (55, 56) maintained the overall integrity of the four clades, with some shuffling between the closely related clades I and II (Fig. S1B).
FIG 3.

Clade-specific differences in metabolism and potential virulence drive genomic differences in C. innocuum strains. (A) Maximum-likelihood tree based on single nucleotide polymorphisms in 524 core genes from 500 replications, colored by clade (node color) and source (circle color) as specified in the legend. (B) Principal-coordinate analysis (PCoA) based on a Bray-Curtis distance matrix of COG gene assignments (presence or absence) generated using Prokka, colored by clade (legend) (PERMANOVA; **, P < 0.001). (C) Relative abundance of major COG categories (color coded in the legend) represented in core, soft-shell core, and accessory genes. (D) Differentially enriched KEGG modules across clades (false-detection rate using an adjusted q value below 0.05), colored by fraction of genomes within each clade.
Genomic differences within C. innocuum and related strains are driven by metabolism.
Principal-coordinate analysis (PCoA) based on Bray-Curtis dissimilarity of presence or absence of COG genes demonstrated significant clustering of strains by clade (P < 0.001, permutational multivariate analysis of variance [PERMANOVA]), with clades I, II, and III closer together than clade IV (Fig. 3B). Overall, the pangenome of C. innocuum displayed ~28,000 gene clusters categorized using the COG database (Fig. 3C). At least 25% of the core genome and 50% of the accessory genes were classified as general functions (R), unknown functions (S), or did not map to the COG database (NA). Genes responsible for basic cellular processes and information storage and processing were equally distributed between the core, soft-shell core, and accessory genomes. Genes involved in metabolism (C, G, E, F, H, I, and P) were highest in the core genome and included genes associated with basic energy-producing pathways like ATP synthesis, gluconeogenesis, tricarboxylic acid (TCA) cycle, urea cycle, or Entner-Duodonoff pathway. From these functions, carbohydrate metabolism (G) held the highest percentage across all gene categories and was increased in the soft-shell core genes.
Interestingly, genes in the mobilome COG category (X) were overrepresented in the accessory genome of C. innocuum. Core mobilome representation included only a single mobilome gene, bacteriophage protein gp37, which forms a fibrous parallel homotrimer at the end of the long tail fibers in bacteriophages (57). Some phage-related genes, such as phage-related holin belonging to COG4824, which also harbors C. difficile TcdE (lysis protein), killer protein of prophage maintenance systems (Doc), predicted transposases (InsQ, InsG, and Tra8), and competence proteins (ComCG), were present in the soft-shell genome. The majority of the mobilome genes were present in the accessory genome, comprising a multitude of predicted transposases, transcription/translation proteins, and phage-related regulatory proteins. This included a plasmid stabilization system protein, ParE, identified across clades I, II, and IV strains, which, in Enterobacteriaceae, confers heat and antibiotic tolerance by maintaining IncI- and IncF-type plasmids and a DNA damage-inducible protein D, which has a role in recombinational DNA damage repair, as seen in Escherichia coli (58, 59).
To identify completeness of metabolic pathways (>75% of total genes), we used Anvi’o for metabolic reconstruction of the strains using the KEGG database. Completed carbohydrate metabolism pathways across all genomes (Fig. S4A) included the pentose phosphate pathway, pyruvate oxidation, glycolysis, glycogen biosynthesis and degradation, Embden-Meyerhof pathway, and ascorbate degradation pathway. All four clades demonstrated incomplete TCA cycle pathways. Several completed amino acid metabolism pathways were identified across all clades (including valine, proline, threonine, tryptophan, leucine, isoleucine, arginine, and ornithine) except for clade III, which also demonstrated a complete module for lysine (Fig. S4B). For lipid metabolism, completed pathways included fatty acid biosynthesis, initiation, and elongation (Fig. S4C).
Complete modules present across C. innocuum clades. Completion of modules associated with carbohydrate (A), amino acid (B), and lipid metabolism (C). Data were obtained by estimating metabolism using Anvi’o (87). Yellow refers to an incomplete module (<75% of the genes required to complete that pathway are present across all strains in their respective clades), and blue refers to a complete module (>75% of the genes required to complete that pathway). Download FIG S4, EPS file, 1.0 MB (1MB, eps) .
Copyright © 2023 Bhattacharjee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We used the Anvi’o functional enrichment tool to identify differentially abundant KEGG modules across clades (P < 0.05). Overall, clade I and II genomes exhibited similar profiles to each other compared to clade III and IV, although differences were observed between clades III and IV genomes (Fig. 3D). Clades I and II shared genes related to biosynthesis of terpenoids and polyketides that were less represented in clades III and IV. The glucuronate (uronate), ascorbate biosynthesis, and ubiquinone biosynthesis pathways were almost exclusively represented within clade IV strains compared to other clades. These pathways consisted of only single KOfam assignments belonging to the pathways UDP-glucose-6-dehydrogenase (EC 1.1.1.22), flavin prenyltransferase (EC 2.5.1.129), and xylulokinase (EC 2.7.1.17).
While module representation across all strains suggests that lipid biosynthesis and utilization are prevalent within C. innocuum (Fig. S3), acylglycerol degradation (triacylglycerol lipase [EC 3.1.1.3]) was observed for strains only in clade III and IV. Our analysis included C. innocuum genomes (SRA accession nos. SRR12535151 and SRR12535143) recently isolated from creeping fat in patients with Crohn’s disease as belonging to clade IV (40). A Helicobacter pylori cagA pathogenicity island signature module (K02283) was also identified as differentially represented in clade IV genomes. This includes the type IV pilus assembly protein CpaF (EC 7.4.2.8).
To identify more specialized polysaccharide metabolism in C. innocuum, we used dbCAN and the CAZy database to identify carbohydrate-active enzymes (CAZymes) (60). Glycoside hydrolases in the GH1 group and GT2.2 glycosyl transferases were the most abundant CAZymes in C. innocuum collectively, albeit with some variability across the four clades (Fig. 4A). GT14, which produces a glycogen-branching protein, was present in all clades, while GT11, a fucosyltransferase, was only present in clade IV. Compared to clade II strains, strains in clade I were more likely to contain glycoside hydrolases involved in alpha- or beta-d-glycosidic bond hydrolysis (GH2, GH43_35, GH65, GH106, GH140, and GH36). GH146, a β-l-arabinofuranosidase that cleaves β-l-Araf bonds in plant pectins, was present in all clade III strains and a subset of clade I (61). The acetyl-mannosamine transferase GT26 was only present in clade III strains. Only three types of carbohydrate esterases (CEs) were identified across all C. innocuum strains, all involved in plant cell degradation, CE6 (acetyl xylan esterase), CE4 (chitin deacetylase), and CE9 (N-acetylglucosamine 6-phosphate deacetylase) (62, 63). Four carbohydrate-binding modules were also observed across all clades, including CBM66, part of the LPXTG cell wall anchor domain-containing protein that degrades fructoside residues in fructans, and CBM48, associated with the GH13 family of CAZymes responsible for degrading starches (64). Other carbohydrate-binding modules, such as CBM50, which is often associated with GH25 to degrade chitin or peptidoglycan (65), and CBM32, involved in galactose and/or lactose metabolism, were observed sporadically across clades I, II, and IV.
FIG 4.

C. innocuum strains display clade-specific carbohydrate-activating enzymes (CAZymes). (A) Number of CAZyme types detected normalized to the total number of genomes within each clade, color coded by clade type. (B) Heatmap of number and type of CAZymes in individual genomes, clustered using Euclidean distance. GT, glycosyltransferase; GH, glycoside hydrolase; CE, carbohydrate esterase; CBM, carbohydrate-binding module.
