


Abstract
Microfold (M) cells reside in the intestinal epithelium of Peyer's patches (PP). Their unique ability to take up and transport antigens from the intestinal lumen to the underlying lymphoid tissue is key in the regulation of the gut-associated immune response. Here, we applied a multi-omics approach to investigate the molecular mechanisms that drive M cell differentiation in mouse small intestinal organoids. We generated a comprehensive profile of chromatin accessibility changes and transcription factor dynamics during in vitro M cell differentiation, allowing us to uncover numerous cell type-specific regulatory elements and associated transcription factors. By using single-cell RNA sequencing, we identified an enterocyte and M cell precursor population. We used our newly developed computational tool SCEPIA to link precursor cell-specific gene expression to transcription factor motif activity in cis-regulatory elements, uncovering high expression of and motif activity for the transcription factor ONECUT2. Subsequent in vitro and in vivo perturbation experiments revealed that ONECUT2 acts downstream of the RANK/RANKL signalling axis to support enterocyte differentiation, thereby restricting M cell lineage specification. This study sheds new light on the mechanism regulating cell fate balance in the PP, and it provides a powerful blueprint for investigation of cell fate switches in the intestinal epithelium.
Graphical Abstract
Graphical Abstract.
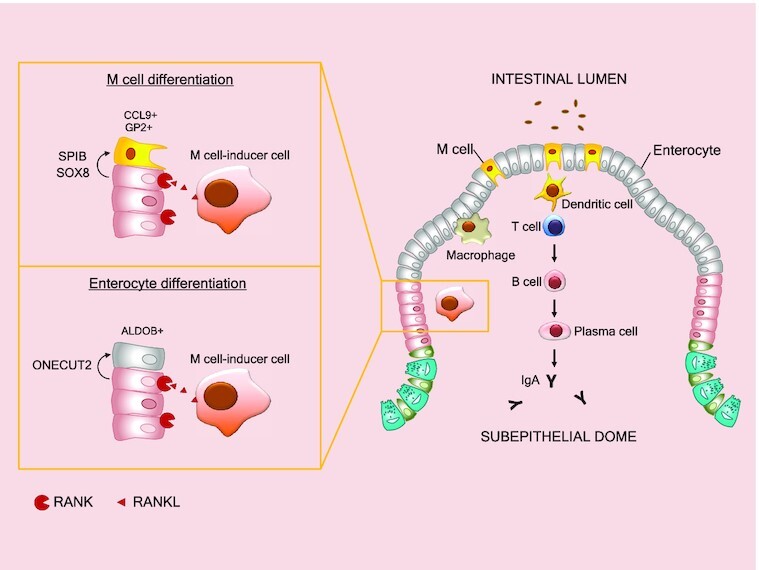
Upon RANKL stimulation enterocytes and microfold cells originate from a common progenitor cell population. Transcription factors SPIB and SOX drive microfold cell differentiation while ONECUT2 promotes enterocyte cell differentiation by inducing transcriptional activation of Hnf4g.
INTRODUCTION
The Peyer's patches (PP) are focal points within the small intestine, constituted by an organized lymphoid follicle and an overlying follicle-associated epithelium (FAE) (1,2). The FAE is a one-cell-thick layer composed mainly of enterocytes and specialized epithelial cells called microfold or M cells (1). M cells are important for enteral uptake and transport of various commensal and pathogenic microorganisms across the epithelial cell layer and to the subepithelial dome (3–6), where immune cells take on a defensive or tolerogenic response (1,7). Like all other cell lineages in the small intestine, M cells derive from intestinal stem cells (ISC) residing in the bottom of the intestinal crypts (8). However, only a small percentage of ISC commit to the M cell lineage, making M cells one of the least abundant cell types in the small intestine (9,10). The polarization of M cells to PP and their low abundance reflect tightly regulated mechanisms restricting the commitment of progenitor cells into M cells, but it is precisely the low number of M cells that makes it difficult to study these mechanisms in vivo.
Intestinal organoids or ‘miniguts’ are in vitro models capable of self-organizing into epithelial structures that molecularly, phenotypically and functionally resemble in vivo intestinal tissue (9,11). Minor modifications of the intestinal organoid culture medium can generate cell type-enriched organoids that can be used to decipher the molecular mechanisms that drive ISC fate (12,13). The receptor activator of nuclear factor κB (RANK) and its ligand (RANKL) were originally described for their key role in the induction of M cell differentiation (14,15). The addition of recombinant RANKL to the conventional intestinal organoid culture medium induces the differentiation of functional M cells in vitro (8), allowing the study of the fundamental signalling regulating M cell lineage specification.
In this study, we used a multi-omics framework on RANKL-treated mouse small intestinal organoids to define the gene-regulatory mechanisms underlying M cell differentiation. Changes in the morphology of organoids upon RANKL treatment were correlated to differences in protein levels, gene expression, histone modification and chromatin accessibility. By integrating bulk and single-cell RNA-sequencing data we were able to untangle complex gene-regulatory networks contributing to the M cell lineage specification in the intestine. Our findings led to the identification of an enterocyte and M cell precursor cluster, where the one cut domain family member 2 (ONECUT2) promotes enterocyte cell differentiation by inducing transcriptional activation of Hnf4g in a RANKL-signalling dependent manner, which in turns restricts differentiation of M cells in organoids and in the mouse small intestine.
MATERIALS AND METHODS
In vivo CSRM617 experiments
For the mouse experiments, all experimental protocols and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Cedars-Sinai Medical Center and the Comparative Medicine Department. All relevant ethical regulations, standards, and norms were rigorously adhered to. Both male and female C57BL/6 mice (5 weeks old) were purchased from Charles River. After arriving, the mice were quarantined for one week prior to the study. Four treatment groups were designed for the in vivo experiments with the ONECUT2 inhibitor CSRM617 (Supplementary Figure S3D). Each treatment group contained three male and three female C57BL/6 mice of 6–6.5 weeks of age. Briefly, six mice received 50 mg kg−1 CSRM617 (Cedars Sinai Medical Center) for 7 days and six mice for 14 days. A recovery group consisted of six mice treated with 50 mg kg−1 CSRM617 (Cedars Sinai Medical Center) for 7 days followed by 7 days treatment with phosphate buffered saline (PBS) and 0.2% dimethyl sulfoxide (DMSO, Sigma-Aldrich). Lastly, a control group treated with PBS and 0.2% DMSO for 7 days was included. All treatments were performed by intraperitoneal injection. Body weight was tracked 3 times per week. For ELISA experiments, faecal samples were collected 30 min before sacrifice of the mice. A total of 100 mg of mice faeces were dissolved in 1 ml of 1× PBS with 1% protease inhibitors (Thermo Scientific) and vortexed for 15 min at 4°C. Supernatant was isolated after centrifugation at 12,000 × g for 10 min and stored at −80°C until IgA measurements. The small intestines of three mice per treatment group (one female and two males) were gently rinsed twice with 1× PBS and collected in individual tubes containing 1.2 ml of RNAlater Stabilization Solution (Invitrogen), and frozen at −80°C until being processed for RNA isolation. The small intestines from the remaining three mice of each treatment group (two female and one male) were collected to prepare separate formalin-fixed paraffin-embedded tissue blocks following the ‘swiss roll’ method (16).
Culture of mouse small intestinal organoids
The mouse small intestinal organoids used in the current study were generated by isolating LGR5 + adult stem cells from the gut of a female Lgr5GFPDTR/+ mouse and culturing them in a well-defined semi-solid culture medium supplemented with essential growth factors (11). The female Lgr5GFPDTR/+ mouse was generated by replacing the first coding exon of Lgr5 with a cassette containing the coding sequence for the enhanced green fluorescent protein (EGFP) linked in frame to the human diphtheria toxin receptor (DTR) gene cDNA (17).
Small intestinal organoids derived from a female Lgr5GFPDTR/+ mouse (17) were cultured as described previously (11). Briefly, organoids were grown embedded in a mix of 90% ice-cold RGF BME Type 2 PathClear (Cultrex, R&D Systems) and 10% Dulbecco's modified Eagle's Medium/Ham's F12 (Gibco), in a humidified atmosphere at 37°C and 5% CO2. For maintenance and control conditions organoids grew in ENR medium which contained advanced Dulbecco's modified Eagle's medium/F12 (Gibco) supplemented with 1× Penicillin–Streptomycin (Gibco), 10 mM HEPES (Gibco), and 1× GlutaMAX Supplement (Gibco), 1× B27 (Gibco), 1.25 mM N‐acetylcysteine (Sigma Aldrich), 50 ng/ml mEGF (Gibco), 10% final volume NOGGIN conditioned medium, 5% final volume R‐SPONDIN1 conditioned medium. For maintenance, organoids were split by mechanical dissociation using a Pasteur pipette every 5 days and the culture medium was refreshed every other day (11). For microfold (M) cell enrichment, organoids were cultured in ENR supplemented with 3 μM CHIR99021 (Axon) and 1.5 mM valproic acid (Sigma) for three days, followed by one day in ENR medium. Afterwards, treatment with 100 ng/ml of recombinant mouse TRANCE (RANKL) (BioLegend) was done for 6 or 9 days, alone or in combination with 40 μM CSRM617 Hydrochloride (Sigma-Aldrich). For EN experiments, RANKL was added to EN medium described previously (12). ENR medium alone or in combination with 40 μM CSRM617 were used as controls. Medium was refreshed every 3 days.
Bulk RNA-sequencing sample preparation
After treatment, organoids from three independent differentiation experiments were harvested using Organoid Harvesting Solution (Cultrex) according to manufacturer's protocol. Pellets were snap frozen and stored at −80°C until being processed for RNA isolation using the RNeasy RNA extraction kit (Qiagen) with DnaseI treatment using RNase-Free DNase (Qiagen). Total RNA (1 μg per replicate) was used for library preparations using the KAPA RiboErase Kit (HMR) (Roche) following manufacturer's protocol. A fragmentation step was carried out for 6.5 min at 94°C and 7 μM NextFlex DNA barcodes (PerkinElmer) were used for adapter ligation. Libraries were amplified using 6 amplification cycles. Library concentration was measured using the dsDNA Fluorescence Quantification Assays (DeNovix), and library size was determined using the BioAnalyzer High Sensitivity DNA Kit (Agilent Technologies) in a BioAnalyzer instrument (Agilent). Sequencing was performed using an Illumina NextSeq 500, and 42-bp paired-end reads were generated.
Bulk ATAC-sequencing sample preparation
After harvesting of organoids from three independent differentiation experiments, pellets were cryopreserved in Recovery Cell Culture Freezing Medium (Gibco) at −80°C until being processed for ATAC-sequencing (ATAC-seq). ATAC-seq libraries were prepared following an adapted protocol from Buenrostro et al. and Corces et al. (18,19). In brief, 50 000 cells per replicate were lysed in 50 μl of ice-cold ATAC Resuspension Buffer (RSB) containing 0.1% IGEPAL (Sigma), 0.1% Tween-20 (Sigma) and 0.01% Digitonin (Sigma) for 3 min on ice. Lysates were resuspended with 1 ml of cold ATAC-RSB containing 0.1% Tween-20 (Sigma) and nuclei were pelleted by centrifugation at 500 rcf for 10 min at 4°C in a fixed angle centrifuge. Nuclei were tagmented for 6 min at 37°C with in-house made Tn5 enzyme while shaking at 650 rpm. The tagmentation reaction was stopped with stop buffer (44 mM EDTA, 131 mM NaCl, 0.3% SDS, and 600 μg/ml proteinase K as final concentration). Genomic DNA was purified using 2× AMPure XP beads (Beckman Coulter) and subsequently amplified by PCR using KAPA HiFi Hotstart Ready Mix and Nextera Index Kit (Illumina) primers. Lastly, a reverse-phase 0.65× AMPure XP beads (Beckman Coulter) DNA purification step was done, followed by 1.5× AMPure XP beads (Beckman Coulter) purification. Library concentration was measured using the dsDNA Fluorescence Quantification Assays (DeNovix), and library size was determined using the BioAnalyzer High Sensitivity DNA Kit (Agilent Technologies). DNA libraries were sequenced with an Illumina NextSeq 500 at a read length of 38 bp.
