

Abstract
Primary CNS histiocytic sarcoma is a rare hematolymphoid malignancy with features of mature histiocytes and carries a poor prognosis. We describe a unique case in which a 50-year-old woman presented with recurrent acute brainstem syndrome, area postrema syndrome, and myelitis with corresponding MRI lesions meeting diagnostic criteria for seronegative NMOSD. Despite initial improvement with steroids and plasma exchange, she experienced recurrent symptoms over ten months referable to new and persistently enhancing lesions. At autopsy, neuropathology revealed a diffusely infiltrative primary CNS histiocytic sarcoma. This case represents a rare clinicoradiologic mimic of NMOSD, underscoring the importance of evaluation for infiltrative diseases in cases of atypical seronegative NMOSD.
Keywords: Primary CNS histiocytic sarcoma, NMOSD mimic, case report
Case Report
A 50-year-old woman presented with right facial pain and numbness and paresthesia of the right first through third fingers which gradually recovered after 3–4 weeks without intervention. She reported a 20-pound unintentional weight loss this same month. Six months later, the symptoms recurred accompanied by acute vertigo, oscillopsia, intractable nausea and vomiting, then diplopia, left facial numbness, ascending, bilateral sensory loss and weakness in her legs, urinary retention, and constipation all within one month. Initial neurologic exam indicated a multifocal brainstem syndrome with nystagmus, left internuclear ophthalmoplegia, diminished left facial sensation, left forehead-sparing facial weakness, and myelopathy characterized by bilateral lower extremity pyramidal pattern weakness, a T5 sensory level, diffuse hyperreflexia with upgoing toes bilaterally, and a wide-based gait requiring front-wheeled walker. Her medical history was non-contributory.
Initial MRI brain and spinal cord showed multifocal T2/FLAIR hyperintense lesions with enhancement including the dorsal pons, area postrema and cervicomedullary junction extending to C2-C3, with a non-enhancing longitudinally extensive T5-T7 thoracic lesion (Figure 1A–C). CSF showed a lymphocytic pleocytosis (WBC 42 cells/μl; 89% lymphocytes), elevated protein (89 mg/dL) and >2 unique CSF oligoclonal bands. Extensive serum and CSF testing were otherwise unremarkable including negative aquaporin 4 (AQP4-IgG), myelin oligodendrocyte glycoprotein (MOG-IgG), and paraneoplastic autoantibodies. CT chest/abdomen/pelvis and whole-body PET CT were negative. Given her clinical presentation of acute brainstem syndrome, area postrema syndrome, and acute transverse myelitis with supportive MRI features of a peri-ependymal brainstem lesion, dorsal medullary lesion, and longitudinally extensive transverse myelitis (LETM) lesion, she met diagnostic criteria for seronegative NMOSD.1