C. innocuum exhibits strain-level variation in substrate use in vitro.
To examine differences in nutrient use across strains, we selected seven representative strains to examine their ability to use distinct carbohydrate sources in vitro. Representative strains were selected from a 99% dereplication cutoff of C. innocuum genomes, which clustered the genomes into seven groups, representing three of the four clades. We assessed both growth and acid production of the strains in minimal media supplemented with single carbohydrates. After 24 h of growth assessment, acid production was assessed using the colorimetric pH indicator bromocresol purple (BCP), which approximates pH changes as a result of fermentation (Fig. S5). Acid production signifying fermentation was determined as low (pH 5.5 to 6.5 and optical density at 588 nm [OD588] of 0.26 to 0.44) or high (pH < 5.5 and OD588 < 0.26).
Absorbance values and pH exhibit a strong correlation in BMCA for bromocresol purple (BCP) assay. (A) Correlation between absorbance values (OD588) of BCP and pH. Dark blue represents the linear regression of absorbance versus pH for rich media (TCCFB) control. Light blue represents the linear regression of absorbance versus pH for basal media (BMCA). An R2 of 0.94 indicates a strong correlation between absorbance (OD588) of BCP to pH in BMCA. (B) Regression equation for BMCA used to predict pH values for strains grown in BMCA containing one carbohydrate source as a predictor of bacterial growth. (C) Actual pH values used to confirm expected values calculated from BMCA linear regression. Download FIG S5, EPS file, 1.3 MB (1.3MB, eps) .
Copyright © 2023 Bhattacharjee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We observed variation in the ability of strains to grow on minimal media supplemented with single carbohydrate sources (Fig. 5). None of the strains could grow on lactose or raffinose, confirming previous observations for C. innocuum (27, 42). In contrast to previous reports (32), salicin did not support growth of any of the strains tested. Most strains demonstrated high growth (defined by both significant increases in OD and high acid production) in glucose and fructose, albeit at various growth rates. Significant growth on glucose and mannose was observed by all but one strain in each, CM647 and CM152, respectively (analysis of variance [ANOVA] on the area under the concentration-time curve [AUC], P < 0.05). Cellobiose, mannitol, and trehalose all supported growth and acid production in most strains (ANOVA, P < 0.05). Maltose supported the growth of only one strain, CM208A (ANOVA, P < 0.05), whereas sorbitol only supported growth of two strains, CM647 and CM220 (ANOVA, P < 0.05).
FIG 5.

C. innocuum exhibits strain-specific differences in nutrient utilization in vitro. (A) Growth curves of strains (n = 7) inoculated into basal medium with Casamino Acids (BMCA) with indicated carbohydrate source over 24 h, measured at OD600. Gray indicates the negative control (strain inoculated into BMCA without addition of carbohydrate). The significance of growth was determined by ANOVA on area under curve per strain (within each sugar type), followed by Tukey’s honestly significant difference (HSD); *, P < 0.05; **, P < 0.005; ***, P < 0.0001. (B) Growth (from OD600) and acid production (from bromocresol purple assay) data were combined to show nutrient utilization patterns in representative strains. High acid production was classified as pH of <5.5 and OD of <0.26; low acid production was classified as pH 5.5 to 6.5 and OD588 of 0.26 to 0.44 (legend). Clade and strain designation indicated by the legend.
Only some of the variable growth aligned with their clade designation. Within clade I, both CM152 and d22_429 followed a similar pattern of acid production (Fig. 5B) and grew at various efficiencies in glucose, fructose, mannitol, and trehalose (Fig. 5A). Within clade IV, both strains CM220 and CM679 grew efficiently in several carbohydrates and more efficiently on sucrose than most strains (ANOVA, P < 0.0001). The most variation was observed in clade II strains, with CM647 consistently displaying minimal growth on most carbohydrates. In contrast, both CM208A and CM151C exhibited some of the highest growth in glucose, cellobiose, and mannose compared to other strains (ANOVA, P < 0.005), but CM208A did not grow in mannitol compared to CM151C (ANOVA, P < 0.005). CM208A also consistently produced less acid than CM151C, except for growth in fructose (Fig. 5B).
C. innocuum exhibits clade-level variation in putative virulence factors and toxins.
An exotoxin has not been identified from C. innocuum despite previous evidence of association with infection (66). Using PathoFact to identify potential virulence (bit score > 50), we observed differential distribution of putative virulence factors across the four clades (Fig. 6A) (67). Overall, clade III strains had lower numbers of virulence factors detected over the other clades, which also lacked the type II toxin-antitoxin system from the YafQ/RelB/ParE family and phage lysis holins. Some factors were present across all clades, including members of hemolysin III, hlyIII and tlyC; NlpC/P60 family, pspA and pspC (identified as entD in the software); and a type III toxin-antitoxin system from ToxN/AbiQ (Fig. 6A). GGGtGRt, tlyC, and hlyIII demonstrated the highest bit scores across all clades, including within the reference strain C. innocuum 14501. While we identified the presence of C. difficile tcdAB-like genes in all clades, a BLAST search across the NCBI databases revealed that they likely belong to the NlpC/P60 family of proteins, either as surface protein pspAC alongside a penicillin-binding protein mrcB or as a glucan-binding domain-containing protein, not yet fully characterized. A PCoA based on the Bray-Curtis dissimilarity distance from the presence or absence of putative virulence genes from C. innocuum strains isolated from either diarrheal patients or healthy controls (n = 119) (46) did not demonstrate clustering based on clinical status (Fig. S7), suggesting that no groups or single putative toxins were associated with disease. Additionally, using functional enrichment for general pathways (Anvi’o) within this genome set did not identify significantly enriched KEGG or COG classes.
FIG 6.

Certain clades of C. innocuum display enhanced potential virulence. (A) Average bit score of select putative secreted and nonsecreted toxins (bit score > 50) across C. innocuum clade, identified using PathoFact. (B) Location of antibiotic resistance genes (AMR), putative virulence factors, and toxins identified using PathoFact (three innermost circles) and genomic islands, identified using IslandViewer 4 (penultimate outer circle; green, forward orientation; brown, reverse; purple, bidirectional orientation).
Predicted presence or absence of antimicrobial resistance genes in C. innocuum. Antimicrobial resistance gene categories identified by PathoFact in C. innocuum clades (black, presence of a gene in the antibiotic resistance gene category in at least one strain). Download FIG S6, EPS file, 0.5 MB (550.1KB, eps) .
Copyright © 2023 Bhattacharjee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
C. innocuum strains isolated from Clostridioides difficile diarrhea patients do not cluster by gene content. Principal-coordinate analysis (PCoA), based on a Bray-Curtis distance matrix, made from the presence or absence of COG gene assignments generated using Prokka shaped by clade and colored by disease association (legend) within C. innocuum strains isolated from C. difficile-diarrheal patients in Cherny et al. (46). PERMANOVA, performed using adonis2 (vegan) in R, indicated significant correlation with clade assignment (**, P < 0.001) but no significant correlation with disease. Download FIG S7, TIF file, 2.8 MB (2.9MB, tif) .
Copyright © 2023 Bhattacharjee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The chromosomal location of virulence and antibiotic resistance genes identified by PathoFact were visualized in conjunction with genomic islands (using IslandViewer 4), with C. innocuum 14501 as a reference (Fig. 6B) (68). Resistance genes against vancomycin glycopeptide (vanRS), tetracycline, as well as several ABC transporters and aminoglycoside genes were identified in all clades. Bacitracin resistance (bcrAC) was found only in clades I and III (Fig. S6). Results from IslandViewer 4 predicted 42 genomic islands, with 113 virulence factors and 7 toxin genes aligning with genomic island locations (Fig. 6B). While none of the hemolysins aligned with genomic island predictions, a type II TA system involving RelE/StbE family of toxin-antitoxin system (with 11 additional genes) and a group of NlpC/P60 glucan-binding proteins (labeled as tcdAB by PathoFact, with 9 additional genes), each aligned with a predicted genomic island.
