

ABSTRACT
A key hallmark of Alzheimer Disease (AD) pathology is the accumulation of tau protein in the form of neurofibrillary tangles across large-scale networks of the human brain cortex. Currently, it is still unclear how tau accumulates within specific cortical systems, and whether in situ genetic traits play a role in this circuit-based propagation progression. In this study, using two independent cohorts of cognitively healthy older participants, we reveal the backbone of tau spreading and its network intersections with high-resolution transcriptomic genetic data. We observed that specific connectomic-genetic gradients exist along the tau spreading network. Particularly, we identified 577 genes that significantly accompany the spatial spreading of tau; a set of genes in which APOE and glutamatergic synaptic genes (e.g. SLC1A2) play a central role. Thus, our study characterizes neurogenetic topological vulnerabilities in distinctive brain circuits of tau spreading and suggests that drug development strategies targeting the gradient expression of this set of genes should be explored to help stop or prevent the accumulation of tau.
One Sentence Summary:
Tau accumulation is related to a gradient gene-expression signature of cell-susceptibility where APOE and SLC1A2 play a key role.
INTRODUCTION
The most common form of dementia, Alzheimer Disease (AD) is one of the biggest public health challenge today. Moreover, its prevalence is expected to double in the coming 20 years increasing its burden on society(1). AD is characterized by the abnormal accumulation of amyloid and tau, which either alone or more probably in combination, might be among the most significant factors of disease progression. Advances in the development of novel high-affinity radiolabels have enabled the study of tau accumulation in vivo(2). Specifically, tau-PET (also named FTP-PET for the specific Flortaucipir tracer) has been reported to detect early local tau pathology in preclinical AD(3), showed good concordance with histopathological data, and accurately recapitulated the Braak neuropathological staging of neurofibrillary tangles (NFT)(2, 4, 5). Such pathological accumulation of tau has been directly linked to longitudinal atrophy(6), and is predictive of memory decline and clinical progression(7). Thus, it is crucial to better understand, preemptively detect, and individually predict tau accumulation in order to treat AD.
The pathological accumulation of amyloid and tau are not randomly distributed but rather follow a stereotypical spatial pattern that follows large-scale networks, suggesting that AD is a network-afflicting brain disease. Accordingly, recent studies have focused not only on signal intensity changes, but also on the large-scale network relationship of molecular binding affinity between distributed brain regions using both dimensionality reduction approaches(8, 9) and high-resolution network analyses(3, 10). However, it is still unknown whether tau accumulation relates to cerebral local in situ genetic traits that might cause or influence the circuit-based propagation of tau. Recent research has focused on analyzing the genetic and metabolic fingerprint that might characterize a regional vulnerability in the most affected areas (11, 12). Concerted efforts have been directed to study the local region properties using post-mortem data, from cellular morphology to cell-type specific gene expression(13, 14). Thus, previous neuroimaging-genetic studies have mainly focused on the spatial relationship between gene expression and diverse neuroimaging measures, but have neglected the connectomic signatures of AD-related pathology or the topographic integration between the large-scale networks underlying tau propagation and the local constitutive genetic expression patterns.
Here, we aimed to characterize the network stereotypical pattern of tau accumulation and the spatial gene expression gradients across the cortical mantle suggesting a regional vulnerability that accompanies tau propagation. Using two independent samples of cognitively unimpaired participants from the Harvard Aging Brain Study (HABS) and Alzheimer's Disease Neuroimaging Initiative (ADNI) cohorts, we first developed a novel graph theory algorithm to obtain the backbone tau-PET network across cortical areas in the aging brain and assess its utility to predict one and two-year tau accumulation. This data-driven strategy led us to identify and analyze regions that encompass the whole continuum of the Braak staging. Thereafter, we studied how the network-wise propagation of tau within the backbone network was related to the local expression of genes using the AHBA high-resolution transcriptome dataset(15). Such analysis allowed us to identify a set of genes, representing the connectomic-genetic gradients of gene expression that characterize the regional vulnerability along the tau spreading network. We then studied the biological significance and potential implications of these protein coding genes based on their genetic functional components, interactomic properties and relationship with NFT-related proteins. The integration of large-scale network information with high-resolution gene expression data allowed us to describe gradients of neurogenetic vulnerability of tau spreading in the human cerebral cortex.
RESULTS
Tau-PET Backbone-Graph Follows Tau Accumulation Patterns of preclinical AD
Using cross-sectional data from a subset of amyloid positive cognitively unimpaired participants from HABS (N=19) and ADNI (N=52) with no follow-up data, we studied the FTP-PET signal changes in preclinical AD across different brain systems using a graph theory approach. We developed a node-aggregation algorithm (NAA, Fig 1-I) that aggregates groups of nodes with converging information, into a set of regions of interest, here referred to as super-nodes, that characterize the system information of in vivo tau accumulation in preclinical AD (online methods). By applying the NAA, we obtained 58 super-nodes encompassing medial and lateral temporal regions, cuneus and precuneus, and posterior cingulate cortex, temporoparietal areas, and portions of the inferior frontal cortex, including areas in the whole spectrum of Braak staging (Fig 2-II). Importantly, we confirmed the robustness of the super-nodes size and localization with a permutation approach using 500 random subsets of 50 participants from the original sample and assessing their overlap with the original network (Fig 2-II). The similarity between the permutations from the subsets were significantly larger compared to two null distributions created from 500 permutations using two different approaches that control for spatial auto-correlation (online methods). When studying the importance of each super-node in the network, as measured by the degree centrality, we identified hub super-nodes with a number of connections over the mean, including the middle temporal gyrus, inferior temporal gyrus and precuneus (Fig 2-III).
Fig 1. Methodological summary.
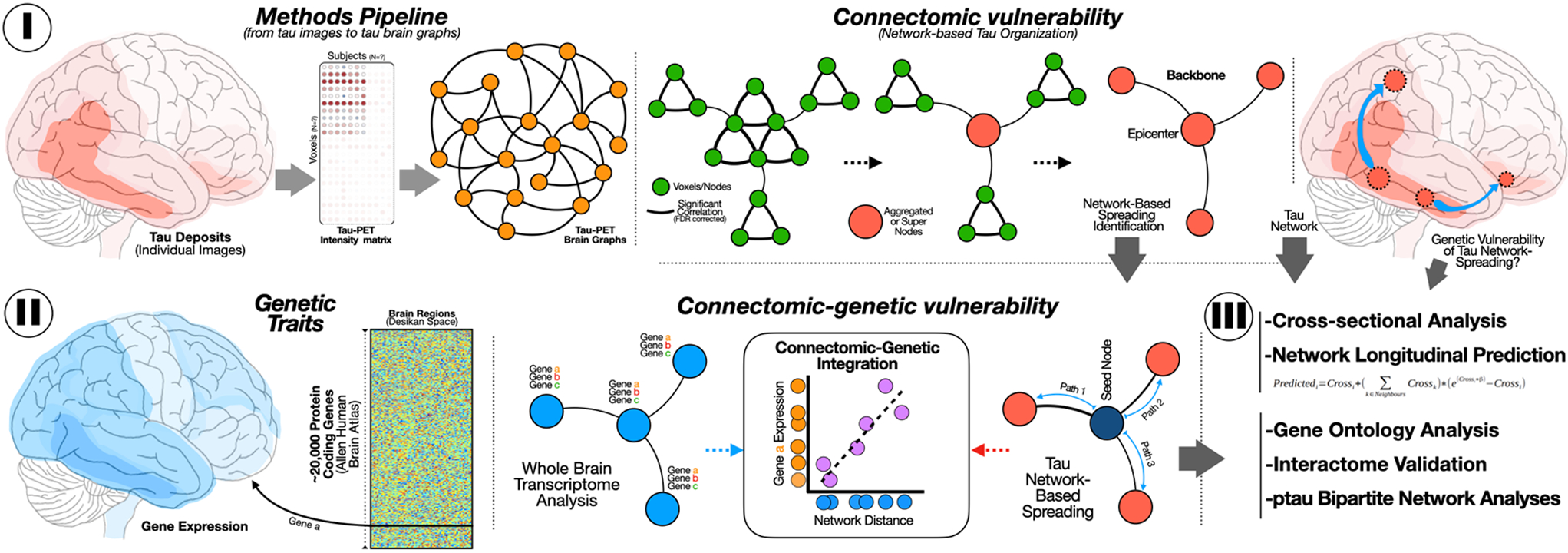
(I) For each pair of voxels of tau-PET (or FTP-PET), we computed the pairwise Pearson correlation to obtain a population connectivity (adjacency) matrix to further apply the NAA to retrieve the tau-PET network backbone. (II) AHBA dataset with transcriptome information from 10,027 genes in 180 regions from the Glasser atlas are projected to the super-nodes from the NAA to investigate the relationship between gene expression and network distance from the left entorhinal.
