

Abstract
DNA polymerase theta (Polθ) is an attractive synthetic lethal target for drug discovery, predicted to be efficacious against breast and ovarian cancers harboring BRCA-mutant alleles. Here, we describe our hit-to-lead efforts in search of a selective inhibitor of human Pol θ (encoded by POLQ). An high throughput screening (HTS) campaign of 350,000 compounds identified an 11 micromolar hit, giving rise to the N2-substituted fused pyrazolo series which was validated by biophysical methods. Structure-based drug design efforts along with optimization of cellular potency and ADME ultimately led to the identification of RP-6685: a potent, selective, and orally bioavailable Polθ inhibitor that showed in vivo efficacy in a HCT116 BRCA2−/− mouse tumor xenograft model.
Keywords: Polθ, BRCA2, Double-strand break repair, PARP-i, POLQ-i, TMEJ, ALT-EJ, MMEJ, DNA repair, cancer, Structure-based Drug Design
Graphical Abstract
INTRODUCTION
DNA double-strand breaks (DSBs) arising during DNA replication or due to exogenous insults including irradiation and chemotherapy are highly deleterious lesions that can induce genome instability and lead to cell death. DSBs are repaired by three major repair pathways, including homologous recombination (HR), non-homologous mediated end-joining (NHEJ), and microhomology mediated end-joining (MMEJ). In G1 phase of the cell cycle DSBs are predominantly repaired by NHEJ at the cost of small insertions and deletions at break sites. DNA end-resection in S and G2 allows accurate repair by HR and relies on BRCA1 and BRCA2. MMEJ is a third mechanistically distinct pathway that is intrinsically mutagenic, whereas breaks sites are often scarred with insertions and deletions. MMEJ is viewed as a back-up pathway being essential in settings when NHEJ or HR is impaired, such as through loss of BRCA1/2.1 Furthermore, increasing evidence suggests that MMEJ is uniquely important for DNA repair during mitosis.2
Human DNA polymerase θ (Polθ) is a unique, large (290kDa) multifunctional A-family DNA polymerase that is required for DSB repair through the MMEJ pathway.3,4 Unique among eurkaryotic polymerases, the N-terminal domain contains an ATPase activity that displaces RPA from single-stand DNA5. The C-terminal domain contains a DNA polymerase activity that is responsible for filling in the gap after annealing of resected DNA ends6,7.
Polθ inhibition is an attractive drug target. Loss of Polθ has been shown to be synthetic lethal in tumor cells with impaired DNA repair resulting from multiple genetic alterations, such as loss of BRCA2 and FANCD2 and synergizes with PARP inhibition in HR deficient cells.3,8,9 It is unclear which domain, helicase-like (hld), polymerase (pol), or the central unstructured region, is the ideal target for drug development. 10,11,12 Recently, selective Polθ inhibitors have been reported that target the polymerase domain13 and a repurposed antibiotic has been shown to inhibit the helicase domain.14
Here we report the discovery and synthesis of novel potent, selective inhibitors of the DNA polymerase activity of Polθ and our efforts to characterize an inhibitor suitable for in vivo proof-of-concept studies.
RESULTS AND DISCUSSION
Early SAR
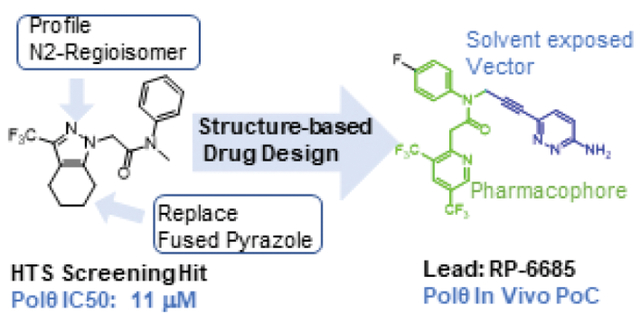
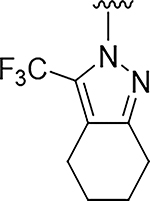
To identify small molecule inhibitor, we carried out a high throughput screen (HTS) of 350,000 compounds (Figure 1). The high-throughput screening strategy employed an enzymatic DNA primer extension assay catalyzed by the Polθ pol domain (aa1819–2590), in which nascent dsDNA was quantified by the picogreen intercalating dye.15 One of the hits obtained was pyrazole 1, with an IC50 of 11μM. Upon close examination by LCMS and NMR we noticed that 1 was a mixture of N-regioisomers, with the N1-regioisomer being the major isomer and the N2-regioisomer present in trace amounts. Resynthesis accomplished in three steps via Claisen condensation chemistry, ester hydrolysis and amide coupling (see Experimental section) afforded a 95:5 mixture of N1: N2 regioisomers. Separation of the regioisomers by silica chromatography or reverse phase preparative HPLC were unsuccessful, as the regioisomers eluted closely together. Separation was ultimately achieved by chiral SFC purification affording the major N1-regioisomer, compound 1, which had an 11 μM IC50 in our primary screening assay (Polθ Picogreen assay). The minor N2-regioisomer, compound 2, had a potency of 0.16 μM and thus served as an attractive starting point for hit-to lead efforts.
Figure 1:

Compound 2 is the starting point for our hit-to lead efforts
Although compound 2 displayed remarkable enzymatic potency for a screening hit, it suffered from metabolic instability in mouse liver microsomes (MLM) (Table 1). With the aim of improving the ADME properties of this hit, we explored the addition of polarity to the fused pyrazole core. Introduction of polar moieties on the fused pyrazole led to basic amine 3, lactam 4, alcohol 5, or sulfone 6 with improved metabolic stability, but complete loss of activity. In parallel we sought to determine the minimal pharmacophore essential for potency. Reducing the size of the fused cyclohexyl to cyclopentyl (7) was well tolerated. However, replacement of the trifluoromethyl moiety by a methyl group (8) led to a significant loss of potency suggesting an important lipophilic interaction with the binding pocket. SAR exploration of the southern portion of the fused pyrazole was conducted to expand our understanding of the binding pocket. Introduction of methyl (9, 10) or gem-dimethyl (11–13) were tolerated, alluding to a lipophilic and somewhat sizeable binding pocket. We prepared the corresponding thioamide 14 of compound 7 which led to a dramatic 40-fold improvement in potency albeit without improvement to metabolic stability.
Table 1:
Fused Pyrazolo Series
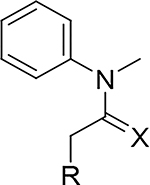
| ||||
|---|---|---|---|---|
|
| ||||
| Compd | R | X | Polθ IC50 (μM)a | Microsomal Stability CL (μL/min/mg) |
|
| ||||
| 2 |
|
O | 0.17 | >555 |
| 3 |
|
O | >100 | 16 |
| 4 |
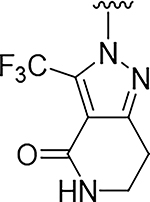
|
O | >100 | 18 |
| 5 |
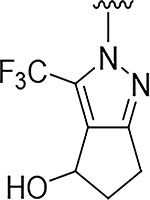
|
O | >100 | 46 |
| 6 |
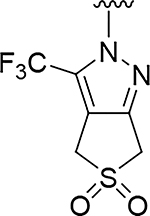
|
O | 43 | 23 |
| 7 |
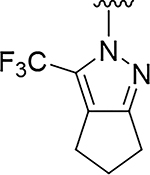
|
O | 0.71 | >555 |
| 8 |
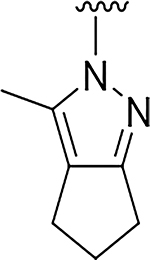
|
O | 10 | 473 |
| 9 |
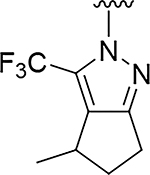
|
O | 0.24 | >555 |
| 10 |
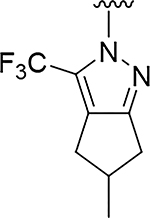
|
O | 0.95 | >832 |
| 11 |
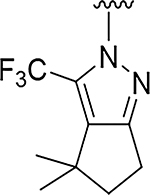
|
O | 0.52 | >555 |
| 12 |
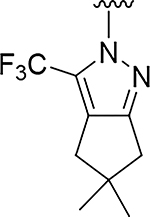
|
O | 0.56 | >555 |
| 13 |
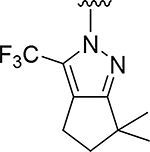
|
O | 2.3 | >832 |
| 14 |
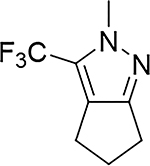
|
S | 0.017 | >555 |
Picogreen assay: 3nM of Polθ enzyme (n=3)
Hit Validation and Kinetics
To better understand the binding properties of this series, we selected compounds 9 and 14 for profiling in biophysical and kinetic assays. Initial evaluation by Differential Scanning Fluorimetry (DSF) did not show any thermal stabilization of WT Polθ induced by compounds 9 or 14 alone (Figure 2). A compound-induced thermal shift was observed only in the presence of a double-stranded DNA substrate providing a 5’ overhang, which mimics a primer-template pair. By inspection of the first derivative plot, 6 degrees of thermal stability was afforded due to binding of both compounds 9 and compound 14, to the enzyme-substrate (ES) complex. This observation validated this series as binders of Polθ and foreshadowed the subsequent results indicating an uncompetitive mechanism of action.
Figure 2:

DSF of Compound 9 and Compound 14 with WT Polθ.
To further investigate the mechanism of inhibition of our Polθ inhibitors, we devised a continuous (kinetic) primer-extension assay to further characterize compound derivatives (see Supporting Info). Compound 14 provided an IC50 of 10 nM in the adapted reaction conditions. To confirm that compound 14 was not titrating the enzyme, a dose-response of 0.5 to 5 nM enzyme was evaluated and no significant variation of the IC50 was observed (Figure 3A). Values for Km and Vmax were determined for dNTPs at different concentrations of compound 14 by fitting the initial reaction velocities to the Michaelis-Menten equation (Figure 3B). Increasing the concentration of compound 14 did not lead to a equivalent increase in KM for dNTPs, while Vmax decreased by nearly 90% in the presence of 40 nM compound 14. In conclusion, these data are inconsistent with competitive inhibition and infer that the compound binds to a site different from the nucleotide binding site. The kinetic data fit most closely with a model of noncompetitive inhibition, meaning that the compound binds equally well to the Polθ ES complex with or without concomitant binding of dNTPs (see Supporting Info Figure S7).
Figure 3: Compound 14’s MOA towards human Polθ.