DISCUSSION
To date, this study marks the most comprehensive characterization of genomic variability within the human gut inhabitant C. innocuum. While C. innocuum has originally been designated a commensal from initial isolation studies (32, 34), it has also been recently associated with various disease states (29, 37–40). We recovered C. innocuum strains from all individuals sampled in this study, suggesting a high prevalence of C. innocuum in the “healthy” human gut. This is further strengthened by the prevalence of C. innocuum in human 16S rRNA gene-based surveys using a custom classifier specific for C. innocuum 16S rRNA sequences, which also identified increased abundance of C. innocuum following antibiotic use. These data, in addition to the consistent association of C. innocuum with disease in culture-based studies, support a closer look at the role of C. innocuum in the gut microbiota.
Our genomic analysis of C. innocuum clarifies some of the functions attributable to C. innocuum colonization of the gut. We identified several complete modules in both carbohydrate and amino acid metabolism within the C. innocuum core genome. These also included multiple genes associated with utilization of saccharides, such as glucose, mannose, fructose, xylose, mannitol, chitin, xylan, and other starches and peptidoglycans, indicating an ability to use plant polysaccharides directly or by-products of polysaccharide degradation by other commensals. Additionally, all C. innocuum strains exhibited several partial and complete modules for lipid metabolism. None of the tested strains were able to grow in lactose, salicin, and raffinose, corroborating descriptions of C. innocuum clinical isolates growth using a Biolog platform (40, 69).
Our genomic comparison also revealed strain-specific diversity in C. innocuum. Both ANI and phylogenetic analysis of C. innocuum genomes demonstrated four distinct clusters. Clades III and IV were, collectively, 90% or less similar to clades I and II, which contained all available C. innocuum reference strains, suggesting that these clades may represent a new erysipelotrichial species related to C. innocuum. Even after exclusion of clade III and IV genomes, the pangenome of C. innocuum remains highly open as assessed by Heap’s law (70). It has been suggested that an open genome may reflect a more sympatric lifestyle, whereby related species interacting with each other can easily share genetic elements (71). While our current study did not specifically focus on identification of mobile elements, most mobilome-related genes were present in the accessory portion of the C. innocuum pangenome, suggesting a high degree of horizontal gene transfer among C. innocuum and the two related species.
The ability to acquire new genes can provide a competitive metabolic advantage in a microbially dense environment. As the nutrient niche theory stipulates, colonization by an invading microbe, pathogenic or commensal, is at least partially dependent on its ability to better utilize nutrients to outcompete extant microbes in that environment (72). For example, coexistence of the highly abundant gut inhabitant Bacteroides thetaiotaomicron is likely possible at least in part due to the diversity of polysaccharide-utilizing loci observed across different strains that allow flexibility in resource utilization (73). We observed coexistence of several C. innocuum strains within a single fecal sample, none of which were 100% identical to each other, and some of which spanned multiple clades within an individual. Overall, the CAZymes observed across C. innocuum were fewer than previously characterized gut commensals (13, 74, 75), with some clade-specific CAZymes. These genome-encoded CAZyme differences between clades and potential new species could indicate niche partitioning to support related strains within the same environment or the ability to localize into distinct gastrointestinal locations.
The realized, or expressed, niche of strain coexistence is likely more complicated (76). Our in vitro growth assays support niche partitioning within the canonical C. innocuum clades I and II at both clade- and strain-specific levels. For example, both CM152 (clade I) and CM151C (clade II), isolated from the same individual, could use fructose, glucose, mannose, and trehalose for growth. Yet the former grew significantly better with fructose, whereas the latter grew better on glucose, mannose, and trehalose. Despite the observation of clade-specific genomic patterns in CAZymes, we did not observe overt clade-specific growth patterns in vitro. This suggests that realized metabolic niche partitioning can occur within an individual beyond the categorical genomic features assessed, emphasizing the importance of regulatory or additional genes that contribute to successful coexistence of related strains. These differences also likely influence or are influenced by other members of the collective microbiota within an individual.
The demonstrated genomic and phenotypic variability observed across the C. innocuum species may also be of clinical importance. C. innocuum is commonly isolated in conjunction with gastrointestinal tissue or fecal clinical samples (35, 40, 66). A recent retrospective study in a Taiwanese clinical cohort isolated C. innocuum rather than C. difficile from patients with C. difficile-like clinical presentation (35). The C. innocuum isolates in this study were reported to cause a range of cytotoxicity and enteropathogenic effects in vitro. Most recently, Cherny et al. reported that C. innocuum isolates from pediatric patients enrolled in C. difficile studies cross-reacted with the enzyme immunoassay (EIA) diagnostic test for C. difficile toxins A and B (66). The study identified a putative C. innocuum toxin EIA cross-reactive factor (ErF) similar to the NlpC/P60 family of toxins in all isolates tested but observed no cytotoxicity. We identified the same putative toxin A/B gene in clades I and II, with a significantly higher similarity score for tcdA/B in clade II genomes. C. innocuum has also been postulated as a potential causative agent of “creeping fat,” an extraintestinal phenomenon correlated with Crohn’s disease (CD) (40). Ha et al. demonstrated that C. innocuum isolated from various human intestinal mucosal locations could translocate into tissue in a mouse model of inflammatory bowel disease (IBD). Our analysis, which included genomes from that study, did not identify clade-specific clustering based on the anatomical site. However, the two clade IV representatives identified as part of this study were both isolated from creeping fat lesions. Yet genomic content, either comprehensively or within a subset of putative virulence factors, did not correlate with disease status (46). Furthermore, genomes associated with disease state (either with C. difficile or in association with “creeping fat” in CD) spanned the four clades identified in this study, demonstrating no definitive “virulent” strain. Together, these data suggest the possibility of C. innocuum or a closely related species as an opportunistic, rather than an absolute, pathogen.
In summary, we demonstrate strain-specific variation of a prevalent gut “commensal” that, until recently, was considered relatively benign in the gut environment. The increased association of C. innocuum with gastrointestinal conditions supports further investigation of the role of C. innocuum in the gut, with an emphasis on identification of novel virulence or invasive factors that might enable C. innocuum to cause disease. Furthermore, our results reveal the importance of understanding strain variation that can be extended to other gut commensals.
MATERIALS AND METHODS
Isolation of C. innocuum.
This study was approved by Clemson University’s Institutional Review Board. Healthy donors were over 18, had not taken antibiotics or been diagnosed with any infections within 6 months, and were not immunocompromised or diagnosed with chronic gastrointestinal conditions. Upon receipt, fecal samples were placed under anaerobic conditions (Coy Laboratory Products, Grass Lake, MI, USA; 85% nitrogen, 10% hydrogen, and 5% carbon dioxide) and streaked onto brain heart infusion (BHI) (77), BHI supplemented with fetal bovine serum (FBS; 50 mL/L BHI), or taurocholate cycloserine-cefoxitin-fructose (TCCFA) (78, 79) by using different streaking strategies. Streaks were incubated at 37°C for at least 24 h and then picked and streaked for purity. Samples were stored at −80°C in 20% glycerol stocks for future in vitro work or DNA extraction (see Text S1 in the supplemental material).
Supplemental materials and methods containing detailed methodology on medium preparation; C. innocuum isolation pipeline; bromocresol purple assay; and whole-genome assembly pipeline, phylogeny, pangenome analysis, CAZyme analysis, and virulence factor analysis. Download Text S1, DOCX file, 0.1 MB (61.8KB, docx) .
Copyright © 2023 Bhattacharjee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DNA extraction and identification of C. innocuum.
