


Abstract
In countless bacterial species, the lifetimes of most mRNAs are controlled by the regulatory endonuclease RNase E, which preferentially degrades RNAs bearing a 5′ monophosphate and locates cleavage sites within them by scanning linearly from the 5′ terminus along single-stranded regions. Consequently, its rate of cleavage at distal sites is governed by any obstacles that it may encounter along the way, such as bound proteins or ribosomes or base pairing that is coaxial with the path traversed by this enzyme. Here, we report that the protection afforded by such obstacles is dependent on the size and persistence of the structural discontinuities they create, whereas the molecular composition of obstacles to scanning is of comparatively little consequence. Over a broad range of sizes, incrementally larger discontinuities are incrementally more protective, with corresponding effects on mRNA stability. The graded impact of such obstacles suggests possible explanations for why their effect on scanning is not an all-or-none phenomenon dependent simply on whether the size of the resulting discontinuity exceeds the step length of RNase E.
INTRODUCTION
Messenger RNA (mRNA) degradation is a mechanism of universal importance for controlling gene expression. By limiting the lifetime of mRNAs, this process determines how many times they can be translated by ribosomes to generate the proteins they encode. mRNA half-lives range from seconds to about an hour in bacteria and from minutes to days in vertebrates, with corresponding effects on protein synthesis.
In Escherichia coli and countless other proteobacteria, RNase E plays a critical role in controlling the lifetimes of most mRNAs (1). This low-specificity endonuclease cuts in single-stranded regions of RNA that are AU-rich (2–6), as does its paralog RNase G, whose much lower cellular concentration precludes it from making more than a minor contribution to mRNA degradation in E. coli (7). The diverse half-lives of mRNAs in vivo might seem difficult to reconcile with the relaxed cleavage-site specificity of RNase E. However, various lines of evidence suggest that their disparate lifetimes are determined not by the mere presence of susceptible cleavage sites, which are ubiquitous in mRNA, but by the ease with which RNase E is able to gain access to them (8). It does so by either of two mechanisms, one direct and the other 5′-end-dependent. The former mechanism involves binding of the enzyme directly to the sites that it cuts, while the latter is a multistep process that involves conversion of the initial 5′-terminal triphosphate to a monophosphate by the RNA pyrophosphohydrolase RppH and the recruitment of RNase E by the resulting 5′ monophosphate, which can accelerate subsequent RNA cleavage at internal sites by as much as 100-fold (9–14). Monophosphorylated 5′ ends can also be generated by endonucleolytic cleavage of triphosphorylated transcripts without the prior removal of 5′-terminal phosphates.
Interestingly, RNase E (as well as RNase G) locates cleavage sites in monophosphorylated RNA not by searching freely in three dimensions but rather by scanning linearly from the 5′ monophosphate along single-stranded RNA segments by a mechanism thought to resemble one-dimensional diffusion (15,16). As a result, its access to those sites is governed by any obstacles that it might encounter along the way, such as a bound protein or ribosome or base pairing that is coaxial with its path (e.g. an RNA pseudoknot or a bound sRNA), each of which creates a large structural discontinuity in single-stranded RNA that can protect distal sites from cleavage and thereby increase protein synthesis. By contrast, RNase E can readily bypass small discontinuities such as that created by a stem-loop orthogonal to its path, suggesting that it migrates along RNA in steps that are more than one nucleotide long. These properties are intrinsic to RNase E and do not require ancillary proteins or an external source of energy such as ATP. However, despite the importance of this process for bacterial pathogenesis, stress responses, and riboswitch mechanisms (15–17), little else is known about the mechanism of scanning or the characteristics of obstacles that govern their ability to protect distal sites from cleavage. For example, it has not been determined whether the capacity of RNase E to bypass obstacles depends on a threshold discontinuity size related to its step length, below which the impact of an obstacle is insignificant and above which it is maximal; nor is it known whether the molecular composition of obstacles influences their efficacy.
We have now investigated the relationship between the size, makeup, and longevity of structural discontinuities and their impact as obstacles to scanning by RNase E. Our findings indicate that the effect of such discontinuities on distal cleavage is graded in proportion to their size, not all-or-none, and that maximizing their influence requires their persistence over time. By contrast, the molecular composition of obstacles to scanning appears to be much less consequential.
MATERIALS AND METHODS
Strains, growth conditions, and RNA extraction
mRNA cleavage and degradation was analyzed in a derivative of the E. coli K-12 strain BW25113 (18) that lacked the chromosomal yeiP gene. Bacterial cultures were grown to log phase at 37°C in MOPS medium (19) containing 0.2% glucose, and total RNA was harvested by hot phenol extraction (15) and stored at –20°C. The effect of retapamulin was examined by adding it (100 μg/ml) to log-phase cultures 5 min before harvesting RNA.
Plasmids
Reporters for testing the efficacy of obstacles to scanning by RNase E in E. coli were all derived from plasmid pAC3 (15), which encodes a modified E. coli yeiP transcript whose extended 5′ UTR comprises a BglII restriction site flanked on both sides by multiple RNase E cleavage sites. The reporter plasmids (Table S1) were constructed by substituting DNA encoding various obstacles for the (AC)3 hexanucleotide located immediately upstream of the BglII site (Table S2).
