


Abstract
Nucleotide excision repair (NER) is critical for removing bulky DNA base lesions and avoiding diseases. NER couples lesion recognition by XPC to strand separation by XPB and XPD ATPases, followed by lesion excision by XPF and XPG nucleases. Here, we describe key regulatory mechanisms and roles of XPG for and beyond its cleavage activity. Strikingly, by combing single-molecule imaging and bulk cleavage assays, we found that XPG binding to the 7-subunit TFIIH core (coreTFIIH) stimulates coreTFIIH-dependent double-strand (ds)DNA unwinding 10-fold, and XPG-dependent DNA cleavage by up to 700-fold. Simultaneous monitoring of rates for coreTFIIH single-stranded (ss)DNA translocation and dsDNA unwinding showed XPG acts by switching ssDNA translocation to dsDNA unwinding as a likely committed step. Pertinent to the NER pathway regulation, XPG incision activity is suppressed during coreTFIIH translocation on DNA but is licensed when coreTFIIH stalls at the lesion or when ATP hydrolysis is blocked. Moreover, ≥15 nucleotides of 5′-ssDNA is a prerequisite for efficient translocation and incision. Our results unveil a paired coordination mechanism in which key lesion scanning and DNA incision steps are sequentially coordinated, and damaged patch removal is only licensed after generation of ≥15 nucleotides of 5′-ssDNA, ensuring the correct ssDNA bubble size before cleavage.
Graphical Abstract
Graphical Abstract.
A ≥15 nucleotide 5′ ssDNA stretch allows for proper coreTFIIH-XPG complex coordination that efficiently unwinds in the presence of ATP but cleaves in its absence or upon damage encounter.
INTRODUCTION
Nucleotide excision repair (NER) removes a broad spectrum of bulky DNA lesions (1–8) caused by UV irradiation, environmental mutagens, chemotherapeutic treatments, and endogenous reactive oxygen stress. Deficiencies in NER, such as those prevalent in xeroderma pigmentosum (XP), are manifested through hypersensitivity to UV light and extreme predisposition to skin cancer (9,10). Additionally, NER deficiencies are linked to developmental defects such as Cockayne syndrome (CS) and trichothiodystrophy (TTD) (11–13). NER is critical for maintaining genomic integrity and preventing DNA damage signaling pathways (14–16). Cells that express catalytically inactive XPG nuclease show an increase in the DNA damage response (DDR), indicating that the lack of a second incision by XPG, as needed to remove the damage containing ssDNA patch, can trigger stress and an increased DDR (17).
NER is classified into global genome NER (GG-NER) and transcription-coupled NER (TC-NER). Although the initial damage recognition step involves different proteins across these two pathways (18,19), NER converges to a common set of factors after TFIIH recruitment (20). TFIIH is composed of a 7-subunit core complex (coreTFIIH), comprising of the ATPases XPB and XPD along with p62, p52, p44, p34 and p8, as well as a 3-subunit cyclin-activated kinase (CAK) module consisting of CDK7, cyclin H and MAT1 (19). XPB is mainly responsible for initial DNA bubble opening due to its dsDNA translocation activity, whereas XPD acts as a lesion-scanning protein due to its 5′ to 3′ ssDNA translocation activity (7,21,22). During NER, the TFIIH holoenzyme is initially recruited upstream of the damage site by XPC and causes the unwinding of a few nucleotides to enable XPA recruitment, which in turn triggers the release of ATPase down-regulator CAK (23). Therefore, XPA is likely required for bubble elongation (24,25). Importantly, the role of bubble opening and bubble length in enabling 5′ incision by XPF-ERCC1 and 3′ incision by XPG remains a central yet enigmatic point of NER regulation. Notably, XPG is recruited early by TFIIH to the damage site during NER, but cuts the DNA last. In fact, even repair synthesis can be initiated before incision by XPG. XPF-ERCC1 cleavage requires the presence but not the catalytic activity of XPG (26). These observations suggest that the initial binding of XPG can play a critical regulatory role in NER, with the catalytic activity being applied at a later stage (26,27). Furthermore, based on cross-linking studies and the TFIIH-DNA structure, XPG is proposed to directly contact XPD (20,24).
To achieve a functional and mechanistic understanding of how molecular components work together for bulky lesion repair, it is important to define specific XPG interactions with TFIIH and their potential roles in regulating NER. Strikingly, bound to 5′ overhang substrates, both XPA and XPG have shown strong stimulatory effects on the helicase activity of coreTFIIH (24). Although XPA stimulates both XPB and XPD translocation activities (24,28), XPG stimulates the XPD translocation activity alone, potentially by lifting the cap domain of XPD to enable easier slotting of ssDNA through XPD. However, direct evidence for this activity is lacking and other facets of this interaction remain elusive. Using these observations as the starting point, we reasoned that single-molecule imaging may help elucidate the mechanisms by which XPG influences TFIIH activity and how TFIIH regulates 5′ incision by XPG.
Therefore, we employed single-molecule imaging to simultaneously monitor the rates of coreTFIIH translocation along ssDNA and of coreTFIIH unwinding of dsDNA. We show that neither XPG nor XPA significantly stimulated the individual rates of these activities, but increased the probability of switching from ssDNA translocation to dsDNA unwinding upon encountering the ss/ds junction. Further, bulk activity assay results show that when coreTFIIH was actively translocating on DNA, XPG nuclease activity was down-regulated. However, inhibiting coreTFIIH translocation activity either by removing ATP or using a non-hydrolyzable ATP analog stimulated XPG nuclease activity. This stimulation is further enhanced when coreTFIIH is stalled by a lesion even in the presence of ATP. Remarkably, stimulation of both helicase and nuclease activities is dependent on the length of ssDNA, with a permissive 5′ overhang length of ∼15–20 nucleotides (nt). Collectively, we have uncovered a paired coordination between coreTFIIH and XPG which accounts for lesion-scanning, efficient strand separation at the 3′ end of the bubble, and precise incision that occurs upon lesion detection alone.
MATERIALS AND METHOD
DNA substrate preparation
The HPLC-purified DNA oligonucleotides (Supplementary Materials Table S1) purchased from Integrated DNA Technologies (IDT, USA) and Sigma-Aldrich (Germany). Substrates were prepared by annealing equimolar ratios of the respective substrates in an annealing buffer (50 mM Tris–HCl pH 8.0, 1 mM EDTA pH 8.0, 100 mM NaCl). The solution was heated to 95°C for 5 min, and slowly cooled to 22°C. Further purification was performed on a non-denaturing PAGE gel and the DNA was eluted using the crush and soak method (29) by incubation in TE buffer (50 mM Tris–HCl pH 8.0, 1 mM EDTA pH 8.0) for 3 h at 22°C. All measurements were performed using freshly prepared substrates that were not subjected to freeze and thaw cycles.
7-subunit human TFIIH cloning, expression and purification
The TFIIH core complex (XPB-preScission-GFP, XPD, p62, p52, p44, p34 and p8) was cloned into a MacroBac vector 438a (30). The protein was expressed in Sf9 cells (Sigma-Aldrich, Germany) supplemented with 1 mM l-cysteine and 0.1 mM ferric ammonium citrate. Based on the fusion of XPB with GFP, coreTFIIH was purified anaerobically using a GFP-nanobody binder (31) covalently linked to agarose-beads (NHS agarose, Pierce/Thermo). The coreTFIIH was eluted with preScission protease; and Superose 6 (10/300) or Hi-Load Superdex 200 (16/600) was used for large-scale purification with 25 mM HEPES pH 7.8, 150 mM NaCl, 50 mM KCl, 3% glycerol and 3 mM β-mercaptoethanol, as performed previously. The eluate containing the TFIIH core complex was confirmed by SDS-PAGE gel after showing a 420 nm absorption peak and was stored at –80°C until use.
Human XPG cloning, expression and purification
The human XPG sequence (P28715-1) was synthesized by Genscript. The sequence was cloned into a pISUMO vector (LifeSensors) to obtain N-terminal double histidine- and SUMO-tagged proteins using the Gibson cloning protocol (32,33). Sf9 cells used for transfection were cultured in ESF 921 medium (Expression Systems). To prepare the baculovirus, bacmid DNA containing the XPG construct was transfected into Sf9 cells using FuGENE® HD (Promega) according to the manufacturer's instructions. Amplification was performed to obtain a solution with a higher viral titer which was referred to as P3. Next, 4 l of the Sf9 suspension culture at a density of 2 × 106 cells/ml was transfected with the aforementioned P3 viral titer. Cells were harvested at 60 h post infection by centrifugation at 5500 ×g for 10 min, followed by resuspension in lysis buffer (50 mM Tris–HCl pH 7.5, 500 mM NaCl, 5 mM β-mercaptoethanol (BME), 0.2% NP-40, 1 mM PMSF, 5% (v/v) glycerol, and an EDTA-free protease inhibitor cocktail tablet for each 50 ml of suspension (Roche, UK)) to a concentration of 3 ml per 1 g of wet cells. The suspension was sonicated and the debris was pelleted by centrifugation at 95 834 ×g for 1 h at 4°C. The supernatant was adjusted to a final concentration containing 20 mM imidazole and was directly loaded onto a HisTrap HP 5 ml affinity column (GE Healthcare) which was previously equilibrated with buffer A (50 mM HEPES–KOH pH 7.9, 750 mM NaCl, 10 mM BME, 10% glycerol, 20 mM imidazole and an EDTA-free protease inhibitor cocktail tablet for each 50 ml of solution). A washing step was performed with 100 ml of buffer A to remove the nonspecifically-bound proteins. Elution was performed with 50 ml of the linear gradient against buffer B (50 mM HEPES–KOH pH 7.9, 750 mM NaCl, 300 mM imidazole, 10 mM BME, 10% glycerol, and an EDTA-free protease inhibitor cocktail tablet for each 50 ml of solution). Protein-rich fractions were pooled, supplemented with SUMO star protease (LifeSensors) to remove the SUMO and histidine tags, leaving the XPG N-terminus starting with residue 2, and the samples were incubated for 16 h at 4°C in dialysis buffer (50 mM HEPES–KOH pH 7.9, 400 mM NaCl, 10 mM BME, 10% glycerol and an EDTA-free protease inhibitor cocktail tablet for each 50 ml of solution). The dialyzed mixture was loaded onto a fresh HisTrap HP 5 ml affinity column and the XPG was collected in the flow-through fraction. The fractions were pooled, concentrated to a volume of 1.5 ml, and loaded onto a HiLoad 16/600 Superdex 200 pg (GE Healthcare) size exclusion chromatography column pre-equilibrated with storage buffer (100 mM HEPES–KOH pH 7.5, 400 mM NaCl, 1 mM DTT, and 10% glycerol). Protein fractions were pooled, flash-frozen, and stored at −80°C.