ChIPmentation sample preparation
Following harvesting, organoids were crosslinked with 1% formaldehyde in 1× PBS shaking at 650 rpm for 10 min at room temperature, and glycine (125 mM final concentration) was added to quench the reaction. After a washing step with 1× ice-cold PBS, cell pellets were snap frozen and stored at −80°C until being processed for ChIPmentation. A total of 250 000 cells per replicate were lysed in a buffer containing 20 mM HEPES pH 7.6, 1% SDS, 1× Protease Inhibitor Cocktails (Roche). Chromatin was sheared using the medium setting of the Biorupter Pico (Diagenode) with 6 cycles (20 s on/30 s off) to achieve fragments between 200 and 600 bp. Samples were then spun at 13 000 rpm for 5 min at room temperature and supernatant containing fragmented chromatin was used for ChIPmentation as described by Schmidl et al. (20) with the modifications published in Lindeboom et al. (12). Briefly, before starting the immunoprecipitation, we saved 1% of the input material of either control or RANKL-treated organoids and stored it at 4°C until further processing (tagmentation). Then, chromatin of each replicate was incubated with 1 μg of antibody against H3K27ac (Diagenode, C15410196, RRID:AB_2637079) or 1 μg of antibody against ONECUT2 (Proteintech, 21916-1-AP, AB_2848180) in dilution buffer (1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris pH 8, 167 mM NaCl, 1× Protease Inhibitor Cocktail (Roche)), rotating overnight at 4°C. Antibody-bound chromatin was purified using protein A and protein G Dynabeads (Invitrogen) followed by two washing steps with increasing concentration of NaCl and one washing step with TE buffer as described before (12). Tagmentation of immunoprecipitated and input chromatin was performed with in-house generated Tn5 enzyme for 10 min at 37°C while shaking at 550 rpm, followed by washing steps with decreasing amounts of NaCl and soap (12). Lastly, samples were de-crosslinked with 20 μg of proteinase K (Sigma) in elution buffer (0.5% SDS, 300 mM NaCl, 5 mM EDTA, 10 mM Tris pH 8) for 1 h at 55°C and 1000 rpm shaking followed by an overnight incubation at 65°C and 1000 rpm shaking. The eluted, tagmented chromatin was treated with 10 μg proteinase K (Sigma) for 1 additional hour at 55°C and purified using 2× AMPure XP beads (Beckman Coulter). The generated libraries were amplified using the KAPA HiFi Hotstart Ready Mix and Nextera Index Kit 1 (i7) and 2 (i5) primers (Illumina). Amplified libraries were purified using a 0.65× AMPure XP beads (Beckman Coulter) followed by a 1.8× AMPure XP beads (Beckman Coulter) purification. Library concentration was measured using the dsDNA Fluorescence Quantification Assays (DeNovix), and library size was determined using the BioAnalyzer High Sensitivity DNA Kit (Agilent Technologies). DNA libraries were sequenced with an Illumina NextSeq 500, and 38-bp paired-end reads were generated.
Single-cell RNA-sequencing sample preparation
After harvesting, organoids were dissociated using TrypLE (Gibco). Viable single cells were sorted in 384-well PCR plates (Bio-Rad) using the Becton Dickinson Aria (BD Biosciences) sorter with a 100 μm nozzle, based on forward and side scatter gating. Prior to sorting, plates were primed with oligos (5‘-3’) containing the T7 promoter, a 5′ Illumina adapter, unique molecular identifiers (UMIs), a unique cell barcode and the oligo-(dT)N for tagging of individual mRNA molecules within each cell (21). Immediately after sorting, plates were centrifuged for 2 min at 1200 × g and 4°C and stored at −80°C until library preparation. Libraries were prepared using a modified CEL-Seq2 protocol (22). In brief, a micro-dispenser machine Nanodrop II (BioNex) was used to dispend in each well the ERCC spike-in control RNAs (1:50 000, Invitrogen), reagents used for the reverse transcription (RT) reaction such as Superscript II (Invitrogen) and RNasein Plus (Promega), and for the second strand synthesis such as Escherichia coli DNA polymerase and E. coli DNA ligase (New England Biolabs). Second strand synthesis was done in the presence of E. coli RNase H (Invitrogen). Double stranded cDNA from each well was pooled together and purified with AMPure XP beads (Beckman Coulter). In vitro transcription was performed overnight according to the Ambion MegaScript IVT kit protocol (Invitrogen), followed by an exonuclease digestion step with EXOI/rSAP-IT (Applied Biosystems). The amplified RNA was later fragmented and cleaned up using AMPure XP beads (Beckman Coulter). Samples were then subjected to library RT using random octamers (21) and to cDNA amplification thereafter using the Phusion High-Fidelity DNA Polymerase (New England Biolabs). A forward primer sequence against the 5′ Illumina adapter (21) and a reverse primer sequence against the octamers overhang incorporated during RT were added to the amplification mix. The reverse primer contained an Illumina 3′ adapter sequence and an index sequence to uniquely identify each library (NextFlex DNA barcodes, PerkinElmer). Libraries were purified and quantified using the dsDNA Fluorescence Quantification Assays (DeNovix) and fragment sizes were assessed with the BioAnalyzer High Sensitivity DNA Kit (Agilent Technologies). The libraries were sequenced on an Illumina NextSeq 500, and 63-bp paired-end reads were generated.
Bulk RNA-sequencing analysis
Paired-end reads were aligned to the mouse mm10 genome using the STAR RNAseq-aligner v2.7.9a (23), with –sjdbOverhang set to 100. Mapped reads were counted with HTSeq-count v0.13.5 (24) and the following settings: -r pos –s reverse –t exon. Normalization and log2-transformation of data was done using the DESeq2 package v1.34.0 (25). Genes with an adjusted P-value <0.05 were considered statistically significant. Expression heatmaps were created with ComplexHeatmap v2.10.0 (26).
Bulk ATAC-sequencing analysis
Preprocessing of reads was done automatically with atac-seq workflow from the seq2science pipeline v.0.3.1 (27). In short, reads were trimmed using fastp v0.20.1 (28) and aligned to the mouse mm10 genome using bwa-mem v0.7.17 (29). Mitochondrial reads, multipmapper reads, mapped reads below a minimum mapping quality of 20 or reads inside the ENCODE blacklist (30) were filtered out. Reads were shifted +4 bp/–5 bp on the positive/negative strand to account for the Tn5 inserting bias. Duplicate reads were removed with picard MarkDuplicates v2.21.2 (‘Picard Toolkit’ 2019, Broad Institute GitHub repository https://broadinstitute.github.io/picard/). Peaks were called using macs2 v2.2.7 (31), with the settings –shift -100 –extsize 200 –nomodel –keep-dup 1 –buffer-size 10 000 in BAM mode. Being paired-end reads, the option macs2_keep_mates was set to true.
ChIPmentation analysis
ChIPmentation sequencing reads were processed with the chip-seq workflow from seq2science v.0.3.1 and v.0.7.2 (27), for histone mark and Onecut2 ChIPmentation experiments, respecitvely, taking the 1% input samples of either control or RANKL-treated organoids as background for peak calling. The workflow of preprocessing, alignment, deduplication and quality filtering was similar to the workflow described for the ATAC-seq analysis, but without shifting sequencing reads in alignment or during peak calling.
For the ONECUT2 ChIPmentation, we used Diffbind (32) (v.3.4.11) to identify differentially bound genomic loci between RANKL- and control-treated organoids. For visualization of the results, we used deeptools (33) to show the ChIPmentation and DNA accessibility signal at shared and RANKL-specific sites as heatmaps or average plots. Genes assigned with GREAT (34) to the top 250 peaks based on the fold change of RANKL over control were used to generate a ONECUT2 ChIP gene set.
Lastly, HNF4G ChIPmentation samples from mouse small intestinal organoids grown in EN medium and control were obtained from GSE114113 (12) and processed with the chip-seq workflow from seq2science (v.0.7.2), without input controls for peak calling. We used GREAT (34) to assign each peak to the nearest gene and subsequently defined a EN-specific HNF4G target gene set by selecting genes near peaks from EN-cultured organoids that were not present in the control-samples, and subsequently selecting the top 250 peaks based on the fold change of the peak over background to generate an HNF4G ChIP gene set.
Integration and analysis of bulk sequencing data
ATAC-seq peaks generated for each replicate by seq2science, for both control and RANKL-treated conditions, were combined in one peak file using the combine_peaks function from the package GimmeMotifs v0.16.0 (35) with the window parameter (-w 4000) (generating roughly 60 000 accessible sites). ChIPmentation alignments, corresponding to the H3k27ac signal, were quantified at these regions using the coverage_table function from GimmeMotifs v0.16.0 (35) with a window of 2 kb. Differential H3k27ac signal (padj < 0.05) was defined with the DESeq2 package v1.34.0 (25) and used as input for the tool gimme maelstrom to detect differential motifs, without filtering out redundant motifs. A z-score cut-off of >1.64 and <−1.64 was used to define enriched motifs in either control or RANKL-treated conditions. Motifs were matched to transcription factors with the gimme motif2factors conversion table provided by GimmeMotifs (35). Only motifs for which the corresponding transcription factor was differentially expressed (Padj < 0.05) in the bulk RNA-seq data were included for visualization in an integrative heatmap, which was generated with ComplexHeatmap v2.10.0 (26).
Gene-regulatory network analysis with ANANSE
The ananse binding command line tool from ANANSE v0.3.0 (36) was used to predict treatment-specific transcription factors binding profiles, using the ATAC-seq and ChIPmentation outputs from seq2science. Enhancer regions were specified with the combined peak summit file containing identified ATAC-seq peaks from both RANKL-treated and control conditions. The function ananse network was run to infer treatment-specific gene-regulatory networks. For this, binding profiles from ananse binding, gene-level transcript-per-million (TPM) data for each condition and the mm10 as the genome assembly were used. The mm10 genome assembly was downloaded from UCSC using genomepy v0.9.1 (37). To obtain a gene-level TPM file, reads in the RNA-seq FASTQ files (.fastq) were quantified using the command line tool Kallisto v0.46.2 (38). The prebuild Kallisto index constructed from the Ensemble reference mouse transcriptome GRCm38 was downloaded from https://github.com/pachterlab/kallisto-transcriptome-indices/releases and used. Using tximport v1.14.2 (39) the expression of all isoforms corresponding the same gene was summed up and identifiers were changed to gene-level. Lastly, the ananse influence function was used to generate a differential gene-regulatory network, based on the difference in the interaction score calculated per factor between RANKL treatment and control conditions. Number of top edges used to define the differential network was set to 500.00 (-i 500_000).
Analysis of single-cell RNA-sequencing data
Raw FASTQ files were demultiplexed, aligned and annotated to the mouse GRCm38 genome indexed with sequences from the ERCC ExFold RNA Spike-In Mixes (invitrogen), and counted using the kallisto|bustools wrapper kb_phython v0.26.3 (38,40). Pre-processing and quality control was performed using the Scater package v1.10.1 (41). To exclude low-quality cells from further analysis, any cell with <500 detected features, <1000 total transcripts, >40% mitochondrial transcripts or >20% ERCC transcripts were removed from the datasets. Normalization, natural log-transformation, scaling (with ‘nCount’ and ‘nFeature’ as variables to regress), and clustering analysis were performed using Seurat v3.1.5 (42). We determined the 2000 highly variable genes using the variance stabilizing transformation (VST) method. Dimensionality reduction was performed by principal component analysis (PCA), and was followed by Louvain clustering using the FindNeighbors function with the top 30 principal components (PCs) and the FindClusters function with a resolution set to 1.6 for the RANKL treatment dataset (21 099 genes across 6636 cells), and the top 20 PCs and a resolution of 1.5 for the dataset of RANKL and CSRM617 combined treatment (9257 genes across 1903 cells). Data was projected in a two-dimensional space using the Uniform Manifold Approximation and Projection (UMAP) method. Label transferring from clusters 6, 12 and 17 from the RANKL treatment dataset (21 099 genes in 820 cells) to clusters 3, 9, 10 and 12 from the dataset of RANKL and CSRM617 combined treatment (9257 genes in 427 cells) was done using the Seurat FindTransferAnchors function using the RANKL treatment dataset selected clusters as a reference.
Trajectory inference
RNA velocity estimation was done using velocyto.R v.0.6 (43). The raw counts corresponding to spliced and unspliced RNA forms for each gene, generated by the kallisto|bustools wrapper and stored as separate assays in the Seurat object, were used by velocyto.R to create an RNA velocity map. The RNA velocity map was then projected as a grid with arrows onto the UMAP space that was generated by Seurat with the function show.velocity.on.embedding.cor.