All isolates were heat extracted at 95°C for 20 min for PCR using Go Taq (Promega; catalog no. M7132) and 8F and 1492R primers to amplify the whole 16S rRNA gene (4). PCR products were cleaned up using) and sent to Eton Biosciences for Sanger sequencing, using EzBioCloud and RDP databases Illustra ExoProStar kit (Cytiva; catalog no. US78210for identification (80, 81). DNA extraction for sequencing was performed from 1.8 mL of overnight culture using the Qiagen DNeasy UltraClean microbial kit (Qiagen; catalog no. 12224-250). Extracted DNA was diluted to 10 ng/μL concentration (Qubit, Life Technologies; catalog no. Q33230) and sent to the Microbial Genome Sequencing Center (MiGS), Pittsburgh, Pennsylvania (https://www.seqcenter.com/), for Illumina sequencing using the NextSeq 2000 platform.
Prevalence of C. innocuum in human 16S rRNA gene-based surveys.
FASTA sequences of full-length 16S rRNA sequences from C. innocuum genomes were formatted for alignment in mothur (43) and aligned using the SILVA database (v132) (82). Previously published sequences from fecal microbiota samples representing healthy (83), hospitalized, and/or septic (84) or antibiotic-exposed (85) individuals were processed in mothur using the Schloss lab standard operating procedure (SOP), aligning to the SILVA database (44) and then classifying to the custom classifier using the classify.seqs command in mothur (cutoff = 95) or directly to the RDP database (v16) for comparison (86). The log10 relative abundance of C. innocuum was plotted in R using the Kruskal-Wallis test in R with a pairwise Wilcoxon rank-sum test for pairwise comparisons between groups.
In vitro growth of C. innocuum.
Representative strains were chosen from all available C. innocuum strains based on 99% dereplication using pyANI in anvi-dereplicate-genomes (52, 87). Strains from freezer stocks were initially streaked onto TCCFA in an anaerobic chamber and incubated at 37°C for 24 h. A single colony was added into 4 mL TCCF broth (TCCFB) and incubated at 37°C for 24 h. We centrifuged 1.8 mL of overnight culture at 6,000 rpm for 5 min. Ten microliters of the resuspended pellet in 1.8 mL of prereduced water was added into wells of a 96-well plate (CoStar) containing 100 μL basal medium (88) with Casamino Acids (BMCA) with or without selected carbohydrate sources at 4% (wt/vol). A positive control of the resuspended strain in TCCF broth and BMCA without strain was included on each plate. The prepared plate was placed into a plate reader (Tecan Sunrise) for growth at 37°C, measuring the optical density at 600 nm (OD600) every 15 min for 24 h. A bromocresol purple (BCP) assay was used on the plate growth to assess pH (Text S1).
Whole-genome assembly and phylogeny.
Full commands are available at https://github.com/SeekatzLab/C.innocuum-diversity and are further described in Text S1. Briefly, raw reads were quality checked and adapter trimmed using Trim Galore! (89) and assembled using SPAdes (90) as optimized with MEGAHIT (91). Quast with MultiQC was used to calculate assembly statistics (Table S1) (92, 93). Average coverage was calculated using Bowtie 2 and SAMtools (94, 95). Prokka was used to annotate assemblies (50). To verify the assembly identity, annotations were run through NCBI BLAST and EzBioCloud. Assemblies were also mapped onto the Genome Taxonomy Database (GTDB) (45) through GTDB-tk using classify_wf (96). Maximum-likelihood trees from the C. innocuum core genome SNP sites were determined by Roary using RaXML 8.2.12 with bootstrapping 500 times. The 16S rRNA maximum-likelihood tree was aligned using Clustal Omega and bootstrapped 500 times by RAxML (97, 98). The amino acid fasta phylogenetic tree was mapped against the PhyloPhlAn database with DIAMOND using PhyloPhlAn (55). Trees were visualized using GraPhlAn (99) or the ITOL web server (https://itol.embl.de/) (100–102).
Pangenome analysis, functional enrichment, average nucleotide identity, and dereplication.
Contigs from SPAdes were reformatted and annotated with the COG and KEGG databases using Anvi’o v7.0 (87). Anvi’o was also used to create and visualize the pangenome, determine average nucleotide identity (ANI), and dereplicate strains within the data set (Text S1). Heap’s law was calculated in R (formulated as n = κNγ, where n is the pangenome size, N is the number of genomes used, and κ and γ are the fitting parameters), and the α parameter was calculated using micropan (70, 103). ANI was computed using the anvi-compute-genome-similarity with pyANI (52). Dereplication between strains was computed using anvi-dereplicate-genomes at 90, 95, 98, 99, 99.9, and 100% similarity threshold. The false-detection rate correction for P values for functional enrichment was applied using the package qvalue from Bioconductor (104) and visualized using dplyr, ggplot2, and readxl packages (105–107). Principal-coordinate analysis and associated PERMANOVA statistics were performed in R using vegan and ape packages (108, 109).
CAZyme and putative virulence.
CAZymes were predicted using dbCAN v2.0.6 (60), using the FASTA nucleotide sequences generated from Prokka for each of the strains. Prokka-generated nucleotide FASTA files (FNA) were processed through PathoFact (67) to predict virulence factors, toxins, and antimicrobial peptides. Genomic islands were predicted in C. innocuum 14501 using the web computational tool IslandViewer 4 (68). Circos was used to visualize chromosomal locations on reference sequence ATCC 14501 (68).
Data availability.
All raw sequence data and associated information have been deposited in the NCBI Sequence Read Archive under BioProject accession no. PRJNA841489. All data analysis from raw sequence processing and additional data tables (containing information on dereplication reports, medium composition for in vitro studies, average number of new genes versus number of genomes, full list of bioinformatics resources, and list of antibiotic resistance genes) for the final manuscript are available at https://github.com/SeekatzLab/C.innocuum-diversity.
ACKNOWLEDGMENTS
We thank the participants who consented to donating their fecal samples to our study. We acknowledge Clemson University for the generous allotment of computing time on Palmetto cluster. This publication was made possible, in part, with support from the Clemson University Genomics and Bioinformatics Facility, which receives support from an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM109094. A.M.S. was supported by grant number K01-DK111794 from the National Institute of Diabetes and Digestive and Kidney Diseases.
We acknowledge the Microbial Genome Sequencing Center (MiGS) for whole-genome sequencing of our C. innocuum strains and Eton Biosciences for Sanger sequencing the amplified 16S rRNA genes.
A.M.S. has received consultation fees from Finch Therapeutics and Rebiotix/Ferring Pharmaceuticals.
D.B., Conceptualization, Data Curation, Formal analysis, Investigation, Methodology, Software, Writing – Original Draft, and Writing – Review and Editing; C.F., Data Curation, Formal analysis, Investigation, Methodology, Validation, and Writing – Original Draft; C.W.-M – Methodology, Investigation, and Writing – Original Draft; A.M.S. – Supervision, Project Administration, Funding Acquisition, Conceptualization, Writing – Original Draft, and Writing – Review and Editing.
Contributor Information
Anna M. Seekatz, Email: aseekat@clemson.edu.