Human XPA cloning, expression and purification
The human XPA sequence was cloned into a PE-SUMO vector backbone and transformed into BL21 (DE3) cells (Sigma-Aldrich). The cells were incubated in LB media until they reached an OD600 of 0.8. Protein expression was induced with 0.1 mM isopropyl‐β‐d‐thiogalactopyranoside (IPTG) for 24 h at 16°C. The cells were harvested by centrifugation at 4°C, and the cell pellet was resuspended in lysis buffer (50 mM HEPES pH 7.5, 750 mM NaCl, 20 mM imidazole, 10 mM β-mercaptoethanol (BME), and 1 mM PMSF). The cells were lysed by a cell disruptor at 30 kPsi and the lysate was subjected to ultracentrifugation. The filtered supernatant was passed through a Ni-NTA column (HisTrap HP 5 ml, GE Healthcare) using Buffer A (50 mM HEPES pH 7.5, 750 mM NaCl, 20 mM imidazole, and 10 mM β-mercaptoethanol) and eluted with linear gradient of Buffer A and 300 mM imidazole. The eluate was supplemented with Sumostar protease (LifeSensors) and left to dialyze in buffer A for 16 h. The mixture was loaded onto a fresh HiTrap HP 5 ml column, and the protein fraction was collected in the flow-through. The collected fractions were pooled, diluted to a lower salt concentration of 300 mM NaCl, and loaded onto a HiLoad 16/600 Superdex 75 pg (GE Healthcare) size-exclusion column. Elution was performed using a gradient of Buffer B (100 mM HEPES pH 7.5, 300 mM NaCl, 10 mM and 10% glycerol) and 1 M NaCl. Elution occurred at approximately 300 mM NaCl. The collected fractions were dialyzed against the storage buffer (20 mM Tris–HCl pH 8.0, 1 mM DTT, 100 mM NaCl and 10% glycerol), flash-frozen and stored at −80°C.
Protocatechuate 3,4-dioxygenase (PCD) cloning, expression and purification
The sequences of the pcaH and pcaG subunits of PCD were codon-optimized and cloned with separate promoters in the pRSF-1b vector. A deca-histidine label was added to the N-terminus of pcaH. The PCD expression clone was transformed into the Escherichia coli BL21 (DE3) strain, and expressed in 2× YT media with 0.2 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) induction at 25°C for 8 h. Ferrous ammonium sulfate was added before induction to a final concentration of 20 mg/l of culture. PCD was purified using Ni-NTA affinity and Superdex-200 pg gel filtration chromatography. The PCD enzyme is a constituent of the oxygen scavenging system used during smFRET assays.
Bulk fluorescence assays
Bulk fluorescence unwinding assays were performed on a spectrofluorometer device (Fluoromax-4, Horiba Jobin Yvon). The sample holder was coupled to a temperature control device (Wavelength Electronics) for precise temperature control. The fluorophore intensity was recorded using the kinetics mode of the instrument. Sample excitation was performed at 620 nm with a 29 nm bandwidth, and the emission was collected at 685 nm using a 25 nm bandwidth. Measurements were performed at 37°C. The quartz cuvette (Hellma Analytics) contained 1 mM ATP and 10 nM DNA, resuspended in buffer (40 mM Tris–HCl pH 7.5, 40 mM KCl, 1 mM DTT, 0.1 mg/ml BSA, 10 mM MgCl2 and 5% (v/v) glycerol). A 3 min incubation was performed to warm the solution at 37°C, in a total volume of 7 ul. Unwinding was initiated by adding 5 ul of protein at a final concentration of 400 nM, diluted in the aforementioned buffer. The protein mixture was kept in an externally heated mantle at the same temperature as the cuvette in the instrument. Data recording was initiated immediately after protein addition. The time of data acquisition varied according to the time required to observe a plateau along the relative fluorescence intensity versus time plot (Supplementary Figure S1A). The integration time was always 0.1 s, and the data was acquired every 1 s. The y-axis data showed counts per second per microampere (CPS/μAmp) units after real-time correction for fluctuations in the source signal and the detector response as an inbuilt function of the instrument. The data were expressed as relative fluorescence set to 100% based on the signal intensity of equimolar amounts of free fluorescent oligo with 400 nM coreTFIIH. We determined the proportional amount of unwound DNA in nM units, where 100% equals 10 nM. Each plotted data point was obtained from the average of three measurements. The plotted error bars represent the standard error of the mean. The initial linear part of each curve was fitted to a linear function in the OriginPro™ Version 2021b software (OriginLab Corporation) as described above. The slope obtained from the fit represents the relative initial reaction rate and has the same units as the slope of the line nM/s (nM·s–1).
Single-molecule FRET experiments
smFRET experiments were performed in a microfluidic flow chamber constructed as described before (29) using a custom built microscope (26). The coverslip surface was passivated and functionalized with biotin-PEG and PEG (1:100, respectively) and subsequently incubated with a 0.03 mg/ml solution of filtered NeutrAvidin (NA) dissolved in PBS buffer. Excess NA was washed-off and the biotinylated substrate was flown for surface tethering. The objective was maintained at 37°C using an objective heater (Bioptechs). The tube containing the solution and the tubing that delivered the solution was maintained at 37°C using an in-house heating mantle. The temperature of the liquid in the tube and the flow cell at the objective-cell interface was 37.0 ± 0.2°C measured using a thermocouple thermometer (Omega Engineering). This precaution ensured that the solution within the flow cell and the incoming solution were at the same temperature and no heat shift-specific photophysical effects are observed. The photostability of fluorophores was improved using a protocatechuic acid/ protocatechuate-3,4-dioxygenase oxygen scavenging system and Trolox. The imaging buffer in the smFRET cleavage experiments was composed of 40 mM Tris–HCl pH 7.5, 40 mM KCl, 10 mM MgCl2, 1 mM DTT, 0.1% BSA and 5% (v/v) glycerol. The Cy3 and Cy5 fluorophores were excited with 532 and 640 nm lasers, respectively, in objective-based Total Internal Reflection Fluorescence (TIRF) mode as described in detail elsewhere (34). The emissions of the donor and the acceptor were split inside a Dualview module. The images of the surface-immobilized DNA were recorded using alternating green (532 nm) and red (640 nm) excitations. The image acquisition was synchronized to the laser excitation by triggering the acousto-optic tunable filter (AOTF) using an Electron Multiplying Charge Coupled Device (EMCCD) camera to prevent photobleaching of the sample when not acquiring data. The recorded data of the immobilized molecules consisted of 2400 frames, each lasting 177 ms. We typically recorded 400 linked molecules in both the donor and acceptor channels. The histogram was constructed by using 100 bins containing the distribution of the states in each molecule against the FRET efficiency (35). Each condition was repeated at least 3 times with a new set of reagents and a freshly prepared functionalized flow cell. The identification of initial FRET states and dye intensities from smFRET time traces was performed using the vbFRET package implemented in iSMS (36,37). Visual inspection of traces and assigning time points for determining the dwell time of certain behaviors were performed using the cursor function in the iSMS software. The quenching phase was identified from the timepoints where the Cy5 signal started to decrease till it reached zero intensity. The unwinding phase was identified from the points where the acceptor reaches zero intensity till donor loss was marked by instantaneous zero signal intensity.
Electrophoretic mobility shift assays (EMSA)
EMSA of coreTFIIH-DNA complexes were performed using different protein concentrations (0, 10, 20, 40, 60, 80, 100, 200, 300 and 400 nM). In a total volume of 12 μl, coreTFIIH and/or XPG were incubated with fluorescently labeled 5′ overhang DNA (5 nM) at 22°C for 10 min in binding buffer (40 mM Tris–HCl pH 7.5, 40 mM KCl, 1 mM DTT, 0.1 mg/ml BSA and 5% (v/v) glycerol). The complexes were separated via 6% native PAGE (Invitrogen) in 1× TBE buffer at room temperature and imaged using the Typhoon Trio Imager (GE Healthcare) at 635 nm. For band quantification, we used the GelQuantNET™ software (Biochemlab Solutions.com). The percentage of the bound substrate was calculated from its contribution to the total fluorescence of the respective lane. The binding constant Kd was calculated using the equation: Y = Max [coreTFIIH]n/(Kdn + [coreTFIIH]n), where Max is the concentration at which 100% of the substrate was bound by the protein, and n is the Hill coefficient.
Polyacrylamide gel electrophoresis cleavage assays
Bulk functional assays were performed to obtain the variable outcomes due to the presence or absence of ATP in the reaction, and to determine relative rates of cleavage at saturating protein concentrations. Fluorophore-labeled 5′ overhang DNA (5 nM) (Supplementary Materials Table S1) and 400 nM coreTFIIH and/or XPG were added under reaction buffer conditions (40 mM Tris–HCl pH 7.5, 40 mM KCl, 1 mM DTT, 0.1 mg/ml BSA and 5% (v/v) glycerol) at 37°C in a final volume of 60 μl. The reaction was started by adding ATP (final concentration of 1 mM) and MgCl2 (final concentration of 10 mM) to the buffer containing DNA, coreTFIIH, and XPG to evaluate increased unwinding. Alternatively, MgCl2 alone was added to evaluate the increased cleavage. Aliquots (5 μl) at various timepoints were pipetted into test tubes containing 5 μl of STOP buffer (50 mM EDTA, 0.2% SDS, 0.5 mg/ml proteinase K, and 40 mM Tris–HCl pH 7.5). The timepoints and total duration of incubation were selected to generate informative curves with the linear region containing at least 5-datapoints, and in the case of coreTFIIH + XPG to generate a complete cleavage curve (Supplementary Figure S4). Next, incubation at 45°C for 30 min was performed to ensure complete deproteination. The reactions were resolved using native PAGE (10% TBE gel, Invitrogen). The gels were imaged using a laser scanner (Typhoon Trio, GE Healthcare) at 635 nm, and the bands were quantified using GelQuant.NET (BiochemlabSolutions.com). The percentage of the cleaved or unwound substrate was calculated as a fraction of the total fluorescence in the respective lane. The relative rate of cleavage was determined from the slope of the linear region from the respective curve. Fitting was performed using the OriginPro™ Version 2021b software (OriginLab Corporation) using the equation y = mx, where m is the slope of the line, and x and y indicate the values on the x and y axes, respectively. Consequently, the linear rate was assigned units of the y/x axes, i.e. nM·s−1. Each data point in the time plot represents an average value obtained from three independent experiments.