For the pseudotime analysis, raw counts of cells from the Seurat clusters 6, 12 and 17 from the RANKL treatment dataset that passed the Scater quality assessment were processed with the R package Monocle3 v1.0.0 (44) with default settings. The preprocess_cds function of Monocle3 normalized by log and scaled the data. The function also calculated a low-dimensional space used as input for dimensionality reduction with a UMAP. With the learn_graph function, Monocle3 used a principal graph-embedding procedure based on the SimplePPT algorithm (45,46) to represent on the UMAP the possible paths cells can take as they develop. The pseudotime was calculated across these paths by setting the furthest away cells from cluster 17 as time zero. Lastly, we used graph_test, a function based on the Moran's statistical test (47) to define the pseudotime-dependent genes and the find_gene_modules function to group these genes into modules based on their co-expression across clusters.
Motif activity prediction with SCEPIA
Using SCEPIA v0.5.1 (https://doi.org/10.5281/zenodo.4892888) a list of transcription factors of interest was established by predicting transcription factor motif activities based on the single-cell RNA-seq data. The normalized and scaled spliced RNA counts and associated annotation generated with Seurat (v3), including highly variable genes and clustering, were exported to an AnnData object in Python v3.9.6. First, H3K27ac profiles were assigned to the single cells using a H3K27Ac reference database. The ENCODE H3K27Ac ChIP-seq database of 121 human cell lines and tissues included in SCEPIA was used as a reference (48,49). The regulatory potential, a summarized, distance-weighted measure of H3K27ac within 100 kb of the gene transcription start site, was calculated for every gene, according to Xu et al. (36) similar to Wang et al. (50). Using the top 2000 highly variable genes from the scRNA-seq data, cells were matched to H3K27Ac profiles using Ridge regression with the normalized and scaled gene expression level as the response variable and the H3K27Ac regulatory potential as the explanatory variable. The output of this regression was a matrix with per single cell a coefficient for each cell type (single cell × cell type matrix). Only the top 50 scoring cell types (ordered by the mean absolute coefficient over the clusters) were kept and from these the top 10 000 highest variable enhancers, based on the genome-wide H3K27Ac data, were selected for motif activity analysis using the Bayesian Ridge method with GimmeMotifs v0.16.1 (35) resulting in a motif x cell type matrix. The predicted motif coefficients (‘motif activity’) for each H3K27Ac reference cell type were combined into single cell motif activities using a dot product of the single cell x cell type matrix and the motif × cell type matrix. Pearson correlations between single cell motif activity and the single cell expression levels of the transcription factors binding the motifs, based on the default database of GimmeMotifs, were calculated, followed by 100,000 random permutations to determine a permutation-based P-value. The permutation-based p-value was combined with the p-value of the correlation using Fisher's method and corrected for multiple testing using the Benjamini-Hochberg correction. From the list of transcription factor-motif combinations, the hits with the highest positive correlation coefficients and an FDR ≤ 0.1 were selected. The gimme logo tool from GimmeMotifs (35) was used to generate motif logos.
Gene-regulatory network analysis with pySCENIC
Raw counts from the Seurat object were used as input for pySCENIC v0.11.0 (51). First, gene-regulatory networks between transcription factors and putative target genes were inferred with the grnboost2 function from the Arboreto package v0.1.6 (52) part of pySCENIC, using a mouse transcription factor database available from the TFCat portal through pySCENIC. The ctx command kept only transcription factor motifs within 10 kb from the target transcription start site (TSS) and pruned indirect targets from dataset based on cis-regulatory cues. For this, the motifs database motifs-v9-nr.mgi-m0.001-o0.0, the cis-target databases mm10__refseq-r80__500bp_up_and_100bp_down_tss.mc9nr and mm10__refseq-r80__10kb_up_and_down_tss.mc9nr were used. As a default option, only transcriptional regulatory units (regulons) with a positive correlation between the expression of a transcription factor and its targets were used for further analysis. The area under the curve (AUC) scores (regulon activity) were calculated for every cell using the aucell function with default options and later averaged per Seurat cluster. Data was visualized using gplots v3.1.1 (53).
Gene-set enrichment analysis
Gene-set enrichment analysis (GSEA) was performed using the fgsea package v1.19.3 (54). Mouse in vivo intestinal cell-type gene sets were extracted from (9) by selecting the top 250 differentially expressed genes with an FDR (Q, max) <0.05 and a mean natural-log fold change >0.5. To create the ONECUT1-, ONETCUT2- and ONECUT3-specific signatures, RNA-seq data from (55) was downloaded using the download-fastq workflow from seq2science and re-analyzed with DESeq2 package v1.34.0 (25). Top 250 differentially expressed genes (Padj < 0.05), unique for each overexpression condition and with a log2 fold change >0.5 over control were used to create the gene sets. Lastly, genes with a Padj <0.05 and a mean natural-log fold change >0.5 were used to create the cluster 12 gene set from the RANKL treatment single-cell dataset. For all experiments, ranked files with log2 fold change expression values (Padj < 1) were subjected to GSEA.
Real-time semi-quantitative RT-qPCR
Mice small intestines were washed with 1X PBS to remove excess of RNAlater (Invitrogen) and 15–20 mg of tissue were lysed with 1 ml of TRIzol (Ambion) and incubated overnight at −20°C. At the next day, tissue was homogenized using RNAse-free pestles, 14G (1.6 mm) and 21G (0.8 mm) needles. Lysates were centrifuged for 5 min at 12 000 × g and 4°C. Supernatant was processed for RNA isolation following TRIzol manufacturer's instructions. After this, a total of 30 μg of isolated total RNA per replicate was purified with the Quick-RNA Microprep Kit (Zymo research) and 1 μg of purified RNA was used for cDNA synthesis using the iScript cDNA Synthesis Kit (Bio-Rad). Real-time PCR analysis was performed using the iQ SYBR Green Supermix (Bio-Rad) in a CFX96 Real-Time system (Bio-Rad). Crossing-point (Cp) values were determined by the CFX Manager software version 3.0 (Bio-Rad). Expression levels of Gapdh were used for normalization. Relative gene expression levels were calculated according to Pfaffl et al. (56). Primers for RT-qPCR can be found in Supplementary Table S8.
Flow-cytometry analysis
After harvesting, organoids were dissociated using TrypLE (Gibco). Organoid cells were counted using the Muse Cell Analyzer (Merck) and a total of 100,000 viable cells per well and in triplicates per treatment were seeded in conic v bottom 96-well plates (greiner bio-one). After washing with 1% PBA buffer (1% Bovine serum albumin (Sigma) in 1× PBS), cells were incubated with rat anti-mouse GP2 antibody (MBL International Corporation, D278-5, RRID:AB 11160946) in a 1:10 dilution in 100 μl of 1% PBA buffer per well. Incubation was done for 30 min at 4°C and was followed by three washing steps with 1% PBA buffer. All washing steps were done by centrifugation of 3 min at 200 × g and 4°C. Cells were lastly fixed with 1% formaldehyde for 20 min at room temperature and in dark. After removal of the formaldehyde solution and one washing step, cells were resuspended in 1% PBA before being analyzed in the MACSQuant Analyzer 10 (Miltenyi Biotec). FITC signal (LGR5+ cells) and PE signal (GP2+ cells) were detected in cell population selected based on forward and side scatter gating, and pseudocolor density plots were generated using FlowJo v10 (BD Biosciences).
Enzyme-linked immunoassay (ELISA)
Measurement of secretory IgA (SIgA) in mouse faeces samples after treatment with the ONECUT2 inhibitor CSRM617 Hydrochloride (Sigma-Aldrich) or PBS control was done using the SIgA Mouse Uncoated ELISA kit (Invitrogen) following manufacturer's instructions. Faecal supernatants were diluted to a final assay concentration of 10 ng/ml. A solution of 2N H2SO4 was used to stop the reaction and the concentration of SIgA was deduced from the standard curve made with the IgA standards provided by the kit.
Live imaging and immunohistochemistry
Whole-mount immunofluorescence staining of organoids was performed in conical bottom polystyrene 12 ml tubes (Greiner). Briefly, after harvesting of organoids, these were transferred to the round-bottom 15 ml tubes pre-coated with 1% PBA. Two washing steps with ice-cold 1× PBS were carried out by gently resuspending the organoids and letting them sit at the bottom of the tubes before discarding supernatant. Organoids were then fixed in 4% formaldehyde for 15 min followed by two washing steps with ice-cold 1× PBS. Permeabilization of organoids was done with 0.5% Triton in 1× PBS for 30 min. This was followed by two washing steps and a quenching step with 20 mM Glycine in 1× PBS for 30 min. Unspecific signal was then blocked with 1% PBA for 30 min. Three washing steps with 1% PBA were done before overnight incubation with primary antibodies rat anti-GP2 (MBL International Corporation, D278-3, RRID:AB_10598188), diluted 1:100 in blocking solution. To prevent organoids from sitting at the bottom of the tube, incubation was carried out in a shaker at 45 rpm. Organoids were then washed three times with 1× PBS and incubated with Alexa Fluor 647 Goat anti-Rat IgG secondary antibody (Invitrogen, A-21247, RRID:AB_141778) diluted 1:400 in blocking solution, for 1.5 h in a shaker at 45 rpm and covered from light. Antibodies were washed out three times with 1× PBS. Staining with methanolic phalloidin (Thermo Fisher Scientific) diluted in blocking solution was performed for 20 min and followed by three washing steps. Finally, nuclear counterstaining with Hoechst (50 μg/ml, brand) was done for 20 min. Organoids were washed three times more and mounted on 22 mm-diameter glass bottom dishes (WillCo Wells) with Ibidi mounting medium (Ibidi). For live microscopy, EDU labelling was performed using the Click-iT EdU Imaging Kits (Invitrogen) and following manufacturer's protocol on organoids embedded in single BME domes, seeded on 8-well chamber slides (Ibidi).
Formalin-fixed paraffin-embedded mice small intestines were sectioned 4 μm-thick and mounted on Superfrost Plus slides (Thermo Fisher Scientific). Deparaffination was performed overnight at 37°C and re-hydration steps were followed by heat-induced epitope retrieval with boiling sodium citrate (pH9) for 10 min. Blocking of endogenous perodixase activity was done with 1% H2O2 for 15 min and was followed by blocking of unspecific binding with 2% PBA for 1 h. GP2 or SPIB primary antibodies (1:100) were incubated overnight in 1% PBA. For detection of GP2 protein, sections were incubated for 30 min with biotinylated Rabbit IgG anti-rat (H + L) secondary antibody (Vector Laboratories, BA-4001, RRID:AB_10015300) 1:200 in 1% PBA and 30 min with an ABC mix solution from the VECTASTAIN Elite ABC Reagent kit (Vector Laboratories, PK-6101, RRID:AB_2336820). To reveal the reaction, sections were incubated with Bright DAB solution (Immunologic) for 5 min followed by 10-s nuclear counterstaining with Hematoxylin, dehydration and mounting with Permount mounting media (Fisher Chemical). Overnight incubation with sheep anti-SPIB antibody (R&D Systems, AF7204, RRID:AB_10995033) diluted 1:100 in blocking solution was followed by a 1-hour incubation with Alexa Fluor 568 Donkey anti-Sheep IgG (H + L) secondary antibody (Invitrogen, A-21099, RRID:AB_2535753) diluted 1:400 in blocking solution. Slides were mounted with Fluoromount-G with DAPI mounting medium (Invitrogen). Except for first antibody incubations, all steps for immunohistochemistry were done at room temperature.
Image analysis
Live-cell and three-dimensional images were captured with a Leica TCS SP8 microscope (Leica). For live imaging the organoids were held at 37°C. Organoids were imaged in XYZ mode using an HCX PL APO 63×/1.2 water immersion objective. Post-acquisition analysis of fluorescence signal was performed manually using ImageJ version 1.53c (57). Bright-field and fluorescence imaging of mouse tissue sections was carried out in the Axio Observer 7 microscope with Sample Finder AI (Zeiss) using a 20×/0.8 objective.
Brightfield images of organoids were fed to ImageJ and processed using the OrgM macro to measure circularity (58) defined by the formula 4π(area/perimeter2). For analysis thresholding mode was enable and watershed mode was disabled. Circularity was plotted for each treatment.
Statistical analysis
Statistical difference in organoid circularity obtained by ImageJ, fluorescence signal detected by Flow cytometry, quantification of immunohistochemistry analysis, relative expression obtained by RT-qPCR or fluorescence signal corresponding to IgA levels were calculated using an unpaired t test in the Prism software version 5.03 (GraphPad). Bars graphs represent the mean ± SEM. The n numbers are provided in figure legends or in the Materials and methods section corresponding to each technique. A P-value of <0.05 was considered statistically significant and P <0.05 was represented by one star (*), P <0.01 was represented by two stars (**) and P <0.001 was represented by three stars (***).