(III) Reliability and validation strategies of connectomic and connectomic-genetic findings.
Fig 2. Generation of the Tau-PET network backbone and its potential to track longitudinal changes.
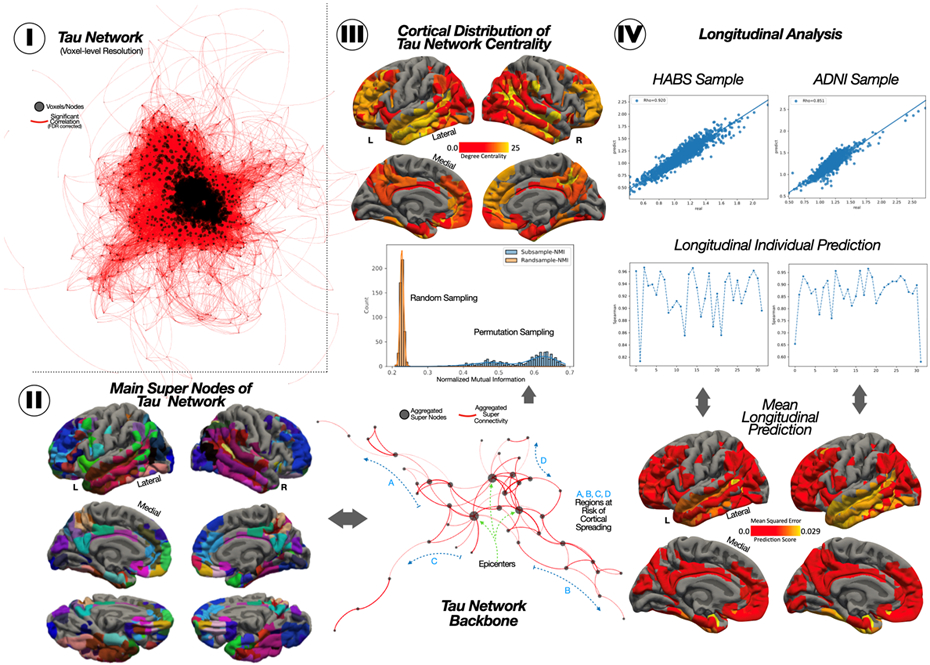
(I) The initial voxel-wise tau-PET network. (II) The 58 nodes resulting from the node-aggregation algorithm (NAA), its network representation, and the histogram resampling the robustness of our results over 500 permutations with a subset of 50 participants. (III) The topographical degree centrality measure of the tau network backbone. (IV) Scatterplot of the association between predicted and observed Tau-PET, the individual correlation for each of the participants, and the MSE error per ROI.
Next, we assessed the accuracy of the prediction of regional tau accumulation in a non-overlapping longitudinal cohort from HABS (N=32; 2-year follow-up) and ADNI (N=32; 1-year follow-up). Compared to other approaches that use the (structural or functional) connectome as a proxy to predict the longitudinal tau accumulation, we instead took advantage of the previously computed FTP-PET backbone graph. Overall, when considering the pool of all super-nodes mean FTP-PET signal from all the participants, we found a high association between the real longitudinal FTP-PET and the predicted FTP-PET, for both the HABS cohort (rho=0.92) and in ADNI (rho=0.85). We also found high associations at the individual level for both cohorts (HABS= 0.92+/−0.04, ADNI=0.87+/−0.08). We then studied the reliability in the regional prediction of tau accumulation, assessing the mean squared error (MSE) between predicted and real FTP-PET signal for each super-node, in each cohort separately. The super-node with the maximum error was the left lateral temporal gyrus, with a MSE value of 0.026 in ADNI and 0.028 in HABS (Fig 2-IV).
Genetic Vulnerability of Tau Network Spreading
Next, we combined high-resolution gene expression data from the AHBA with the previously computed FTP-PET backbone network to identify which genes showed a gradient of transcriptomic expression related to the network-based tau spreading. For each super-node, we computed the network distance to the left entorhinal, where those regions with shorter distance regions would be next regions to be affected by tau spreading (online methods). Importantly, we found that the network distance map computed between the entorhinal seed and the other backbone nodes, resamples the pattern of Braak staging (Fig 3-I), where Braak III-IV regions were closer to the seed compared to Braak V-VI regions. When we assessed the association between each super-node distance from the seed and the transcriptome expression from the 10,027 genes in the AHBA, we identified a set of 744 statistically significant associations (Fig 3-II; FDR < 0.001). The expression of 414 of the genes were positively associated with network-based distance to seed with a Spearman rho>0.55, whereas 330 genes were negatively associated with network-based distance with a Spearman rho < −0.56. Of note, all obtained genes remained significant when applying a permutation-based approach where the backbone FTP-PET network links where randomly interchanged to generate random maps of different spreading patterns, which suggests that the relationship between the gradient of gene expression and the network-wise distance was dependent on the FTP-PET backbone structure (Suppl. Material 1). Among the 744 genes, 167 could not be validated with the transcriptome data of the ROS/MAP participants – a cohort with bulkRNA sequencing of more than 700 participants in the continuum of AD-, and were thus excluded from further analyses. From the resulting 577 genes, 122 were significantly differentially expressed when comparing HC vs AD in the ROS/MAP cohort.
Fig 3. Connectomic-genetic gradient within the Tau-PET network.
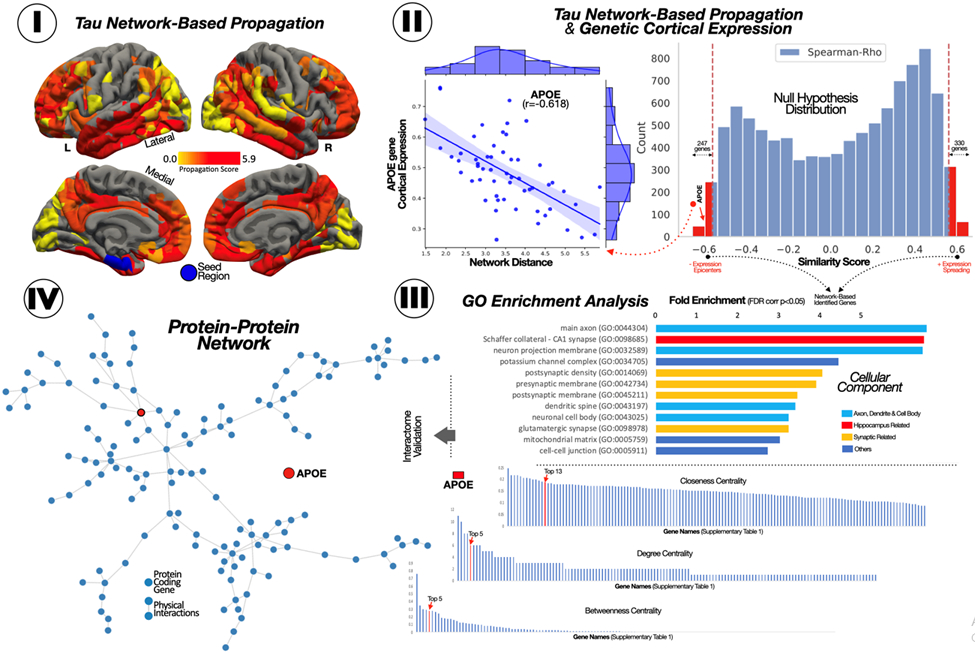
(I) Surface representation of the network-based distance of each super-node to the left entorhinal node. (II) Histogram of the association between gene expression and network-based distance for 10,027 genes and the scatterplot for APOE. (III) Cellular component overrepresentation based on Panther-GO. (IV) Gene-gene network based on physical interaction and the centrality measure for its genes.