Model building
To model XPB and XPD, we used the existing TFIIH-XPA-DNA cryo-EM density data (24). The missing loop regions of XPB and XPD were modeled into the EM density. Using the existing XPG crystal structure and AlphaFold2 (38,39), XPG was placed based on XPG incision relative to the lesion (40,41) and previous TFIIH-XPG cross-linking data (24). The DNA was modeled based on the existing TFIIH-XPA-DNA cryo-EM density data (24) and the FEN1-substrate DNA structure (42) (PDB ID 5UM9). Extra nucleotides on the lesion-containing strand were accommodated before and after the XPD ssDNA binding interface without steric hindrance. Our model is available in ModelArchive at https://modelarchive.org/doi/10.5452/ma-mrlg5.
RESULTS
XPG and XPA stimulate dsDNA unwinding by coreTFIIH in bulk
To delve into factors underlying DNA, XPG, XPA and coreTFIIH interactions, we evaluated the dsDNA unwinding activity of XPD in coreTFIIH (Figure 1 Ai, Aii) by using a substrate containing a 5′ overhang that was 26 nt in length. We analyzed dsDNA unwinding activity on a native gel using Cy5 attached to the displaced oligo (Figure 1Ai). In another experiment, we attached a quencher-fluorophore system (OnyxQ/Cy5) to track DNA unwinding quantitatively, by measuring the increase in Cy5 fluorescence signal intensity upon DNA unwinding, as described previously (Figure 1B) (24). We observed strong stimulation of coreTFIIH unwinding activity by XPG and XPA, consistent with previous results (Figure 1B). CoreTFIIH alone showed a relative unwinding rate of 0.040 ± 0.002 nM· s−1, and the rates with XPA or XPG were 0.140 ± 0.005 nM·s−1 and 0.450 ± 0.010 nM·s−1, respectively (Figure 1B; Supplementary Figure S1). Adding XPA with XPG did not further increase stimulation beyond the ∼11-fold difference observed when XPG alone was added to coreTFIIH.
Figure 1.

coreTFIIH unwinding activity is stimulated by XPG, XPA, and longer 5′ overhangs. (Ai) Unwinding activity of coreTFIIH visualized on a native gel using a 5′ overhang substrate after 5 min of incubation at 37°C in the presence of 1 mM ATP. (Aii) Representative Comassie blue stained gel of the final size exclusion column (SEC) load and elution fractions. All coreTFIIH subunits are labelled next to their respective band. The lowest band for P8 stains poorly on a regular basis due to a low number of aromatic residues. (B) Fluorescence intensity time plots showing coreTFIIH unwinding activity in the presence of XPA and/or XPG. Values obtained from linear fits of the linear region are color-coded. (C) Relative unwinding rates are plotted against the 5′ overhang length. Units of the Y-axis (nM) over the X-axis (s) were assigned to the slope (nM·s−1). All data points include the average of three independent experiments. Error bars indicate the standard error of the mean (SEM). See also Supplementary Figure S1.
Next, we investigated if the stimulation of coreTFIIH activity by XPG and XPA was dependent on the length of the 5′ overhang. The XPD footprint was previously reported to be ∼20 nt in length (43,44), with more recent structural data showing occlusion of only 10–11 nt (20,24). We used substrates with 5, 10, 15, 20, 26 and 40 nt 5′ overhangs (Figure 1C) under four different conditions: with coreTFIIH, coreTFIIH + XPG, coreTFIIH + XPA, and coreTFIIH + XPG + XPA. The observed activity was highest for the 20, and 26 nt overhangs across all conditions (Figure 1C; Supplementary Figure S1). We observed a positive correlation between overhang length and the relative unwinding rate with coreTFIIH (Supplementary Figure S1A). The addition of XPA and/or XPG boosted the unwinding rate across all overhang lengths examined. The maximum stimulation was observed with the addition of XPA (∼4-fold) and XPG (∼11-fold) (Supplementary Figure S1). Simultaneous addition of XPG and XPA resulted in no further stimulatory effects across all overhangs except for the 5 nt length (Supplementary Figure S1A). Thus, the results provide evidence for a clear preference of the coreTFIIH-XPG complex for 20 ± 5 nt long 5′ ssDNA to achieve an optimal unwinding rate. These results show that NER downregulates the 5′ to 3′ helicase activity/processivity of coreTFIIH-XPG until a stretch of 5′ overhang DNA of sufficient length is generated.
We also tested the effect of shortening the dsDNA on the unwinding rate by comparing a 40ss/24ds with a 40ss/18ds substrate. Structural comparison to the superfamily of 5′-nucleases to which XPG belongs (45,46) indicated that XPG may interact with ∼14 base pair dsDNA. Despite the ∼77°C melting temperature of both duplexes, the shorter duplex showed faster unwinding across all conditions (Supplementary Figure S1E).
XPG and XPA do not change the ssDNA translocation and dsDNA unwinding rates
Next, we used single-molecule imaging to simultaneously monitor the rates of coreTFIIH translocation on ssDNA and unwinding of dsDNA to analyze the stimulation observed by XPG and XPA in bulk assays. We relied on the quenching property of the iron-sulfur cluster binding domain (FeS CBD, henceforth referred to as FeS cluster) (43,44,47,48) to determine the position of coreTFIIH on ssDNA over time, while simultaneously monitoring the unwinding activity via fluorescence resonance energy transfer (FRET), as explained below.
The Cy5 fluorophore (acceptor) was positioned at the 3’ end of the non-translocating strand, while the Cy3 (donor) was positioned at the 3’ end of the translocating strand (Figure 2Ai). This arrangement allowed us to follow coreTFIIH translocation on ssDNA as it approaches the duplex through gradual quenching of Cy5. Once in the duplex, unwinding was followed by the simultaneous recovery of Cy5 intensity and gradual quenching of Cy3, as well as by the change in FRET efficiency between the two fluorophores (Figure 2Aii). The lengths of the 5′ overhang and duplex regions in our 40ss/18ds substrate enabled us to assume a nearly linear relationship between coreTFIIH-fluorophore distance and fluorophore intensity through quenching, as established in previous studies (43,44,48). Finally, complete strand separation was detected through the abrupt loss of the Cy3 signal resulting in a full-amplitude FRET efficiency time trace (Figure 2Aiii). Consequently, we determined the time spent by coreTFIIH on ssDNA and dsDNA, referred to as the quenching and unwinding phases, respectively (Figure 2A, B). A single quenching or unwinding phase indicates a burst event that did not have large backtracking steps, which could be discerned visually from the inherent noise in the traces obtained. Such behavior is considered processive and was used for rate determination (Figure 2Ai, B). Therefore, the length of the quenching phase was divided by 40 nt to obtain the dwell time of coreTFIIH per nucleotide. Similarly, the unwinding phase was divided by 18 base pairs (bp) to determine the dwell time required for unwinding a single base pair. The inverse of these values represents the translocation velocity on ssDNA and the unwinding velocity of dsDNA.
Figure 2.

XPG increases the success of unwinding by reducing abortive translocation at the ss/ds junction. (A) (i) Schematic of the 40ss/18ds substrate with donor (Cy3) and acceptor (Cy5) fluorophores positioned at the 3′ ends and tethered to the functionalized surface via a biotin-neutravidin-biotin link. coreTFIIH subunits XPB, p62, p52, p44, p32, and p8 are shown in light gray. The XPD ATPase is colored blue. The Cy5 and Cy3 fluorophores are colored red and green, respectively. (ii) Schematic representation of the quenching (pink shade) and unwinding (blue shade) phases. coreTFIIH translocation in the 3′ direction along the ssDNA overhang induces Cy5 quenching. During unwinding, coreTFIIH separates the strands of the duplex in the 5′ to 3′ direction as observed through variable quenching of both fluorophores and changes in their FRET E status. (iii) Example trace of a single quenching event followed by an unwinding event. Donor emission intensity due to donor excitation (Dem-Dex), and acceptor emission intensity due to acceptor excitation (Aem-Aex) are overlaid in the upper panel to showcase the anti-correlated behavior during unwinding. (B) Example trace containing multiple quenching events of the acceptor before a single unwinding event. (C) The rate of translocation on ssDNA (nt/s) was calculated from the duration of the quenching phase, along with the rate of strand separation (bp/s) calculated from the duration of the unwinding phase. Different conditions using coreTFIIH alone, or in combination with XPA, XPG, and XPA + XPG are labeled below the plot along with the mean and standard deviation (SD) of the Gaussian distribution to which the data points were fitted. Each dot in the plot represents the rate of a single quenching phase (magenta) that directly preceded the unwinding phase (cyan). After donor loss, oscillation of the FRET E signal between 0 and 1 in the bottom panel reflects trace emissions of Cy5 which periodically cross the detection threshold. These trace emissions are due to a combination of factors such as auto florescence and spectral overlap of the donor and acceptor, and channel leakage. (D) Plot of the number of quenching events that precede an unwinding event. Each dot represents the number of events for a separate trace. N represents the number of dots in each lane of the plots in panels C and D. The mean is indicated with black dots, whereas SD is shown with error bars. The mean, SD values, and the respective data points in the plot are color-coded for easier comparison. See also Supplementary Figures S2 and S3.
In the absence of proteins, the donor and acceptor intensities remained relatively constant at a FRET value of ∼0.3 (Supplementary Figure S2A, B), whereas in their presence the aforementioned quenching and FRET changes were distinguishable (Figure 2Aiii, B). The rate of coreTFIIH translocation and unwinding was 20.7 ± 10.7 nt·s−1 and 12.3 ± 4.6 bp·s−1, respectively; these values are 2-fold greater compared to previous studies (43,44). The rate of coreTFIIH translocation changed modestly regardless of whether XPA alone, XPG alone, or XPG and XPA together were present (Figure 2C). More importantly, the small variances in the velocities observed in the smFRET measurements cannot explain the ∼4–12-fold stimulation by XPA and XPG observed in the bulk assays (Figure 1; Supplementary Figure S1).