Kinetic parameters were determined using a real time kinetic assay. (A) shows the inhibition of Polθ by compound 14 for concentrations of the enzyme varying from 5 to 0.5 nM. (B) shows dNTP dependency for the enzyme at various concentrations of compound 14. It displays the dependency of the enzyme velocity on various inhibitor (0 to 40 nM) and dNTP concentrations.
X-ray crystallography reveals the binding mode of compound 14
Compound 14 proved to be an excellent ligand for X-ray crystallography studies of the Polθ polymerase domain. Co-crystals of compound 14 bound to the polymerase domain of Polθ in complex with dsDNA and dideoxynucleotide were readily obtained but diffracted poorly (~4Å). To increase the diffraction quality of protein crystals, we engineered truncations in the protein expression construct, by deleting five flexible loops that were not resolved in previously published structures (see Supporting info for details). The crystal structure of Polθ bound to compound 14 was phased by molecular replacement, utilizing the previously published structure16, and refined at 2.59 Å resolution (PDB 8E23, see Table S1 for data collection and refinement statistics). Compound 14 was unambiguously modeled binding to an allosteric site (Figure S1A) far away from any of the five flexible loops removed during construct engineering, thus validating its use for high resolution SBDD. Because the prior mechanism-of-action studies predicted allosteric inhibition, resolving compound 14 adeptly bound to Polθ provided a satisfying result and foundation for iterative, rational optimizations.
The structure of the engineered Polθ pol domain displays a canonical PolI-family architecture consisting of exonuclease, palm, fingers and thumb subdomains (Figure 4A). Polθ is devoid of exonuclease activity but retains an inactivated exonuclease-like fold. In the pol active site, clamped by the palm, fingers and thumb subdomains, lies a 5’ overhang double stranded DNA substrate and an incoming ddGTP molecule positioned for addition to the elongating DNA. The primer strand was chain-terminated by enzymatic incorporation of a prior ddGTP molecule during crystallization, thus trapping the assembled holoenzyme prior to covalent incorporation of the bound ddGTP.17 Notably, compound 14 binds to the fingers subdomain opposite to the binding position of the incoming ddGTP. Overall, the structure of the polymerase, DNA and ddGTP components are highly similar to those reported previously, with the exception of localized perturbations to the fingers subdomain induced by binding to compound 14 as noted below.16
Figure 4. The crystal structure of compound 14 bound to an engineered construct of the polymerase domain of PolƟ.


A. Ribbon representation of the polymerase domain of Polθ bound to DNA, ddGTP, and compound 14 (PDB 8E23). B. Detailed crosseye stereo view of compound 14 bound to the fingers subdomain of PolƟ. PolƟ backbone is shown as ribbon with binding pocket side chains shown as sticks. Carbon, oxygen, nitrogen, and sulfur, atoms are coloured royal blue, red, blue, and yellow. Compound 14 is shown as sticks with carbon oxygen, and nitrogen atoms coloured cyan, red, blue.C. A LigPlot of the interaction of 14 with PolƟ. D. Surface view of compound 14 bound to the fingers subdomain PolƟ. Protein surface is coloured grey with protein and compound 14 atoms coloured as in B. E. Molecular model of the binding of compound 15 to Polθ. Flexible ligand alignment of 15 was performed on the 14 co-crystal structure, followed by energy minimization of 15 in MOE. Compound 15 is shown as sticks with orange carbon atoms and solvent-accessible surface shown as an orange mesh. Compound 14 is shown as sticks with cyan carbon atoms and solvent-accessible surface shown as a cyan mesh.
Compound 14 nestles in a near-fully enclosed pocket composed entirely of fingers α-helices (Figure 4B). The binding pocket is highly hydrophobic in nature, lined by numerous lipophilic side chains as shown in Figure 4B, 4C. The binding affinity and specificity of compound 14 appear to be achieved by hydrophobic interactions and shape complementarity, with no strong polar interactions evident within the pocket. Exposure of compound 14 to solvent is observed at opposite ends of the binding pocket. The two exit paths are separated by the side chains of Arg 2419 and Glu 2365, which engage in a favorable salt bridge that clamps compound 14 into its binding pocket (Figure 4D).
The α-helical finger domain of DNA pols undergoes drastic conformational changes during the catalytic cycle of DNA synthesis.18 In one extreme conformation, termed the closed conformation, the fingers clamps on the DNA substate and incoming dNTP. As exemplified by a Polθ crystal structure corresponding to a closed state (PDB=4X0P) (Figure S2A), helix αO plays a critical role in direct interactions with the incoming ddNTP. Once the fingers have locked the ddNTP in the insertion site, nucleophilic attack from the 3’ hydroxyl of the elongating DNA onto the α-phosphate of the incoming dNTP results in the extension of the primer DNA by 1 base. The fingers subdomain then opens to allow translocation of the DNA and diffusion of the pyrophosphate leaving group. This conformation, exemplified by a Polθ crystal structure corresponding to an open state (PDB=6XBU) (Figure S2B), involves distortions to the fingers subdomain including major movements of helices O, O1, and N with respect to the nucleotide insertion site (Figure S2C).19 Helix αO, in particular, undergoes a large shift that opens the insertion site for the next incoming nucleotide (Figure S2C).
As revealed by superpositions onto previously determined open and closed state structures of Polθ, the binding of compound 14 to Polθ appears to lock the fingers domain in the closed state (Figure 5A, 5B, 5C). We hypothesize that this may freeze the catalytic cycle by trapping the closed ternary complex and preventing the essential transition to an open state. While not fully preexisting, a near complete binding pocket for compound 14 is evident in the closed state structure (Figure S3A) and can be more completely induced by rotamer flips to the sidechains of Tyr 2412 and Phe 2416. In contrast, the binding pocket for compound 14 is completely occluded (in particular by helix αO) in the open conformation of the fingers subdomain (Figure S3B). As the closed state of the fingers subdomain is stabilized by the binding of DNA substrate, this may explain the observed dependency on DNA for the binding of compound 14 to Polθ.18
Figure 5. Compound 14 binds to a closed conformation of the fingers subdomain of PolƟ.