Analysis of 5′ UTR cleavage
To resolve the various decay intermediates of interest by electrophoresis, the reporter transcripts were first subjected to site-specific cleavage with DZyeiP69, a 10–23 deoxyribozyme (20) that cuts in yeiP codon 12 (Table S3) (21). This was accomplished by combining total cellular RNA (10 μg) with 600 pmol of DZyeiP69 in a total volume of 36 μl, heating the mixture to 85°C for 5 min, and then slowly cooling it to room temperature. A buffer containing 500 mM Tris–HCl (pH 7.5), 100 mM MgCl2, and 100 mM dithiothreitol (4 μl) was added, and deoxyribozyme-catalyzed cleavage was allowed to proceed for 4 hr at 37°C. The reaction was quenched with 3 mM EDTA (210 μl), and the products were recovered by ethanol precipitation, separated by electrophoresis on a 7.5% polyacrylamide–8 M urea gel, and examined by northern blotting. In each case, RNA was electroblotted from the gel to an Immobilon-Ny+ membrane (MilliporeSigma) and probed with a radiolabeled oligonucleotide (JR279, Supplementary Table S3) that annealed to the reporter between the 3′-most 5′ UTR cleavage site and the site of deoxyribozyme cleavage. Radioactive bands were visualized on an Amersham Typhoon Trio or Typhoon FLA 9500 IP imager (GE Healthcare) and quantified by using ImageQuant TL software (GE Healthcare).
Analysis of mRNA decay rates
Reporter mRNA half-lives were measured in log-phase cultures of E. coli by inhibiting RNA synthesis and then monitoring degradation. At time intervals after adding rifampicin (200 μg/ml) to block further transcription initiation, culture samples were chilled rapidly to 0°C, and total RNA was extracted. Reporter mRNA decay was analyzed by subjecting equal amounts of total RNA from each sample (10 μg) to site-specific cleavage with DZyeiP54, which cuts in yeiP codon 7 (Supplementary Table S3), and then examining the cleavage products by gel electrophoresis (7.5% polyacrylamide–8 M urea) and northern blotting with a radiolabeled probe (JR275, Supplementary Table S3) that annealed between the 3′-most 5′ UTR cleavage site and the site of deoxyribozyme cleavage. Radioactive bands were visualized and quantified as described above, and mRNA half-lives were calculated by linear regression analysis of band intensities.
RESULTS
Incremental effect of enlarging discontinuities created by protein binding
Previous studies in E. coli and in vitro indicate that RNase E locates cleavage sites in monophosphorylated RNA by scanning linearly from the 5′ end along single-stranded regions of RNA (15,16). As a result, cleavage at those sites can be impeded by any obstacles to scanning that this endonuclease encounters along the way. These conclusions were reached initially by examining the effect of inserting a variety of potential obstacles into the unstructured 5′ untranslated region (UTR) of AC3 mRNA, a reporter transcript whose rapid degradation in E. coli is RppH-dependent (15). This 81-nt 5′ UTR contains several RNase E cleavage sites well upstream of the ribosome-binding site and protein-coding region. Effective obstacles to scanning were found to increase the molar ratio of decay intermediates generated by 5′ UTR cleavage upstream of the obstacle versus downstream of the obstacle and to prolong the lifetime of the shortest upstream cleavage product by inhibiting further degradation (15). Tests of the effect of such obstacles in other RNA contexts corroborated these findings.
One such obstacle was created by introducing a binding site for TRAP, the tryptophan operon RNA-binding attenuation protein of Bacillus subtilis (22). This protein comprises 11 identical subunits that assemble to form a ring. In the presence of tryptophan, it binds tightly to RNA sites comprising tandem G/UAGNN pentanucleotide repeats. In the complex of TRAP with 11 such repeats, the RNA encircles the protein ring, with one pentanucleotide repeat bound to each TRAP subunit, such that the sites where RNA enters and exits the complex are in close spatial proximity to one other (23). RNAs containing as few as five repeats also bind TRAP tightly (24).
We have previously reported that, when a truncated TRAP-binding site comprising eight tandem GAGNN repeats was inserted into AC3 and the decay of the resulting transcript (TR8) was examined in E. coli cells engineered to produce TRAP, the ratio of cleavage upstream versus downstream of the site of insertion increased more than 25-fold (Figure 1) and the half-life of the shortest upstream cleavage product increased 8-fold (15). Because TRAP binding to TR8 is expected to create a 69-Å discontinuity within an otherwise unstructured 5′ UTR, the length of each step taken by RNase E as it scans RNA presumably is less than 69 Å. By contrast, TRAP binding to a reporter (TR11) containing 11 tandem GAGNN repeats prolonged the half-life of the shortest upstream cleavage product only 1.5-fold (15), apparently because the resulting structural discontinuity was too small to prevent it from being bypassed by RNase E.
Figure 1.

Protective effect of discontinuities created by TRAP binding. (A) 5′ UTR of reporter mRNAs containing a TRAP-binding site. Arrows, RNase E cleavage sites; gray ring, TRAP 11-mer; white rectangle with a jagged edge, beginning of the protein coding region; broad black line, (AC)3 spacer. (B) Cleavage within the 5′ UTR of reporter mRNAs containing a TRAP-binding site. Isogenic strains of E. coli containing each reporter mRNA and TRAP were grown to mid-log phase in the presence of tryptophan. Total RNA was extracted, and equal amounts were analyzed by northern blotting to detect cleavage within the 5′ UTR. The blot was probed with a radiolabeled oligonucleotide complementary to the coding region. M, boundary marker between the upstream and downstream cleavage sites. (C) Relative abundance of 5′ UTR cleavage products. The sum of the intensities of the bands in panel (B) resulting from cleavage upstream of the TRAP-binding site or (AC)3 spacer (A + B + C) was divided by the corresponding sum for 5′ UTR cleavage downstream (V + W + X + Y + Z). Each value is the average of three biological replicates. Error bars correspond to standard deviations.