XPG and XPA enhance switching from ssDNA translocation to dsDNA unwinding
The data from single-molecule analysis showed that the enhanced translocation rate of individual coreTFIIH molecules was not the cause of stimulation observed in bulk assays. However, bulk assays represent an ensemble activity of individual rates of ssDNA translocation, dsDNA unwinding, and their respective efficiencies. This makes bulk-derived rates inadequate to unravel the mechanistic basis of the observed stimulation. Pertinent to the latter, the single-molecule trajectories revealed that there were multiple quenching phase events per single unwinding event, suggesting that frequent abortion of translocation occurred at the ss/ds junction (Figure 2B). We observed complete unwinding on our functionalized surface by the end of the ∼7 min acquisition period in all conditions (Supplementary Figure S2D), indicating that complete unwinding was achieved with time. Moreover, the difference in the number of traces where unwinding was achieved in the first 20 s, an estimate of the initial unwinding rate in smFRET, reflected the degree of stimulation with XPA and XPG seen in bulk (Figure 1A; Supplementary Figure S2A). Therefore, the stimulation effects observed in the bulk analysis are possibly reflected in smFRET, as reported by a decrease in number of aborted events. Thus, the stimulation observed in the bulk experiments may be a consequence of change in the propensity of abortion when switching from ssDNA translocation to dsDNA unwinding at the ss/ds junction. Due to spatial and temporal resolution limits, and the inability to control transient interactions between DNA and protein particles during flow, we were unable to confidently interpret smaller fluctuations in the traces obtained. Therefore, we counted the number of full-amplitude (100% to 0% intensity) quenching events before a single unwinding event (Figure 2B; Supplementary Figure S3). The number of non-processive quenching (NPQ) events was ∼8 on average, but decreased to ∼5 in the presence of XPA (Figure 2D). In the presence of XPG or XPG + XPA, the number of NPQ events was further decreased and coreTFIIH required ∼1.5 translocation attempts on average before commitment to unwinding. In remarkable contrast to the abortive nature of the quenching events (ssDNA translocation), we were unable to reliably identify a single trace where dsDNA unwinding was initiated and subsequently aborted, suggesting that unwinding is a committed step. Previously, partial unwinding has been observed (43,44), but differences in the results obtained may arise due to the presence of lower ATP levels, lower temperature, and performing the experiments with XPD alone, in contrast with the 7-subunit coreTFIIH examined herein.
Thus, coreTFIIH on its own cannot unwind dsDNA efficiently in most instances. In several cases, coreTFIIH appears to dissociate on encountering the ss/ds junction, as indicated by the sharp recovery of Cy5 fluorescence on reaching 0% intensity (Figure 2B, quenching phases). Therefore, efficient 5′ to 3′ unwinding during NER may be strongly licensed by the simultaneous presence of coreTFIIH and XPG, and to some extent by the presence of XPA.
ATP hydrolysis controls the unwinding-to-cleavage switch in coreTFIIH-XPG
As XPG stimulates coreTFIIH unwinding, it is possible that coreTFIIH stimulates XPG incision. We next tested for XPG incision activity in the presence of coreTFIIH on the 40ss/18ds substrate. We resolved the unwinding (18 nt ssDNA) and 5′ overhang cleavage (17nt/18 nt dsDNA) products on a native gel after bulk cleavage assays. In the presence of ATP, equimolar concentrations of XPG stimulated the helicase activity of coreTFIIH (lane 3 versus lane 8, Figure 3A), consistent with the results of our fluorescence-based helicase assays (Figure 1). In the presence of XPG alone, we observed a small but detectable amount of dsDNA product that was consistent with 5′ nuclease incision of the substrate (lanes 5–6; Figure 3A). Interestingly, in the absence of ATP, coreTFIIH strongly stimulated XPG cleavage of the 5′ overhang (lanes 5 and 7; Figure 3A). In the presence of coreTFIIH and ATP, XPG incision activity was absent, and the unwinding product alone was detected (lane 8; Figure 3A). These findings indicated that the coreTFIIH-XPG complex has an ATP switch that controls efficient unwinding or cleavage. To test for ATP-hydrolysis dependency of the switch, we used the non-hydrolyzable ATP analog AMPcPP instead of ATP. Our results showed that coreTFIIH enhanced cleavage activity similar to no ATP conditions (lane 9; Figure 3A).
Figure 3.
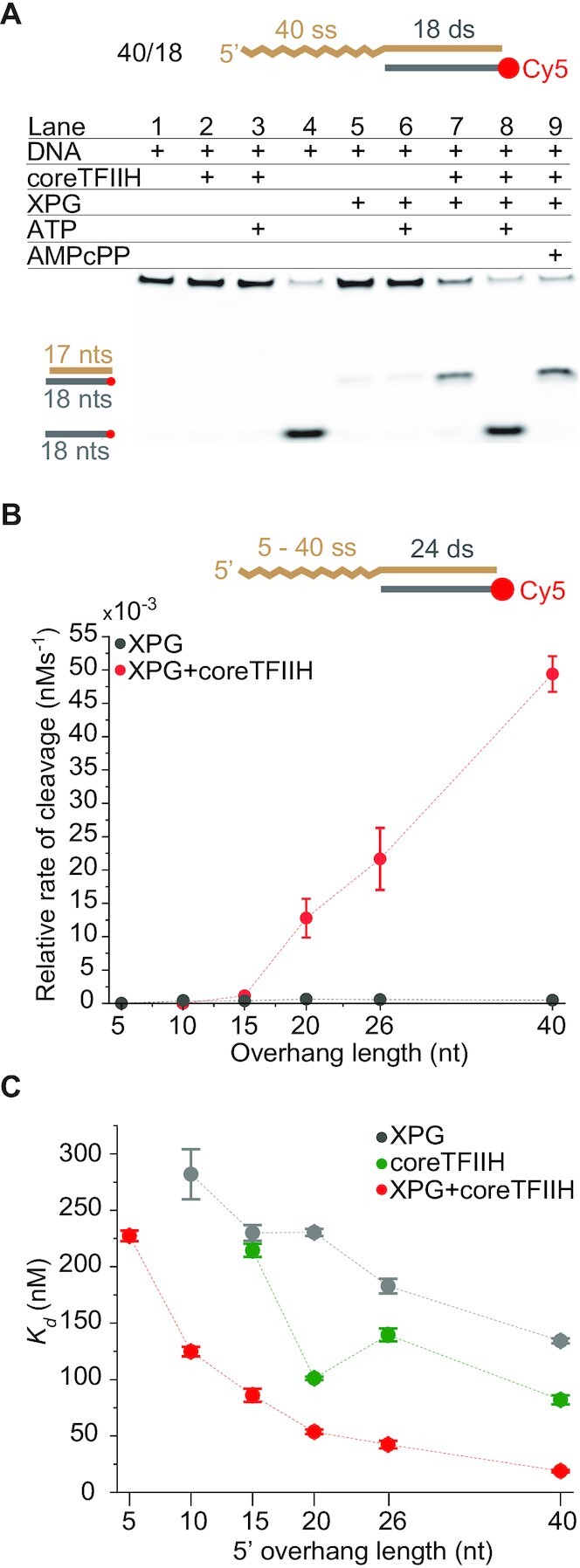
Mutually exclusive regulation of coreTFIIH and XPG is mediated by ssDNA length. (A) The unwinding or cleavage outcomes were assessed via 2 min incubation bulk reactions evaluated using 10% TBE gels. The presence of a component is indicated with a plus sign. Lane 4 contains DNA alone heated for 10 min at 95°C before gel loading. (B) A plot for the relative cleavage rate versus the 5′ overhang length. The rates were obtained from PAGE bulk cleavage assay results. (C) A plot of Kd values obtained from EMSA with coreTFIIH, XPG, and coreTFIIH + XPG for different 5′ overhang lengths. For panels A and B, the concentration of each protein and DNA was 400 and 5 nM, respectively. The data points in the panels represent the mean of three independent experiments and the error bars represent the standard error of the mean (SEM). See also Supplementary Figures S4 and S5.
Next, we asked whether XPG stimulation depends on the 5′ overhang length in a manner similar to the coreTFIIH unwinding activity. We incubated XPG with coreTFIIH under conditions that favor incision, i.e. without ATP. As shown in Figure 1, the helicase activity of coreTFIIH + XPG was greatly reduced on short overhangs compared to longer overhangs (Figure 1B). Therefore, we evaluated the XPG cleavage rate on substrates with 5, 10, 15, 20, 26 and 40 nt 5′ overhangs. For the XPG-only control, the rate of product generation showed a weak correlation with overhang length (Supplementary Figure S4A). However, for the coreTFIIH + XPG condition, there was a strong positive correlation between incision activity and overhang length (Figure 3B). In the presence of coreTFIIH, stimulation of the XPG incision activity was not observed for the 5 and 10 nt substrates, and was only 3-fold for the 15 nt substrate (Figure 3B; Supplementary Figure S4A). For overhangs of 20, 26 and 40 nt, the incision rate was stimulated ∼21-, ∼38- and ∼103-fold, respectively. Altogether, the data indicate that coreTFIIH regulates the relative cleavage of XPG by sensing the length of ssDNA.
Thus, binding of coreTFIIH and specifically its XPD subunit to a stretch of ∼20 nt of ssDNA is essential for coordinated XPD-XPG interaction, which leads to enhanced functionality of both enzymes. Importantly, we obtained ∼100% of the cleaved product within 10 min using coreTFIIH and XPG (Supplementary Figure S4D). This rate is much faster than XPG-dependent cleavage of the splayed arm (Y-substrate) or the bubble substrates observed under comparable conditions (46,49). Additionally, although the unwinding rate for coreTFIIH-XPG reached a peak at 20–26 nt overhangs, the cleavage peak was at 40 nt. The extra length may enable increased flexibility at the ss/ds junction while coreTFIIH is still holding the ssDNA. This consideration suggests that protein-protein interactions are key for XPG activation.