Interactome and Gene Ontology Analyses
To assess the biological meaning of our 577 tau network-related genes (TNG), we focused on their molecular physical interactions and cellular component overrepresentations (Fig 3). The Panther GO analysis revealed a set of enriched genes involved in neuron structure (main axon, dendritic spine, and neuronal cell body), the synapsis (postsynaptic density and presynaptic membrane) and the Schaffer collateral in the hippocampi (FWE < 0.05; Fold Enrichment > 2). We then analyzed the importance of each gene within the TNG-interactome network using a curated set of gene-gene physical interaction from Genemania (online methods). We found that MAPK3 (top1) and APOE (top6) had a central role in these genetic interactions, being the genes with higher degree, betweenness and closeness centrality (Suppl Table 1). We validated our results studying the relationship between our 577 TNG and the 74 pTau-interactome genes recently described by Drummond and colleagues(16), computing a bi-partite network to display the relationship between those two sets of genes (online methods). We found that most of the TNG were associated with pTau-interactome genes (Fig 4-I). Importantly, these associations were non-random, as shown in the ad-hoc permutation-based analyses (Fig 4-II, online methods), suggesting a high affinity between the TNG and pTau-interactome genes, compared to random neuro-genes. Specifically, the mean degree and the unique number of genes interconnecting the TNG to the pTau-interactome were significantly higher (a mean degree of 0.8 and 108 unique genes compared to lower than 0.3 mean degree and lower than 65 unique genes for the random permutations). We did not find that these associations were driven by a group of genes in any specific cellular component GO-term. We also studied which TNG were more directly related to more protein expression in NFT, as measured in Drummond et al. We found that APOE and SLC1A2, the two only TNG related to MAPT, had a strong influence in the level of NFT protein expression (Fig 4).
Fig 4. TNG are associated to pTau interactome genes.
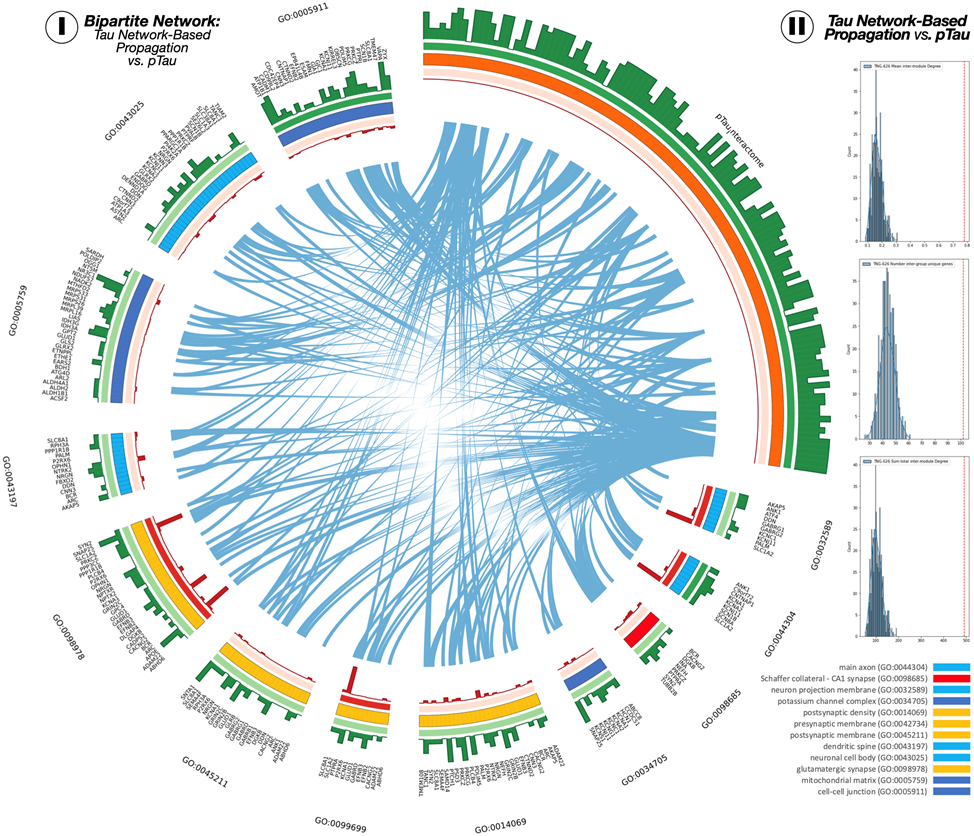
(I) The circular layout of the bipartite network. Links between the TNG and pTau interactome sets are shown in blue. Green bars represent the gene degree within each set, and red bars represent the amount of protein expressed in NFT of the pTau interactome connected to each TNG. (II) Network centrality measures of mean intermodule degree (top), number of unique genes connected between modules (middle), and total intermodule degree. Red dashed lines represent the score from TNG-pTau interactome circular layout. Blue histogram distributions are obtained from 1000 permutations.
DISCUSSION
In this study, using two independent cohorts of cognitively unimpaired older participants (HABS and ADNI), we characterized the in vivo accumulation of tau pathology in large-scale networks using a novel graph theory approach and demonstrated its potential to be used as a proxy for the prediction of longitudinal tau accumulation in preclinical AD. Moreover, we integrated the tau large-scale network information with high-resolution transcriptomic genetic data to characterize the gradient of neurogenetic vulnerability related to the spreading of tau across brain circuits. We found that 577 genes specifically predispose the spread of tau; a set of genes in which APOE and glutamatergic synaptic genes (SLC1A2) have central roles.
Our study integrated high-resolution cortical gene expression information with neuroimaging network data to investigate the underlying biological pathways of AD-related pathology spreading. Several recent studies have focused on assessing the relationship between gene expression and both imaging signal intensity(17, 18) and statistical differences(19, 20). For example, Grothe and collaborators (2018) showed that the topographic expression of APP and MAPT was related to amyloid accumulation and neurodegeneration -atrophy- in AD, respectively. Our group expanded this topic of researcher by comparing tau and amyloid propagation maps and gene expression profiles of AD-related genes(10). The present study develops an innovative step forward: we investigated the intrinsic genetic vulnerability of AD pathology networks associated to stereotypical pattern of tau accumulation. To this aim, we developed a method to integrate gene expression data into the FTP-PET network to study the association between them. Specifically, instead of assessing the relationship between signal intensity and gene expression, we integrated genetic information and network-based distances from the entorhinal cortex to all the subsequent regions, to evaluate if the network spreading of tau accumulation was related to gradients of gene expression.