Robert A. Britton, Baylor College of Medicine
REFERENCES
- 1.Guiot HFL. 1982. Role of competition for substrate in bacterial antagonism in the gut. Infect Immun 38:887–892. doi: 10.1128/iai.38.3.887-892.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sweeney NJ, Klemm P, McCormick BA, Moller-Nielsen E, Utley M, Schembri MA, Laux DC, Cohen PS. 1996. The Escherichia coli K-12 gntP gene allows E. coli F-18 to occupy a distinct nutritional niche in the streptomycin-treated mouse large intestine. Infect Immun 64:3497–3503. doi: 10.1128/iai.64.9.3497-3503.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson KH, Perini F. 1988. Role of competition for nutrients in suppression of Clostridium difficile by the colonic microflora. Infect Immun 56:2610–2614. doi: 10.1128/iai.56.10.2610-2614.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duncan SH, Louis P, Thomson JM, Flint HJ. 2009. The role of pH in determining the species composition of the human colonic microbiota. Environ Microbiol 11:2112–2122. doi: 10.1111/j.1462-2920.2009.01931.x. [DOI] [PubMed] [Google Scholar]
- 5.Rivera-Chavez F, Zhang LF, Faber F, Lopez CA, Byndloss MX, Olsan EE, Xu G, Velazquez EM, Lebrilla CB, Winter SE, Baumler AJ. 2016. Depletion of butyrate-producing clostridia from the gut microbiota drives an aerobic luminal expansion of Salmonella. Cell Host Microbe 19:443–454. doi: 10.1016/j.chom.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garcia-Gutierrez E, Mayer MJ, Cotter PD, Narbad A. 2019. Gut microbiota as a source of novel antimicrobials. Gut Microbes 10:1–21. doi: 10.1080/19490976.2018.1455790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Umu OC, Bauerl C, Oostindjer M, Pope PB, Hernandez PE, Perez-Martinez G, Diep DB. 2016. The potential of class II bacteriocins to modify gut microbiota to improve host health. PLoS One 11:e0164036. doi: 10.1371/journal.pone.0164036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Human Microbiome Project Consortium. 2012. Structure, function and diversity of the healthy human microbiome. Nature 486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson AJ, Vangay P, Al-Ghalith GA, Hillmann BM, Ward TL, Shields-Cutler RR, Kim AD, Shmagel AK, Syed AN, Walter J, Menon R, Koecher K, Knights D, Personalized Microbiome Class Students . 2019. Daily sampling reveals personalized diet-microbiome associations in humans. Cell Host Microbe 25:789–802.e5. doi: 10.1016/j.chom.2019.05.005. [DOI] [PubMed] [Google Scholar]
- 10.Sommer F, Backhed F. 2013. The gut microbiota–masters of host development and physiology. Nat Rev Microbiol 11:227–238. doi: 10.1038/nrmicro2974. [DOI] [PubMed] [Google Scholar]
- 11.Guo X, Li S, Zhang J, Wu F, Li X, Wu D, Zhang M, Ou Z, Jie Z, Yan Q, Li P, Yi J, Peng Y. 2017. Genome sequencing of 39 Akkermansia muciniphila isolates reveals its population structure, genomic and functional diversity, and global distribution in mammalian gut microbiotas. BMC Genomics 18:800. doi: 10.1186/s12864-017-4195-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Becken B, Davey L, Middleton DR, Mueller KD, Sharma A, Holmes ZC, Dallow E, Remick B, Barton GM, David LA, McCann JR, Armstrong SC, Malkus P, Valdivia RH. 2021. Genotypic and phenotypic diversity among human isolates of Akkermansia muciniphila. mBio 12:e00478-21. doi: 10.1128/mBio.00478-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karcher N, Pasolli E, Asnicar F, Huang KD, Tett A, Manara S, Armanini F, Bain D, Duncan SH, Louis P, Zolfo M, Manghi P, Valles-Colomer M, Raffaeta R, Rota-Stabelli O, Collado MC, Zeller G, Falush D, Maixner F, Walker AW, Huttenhower C, Segata N. 2020. Analysis of 1321 Eubacterium rectale genomes from metagenomes uncovers complex phylogeographic population structure and subspecies functional adaptations. Genome Biol 21:138. doi: 10.1186/s13059-020-02042-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seong CN, Kang JW, Lee JH, Seo SY, Woo JJ, Park C, Bae KS, Kim MS. 2018. Taxonomic hierarchy of the phylum Firmicutes and novel Firmicutes species originated from various environments in Korea. J Microbiol 56:1–10. doi: 10.1007/s12275-018-7318-x. [DOI] [PubMed] [Google Scholar]
- 15.Gibiino G, Lopetuso LR, Scaldaferri F, Rizzatti G, Binda C, Gasbarrini A. 2018. Exploring Bacteroidetes: metabolic key points and immunological tricks of our gut commensals. Dig Liver Dis 50:635–639. doi: 10.1016/j.dld.2018.03.016. [DOI] [PubMed] [Google Scholar]
- 16.Wexler AG, Goodman AL. 2017. An insider's perspective: Bacteroides as a window into the microbiome. Nat Microbiol 2:17026. doi: 10.1038/nmicrobiol.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ludwig W, Schleifer K, Whitman W. 2009. Class III. Erysipelotrichia class nov. In De Vos P, Garrity GM, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer K-H, Whitman WB (ed), Bergey's manual of systematic bacteriology. Volume 3, the Firmicutes, 2nd ed. Springer, New York, NY. [Google Scholar]
- 18.Ludwig W, Schleifer K-H, Whitman WB. 2009. Revised road map to the phylum Firmicutes, p 1–13. In De Vos P, Garrity GM, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer K-H, Whitman WB (ed), Bergey's manual of systematic bacteriology. Volume 3, the Firmicutes, 2nd ed. Springer, New York, NY. [Google Scholar]
- 19.Garrity GM, Bell JA, Lilburn T. 2005. The revised road map to the manual, p 159–187. In Brenner DJ, Krieg NR, Staley JT, Garrity GM (ed), Bergey’s manual of systematic bacteriology. Volume 2, the Proteobacteria, part A, introductory essays. Springer, Boston, MA. [Google Scholar]
- 20.Jensen GB, Hansen BM, Eilenberg J, Mahillon J. 2003. The hidden lifestyles of Bacillus cereus and relatives. Environ Microbiol 5:631–640. doi: 10.1046/j.1462-2920.2003.00461.x. [DOI] [PubMed] [Google Scholar]
- 21.Bhattacharjee D, McAllister KN, Sorg JA. 2016. Germinants and their receptors in clostridia. J Bacteriol 198:2767–2775. doi: 10.1128/JB.00405-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rinninella E, Raoul P, Cintoni M, Franceschi F, Miggiano GAD, Gasbarrini A, Mele MC. 2019. What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms 7:14. doi: 10.3390/microorganisms7010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Danilova I, Sharipova M. 2020. The practical potential of bacilli and their enzymes for industrial production. Front Microbiol 11:1782. doi: 10.3389/fmicb.2020.01782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, Kim S, Fritz JV, Wilmes P, Ueha S, Matsushima K, Ohno H, Olle B, Sakaguchi S, Taniguchi T, Morita H, Hattori M, Honda K. 2013. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 25.Mathewson ND, Jenq R, Mathew AV, Koenigsknecht M, Hanash A, Toubai T, Oravecz-Wilson K, Wu SR, Sun Y, Rossi C, Fujiwara H, Byun J, Shono Y, Lindemans C, Calafiore M, Schmidt TM, Honda K, Young VB, Pennathur S, van den Brink M, Reddy P. 2016. Gut microbiome–derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat Immunol 17:505–513. doi: 10.1038/ni.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davis JJ, Xia F, Overbeek RA, Olsen GJ. 2013. Genomes of the class Erysipelotrichia clarify the firmicute origin of the class Mollicutes. Int J Syst Evol Microbiol 63:2727–2741. doi: 10.1099/ijs.0.048983-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cherny KE, Muscat EB, Reyna ME, Kociolek LK. 2021. Clostridium innocuum: microbiological and clinical characteristics of a potential emerging pathogen. Anaerobe 71:102418. doi: 10.1016/j.anaerobe.2021.102418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shin JH, Sim M, Lee JY, Shin DM. 2016. Lifestyle and geographic insights into the distinct gut microbiota in elderly women from two different geographic locations. J Physiol Anthropol 35:31. doi: 10.1186/s40101-016-0121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaakoush NO. 2015. Insights into the role of Erysipelotrichaceae in the human host. Front Cell Infect Microbiol 5:84. doi: 10.3389/fcimb.2015.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Y, Wu J, Li JV, Zhou NY, Tang H, Wang Y. 2013. Gut microbiota composition modifies fecal metabolic profiles in mice. J Proteome Res 12:2987–2999. doi: 10.1021/pr400263n. [DOI] [PubMed] [Google Scholar]