XPG and coreTFIIH show improved binding to overhang substrates when present in tandem
Reportedly (46,50), XPG alone does not engage its 5′ overhang substrate effectively. Previous studies report that no binding occurs in the absence of the 3′ overhang (50). To determine the dissociation constant (Kd), we measured XPG activity using higher concentrations than previously tested (46) (Figure 3C; Supplementary Figure S5). Interestingly, the Kd values of XPG decrease with an increase in ssDNA overhang length, as analogously observed for DNA bubbles (49). The 40 nt overhang substrate had the lowest Kd at ∼134 nM. The same trend was observed with coreTFIIH except that the Kd values were lower than with XPG for all substrates. The lowest Kd of ∼82 nM for coreTFIIH was obtained with the 40 nt overhang. Compared to the individual proteins, the Kd values with the coreTFIIH-XPG complex were decreased significantly at ∼19 nM for the 40 nt overhang. In fact, The Kd values for all overhang lengths showed a 2–7-fold decrease, indicating that the coreTFIIH-XPG complex bound all overhang lengths with higher efficiency than coreTFIIH or XPG alone.
The lower Kd values of coreTFIIH-XPG compared to coreTFIIH or XPG alone may be explained by the smFRET results. coreTFIIH alone shows a fast off rate at the ss/ds junction. With XPG, coreTFIIH unwinds efficiently, indicating more stable binding and a slower off rate at the ss/ds junction. The ∼4–12-fold stimulation of the unwinding rate (Figure 1; Supplementary Figure S1) is modestly greater than the ∼2–7-fold stimulation of binding efficiency (Figure 3C; Supplementary Figure S5). This result suggests that XPG stimulates coreTFIIH unwinding by enhancing both the binding to the DNA substrate and the transitioning from ssDNA translocation to dsDNA unwinding at the ss/ds interface. The ∼100-fold increase in the relative XPG cleavage rate, relative to the 7-fold decrease in Kd, suggests a direct post-binding stimulation of XPG catalytic activity by coreTFIIH.
Damage encounter signals an unwinding-to-cleavage switch in coreTFIIH-XPG
The stimulation of XPG by coreTFIIH in the absence of ATP implies the existence of a mechanism that regulates the second incision by XPG during NER. However, these findings have limited relevance under NER conditions in vivo where ATP is always present. Therefore, we reasoned that coreTFIIH stalling and/or disruption of the ATP hydrolysis cycle acts as a regulator of the mutually exclusive functions of coreTFIIH and XPG. Notably, the XPD ATPase activity is disrupted when the lesion becomes sterically stuck between the FeS and Arch domains (51,52). Thus, the disruption of coreTFIIH conformational changes by a bulky lesion may serve as the atomistical trigger for XPG incision.
To test this model, we designed two substrates with a damage-mimicking cholesterol moiety in the ssDNA overhang at positions 4 (Chol4) or 11 (Chol11) relative to the ss/ds junction (Figure 4A; Supplementary Figure S4C). The two different lesion locations helped evaluate the stringency effect of ssDNA length that spans the lesion and the ss/ds junction. As expected, both substrates showed no unwinding due to the damage, despite the presence of ATP (Figure 4A; Supplementary Figure S4C). Importantly, coreTFIIH addition stimulated XPG cleavage for both substrates close to the ss/ds junction. Therefore, we quantified the relative cleavage rates on the aforementioned substrates. Cleavage rates of Chol4 and Chol11 in the presence of coreTFIIH were ∼122- and ∼94-fold faster than without coreTFIIH, respectively. In the presence of ATP, the cleavage rates of coreTFIIH-XPG were increased to ∼640- and ∼715-fold for Chol4 and Chol11, respectively, relative to the condition without coreTFIIH (Figure 4B, Supplementary Figure S4A, B). Hence, in the presence of DNA damage and ATP, XPG alone displayed the lowest cleavage activity, while the addition of both coreTFIIH and XPG together displayed the highest cleavage activity. The aforementioned effects were comparable in Chol4 and Chol11 suggesting that the footprint of the coreTFIIH-XPG complex along the ssDNA between the lesion and the duplex can vary significantly.
Figure 4.
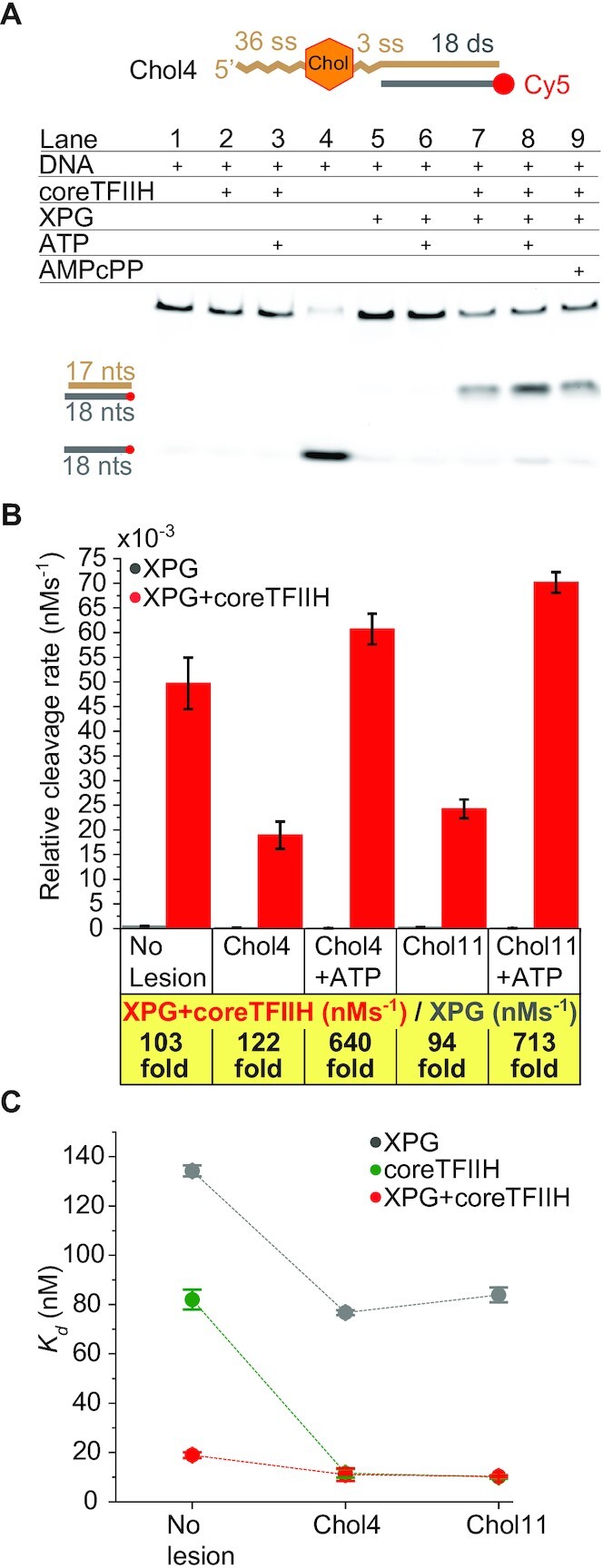
Lesion recognition induces the unwinding-to-cleavage switching of coreTFIIH-XPG. (A) The presence of a component is indicated with a plus sign. Lane 4 contains DNA heated for 10 min at 95°C before gel loading. The DNA substrate contains a cholesterol moiety at position 4 in the ssDNA overhang. The concentration of each protein and DNA was 400 and 5 nM, respectively. (B) Comparison of the relative rates of cleavage for the lesion-free 40ss/18ds substrate and with cholesterol moieties at positions 4 or 11 on the ssDNA overhang, starting from the ss/ds junction. A comparison of Chol 4 and Chol 11 in the presence of 1 mM ATP is shown. The nearest-integer fold difference of the rates using XPG or coreTFIIH + XPG is provided below the graph. (C) Comparison of Kd values for the lesion-free and lesion-bearing substrates with XPG, coreTFIIH and coreTFIIH + XPG. The data points represent the mean values from three different experiments, and the error bars represent the standard error of the mean (SEM). See also Supplementary Figures S4 and S5.
Altogether, the presence of ATP and a lesion on a long ssDNA overhang can suppress the incision activity by XPG alone. However, in the presence of coreTFIIH, the same conditions result in strong stimulation of XPG incision. This co-dependence prompted us to re-check the binding affinity of our complex to the respective substrates. Similar to the lesion-free substrate, coreTFIIH-XPG has a 7-fold/8-fold lower Kd value for Chol4/Chol11 than XPG alone. CoreTFIIH provides the dominant DNA binding interface, as the Kd values for lesion-containing substrates for coreTFIIH alone and coreTFIIH-XPG are similar (Figure 4C; Supplementary Figure S5). Given the ∼94–122-fold stimulation in cleavage activity, but ∼7–8-fold stimulation of binding, the stimulation of XPG cleavage activity by coreTFIIH is likely caused mostly by post-binding processes.
DISCUSSION
Lesion scanning, repair licensing, and DNA incision steps are complex structural mechanisms required to control DDR for genome integrity, as observed for many nucleases (42,53,54) and for dsDNA break repair pathways (55–57). NER must similarly be strictly coordinated because of the toxicity and mutagenicity of its intermediates and the plurality of its protein components. This coordination requires sequential protein exchange, and the regulatory processes governing the pathway have remained unclear. We have combined single-molecule analysis and bulk cleavage assays to unveil key mechanisms that enable sequential regulation of NER by coupling TFIIH and XPG for bubble opening and the second incision.
Our results show that XPG decreased the number of abortive translocation events (iterative sharp recovery of Cy5 fluorescence upon reaching 0% intensity) by coreTFIIH and requires a 5′ overhang of more than 15 nt (Figure 2D). These findings are critical to understanding the regulation of bubble opening by TFIIH. During incision, XPG interacts with 12 nt of the DNA duplex 3′ to the junction, and with 3 nt of 5′ ssDNA at the junction (50). Analogous to other members of the 5’ nuclease superfamily (FEN1, EXO1, GEN1) (45,58), specific XPG structures can act as a wedge at the ss/ds junction to facilitate a helicase-like strand separation, except as additionally motorized by the XPD ssDNA translocase activity. This XPG wedge in itself may explain how XPG reduces abortive translocation events and facilitates the transition from ssDNA translocation to dsDNA unwinding. The ssDNA occluded by XPD during unwinding is only 10–11 nt long (20,24). Additional dynamic interactions of XPD with DNA, without completely burying the DNA within the protein, can potentially increase the observable footprint of XPD-DNA interactions to ∼20 nt (43,44). This is the ssDNA length at which we observed the highest unwinding rates (Figure 1C). Therefore, the enhancement of function observed with coreTFIIH-XPG likely requires coreTFIIH to occlude a part of the ssDNA and wrap another part. Specifically, during NER, the excised oligonucleotide portion located 5′ of the damage site is 19–20 nt long (59). The occurrence of this length supports the argument that the 15–20 nucleotide bubble size is principally required for optimizing the formation of the critical enabling coreTFIIH-XPG complex.