In principle, the step length of RNase E might define a critical obstacle size, below which bypass is easy and above which bypass is nearly impossible. If so, the propensity of obstacles to be bypassed might be expected to undergo an abrupt transition at that critical length. To test this possibility, we examined the effect of discontinuities of various other sizes by inserting TRAP-binding sites comprising ten (TR10), nine (TR9) or seven (TR7) tandem GAGNN repeats, resulting in discontinuities of 35, 54 or 78 Å respectively upon TRAP binding (as estimated from the published X-ray crystal structure of TRAP bound to eleven such repeats (23)). The ratio of cleavage upstream versus downstream of the TRAP-binding site in TR11, TR10, TR9, TR8 and TR7 was found to increase steadily as a function of the size of the discontinuity before plateauing for obstacles ≥69 Å in length (Figure 1; Supplementary Table S4). No threshold discontinuity size of critical importance was evident from these experiments.
Incremental effect of enlarging discontinuities caused by pseudoknot formation
Scanning by RNase E can be impeded not only by a bound protein but also by base pairing that is coaxial with the path from the 5′ monophosphate to downstream cleavage sites, such as would result from sRNA binding or the formation of an H-type pseudoknot (15,16). Previously, we have shown that a VPK pseudoknot containing 11 coaxially aligned base pairs (25) poses a modest impediment to scanning (16). We next examined two additional pseudoknots derived from beet western yellows virus (BWYV) or human telomerase RNA (hTRΔU), which contained 8 or 15 coaxial base pairs, respectively (26,27) (Figure 2A, B). As observed for TRAP binding, the efficacy of these three pseudoknots as obstacles to scanning increased as a function of their length, as judged either by the ratio of cleavage upstream versus downstream of each pseudoknot (Figure 2C, D; Supplementary Table S5) or by the lifetime of the shortest upstream cleavage product (Figure 3; Supplementary Table S6).
Figure 2.

Protective effect of discontinuities created by pseudoknots. (A) 5′ UTR of reporter mRNAs containing a pseudoknot. Arrows, RNase E cleavage sites; white rectangle with a jagged edge, beginning of the protein coding region. (B) Secondary structure of the BWYV, VPK, hTRΔU and SARS-CoV-2 pseudoknots (25–27,32). (C) Cleavage within the 5′ UTR of reporter mRNAs containing a pseudoknot between the upstream and downstream cleavage sites. Equal amounts of total cellular RNA from isogenic strains of E. coli containing each reporter mRNA were analyzed by northern blotting to detect cleavage within the 5′ UTR. The blot was probed with a radiolabeled oligonucleotide complementary to the coding region. M, boundary marker between the upstream and downstream cleavage sites. (D) Relative abundance of 5′ UTR cleavage products. The sum of the intensities of the bands in panel (C) resulting from cleavage upstream of the pseudoknot or (AC)3 spacer was divided by the corresponding sum for 5′ UTR cleavage downstream. Each value is the average of three biological replicates. Error bars correspond to standard deviations.
Figure 3.

Effect of pseudoknots on the decay rates of reporter mRNAs and their upstream cleavage products. (A) Northern blots. Transcription was arrested by adding rifampicin to E. coli cells that contained each reporter (AC3, BWYV, VPK, hTRΔU, or SARS-CoV-2), and equal amounts of total cellular RNA isolated at time intervals thereafter were examined by northern blotting. Only bands representing the intact transcript and upstream cleavage products (A, B and C) are shown. (B) Graphs. The remaining percentage of the intact transcript (top) and cleavage product C (bottom) was plotted semilogarithmically for each reporter as a function of time after blocking further RNA synthesis. Representative experiments are shown.
To rule out the alternative possibility that the diverse effects of the various pseudoknots resulted instead from sequence differences, we investigated the consequence of increasing the length of the VPK pseudoknot. This was accomplished by extending one of its component stems by 2–4 bp while also lengthening the corresponding loop by an equivalent number of nucleotides to avoid strain that might otherwise prevent the additional base pairs from forming (VPK+2, VPK+3, VPK+4). Doing so increased the ratio of upstream versus downstream cleavage products by up to three-fold while also prolonging the lifetime of the shortest upstream cleavage product (Figure 4; Supplementary Tables S7 and S8). By contrast, inserting four unpaired nucleotides immediately downstream of the VPK pseudoknot so as to increase the spacing between the cleavage sites surrounding the pseudoknot by a comparable amount without enlarging the pseudoknot itself (VPK-AC2) had a negligible effect, as did lengthening the pseudoknot loop without also adding base pairs to the adjacent stem (Supplementary Figure S1). We conclude that the degree of protection afforded by a pseudoknot depends incrementally on the number of coaxial base pairs that it contains.
Figure 4.