Figure 1.
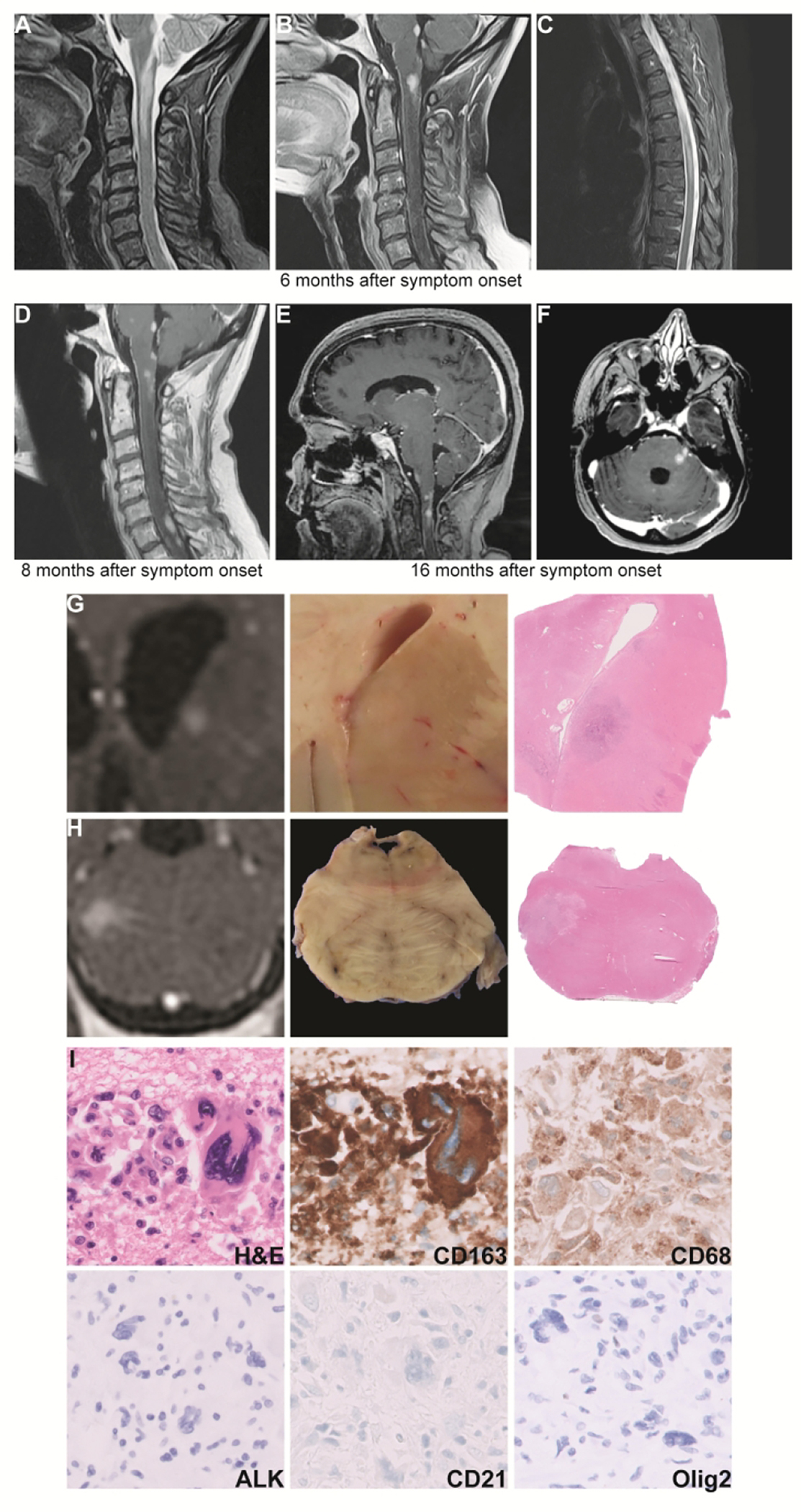
Neuroimaging: MRI at initial hospitalization, 6 months after symptom onset, demonstrates a longitudinally extensive enhancing lesion at the cervicomedullary junction with serpiginous enhancement extending into the area postrema, an enhancing lesion in the inferior dorsal pons, and leptomeningeal enhancement of the cervical cord (T2-weighted image, A; T1-weighted post-contrast image, B). There was also a non-enhancing T2-hyperintense longitudinally extensive T5-T7 lesion (C). Not shown are additional enhancing lesions in the left lateral pons and trigeminal nerve root, left midbrain, and peri-ependymal region along occipital horn of left lateral ventricle. MRI 8 months after symptom onset shows new right paramedian pontine and cervical and thoracic spinal cord lesions with persistent enhancement of previous lesions (T1 post-contrast image, D). MRI 16 months after symptom onset shows multiple new enhancing lesions including in the bilateral caudate heads, splenium of corpus callosum, midbrain, ventral medulla, and cerebellar vermis (E). A persistently enhancing left middle cerebellar peduncle lesion was the target of the patient’s second brain biopsy (F). Gross and microscopic characterization of enhancing lesions: (A), Coronal T1-weighted post-contrast MRI demonstrating enhancing lesion in the caudate head with corresponding gross photo showing faint pallor and low-power microscopy showing a cellular lesion. (B), Axial T1-weighted post-contrast MRI of enhancing lesion at the left pons near the trigeminal nerve root with corresponding gross photo and low-power microscopic image showing loss of gray-white distinction. (C), High-power H&E and immunohistochemistry show an infiltration of large, pleomorphic CD163 and CD68 positive cells that are negative for ALK-1, CD21, and Olig2. Not shown are positive lysozyme and negative CD30, CD34, CD117, CD1a, CD21, Langerin, CD3, CD4, mixed cytokeratins AE1/ CAM 5.2, HMB45, MiTF, S100, Olig2, GFAP, and BRAF V600E. Immunohistochemistry for CD163 corresponds to the same field as the H&E, whereas other images are from different fields. Radiology images have been rotated or flipped to match gross images with true left on the left of the image and vice versa.