RESULTS
Generation of M cell-enriched mouse small intestinal organoids
In vivo, RANKL is expressed by MAdCAM-1− mesenchymal cells beneath the FAE, functioning as M cell inducers by directly interacting with RANK-expressing progenitor epithelial cells (59,60). To mimic this mechanism in vitro, organoid culture medium was supplemented with recombinant RANKL for 6 days. Upon treatment, organoids acquired a cyst-like phenotype with wide, expanded villi regions and expression of glycoprotein 2 (GP2) in the apical membrane of a few cells (Figures 1A and B). GP2 is an endocytic receptor for microorganisms and a marker for mature M cells (9,61). Our mouse small intestinal organoids carry a GFP reporter cassette at the locus of the leucine-rich repeat G-protein-coupled receptor (Lgr5) gene (17), a marker for ISC (62), which allowed us to visualize LGR5+ ISC in our organoid cultures. Concomitant to the increase in GP2 signal, RANKL treatment led to a reduction of LGR5 expression when compared to control organoids growing in conventional organoid culture medium (Figure 1B). To evaluate the efficiency of M cell enrichment, flow-cytometry analysis was performed. In control conditions, the number of ISC and M cells was roughly 20% and <0.1%, respectively (Figure 1C). Treatment with RANKL achieved an average 4x increase in the number of M cells consistent with what is found in vivo (9,63) and an almost 50x decrease of ISCs (Figure 1C).
Figure 1.

Generation of M cell-enriched mouse small intestinal organoids. (A) Brightfield images of control and M cell-enriched (RANKL-treated) organoids, next to graphical representation of their cell type composition. Microscope magnification is depicted. Graph shows the quantification of organoid circularity (AU, arbitrary units) and mean ± SEM. Control n = 50 and RANKL n = 51. Unpaired t test P < 0.001 is represented by (***). (B) Nucleus (Hoechst), F-actin (Phalloidin) and GP2 staining and detection of LGR5 signal in control and RANKL-treated organoids. Microscope magnification is depicted. On the right, a zoom in image from the RANKL condition is displayed. (C) Representative density plots depict fluorescent intensities in the FITC (LGR5) and PE (GP2) channels, detected by flow cytometry. The percentage for each gate population is described. Bar graph represents the mean ± SEM of the fold change from at least five differentiation experiments. (D) Heatmap showing the relative change in mRNA expression upon RANKL treatment compared to control. Known markers of M cells and other intestinal cell lineages are displayed. Rows show Z scores of normalized, log2-transformed values from significantly changing genes (Padj < 0.05). (E) Gene-set enrichment analysis showing in vivo gene signatures of mouse small intestine cell lineages from Haber et al. (9) enriched in RANKL-treated organoids compared to control organoids (dark grey). Normalized enrichment score (NES) and padj values are described. Signatures enriched in control compared to RANKL condition (light grey) have a negative NES. (F) Gene-set enrichment analysis showing gene ontology (GO) terms significantly enriched (Padj < 0.05) in control (light grey) and RANKL-treated organoids (dark grey).
In line with these findings, other known markers of M cells such as Anxa5, Ccl20, Pglyrp1, Marcksl1, Ccl6, Gp2, Tnfaip2, Rac2 and Ccl9 (9,61,64) were significantly upregulated in the RANKL conditions at a transcriptome level, while markers of ISC and other intestinal cell types showed opposite dynamics (Figure 1D). To evaluate whether our in vitro model recapitulated in vivo tissue, whole-transcriptome changes induced upon RANKL-treatment were compared to cell type-specific intestinal signatures generated from a recently published single-cell survey of the mouse small intestine (9). The M cell and the Tuft cell gene signatures were significantly enriched under RANKL conditions, while the Paneth cell, ISC and Goblet cell signatures were enriched in control conditions (Figure 1E). In addition, the M cell-enriched organoids were significantly enriched for genes involved in immune cell regulation but depleted in those associated with cell cycle and DNA replication (Figure 1F), reflecting the physiological role of M cells. Together, these findings support the use of our in vitro model for the study of intestinal M cell differentiation with the potential of generating data of in vivo relevance.
RANKL-induced transcriptional regulation
To investigate the molecular drivers of M cell differentiation, we focussed our next analyses on transcription factors. A prerequisite for transcription factor-mediated activity is an accessible chromatin, which is characterized by the presence of the H3K27ac modification, which marks active enhancers and, to a lesser extent, active promoters (65). We used ATAC-sequencing and ChIPmentation to profile chromatin accessibility and the H3K27ac modification in the control and RANKL-treated organoid cultures. A total of 55 055 accessible sites were detected in the RANKL and control sequencing datasets, which were used to identify genomic locations that undergo a significant change in H3K27ac levels between both conditions. At these sites, we identified motifs of 93 and 123 differentially expressed transcription factors (Padj < 0.05) enriched in the RANKL and control conditions, respectively (Figure 2A). Among those enriched in RANKL conditions we found Relb, Spib and Sox8, known drivers of M cell differentiation (66–68), which further supports an active regulatory role for these 216 differentially expressed transcription factors (Figure 2A).
Figure 2.

RANKL-induced transcriptional regulation. (A) Heatmap displaying enrichment of transcription factor motifs detected at accessible genomic locations (ATAC-sequencing data) that display significant dynamics of histone modification H3K27ac (ChIPmentation data, Padj < 0.05) between RANKL-treated and control organoids. Only motifs of transcription factors differentially expressed (padj < 0.05) between RANKL-treated and control organoids from the bulk RNA-sequencing data in Figure 1D are included. Rows show z scores of motif enrichment with a cut-off of >1.64 and ←1.64. (B) UMAP embedding of single cell transcriptome from RANKL-treated organoids. Each dot represents a single cell. Cell colours represent cluster identity. Next, highlighted on black the partitions corresponding to cell populations expressing markers of the absorptive or secretory lineages, or markers of intestinal stem cells (ISC) and transit-amplifying (TA) cells. (C) UMAP embedding overlay showing the harvesting time points during differentiation. Next, normalized and natural log-transformed expression of Id3, Lgr5 and Mki67. (D) Velocity projections on UMAP embedding coloured as in (B). Below, normalized and natural log-transformed expression of top markers from cluster 17 (Pglyrp2) and cluster 12 (Anxa5). Velocity projections are shown. (E) Heatmap showing row z scores of regulon activity (area under the curve scores) averaged per each cell population as analysed by pySCENIC. On red, regulons of transcription factors for which an active motif was found in (A). Group of interest 1 (GOI1) and 2 (GOI2) are shown.
To obtain cell type-specific transcriptional regulation profiles across time, single-cell RNA-sequencing analysis was performed in RANKL-treated organoids harvested at different time points during treatment. In total, 6636 cells were grouped into 20 clusters, which expressed markers from the absorptive or secretory lineages, or markers distinctive of ISC and daughter transit-amplifying cells (TA) (Figure 2B, Supplementary Figure S1A and Supplementary Table S1). Time seemed to separate cell clusters into two main partitions (Figure 2C). One partition, with cells generated within the first five days of treatment, included cells expressing absorptive lineage makers (cluster 12, 13 and 17) and clusters expressing Id3, a marker for TA cells (cluster 2, 3, 7 and 10). Cluster 12 expressed known M cell markers such as Anxa5, Pglyrp1, Ccl9, Serpinb1a, Rac2 and Spib (Supplementary Figure S2A and Supplementary Table S1). The second time partition was constituted by cells originating from day 6 to day 9 (experimental end point), marked by the recovery of Lgr5 expression and an increase in the number of secretory cells in clusters 8, 15 and 19 (Figure 2C and Supplementary Figure S2B). Mki67, a marker of cell proliferation, seemed to be evenly distributed between cells from both partitions. In agreement with this, EdU labelling on RANKL-treated organoids showed that day 3 M cell-enriched organoids displayed a high number of LGR5−, proliferative cells (EdU-labelled), whereas day 6 organoids had proliferative and LGR5+ cells solely at crypt bottoms (Supplementary Figure S2C).
This time-resolved single-cell transcriptome data was suitable for RNA velocity calculations (43), an analysis in which spliced and unspliced RNA abundance is used to infer a ‘direction of change’ and predict the future state for each cell in a two-dimensional space. With the aim of unravelling the mechanisms driving M cell differentiation, we further narrowed analyses to cell clusters generated within the first five days of RANKL treatment. RNA velocity results suggested that day 0 Lgr5+/Mki67+ cells (cluster 14) generated Lgr5−/Mki67+ cells characterised by high Ccnb2 expression (cluster 3) and that these cells turned in time into non-proliferative Id3+ cells (i.e. cluster 2) (Figure 2D and Supplementary Table S1). This process resembled the generation of TA cells from ISC observed in vivo (69). Interestingly, a subset of cluster 17 cells, originated on day 1 from day 0 cluster 13, was shown as a precursor of two cell populations, the Anxa5+ M cells (cluster 12), and a second cell population of cluster 17 that expressed Pglyrp2, a marker of FAE enterocytes (70) (Figure 2D and Supplementary Table S1). In agreement with these findings, clusters originating from cluster 13 (hereafter referred to as group of interest 1 or GOI1) displayed transcriptional regulatory units or ‘regulons’ (71) for the transcription factors NFKB2, RELB, SPIB and SOX8, well-established drivers of M cell differentiation, and regulons for HNF4G, ZBTB7B, RXRA and RARA, regulators of the absorptive lineage (12,13,72) (Figure 2E). In contrast, the group of interest 2 (GOI2) which originated from cluster 14 presented regulons of transcription factors previously associated with cell cycle control and crypt-villus maintenance such as MYC, E2F1 and E2F3 (73), and TEAD2, TEAD4 and SOX9 which regulate TA expansion (74,75). A total of 358 regulons supported the identity of cells from GOI1 and GOI2, from which a motif for 56 of these transcription factors was found within an accessible genomic region and significantly enriched by H3K27ac upon RANKL treatment (Padj < 0.05) based on our bulk integrative analysis (Figure 2E, highlighted in red). Collectively, our bulk and single-cell sequencing data defined the transcriptional and cellular responses to RANKL treatment in our organoid model and allowed the identification of a proposed M cell precursor cell population in cluster 17.
ONECUT2 signalling in M cell precursor cell population
To predict transcription factor motif activity from single cell RNA-sequencing data, we designed a novel computational tool called SCEPIA (Single Cell Epigenome-based Inference of Activity) (76). By using computationally inferred epigenomes of single cells, SCEPIA can identify transcription factors that determine cellular states. In total, 81 transcription factors showed substantial motif activity within our single cell dataset. Among these, we found transcription factors known for their function in different intestinal cell types (9) like Pax4 and Neurog3 in cells of Enteroendocrine cell identity (cluster 15), Runx1 in Tuft cells (cluster 19), or Spib in M cells (cluster 12) (Supplementary Table S2). From the transcription factors highly expressed within GOI1 (cluster 6, 12, 13 and 17) Hnf4g, Klf5, Onecut2 and Tcf7l2 were the top four with the highest positive correlation between transcript expression and motif activity (Figure 3A and Supplementary Table S2). The hepatocyte nuclear factor 4 gamma (HNF4G) is a key driver of enterocyte cell differentiation (12,72). The krüppel-like factor 5 (KLF5) is a zinc-finger transcription factor expressed in both ISC and TA cells (77–79) while the one cut domain family member 2 (ONECUT2) transcription factor is decreased in LGR5+ ISC (80). The transcription factor 7-like 2 (TCF7L2) is a known downstream effector of the WNT pathway, expressed along the entire crypt-villus axis (81). All four transcription factors have been shown to be essential for proper differentiation of the embryonic intestinal epithelium (77,82–85), but their role in the modulation of gene expression during M cell differentiation was thus far unknown.
Figure 3.