- 31.Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. 2009. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 1:6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith LD, King E. 1962. Clostridium innocuum, sp. N., a spore-forming anaerobe isolated from human infections. J Bacteriol 83:938–939. doi: 10.1128/jb.83.4.938-939.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yutin N, Galperin MY. 2013. A genomic update on clostridial phylogeny: Gram-negative spore formers and other misplaced clostridia. Environ Microbiol 15:2631–2641. doi: 10.1111/1462-2920.12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mohr C, Heine WE, Wutzke KD. 1999. Clostridium innocuum: a glucoseureide-splitting inhabitant of the human intestinal tract. Biochim Biophys Acta 1472:550–554. doi: 10.1016/s0304-4165(99)00162-2. [DOI] [PubMed] [Google Scholar]
- 35.Chia JH, Feng Y, Su LH, Wu TL, Chen CL, Liang YH, Chiu CH. 2017. Clostridium innocuum is a significant vancomycin-resistant pathogen for extraintestinal clostridial infection. Clin Microbiol Infect 23:560–566. doi: 10.1016/j.cmi.2017.02.025. [DOI] [PubMed] [Google Scholar]
- 36.Chia JH, Wu TS, Wu TL, Chen CL, Chuang CH, Su LH, Chang HJ, Lu CC, Kuo AJ, Lai HC, Chiu CH. 2018. Clostridium innocuum is a vancomycin-resistant pathogen that may cause antibiotic-associated diarrhoea. Clin Microbiol Infect 24:1195–1199. doi: 10.1016/j.cmi.2018.02.015. [DOI] [PubMed] [Google Scholar]
- 37.Cutrona AF, Watanakunakorn C, Schaub CR, Jagetia A. 1995. Clostridium innocuum endocarditis. Clin Infect Dis 21:1306–1307. doi: 10.1093/clinids/21.5.1306. [DOI] [PubMed] [Google Scholar]
- 38.Castiglioni B, Gautam A, Citron DM, Pasculle W, Goldstein EJ, Strollo D, Jordan M, Kusne S. 2003. Clostridium innocuum bacteremia secondary to infected hematoma with gas formation in a kidney transplant recipient. Transpl Infect Dis 5:199–202. doi: 10.1111/j.1399-3062.2003.00037.x. [DOI] [PubMed] [Google Scholar]
- 39.Crum-Cianflone N. 2009. Clostridium innocuum bacteremia in a patient with acquired immunodeficiency syndrome. Am J Med Sci 337:480–482. doi: 10.1097/MAJ.0b013e31819f1e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ha CWY, Martin A, Sepich-Poore GD, Shi B, Wang Y, Gouin K, Humphrey G, Sanders K, Ratnayake Y, Chan KSL, Hendrick G, Caldera JR, Arias C, Moskowitz JE, Ho Sui SJ, Yang S, Underhill D, Brady MJ, Knott S, Kaihara K, Steinbaugh MJ, Li H, McGovern DPB, Knight R, Fleshner P, Devkota S. 2020. Translocation of viable gut microbiota to mesenteric adipose drives formation of creeping fat in humans. Cell 183:666–683.e17. doi: 10.1016/j.cell.2020.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnston NC, Goldfine H, Fischer W. 1994. Novel polar lipid composition of Clostridium innocuum as the basis for an assessment of its taxonomic status. Microbiology (Reading) 140:105–111. doi: 10.1099/13500872-140-1-105. [DOI] [PubMed] [Google Scholar]
- 42.Alexander CJ, Citron DM, Brazier JS, Goldstein EJ. 1995. Identification and antimicrobial resistance patterns of clinical isolates of Clostridium clostridioforme, Clostridium innocuum, and Clostridium ramosum compared with those of clinical isolates of Clostridium perfringens. J Clin Microbiol 33:3209–3215. doi: 10.1128/jcm.33.12.3209-3215.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parks DH, Chuvochina M, Rinke C, Mussig AJ, Chaumeil PA, Hugenholtz P. 2022. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res 50:D785–D794. doi: 10.1093/nar/gkab776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cherny KE, Muscat EB, Balaji A, Mukherjee J, Ozer EA, Angarone MP, Hauser AR, Sichel JS, Amponsah E, Kociolek LK. 2022. Association Between Clostridium innocuum and antibiotic-associated diarrhea in adults and children: a cross-sectional study and comparative genomics analysis. Clin Infect Dis ciac483. doi: 10.1093/cid/ciac483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cherny KE, Ozer EA, Kochan TJ, Johnson S, Kociolek LK. 2020. Complete genome sequence of Clostridium innocuum strain LC-LUMC-CI-001, isolated from a patient with recurrent antibiotic-associated diarrhea. Microbiol Resour Announc 9:e00365-20. doi: 10.1128/MRA.00365-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cherny KE, Ozer EA, Kochan TJ, Kociolek LK. 2020. Complete genome sequence of Clostridium innocuum strain ATCC 14501. Microbiol Resour Announc 9:e00452-20. doi: 10.1128/MRA.00452-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garzetti D, Brugiroux S, Bunk B, Pukall R, McCoy KD, Macpherson AJ, Stecher B. 2017. High-quality whole-genome sequences of the oligo-mouse-microbiota bacterial community. Genome Announc 5:e00758-17. doi: 10.1128/genomeA.00758-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 51.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, Fookes M, Falush D, Keane JA, Parkhill J. 2015. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31:3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pritchard L, Glover RH, Humphris S, Elphinstone JG, Toth IK. 2016. Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal Methods 8:12–24. doi: 10.1039/C5AY02550H. [DOI] [Google Scholar]
- 53.Konstantinidis KTT, Tiedje JM. 2005. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci USA 102:2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jain C, Rodriguez RL, Phillippy AM, Konstantinidis KT, Aluru S. 2018. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun 9:5114. doi: 10.1038/s41467-018-07641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Asnicar F, Thomas AM, Beghini F, Mengoni C, Manara S, Manghi P, Zhu Q, Bolzan M, Cumbo F, May U, Sanders JG, Zolfo M, Kopylova E, Pasolli E, Knight R, Mirarab S, Huttenhower C, Segata N. 2020. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat Commun 11:2500. doi: 10.1038/s41467-020-16366-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Segata N, Bornigen D, Morgan XC, Huttenhower C. 2013. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat Commun 4:2304. doi: 10.1038/ncomms3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bartual SG, Garcia-Doval C, Alonso J, Schoehn G, van Raaij MJ. 2010. Two-chaperone assisted soluble expression and purification of the bacteriophage T4 long tail fibre protein gp37. Protein Expr Purif 70:116–121. doi: 10.1016/j.pep.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 58.Kamruzzaman M, Iredell J. 2019. A ParDE-family toxin antitoxin system in major resistance plasmids of Enterobacteriaceae confers antibiotic and heat tolerance. Sci Rep 9:9872. doi: 10.1038/s41598-019-46318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Uranga LA, Balise VD, Benally CV, Grey A, Lusetti SL. 2011. The Escherichia coli DinD protein modulates RecA activity by inhibiting postsynaptic RecA filaments. J Biol Chem 286:29480–29491. doi: 10.1074/jbc.M111.245373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, Busk PK, Xu Y, Yin Y. 2018. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 46:W95–W101. doi: 10.1093/nar/gky418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saito K, Viborg AH, Sakamoto S, Arakawa T, Yamada C, Fujita K, Fushinobu S. 2020. Crystal structure of beta-L-arabinobiosidase belonging to glycoside hydrolase family 121. PLoS One 15:e0231513. doi: 10.1371/journal.pone.0231513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Low KE, Xing X, Moote PE, Inglis GD, Venketachalam S, Hahn MG, King ML, Tetard-Jones CY, Jones DR, Willats WGT, Slominski BA, Abbott DW. 2020. Combinatorial glycomic analyses to direct CAZyme discovery for the tailored degradation of canola meal non-starch dietary polysaccharides. Microorganisms 8:1888. doi: 10.3390/microorganisms8121888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chang HX, Yendrek CR, Caetano-Anolles G, Hartman GL. 2016. Genomic characterization of plant cell wall degrading enzymes and in silico analysis of xylanases and polygalacturonases of Fusarium virguliforme. BMC Microbiol 16:147. doi: 10.1186/s12866-016-0761-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shen J, Zheng L, Chen X, Han X, Cao Y, Yao J. 2020. Metagenomic analyses of microbial and carbohydrate-active enzymes in the rumen of dairy goats fed different rumen degradable starch. Front Microbiol 11:1003. doi: 10.3389/fmicb.2020.01003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ameri R, Garcia JL, Derenfed AB, Pradel N, Neifar S, Mhiri S, Mezghanni M, Jaouadi NZ, Barriuso J, Bejar S. 2022. Genome sequence and carbohydrate active enzymes (CAZymes) repertoire of the thermophilic Caldicoprobacter algeriensis TH7C1(T). Microb Cell Fact 21:91. doi: 10.1186/s12934-022-01818-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cherny KE, Balaji A, Mukherjee J, Goo YA, Hauser AR, Ozer E, Satchell KJF, Bachta KER, Kochan TJ, Mitra SD, Kociolek LK. 2022. Identification of Clostridium innocuum hypothetical protein that is cross-reactive with C. difficile anti-toxin antibodies. Anaerobe 75:102555. doi: 10.1016/j.anaerobe.2022.102555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.de Nies L, Lopes S, Busi SB, Galata V, Heintz-Buschart A, Laczny CC, May P, Wilmes P. 2021. PathoFact: a pipeline for the prediction of virulence factors and antimicrobial resistance genes in metagenomic data. Microbiome 9:49. doi: 10.1186/s40168-020-00993-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bertelli C, Laird MR, Williams KP, Simon Fraser University Research Computing G, Lau BY, Hoad G, Winsor GL, Brinkman FSL, Simon Fraser University Research Computing Group . 