Effect of extending the VPK pseudoknot. (A) Pseudoknot secondary structures. In VPK+2, VPK+3 and VPK+4, two, three, or four base pairs, respectively, were added to the VPK pseudoknot, together with two, three, or four unpaired loop nucleotides. In VPK-AC2, four unpaired nucleotides (ACAC) were added immediately downstream of an unmodified VPK pseudoknot. (B) Cleavage within the 5′ UTR of reporter mRNAs containing a pseudoknot between the upstream and downstream sites. Equal amounts of total cellular RNA from isogenic strains of E. coli containing each reporter mRNA were analyzed by northern blotting to detect cleavage within the 5′ UTR. The blot was probed with a radiolabeled oligonucleotide complementary to the coding region. M, boundary marker between the upstream and downstream cleavage sites. (C) Relative abundance of 5′ UTR cleavage products. The sum of the intensities of the bands in panel (B) resulting from cleavage upstream of the pseudoknot was divided by the corresponding sum for 5′ UTR cleavage downstream. Each value is the average of three biological replicates. Error bars correspond to standard deviations. (D) Decay rates of reporter mRNAs and their upstream cleavage products. (Left) Transcription was arrested by adding rifampicin to E. coli cells that contained VPK, VPK+4 or VPK-AC2 mRNA, and equal amounts of total cellular RNA isolated at time intervals thereafter were examined by northern blotting. Only bands representing the intact transcript and upstream cleavage products (A, B and C) are shown. (Right) The remaining percentage of the intact transcript (top) and cleavage product C (bottom) was plotted semilogarithmically as a function of time after blocking further RNA synthesis. Representative experiments are shown.
A long pseudoknot from SARS-CoV-2 (Figure 2B) was similarly effective at blocking scanning by RNase E. This pseudoknot acts in cis to promote translational frameshifting in virus-infected cells (28), where it is thought to be in equilibrium with an alternative RNA conformation comprising a pair of tandem hairpins whose loops base pair with (‘kiss’) one another (29). Lacking several 3′ flanking nucleotides needed to form the second of those stem-loops, the minimal SARS-CoV-2 RNA element that we first tested in E. coli was expected to form a stable pseudoknot, and its 15 coaxial base pairs proved to be a potent impediment to scanning (Figures 2C, D and 3). As expected, its efficacy as an obstacle was diminished somewhat by adding back the missing 3′ nucleotides, which enabled the tandem stem-loops to begin competing conformationally with the pseudoknot, and was nearly abolished by artificially extending the base-paired region of the second RNA hairpin so as to lock in the tandem stem-loops conformation (Supplementary Figures S2 and S3; Supplementary Table S9). Indeed, by largely replacing the coaxial pseudoknot with a pair of orthogonal stem-loops, the latter modification reduced the ratio of upstream to downstream cleavage products from 31 to just 2.
Figure 5.
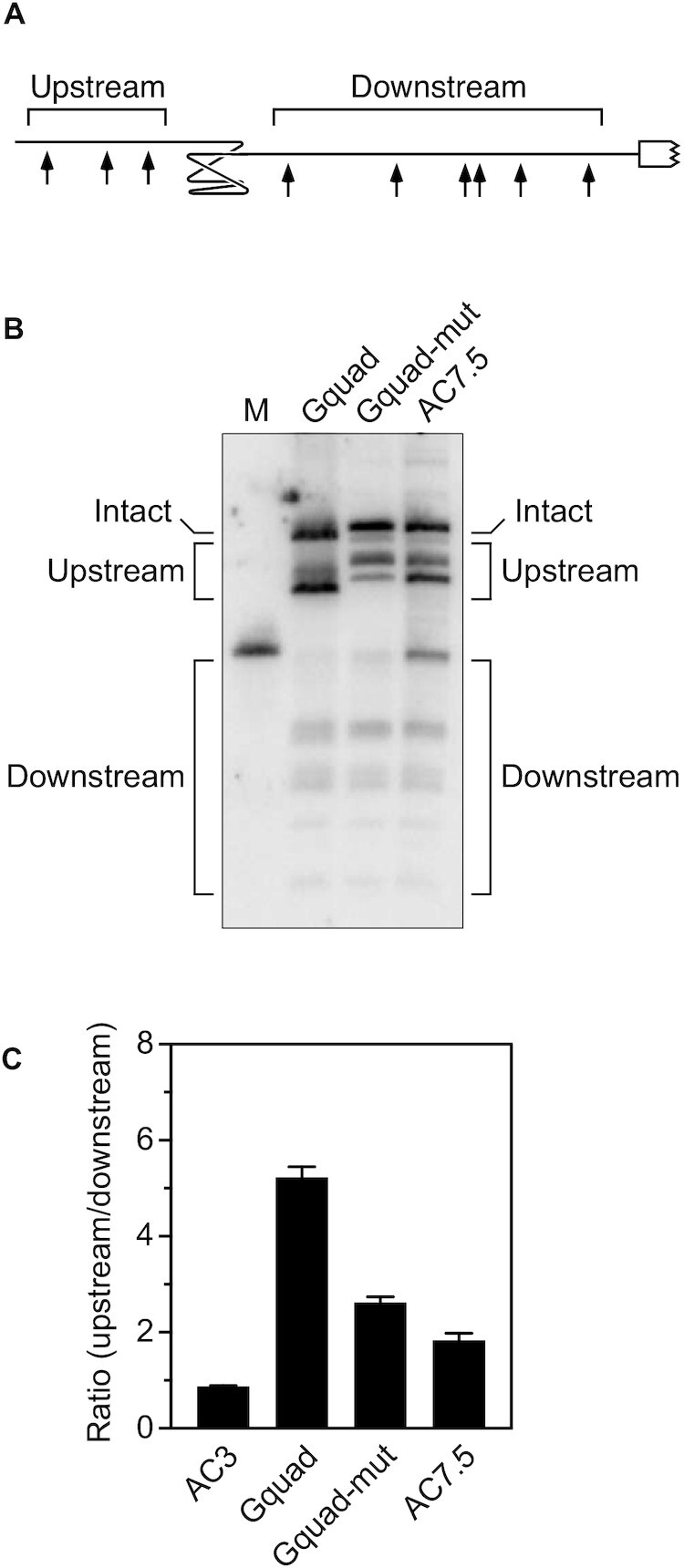
Protective effect of a G-quadruplex. (A) 5′ UTR of a reporter mRNA containing a G-quadruplex. Arrows, RNase E cleavage sites; white rectangle with a jagged edge, beginning of the protein coding region. (B) Cleavage within the 5′ UTR of reporter mRNAs containing a G-quadruplex (Gquad: GGGUGGGUGGGUGGG) or either of two other pentadecanucleotides (Gquad-mut: CGGUGCGUGGCUCGG; AC7.5: ACACACACACACACA) between the upstream and downstream sites. Equal amounts of total cellular RNA from isogenic strains of E. coli containing each reporter mRNA were analyzed by northern blotting to detect cleavage within the 5′ UTR. The blot was probed with a radiolabeled oligonucleotide complementary to the coding region. M, boundary marker between the upstream and downstream cleavage sites. (C) Relative abundance of 5′ UTR cleavage products. The sum of the intensities of the bands in panel (B) resulting from cleavage upstream of the G-quadruplex, pentadecanucleotide, or (AC)3 spacer was divided by the corresponding sum for 5′ UTR cleavage downstream. Each value is the average of three biological replicates. Error bars correspond to standard deviations.