A. Detailed view of the fingers subdomain of Polθ bound to DNA, ddGTP, and compound 14 (PDB 8E23).. Atom colouring as in Figure 4. B. Superposition of the compound 14-Polθ co-crystal structure with the structure of Polθ in a closed fingers conformation (PDB =4X0Q) C. Superposition of the compound 14-Polθ co-crystal structure with the structure of Polθ in an open fingers conformation (PDB =6XBU)
SBDD-enabled SAR
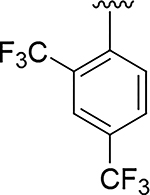
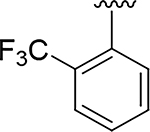
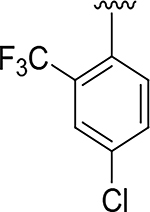
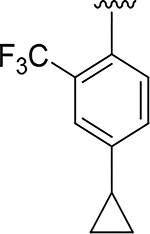
The expanded understanding of the binding mode of compound 14 provided the opportunity to employ structure-based drug design to address the metabolic and synthetic challenges of the fused pyrazole series of inhibitors. We first explored the possibility of replacing the fused pyrazole pharmacophore with various substituted phenyls or heteroaryls, as demonstrated in Table 2. With prior knowledge from the fused-pyrazole series that (a) the ortho trifluoromethyl was critical for potency, and that (b) the southern portion was highly lipophilic, we first designed compound 15 which also contained a second suitably positioned 4-trifluoromethyl. Based on molecular modelling (flexible ligand alignment of 15 to the 14 co-crystal structure, followed by energy minimization of 15 in MOE), the phenyl with the trifluoromethyl group in the para position of compound 15 displayed better surface complementarity than the corresponding region of compound 14, and the para trifluoromethyl of 15 was also predicted to form a favorable electrostatic interaction with the sulfur of Cys2386 (Figure 4E). Compound 15 exhibited a 10-fold improvement in potency over hit compound 2. Furthermore, this aryl replacement enabled a more efficient exploration of the SAR due to the tractability of the chemistry and elimination of the regiochemistry problems associated with the fused pyrazole. The importance of the para trifluoromethyl was underlined by compound 16, which lacks the ability to sufficiently fill the lipophilic pocket, resulting in a dramatic 700-fold loss in potency compared to compound 15. A chloro (17) or cyclopropyl (18) substituent was able to fill this pocket, but not as effectively as the rationally designed compound 15. Attempts to decrease the lipophilicity of 15 (clogP = 4.6) by introducing additional polar substituents on the southern phenyl core (data not shown) were detrimental to potency. However, the heterocyclic pyridine 19 and N-linked pyridone 20 were able to preserve double digit nanomolar potency while dramatically reducing the clogP by 1 or 2 log units, respectively. These heterocycles would become critically important to the optimization of drug-like properties in subsequent derivatives.
Table 2:
Surrogates of the Fused Pyrazole
| |||
|---|---|---|---|
|
| |||
| Compd | R | Polθ IC50 (μM)a | cLogP |
|
| |||
| 15 |
|
0.014 | 4.6 |
| 16 |
|
9.2 | 3.7 |
| 17 |
|
0.121 | 4.3 |
| 18 |
|
0.040 | 4.5 |
| 19 |
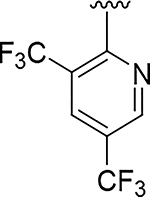
|
0.065 | 3.8 |
| 20 |
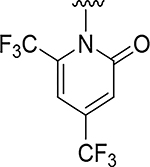
|
0.058 | 2.4 |
Picogreen assay: 3nM of Polθ enzyme, (n=3)
Building on the success of compound 15, we next focused on the amide nitrogen substituents (Table 3). Our goals were to (i) improve metabolic stability through phenyl substitution or replacement, (ii) evaluate if polarity could be tolerated in the northern portion, and (iii) explore the possibility of extending the N-methyl group into the solvent-exposed area shown in the compound 14 co-crystal structure (Figure 4C). Swapping the phenyl for pyridyl led to a significant loss of potency (21–23), as was the case with introducing polar substituents on the phenyl ring (data not shown). These observations were consistent with the hydrophobic nature of the binding pocket. Metabolite identification studies in mouse and human microsomes with 15 indicated that N-demethylation and hydroxylation of the phenyl ring were the major routes of metabolism. Introduction of a fluorine atom (24) increased microsomal stability and eliminated the phenyl ring hydroxylation leaving demethylation as the only major metabolite. Microsomal stability was further improved by replacing the methyl hydrogens with deuterium (25). Substitution of the methyl with a nitrile (26) showed that polarity was tolerated at this position by extending into the solvent-exposed area, as predicted by the observation of a solvent portal in the compound 14 co-crystal structure (Figure 4B and 4C).
Table 3:
Surrogates of the N-methyl Aniline
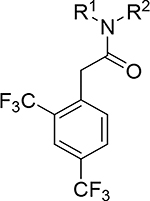
| |||||
|---|---|---|---|---|---|
|
| |||||
| Cmpd | R1 | R2 | Polθ IC50 (μM)a | clogP | Mouse Microsomal stability CL (μL/min/mg) |
|
| |||||
| 15 |
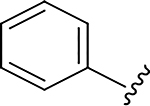
|
Me | 0.012 | 4.6 | >555 |
| 21 |
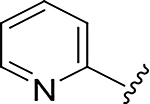
|
Me | 2.7 | 4.0 | >832 |
| 22 |
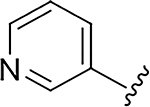
|
Me | 3.2 | 3.4 | 216 |
| 23 |
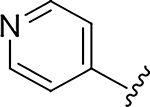
|
Me | 2.2 | 3.4 | 56 |
| 24 |
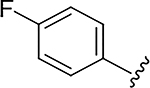
|
Me | 0.017 | 4.8 | 125 |
| 25 |
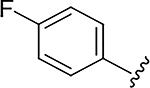
|
CD3 | 0.019 | 4.8 | 52 |
| 26 |
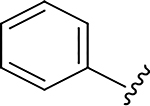
|
CH2CN | 0.042 | 4.2 | 115 |
Picogreen assay: 3nM of Polθ enzyme, (n=3)
Exploiting this access to solvent via a propargyl or propargyl amide (27 and 28) provided handles for late-stage functionalization and had dramatically improved potencies over 15 (Table 4). Functionalization of the propargyl amide with various heterocycles (29–31) resulted in sub-nanomolar biochemical potencies, with compound 31 exhibiting an exquisite enzymatic potency of 68 picomolar. Functionalization of the propargyl with heterocycles was also promising, providing compounds 32–37 which exhibited low-nanomolar biochemical potencies and improved clearance in mouse liver microsomes.
Table 4:
Exploring the Solvent-Exposed Vector
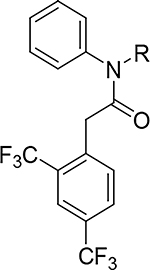
| |||
|---|---|---|---|
|
| |||
| Compd | R | Polθ IC50 (μM)a | Mouse Microsomal Stability CL(μL/min/mg) |
|
| |||
| 27 |
|
0.014 | 597 |
| 28 |
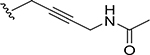
|
0.00082 | >832 |
| 29 |
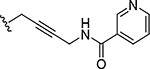
|
0.00020 | 492 |
| 30 |
|
0.00040 | 469 |
| 31 |
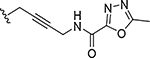
|
0.000068 | 614 |
| 32 |
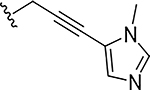
|
0.0012 | 57 |
| 33 |
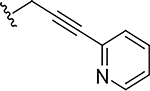
|
0.0029 | 64 |
| 34 |
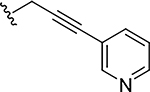
|
0.0025 | 110 |
| 35 |
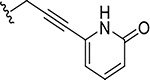
|
0.00051 | 39 |
| 36 |
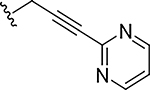
|
0.0039 | 105 |
| 37 |
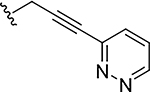
|
0.0014 | 31 |
Picogreen assay: 0.3nM of Polθ enzyme, (n=3)
The co-crystal structure of compound 37 bound to PolƟ (PDB 8E24, Figure 6A,6B), refined at 2.34Å resolution, provided insight into the improvement in enzymatic potency relative to compound 14. The pyridazine ring of 37 that extends out from one end of the solvent exit channel displays favorable π-π stacking with the sidechain of Tyr2412. This compliments the preexisting favorable π-π T-stacking interaction of the Tyr2412 side-chain with the benzene group of 37 and 14. A carbon atom in the pyridazine has favorable electrostatics with the carboxylate of Glu2365, while the carbonyl of the amide has favorable electrostatics with the guanidinium group of Arg2419, thus stabilizing both side-chains of the Glu-Arg salt bridge that separates the two portals to solvent and that clamps compounds 37 and 14 into the binding pocket. The hydrophobic 2,4-bis(trifluoromethyl)phenyl core of 37 better fills the hydrophobic cavity relative to the 3-(trifluoromethyl)-cyclopentyl fused pyrazole core of 14 (as observed for 15 in Figure 4E). One fluorine of the para CF3 also forms favorable electrostatics with the sulfur atom in Cys2386, as predicted. The alkyne withdraws electrons from the methylene linker, making that methylene group more partially positive, which enables a stronger electrostatic interaction with the backbone carbonyl of Tyr2412.
Figure 6: The Crystal structure of Compound 37 bound to PolƟ.