2017. IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res 45:W30–W35. doi: 10.1093/nar/gkx343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ghimire S, Wongkuna S, Scaria J. 2020. Description of a new member of the family Erysipelotrichaceae: Dakotella fusiforme gen. nov., sp. nov., isolated from healthy human feces. PeerJ 8:e10071. doi: 10.7717/peerj.10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tettelin H, Riley D, Cattuto C, Medini D. 2008. Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol 11:472–477. doi: 10.1016/j.mib.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 71.Diene SM, Merhej V, Henry M, El Filali A, Roux V, Robert C, Azza S, Gavory F, Barbe V, La Scola B, Raoult D, Rolain JM. 2013. The rhizome of the multidrug-resistant Enterobacter aerogenes genome reveals how new “killer bugs” are created because of a sympatric lifestyle. Mol Biol Evol 30:369–383. doi: 10.1093/molbev/mss236. [DOI] [PubMed] [Google Scholar]
- 72.Freter R, Brickner H, Botney M, Cleven D, Aranki A. 1983. Mechanisms that control bacterial populations in continuous-flow culture models of mouse large intestinal flora. Infect Immun 39:676–685. doi: 10.1128/iai.39.2.676-685.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pudlo NA, Urs K, Kumar SS, German JB, Mills DA, Martens EC. 2015. Symbiotic human gut bacteria with variable metabolic priorities for host mucosal glycans. mBio 6:e01282-15. doi: 10.1128/mBio.01282-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Os P, Martin JC, Lawley TD, Browne HP, Harris HMB, Bernalier-Donadille A, Duncan SH, O'Toole PW, Ps K, Jf H. 2016. Polysaccharide utilization loci and nutritional specialization in a dominant group of butyrate-producing human colonic Firmicutes. Microb Genom 2:e000043. doi: 10.1099/mgen.0.000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.El Kaoutari A, Armougom F, Gordon JI, Raoult D, Henrissat B. 2013. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat Rev Microbiol 11:497–504. doi: 10.1038/nrmicro3050. [DOI] [PubMed] [Google Scholar]
- 76.Pereira FC, Berry D. 2017. Microbial nutrient niches in the gut. Environ Microbiol 19:1366–1378. doi: 10.1111/1462-2920.13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sorg JA, Dineen SS. 2009. Laboratory maintenance of Clostridium difficile. Curr Protoc Microbiol 12:9A.1.1–9A.1.10. doi: 10.1002/9780471729259.mc09a01s12. [DOI] [PubMed] [Google Scholar]
- 78.George WL, Sutter VL, Citron D, Finegold SM. 1979. Selective and differential medium for isolation of Clostridium difficile. J Clin Microbiol 9:214–219. doi: 10.1128/jcm.9.2.214-219.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wilson KH, Silva J, Fekety FR. 1981. Suppression of Clostridium difficile by normal hamster cecal flora and prevention of antibiotic-associated cecitis. Infect Immun 34:626–628. doi: 10.1128/iai.34.2.626-628.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yoon SH, Ha SM, Kwon S, Lim J, Kim Y, Seo H, Chun J. 2017. Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol 67:1613–1617. doi: 10.1099/ijsem.0.001755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Daquigan N, Seekatz AM, Greathouse KL, Young VB, White JR. 2017. High-resolution profiling of the gut microbiome reveals the extent of Clostridium difficile burden. NPJ Biofilms Microbiomes 3:35. doi: 10.1038/s41522-017-0043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rao K, Seekatz A, Bassis C, Sun Y, Mantlo E, Bachman MA. 2020. Enterobacterales infection after intestinal dominance in hospitalized patients. mSphere 5:e00450-20. doi: 10.1128/mSphere.00450-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.MacPherson CW, Mathieu O, Tremblay J, Champagne J, Nantel A, Girard SA, Tompkins TA. 2018. Gut bacterial microbiota and its resistome rapidly recover to basal state levels after short-term amoxicillin-clavulanic acid treatment in healthy adults. Sci Rep 8:11192. doi: 10.1038/s41598-018-29229-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM. 2014. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 42:D633–D642. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eren AM, Esen OC, Quince C, Vineis JH, Morrison HG, Sogin ML, Delmont TO. 2015. Anvi'o: an advanced analysis and visualization platform for 'omics data. PeerJ 3:e1319. doi: 10.7717/peerj.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tanner RS. 2007. Cultivation of bacteria and fungi, p 69–78. In Hurst CJ, Crawford RL, Garland JL, Lipson DA, Mills AL, Stetzenbach LD (ed), Manual of environmental microbiology. ASM Press, Washington, DC. [Google Scholar]
- 89.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 90.Prjibelski A, Antipov D, Meleshko D, Lapidus A, Korobeynikov A. 2020. Using SPAdes de novo assembler. Curr Protoc Bioinformatics 70:e102. doi: 10.1002/cpbi.102. [DOI] [PubMed] [Google Scholar]
- 91.Li D, Liu CM, Luo R, Sadakane K, Lam TW. 2015. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31:1674–1676. doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 92.Mikheenko A, Prjibelski A, Saveliev V, Antipov D, Gurevich A. 2018. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34:i142–i150. doi: 10.1093/bioinformatics/bty266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ewels P, Magnusson M, Lundin S, Käller M. 2016. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32:3047–3048. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Langmead B, Wilks C, Antonescu V, Charles R. 2019. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 35:421–432. doi: 10.1093/bioinformatics/bty648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, Li H. 2021. Twelve years of SAMtools and BCFtools. Gigascience 10:giab008. doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chaumeil PA, Mussig AJ, Hugenholtz P, Parks DH. 2019. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 1925–1927. doi: 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sievers F, Higgins DG. 2018. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci 27:135–145. doi: 10.1002/pro.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Asnicar F, Weingart G, Tickle TL, Huttenhower C, Segata N. 2015. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 3:e1029. doi: 10.7717/peerj.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yu G. 2020. Using ggtree to visualize data on tree-like structures. Curr Protoc Bioinformatics 69:e96. doi: 10.1002/cpbi.96. [DOI] [PubMed] [Google Scholar]
- 101.Wang LG, Lam TT, Xu S, Dai Z, Zhou L, Feng T, Guo P, Dunn CW, Jones BR, Bradley T, Zhu H, Guan Y, Jiang Y, Yu G. 2020. Treeio: an R package for phylogenetic tree input and output with richly annotated and associated data. Mol Biol Evol 37:599–603. doi: 10.1093/molbev/msz240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Letunic I, Bork P. 2021. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49:W293–W296. doi: 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Snipen L, Liland KH. 2015. micropan: an R-package for microbial pan-genomics. BMC Bioinformatics 16:79. doi: 10.1186/s12859-015-0517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shaiber A, Willis AD, Delmont TO, Roux S, Chen LX, Schmid AC, Yousef M, Watson AR, Lolans K, Esen OC, Lee STM, Downey N, Morrison HG, Dewhirst FE, Mark Welch JL, Eren AM. 2020. Functional and genetic markers of niche partitioning among enigmatic members of the human oral microbiome. Genome Biol 21:292. doi: 10.1186/s13059-020-02195-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wickham H. 2016. ggplot2: elegant graphics for data analysis. https://ggplot2.tidyverse.org.