Interestingly, in the presence of ATP, coreTFIIH-XPG interaction facilitates enhanced unwinding (Figure 3A), whereas it facilitates greatly enhanced cleavage in the absence of ATP. On testing how different lengths of ssDNA affect cleavage, we observed that short overhangs can modestly suppress cleavage, whereas overhangs of 15 nt and longer can greatly stimulate cleavage (Supplementary Figure S4A). These findings reveal an effective self-regulatory mechanism that senses the generation of a bubble of the correct size before cleavage can be performed by the coreTFIIH-XPG complex. This mechanism is supported by our results that show the stimulatory effects reach up to ∼700-fold in the presence of ATP on a damaged substrate (Figure 4B; Supplementary Figure S4B).
In the presence of a non-hydrolyzable ATP analog, the coreTFIIH-XPG complex cleaved a damage-free substrate almost as well as a damage-bearing substrate in the presence of ATP (Figure 4A; Supplementary Figure S4C). Therefore, damage encounter per se does not enable the stimulation of cleavage. However, stalling or disruption of ATP hydrolysis can trigger efficient incision. XPD-AMPcPP may have a conformation that resembles damage-bound XPD, because ATP-analogs are bound with slight structural variations compared to ATP (60). We cannot exclude the possibility that a disrupted ATP hydrolysis cycle of XPB may also act as a trigger of XPG incision. Such a scenario is however harder to envision as XPB is not directly involved in probing the DNA for the presence of a damage. Therefore, a damage-induced disruption of the ATP hydrolysis cycle is likely to involve XPD and not XPB. Furthermore, the reported interaction of XPD and XPG would support a direct incision trigger by XPD-XPG that is likely more efficient and better regulated than an indirect XPB-induced incision trigger. Thus, we propose a novel and crucial mechanistic switch in NER involving a nuclease (XPG) that supports a helicase in DNA unwinding, and a helicase (XPD) that supports a nuclease for cleavage activity. The underlying molecular characteristics of this switch were identified using singe-molecule experiments interpreted in the context of structural complexes. Given the crucial role of XPG incision in NER and in preventing increased DDR, we evaluated the potential molecular interactions that can enforce XPG incision regulation.
In particular, we evaluated three scenarios in which cleavage by coreTFIIH-XPG can occur: (i) a lesion-free substrate without ATP, (ii) a lesion-bearing substrate with added ATP and (iii) a lesion-free substrate with added AMPcPP (Figure 4; Supplementary Figure S4). Interestingly, the incision placement was conserved in all three cases. Therefore, the XPG cut position is determined by the duplex portion of all three substrates which is identical among them. During incision, 12 of the 15 nucleotides that interact with XPG are found within the duplex portion (46,50). The results of the smFRET analysis showed that coreTFIIH preferentially binds at the 5′ end of ssDNA, which supports and extends prior observations for XPD alone (43,44,52). This may be because ssDNA is condensed, offering limited access to nucleotides further into the ssDNA overhang, or due to intrinsic affinity of the XPD HD2 domain towards the 5′ end of DNA. The lack of differences in the results obtained with Chol4 and Chol11, and the absence of multiple incisions in the damage-free substrate, indicate the presence of a specific mechanism for DNA handling. The ssDNA between XPD and XPG may be potentially extruded in a loop or other compacted structures. Notably, loop extrusion enables direct protein-protein interactions when handling distant portions of DNA. An extrusion mechanism would also be advantageous during NER where ssDNA generated behind XPB may be looped, allowing direct protein-protein communication at the 5′ and 3′ ends of the bubble. This particular hypothesis where bubble DNA is highly looped to allow proximity and communication of the 5’ and 3’ bubble ends is supported by a recent study of XPA and RPA(61). Therefore, we uncovered different probable and testable scenarios for the handling of the 5’ overhang DNA by coreTFIIH-XPG dependent on ATP availability (Figure 5A–C). The looped-out DNA may act as a structural hallmark that is necessary for XPG incision as it bends ssDNA at a sharp angle, relative to the dsDNA. This represents a structural feature of the substrate required for scissile phosphate positioning in the active site of XPG, and is critical for incision, as in other structure-specific 5′ nucleases (42,45,46,53,62).
Figure 5.

Structural models that rationalize coreTFIIH-XPG activity on the ss/ds junction based on existing structural information and our experimental results and analyses. Initial binding to the substrate proceeds via the 5′ end by coreTFIIH which has a higher affinity for the substrate compared to XPG. For simplicity, the XPD domain of coreTFIIH alone is represented (green) along with XPG (purple). XPD recruits XPG via DNA-mediated protein-protein interactions. The ssDNA between XPD and XPG is highly condensed or extruded to enable direct XPD-XPG interactions even if XPG is located at the duplex. (A) The absence of ATP precludes translocation which enables access and flexibility at the junction, resulting in incision. (B) In the presence of ATP, DNA is pulled through XPD by the action of ATP hydrolysis. The ATP-hydrolyzing/translocating XPD prevents incision by XPG. Once all of the ssDNA is translocated, the complex begins unwinding which proceeds to completion in the absence of a lesion. (C) In the presence of a lesion, the XPD-XPG complex translocates until stopped by the lesion. This stalling and the potential disruption of ATP-hydrolysis trigger XPG incision. (D) A model of XPB (blue), XPD (green), and XPG (purple) interactions at the opened bubble substrate. The non-translocating strand is shown when bound by XPB, and the rest is omitted as it would require Replication protein A (RPA) docking. The translocating strand is shown in gold and black. The two paths show two possibilities of looping DNA out (arrows), to enable efficient DNA handling and XPD-XPG interaction. The model showcases the sharp bending of the 3’ duplex by XPG which enables active site positioning while maintaining XPD-XPG contacts.
To evaluate the concept of DNA bending, we modeled the path of the DNA in a bubble structure with 25 unpaired nucleotides in a minimal system containing the XPB-XPD-XPG catalytic domain (Figure 5D), which was based on a cryoEM model of the coreTFIIH NER complex (24), the XPG crystal structure (46), the FEN1-substrate DNA crystal structure (42), and cross-linking data (24). The XPB-XPD-XPG model accounts for known specific structural requirements: (i) XPB positioning at the 5′ duplex, (ii) XPD positioning at the ssDNA and (iii) XPG at the 3′ duplex, with the active site of XPG located in a position to cut 1 nt into the duplex. In this complex, the wedge separating strands for XPD would serve as the hydrophobic wedge in XPG helix 2. Based on biochemical data (40,51,63), we predict that 6 nt of ssDNA may be located between XPG and the site where the lesion would be blocked from passing through XPD. Based on the DNA position and cross-linking between XPD and XPG (24), gateway helices (residues 78–126 and 733–759) above the active site of XPG make extensive contact with the XPD arch domain (residues 257–280 and 384–393). To account for the difference between the biochemically established size of the NER bubble (40,51,63) and the smaller footprint of ssDNA that passes through XPD, the ssDNA on the lesion strand must loop out. Such looping may occur without steric hindrance in two places: in the space between XPB and XPD (Loop A, Figure 5D), or between XPG and XPD (Loop B, Figure 5D). Furthermore, both potential ssDNA paths may occur during different stages of NER. The presence of a loop on the 5′ side of the NER bubble may facilitate incision by XPF/ERCC1. Conversely, the creation of a loop on the 3′ side of the NER bubble may enable incision by XPG. Thus, our model introduces testable hypotheses to rationalize the sequential nature of nuclease excision in NER. We also acknowledge the recently published model (61), which proposes looping of the undamaged strand that involves all nucleotides of the bubble, as opposed to the rather local looping we present in our model. Both models however posit the notion of a sharp bend at the 3’ end of the bubble, which could be a structural hallmark necessary for incision by XPG.
Interestingly, the invariability of the cut position has mechanistic implications, whether achieved by XPG alone or in tandem with coreTFIIH (Figure 3A). The data indicates that coreTFIIH-XPG always unwinds our substrate to completion in the absence of lesion, and that an actively ATP-hydrolyzing coreTFIIH prevents XPG cleavage. The unwinding-to-completion hypothesis is supported by our smFRET results as we did not identify any rezipping events. Yet, transient stalling and re-zipping after short unwinding events may occur, as the data lacked the temporal or spatial resolution to unambiguously identify rezipping events from inherent noise. We hypothesize that when XPG is bound to coreTFIIH, it becomes poised for incision but is barred by the movement of DNA and the ATPase lobes of XPD.
Different mechanisms of DNA entry pore ordering in XPD may depend on the presence of a normal versus a damaged nucleotide (64). Hence, one way of triggering XPG by XPD may involve XPG encountering two different XPD conformations that arise from whether XPD encounters a damage or not. Additionally, our findings indicate that AMPcPP triggers enhanced incision of XPG in the presence of coreTFIIH, suggesting that it is not damage-recognition per se, but rather a stalled XPD that triggers the cut. We and others have shown that in the 5’ nuclease superfamily, FEN1 and EXO1 have two substrate DNA binding modes, with initial binding with the scissile phosphodiester not positioned to be incised that is followed by the movement of the substrate DNA into a catalytically active position near the bound divalent cations upon validating its features (42,53,65–68). Perhaps when the DNA is being moved by coreTFIIH, the DNA is bound in the non-catalytic position. Only when DNA is no longer moving, can XPG distort the dsDNA to move the scissile phosphodiester into catalytic position. If so, in cells where ATP is present, XPG incision is suppressed to ensure incision only occurs when damage is encountered and XPD is stalled.
Thus, we have identified a unique mechanistic ensemble involving a helicase and a nuclease that promote mutual functioning in a sequential, mutually exclusive, and ssDNA length-dependent manner. This functional versatility is reminiscent of Dna2 which works as a helicase and nuclease (69), but is unique in the specific way it achieves either function. One reason for this uniqueness may relate to the fact that the same coreTFIIH-XPG complex may act in various cellular pathways. Given the emerging evidence of XPG involvement in chromatin looping, DNA demethylation, and TFIIH stabilization during transcription (70,71), elucidating the mechanistic nature of the coreTFIIH-XPG complex may have profound biological significance beyond NER.