A Crosseye stereo view of compound 37 bound to PolQ (PDB 8E24). PolƟ backbone is shown as ribbon with side chains shown as sticks. Carbon, oxygen, nitrogen, and sulfur, atoms are coloured royal blue, red, blue, and yellow, respectively. Compound 37 is shown as sticks with carbon oxygen, nitrogen, and florine atoms coloured cyan, red, blue and grey, respectively. Dashed lines highlight a hydrogen bond interaction.B. A LigPlot of the interaction of 37 with PolƟ.
In early SAR the southern bis-trifluoromethyl phenyl core was held constant (as exemplified in Tables 3, 4). We had noted previously that the introduction of nitrogen in the bis-trifluoromethyl aryl core lowered the logP (Compound 19). This impacted mouse plasma protein binding (PPB) where compound 19 had a higher unbound fraction than compound 15 (fu =0.06 vs fu =0.005). We therefore switched our SAR efforts to elaborate this bis-trifluoromethyl pyridyl core to further our goal of identifying candidates suitable for in vivo mouse xenograft studies.
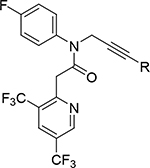
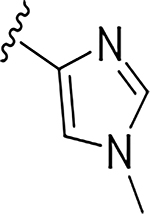
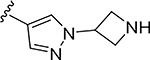
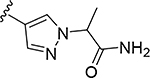
More extensive SAR exploration on the propargyl potency vector was conducted by introducing basic amine and polar functionality on various 5-and 6-membered heterocycles (Table 5). We chose to drive our SAR strategy based on free exposure (total exposure in mouse plasma corrected for PPB) by evaluating our more metabolically stable compounds (CL<30 μL/min/mg in MLM) in mouse oral pharmacokinetic (PK) studies. Compounds were rank ordered by free exposure (Free Average Concentration) to identify the best candidate for further profiling in xenograft studies. Compound 38 had low free exposure due to low AUC (area under the PK curve) and low free fraction. Introducing a basic amine on pyrazole 39 led to a 3-fold improvement in both AUC and free fraction. Adding polar appendages on the pyrazole, as in compounds 40–43, resulted in improved free exposures. Conversely, methoxy-substituted compounds 44–45 were notably inferior, largely due to their low free fraction. Structurally similar amino heteroaryl compounds 46, 47, and RP-6685 showed superior profiles, with compound RP-6685 showing the best free exposure, leading it to be chosen for further profiling.
Table 5:
In Vivo Free Exposure
| |||||
|---|---|---|---|---|---|
|
| |||||
| Cmpd | R | Polθ IC50 (μM)a | AUC PO (μM*hr)b | PPB (fu)c | Free Caveraged (μM) |
|
| |||||
| 38 |
|
0.0090 | 5.31 | 0.018 | 0.003 |
| 39 |
|
0.00075 | 14.1 | 0.051 | 0.029 |
| 40 |
|
0.00099 | 27.0 | 0.044 | 0.049 |
| 41 |
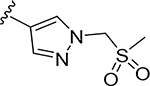
|
0.00091 | 76.3 | 0.019 | 0.060 |
| 42 |
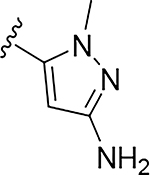
|
0.0017 | 35.6 | 0.044 | 0.065 |
| 43 |
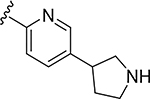
|
0.00072 | 38 | 0.019 | 0.030 |
| 44 |
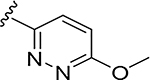
|
0.0049 | 27.7 | 0.006 | 0.006 |
| 45 |
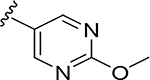
|
0.0021 | 7.0 | 0.013 | 0.003 |
| 46 |
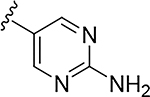
|
0.0016 | 24.7 | 0.015 | 0.081 |
| 47 |
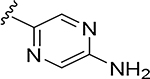
|
0.0042 | 108 | 0.018 | 0.081 |
| RP-6685 |
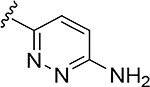
|
0.0058 | 49.7 | 0.052 | 0.107 |
Picogreen assay: 0.3nM of Polθ enzyme, (n=3); PO:
20mg/kg, in either 0.5%MC/0.02%SLS or 5% NMP:95% (10% VitE TPGS).
PPB by equilibrium dialysis, fraction unbound.
Free Caverage = AUC*fu/24
Characterization of RP-6685
RP-6685 displayed exquisite selectivity for Polθ, being inactive on several other human DNA polymerases, namely Pols α, ε, γ, λ, μ (Table 6). These results were consistent with Pols α, ε, and λ and μ belonging to different classes of polymerases with markedly different sequence homology. Polγ, like Polθ, belongs to the family A sequence class but shares very limited homology (i.e., < 20% sequence identity with Polθ).
Table 6:
Potency, Selectivity, ADME Profilea of RP-6685
| FL Polθ IC50 (μM)a | Pols α, ε, γ, ν. (μM) | DSB HCT116 WT IC50 (μM) | TLR IC50 (μM) | Proliferation HCT116 BRCA2−/− / BRCA2+/+ IC50 (μM) | MDCK-WT Permeability Papp (Efflux ratio) | Mouse PK CL(mL/min/kg) / Vdss (L/kg) / t1/2 (h) / F (%)b | PPB (fu)c |
|---|---|---|---|---|---|---|---|
|
| |||||||
| 0.00055 | >100 | 0.45 | 0.94 | 0.32 / >15 | 6.2 (1.4) | 36.8 / 1.1 / 0.4 / 66 | 0.052 |
aa 1–2590
iv/po dosing in CD1 mice (vehicle: 5% NMP:95%(10% VitE TPGS) iv, 2.5mg/kg; PO, 2.5mg/kg..
PPB by rapid equilibrium dialysis. All reported enzymatic and cellular potencies are the mean of n=3.
To further validate the activity of our improved inhibitors, full-length Polθ (aa 1–2590)20,21,22 (see Supporting Info) was produced in HEK293 cells. Both ATPase and polymerase activities were detected in this enzyme. RP-6685 was extremely potent with an IC50 of 550 pM against the pol activity of full-length Polθ and inactive on the ATPase activity.
We developed a cell-based assay to measure Polθ-mediated repair events at a CRISPR/Cas9 induced break at the AAVS1 locus in HCT116 cells in which the repair can proceed by either NHEJ or MMEJ 22(see Supporting Info). Knockdown of Polθ or knock-in polymerase-dead Polθ of results in ablation of MMEJ with corresponding increase in NHEJ-mediated repair (Figure 7).24 When tested in this DSB repair of AAVS1 loci, RP-6685 exhibited a potency of 0.5 μM, thus providing evidence for on-target activity. RP-6685 was also profiled in the Traffic-Light Reporter assay (TLR) which is a fluorescent-based assay for Polθ inhibition in a HEK293 LIG4−/− cell line (Figure 8A).3 This assay measures both MMEJ and Homologous Recombination (HR) in HEK293 cells unable to repair DSBs by NHEJ. Treatment of cells with RP-6685 provided a dose-dependent decrease in MMEJ comparable to what was observed when these cells were transfected with siRNAs targeting Polθ (Figure 8B).
Figure 7:

DSB assay. A) Type of DNA repair observed at AAVS1 cut site. Non-homologous end-joining (open bars) and microhomology-mediated end-joining (filled bars). B) Inhibition of MMEJ-mediated DNA repair by RP-6685.
Figure 8:

A) TLR schematic. B) TLR assay with Ctrl siRNA and PolQ siRNA. C) Inhibition of Alt-EJ-mediated DNA repair by RP-6685.
We tested RP-6685 in a cell proliferation assay employing isogenic HCT116 BRCA2−/− and BRCA2+/+ cell lines. HCT116 colorectal cancer cells provide one of the few available genetic backgrounds that tolerate knock-out of the BRCA2 gene. Micromolar-range dosing of RP-6685 was required to inhibit proliferation of HCT116 BRCA2−/− cells, resulting in a large shift in cellular potency relative to the low nanomolar range IC50 reported for the in vitro picogreen assay. To better understand the potency drop we established a primer extension assay where the inhibitors are non-competitive with respect to nucleotide utilization and by DSF experiments that the DNA must be bound prior to inhibitor binding consistent with an uncompetitive mechanism in terms of the DNA substrate.13 Our structural analysis shows that this class of inhibitors cannot bind the finger-open conformation of Polθ, consistent with the scenario where the enzyme must first engage with the DNA substrate prior to inhibition by the drug. Additional studies will be necessary to better understand why RP-6685 and similar compounds are highly potent against the enzyme yet have attenuated effect when killing BRCA2 −/− cells culture.
In-Vivo profiling
RP-6685 displayed a favorable PK profile with good oral bioavailability in mouse. Consequently, RP-6685 was selected for further evaluation as a proof-of-concept molecule in isogenic HCT116 BRCA2+/+ and BRCA2 −/− xenograft tumor models (Figure 9). RP-6685 was administered orally at the maximum tolerated dose of 80 mg/kg BID for 21 days. As expected from the low potency based on in vitro assays, we observed no tumor growth inhibition in RP-6685-treated mice bearing BRCA2+/+ HCT116 tumors (Figure 9A). Instead, in the context of BRCA2-deficient HCT116 model, RP-6685-treated mice showed tumor regression during the first 8 days of treatment, reaching statistical difference with the vehicle control group at Day 8. This initial anti-tumor activity was not sustained, and the difference between vehicle and RP-6685-treated mice was not statistically significant by Day 21 (Figure 9B). RP-6685 was well tolerated in the nude mice throughout the study, with only mild clinical signs and no significant body weight loss (Figure 9C, 9D). The pharmacokinetics of RP-6685 were measured at Day 1 and 7 (Figure 10, 9B) leading to the observation that RP-6685 exposure declined by 55 – 67% over the week of dosing, possibly due to autoinduction of metabolism. This resulted in an estimated change in sustained levels above the IC50 for HCT116 viability from 23 hours at Day 1 to only 6 hours by Day 7. The xenograft HCT116 BRCA2 −/− tumors were harvested and dissociated, the cells were counted, plated, and processed for PD (pharmacodynamics) markers by immunofluorescence to assess target engagement (see Supporting Info). RP-6685 treated cells isolated from tumors showed a modest trend towards increased micronuclei and γH2AX, which are hallmarks of DNA damage (supporting information).
Figure 9: The Efficacy and Tolerability of RP-6885 in an Isogenic HCT116wt and BRCA2−/− Xenograft Model.

Mice implanted with either HCT116 or HCT116 (BRCA2 −/−) tumor cells were treated orally BID with Vehicle or RP-6685 at 80 mg/kg for 21 days. A, B Mean tumor volume; C, D body weight change ± SEM ( n = 8 mice per group). * Statistical significance relative to vehicle control at days 8 and 21 was established by unpaired t-test with Welch’s correction (GraphPad Prism v9). *P < 0.05
Figure 10. The pharmacokinetics of RP-6685 in CD-1 nude mice on days 1 and 7 of oral administration in CD-1 nude mice.