- 106.Wickham H, François R, Henry L, Müller K. 2022. dplyr: a grammar of data manipulation. https://dplyr.tidyverse.org.
- 107.Wickham H, Bryan J. 2022. readxl: read nnxcel files. https://readxl.tidyverse.org.
- 108.Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Henry M, Stevens H, Szoecs E, Wagner H. 2020. vegan: community ecology package. https://CRAN.R-project.org/package=vegan.
- 109.Paradis E, Schliep K. 2019. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35:526–528. doi: 10.1093/bioinformatics/bty633. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Excel sheet containing genome metadata. Download Table S1, XLSX file, 0.05 MB (48.8KB, xlsx) .
Copyright © 2023 Bhattacharjee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Maximum-likelihood trees of C. innocuum based on full-length 16S rRNA gene- and PhyloPhlAn-selected markers. (A) Maximum-likelihood tree of full-length 16S rRNA generated by RAxML, aligned in Clustal Omega (99). (B) Maximum-likelihood tree of C. innocuum genomes based on 400 selected markers generated using PhyloPhlAn (500 bootstraps). The C. difficile genomes (CD630 and R20291) were used as an outgroup for both. Download FIG S1, EPS file, 1.4 MB (1.4MB, eps) .
Copyright © 2023 Bhattacharjee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The pangenome of C. innocuum and related species is open. (A) Total number of genes as a function of number of all included genomes within the four clades (n = 232). Yellow-colored solid line represents the average number of total genes from five subsamplings, with transparent yellow as error bars. The function follows Heap’s law (top left corner). The alpha is from Heap’s law estimate, run over 500 iterations using micropan in R (P < 2e-16). (B) Total number of unique genes as a function of number of genomes (P < 2e-16). Download FIG S2, EPS file, 1.4 MB (1.5MB, eps) .
Copyright © 2023 Bhattacharjee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The pangenome for canonical C. innocuum strains is open. (A) Total number of genes as a function of number of genomes within clades I and II (n = 193). Yellow-colored boxplots represent the average number of total genes from five subsamplings. The function follows Heap’s law (top left corner). The alpha is from Heap’s law estimate, run over 500 iterations using micropan in R (P < 2e-16). (B) Total number of unique genes as a function of number of genomes (P < 2e-16). (C) Core, soft-shell core, and accessory/singleton gene representation of the pangenome in clade I (green) or clade II (yellow) C. innocuum strains. Download FIG S3, TIF file, 2.7 MB (2.8MB, tif) .
Copyright © 2023 Bhattacharjee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Complete modules present across C. innocuum clades. Completion of modules associated with carbohydrate (A), amino acid (B), and lipid metabolism (C). Data were obtained by estimating metabolism using Anvi’o (87). Yellow refers to an incomplete module (<75% of the genes required to complete that pathway are present across all strains in their respective clades), and blue refers to a complete module (>75% of the genes required to complete that pathway). Download FIG S4, EPS file, 1.0 MB (1MB, eps) .
Copyright © 2023 Bhattacharjee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Absorbance values and pH exhibit a strong correlation in BMCA for bromocresol purple (BCP) assay. (A) Correlation between absorbance values (OD588) of BCP and pH. Dark blue represents the linear regression of absorbance versus pH for rich media (TCCFB) control. Light blue represents the linear regression of absorbance versus pH for basal media (BMCA). An R2 of 0.94 indicates a strong correlation between absorbance (OD588) of BCP to pH in BMCA. (B) Regression equation for BMCA used to predict pH values for strains grown in BMCA containing one carbohydrate source as a predictor of bacterial growth. (C) Actual pH values used to confirm expected values calculated from BMCA linear regression. Download FIG S5, EPS file, 1.3 MB (1.3MB, eps) .
Copyright © 2023 Bhattacharjee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Predicted presence or absence of antimicrobial resistance genes in C. innocuum. Antimicrobial resistance gene categories identified by PathoFact in C. innocuum clades (black, presence of a gene in the antibiotic resistance gene category in at least one strain). Download FIG S6, EPS file, 0.5 MB (550.1KB, eps) .
Copyright © 2023 Bhattacharjee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
C. innocuum strains isolated from Clostridioides difficile diarrhea patients do not cluster by gene content. Principal-coordinate analysis (PCoA), based on a Bray-Curtis distance matrix, made from the presence or absence of COG gene assignments generated using Prokka shaped by clade and colored by disease association (legend) within C. innocuum strains isolated from C. difficile-diarrheal patients in Cherny et al. (46). PERMANOVA, performed using adonis2 (vegan) in R, indicated significant correlation with clade assignment (**, P < 0.001) but no significant correlation with disease. Download FIG S7, TIF file, 2.8 MB (2.9MB, tif) .
Copyright © 2023 Bhattacharjee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supplemental materials and methods containing detailed methodology on medium preparation; C. innocuum isolation pipeline; bromocresol purple assay; and whole-genome assembly pipeline, phylogeny, pangenome analysis, CAZyme analysis, and virulence factor analysis. Download Text S1, DOCX file, 0.1 MB (61.8KB, docx) .
Copyright © 2023 Bhattacharjee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
All raw sequence data and associated information have been deposited in the NCBI Sequence Read Archive under BioProject accession no. PRJNA841489. All data analysis from raw sequence processing and additional data tables (containing information on dereplication reports, medium composition for in vitro studies, average number of new genes versus number of genomes, full list of bioinformatics resources, and list of antibiotic resistance genes) for the final manuscript are available at https://github.com/SeekatzLab/C.innocuum-diversity.