ONECUT2 signalling in M cell precursor cluster. (A) On the left, UMAP embedding overlay showing three partitions, group of interest 1 (GOI1), 2 (GOI2) and 3 (GOI3). On the right, top four transcription factors expressed within GOI1, with the highest positive correlation between transcript factor expression and motif activity. The logos for the motif of HNF4G (GM.5.0.Nuclear_receptor.0068), KLF5 (GM.5.0.C2H2_ZF.0006), ONECUT2 (GM.5.0.CUT_Homeodomain.0005) and TCF7L2 (GM.5.0.Mixed.0092) are displayed. (B) ONECUT2 ChIPmentation signal and DNA accessibility (from ATAC sequencing) at shared or RANKL-specific ONECUT2 binding sites. From ChIPmentation, heatmap shows one representative replicate for each condition as well as input signal for RANKL and control samples. (C) Gene-regulatory network of Onecut2 drivers predicted by ANANSE. Interaction scores (arbitrary cut-off ≥0.83) are depicted. (D) Trajectory plot overlaid with cluster identity of individual cells from RANKL-treated clusters in GOI1. Three major branches are shown along the plot. On the right, same trajectory plot with cells ordered along a pseudotime. Below, log10 expression values of Onecut2 and top genes from each branching point are shown. (E) Co-regulated modules of differentially expressed genes by cells from RANKL-treated clusters in GOI1. The transcription factors predicted by ANANSE as Onecut2 drivers and ONECUT2 targets, that are differentially expressed (Padj < 0.05) by cells from RANKL-treated clusters within GOI1 and defined as pseudotime-dependent genes are highlighted.
To define the context for HNF4G-, KLF5-, ONECUT2- and TCF7L2-mediated transcriptional regulation, a differential gene-regulatory network was built using ANANSE (36). Different to our integrative approach, ANANSE takes into account the transcription factor target gene expression in addition to transcription factor expression itself and H3K27ac and chromatin accessibility profiles to build a differential gene-regulatory network in RANKL over control conditions. A total of 15371 genes (369 drivers and 15002 target genes) constituted the gene-regulatory networks for HNF4G, KLF5, ONECUT2 and TCF7L2 (Supplementary Table S3). Interestingly, upon RANKL treatment, ANANSE identified ONECUT2-binding motifs enriched in active regulatory elements of both Tcf7l2 and Klf5 (Supplementary Table S3), proposing ONECUT2 as an upstream regulator of TCF7L2 and KLF5-mediated transcriptional activity.
ONECUT2 forms part of the cut homeobox family of transcription factors and in mammals this family also includes ONECUT1 and ONECUT3 proteins (86–88). These three proteins have been shown to have a unique as well as an overlapping transcriptional output (55). To determine whether the enrichment for the ONECUT2-binding motifs determined by SCEPIA and ANANSE was specific for the ONECUT2 transcription factor, we performed a gene-set enrichment analysis (GSEA) with signatures unique for each cut homeobox protein. Only the ONECUT2 gene signature was shown to be significantly enriched upon RANKL treatment (Supplementary Figure S2D). Leading-edge (highly enriched) genes from the ONECUT2-specific signature encode proteins associated with the epithelial membrane, as well as transport and metabolism (Supplementary Table S4), in line with previous reports about the ONECUT2 signalling in the mouse small intestine (83).
We then performed ChIPmentation using a ONECUT2-specific antibody. We identified 1008 ONECUT2 binding sites that were shared among conditions and 7261 ONECUT2 binding sites specific to the RANKL condition (FDR < 0.05) (Figure 3B and Supplementary Figure S2E). Only 7 ONECUT2 binding sites were specific to control conditions (Supplementary Table S5). Consistent with its role in transcription regulation, ONECUT2 peaks were found mostly in accessible genomic regions as determined by ATAC sequencing (Figure 3B and Supplementary Figure S2E). Upon RANKL treatment, ONECUT2 was found significantly bound (FDR < 0.05) to chromatin regions upstream of 4370 genes, which included Tcf7l2, Klf5 and Hnf4g and 167/364 ANANSE-predicted ONECUT2 target genes (Supplementary Table S5). This result places ONECUT2 as a master regulator in the GOI1. ANANSE also identified motifs for 21 transcription factors within Onecut2 enhancer regions, among which those for RELB and NFKB2 binding, the latter having the highest predicted interaction score (Figure 3C). Consistently, Onecut2 was found part of the regulon of RELB (Supplementary Table S6). Heterodimer formation between RELB and the NFKB2 p52 subunit is known to be required for transcriptional activation of Spib and Sox8 (64), proposing the involvement of the RELB-p52 dimer in the regulation of Onecut2 expression.
To observe the process of M cell lineage commitment in more detail, we performed an unsupervised trajectory analysis in the RANKL-treated cell populations from the GOI1. Single cells were ordered based on transcriptome expression and plotted as a function of pseudotime (44). This analysis confirmed the results obtained by RNA velocity, where a subset of cells from cluster 17 was shown as progenitor cells in the GOI1 (Figure 3D). The analysis also showed that Klf5, Spib and Hnf4g, which characterized cluster 6, 12 and the second subset of cells from cluster 17, respectively, are transcription factors expressed in a pseudotime-dependent manner (Figure 3D). We next investigated how other genes from the Onecut2 regulatory network organized along the differentiation trajectories. For this, we included the ONECUT2 targets determined by ChIPmentation and ANANSE-predicted Onecut2 drivers, and only transcription factors that were differentially expressed (Padj < 0.05) within RANKL-treated cells of the GOI1. Including Onecut2, the expression of 26 transcription factors was found to be pseudotime-dependent (Supplementary Table S7). To illustrate their contribution to each pseudotime branch, co-expressed genes were further grouped into modules that co-regulate across the cell clusters (Figure 3E and Supplementary Table S7). Spib, Nfe2l1 and Pparg were assigned to modules of genes expressed at high levels in cluster 12, while Klf5, Id2, Klf10, Bhlhe40, Nr3c1, Pten and Isx were part of modules of genes highly expressed in cluster 6. Creb3l2, Creb3l3, Hnf4g and other genes belonging to the ONECUT2 transcriptional output were assigned to cluster 17 modules. Remarkably, Nfkb2, Relb and Onecut2 shared the same expression module (Figure 3E and Supplementary Table S7), with genes in common upregulated in cluster 17, supporting the proposed regulatory mechanism of Onecut2 expression by the RELB/p52 axis. Collectively, these analyses showed that precursor cells within cluster 17 give rise to cluster 12 and a second cell population of cluster 17 which based on their transcriptome profile correspond to an M cell and an enterocyte cell population, respectively, and proposed ONECUT2 as an upstream regulator of this process.
ONECUT2 restricts M cell lineage specification
To investigate the functional consequences of the ONECUT2 protein on M cell differentiation, organoid cultures were treated with the ONECUT2 inhibitor CSRM617. This compound was designed to specifically bind to the ONECUT2 homeodomain (HOX), thereby inhibiting DNA binding and ONECUT2-mediated transcriptional activation (89). CSRM617 alone had little, if any, effect on the morphology of organoids when compared to control conditions (Figure 4A). In contrast, addition of CSRM617 to RANKL medium greatly affected the phenotype previously observed with RANKL treatment, resulting in small-lumen organoids with angular crypts (Figure 4A). Consistent with the observed phenotype, CSRM617 alone did not affect much the transcriptome profile, compared to control conditions (Supplementary Figure S3A). Co-treatment of RANKL and CSRM617 compared to RANKL alone led to a significant downregulation (padj < 0.05) of 4317 genes, which included 1150 ONECUT2 target genes determined by ChIPmentation (Figure 4B). Together, these results indicate that ONECUT2 signalling in mouse small intestinal organoids is RANKL-dependent.
Figure 4.

ONECUT2 restricts M cell lineage specification in vitro. (A) Bright-field images of control organoids or those treated with RANKL, ONECUT2 inhibitor CSRM617 or a combination of both. Microscope magnification is depicted. Below, organoid circularity quantification graph (AU, arbitrary units) and mean ± SEM. Control n = 40, CSRM617 n = 35, RANKL n = 33 and RANKL + CSRM617 n = 70. (B) Heatmap showing the relative mRNA expression of transcripts upon co-treatment with RANKL and CSRM617, compared to RANKL alone. Rows show z scores of normalized, log2-transformed values from significantly changing genes (Padj < 0.05). (C) Bar graph representing the mean ± SEM of at least four differentiation experiments. Below, density plots depicting fluorescent intensities in FITC (LGR5) and PE (GP2) channels, determined by flow cytometry. Percentage of gated populations is described. (D) Gene-set enrichment analysis (GSEA) showing RANKL-treated cluster 12-specific gene signature, enriched in organoids treated as in (A), compared to control. Dotplot graph shows normalized enrichment score (NES, colour) and FDR (size). (E) Label transferring from RANKL-treated cell clusters within GOI1 to cells co-treated with RANKL and CSRM617. Bar graph (right) shows the fold change (FC) number of cells with RANKL clusters’ identity (RANKL + CSRM617 over RANKL). (F) GSEA showing the HNF4G- and ONECUT-ChIP signatures, enriched in organoids growing in EN medium, compared to control medium. NES and padj values are described. (G) Relative mRNA expression of Onecut2, Hnf4g, Klf5 and Klf6 detected in organoids growing on EN medium alone or with RANKL. (H) Ratio of LGR5+ or GP2+ cell numbers in organoids growing on EN medium alone or with RANKL or regular RANKL medium over control, determined by flow cytometry. Bar graph representing the mean ± SEM of three differentiation experiments. For (A), (C), (G) and (H), unpaired t test P <0.05 is represented by (*), P <0.01 by (**) and P <0.001 by (***). (I) RANK/RANKL signalling is essential to maintain a proper balance between M cell and enterocyte cell differentiation.
Given the fact that Onecut2 was shown as a master regulator of GOI1, we hypothesized that inhibition of ONECUT2 genomic functions would have a negative effect on M cell differentiation. Strikingly, however, co-treatment with CSRM617 and RANKL resulted in an increase in the number of GP2+ cells (Figure 4C). Effectively, co-treatment with CSRM617 resulted in a significant enrichment of the overall M cell signature from our single-cell dataset (Figure 4D). These results indicated that ONECUT2 exerts an inhibitory effect in RANKL-induced M cell differentiation in vitro.
We next assessed the transcriptome profile of RANKL-treated cell populations from GOI1 in single cells from organoids co-treated with RANKL and CSRM617 (Supplementary Figure S3B). The transcriptome profile of cluster 17 cells was almost completely lost in the co-treatment single-cell dataset (Figure 4E), possibly reflecting an absolute induction toward the M cell lineage and supporting the role of ONECUT2 in the maintenance of cluster 17. The co-treatment also resulted in the loss of 147 regulons that were previously identified in RANKL-alone conditions, 48 of them corresponded to ONECUT2 target genes determined by ChIPmentation (Supplementary Figure S3C, highlighted on red). The lost regulons also included transcription factors characteristic of GOI2, a group of cell clusters largely composed by Mki67+/Lgr5− proliferative cells (Figures 2C and E, and Supplementary Figures S2C and S3C). This is in agreement with the loss of the cystic phenotype that was observed in organoids subjected to co-treatment with RANKL and CSRM617 (Figure 4A) and stresses a switch in cellular programs, from a balanced proliferative/differentiated state to an overdriven differentiation program.
The induction of M cell differentiation observed upon ONECUT2 inhibition was accompanied by a reduction in the number of enterocyte cluster 17 cells. Because enterocytes and M cells derive from the same precursor cell population, we wondered whether the induction of enterocyte cell differentiation would then reduce M cell numbers. In a previous study, we established the culture medium conditions for the enrichment of enterocytes in mouse small intestinal organoids, the so-called EN medium (12). Interestingly, the HNF4G but not ONECUT2 signature was found significantly upregulated in EN conditions (Figure 4F). Remarkably, the addition of RANKL to EN medium upregulated the levels of Onecut2, Hnf4g, and ONECUT2 targets Klf5 and Klf6 (Figures 4G), and markedly lowered the number of M cells compared to regular RANKL medium (Figure 4H). These experiments demonstrated that the ONECUT2 induction of enterocyte differentiation and inhibition on M cell lineage specification in vitro is dependent on the RANK/RANKL signalling axis (Figure 4I).