RP-6685 was administered BID (at time 0 and 8h) to n=3 CD-1 nude mice at the indicated doses and whole blood sampled at various time points on Day 1 and Day 7 after time 0. RP-6685 free plasma concentrations were determined taking into consideration plasma protein binding and the blood/plasma ratio and are indicated as mean ± SEM at each time point. The AUC was calculated using WinNonLin. The dashed line indicates the IC50 of RP-6685 in an Incucyte viability assay with HCT-116 (BRCA2 −/−) tumor cells.
Synthesis
The synthetic route for compounds 2, 3, 6, 7, 9–13 (Table 1) is shown in Scheme 1. A one-pot, 3-component Claisen condensation of the substituted cyclic ketone, ethyl 2,2,2-trifluoroacetate and ethyl 2-hydrazinoacetate25 followed by ester hydrolysis provided a mixture of N1 and N2 regioisomers (typically 1:9 ratio) which was used without further purification. T3P- or HATU-mediated amide coupling with methyl aniline, followed by either silica chromatography or reverse phase preparative HPLC afforded the desired N2-regioisomer. The N2 regioisomers could easily be distinguished from the undesired N1-regioisomer by distinct chemical shifts in 19F and methylene protons in the 1H NMR spectra. To that end we prepare the N1-regioisomer of compound 7 (compound 51) via a different methodology in 4 steps shown in Scheme 2. Claisen condensation of 1-cyclopentyl-2,2,2-trifluoroethan-1-one with hydrazine hydrate afforded 2-(2,2,2-trifluoroacetyl) cyclopentan-1-one which could then be alkylated to give exclusively the N1-regioisomer. Ethyl ester hydrolysis, followed by amide coupling afforded compound 51. Compound 4 was accessed via Beckman according to Scheme 3.26 Compound 5 was prepared according to Scheme 4 following described literature procedures. 26 Oxidation of Int-48 provided ketones Int-52a and Int-52b, easily separable by silica chromatography. Ester hydrolysis, amide coupling followed by reduction of the ketone afforded compound 5. Compound 8 was prepared as shown in Scheme 5. Claisen condensation of 2-acetylcyclopentan-1-one with ethyl hydrazino acetate afforded N-regioisomers, easily separable by silica chromatography, followed by ester hydrolysis and amide coupling afforded Compound 8. Compound 14 was easily prepared in one-step by reacting compound 7 with Lawesson reagent. Compounds 15–26 (Tables 2,3) were easily accessed via in one step via T3P- or HATU-mediated amide coupling. The synthetic route of compounds 32–37 (Tables 4) are shown in Scheme 6. Deprotonation of aniline followed by SN2 substitution with propargyl bromide afforded intermediates Int-55, Int-56 in gram quantities. T3P mediated coupling with 2-[2,4-bis(trifluoromethyl)phenyl]acetic acid was high yielding and afforded intermediates 27, Int-57 which could be functionalized late-stage via Sonogashira coupling.27 The synthetic route of compounds 28–31 (Tables 4) is shown in Scheme 7 were prepared by amide coupling of Int-59. Int-59 was prepared via displacement of the mesylate with sodium azide, followed by azide reduction. Compounds 38, 40–42, 44–47, RP-6685 (Table 6) were prepared as shown in Scheme 8 via late-stage functionalization using Sonogashira coupling of Int-63. Int-63 was prepared in five steps: installation of malonate via SNAr on commercially available 2-chloro-3,5-bis(trifluoromethyl)pyridine, followed by TFA-mediated decarboxylation, methyl ester hydrolysis and amide coupling affording Int-63 in gram quantities. A more convergent multi-gram route for the preparation of RP-6685 is shown in Scheme 9.
Scheme 1.

(a) LDA, THF or ether, −78 °C, 30 min then ethyl trifluoroacetate 2h at rt; (b) ethyl 2-hydrazinoacetate, isopropanol or toluene, 75 °C, 2h;(c) LiOH, H2O, 70 °C, 1h (d) N-Me aniline, DIPEA, HATU or T3P, DMF or THF rt.
Scheme 2.

(a) Hydrazine, Ethanol, 120 °C microwave, 120 °C (b) NaH (60%), Ethyl bromoacetate, DMF, 0 °C to rt, 1h; (c) LiOH, MeOH, THF, H2O, rt 3h (d) N-Me aniline, T3P, DIPEA, THF, 70 °C, 1h.
Scheme 3.

(a) methane sulfonic acid, NaN3, rt; (b) LiOH, MeOH, THF, H2O, rt 3h; (c) N-Me aniline, HATU, DIPEA, DMF, rt.
Scheme 4.

(a) CrO3, AcOH, o/w; (b) N-Me aniline, HATU, DIPEA, DMF, rt; (c) LiOH, MeOH, THF, H2O, rt 3h; (d) NaBH4, DCM, isopropanol, rt.
Scheme 5.

(a) ethyl 2-hydrazinoacetate, ethanol, 75 °C, 12h; (b) LiOH, MeOH, THF, H2O, rt 3h; (c) N-Me aniline, T3P, DIPEA, THF, 80 °C, 6h.
Scheme 6.

(a) NaH, 30 min rt, DMF; (b) propargyl bromide, 2h rt; (c) 2-[2,4-bis(trifluoromethyl)phenyl]acetic acid, T3P, DIPEA, THF rt, 1h; (d) Aryl bromide, Pd(PPh3)4, CuI, TEA, DMF, 70 °C, 4h.
Scheme 7.

(a) 2-[2,4-bis(trifluoromethyl)phenyl]acetic acid, 4-anilinobut-2-yn-1-ol, T3P, DIPEA, THF rt, 1h; (b) 4-methylbenzenesulfonyl chloride, N,N-diethylethanamine N,N-dimethylpyridin-4-amine, DCM, 0 °C; (c) sodium azide, DMF, rt; (d) Ph3P, THF, rt (e) carboxylic acid, DIPEA, HATU or T3P, DMF or THF rt.
Scheme 8.

(a) tert-Butyl methyl malonate, NaH, 1h, rt, THF; (b) 2-chloro-3,5-bis(trifluoromethyl)pyridine, 60 °C, o/n (c) TFA / DCM, 2h rt; (d) LiOH, Dioxane/MeOH/H2O 80 °C, 20min; (e) Int-56, T3P, DIPEA, THF rt, 1h; (f) Aryl bromide, Pd(PPh3)4, CuI, TEA, DMF, 70 °C, 4h
Scheme 9:

(a) Aryl bromide, Pd(PPh3)4, CuI, TEA, DMF, 70 °C, 4h; (b) 62, T3P, DIPEA, THF rt, 1h; (c) DCM/TFA, rt.
CONCLUSIONS
Starting with an 11 μM HTS screening hit, SBDD enabled by co-crystal structures and molecular modeling allowed us to design potent, selective, and bioavailable inhibitors of Polθ polymerase. Among them, RP-6685 was further profiled in proof-of-concept in vivo mouse HCT116 BRCA2−/− xenograft studies and displayed encouraging efficacy. This work demonstrates the potential for inhibitors targeting the polymerase activity of Polθ as candidates for further clinical development, but also highlights the challenges associated with this class of inhibitors. In particular, the large shift between enzymatic and cellular assays may be an inherent property of binding to the allosteric site induced by interaction with dsDNA breaks. Further work to better understand these challenges is underway.
EXPERIMENTAL SECTION
Chemistry
Compounds 1–47, and RP-6685 are >95% pure by hplc analysis. Solvents and reagents were obtained from commercial suppliers and were used without further purification. UPLCMS analyses for reaction monitoring were performed on a Waters Acquity-H UPLC Class system using an Acquity UPLC HSS C18 2.1×30mm column eluting with a gradient (1.86 min) of acetonitrile (15% to 98%) in water (both containing 0.1% formic acid) using electrospray ionization (ESI). Prep-HPLC separations were performed on a Teledyne Isco Combi Flash EZ Prep system using either Phenomenex Gemini® 5 μm NX-C18 110Å 150 × 21.2 mm column at a flow of 40 mL/min over 12 min (<100 mg or multiple injections of <100 mg) unless otherwise specified or HP C18 RediSep Rf gold column (>100 mg) eluting with a gradient of acetonitrile in water (both containing 0.1% formic acid) unless otherwise specified. Purifications by silica gel chromatography were performed on a Teledyne Isco Combi Flash Rf system using RediSep Rf silica gel columns. Purity of final compounds was assessed by injection of a small aliquot on a Waters Acquity-H UPLC® Class system using an Acquity® UPLC BEH C18 2.1×50 mm column eluting with a gradient (7 min) of acetonitrile (2% to 98%) in water (both containing 0.1% formic acid). Magnetic resonance (NMR) spectra were obtained on a Varian 400 MHz NMR spectrometer with an Oxford NMR AS400 magnet and are referenced in ppm relative to the residual solvent peak in the indicated solvent. for 1H NMR spectra, multiplicities, coupling constants in hertz, and numbers of protons are indicated parenthetically. HRMS Samples were chromatographed using a Waters Acquity H-class UPLC system by employing a 4-minute aqueous gradient from 15 to 90% acetonitrile with 0.1% formic acid. High resolution mass spectra were collected using a Waters Xevo G2 Q-tof mass spectrometer operated in positive mode. A lockspray solution containing leucine enkephalin was used to maintain mass accuracy during analysis. Calibration was performed according to the manufacturer’s guidelines and the mass accuracy was determined within 5 ppm of the theoretical exact mass.
Synthesis of RP-6685 (Scheme 9)
O1-tert-Butyl O3-methyl 2-[3,5-bis(trifluoromethyl)-2-pyridyl]propanedioate (Int-60).
To a solution of tert-Butyl methyl malonate (11.17 g, 64.1 mmol, 10.8 mL) in dry THF (120 mL) at rt was added sodium hydride (4.36 g, 108.8 mmol, 60% purity) portion-wise. The reaction mixture was stirred at rt for 1 hr prior to addition of 2-chloro-3,5-bis(trifluoromethyl)pyridine (10.0 g, 40.0 mmol) dropwise. The reaction was stirred at 60 °C overnight. The reaction mixture was cooled to rt, poured in aqueous saturated NH4Cl and extracted with 3 times with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel chromatography eluting with a gradient of 5–100% EtOAc/heptane. The relevant fractions were combined and concentrated in vacuo to give Int-60 (13.2 g, 85% yield) as a colorless oil that crystallizes upon refrigeration. ESI MS m/z 332.0 [M+ H-tBu]+. 1H NMR (400 MHz, Chloroform-d) δ 9.05 (s, 1H), 8.21 (s, 1H), 5.19 (s, 1H), 3.81 (s, 3H), 1.47 (s, 9H).
Methyl 2-[3,5-bis(trifluoromethyl)-2-pyridyl]acetate (Int-61).
To a solution of Int-60 (13.2 g, 34.0 mmol) in dry DCM (120 mL) at rt was added trifluoroacetic acid (26.2 mL, 340.8 mmol) dropwise. The reaction mixture was stirred at rt for 4 h. The volatiles were removed in vacuo (bath at 20 °C) and the yellow oil was diluted with DCM (100 mL), a saturated aqueous solution of NaHCO3 was added and the biphasic mixture was stirred vigorously at rt for 5 min. The organic phase was dried over MgSO4, filtered and volatiles removed in vacuo to afford 61 as a yellow oil. ESI MS m/z 288.0 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.29 – 9.25 (m, 1H), 8.64 – 8.59 (m, 1H), 4.17 (s, 2H), 3.65 (s, 3H).
2-[3,5-Bis(trifluoromethyl)-2-pyridyl]acetic acid (Int-62).
To a cold solution of Int-61 (12.8 g, 44.5 mmol) in Dioxane (78 mL), MeOH (39 mL) and H2O (39 mL) at 0 °C was added lithium hydroxide monohydrate, 98% (3.74 g, 89.1 mmol) in one portion. The reaction mixture was stirred at 60 °C for 10 min. The solution was cooled down to rt and volatiles are removed in vacuo. Water (52 mL) was added and the residue was suspended (sonication) prior to cooling down the solution to 0 °C. Formic acid, 97% (5.13 g, 111.4 mmol, 4.20 mL) was added dropwise at 0 °C. The resulting precipitate was collected by filtration, washed with water (x3) and dried in vacuo to afford Int-62 (8.9 g, 73% yield) as a white solid. ESI MS m/z 274.0 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 12.86 (s, 1H), 9.24 (s, 1H), 8.57 (d, J = 2.7 Hz, 1H), 4.04 (d, J = 2.9 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −60.07, −61.02.
2-[3,5-Bis(trifluoromethyl)-2-pyridyl]-N-(4-fluorophenyl)-N-prop-2-ynyl-acetamide (Int-63).
To a mixture of 2-[3,5-bis(trifluoromethyl)-2-pyridyl]acetic acid (2.08 g, 7.60 mmol), 4-fluoro-N-prop-2-ynyl-aniline (1.13 g, 7.60 mmol), DIPEA (1.96 g, 15.1 mmol, 2.65 mL) in THF (10 mL) was added T3P (9.67 g, 15.1 mmol, 9.04 mL, 50% purity) and the reaction stirred at 80 °C for 30 min. The reaction mixture was diluted with sat. sodium bicarbonate and extracted with ethyl acetate. The organic was evaporated in vacuo and purified by silica chromatography eluting with 5–60% ethyl acetate:hexanes. The relevant fractions were evaporated to give Int-63 (2.20 g, 71% yield). ESI MS m/z 500.1 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.21 (d, J = 2.2 Hz, 1H), 8.50 (d, J = 2.2 Hz, 1H), 7.57 – 7.41 (m, 2H), 7.33 (t, J = 8.7 Hz, 2H), 4.46 (d, J = 2.5 Hz, 2H), 3.84 (s, 2H), 3.20 (t, J = 2.5 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ −60.17, −60.91, −112.96. 13C NMR (101 MHz, DMSO-d6) δ 167.70, 163.27, 160.83, 158.52, 149.51, 137.40 (d, J = 2.9 Hz), 132.81 (d, J = 4.6 Hz), 131.07 (d, J = 9.0 Hz), 127.40, 125.10 – 122.71 (m), 121.96 (d, J = 3.5 Hz), 117.01 (d, J = 22.8 Hz), 79.55, 75.44, 41.94 (d, J = 2.0 Hz), 38.19.
4-Fluoro-N-prop-2-ynyl-aniline (Int-56).
To a mixture of 4-fluoroaniline (24.89 g, 224 mmol, 21.5 mL) in DMF (50 mL) was added sodium hydride (9.41 g, 235 mmol, 60% purity) and stirred for 30 min at rt. To this was added 3-bromoprop-1-yne (34.97 g, 235 mmol, 40.2 mL) and the reaction stirred at rt for 2h. The reaction mixture was diluted with sat. NaHCO3 and suction filtered to give yellow solid. The filtrate was extracted with ethyl acetate. The organic was washed with sat. ammonium chloride. The combined organics were evaporated and silica eluting with 20% ethyl acetate:hexanes. The relevant fraction was combined to give Int-56 (10.73 g, 32% yield). ESI MS m/z 150.1 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 7.00 – 6.82 (m, 2H), 6.66 – 6.47 (m, 2H), 5.83 (t, J = 6.3 Hz, 1H), 3.78 (dd, J = 6.2, 2.4 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −128.82. 13C NMR (101 MHz, DMSO-d6) δ 170.74, 156.33, 154.02, 144.85 (d, J = 1.9 Hz), 115.57 (d, J = 22.1 Hz), 114.06 (d, J = 7.3 Hz), 82.59, 73.36, 32.97.
tert-Butyl N-[6-[3-(4-fluoroanilino)prop-1-ynyl]pyridazin-3-yl]carbamate (Int-66).
Nitrogen was bubbled through a solution of 4-fluoro-N-prop-2-ynyl-aniline Int-56 (7 g, 46.9 mmol), tert-Butyl N-(6-chloropyridazin-3-yl)carbamate (12.93 g, 56.3 mmol) in dry DMF (90 mL) while sonicating for 15 min. Copper(I) iodide (893 mg, 4.69 mmol) and palladium (0) tetrakis(triphenylphosphine) (2.71 g, 2.35 mmol) were then added followed by triethylamine (9.50 g, 93.8 mmol, 13.0 mL). The reaction mixture was stirred at 70 °C for 4 h. The reaction mixture was cooled to rt, poured in aq. saturated NH4Cl and extracted with EtOAc (3x). The combined organic layers were washed with brine (x3), dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica chromatography eluting with a gradient of 20% to 100% EtOAc:heptanes. The relevant fractions were combined and concentrated in vacuo to afford Int-66 (9.15 g, 56% yield) as a brown solid. ESI MS m/z 343.1 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.60 (s, 1H), 7.99 (d, J = 9.2 Hz, 1H), 7.59 (d, J = 9.3 Hz, 1H), 6.93 (t, J = 8.9 Hz, 2H), 6.65 (dd, J = 9.0, 4.6 Hz, 2H), 6.03 (t, J = 6.3 Hz, 1H), 4.13 (d, J = 6.2 Hz, 2H), 1.43 (s, 9H). 19F NMR (376 MHz, DMSO-d6) δ −128.59 (tt, J = 8.9, 4.6 Hz).
tert-Butyl N-[6-[3-(N-[2-[3,5-bis(trifluoromethyl)-2-pyridyl]acetyl]-4-fluoro-anilino)prop-1-ynyl]pyridazin-3-yl]carbamate (67).