In contrast to the consensus in the literature that tau spreads in a prion-like manner, recent findings have cast doubt on the notion that all tau accumulation is only driven by cell-cell transmission. It has been suggested that tau accumulation could be a result of both spreading and local amplification/phosphorylation, in addition to local vulnerability(21, 22). For example, Meisl and colleagues (2021) have recently shown that the tau accumulation rate in animal models is more related to local amplification than seed spreading. In this sense, several studies have suggested a specific cellular/molecular vulnerability driving (or modulating) the downstream neurodegeneration (and tau spreading) process(13, 23, 24). However, most studies have only assessed whether local regional vulnerability (i.e. study if the cellular-composition, genetic and molecular environment for a specific region) present such selectivity to accumulate tau. Thus, when integrating global-brain information, such genetic vulnerabilities have been studied using intensity maps, which do not account for biological spreading or the network-wise nature of tau accumulation. In the present study, we studied the gradient of change in the gene expression from healthy young individuals across several brain regions, in conjunction with the network of tau deposition, to disentangle the gradient genetic fingerprint of regional vulnerability leading to tau spread. We observed that the set of TNG is overrepresented by various genetic pathways suggesting regional vulnerability, such as glutamatergic synaptic ontologies. Previous research has described that glutamatergic excitatory neurons are more vulnerable to AD(25), and conversely, that the pathways of genes overexpressed in such neurons are strongly related to tau accumulation(13). Overall, our results extend the idea -also recently pointed by Meisl and colleagues(22)- where different regional gene expression profiles are associated to local accumulation of tau and its stereotypical temporality of spreading. We found a key role of two genes in that process: SLC1A2 and APOE. SLC1A2 is localized in the neuron and glia membrane and its main function is to clear glutamate from the synaptic cleft. The association between SLC1A2 and tau is in agreement with the hypothesis that excitatory neurons are more vulnerable to tau pathology. Several studies have also pointed to its importance in cognitive decline where lower expression of this gene in astrocytes and neurons was associated to worse cognitive performance(26). Additional studies using animal models and bioinformatic approaches have also identified SLC1A2 as a potential candidate for drug development in AD (27).
Whereas historically APOE has been more associated with amyloid pathology rather than with tau, we found that it has a central role in our TNG interactome, highlighting its potential relevance in the vulnerability of tau accumulation as well(10). Moreover, our bipartite network analyses showed that APOE has a high indirect influence in the expression of proteins in NFT, due to its physical interaction with MAPT. Several recent studies have found a direct association between ApoE4 and the proliferation of tau in animal models(28). In fact, the removal/reduction in ApoE, specifically ApoE expressed on astrocytes, has been shown to decrease tau accumulation and decrease tau-mediated neurodegeneration(29, 30). Such cellular vulnerability has also been recently reported in human neurons where ApoE drive selective neurodegeneration(31). Importantly, both APOE and SLC1A2 showed a negative correlation between their expression and FTP-PET network-based distance, suggesting that early areas to accumulate tau might be more vulnerable due to higher basal expression of both genes, where a transcriptome alteration of their expression might have a stronger impact in tau accumulation. Our results would suggest that the stereotypical pattern of the spread of tau follow the gradient of expression of APOE and SLC1A2, where a pathological alteration of its expression could affect more severely, and earlier, those brain regions with higher expression and network-wise proximity to the entorhinal cortex. Importantly, the gradient profile that characterizes our findings points to specific proteomics that could be targeted depending on the different status of tau accumulation. Our findings suggest novel molecular pathways that might be used to develop drugs to stop the spreading of tau in cortical systems. Specifically, APOE has been proposed as a potential candidate to develop effective therapies in AD(32). Our results highlight the importance of accounting by the extension of tau accumulation for both participant recruitment and when evaluating the efficacy for APOE-targeted drugs. On the one hand, participants with high load of tau pathology in Braak V-VI might not benefit from an ApoE treatment, since its expression is low in those areas. On the other hand, such therapies have the potential to stop tau pathology when its present only in early-Braak areas. Further work using drug discovery approaches using machine learning models(33) can identify the most promising protein candidates, from the provided set of genes, to develop different drugs depending on the topography of tau accumulation of each participant.
Our investigations used network information to obtain a robust backbone representation of the FTP-PET network, aggregating groups of nodes which share connectivity profiles. As a result, we obtained a low dimensional connectome that overcomes previous limitation when studying topological and temporal tau alterations using high-dimensionality PET network studies, such as redundant link information (caused by inherent local smoothing of the data) and extensive computational resources. Notably, our method identified, using a sample of amyloid positive cognitively unimpaired individuals, a pattern of co-accumulation in both early Braak stages (e.g., entorhinal, or inferior temporal)(34) and neocortical regions affected at late Braak stages(2) suggesting that slight increases in FTP-PET in Braak V/VI might also be meaningful in early stages of the disease. We also propose that the backbone FTP-PET network might be a reliable proxy to predict longitudinal tau accumulation. Strongly grounded on the prion-like spreading of tau, several studies in the literature have proposed various propagation models to accurately predict longitudinal tau accumulation based on the structural or functional connectomes(35-38). Contrary to such approaches, we explored the possibility to first, use data-driven backbone-nodes instead of cortical parcellation atlas - which are not generated from and for FTP-PET data - and second, use the backbone FTP-PET connectome as a proxy for the spreading of tau, instead of focusing on the structural or functional connectome. We acknowledge that our approach is more direct compared to sophisticated frameworks that model the prion-like spread of tau over the structural connectome accounting for different factors such as tau aggregation and diffusion rates(21). Moreover, updated models from the same group have also shown that accounting by local (genetic) pathological factors, such as reactive microglia, might improve the prediction of longitudinal tau accumulation(39). We hypothesize that, despite being more simplistic, our tau backbone graph approach used to predict longitudinal tau might intrinsically include the aforementioned parameters since its computed using a data-driven approach based directly on tau-PET data, and allow to accurately predict longitudinal accumulation with a more straightforward model. In this sense, we obtained high significant correspondence between the real and predicted longitudinal tau accumulation both at individual level and at region of interest level. Further work is nonetheless needed to validate this model in later stages of the disease, where the amount of accumulation of tau per year is increased.
Overall, our analyses identified a gradient gene-expression signature of cell-susceptibility to accumulate tau along the different Braak areas. Our results highlight the key role of neuron- and synapse-related genes, such as APOE and SLC1A2, in the stereotypical pattern of tau spread. Moreover, the multi-genetic findings presented in this study support recent views about the different patterns of spreading of tau, where substantial individual differences in the topological gene expression profile and its local vulnerability could explain the different pathways through which tau would propagate.
Study Design
Our main objective was to investigate the genetic fingerprints related to regional vulnerability along the brain systems of tau accumulation. We hypothesized that the gradual change in the cortical expression of genes will be related to the stereotypical pattern of tau accumulation along the human brain. To test this, we included a total of 490 participants from two large studies. We included 145 participants from our cohort, the Harvard Aging Brain Study (HABS, https://habs.mgh.harvard.edu/), a cohort of cognitive aging and preclinical AD recruited from the community conducted at Massachusetts General Hospital (MGH); and 345 cognitively unimpaired participants from the Alzheimer’s Disease Neuroimaging Initiative (ADNI; http://adni.loni.usc.edu/), a multi-center study designed to accelerate the discovery of biomarkers indicating progression of Alzheimer’s disease pathology. ADNI was launched in 2003 as a public– private partnership led by principal investigator M. W. Weiner. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), PET, other biological markers and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early-onset AD. For up-to-date information, see http://adni.loni.usc.edu.
At study entry, all the included participants were assessed as cognitively clinically normal, with clinical dementia rating value of zero. Participants were included based on the following inclusion criteria: i) baseline tau-PET, ii) baseline amyloid-PET and, iii) a structural T1-weighted MRI. We tested demographic study differences using the Mann-Whitney U Test and chi square test.
Structural MRI
Structural 3D T1-weighted were acquired in 3 Tesla scanner, using a magnetization-prepared rapid-acquisition gradient-echo (MPRAGE) sequence. The T1-weighted MPRAGE structural images had a resolution of at least 1.3 x 1.3 x 1.3 mm voxels. All the T1-weighted images were preprocessed with Freesurfer v6.0 (https://surfer.nmr.mgh.harvard.edu/) as previously described(40).