DATA AVAILABILITY
No original code was written in the creation of this report. Any additional information required to reanalyze the data reported in this paper is available from Samir M. Hamdan (Samir.hamdan@kaust.edu.sa) upon request.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Goran Kokić and Dr Patrick Cramer for graciously providing a construct for core-TFIIH expression for comparison and benchmarking with our coreTFIIH construct during initial studies.
Author contributions: C.T. purified coreTFIIH. A.B. and M.T. purified XPG. A.B. purified the protocatechuate 3,4-dioxygenase. A.B. performed smFRET and bulk fluorescence experiments. A.B., G.Y., and L.A. performed the PAGE and EMSA experiments. A.B. and M.A.S. processed the smFRET data. J.Y., C.Y. and I.I. modeled the DNA bubble with XPB-XPD-XPG. A.B. and S.M.H. designed the research plan and wrote the article. I.I., S.T., and J.A.T. provided critical input for experimental design and data analysis, and contributed to the manuscript. All authors discussed the results and were involved in the editing of the manuscript.
Contributor Information
Amer Bralić, Bioscience Program, Biological and Environmental Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia.
Muhammad Tehseen, Bioscience Program, Biological and Environmental Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia.
Mohamed A Sobhy, Bioscience Program, Biological and Environmental Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia.
Chi-Lin Tsai, Department of Molecular and Cellular Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Lubna Alhudhali, Bioscience Program, Biological and Environmental Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia.
Gang Yi, Bioscience Program, Biological and Environmental Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia.
Jina Yu, Department of Chemistry, Georgia State University, Atlanta, GA 30302 USA; Center for Diagnostics and Therapeutics, Georgia State University, Atlanta, GA, 30302, USA.
Chunli Yan, Department of Chemistry, Georgia State University, Atlanta, GA 30302 USA; Center for Diagnostics and Therapeutics, Georgia State University, Atlanta, GA, 30302, USA.
Ivaylo Ivanov, Department of Chemistry, Georgia State University, Atlanta, GA 30302 USA; Center for Diagnostics and Therapeutics, Georgia State University, Atlanta, GA, 30302, USA.
Susan E Tsutakawa, Molecular Biophysics and Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA.
John A Tainer, Molecular Biophysics and Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA; Department of Molecular and Cellular Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA; Department of Cancer Biology, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Samir M Hamdan, Bioscience Program, Biological and Environmental Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
King Abdullah University of Science and Technology through Competitive Research Award [URF/1/3764–01-01 (CRG7) to S.M.H. and J.A.T.]; NCI [P01 CA092584 to J.A.T., S.T., I.I., in part]; NCI [R35 CA220430 to J.A.T.]; CPRIT [RP180813 to J.A.T.]; NIEHS [R01ES032786 to S.T., I.I.]; Robert A. Welch Chemistry Chair (to J.A.T.); NSF MCB (to I.I.) Funding for open access charges were provided by the King Abdullah University of Science and Technology through URF/1/3764–01-01 (CRG7).
Conflicts of interest statement. None declared.
REFERENCES
- 1. Reardon J.T., Sancar A.. Recognition and repair of the cyclobutane thymine dimer, a major cause of skin cancers, by the human excision nuclease. Genes Dev. 2003; 17:2539–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wakasugi M., Sancar A.. Assembly, subunit composition, and footprint of human DNA repair excision nuclease. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:6669–6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sancar A. DNA excision repair. Annu. Rev. Biochem. 1996; 65:43–81. [DOI] [PubMed] [Google Scholar]
- 4. Matsunaga T., Park C.H., Bessho T., Mu D., Sancar A.. Replication protein A confers structure-specific endonuclease activities to the XPF-ERCC1 and XPG subunits of human DNA repair excision nuclease. J. Biol. Chem. 1996; 271:11047–11050. [DOI] [PubMed] [Google Scholar]
- 5. Svoboda D.L., Taylor J.S., Hearst J.E., Sancar A.. DNA repair by eukaryotic nucleotide excision nuclease. Removal of thymine dimer and psoralen monoadduct by HeLa cell-free extract and of thymine dimer by Xenopus laevis oocytes. J. Biol. Chem. 1993; 268:1931–1936. [PubMed] [Google Scholar]
- 6. Huang J.C., Svoboda D.L., Reardon J.T., Sancar A.. Human nucleotide excision nuclease removes thymine dimers from DNA by incising the 22nd phosphodiester bond 5' and the 6th phosphodiester bond 3' to the photodimer. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:3664–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Evans E., Moggs J.G., Hwang J.R., Egly J.M., Wood R.D.. Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 1997; 16:6559–6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shivji M.K., Podust V.N., Hübscher U., Wood R.D.. Nucleotide excision repair DNA synthesis by DNA polymerase epsilon in the presence of PCNA, RFC, and RPA. Biochemistry. 1995; 34:5011–5017. [DOI] [PubMed] [Google Scholar]
- 9. Scharer O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013; 5:a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gillet L.C., Schärer O.D.. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev. 2006; 106:253–276. [DOI] [PubMed] [Google Scholar]
- 11. DiGiovanna J.J., Kraemer K.H.. Shining a light on xeroderma pigmentosum. J. Invest. Dermatol. 2012; 132:785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bukowska B., Karwowski B.T.. Actual state of knowledge in the field of diseases related with defective nucleotide excision repair. Life Sci. 2018; 195:6–18. [DOI] [PubMed] [Google Scholar]
- 13. Lehmann A.R. DNA repair-deficient diseases, xeroderma pigmentosum, cockayne syndrome and trichothiodystrophy. Biochimie. 2003; 85:1101–1111. [DOI] [PubMed] [Google Scholar]
- 14. Giannattasio M., Lazzaro F., Longhese M.P., Plevani P., Muzi-Falconi M.. Physical and functional interactions between nucleotide excision repair and DNA damage checkpoint. EMBO J. 2004; 23:429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marini F., Nardo T., Giannattasio M., Minuzzo M., Stefanini M., Plevani P., Muzi Falconi M.. DNA nucleotide excision repair-dependent signaling to checkpoint activation. Proc. Natl Acad. Sci. U.S.A. 2006; 103:17325–17330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marti T.M., Hefner E., Feeney L., Natale V., Cleaver J.E.. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc. Natl Acad. Sci. U.S.A. 2006; 103:9891–9896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Overmeer R.M., Moser J., Volker M., Kool H., Tomkinson A.E., van Zeeland A.A., Mullenders L.H., Fousteri M.. Replication protein a safeguards genome integrity by controlling NER incision events. J. Cell Biol. 2011; 192:401–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Volker M., Moné M.J., Karmakar P., van Hoffen A., Schul W., Vermeulen W., Hoeijmakers J.H., van Driel R., van Zeeland A.A., Mullenders L.H.. Sequential assembly of the nucleotide excision repair factors in vivo. Mol. Cell. 2001; 8:213–224. [DOI] [PubMed] [Google Scholar]
- 19. Araújo S.J., Tirode F., Coin F., Pospiech H., Syväoja J.E., Stucki M., Hübscher U., Egly J.-M., Wood R.D.. Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH, and modulation by CAK. Genes Dev. 2000; 14:349–359. [PMC free article] [PubMed] [Google Scholar]
- 20. Tsutakawa S.E., Tsai C.L., Yan C., Bralić A., Chazin W.J., Hamdan S.M., Schärer O.D., Ivanov I., Tainer J.A.. Envisioning how the prototypic molecular machine TFIIH functions in transcription initiation and DNA repair. DNA Repair (Amst.). 2020; 96:102972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tapias A., Auriol J., Forget D., Enzlin J.H., Schärer O.D., Coin F., Coulombe B., Egly J.M.. Ordered conformational changes in damaged DNA induced by nucleotide excision repair factors. J. Biol. Chem. 2004; 279:19074–19083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fishburn J., Tomko E., Galburt E., Hahn S.. Double-stranded DNA translocase activity of transcription factor TFIIH and the mechanism of RNA polymerase II open complex formation. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:3961–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Coin F., Oksenych V., Mocquet V., Groh S., Blattner C., Egly J.M.. Nucleotide excision repair driven by the dissociation of CAK from TFIIH. Mol. Cell. 2008; 31:9–20. [DOI] [PubMed] [Google Scholar]
- 24. Kokic G., Chernev A., Tegunov D., Dienemann C., Urlaub H., Cramer P.. Structural basis of TFIIH activation for nucleotide excision repair. Nat. Commun. 2019; 10:2885–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van Eeuwen T., Shim Y., Kim H.J., Zhao T., Basu S., Garcia B.A., Kaplan C.D., Min J.-H., Murakami K.. Cryo-EM structure of TFIIH/Rad4–Rad23–Rad33 in damaged DNA opening in nucleotide excision repair. Nat. Commun. 2021; 12:3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Staresincic L., Fagbemi A.F., Enzlin J.H., Gourdin A.M., Wijgers N., Dunand-Sauthier I., Giglia-Mari G., Clarkson S.G., Vermeulen W., Schärer O.D.. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 2009; 28:1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ito S., Kuraoka I., Chymkowitch P., Compe E., Takedachi A., Ishigami C., Coin F., Egly J.M., Tanaka K.. XPG stabilizes TFIIH, allowing transactivation of nuclear receptors: implications for cockayne syndrome in XP-G/CS patients. Mol. Cell. 2007; 26:231–243. [DOI] [PubMed] [Google Scholar]
- 28. Kappenberger J., Koelmel W., Schoenwetter E., Scheuer T., Woerner J., Kuper J., Kisker C.. How to limit the speed of a motor: the intricate regulation of the XPB ATPase and translocase in TFIIH. Nucleic Acids Res. 2020; 48:12282–12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sobhy M.A., Bralić A., Raducanu V.-S., Tehseen M., Ouyang Y., Takahashi M., Rashid F., Zaher M.S., Hamdan S.M.. Single-Molecule förster resonance energy transfer methods for real-time investigation of the holliday junction resolution by GEN1. J. Vis. Exp. 2019; 151:e60045. [DOI] [PubMed] [Google Scholar]