To a solution of 2-[3,5-bis(trifluoromethyl)-2-pyridyl]acetic acid 62 (7.3 g, 26.7 mmol) and tert-Butyl N-[6-[3-(4-fluoroanilino)prop-1-ynyl]pyridazin-3-yl]carbamate 66 (9.15 g, 26.7 mmol) in dry THF (91 mL) at 0 °C was added DIPEA (10.36 g, 80.1 mmol, 13.9 mL) dropwise at rt followed by T3P (25.51 g, 40.0 mmol, 23.8 mL, 50% purity). The reaction mixture was stirred at 0 °C for 15 min then 1 h at rt. The reaction mixture was evaporated in vacuo and the residue purified by silica chromatography eluting with a gradient of 10–60% EtOAc:heptanes. The relevant fractions were combined and concentrated in vacuo to afford 67 (13.03 g, 81% yield) as a brown solid. ESI MS m/z 598.1 [M+H]+. 1H NMR (400 MHz, Chloroform-d) δ 9.00 (d, J = 2.1 Hz, 1H), 8.18 (d, J = 9.3 Hz, 1H), 8.13 (d, J = 2.1 Hz, 1H), 7.70 (s, 1H), 7.43 (dd, J = 8.9, 4.7 Hz, 3H), 7.17 (t, J = 8.5 Hz, 2H), 4.79 (s, 2H), 3.90 (s, 2H), 1.54 (s, 10H).
N-[3-(6-Aminopyridazin-3-yl)prop-2-ynyl]-2-[3,5-bis(trifluoromethyl)-2-pyridyl]-N-(4-fluorophenyl)acetamide (RP-6685).
To a solution of 67 (13.03 g, 21.8 mmol) in dry DCM (100 mL) was added trifluoroacetic acid (33.6 mL, 436 mmol,) dropwise. The reaction mixture was stirred at rt for 2 hrs. The mixture was evaporated in vacuo. The brown oily residue was dissolved in EtOAc (200 mL) and sat. aq. NaHCO3 was added carefully (gas evolution). The organic phase was washed with NaOH 2N (50 mL; reaching pH= 9), brine, dried over MgSO4, filtered and concentrated in vacuo. The light yellow solid was recrystallized in EtOAc. The solids were filtered, washed with a minimum of EtOAc and dried in vacuo. The solid was dissolved in MeCN (300 mL)/Nanopure water (300 mL) and lyophilized to RP-6685 (9.54 g, 87% yield, 99.9% purity) as a white solid. ESI MS m/z 498.2. [M+H]+. 1H NMR (400 MHz, Chloroform-d) δ 9.05 – 8.92 (m, 1H), 8.12 (d, J = 2.1 Hz, 1H), 7.72 – 7.62 (m, 0H), 7.54 (dtt, J = 8.6, 3.4, 1.7 Hz, 0H), 7.46 – 7.35 (m, 2H), 7.23 – 7.07 (m, 3H), 6.67 (d, J = 9.1 Hz, 1H), 5.13 (s, 2H), 4.76 (s, 2H), 3.89 (s, 2H).
Biological Assays
Polθ Picogreen assay (Primary Screening assay).
All IC50s are the mean of n=3. The Polθ enzyme was incubated with a 10-point concentration response of inhibitors for 15 minutes at 37 oC in the following buffer: 10 mM Tris Cl pH 8.0, 50 mM NaCl glycerol, 10 mM MgCl2, 1mM DTT and 0.01% Tween-20. Following pre-incubation with inhibitors, DNA+dNTP solution was added. The microplate was centrifuged for 10 sec., incubated 120 min at 37 oC. The reaction was stopped by addition of 15 μL picogreen / stop solution. After 5 min, the fluorescence intensity was measured. The % inhibition of compounds was calculated using (DMSO + Polθ) and (DMSO no Polθ) control wells. IC50s and % at top plateau were determined for each compound. The final concentration of PolQ in 15 μL reaction volume was 0.3 nM, annealed DNA oligos were 6 nM, and dNTP were 10 μM. The DNA was made by annealing 2 different oligos containing the following sequence of nucleotides:
5’ CAC TGA CTG TAT GAT G 3’
5’ACTATCAAAACTATCAAATGGACTATCAAAACTATCAAATGCTATACAGGAAAAAAAAATGAGTTATTAATCTTCATCATACAGTCAGTG
The oligos are annealed by heating at 95 °C for 5 minutes in the following buffer (10 mM Tris-HCl pH7.5, 50 mM NaCl, 1 mM EDTA) and cooled to RT.
Traffic-light Reporter Assay (TLR)
Day 1, 293T-Lig4−/−TLR1.1 cells (NHEJ-deficient donated from the Sfeir Lab)1 were seeded in poly-d-lysine coated 96-well plates (100 μL cells/well). Cells were incubated in a tissue culture incubator (5% CO2, 37 °C) for 4 to 6 hours prior to addition of Polθ inhibitors using a TECAN D300e instrument. The next day, cells were transduced with 100 μL of media containing 10% lentivirus solution and 8 μg/mL polybrene in presence of PolQ inhibitors. 24h later, media was replaced with fresh media containing PolQ inhibitors and the cells were incubated for an additional 96 h (5% CO2, 37 °C). At the end of the incubation period, the cells were collected, fixed with 2% paraformaldehyde and analyzed by flow cytometry (GFP and mCherry). Data analysis was done with FloJo software.
Cell culture.
293T-Lig4−/−TLR1.1 cells were a gift from A. Sfeir. They were grown in DMEM (Hyclone SH30243.02) supplemented with 10% FBS (Avantor Cat 97068–085) and 1% Penicillin/Streptomycin (Corning cat 30–001-CI). HCT116 BRCA2 +/+ and HCT116 BRCA2 −/− clone 46 were purchased from ATCC (Cat CCL-247) and ECACC General Cell Collection (Cat 18061301) respectively. They were grown in culture in McCoy’s media (Hyclone SH30200.01) supplemented with 10% FBS and 1% Penicillin/Streptomycin. Cells were routinely tested for mycoplasma and murine pathogens and were used at passages of <20 from the initial source vial from supplier.
Cell proliferation assays.
HCT116 BRCA2 +/+ (400 cells/well) and HCT116 BRCA2 −/− (750 cells/well) cells were seeded in a 96-well plate (Costar 3595) and incubated overnight in a tissue culture incubator (5% CO2, 37 °C). The next day, PolQ or PARP inhibitor (control) compounds were added using a TECAN D300 instrument. DMSO concentration was kept below 0.2% final. Media was replenished with fresh compounds every 3 or 4 days. The assay duration was 7 days for BRCA2 +/+ and 12–14 days for BRCA2 −/− cells (approximately 6 population doublings). Cellular growth was monitored using the Incucyte® Live-Cell Analysis System to capture phase contrast images. Plates were scanned daily with 4x objective or at the end of experiment when DMSO-treated wells reached 90% confluency. Cellular confluency was determined using Incucyte® Cell-by-Cell Analysis Software Module and dose-response curves calculated using GraphPad Prism software
Cas9-Induced Double-Strand DNA Break assay (DSB).
Alt-R® S.p. Cas9 Nuclease V3 (IDT cat 1081059) (40 μg) and AAVS1 sgRNA (GGGGCCACTAGGGACAGGAT) (240 pmol) were combined to allow the formation of complex for 15 min at RT. Electroporation Enhancer (IDT cat 1075916) (0.2 nmol) was added at the end of incubation period. HCT116 WT cells (1×106) were nucleofected with Cas9/sgRNA complex using Lonza 4D nucleofector instrument (SE buffer, EN113 program) and seeded in a 96-well plate (Costar 3595) at 35,000 cells/well. DNA-PK inhibitor AZD7648 (Chemitek cat CT-A7648) (final concentration 0.3 μM) and PolQ inhibitors were added using a TECAN D300 instrument. Cells were incubated for 24h in a tissue culture incubator (5% CO2, 37 °C). The next day, the culture media was removed, and cells disrupted with lysis buffer for 10 min at 75 °C (Sigma # XNAB2). PCR reactions around breaking site were carried out with 1/10 of cell lysis and Phusion hot start flex DNA pol (New England Bioloabs Cat# M0535L). PCR samples were sent to Genome Quebec for Sanger DNA sequencing (https://www.genomequebec.com). Sequencing data were analyzed using the TIDE software (https://tide.nki.nl). The PolQ-mediated MMEJ signature is represented by −5 and −12bp indels. PolQ inhibitor dose-response curves were calculated using GraphPad Prism software.
Mouse Pharmacokinetic Studies.
All procedures performed on animals were in accordance with regulations and established guidelines at Repare Therapeutics, with Institutional Animal Care Committee-approved protocols. Female CD1 mice (20–30g) were administered with the test compound at a dose of 1 mg/kg intravenously using a solution formulation. Oral bioavailability was determined at doses 2.5 or 20 mg/kg using either a solution or suspension formulation. The blood samples for the IV experiment were collected at pre-dose, 5, 15, 30 min, 1, 2, 4, 8 and 24 h time points. The blood samples for the PO experiment were collected at pre-dose, 15, 30 min, 1, 2, 4, 6, 8 and 24 h time points. Microsampled whole blood was collected for the mouse pharmacokinetic determinations using a previously described method (ref: Bateman, K. P.; Castonguay, G.; Xu, L.; Rowland, S.; Nicoll-Griffith, D. A.; Kelly, N.; Chan, C.-C. J. Chromatogr. B 2001, 754, 245–251.) All samples were quantified using a reversed-phase liquid chromatography gradient coupled to electrospray mass spectrometry operated in positive mode. PK parameters were calculated using non-compartmental analysis.
In Vivo Studies: HCT116 Cell line-derived xenograft
In vivo studies involving the HCT116 and HCT-116 BRCA2 −/− cell-derived xenografts in female CD1 Nude mice (Charles River lab, 8 mice per group) were performed at Repare Therapeutics, with Institutional Animal Care Committee-approved protocols. 2×10^7 HCT116 BRCA2 −/− cells were resuspended in sterile PBS and injected subcutaneously. Tumor growth was monitored by caliper measurements to calculate the tumor volume (TV) using the formula: Tumor volume = 0.52 × L × W2. Tumor growth inhibition (%TGI) was defined as: %TGI= ((TVvehicle/last – TVvehicle/day0) − (TVtreated/last – TVtreated/day0)) / (TVvehicle/last – TVvehicle/day0) ×100. Bodyweight change was represented as change in BW using the formula: %BW change = (BWlast- BWday0) / BWday0 ×100. BW change was calculated based on individual BW changes relative to Day 0. Statistical significance relative to vehicle control or other test groups was established using the unpaired t-test with Welch’s correction (GraphPad Prism v9.0). Mouse pharmacokinetics to assess representative PK levels in the efficacy study were evaluated in a satellite cohort of CD-1 nude mice (n=3) treated with 50 or 100 mg/kg RP-6685 BID. Whole blood was collected pre-dosing at time 0 and, 1,2,4, 8 hrs post the morning dose, then dosed again at 8 h later and blood collected at 10, 12 and 24h on Days 1 and 7. RP-6685 was quantified as indicated below and compound levels converted to free plasma using an Fu of 0.5 and a blood/plasma ratio of 0.6.
In Vivo Studies: Pharmacokinetics (PK) and pharmacodynamic (PD) markers