Tau PET
Tau burden was measured in both cohorts using the Flortaucipir (FTP-PET) tracer (formerly AV1451 or T807). Acquisition parameters for each study have been described elsewhere(2). FTP was preprocessed using previously published in-house pipelines(10). Briefly, we computed the SUVr parametric maps normalizing the FTP intensity by the mean cerebellar grey matter intensity. We co-registered the FTP map to T1 using Freesurfer’s mri_coreg. We computed the T1-MNI registration using SPM12. We then normalized the FTP-PET SUVr maps to the MNI space concatenating the PET-T1 and the T1-MNI registration. All FTP-PET maps were down-sampled at the normalized space to 8-mm isotropic voxel to study the high-dimensional data without computational limitations. For subsequent analyses, and to discard cortical brain regions with off-target FTP-PET binding, we select only those voxels were the fitting of the FTP-PET signal for the whole sample of participants with a 2-component Gaussian Mixture Model outperformed the fitting of a single Gaussian, using the Akaike Information Criteria to compare between both models(35) [Suppl. Fig 2]. This is founded in the idea that if a certain voxel is strongly contaminated by off-target binding, partial volume effect, or none of the included participants do present abnormal AD-related binding, data should show a skewed distribution (one gaussian). Voxels following this pattern were excluded for subsequent analyses. After visual inspection on the raw FTP-PET intensity image, and FTP-PET to T1 registration, we excluded 65 participants due to miss-alignment during the registration and low SNR quality of the raw FTP-PET images.
Amyloid PET
HABS participants were injected with 10-15 mCi 11C-PiB intravenously as a bolus and followed immediately by a 60-min dynamic PET scan in 3-D mode. PiB-PET images were co-registered to T1-MRI using mri_coreg from Freesurfer. Dynamic PIB-PET was modeled with a Logan model, using the cerebellar GM as reference region to generate parametric Logan DVR images. For each participant, we computed the amyloid burden as the mean from a cortical composite including frontal, lateral temporal, and parietal, and retrosplenial (FLR) regions as defined using the Desikan-Killiany atlas, as in previous studies(2). PIB-PET positivity was computed using a threshold of Logan DVR > 1.2, previously computed in the HABS sample derived from a Gaussian mixture modeling(41). For ADNI participants, we download the amyloid burden composite directly from their webpage (12 Feb 2020). Details for FBP-PET and FBB-PET acquisitions are described elsewhere (http://adni.loni.usc.edu/wp-content/uploads/2012/10/ADNI3_PET-Tech-Manual_V2.0_20161206.pdf). Briefly, FBP-PET and FBB-PET images were co-registered to the T1-MRI Freesurfer processed image and computed the weighted mean inside a cortical summary region that is made up of frontal, anterior/posterior cingulate, lateral parietal, lateral temporal regions, normalized by the signal intensity of the whole cerebellum to obtain an amyloid burden FBP-PET and FBB-PET SUVr scalar value(42). We used a previous validated threshold of FBP-PET > 1.11 and FBB-PET > 1.08 to assess amyloid positivity. We end up with 136 amyloid positive participants across the two samples.
Tau-PET and Backbone Graphs
Contrary to conventional intensity-based PET imaging studies, recent methodological advances have allowed the study of FTP-PET signals across different brain systems using high-dimensional network-based approaches(3, 10). Due to the intrinsic shared properties, as well as local gaussian smoothing of the PET signal, nodes in high-dimensional networks might be interrelated between them, sharing link information, and including redundancy in the analyses which result into technical caveats (such as computational time or difficulties to get significant results due to high-dimensional multiple comparisons corrections). In the present study, we developed a novel node-aggregation algorithm (NAA) with the intention of i) integrating all the converging information and ii) obtaining a backbone or minimal graph characterization of the FTP-PET network. Moreover, such an approach will result in a set of meaningful data-driven ROIs for tau uptake, compared to conventional atlas-based approaches, where the delineation of the ROIs might be mining-less for the study of in vivo tau pathology.
Computation of high-dimensional FTP-PET association matrix (or connectivity matrix) has been explained in detail elsewhere(10). Briefly, for each pair of voxels within the previously obtained FTP-PET grey matter mask, we computed the Pearson r correlation and its corresponding p-value using the cross-sectional sample of participants (N=71). We corrected for multiple comparisons using a threshold of FDR q < 0.005 and selected the top 20% correlations. The resulting association matrix was used as input to the NAA. The aim of NAA is to iteratively identify the set of nodes with maximum shared information and aggregate them. Thus, we first computed the number of 2-simplex (i.e., set of 3 nodes interconnected) for each node of the high-dimensional matrix using equation 1:
| (1) |
where N is the total number of nodes, A is the voxel-wise adjacency matrix and j,k,i are nodes. After computing the number of 2-simplex per voxel, we, searched for the node with highest number of 2-simplex and aggregate all the interconnected nodes into a new super-node (Fig 1). We repeated the aggregation of nodes iteratively, using the original adjacency matrix, till there is not any set of 2-simplex in the network. Afterwards, we computed the edge/link between two super-nodes as the mean of the correlation between all the nodes that are part of the super-nodes, which resulted into the minimal FTP-PET network. We studied the resulting minimal network computing each super-node degree as the sum of weighted links that arise from each super-node (Fig 2). For visualization purposes, we projected the set of super-nodes and the degree map to Freesurfer fsaverage standard space using mri_vol2surf.
Robustness of tau backbone graph
We studied the stability of the resulting network with a permutation approach by re-running the algorithm 500 times in a subset of 50 random participants and computed the spatial normalized Mutual Information (NMI) comparing the permuted-sample vs original sample network. To estimate the significance of our findings, we compared the sub-sample permutation results with two different null models that account for spatial auto-correlation. First, at the subject-level, using BrainSmash(43), we computed for each of the 71 participants, a surrogate map which preserve the spatial variance, spatial auto-correlation and global levels of SUVr intensity. From the obtained surrogate maps, we re-compute the tau backbone network running the NAA algorithm and compute the NMI between the network driven by the surrogate maps and the original network. This process was repeated 500 times. Second, we used a spin rotation approach(44) to randomly rotate 500 times (in the surface sphere space) the original backbone graph and computed the NMI between the rotated map and the original network for each one of the 500 permutations. These two frameworks allowed us to compare the results from the sub-sample permutation approach with two different null-models accounting by the spatial auto-correlation.
Longitudinal Individual Prediction of tau-PET Accumulation
A subset of 64 participants had longitudinal FTP-PET data (32 HABS participants at 2 years, 32 ADNI participants at 1 year follow-up). We designed a propagation model grounded on recent in vivo findings(38, 45), but instead of using structural or functional connectome information to define the connectivity between different regions, we used as a propagation skeleton the FTP-PET network driven by the NAA approach. We defined the longitudinal model propagation as:
| (2) |
where j refers to super-node, p to participant, A is the adjacency matrix, crossTau is the cross-sectional mean FTP-PET on super-node j and participant p, and BETA is the regularization parameter. We estimated the BETA parameter using the Powell optimizer as implemented in the SciPy python package. In brief, our model will predict the accumulation of longitudinal tau based on their network neighbor’s tau, where regions with high-uptake communities will accumulate faster. We performed three statistical analyses to evaluate the performance of our model to predict longitudinal data. For each cohort, we first computed the Spearman Rho between the predicted and real longitudinal FTP-PET pooling all the super-nodes from all the participants together. We then estimated the intra-participant agreement using the Spearman Rho coefficient to evaluate individually the accuracy of the model. Finally, we assessed the accuracy of the prediction on a regional basis, computing the mean standard error (MSE) for each one of the super-nodes, displaying the values in the cortical surface.