ONECUT2 limits M cell number in peyer's patches
To investigate the physiological relevance of our findings in vivo, mice were treated with CSRM617 as described in Supplementary Figure S3D. Because RANKL is naturally highly expressed within the PP of the small intestine (59,60), we assessed the protein expression of M cell markers in this tissue. Reassuringly, treatment with CSRM617 significantly increased the number of SPIB+ and GP2+ cells in the PP, compared to control treatment (Figure 5A). In control mice sections, in line with previous reports (90), nuclear SPIB was observed mostly in cells from the FAE crypts while GP2 signal was detected in cells at the top of the PP dome (Figure 5A). Interestingly, in CSRM617 conditions SPIB+ cells were found localized in both the FAE crypts and the PP domes. This displacement is possibly caused by the increase in the number of SPIB+ cells (Figure 5A). Consistently, treatment induced an upregulation of M cell markers Spib, Gp2 and Tnfaip2 in the small intestine tissue, all of which increased in time (Figure 5B). While the expression of the transcription factor and M cell driver Spib was significantly downregulated immediately after termination of the treatment, mature M cell markers Gp2 and Tnfaip2 (9) remained expressed at levels higher than in control conditions even after a week of recovery from treatment, likely reflecting the turnover of M cells (Figure 5B). Treatment achieved downregulation of ONECUT2-targeted genes Klf5, Klf6, Hoxb8, Foxn3 and Tfcp2l1, similar to the effect observed in organoids (Figure 5C). ONECUT2 inhibition also led to a reduction in the levels of Hnf4g (Figure 5C) but did not affect the transcript levels of markers from other cell types in the small intestine such as Dclk1 (Tuft), Defa24 (Paneth) or Reg4 (Enteroendocrine) (Supplementary Figure S3E), reiterating on the RANKL-dependent role of ONECUT2 to cells of the absorptive lineage.
Figure 5.

ONECUT2 regulates M cell number and mucosal immunity in vivo. (A) Immunohistochemistry analysis for GP2 and SPIB protein expression in Peyer's patches of small intestine sections from mice treated with PBS control or CSRM617 for 14 days. Nuclear counterstaining with hematoxylin (bright field) or DAPI (fluorescence) is shown. Scale bars are annotated. Dotted rectangles show amplified areas. Yellow arrow heads show GP2+ cells and red arrow heads mark autofluorescence signal. Quantification of SPIB+ cells from n = 6 Peyer's patches sections from mice treated as in (A). (B) RT-qPCR analysis showing relative mRNA expression of M cell markers Spib, Gp2 and Tnfaip2 in small intestine sections from mice treated as described in Supplementary Figure S3D. Unpaired t test P <0.05 is represented by (*) and P <0.01 by (**). (C) Relative mRNA expression of ONECUT2-targeted genes Klf5, Klf6, Hoxb8, Foxn3, Tfcp2l1 and Hnf4g detected in tissue described in (B). Unpaired t test P <0.05 is represented by (*), P <0.01 by (**) and P <0.001 by (***). (D) Secretory IgA (SIgA) levels in faecal samples from mice treated as described in (B). Unpaired t test P <0.05 is represented by (*). (E) Relative mRNA expression of markers from IgA+ B cells Ccr9 and Itgb7 detected in tissue from (B).
The organized lymphoid follicles underlying the PP are the main source of secretory IgA (SIgA) (7), the body's first line of defence in response to toxins and bacterial and viral infections (91). The unique ability of M cells to take up and transport antigens from the intestinal lumen to the underlying lymphoid tissue is the first step in the production of SIgA by IgA+ B cells (92,93). Changes in the levels of SIgA directly reflect variations in M cell numbers and their transcytosis and antigen-presenting capabilities (15,93–95). ELISA experiments for targeted detection of mouse IgA in faecal samples of mice treated with CSRM617 showed a time-dependent increase in the levels of SIgA, when compared to control samples (Figure 5D). SIgA levels dropped again a week after receiving control treatment (Figure 5D), consistent with the expression levels of M cell markers discussed above. SIgA levels also correlated with the expression of Ccr9 and Itgb7, markers of IgA+ B cells (96) (Figure 5E). The weight of mice treated with CSRM617 was comparable to that of littermate controls and no difference was observed between genders (Supplementary Figure S3F). Based on these observations, we conclude that ONECUT2 restricts the M cell number in the PP, indirectly regulating B cell functions and SIgA production.
DISCUSSION
In this study, we used multiple omics platforms and diverse computational tools to define the molecular landscape of M cell-enriched mouse small intestinal organoids in an unbiased, comprehensive manner. By combining the strengths of bulk sequencing methods (e.g. small input material, sequencing depth) and single-cell approaches (e.g. characterisation of small populations, trajectory inference tools) to generate time-resolved data, we untangled complex gene-regulatory networks that contribute to the M cell lineage specification.
Our integrative approach revealed that ONECUT2 acts as a negative regulator of M cell differentiation in vitro and in vivo, by supporting enterocyte differentiation in a RANKL-dependent manner. Under RANKL conditions, blockage of ONECUT2-mediated transcriptional activity led to an increase in the number of M cells (Spib+, Anxa5+, Rac2+) and a decrease in the enterocytes cell population (Hnf4g+, Pglyrp2+, Alpi+). A low M cell number has been proposed to be needed for the maintenance of proper intestinal barrier function and to prevent an over-reactive immune response (97), and regulating ONECUT2 expression could be one of the mechanisms ensuring this balance. The elevated expression of IgA+ B cell markers and high levels of intestinal SIgA we observed in vivo upon ONECUT2 inhibition supports this hypothesis. Moreover, the expression of Klf5, a transcription factor predicted to be target of ONECUT2 has been shown to protect mice against murine colitis by activating the JAK-STAT signalling pathway (98). On the other hand, too low M cell numbers also compromise the mucosal immune response. The density of mature M cells in the FAE of aged mice is significantly lower than in young mice (99). Consequently, aged mice PP are deficient in their ability to promote transcytosis of luminal antigen particles across the FAE and to recruit B cells to subepithelial domes (99). Based on its capacity to induce M cell differentiation, CSRM617 could be used to revert the age-related decline in mucosal immune response.
Since their discovery in 1974 (100), there have been many questions regarding the mechanisms supporting and restricting M cell differentiation in the small intestine (101). The establishment of the key role that RANK/RANKL signalling plays in M cell differentiation has allowed the development of in vitro and in vivo models that have helped us answering some of these questions. The RANKL-dependent ONECUT2 signalling we described provide the first insight into the mechanisms that negatively regulate M cell differentiation in the small intestine. Single-cell RNA sequencing analysis in RANKL-treated small intestinal organoids showed us that at a transcriptome level M cells share a lot of similarities with enterocytes, similarities that separate these two cell types from cells of the secretory lineage. This is consistent with the transcriptome profiles observed in the FAE of the mouse small intestine (9) and supports the commonly accepted theory that enterocytes and M cells have a common precursor. Our study represents the first piece of evidence showing that upon RANKL stimulation both cell types originate from a common progenitor cell population.
Onecut2 was one of the transcription factors with the highest expression and motif activity in the cell population precursor for M cells and enterocytes. Expression of Onecut2 was correlated with that of Nfkb2 and Relb, transcription factors responsible for transcriptional activation of Spib and Sox8 (64) and which expression in the small intestine are restricted to follicle-associated crypts (102). Motifs for both NFKB2 and RELB were found within Onecut2 enhancers, supporting a potential role for RELB-p52 dimer in the transcriptional regulation of Onecut2.
We have created a new computational tool we named SCEPIA (76) and validated it for its capacity to predict transcription factor motif activity from single cell RNA-sequencing data, to identify those transcription factors, like ONECUT2, that are determinant of cellular states. Likewise, we have generated several resource datasets that can be used to perform different analyses, for instance, to uncover additional regulatory axes underlying RANKL signalling, or as reference datasets of the chromatin remodelling and transcriptional responses to ONECUT2-transcription factor activity. In human and mouse, ONECUT2 is highly expressed in the liver, pancreas, small intestine and the nervous system. In addition, ONECUT2 expression is significantly associated with poor clinical outcome in diverse type of cancers such as prostate, breast, gastric, colon, clear cell renal, brain, and lung cancer (89). The systemic administration of the ONECUT2 inhibitor CSRM617 in mice, described in this study, generated tissue resources that can be used to study the many diverse roles of the ONECUT2 in different tissues and in the context of health and disease.
Limitations of study
Mouse small intestinal organoids are a powerful and versatile in vitro model, which resembles the in vivo intestinal epithelium at a cellular, molecular and functional level, thereby facilitating the study of complex biological processes such as stem cell renewal and cell differentiation. Though organoids typically consist of several distinct epithelial cell types, these cultures lack other non-epithelial cells (i.e. stromal, mesenchymal or immune cells), which represent a source of factors that can be important for the cellular maturation and/or differentiation of epithelial cells. For M cells, beside MAdCAM-1− mesenchymal cells, which express RANKL, lympho-epithelial cell interactions such as those with B cells have been shown to play a role in the induction of M cell differentiation (103,104). In addition, it has been shown that M cell differentiation can be induced in response to external stimuli such as a pathogen infection (102) but incorporating all these factors in a culture with organoids can be challenging. Thus, it remains to be answered whether pathogen-induced signalling would overrule inhibitory M cell differentiation signals such as those mediated by ONECUT2, uncovered in this study.
CONCLUSIONS
In this study, we have shown that the ONECUT2 transcription factor acts downstream of RANK/RANKL signalling to orchestrate enterocyte and M cell lineage specification in vitro and in vivo. Our perturbation studies demonstrated that ONECUT2 in fact restricts M cell differentiation within the FAE which has an impact in mucosal immunity as the levels of SIgA, the body's first line immune defence, is a direct consequence of M cell function.
While the focus of this study resulted in the discovery of a transcription factor as a regulator of intestinal enterocyte and M cell differentiation, the computational workflows described here can serve as an outline for future endeavours aimed at deciphering gene expression regulation of stem cell fate and differentiation in the intestine and other multicellular tissues. Over the last years, different computational tools have been designed to model complex gene-regulatory networks and to facilitate the integration of multiple data types (i.e. transcriptome, genome) of different resolutions (i.e. bulk, single-cell). Though each tool is informative on its own, we found that the integration of all these was essential for the elaboration of the conclusions presented in this study.
DATA AVAILABILITY
All sequencing data generated in this study (i.e. FASTQ files) as well as processed data (i.e. raw and normalized counts) have been deposited at GEO and are publicly available in the SuperSeries record GSE194184 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE194184 which links to SubSeries for ATAC-sequencing (GSE194183; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE194183), ChIP-sequencing (GSE194182; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE194182), bulk RNA-sequencing (GSE194177; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE194177) and single-cell RNA-sequencing data (GSE194178; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE194178).
The code to carry out the analysis described in this study with SCEPIA (76) v0.5.1 is available at Zenodo DOI: 10.5281/zenodo.7420128 (https://zenodo.org/record/7420128).
Any additional information required to reanalyse the data reported in this paper is available from Dr. Michiel Vermeulen (michiel.vermeulen@science.ru.nl) upon reasonable request.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank the members of the Vermeulen laboratory for valuable suggestions and troubleshooting. We thank Marijke Baltissen and Lieke Lamers from the Sequencing facility from the department of Molecular Biology from Radboud University (RU) for their help with next generation sequencing, also Rob Woestenenk and Tom van Oorschot from the Radboud Technology Center for Flow Cytometry, and Laura Wingens from the department of Molecular Developmental Biology at RU for their training and assistance with fluorescence-activated cell sorting and flow cytometry. We would like to thank Marieke Willemse from the Radboud Technology Center for Microscopy for the training and assistance with microscopy. The authors also thank the Pathology laboratory at the Cedars Sinai Medical Center and Birgitte Walgreen from the department of Rheumatology at Radboudumc for the technical support with tissue preparation and immunohistochemistry, and Yanpeng Xing from the laboratory of Michael Jung at the University of California, Los Angeles, who synthesized the CSRM617 compound for the in vivo studies. Lastly, the authors thank Jos Smits and the members from the van Heeringen group at RU for their support and fruitful discussions.
Author contributions: M.V.L.V. and M.V. conceived the study and contributed to the experimental design. M.V.L.V., H.N., A.M. and C.Q. performed experiments. M.V.L.V., H.N., R.R.S. and Y.Q. performed data analysis. C.Q. and M.R.F. carried out mice treatment. S.J.v.H. created the computational tool SCEPIA. M.V.L.V. prepared the manuscript. H.N., R.R.S., Y.Q., C.Q., A.M., G.J.C.V., M.R.F., S.J.v.H. and M.V. revised and approved the final version of the manuscript.
Contributor Information
Maria V Luna Velez, Department of Molecular Biology, Radboud University Nijmegen, Faculty of Science, Radboud Institute for Molecular Life Sciences, Oncode Institute, Nijmegen 6525 AJ, The Netherlands.
Hannah K Neikes, Department of Molecular Biology, Radboud University Nijmegen, Faculty of Science, Radboud Institute for Molecular Life Sciences, Oncode Institute, Nijmegen 6525 AJ, The Netherlands.
Rebecca R Snabel, Department of Molecular Developmental Biology, Radboud University Nijmegen, Faculty of Science, Radboud Institute for Molecular Life Sciences, Nijmegen 6525 AJ, The Netherlands.