Protective effect of a G-quadruplex
Another potential source of coaxial structure in some cellular RNAs may be the presence of a G-quadruplex. Prior studies in vitro have shown that the pentadecanucleotide G3UG3UG3UG3 (Gquad) folds stably as a parallel-stranded G-quadruplex comprising three stacked G tetrads, each surrounding a monovalent cation, and three one-nucleotide loops (30). By creating a 16-Å structural discontinuity (31), this G-quadruplex resulted in a five-fold increase in the ratio of upstream versus downstream cleavage products when inserted into AC3 (Figure 5; Supplementary Table S10). By comparison, inserting either of two other pentadecanucleotides unable to form a G-quadruplex (Gquad-mut or AC7.5) increased this ratio only about two-fold.
Correlation between obstacle size and efficacy
To better understand the relationship between the size of a discontinuity and its efficacy as an obstacle to scanning, we compared the fraction of 5′ UTR cleavage products cut downstream of obstacles of various types to the size of the discontinuity that they each create. Included in this analysis were five obstacles resulting from TRAP binding and eight others comprising a pseudoknot or G-quadruplex. In each case, the size of the discontinuity was estimated from a published X-ray or NMR structure (23,25–27,31,32) or by modeling a double-helical extension of such a structure (in the case of VPK+2, VPK+3 and VPK+4). Despite the diversity of the obstacles that were compared, a clear correlation was observed between the size of a discontinuity and its effectiveness as an impediment to scanning (Figure 6). Discontinuities caused by protein binding were slightly (<2-fold) more protective than those of a similar size caused by RNA base pairing. The accessibility of the downstream cleavage sites declined steadily as a function of obstacle size for discontinuities between 10 and 50 Å and then plateaued at a minimum level for those larger than 50 Å. We conclude that the degree of protection afforded by an obstacle to scanning depends primarily on the size of the discontinuity that it creates and not its molecular composition and that there is no apparent threshold size that must be exceeded for such discontinuities to have a significant impact on distal cleavage.
Figure 6.
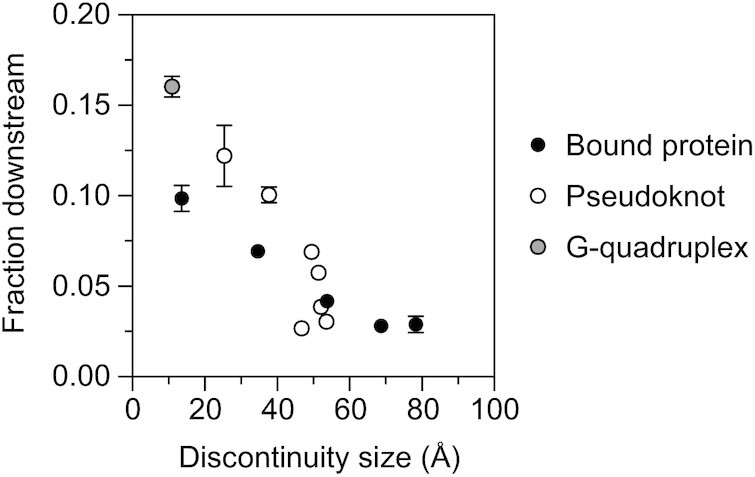
Correlation between the size and protective effect of discontinuities. The fraction of 5′ UTR cleavage products cut downstream of various protein (black circle) and RNA (white or gray circle) obstacles (1/[R+ 1], where R is the ratio of upstream versus downstream cleavage products graphed in Figures 1-2 and 4-5) was plotted as a function of the size of the discontinuity that they each create, as estimated from empirically determined structures (23,25–27,31,32) and predicted structures generated by using an ideal RNA double helix and PyMOL to model the extensions in VPK+2, VPK+3 and VPK+4. Each value is the average of three independent measurements. Error bars (not always visible) correspond to standard deviations.