Network-Brain-Gene Association Relationship
In the present study we have developed a novel approach that integrates spatial high-resolution gene expression within the network structure of tau accumulation. This strategy brings a novel framework to incorporate gene expression data into brain connectivity circuits, rather than just investigate spatial overlaps between transcriptomic and neuroimaging phenotypes. The rationale behind such an approach is to study whether the network of tau spreading from an initial seed point (entorhinal) is also reflected in a gradient of constitutive gene expression within the network, reflecting brain vulnerability to tau propagation. We used a surface anatomical transformation of the cortical transcription profiles of 10027 protein-coding genes that fullfilled a quality control filter, based on 58,692 measurements of gene expression in 3,702 brain samples obtained from the left hemisphere of 6 adult human participants of the AHBA(46, 47). Gene expression was averaged within 180 cortical areas from the Glasser et al atlas(48). Based on previous reports of hemisphere-symmetry in the cortical gene expression, we mirrored the left hemisphere gene expression to the right hemisphere. Since our NAA approach creates data-driven ROIs that do not match the Glasser parcellation, we computed the gene expression within each super-node as the weighted average of each region from the Glasser atlas that co-localized with each super-node, resulting into a gene expression matrix of 58 regions (the number of super-nodes in the backbone FTP-PET network) by 10,027 genes. To assess the network-based spreading, we selected as seed the backbone node with the highest overlap with the entorhinal cortex, namely the left hemisphere entorhinal, previously identified as a starting point of tau-pathology spreading in AB positive individuals(34), and we computed the distance between nodes using the inverse weight of the links, and based on Dijsktra algorithm, which resulted in a single value of network distance from each super-node to the entorhinal. Importantly, we selected this region as seed, since we wanted to evaluate the gradual change of gene expression across the different Braak staging patterns. Thus, we obtained a network distance map where regions (network-wise) closer to the entorhinal mapped to Braak III-IV areas, whereas nodes far away belong to Braak V-VI areas. Then, for each gene, we computed the Spearman Rho coefficient between gene expression and network distance. We selected the significant correlations using a Benjamin-Hochberg FDR q < 0.001. Moreover, we studied if the pattern of genes significantly associated with distance from the entorhinal were network-topography dependent, generating a null-model computation of the distance and the calculation of the Spearman Rho after permuting randomly the super-node edges 1000 times.
Bulk RNAseq Differential Expression
We included bulkRNAseq gene expression derived from the dorsolateral prefrontal cortex of 792 participants from the ROS/MAP cohort. A detailed description of the cohort and patient characteristics can be found elsewhere(49). Participants were categorized as HC (N=260), mild cognitive impaired (N=206) or AD (N=326) based on clinical status. Details on sample collections, tissue and RNA preparation and quality control are provided in previously published work(49). We downloaded the “.bam” files for each participant data from the ROS/MAP repository (https://adknowledgeportal.synapse.org/Explore/Studies/DetailsPage?Study=syn3219045). To assess the deferentially expressed (DE) genes set, we compared gene expression from 55765 genes of HC against AD using DESeq2(50), controlling by age at death, sex, education, library preparation and library batch and extra batch effects as detected by the SVAseq package(51). We considered significant genes with p-value after correction with FDR q < 0.05, as usually done in DE analyses(52).
Tau Network-Based Genes Interactome and Gene Ontology Analysis
We characterize the biological meaning of gene sets from the tau network-based genes (TNG) using Gene Ontology (GO) cellular component profile (Panther DB; default set of parameters(53)). Then, we analyzed the relationship between TNG imputing their interactome using Genemania(54) which returned the gene-gene network based on a curated list of gene physical interaction. From the resulting network, we compute the relevance of each gene computing three centrality measures: closeness, degree and betweenness using Cytoscape(55).
Tau Network-Based Genes and NFT Bipartite Network
Finally, we investigate the relationship between our TNG results (577 genes) and an overlapping NFT-pTau affinity purification-mass spectrometry derived profile that has recently been reported as the pTau-interactome (74 genes)(16). First, using Genemania, we create the bi-partite network of the relationship between the TNG and the pTau-interactome genes. We studied the relationship between the two sets of genes using three measures: i) mean inter-set degree, ii) number of genes inter-connecting from each gene-set and iii) the total sum degree. To estimate their significance, we generated a null distribution of these parameters using a random set of of neuro-genes obtained from the bulkRNAseq data from the ROS/MAP cohort: 577 TNG vs. 74 random genes. Finally, using the information of the amount of protein expressed by each gene of the pTau-interactome in NFTs, as published in Drummond et al, we computed the importance of each of the TNG as the mean of expressed protein for each of its connected genes in the pTau-interactome. This is grounded in the idea that one gene in the TNG might be more relevant to tau pathology if it is related to a gene that does express a lot of proteins in the NFT. We used CIRCOS (56) to visualize the inter-group links, the intra-group degree (green histogram), the GO-term mean inter-set degree and the protein-based importance of each gene (red-histogram). For visualization purposes, we only plot genes present in the GO-terms, which resulted in some genes being displayed more than once.
Supplementary Material
Table 1. Participant characteristics.
Demographics of amyloid positive participants included in the study
| HABS | ADNI | |
|---|---|---|
| Sample (N) | 51 | 85 |
| Sample long (N) | 32 | 32 |
| Age (median, IQR) | 79.2 [73.7-82.9] | 74.8 [69.6-79.1] |
| Sex (female %) | 62.74 | 57.64 |
| metaROI FTP-PET, SUVr (median, IQR) | 1.15 [1.11-1.22] | 1.19 [1.15-1.25] |
Acknowledgments:
We thank the investigators and staff of the Harvard Aging Brain Study, Massachusetts Alzheimer’s Disease Research Center, the individual research participants, and their families and caregivers. We also thank the PET Core and Gordon Center for Medical Imaging of the MGH, the Harvard Center for Brain Science Neuroimaging Core and the Athinoula A. Martinos Center for biomedical imaging support.
Funding:
US National Institute of Aging grant R01AG061811 to JS
US National Institute of Aging grant R01AG061445 to JS
US National Institute of Aging grant R01-AG061083 to JS and PV
US National Institute of Aging grant R01-AG027435-S1 to KAJ and RAS
US National Institute of Aging grant P50-AG00513421 to KAJ and RAS
US National Institute of Aging grant R01-AG046396 KAJ and RAS
US National Institute of Aging grant P01-AG036694 KAJ and RAS
US National Institute of Aging grant P41-EB022544 to GE
Institute of Health Carlos III grant FI18/00275 to VM
Institute of Health Carlos III grant CP20/00038 to AB
CIBERNED – Mobility grant to VM
Footnotes
Competing interests:
Authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. The HABS project is committed to publicly releasing its data. Baseline data are already available online at http://nmr.mgh.harvard.edu/lab/ harvardagingbrain/data. Follow-up data of the HABS data, including the data used this manuscript, are now available by request, pending approval of a data request and agreement to abide by the HABS online data use agreement. ADNI data is available online after signing a data use agreement. All code used in this study is available upon request to the corresponding author.