She received 5 days of 1 g IV methylprednisolone then 60 mg oral prednisone daily, tapered by 10 mg every week with initial improvement in her vertigo and diplopia. Two months later, she experienced recurrence of vertigo, diplopia, and bilateral arm and leg sensory loss and weakness with urinary retention and was found to have new right paramedian pontine and cervical and thoracic spinal cord lesions with persistent enhancement of previous lesions (Figure 1D). She received 5 days of 1250 mg oral prednisone and was re-admitted for plasma exchange with improvement in diplopia, leg sensation, gait, and urinary symptoms. Over the ensuing 8 months she underwent repeated investigations for alternative infiltrative or malignant disease including three additional lumbar punctures, CT chest/abdomen/pelvis, mammogram, pelvic ultrasound, whole-body PET-CT, bone marrow biopsy and two brain biopsies all of which were negative for malignancy in the sampled tissue. Her disease progressed clinically and radiographically (Figure 1E, F) despite escalating immunotherapy including maintenance prednisone (30–50 mg daily), rituximab 1g IV, mycophenolate mofetil (500 mg twice daily, dose limited by severe nausea/fatigue) and cyclophosphamide (500–600 mg/m2, 3 cycles). Sixteen months after her initial presentation the patient and family pursued hospice care, and she died at home three days after hospital discharge.
A complete general autopsy found no evidence of systemic malignancy. Gross neuropathologic exam identified correlates of the enhancing MRI lesions (Figure 2A, B) and multifocal areas of periventricular white matter softening. Microscopic evaluation (Figure 2C) showed multiple infiltrative foci of large, pleomorphic malignant cells positive for expression of histiocytic markers (CD163, CD68, lysozyme) with no expression of dendritic, epithelial, melanocytic, myeloid, or glial markers consistent with primary CNS histiocytic sarcoma. Tumor-only next-generation sequencing found pathogenic mutations in BRAF, CDKN2A, KRAS, and TP53, which are common in histiocytic sarcoma.2
Discussion
This patient initially met diagnostic criteria for seronegative NMOSD, however the ultimate diagnosis at autopsy was primary CNS histiocytic sarcoma. Primary CNS histiocytic sarcoma is extremely rare, with only 32 previously reported cases.3 Diagnosis is challenging given the lack of distinctive clinical or radiographic presentation, poor accessibility to CNS tissue for biopsy, and overlapping pathologic characteristics of histiocytic infiltrates in inflammatory brain lesions including demyelinating disease.4, 5 The majority of cases present with solitary or multiple mass lesions and an aggressive course that is uniformly fatal. One previous case described primary CNS histiocytic sarcoma mimicking tumefactive multiple sclerosis.5 Our case more closely mimicked NMOSD and met diagnostic criteria early in the clinical course. Clues arguing against NMOSD at initial presentation were her concomitant weight loss and presence of subtle leptomeningeal enhancement, though leptomeningeal enhancement has also been reported in NMOSD.6 Ultimately it was the persistently enhancing lesions over 10 months that raised our concerns for an infiltrative process. Enhancement of NMOSD lesions typically resolves by 2 months and always after 6 months.7
This case underscores the imperative to consider (and reconsider) infiltrative malignancies in cases of presumed, treatment-refractory NMOSD and highlights a unique clinical and radiographic presentation of rare primary CNS histiocytic sarcoma.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Consent
Written informed consent for patient information and images to be published was provided by the patient’s legally authorized representative.
References
- 1.Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015;85:177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shanmugam V, Griffin GK, Jacobsen ED, Fletcher CDM, Sholl LM, Hornick JA-O. Identification of diverse activating mutations of the RAS-MAPK pathway in histiocytic sarcoma. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi E, Sakakibara A, Tsuzuki T, Nakamura S. Case of primary central nervous system histiocytic sarcoma with prominent proliferation of histiocytic cells between the trabeculae of reactive glial cells. Neuropathology 2018;38:609–618. [DOI] [PubMed] [Google Scholar]
- 4.Hung YP, Qian X. Histiocytic Sarcoma. Archives of pathology & laboratory medicine 2020;144:650–654. [DOI] [PubMed] [Google Scholar]
- 5.So H, Kim SA, Yoon DH, et al. Primary Histiocytic Sarcoma of the Central Nervous System. Cancer Res Treat 2014;47:322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asgari N, Flanagan EP, Fujihara K, et al. Disruption of the leptomeningeal blood barrier in neuromyelitis optica spectrum disorder. Neurology - Neuroimmunology Neuroinflammation 2017;4:e343–e343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sechi E, Krecke KN, Messina SA, et al. Comparison of MRI Lesion Evolution in Different Central Nervous System Demyelinating Disorders. Neurology 2021;97:e1097–e1109. [DOI] [PMC free article] [PubMed] [Google Scholar]