Importance of obstacle persistence
Previously, we have shown that ribosome binding to an upstream open reading frame (uORF) can impede scanning by RNase E and that the efficacy of such an obstacle depends on the strength of the ribosome binding site, as inferred from its complementarity to 16S ribosomal RNA (15). To ascertain whether the impediment to scanning is also dependent on the length of the uORF, which could determine whether the uORF can accommodate more than one ribosome at a time, we compared the upstream versus downstream cleavage ratio for uORFs containing 6, 12 or 24 codons (Figure 7A). Regardless of the strength of the ribosome binding site (SD-med = AGGA, SD-high = AGGAG, SD-ultra = UAAGGAGG), increasing the length of the uORF by a factor of 2 or 4 had only a modest effect (Figure 7B, C; Supplementary Table S11).
Figure 7.
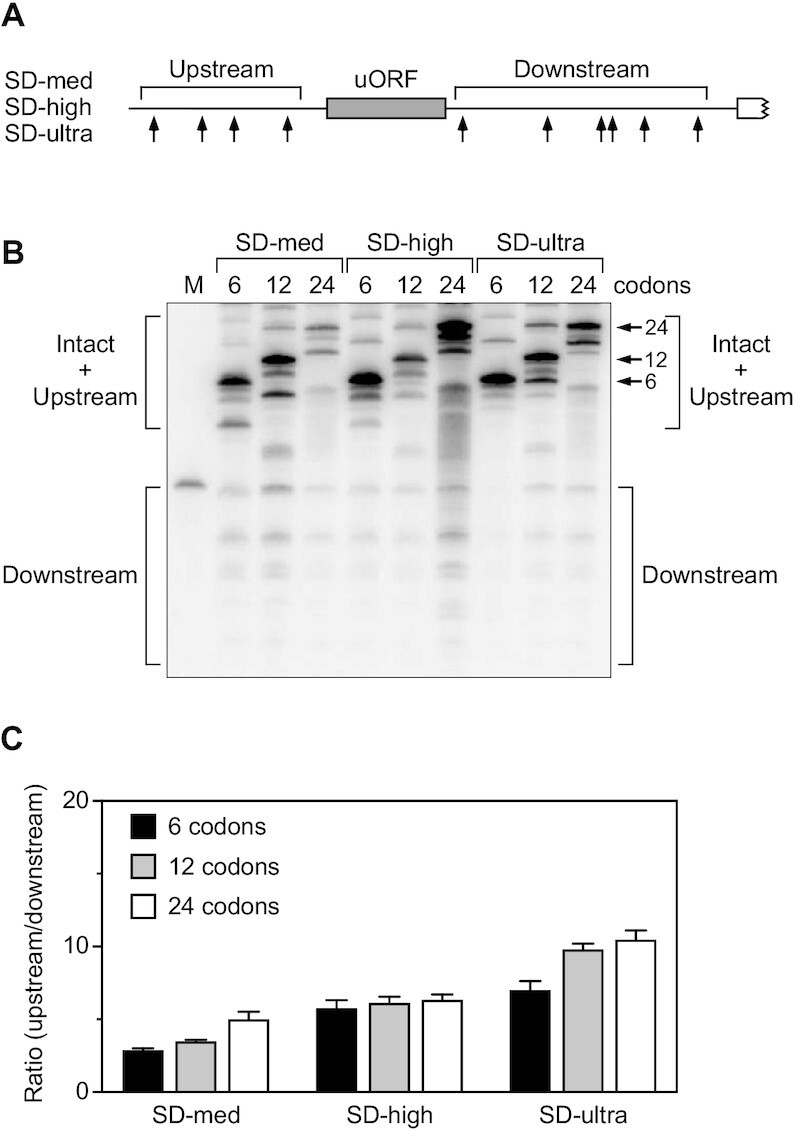
Effect of uORF length. (A) 5′ UTR of reporter mRNAs containing a uORF (SD-med, SD-high, or SD-ultra). Arrows, cleavage sites; gray rectangle, uORF; white rectangle with a jagged edge, beginning of the principal open reading frame. (B) Cleavage within the 5′ UTR of reporter mRNAs containing a 6-, 12- or 24-codon uORF preceded by any of three Shine-Dalgarno elements (SD-med = AGGA, SD-high = AGGAG or SD-ultra = UAAGGAGG) between the upstream and downstream sites. Equal amounts of total cellular RNA from isogenic strains of E. coli containing each reporter mRNA were analyzed by northern blotting to detect cleavage within the 5′ UTR. The blot was probed with a radiolabeled oligonucleotide complementary to the principal open reading frame. Arrows identify bands corresponding to the intact transcripts. M, boundary marker between the upstream and downstream cleavage sites. Faint bands between the upstream and downstream regions resulted from infrequent cleavage within the uORF. (C) Relative abundance of 5′ UTR cleavage products. The sum of the intensities of the bands in panel (B) resulting from cleavage upstream of the uORF was divided by the corresponding sum for 5′ UTR cleavage downstream. Each value is the average of three biological replicates. Error bars correspond to standard deviations.