References
- 1.Zang Z, Lin P, Levey A, Monetary Costs of Dementia in the United States To, N. Engl. J. Med 369, 487–489 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Johnson KA, Schultz A, Betensky RA, Becker JA, Sepulcre J, Rentz D, Mormino E, Chhatwal J, Amariglio R, Papp K, Marshall G, Albers M, Mauro S, Pepin L, Alverio J, Judge K, Philiossaint M, Shoup T, Yokell D, Dickerson B, Gomez-Isla T, Hyman B, Vasdev N, Sperling R, Tau positron emission tomographic imaging in aging and early Alzheimer disease, Ann. Neurol 79, 110–119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sepulcre J, Schultz A, Sabuncu M, Gomez-Isla T, Chhatwal J, Becker A, Sperling R, Johnson K, In vivo Tau, Amyloid and Grey Matter Profiles in the Aging Brain, J. Neurosci, 7364–7374 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schöll M, Lockhart SN, Schonhaut DR, O’Neil JP, Janabi M, Ossenkoppele R, Baker SL, Vogel JW, Faria J, Schwimmer HD, Rabinovici GD, Jagust WJ, PET Imaging of Tau Deposition in the Aging Human Brain, Neuron 89, 971–982 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marquié M, Siao Tick Chong M, Antón-Fernández A, Verwer EE, Sáez-Calveras N, Meltzer AC, Ramanan P, Amaral AC, Gonzalez J, Normandin MD, Frosch MP, Gómez-Isla T, [F-18]-AV-1451 binding correlates with postmortem neurofibrillary tangle Braak staging, Acta Neuropathol. 134, 619–628 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.La Joie R, V Visani A, Baker SL, Brown JA, Bourakova V, Cha J, Chaudhary K, Edwards L, Iaccarino L, Janabi M, Lesman-Segev OH, Miller ZA, Perry DC, O’Neil JP, Pham J, Rojas JC, Rosen HJ, Seeley WW, Tsai RM, Miller BL, Jagust WJ, Rabinovici GD, Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET., Sci. Transl. Med 12, 1–13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sperling RA, Mormino EC, Schultz AP, Betensky RA, Papp KV, Amariglio RE, Hanseeuw BJ, Buckley R, Chhatwal J, Hedden T, Marshall GA, Quiroz YT, Donovan NJ, Jackson J, Gatchel JR, Rabin JS, Jacobs H, Yang HS, Properzi M, Kirn DR, Rentz DM, Johnson KA, The impact of amyloid-beta and tau on prospective cognitive decline in older individuals, Ann. Neurol 85, 181–193 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoenig MC, Bischof GN, Seemiller J, Hammes J, Kukolja J, Onur ÖA, Jessen F, Fliessbach K, Neumaier B, Fink GR, Van Eimeren T, Drzezga A, Networks of tau distribution in Alzheimer’s disease, Brain 141, 568–581 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Ossenkoppele R, Iaccarino L, Schonhaut DR, Brown JA, La Joie R, O’Neil JP, Janabi M, Baker SL, Kramer JH, Gorno-Tempini ML, Miller BL, Rosen HJ, Seeley WW, Jagust WJ, Rabinovici GD, Tau covariance patterns in Alzheimer’s disease patients match intrinsic connectivity networks in the healthy brain, NeuroImage Clin. 23, 101848 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sepulcre J, Grothe MJ, Uquillas O, Ortiz-terán L, Diez I, Yang H, Jacobs HIL, Hanseeuw BJ, Li Q, El-fakhri G, Sperling RA, Johnson KA, Neurogenetic contributions to amyloid beta and tau spreading in the human cortex, Nat. Med (2018), doi: 10.1038/s41591-018-0206-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mrdjen D, Fox EJ, Bukhari SA, Montine KS, Bendall SC, Montine TJ, The basis of cellular and regional vulnerability in Alzheimer’s disease, Acta Neuropathol. 138, 729–749 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu H, Hardy J, Duff KE, Selective vulnerability in neurodegenerative diseases, Nat. Neurosci 21, 1350–1358 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leng K, Li E, Eser R, Piergies A, Sit R, Tan M, Neff N, Li SH, Rodriguez RD, Suemoto CK, Leite REP, Ehrenberg AJ, Pasqualucci CA, Seeley WW, Spina S, Heinsen H, Grinberg LT, Kampmann M, Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease, Nat. Neurosci 24, 276–287 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai L-H, Single-cell transcriptomic analysis of Alzheimer’s disease, Nature 2 (2019), doi: 10.1038/s41586-019-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen EH, Overly CC, Jones AR, The Allen Human Brain Atlas: comprehensive gene expression mapping of the human brain., Trends Neurosci. 35, 711–714 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Drummond E, Pires G, Macmurray C, Askenazi M, Nayak S, Bourdon M, Safar J, Ueberheide B, Wisniewski T, Phosphorylated tau interactome in the human Alzheimer’s disease brain, Brain , 1–15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Acosta D, Powell F, Zhao Y, Raj A, Regional vulnerability in Alzheimer’s: The role of cell-autonomous and transneuronal processes, Alzheimer’s Dement. , 1–13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seidlitz J, Nadig A, Liu S, Bethlehem RAI, Vértes PE, Morgan SE, Váša F, Romero-Garcia R, Lalonde FM, Clasen LS, Blumenthal JD, Paquola C, Bernhardt B, Wagstyl K, Polioudakis D, de la Torre-Ubieta L, Geschwind DH, Han JC, Lee NR, Murphy DG, Bullmore ET, Raznahan A, Transcriptomic and cellular decoding of regional brain vulnerability to neurogenetic disorders, Nat. Commun 11, 1–14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diez I, Sepulcre J, Neurogenetic profiles delineate large-scale connectivity dynamics of the human brain, Nat. Commun 9, 3876 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grothe MJ, Sepulcre J, Gonzalez-Escamilla G, Jelistratova I, Schö M, Hansson O, Teipel SJ, Molecular properties underlying regional vulnerability to Alzheimer’s disease pathology8 for the Alzheimer’s Disease Neuroimaging Initiative*, , 1–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raj A, Tora V, Gao X, Cho H, Choi JY, Ryu Y, Lyoo C, Franchi B, Combined Model of Aggregation And Network Diffusion Recapitulates Alzheimer’s Regional Tau-PET, Brain Connect. , 1–42 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meisl G, Hidari E, Allinson K, Rittman T, DeVos SL, Sanchez JS, Xu CK, Duff KE, Johnson KA, Rowe JB, Hyman BT, Knowles TPJ, Klenerman D, In vivo rate-determining steps of tau seed accumulation in Alzheimer’s disease, Sci. Adv 7 (2021), doi: 10.1126/sciadv.abh1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freer R, Sormanni P, Vecchi G, Ciryam P, Dobson CM, Vendruscolo M, A protein homeostasis signature in healthy brains recapitulates tissue vulnerability to Alzheimer’s disease, Sci. Adv 2, 1–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cornblath EJ, Li HL, Changolkar L, Zhang B, Brown HJ, Gathagan RJ, Olufemi MF, Trojanowski JQ, Bassett DS, Lee VMY, Henderson MX, Computational modeling of tau pathology spread reveals patterns of regional vulnerability and the impact of a genetic risk factor, Sci. Adv , 1–16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arnsten AFT, Datta D, Del Tredici K, Braak H, Hypothesis : Tau pathology is an initiating factor in sporadic Alzheimer ’ s disease, Alzheimers. Dement , 1–10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma A, Kazim SF, Larson CS, Ramakrishnan A, Gray JD, McEwen BS, Rosenberg PA, Shen L, Pereira AC, Divergent roles of astrocytic versus neuronal EAAT2 deficiency on cognition and overlap with aging and Alzheimer’s molecular signatures, Proc. Natl. Acad. Sci. U. S. A 116, 21800–21811 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foster JB, Lashley R, Zhao F, Wang X, Kung N, Askwith CC, Lin L, Shultis MW, Hodgetts KJ, Lin CLG, Enhancement of tripartite synapses as a potential therapeutic strategy for Alzheimer’s disease: A preclinical study in rTg4510 mice, Alzheimer’s Res. Ther 11, 1–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jablonski AM, Warren L, Usenovic M, Zhou H, Sugam J, Parmentier-Batteur S, Voleti B, Astrocytic expression of the Alzheimer’s disease risk allele, ApoEε4, potentiates