- 30. Gradia S.D., Ishida J.P., Tsai M.S., Jeans C., Tainer J.A., Fuss J.O.. MacroBac: new technologies for robust and efficient large-scale production of recombinant multiprotein complexes. Methods Enzymol. 2017; 592:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kirchhofer A., Helma J., Schmidthals K., Frauer C., Cui S., Karcher A., Pellis M., Muyldermans S., Casas-Delucchi C.S., Cardoso M.C.et al.. Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol. 2010; 17:133–138. [DOI] [PubMed] [Google Scholar]
- 32. Gibson D.G., Smith H.O., Hutchison C.A.,3rd, Venter J.C., Merryman C.. Chemical synthesis of the mouse mitochondrial genome. Nat. Methods. 2010; 7:901–903. [DOI] [PubMed] [Google Scholar]
- 33. Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A.,3rd, Smith H.O.. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009; 6:343–345. [DOI] [PubMed] [Google Scholar]
- 34. Sobhy M.A., Elshenawy M., Takahashi M., Whitman B., Walter N., Hamdan S.. Versatile single-molecule multi-color excitation and detection fluorescence setup for studying biomolecular dynamics. Rev. Sci. Instrum. 2011; 82:113702. [DOI] [PubMed] [Google Scholar]
- 35. Holden S.J., Uphoff S., Hohlbein J., Yadin D., Le Reste L., Britton O.J., Kapanidis A.N.. Defining the limits of single-molecule FRET resolution in TIRF microscopy. Biophys. J. 2010; 99:3102–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bronson J.E., Fei J., Hofman J.M., Gonzalez R.L. Jr, Wiggins C.H. Learning rates and states from biophysical time series: a bayesian approach to model selection and single-molecule FRET data. Biophys. J. 2009; 97:3196–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Preus S., Noer S.L., Hildebrandt L.L., Gudnason D., Birkedal V.. iSMS: single-molecule FRET microscopy software. Nat. Methods. 2015; 12:593–594. [DOI] [PubMed] [Google Scholar]
- 38. Varadi M., Anyango S., Deshpande M., Nair S., Natassia C., Yordanova G., Yuan D., Stroe O., Wood G., Laydon A.et al.. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022; 50:D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A.et al.. Highly accurate protein structure prediction with alphafold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Evans E., Fellows J., Coffer A., Wood R.D.. Open complex formation around a lesion during nucleotide excision repair provides a structure for cleavage by human XPG protein. EMBO J. 1997; 16:625–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Constantinou A., Gunz D., Evans E., Lalle P., Bates P.A., Wood R.D., Clarkson S.G.. Conserved residues of human XPG protein important for nuclease activity and function in nucleotide excision repair. J. Biol. Chem. 1999; 274:5637–5648. [DOI] [PubMed] [Google Scholar]
- 42. Tsutakawa S.E., Thompson M.J., Arvai A.S., Neil A.J., Shaw S.J., Algasaier S.I., Kim J.C., Finger L.D., Jardine E., Gotham V.J.B.et al.. Phosphate steering by flap endonuclease 1 promotes 5′-flap specificity and incision to prevent genome instability. Nat. Commun. 2017; 8:15855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Spies M. Two steps forward, one step back: determining XPD helicase mechanism by single-molecule fluorescence and high-resolution optical tweezers. DNA Repair (Amst.). 2014; 20:58–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Qi Z., Pugh R.A., Spies M., Chemla Y.R.. Sequence-dependent base pair stepping dynamics in XPD helicase unwinding. Elife. 2013; 2:e00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Grasby J.A., Finger L.D., Tsutakawa S.E., Atack J.M., Tainer J.A.. Unpairing and gating: sequence-independent substrate recognition by FEN superfamily nucleases. Trends Biochem. Sci. 2012; 37:74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tsutakawa S.E., Sarker A.H., Ng C., Arvai A.S., Shin D.S., Shih B., Jiang S., Thwin A.C., Tsai M.-S., Willcox A.et al.. Human XPG nuclease structure, assembly, and activities with insights for neurodegeneration and cancer from pathogenic mutations. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:14127–14138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Finkelstein I.J., Greene E.C.. XPD helicase speeds through a molecular traffic jam. Mol. Cell. 2009; 35:549–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Honda M., Park J., Pugh R.A., Ha T., Spies M.. Single-molecule analysis reveals differential effect of ssDNA-binding proteins on DNA translocation by XPD helicase. Mol. Cell. 2009; 35:694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sarker A.H., Tsutakawa S.E., Kostek S., Ng C., Shin D.S., Peris M., Campeau E., Tainer J.A., Nogales E., Cooper P.K.. Recognition of RNA polymerase II and transcription bubbles by XPG, CSB, and TFIIH: insights for transcription-coupled repair and cockayne syndrome. Mol. Cell. 2005; 20:187–198. [DOI] [PubMed] [Google Scholar]
- 50. Hohl M., Thorel F., Clarkson S.G., Schärer O.D.. Structural determinants for substrate binding and catalysis by the structure-specific endonuclease XPG. J. Biol. Chem. 2003; 278:19500–19508. [DOI] [PubMed] [Google Scholar]
- 51. Li C.L., Golebiowski F.M., Onishi Y., Samara N.L., Sugasawa K., Yang W.. Tripartite DNA lesion recognition and verification by XPC, TFIIH, and XPA in nucleotide excision repair. Mol. Cell. 2015; 59:1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cheng K., Wigley D.B.. DNA translocation mechanism of an XPD family helicase. Elife. 2018; 7:e42400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rashid F., Harris P.D., Zaher M.S., Sobhy M.A., Joudeh L.I., Yan C., Piwonski H., Tsutakawa S.E., Ivanov I., Tainer J.A.et al.. Single-molecule FRET unveils induced-fit mechanism for substrate selectivity in flap endonuclease 1. Elife. 2017; 6:e21884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tsutakawa S.E., Lafrance-Vanasse J., Tainer J.A.. The cutting edges in DNA repair, licensing, and fidelity: DNA and RNA repair nucleases sculpt DNA to measure twice, cut once. DNA Repair (Amst.). 2014; 19:95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen S., Lee L., Naila T., Fishbain S., Wang A., Tomkinson A.E., Lees-Miller S.P., He Y.. Structural basis of long-range to short-range synaptic transition in NHEJ. Nature. 2021; 593:294–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Syed A., Tainer J.A.. The MRE11–RAD50–NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu. Rev. Biochem. 2018; 87:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ye Z., Xu S., Shi Y., Bacolla A., Syed A., Moiani D., Tsai C.-L., Shen Q., Peng G., Leonard P.G.. GRB2 enforces homology-directed repair initiation by MRE11. Sci. Adv. 2021; 7:eabe9254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Finger L.D., Atack J.M., Tsutakawa S., Classen S., Tainer J., Grasby J., Shen B.. The wonders of flap endonucleases: structure, function, mechanism and regulation. Subcell. Biochem. 2012; 62:301–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li W., Adebali O., Yang Y., Selby C.P., Sancar A.. Single-nucleotide resolution dynamic repair maps of UV damage in Saccharomyces cerevisiae genome. Proc. Natl. Acad. Sci. 2018; 115:E3408–E3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lacabanne D., Wiegand T., Wili N., Kozlova M.I., Cadalbert R., Klose D., Mulkidjanian A.Y., Meier B.H., Böckmann A.. ATP analogues for structural investigations: case studies of a DnaB helicase and an ABC transporter. Molecules. 2020; 25:5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kim M., Kim H.-S., D’Souza A., Gallagher K., Jeong E., Topolska-Wós A., Ogorodnik Le Meur K., Tsai C.-L., Tsai M.-S., Kee M.et al.. Two interaction surfaces between XPA and RPA organize the preincision complex in nucleotide excision repair. Proc. Natl. Acad. Sci. 2022; 119:e2207408119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zaher M.S., Rashid F., Song B., Joudeh L.I., Sobhy M.A., Tehseen M., Hingorani M.M., Hamdan S.M.. Missed cleavage opportunities by FEN1 lead to okazaki fragment maturation via the long-flap pathway. Nucleic Acids Res. 2018; 46:2956–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mathieu N., Kaczmarek N., Ruthemann P., Luch A., Naegeli H.. DNA quality control by a lesion sensor pocket of the xeroderma pigmentosum group d helicase subunit of TFIIH. Curr. Biol. 2013; 23:204–212. [DOI] [PubMed] [Google Scholar]
- 64. Fu I., Mu H., Geacintov N.E., Broyde S.. Mechanism of lesion verification by the human XPD helicase in nucleotide excision repair. Nucleic. Acids. Res. 2022; 50:6837–6853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shi Y., Hellinga H.W., Beese L.S.. Interplay of catalysis, fidelity, threading, and processivity in the exo- and endonucleolytic reactions of human exonuclease I. Proc. Natl Acad. Sci. USA. 2017; 114:6010–6015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Orans J., McSweeney E.A., Iyer R.R., Hast M.A., Hellinga H.W., Modrich P., Beese L.S.. Structures of human exonuclease 1 DNA complexes suggest a unified mechanism for nuclease family. Cell. 2011; 145:212–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tsutakawa S.E., Classen S., Chapados B.R., Arvai A.S., Finger L.D., Guenther G., Tomlinson C.G., Thompson P., Sarker A.H., Shen B.et al.. Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell. 2011; 145:198–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liu R., Qiu J., Finger L.D., Zheng L., Shen B.. The DNA–protein interaction modes of FEN-1 with gap substrates and their implication in preventing duplication mutations. Nucleic Acids Res. 2006; 34:1772–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zheng L., Meng Y., Campbell J.L., Shen B.. Multiple roles of DNA2 nuclease/helicase in DNA metabolism, genome stability and human diseases. Nucleic Acids Res. 2019; 48:16–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Le May N., Mota-Fernandes D., Vélez-Cruz R., Iltis I., Biard D., Egly J.M. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol. Cell. 2010; 38:54–66. [DOI] [PubMed] [Google Scholar]
- 71. Le May N., Fradin D., Iltis I., Bougnères P., Egly J.M. XPG and XPF endonucleases trigger chromatin looping and DNA demethylation for accurate expression of activated genes. Mol. Cell. 2012; 47:622–632. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No original code was written in the creation of this report. Any additional information required to reanalyze the data reported in this paper is available from Samir M. Hamdan (Samir.hamdan@kaust.edu.sa) upon request.