Despite the large footprint of a ribosome bound to RNA (33), the protection of distal cleavage sites by these uORFs was smaller than for discontinuities created by a large pseudoknot or by bound TRAP, even for uORFs with a strong ribosome binding site such as SD-high or SD-ultra. In principle, the smaller than expected impact of the uORFs might be a consequence of the transient nature of ribosome binding, as even tightly bound ribosomes soon migrate away from their initial binding site as they begin translation, eventually dissociating from the mRNA altogether when they reach a termination codon. To test this hypothesis, we compared the efficacy of the six-codon SD-high uORF as an obstacle to scanning in the presence or absence of retapamulin, an antibiotic that blocks protein synthesis by trapping ribosomes at the site of translation initiation (34). Treating cells with this antibiotic significantly increased the ability of SD-high to protect 5′ UTR cleavage sites downstream of the 6-codon uORF, presumably by locking initiating ribosomes in place (Figure 8; Supplementary Table S12). By contrast, retapamulin had little or no effect on the cleavage ratio for VPK, which lacks a uORF between the upstream and downstream cleavage sites but has the same principal open reading frame distal to the downstream sites as SD-high. These findings suggest that the efficacy of uORFs as obstacles to scanning is limited by the brevity of their association with ribosomes.
Figure 8.
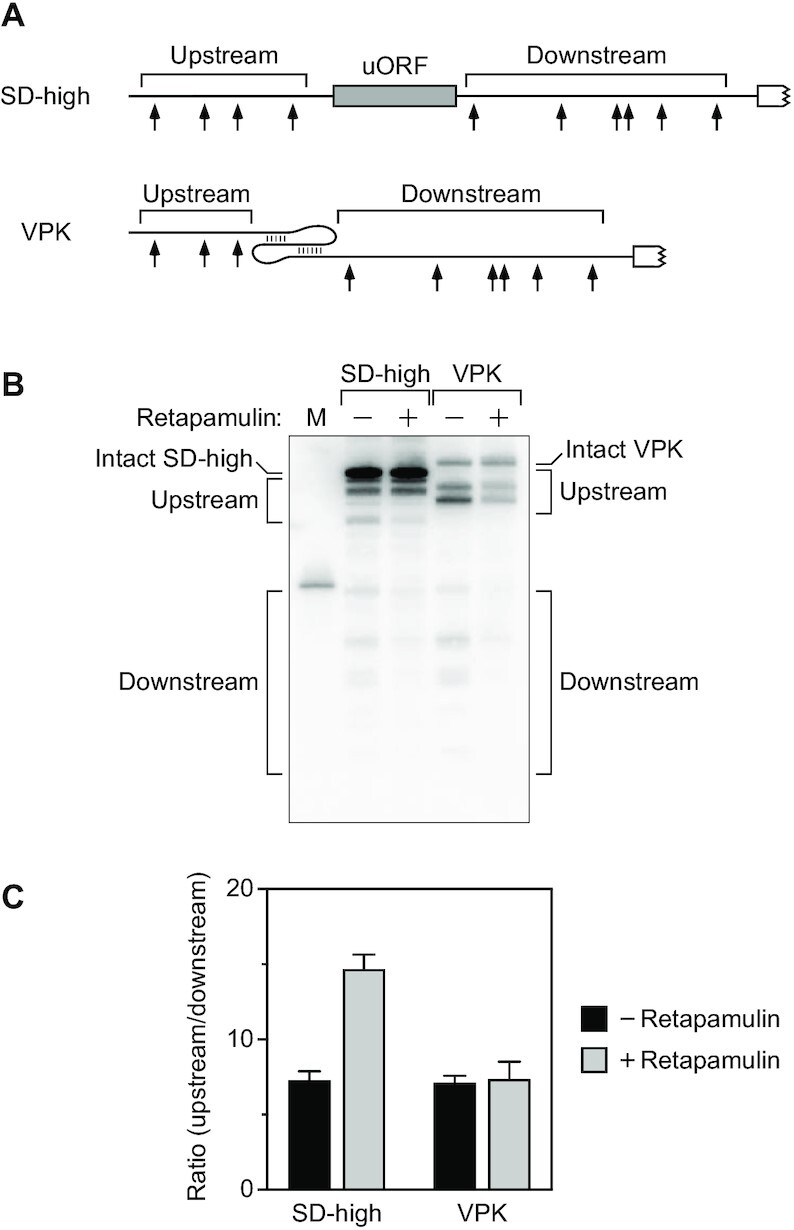
Effect of retapamulin. (A) 5′ UTR of reporter mRNAs containing a uORF (SD-high with six codons) or a VPK pseudoknot. Arrows, cleavage sites; gray rectangle, uORF; white rectangle with a jagged edge, beginning of the principal open reading frame. (B) Cleavage within the 5′ UTR of the reporter mRNAs illustrated in panel (A). Total RNA was extracted from isogenic strains of E. coli containing each reporter before or 5 min after treating the cells with retapamulin, and equal amounts of RNA were analyzed by northern blotting to detect cleavage within the 5′ UTR. M, boundary marker between the upstream and downstream cleavage sites. (C) Relative abundance of 5′ UTR cleavage products. The sum of the intensities of the bands in panel (B) resulting from cleavage upstream of the obstacle was divided by the corresponding sum for 5′ UTR cleavage downstream. Each value is the average of three biological replicates. Error bars correspond to standard deviations.
DISCUSSION
A great deal of evidence indicates that the diverse lifetimes of proteobacterial mRNAs are determined primarily by the ease with which the low-specificity endonuclease RNase E can gain access to its many cleavage sites within each transcript. In monophosphorylated RNA, this enzyme locates such sites by scanning linearly from the 5′ terminus along RNA segments that are single-stranded (15). Consequently, the rate at which RNase E cuts at those sites is governed not only by its ability to initially bind the monophosphorylated 5′ end but also by any obstacles that it may encounter as it scans downstream, with attendant effects on mRNA lifetimes and protein synthesis. Our findings now show that the impediment to scanning posed by such obstacles depends on both their size and their persistence. Obstacles that are large and enduring are more impactful than those that are small or transitory. By contrast, the molecular composition of such an obstacle, be it a bound protein or coaxially base-paired RNA, is much less consequential.