neuronal tau pathology in multiple preclinical models, Sci. Rep 11, 1–18 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Litvinchuk A, Huynh TPV, Shi Y, Jackson RJ, Finn MB, Manis M, Francis CM, Tran AC, Sullivan PM, Ulrich JD, Hyman BT, Cole T, Holtzman DM, Apolipoprotein E4 Reduction with Antisense Oligonucleotides Decreases Neurodegeneration in a Tauopathy Model, Ann. Neurol (2021), doi: 10.1002/ana.26043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang C, Xiong M, Gratuze M, Artyomov M, Ulrich JD, Holtzman DM, Wang C, Xiong M, Gratuze M, Bao X, Shi Y, Andhey PS, Manis M, Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia Article Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration, Neuron , 1–18 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zalocusky KA, Najm R, Taubes AL, Hao Y, Yoon SY, Koutsodendris N, Nelson MR, Rao A, Bennett DA, Bant J, Amornkul D. eun J., Xu Q, An A, Cisne-Thomson O, Huang Y, Neuronal ApoE upregulates MHC-I expression to drive selective neurodegeneration in Alzheimer’s disease, Nat. Neurosci 24, 786–798 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu CC, Kanekiyo T, Xu H, Bu G, Apolipoprotein e and Alzheimer disease: Risk, mechanisms and therapy, Nat. Rev. Neurol 9, 106–118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez S, Hug C, Todorov P, Moret N, Boswell SA, Evans K, Zhou G, Johnson NT, Hyman BT, Sorger PK, Albers MW, Sokolov A, Machine learning identifies candidates for drug repurposing in Alzheimer’s disease, Nat. Commun 12 (2021), doi: 10.1038/s41467-021-21330-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanchez JS, Becker JA, Jacobs HIL, Hanseeuw BJ, Jiang S, Schultz AP, Properzi MJ, Katz SR, Beiser A, Satizabal CL, O’Donnell A, DeCarli C, Killiany R, El Fakhri G, Normandin MD, Gómez-Isla T, Quiroz YT, Rentz DM, Sperling RA, Seshadri S, Augustinack J, Price JC, Johnson KA, The cortical origin and initial spread of medial temporal tauopathy in Alzheimer’s disease assessed with positron emission tomography, Sci. Transl. Med 13, eabc0655 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vogel JW, Iturria-Medina Y, Strandberg OT, Smith R, Levitis E, Evans AC, Hansson O, Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease, Nat. Commun 11, 2612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Franzmeier N, Dewenter A, Frontzkowski L, Dichgans M, Rubinski A, Neitzel J, Smith R, Strandberg O, Ossenkoppele R, Buerger K, Duering M, Hansson O, Ewers M, Patient-centered connectivity-based prediction of tau pathology spread in Alzheimer’s disease., Sci. Adv 6 (2020), doi: 10.1126/sciadv.abd1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raj A, Graph Models of pathology spread in Alzheimer’s Disease: An Alternative to Conventional Graph Theoretic Analysis, Brain Connect. , 1–61 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang F, Chowdhury SR, Jacobs HIL, Sepulcre J, Wedeen VJ, Johnson KA, Dutta J, Longitudinal predictive modeling of tau progression along the structural connectome, Neuroimage 237, 118126 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anand C, Maia P, Torok J, Mezias C, Raj A, The Effects of Microglia on Tauopathy Progression Can Be Quanti ed Using Nexopathy in Silico ( Nexis ) Models The effects of microglia on tauopathy progression can be quantified using Nexopathy in silico ( Nex is ) models, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Montal V, Vilaplana E, Pegueroles J, Bejanin A, Alcolea D, Carmona-Iragui M, Clarimon J, Levin J, Cruchaga C, Graf-Radford N, Noble JM, Lee J-H, Allegri R, Karch CM, Laske C, Schofield PR, Salloway S, Ances B, Benzinger T, McDade E, Bateman R, Blesa R, Sanchez-Valle R, Lleo A, Fortea J, Biphasic cortical macro and microstructural changes in autosomal dominant Alzheimer disease, Alzheimer’s Dement. in press, 1–11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mormino EC, Betensky RA, Hedden T, Schultz AP, Ward A, Huijbers W, Rentz DM, Johnson KA, Sperling RA, Amyloid and APOE ε4 interact to influence short-term decline in preclinical Alzheimer disease, Neurology 82, 1760–1767 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Landau SM, Breault C, Joshi AD, Pontecorvo M, a Mathis C, Jagust WJ, a Mintun M, Amyloid-β imaging with Pittsburgh compound B and florbetapir: comparing radiotracers and quantification methods., J. Nucl. Med 54, 70–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burt JB, Helmer M, Shinn M, Anticevic A, Murray JD, Generative modeling of brain maps with spatial autocorrelation, Neuroimage 220, 117038 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Alexander-Bloch A, Shou H, Liu S, Satterthwaite TD, Glahn DC, Shinohara RT, Vandekar SN, Raznahan A, On testing for spatial correspondence between maps of human brain structure and function, Neuroimage (2018), doi: 10.1016/j.neuroimage.2018.05.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jack CR, Wiste HJ, Weigand SD, Therneau TM, Lowe VJ, Knopman DS, Botha H, Graff-Radford J, Jones DT, Ferman TJ, Boeve BF, Kantarci K, Vemuri P, Mielke MM, Whitwell J, Josephs K, Schwarz CG, Senjem ML, Gunter JL, Petersen RC, Predicting future rates of tau accumulation on PET, Brain 143, 3136–3150 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diez I, Sepulcre J, Unveiling the neuroimaging-genetic intersections in the human brain., Curr. Opin. Neurol 34, 480–487 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arnatkevic A, Fulcher BD, Fornito A, A practical guide to linking brain-wide gene expression and neuroimaging data, Neuroimage (2019), doi: 10.1016/j.neuroimage.2019.01.011. [DOI] [PubMed] [Google Scholar]
- 48.Glasser MF, Coalson TS, Robinson EC, Hacker CD, Harwell J, Yacoub E, Ugurbil K, Andersson J, Beckmann CF, Jenkinson M, Smith SM, Van Essen DC, A multi-modal parcellation of human cerebral cortex, Nature 536, 171–178 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Jager PL, Ma Y, McCabe C, Xu J, Vardarajan BN, Felsky D, Klein HU, White CC, Peters MA, Lodgson B, Nejad P, Tang A, Mangravite LM, Yu L, Gaiteri C, Mostafavi S, Schneider JA, Bennett DA, Data descriptor: A multi-omic atlas of the human frontal cortex for aging and Alzheimer’s disease research, Sci. Data 5, 1–13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, Genome Biol. 15, 1–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leek JT, Svaseq: Removing batch effects and other unwanted noise from sequencing data, Nucleic Acids Res. 42, e161 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dols-Icardo O, Montal V, Sirisi S, López-Pernas G, Cervera-Carles L, Querol-Vilaseca M, Muñoz L, Belbin O, Alcolea D, Molina-Porcel L, Pegueroles J, Turón-Sans J, Blesa R, Lleó A, Fortea J, Rojas-García R, Clarimón J, Motor cortex transcriptome reveals microglial key events in amyotrophic lateral sclerosis, Neurol Neuroimmunol Neuroinflam 7, 1–12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mi H, Muruganujan A, Thomas PD, PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees, Nucleic Acids Res. 41, 377–386 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q, GeneMANIA: A real-time multiple association network integration algorithm for predicting gene function, Genome Biol. 9 (2008), doi: 10.1186/gb-2008-9-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.1 Shannon Paul, 1 Markiel Andrew, 2 Ozier Owen, 2 Baliga Nitin S., 1 Wang Jonathan T., 2 Ramage Daniel, 2 Amin Nada, 5 Schwikowski Benno, 1, 5 and Ideker Trey2, 3, 4, Cytoscape: A Software Environment for Integrated Models, Genome Res. 13, 426 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA, Circos: An information aesthetic for comparative genomics, Genome Res. 19, 1639–1645 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.