Mouse whole blood was collected at 0.5, 1, 3, 8, and 24 hours post dose by tail snip and diluted 1:3 with 0.1 M citrate buffer. Plasma was collected from whole blood drawn by cardiac puncture at 1 and 3 hours post dose into tripotassium (K3) EDTA tubes (Sarstedt Cat# 41.1504.105), then centrifuged at 12,000 relative centrifugal force for 10 minutes at 4°C. The concentration of RP-3500 in whole blood and plasma was determined by high-performance liquid chromatography-mass spectrometry (see Supplementary Methods). Excised tumors were cut into fragments, then flash-frozen for protein extract or preserved in 10% formalin. Frozen fragments were homogenized in lysis buffer (Mesoscale discovery #R60TX-2) with protease and phosphatase inhibitors (ThermoFisher #78437, ThermoFisher #78420) using 2.8 mm ceramic bead-containing tubes (OMNI #19-628) and a Bead Ruptor 24 (OMNI international).
Supplementary Material
ACKNOWLEDGEMENTS:
We would like to extend our gratitude to Ono Pharmaceuticals for their financial and scientific support. We are grateful for chemistry support of Piramal Pharma in particular the following chemists who have prepared compounds described in this work: Jiten Thakkar, Saurabh Vijay, Payal Kharadi, Ayyappa Naidu, Piyush Kalaria, Vaibhav Dhamane, Avinash Patil, Pravin Gavande, Manohar Ladani, Prexa Kachhiya, Yogesh Goriya, Nilesh Parmar, Vinod Vishwapathi. This work was supported in part by Canadian Institutes of Health Research grants FDN-143277, PJT-178026, and PJT-180338, to F.S. and was based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Eiger 16M detector on 24-ID-E is funded by a NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We are grateful for the support, training, and custom tools provided by the Chemical Computing Group, Inc., Montreal, Quebec, Canada.28 We thank Agnel Sfeir’s lab for consultation and 293T-Lig4−/−TLR1.1 cells (TLR assay)Lig4. We thank Pascal Turcotte (AdMare BioInnovations, 7171 Frederick-Banting, Montréal, QC, H4S 1Z9, Canada) for SFC separations.
ABBREVIATIONS USED
- DIPEA
diisopropyl ethyl amine
- DSB
double strand break
- iv
intravenous
- MDCK
Madin-Darby canine kidney cell
- MS
mass spectrometry
- po
per os (oral)
- SAR
structure activity relationship
- T3P
Propyl phosphonic anhydride solution
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at:
Synthetic procedures for compounds 1–47, and RP-6685; experimental details for in vitro and in vivo studies; additional crystallographic data for compounds 14 and 37; HPLC traces for compounds 1–47, and RP-6685; 1H NMR, 13C NMR, 19F, HRMS for RP-6685. (PDF)
Molecular formula strings, biological, ADME data (CSV)
Accession Codes
Atomic coordinates for the X-ray structures of compound 14 (PDB 8E23) and compound 37 (PDB 8E24) bound to Polθ are available from the RCSB Protein Data Bank. Authors will release the atomic coordinates upon article publication.
REFERENCES
- (1).Sfeir A; Symington LS Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway. Trends Biochem Sci. 2015, 40 (11):701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Llorens-Agost M; Ensminger M; Le HP.; Gawai, A.; Liu, J.; Cruz-García, A.; POLθ-mediated end joining is Restricted by RAD52 and BRCA2 until the Onset of Mitosis. Nat Cell Biol. 2021, 23(10):1095–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Mateos-Gomez PA; Gong F; Nair N; Miller KM; Lazzerini-Denchi E; and Sfeir A Mammalian Polymerase Theta Promotes Alternative-NHEJ and Suppresses Recombination. Nature, 2015, 518 (7538): 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Brambati A; Barry RM; Sfeir A DNA Polymerase Theta (Polθ) - An Error-Prone Polymerase Necessary for Genome Stability. Curr. Opin. Genet. Dev. 2020. 02; 60: 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mateos-Gomez PA; Kent T; Deng SK; McDevitt S; Kashkina E; Hoang TM; Pomerantz RT; Sfeir A The Helicase Domain of Polθ Counteracts RPA to Promote Alt-NHEJ. Nat. Struct. Mol. Biol. 2017, 24(12):1116–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Yousefzadeh MJ; Wyatt DW; Takata KI; Mu Y; Hensley SC; Tomida J; Bylund GO; Doublié S; Johansson E; Ramsden DA; McBride KM; Wood RD Mechanism of Suppression of Chromosomal Instability by DNA Polymerase POLQ. PLoS Genetics. 2014, 10(10): 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wyatt DW; Feng W; Conlin MP; Yousefzadeh MJ; Roberts SA; Mieczkowski P Essential Roles for Polymerase θ-Mediated End Joining in the Repair of Chromosome Breaks. Mol. Cell. 2016, August 18;63(4):662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ceccaldi R; Liu JC; Amunugama R; Hajdu I; Primack B; Petalcorin MI Homologous-recombination-deficient Tumours are Dependent on Polθ-mediated Repair. Nature. 2015, 518(7538): 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dai CH; Chen P; Li J; Lan T; Chen YC; Qian H; Chen K; Li MY Co-inhibition of Pol θ and HR Genes Efficiently Synergize with Cisplatin to Suppress Cisplatin-resistant Lung Cancer Cells Survival. Oncotarget. 2016, 7(40): 65157–65170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zahn KE; Jensen RB Polymerase θ Coordinates Multiple Intrinsic Enzymatic Activities during DNA Repair. Genes (Basel). 2021, 12 (1310):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Caracciolo D; Riillo C; Di Martino MT; Tagliaferri P; Tassone P Alternative Non-Homologous End-Joining: Error-Prone DNA Repair as Cancer’s Achilles’ Heel. Cancers (Basel). 2021, 13, 1392: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Patel PS; Algouneh A; Hakem R Exploiting Synthetic Lethality to Target BRCA1/2-deficient Tumors: Where We Stand. Oncogene. 2021, 40 (17): 3001–3014.k [DOI] [PubMed] [Google Scholar]
- (13).Zatreanu D; Robinson HMR; Alkhatib O; Boursier M; Finch H; Geo L; Grande D; Grinkevic V; Heald R, A.; Langdon S; Majithiya J; McWhirter C; Martin N,MB; Moore S; Neves J; Rajendra E; Ranzani M; Schaedler T; Stockley M; Wiggins K; Brough R; Sridhar S; Gulati A; Shao N; Badder LM; Novo D; 2, Knight E,G; Marlow R; Haider S; Callen E; 5, Hewitt G; Schimmel J; Prevo R;Alli C; Ferdinand A; Bell C; Blencowe P; Bot C; Calder M; Charles M; Curry J; Ekwuru T; Ewings K; Krajewski W; MacDonald E; McCarron H; Pang L; Pedder C; 9, Rigoreau L; Swarbrick M; Wheatley E; Willis S; Wong A,C; Nussenzweig A; Tijsterman M; Tutt A; Boulton SJ; Higgins G,S; Pettitt S,J; Smith G,C,M;Lord C,J. Pol θ Inhibitors Elicit BRCA-gene Synthetic Lethality and Target PARP Inhibitor Resistance. Nature Communications. 2021, 12:3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhou J; Gelot C; Pantelidou C; Li A; Yücel H; Davis RE, Farkkila A, Kochupurakkal B, Syed A, Shapiro GI, Tainer JA, Blagg BSJ, Ceccaldi R, D’Andrea AD A first-in-class Polymerase Theta Inhibitor Selectively Targets Homologous-Recombination-Deficient Tumors. Nat Cancer. 2021, 2(6):598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Singer VL; Jones LJ; Yue ST and Haugland RP Characterization of PicoGreen Reagent and Development of a Fluorescence-Based Solution Assay for Double-Stranded DNA Quantitation. Analytical Biochemistry. 1997, 249: 228 – 238. [DOI] [PubMed] [Google Scholar]
- (16).Zahn KE; Averill AM; Aller P; Wood RD; Doublié S Human DNA Polymerase θ Grasps the Primer Terminus to Mediate DNA Repair. Nat. Struct. Mol. Biol. 2015, 22(4):304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Doublié S; Tabor S; Long AM; Richardson CC; Ellenberger T Crystal Structure of a Bacteriophage T7 DNA Replication Complex at 2.2 A Resolution. Nature. 1998, 391(6664):251–258. [DOI] [PubMed] [Google Scholar]
- (18).Joyce CM; Potapova O; Delucia AM; Huang X; Basu VP; Grindley ND Fingers-closing and Other Rapid Conformational Changes in DNA Polymerase I (Klenow fragment) and Their Role in Nucleotide Selectivity. Biochemistry. 2008, 47(23):6103–6116. [DOI] [PubMed] [Google Scholar]
- (19).Chandramouly G; Zhao J; McDevitt S; Rusanov T; Hoang T; Borisonnik N; Treddinick T; Lopezcolorado FW; Kent T; Siddique LA; Mallon J; Huhn J; Shoda Z; Kashkina E; Brambati A; Stark JM; Chen XS; Pomerantz RT Polθ Reverse Transcribes RNA and Promotes RNA-templated DNA Repair. Sci. Adv. 2021, 7: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Durocher Y; Perret S; Kamen A High-level and high-throughput Recombinant Protein Production by Transient Transfection of Suspension Growing Human 293-EBNA1 cells. Nucleic Acids Res. 2002, 30, E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).L’Abbé D; Bisson L; Gervais C; Grazzini E; Durocher Y Transient Gene Expression in Suspension HEK293-EBNA1. Cells. Methods Mol. Biol. 2018, 1850, 1–16. [DOI] [PubMed] [Google Scholar]
- (22).Seki M; Masutani C; Yang LW; Schuffert A; Iwai S; Bahar I; Wood RD High-efficiency bypass of DNA damage by human DNA polymerase Q. EMBO J., 2004, 23, 4484–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Leeman JE; Li Y; Bell A; Hussain SS; Majumdar R; Rong-Mullins. X; Blecua P; Damerla R; Narang H; Ravindran PT; Lee NY; Riaz N; Powell SN; Higginson DS Human Papillomavirus 16 Promotes Microhomology-mediated End-joining. Proc. Natl. Acad. Sci. U S A. 2019, 22;116(43): 21573–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Higginson DS; Hussain S,S; Compositions and Methods for Monitoring DNA Repair. WO 2019/246553 Al [Google Scholar]
- (25).Gosselin F; O’Shea PD; Webster RA; Reamer RA; Tillyer RD; Grabowski EJJ Highly Regioselective Synthesis of 1-Aryl-3,4,5-Substituted Pyrazoles. Synlett. 2006, 19, 3267–3270. [Google Scholar]
- (26).Bondy SS; Cannizzaro CE; Chou C-H; Halcomb RL; Hu YE; Link JO; Liu Q; Schroeder, Scott D.; Tse WC; Zhang JR. Compounds for the Treatment of HIV. WO2013/6738, 2013, A1 [Google Scholar]
- (27).Sonogashira K Development of Pd-Cu Catalyzed Cross-coupling of Terminal Acetylenes with sp2-Carbon Halides. J. Organomet. Chem. 2002, 653 (1–2): 46–49 [Google Scholar]
- (28).MOE: Molecular Operating Environment (MOE), 2019.January; Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.