Yarah Quint, Department of Molecular Biology, Radboud University Nijmegen, Faculty of Science, Radboud Institute for Molecular Life Sciences, Oncode Institute, Nijmegen 6525 AJ, The Netherlands.
Chen Qian, Department of Surgery, Division of Cancer Biology, Cedars-Sinai Medical Center, Los Angeles, CA 90048, USA.
Aniek Martens, Department of Molecular Biology, Radboud University Nijmegen, Faculty of Science, Radboud Institute for Molecular Life Sciences, Oncode Institute, Nijmegen 6525 AJ, The Netherlands.
Gert Jan C Veenstra, Department of Molecular Developmental Biology, Radboud University Nijmegen, Faculty of Science, Radboud Institute for Molecular Life Sciences, Nijmegen 6525 AJ, The Netherlands.
Michael R Freeman, Department of Surgery, Division of Cancer Biology, Cedars-Sinai Medical Center, Los Angeles, CA 90048, USA.
Simon J van Heeringen, Department of Molecular Developmental Biology, Radboud University Nijmegen, Faculty of Science, Radboud Institute for Molecular Life Sciences, Nijmegen 6525 AJ, The Netherlands.
Michiel Vermeulen, Department of Molecular Biology, Radboud University Nijmegen, Faculty of Science, Radboud Institute for Molecular Life Sciences, Oncode Institute, Nijmegen 6525 AJ, The Netherlands.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
ERC Consolidator Grant [771059 to M.V.]; Vermeulen lab is part of the Oncode Institute, which is partly funded by the Dutch Cancer Society (to K.W.F.); In vivo studies were funded by the NCI [1R01CA220327]; US Department of Defense [PC180541]. Funding for open access charge: ERC Consolidator Grant [771059 to M.V.].
Conflict of interest statement. CSRM617 is under review for patent protection submitted by Cedars Sinai Medical Center (application number: 17/145,152).
REFERENCES
- 1. Kraehenbuhl J.P., Neutra M.R.. Epithelial M cells: differentiation and function. Annu. Rev. Cell Dev. Biol. 2000; 16:301–332. [DOI] [PubMed] [Google Scholar]
- 2. Corr S.C., Gahan C.C.G.M., Hill C.. M-cells: origin, morphology and role in mucosal immunity and microbial pathogenesis. FEMS Immunol. Med. Microbiol. 2008; 52:2–12. [DOI] [PubMed] [Google Scholar]
- 3. Amerongen H.M., Weltzin R., Farnet C.M., Michetti P., Haseltine W.A., Neutra M.R.. Transepithelial transport of HIV-1 by intestinal M cells: a mechanism for transmission of AIDS. J. Acquir. Immune Defic. Syndr. 1991; 4:760–765. [PubMed] [Google Scholar]
- 4. Amerongen H.M., Weltzin R., Mack J.A., Winner L.S., Michetti P., Apter F.M., Kraehenbuhl J.P., Neutra M.R.. M cell-mediated antigen transport and monoclonal IgA antibodies for mucosal immune protection. Ann. N. Y. Acad. Sci. 1992; 664:18–26. [DOI] [PubMed] [Google Scholar]
- 5. Neutra M.R., Frey A., Kraehenbuhl J.P.. Epithelial M cells: gateways for mucosal infection and immunization. Cell. 1996; 86:345–348. [DOI] [PubMed] [Google Scholar]
- 6. Gonzalez-Hernandez M.B., Liu T., Payne H.C., Stencel-Baerenwald J.E., Ikizler M., Yagita H., Dermody T.S., Williams I.R., Wobus C.E.. Efficient norovirus and reovirus replication in the mouse intestine requires microfold (M) cells. J. Virol. 2014; 88:6934–6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fagarasan S., Honjo T.. Regulation of IgA synthesis at mucosal surfaces. Curr. Opin. Immunol. 2004; 16:277–283. [DOI] [PubMed] [Google Scholar]
- 8. de Lau W., Kujala P., Schneeberger K., Middendorp S., Li V.S.W., Barker N., Martens A., Hofhuis F., DeKoter R.P., Peters P.J.et al.. Peyer's patch M cells derived from Lgr5(+) stem cells require SpiB and are induced by RankL in cultured ‘miniguts. Mol. Cell. Biol. 2012; 32:3639–3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haber A.L., Biton M., Rogel N., Herbst R.H., Shekhar K., Smillie C., Burgin G., Delorey T.M., Howitt M.R., Katz Y.et al.. A single-cell survey of the small intestinal epithelium. Nature. 2017; 551:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hase K., Ohshima S., Kawano K., Hashimoto N., Matsumoto K., Saito H., Ohno H.. Distinct gene expression profiles characterize cellular phenotypes of follicle-associated epithelium and M cells. DNA Res. 2005; 12:127–137. [DOI] [PubMed] [Google Scholar]
- 11. Sato T., Vries R.G., Snippert H.J., van de Wetering M., Barker N., Stange D.E., van Es J.H., Abo A., Kujala P., Peters P.J.et al.. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009; 459:262–265. [DOI] [PubMed] [Google Scholar]
- 12. Lindeboom R.G., van Voorthuijsen L., Oost K.C., Rodríguez-Colman M.J., Luna-Velez M.V, Furlan C., Baraille F., Jansen P.W., Ribeiro A., Burgering B.M.et al.. Integrative multi-omics analysis of intestinal organoid differentiation. Mol. Syst. Biol. 2018; 14:e8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wester R.A., van Voorthuijsen L., Neikes H.K., Dijkstra J.J., Lamers L.A., Frölich S., van der Sande M., Logie C., Lindeboom R.G.H., Vermeulen M.. Retinoic acid signaling drives differentiation toward the absorptive lineage in colorectal cancer. Iscience. 2021; 24:103444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Taylor R.T., Patel S.R., Lin E., Butler B.R., Lake J.G., Newberry R.D., Williams I.R.. Lymphotoxin-independent expression of TNF-related activation-induced cytokine by stromal cells in cryptopatches, isolated lymphoid follicles, and Peyer's patches. J. Immunol. 2007; 178:5659–5667. [DOI] [PubMed] [Google Scholar]
- 15. Knoop K.A., Kumar N., Butler B.R., Sakthivel S.K., Taylor R.T., Nochi T., Akiba H., Yagita H., Kiyono H., Williams I.R.. RANKL is necessary and sufficient to initiate development of antigen-sampling M cells in the intestinal epithelium. J. Immunol. 2009; 183:5738–5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moolenbeek C., Ruitenberg E.J.. The ‘Swiss roll’: a simple technique for histological studies of the rodent intestine. Lab. Anim. 1981; 15:57–59. [DOI] [PubMed] [Google Scholar]
- 17. Tian H., Biehs B., Warming S., Leong K.G., Rangell L., Klein O.D., de Sauvage F.J.. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature. 2011; 478:255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J.. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013; 10:1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B.et al.. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017; 14:959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schmidl C., Rendeiro A.F., Sheffield N.C., Bock C.. ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nat. Methods. 2015; 12:963–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gerlach J.P., van Buggenum J.A.G., Tanis S.E.J., Hogeweg M., Heuts B.M.H., Muraro M.J., Elze L., Rivello F., Rakszewska A., van Oudenaarden A.et al.. Combined quantification of intracellular (phospho-)proteins and transcriptomics from fixed single cells. Sci. Rep. 2019; 9:1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hashimshony T., Senderovich N., Avital G., Klochendler A., de Leeuw Y., Anavy L., Gennert D., Li S., Livak K.J., Rozenblatt-Rosen O.et al.. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016; 17:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Anders S., Pyl P.T., Huber W.. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gu Z., Eils R., Schlesner M.. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016; 32:2847–2849. [DOI] [PubMed] [Google Scholar]
- 27. van der Sande M., Frölich S., Smits J., van Heeringen S.. seq2science (v0.3.1). 2020; Zenodo 10.5281/zenodo.4246844. [DOI]
- 28. Chen S., Zhou Y., Chen Y., Gu J.. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018; 34:i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013; arXiv doi:26 May 2013, preprint: not peer reviewedhttps://arxiv.org/abs/1303.3997.
- 30. Amemiya H.M., Kundaje A., Boyle A.P.. The ENCODE blacklist: identification of problematic regions of the genome. Sci. Rep. 2019; 9:9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W.et al.. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ross-Innes C.S., Stark R., Teschendorff A.E., Holmes K.A., Ali H.R., Dunning M.J., Brown G.D., Gojis O., Ellis I.O., Green A.R.et al.. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012; 481:389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G.. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010; 28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bruse N., van Heeringen S.J.. GimmeMotifs: an analysis framework for transcription factor motif analysis. 2018; bioRxiv doi:20 November 2018, preprint: not peer reviewed 10.1101/474403. [DOI]
- 36. Xu Q., Georgiou G., Frölich S., van der Sande M., Veenstra G.J.C., Zhou H., van Heeringen S.J.. ANANSE: an enhancer network-based computational approach for predicting key transcription factors in cell fate determination. Nucleic Acids Res. 2021; 49:7966–7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van Heeringen S.J. genomepy: download genomes the easy way. 2017; Zenodo 10.5281/zenodo.831969. [DOI]
- 38. Bray N.L., Pimentel H., Melsted P., Pachter L.. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016; 34:525–527. [DOI] [PubMed] [Google Scholar]
- 39. Soneson C., Love M.I., Robinson M.D.. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research. 2015; 4:1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Melsted P., Booeshaghi A.S., Liu L., Gao F., Lu L., Min K.H. (Joseph), da Veiga Beltrame E., Hjörleifsson K.E., Gehring J., Pachter L.. Modular, efficient and constant-memory single-cell RNA-seq preprocessing. Nat. Biotechnol. 2021; 39:813–818. [DOI] [PubMed] [Google Scholar]
- 41. McCarthy D.J., Campbell K.R., Lun A.T.L., Wills Q.F.. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics. 2017; 33:1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M. 3rd, Hao Y., Stoeckius M., Smibert P., Satija R.. Comprehensive Integration of Single-Cell Data. Cell. 2019; 177:1888–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. La Manno G., Soldatov R., Zeisel A., Braun E., Hochgerner H., Petukhov V., Lidschreiber K., Kastriti M.E., Lönnerberg P., Furlan A.et al.. RNA velocity of single cells. Nature. 2018; 560:494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cao J., Spielmann M., Qiu X., Huang X., Ibrahim D.M., Hill A.J., Zhang F., Mundlos S., Christiansen L., Steemers F.J.et al.. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019; 566:496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mao Q., Yang L., Wang L., Goodison S., Sun Y.. SimplePPT: a Simple Principal Tree Algorithm. Proceedings of the 2015 SIAM International Conference on Data Mining (SDM). 2015; Society for Industrial and Applied Mathematics; 792–800.Proceedings. [Google Scholar]
- 46. Mao Q., Wang L., Tsang I.W., Sun Y.. Principal graph and structure learning based on reversed graph embedding. IEEE Trans. Pattern Anal. Mach. Intell. 2017; 39:2227–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moran P.A.P. Notes on continuous stochastic phenomena. Biometrika. 1950; 37:17–23. [PubMed] [Google Scholar]
- 48. ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Davis C.A., Hitz B.C., Sloan C.A., Chan E.T., Davidson J.M., Gabdank I., Hilton J.A., Jain K., Baymuradov U.K., Narayanan A.K.et al.. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018; 46:D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang S., Zang C., Xiao T., Fan J., Mei S., Qin Q., Wu Q., Li X., Xu K., He H.H.et al.. Modeling cis-regulation with a compendium of genome-wide histone H3K27ac profiles. Genome Res. 2016; 26:1417–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van de Sande B., Flerin C., Davie K., De Waegeneer M., Hulselmans G., Aibar S., Seurinck R., Saelens W., Cannoodt R., Rouchon Q.et al.. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 2020; 15:2247–2276. [DOI] [PubMed] [Google Scholar]
- 52. Moerman T., Santos Aibar, BravoGonzález-Blas S., Simm C., Moreau J., Aerts Y., Aerts S. GRNBoost2 and Arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics. 2019; 35:2159–2161. [DOI] [PubMed] [Google Scholar]
- 53. Warnes G., Bolker B., Bonebakker L., Gentleman R., Huber W., Liaw A., Lumley T., Mächler M., Magnusson A., Möller S.. gplots: various R programming tools for plotting data. 2005;
- 54. Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A.. Fast gene set enrichment analysis. 2021; bioRxiv doi:01 February 2021, preprint: not peer reviewed 10.1101/060012. [DOI]
- 55. van der Raadt J., van Gestel S.H.C., Nadif Kasri N., Albers C.A.. ONECUT transcription factors induce neuronal characteristics and remodel chromatin accessibility. Nucleic Acids Res. 2019; 47:5587–5602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001; 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schneider C.A., Rasband W.S., Eliceiri K.W.. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012; 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cai E., Thomas R.A.. OrgM: a Fiji macro for automated measurement of object area, diameter and roundness from bright field images. 2019;
- 59. Nagashima K., Sawa S., Nitta T., Tsutsumi M., Okamura T., Penninger J.M., Nakashima T., Takayanagi H.. Identification of subepithelial mesenchymal cells that induce IgA and diversify gut microbiota. Nat. Immunol. 2017; 18:675–682. [DOI] [PubMed] [Google Scholar]
- 60. Nagashima K., Sawa S., Nitta T., Prados A., Koliaraki V., Kollias G., Nakashima T., Takayanagi H.. Targeted deletion of RANKL in M cell inducer cells by the Col6a1-Cre driver. Biochem. Biophys. Res. Commun. 2017; 493:437–443. [DOI] [PubMed] [Google Scholar]
- 61. Kobayashi A., Donaldson D.S., Kanaya T., Fukuda S., Baillie J.K., Freeman T.C., Ohno H., Williams I.R., Mabbott N.A.. Identification of novel genes selectively expressed in the follicle-associated epithelium from the meta-analysis of transcriptomics data from multiple mouse cell and tissue populations. DNA Res. 2012; 19:407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Barker N., van Es J.H., Kuipers J., Kujala P., van den Born M., Cozijnsen M., Haegebarth A., Korving J., Begthel H., Peters P.J.et al.. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007; 449:1003–1007. [DOI] [PubMed] [Google Scholar]
- 63. Boonekamp K.E., Dayton T.L., Clevers H.. Intestinal organoids as tools for enriching and studying specific and rare cell types: advances and future directions. J. Mol. Cell Biol. 2020; 12:562–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kobayashi N., Takahashi D., Takano S., Kimura S., Hase K.. The Roles of Peyer's Patches and Microfold Cells in the Gut Immune System: relevance to Autoimmune Diseases. Front. Immunol. 2019; 10:2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A.et al.. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kanaya T., Hase K., Takahashi D., Fukuda S., Hoshino K., Sasaki I., Hemmi H., Knoop K.A., Kumar N., Sato M.et al.. The Ets transcription factor Spi-B is essential for the differentiation of intestinal microfold cells. Nat. Immunol. 2012; 13:729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kimura S., Yamakami-Kimura M., Obata Y., Hase K., Kitamura H., Ohno H., Iwanaga T.. Visualization of the entire differentiation process of murine M cells: suppression of their maturation in cecal patches. Mucosal Immunol. 2015; 8:650–660. [DOI] [PubMed] [Google Scholar]
- 68. Kimura S., Kobayashi N., Nakamura Y., Kanaya T., Takahashi D., Fujiki R., Mutoh M., Obata Y., Iwanaga T., Nakagawa T.et al.. Sox8 is essential for M cell maturation to accelerate IgA response at the early stage after weaning in mice. J. Exp. Med. 2019; 216:831–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Potten C.S. Stem cells in gastrointestinal epithelium: numbers, characteristics and death. Philos. Trans. R. Soc. Lond. Ser. B, Biol. Sci. 1998; 353:821–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lo D., Tynan W., Dickerson J., Mendy J., Chang H.-W., Scharf M., Byrne D., Brayden D., Higgins L., Evans C.et al.. Peptidoglycan recognition protein expression in mouse Peyer's Patch follicle associated epithelium suggests functional specialization. Cell. Immunol. 2003; 224:8–16. [DOI] [PubMed] [Google Scholar]
- 71. Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J.et al.. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017; 14:1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen L., Toke N.H., Luo S., Vasoya R.P., Fullem R.L., Parthasarathy A., Perekatt A.O., Verzi M.P.. A reinforcing HNF4-SMAD4 feed-forward module stabilizes enterocyte identity. Nat. Genet. 2019; 51:777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu H., Tang X., Srivastava A., Pécot T., Daniel P., Hemmelgarn B., Reyes S., Fackler N., Bajwa A., Kladney R.et al.. Redeployment of Myc and E2f1-3 drives Rb-deficient cell cycles. Nat. Cell Biol. 2015; 17:1036–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Guillermin O., Angelis N., Sidor C.M., Ridgway R., Baulies A., Kucharska A., Antas P., Rose M.R., Cordero J., Sansom O.et al.. Wnt and Src signals converge on YAP-TEAD to drive intestinal regeneration. EMBO J. 2021; 40:e105770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li Q., Sun Y., Jarugumilli G.K., Liu S., Dang K., Cotton J.L., Xiol J., Chan P.Y., DeRan M., Ma L.et al.. Lats1/2 Sustain Intestinal Stem Cells and Wnt Activation through TEAD-Dependent and Independent Transcription. Cell Stem Cell. 2020; 26:675–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. van Heeringen S.J. vanheeringen-lab/scepia: version 0.5.1 (0.5.1). 2021; Zenodo 10.5281/zenodo.4892888. [DOI]
- 77. Kim C.-K., He P., Bialkowska A.B., Yang V.W.. SP and KLF transcription factors in digestive physiology and diseases. Gastroenterology. 2017; 152:1845–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nandan M.O., Yang V.W.. The role of Krüppel-like factors in the reprogramming of somatic cells to induced pluripotent stem cells. Histol. Histopathol. 2009; 24:1343–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nandan M.O., Ghaleb A.M., Bialkowska A.B., Yang V.W.. Krüppel-like factor 5 is essential for proliferation and survival of mouse intestinal epithelial stem cells. Stem Cell Res. 2015; 14:10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Nigmatullina L., Norkin M., Dzama M.M., Messner B., Sayols S., Soshnikova N.. Id2 controls specification of Lgr5(+) intestinal stem cell progenitors during gut development. EMBO J. 2017; 36:869–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gregorieff A., Pinto D., Begthel H., Destrée O., Kielman M., Clevers H.. Expression pattern of Wnt signaling components in the adult intestine. Gastroenterology. 2005; 129:626–638. [DOI] [PubMed] [Google Scholar]
- 82. McConnell B.B., Yang V.W.. Mammalian Krüppel-like factors in health and diseases. Physiol. Rev. 2010; 90:1337–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dusing M.R., Maier E.A., Aronow B.J., Wiginton D.A.. Onecut-2 knockout mice fail to thrive during early postnatal period and have altered patterns of gene expression in small intestine. Physiol. Genomics. 2010; 42:115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Korinek V., Barker N., Moerer P., van Donselaar E., Huls G., Peters P.J., Clevers H.. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat. Genet. 1998; 19:379–383. [DOI] [PubMed] [Google Scholar]
- 85. Chen L., Toke N.H., Luo S., Vasoya R.P., Aita R., Parthasarathy A., Tsai Y.-H., Spence J.R., Verzi M.P.. HNF4 factors control chromatin accessibility and are redundantly required for maturation of the fetal intestine. Development. 2019; 146:dev179432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lemaigre F.P., Durviaux S.M., Truong O., Lannoy V.J., Hsuan J.J., Rousseau G.G.. Hepatocyte nuclear factor 6, a transcription factor that contains a novel type of homeodomain and a single cut domain. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:9460–9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Vanhorenbeeck V., Jacquemin P., Lemaigre F.P., Rousseau G.G.. OC-3, a novel mammalian member of the ONECUT class of transcription factors. Biochem. Biophys. Res. Commun. 2002; 292:848–854. [DOI] [PubMed] [Google Scholar]
- 88. Kropp P.A., Gannon M.. Onecut transcription factors in development and disease. Trends Dev. Biol. 2016; 9:43–57. [PMC free article] [PubMed] [Google Scholar]
- 89. Rotinen M., You S., Yang J., Coetzee S.G., Reis-Sobreiro M., Huang W.-C., Huang F., Pan X., Yáñez A., Hazelett D.J.et al.. ONECUT2 is a targetable master regulator of lethal prostate cancer that suppresses the androgen axis. Nat. Med. 2018; 24:1887–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Nakamura Y., Kimura S., Hase K.. M cell-dependent antigen uptake on follicle-associated epithelium for mucosal immune surveillance. Inflamm. Regen. 2018; 38:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Corthésy B. Multi-faceted functions of secretory IgA at mucosal surfaces. Front. Immunol. 2013; 4:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Macpherson A.J., Uhr T.. Compartmentalization of the mucosal immune responses to commensal intestinal bacteria. Ann. N. Y. Acad. Sci. 2004; 1029:36–43. [DOI] [PubMed] [Google Scholar]
- 93. Rios D., Wood M.B., Li J., Chassaing B., Gewirtz A.T., Williams I.R.. Antigen sampling by intestinal M cells is the principal pathway initiating mucosal IgA production to commensal enteric bacteria. Mucosal Immunol. 2016; 9:907–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Martinoli C., Chiavelli A., Rescigno M.. Entry route of Salmonella typhimurium directs the type of induced immune response. Immunity. 2007; 27:975–984. [DOI] [PubMed] [Google Scholar]
- 95. Hashizume T., Togawa A., Nochi T., Igarashi O., Kweon M.-N., Kiyono H., Yamamoto M.. Peyer's patches are required for intestinal immunoglobulin A responses to Salmonella spp. Infect. Immun. 2008; 76:927–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wilmore J.R., Gaudette B.T., Gómez Atria D., Rosenthal R.L., Reiser S.K., Meng W., Rosenfeld A.M., Luning Prak E.T., Allman D. IgA plasma cells are long-lived residents of gut and bone marrow that express isotype- and tissue-specific gene expression patterns. Front. Immunol. 2021; 12:791095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kucharzik T., Lügering N., Rautenberg K., Lügering A., Schmidt M.A., Stoll R., Domschke W.. Role of M cells in intestinal barrier function. Ann. N. Y. Acad. Sci. 2000; 915:171–183. [DOI] [PubMed] [Google Scholar]
- 98. Tetreault M.-P., Alrabaa R., McGeehan M., Katz J.P.. Krüppel-like factor 5 protects against murine colitis and activates JAK-STAT signaling in vivo. PLoS One. 2012; 7:e38338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kobayashi A., Donaldson D.S., Erridge C., Kanaya T., Williams I.R., Ohno H., Mahajan A., Mabbott N.A.. The functional maturation of M cells is dramatically reduced in the Peyer's patches of aged mice. Mucosal Immunol. 2013; 6:1027–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Owen R.L., Jones A.L.. Epithelial cell specialization within human Peyer's patches: an ultrastructural study of intestinal lymphoid follicles. Gastroenterology. 1974; 66:189–203. [PubMed] [Google Scholar]
- 101. Kanaya T., Williams I.R., Ohno H.. Intestinal M cells: tireless samplers of enteric microbiota. Traffic. 2020; 21:34–44. [DOI] [PubMed] [Google Scholar]
- 102. Tahoun A., Mahajan S., Paxton E., Malterer G., Donaldson D.S., Wang D., Tan A., Gillespie T.L., O’Shea M., Roe A.J.et al.. Salmonella transforms follicle-associated epithelial cells into M cells to promote intestinal invasion. Cell Host Microbe. 2012; 12:645–656. [DOI] [PubMed] [Google Scholar]
- 103. Golovkina T.V, Shlomchik M., Hannum L., Chervonsky A.. Organogenic role of B lymphocytes in mucosal immunity. Science. 1999; 286:1965–1968. [DOI] [PubMed] [Google Scholar]
- 104. Kernéis S., Bogdanova A., Kraehenbuhl J.P., Pringault E.. Conversion by Peyer's patch lymphocytes of human enterocytes into M cells that transport bacteria. Science. 1997; 277:949–952. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data generated in this study (i.e. FASTQ files) as well as processed data (i.e. raw and normalized counts) have been deposited at GEO and are publicly available in the SuperSeries record GSE194184 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE194184 which links to SubSeries for ATAC-sequencing (GSE194183; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE194183), ChIP-sequencing (GSE194182; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE194182), bulk RNA-sequencing (GSE194177; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE194177) and single-cell RNA-sequencing data (GSE194178; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE194178).
The code to carry out the analysis described in this study with SCEPIA (76) v0.5.1 is available at Zenodo DOI: 10.5281/zenodo.7420128 (https://zenodo.org/record/7420128).
Any additional information required to reanalyse the data reported in this paper is available from Dr. Michiel Vermeulen (michiel.vermeulen@science.ru.nl) upon reasonable request.