The ease with which RNase E can scan past small discontinuities in single-stranded RNA, such as a stem–loop orthogonal to its path (15), indicates that this endonuclease traverses RNA in steps greater than one nucleotide in length. If so, one might expect discontinuities smaller than the step length to have a negligible effect on distal cleavage and those larger than the step length to pose an insurmountable obstacle. However, our data provide no evidence for a threshold discontinuity size below which the impact of an obstacle is insignificant and above which it is maximal, a key new finding. Instead, the effect of obstacle size on downstream cleavage is graded over a broad range of distances, such that incrementally larger discontinuities are incrementally more capable of protecting distal sites from cleavage. By contrast, the protective effect of discontinuities greater than ∼50 Å is invariant. This maximum degree of protection of downstream cleavage sites (corresponding to an upstream/downstream ratio of ∼34 for the very large discontinuities in TR7, TR8, hTRΔU, and SARS-CoV-2) may be limited by the slow rate at which RNase E is able to access distal sites directly without first binding to the 5′ end and may therefore underrepresent the magnitude of the greatest possible impediment to scanning.
How can a graded response to the size of obstacles (rather than an all-or-none response) be reconciled with the concept of a step length? One potential explanation is that the step length of RNase E may be variable rather than fixed, perhaps due in part to the conformational flexibility of RNA. If so, a greater fraction of the range of possible step lengths would exceed the size of a small discontinuity than would exceed the size of a larger discontinuity, thereby impacting the ease with which an obstacle can be bypassed by determining the likelihood that a step of random length would be sufficiently large to overcome it. Another possible rationalization is the probabilistic effect of random RNase E take-off points upstream of an obstacle, such that bypass would require the step length to exceed not just the size of the discontinuity but additionally the variable distance from diverse take-off points to the near side of the discontinuity. A third possibility is that the activation energy required for RNase E to overcome obstacles could depend on their size if reaching past larger discontinuities entails more conformational strain and/or lost entropy.
Other interpretations of the effect of obstacle size appear to be inconsistent with our findings. For example, the greater protective effect of larger obstacles cannot be attributed to the increased separation of the flanking cleavage sites in view of the greater protection observed for VPK+4 versus VPK-AC2, in which the distance between the upstream and downstream cleavage sites is equal, and the similar protection observed for the uORFs of SD6, SD12, and SD24, in which that distance differs by up to fourfold. Likewise, the disparate efficacies of related obstacles that differ in size cannot merely be a consequence of differences in their persistence, as TRAP has a greater protective effect when bound to TR7 versus TR11 despite its lower affinity for the former, which contains only seven tandem GAGNN repeats (versus eleven).
Nevertheless, obstacles to scanning must persist to deter distal cleavage. For example, the protection provided by bound ribosomes is less than their large size would suggest, an apparent consequence of their dissociation from mRNA upon encountering a termination codon. As a result, distal protection by the six-codon uORF of SD-high can be increased by treating cells with retapamulin to freeze ribosomes at sites of translation initiation. No such effect of retapamulin was observed for VPK, which lacks a uORF but has the same cleavage sites and principal open reading frame as SD-high. The presence of cleavage sites outside the expected footprints of ribosomes bound to the two translation initiation sites of SD-high (33) ensured that the effect of retapamulin would not be obscured by direct masking of every potential upstream or downstream cleavage site.
The finding that a G-quadruplex can protect distal cleavage sites is the first evidence that RNA secondary structure other than a base-paired duplex can impede scanning by RNase E. Whereas the four strands of G-quadruplexes composed of DNA can adopt either a parallel, anti-parallel, or hybrid topology, the strands of RNA G-quadruplexes like the one tested here (G3UG3UG3UG3) are almost exclusively parallel (30,35). As a result, this G-quadruplex is able to obstruct scanning because the discontinuity that it creates is coaxial with the path traversed by RNase E as it scans RNA. Naturally occurring G-quadruplexes appear to be widespread in bacterial transcripts, including mRNAs implicated in pathogenesis (36).
Among the largest coaxially base-paired obstacles that we have examined is a branched HL-type pseudoknot derived from SARS-CoV-2 RNA. In its natural viral context, this pseudoknot facilitates translational frameshifting (28), but it can adopt other, presumably inactive conformations as a result of base-pairing interactions with flanking regions (29). The most prominent of those alternative conformations comprises two tandem stem-loops whose loop nucleotides base pair with (‘kiss’) one another (Figure S2B). Our data show that, in the context of a bacterial reporter transcript, the minimal SARS-CoV-2 pseudoknot is very effective at protecting downstream sites from cleavage by RNase E. Adding back nine natural 3′-flanking nucleotides complementary to a nearby segment of the pseudoknot modestly reduces the protective effect of this RNA element, consistent with a conformational equilibrium in which the pseudoknot is slightly favored over a pair of kissing stem-loops. More extensive (and unnatural) sequence changes that strongly reinforce the tandem stem-loops at the expense of the pseudoknot virtually abolish protection of distal cleavage sites, as expected for base pairing that is now entirely orthogonal (stems) or peripheral (kissing loops) to the path traversed by RNase E. These findings illustrate the significant effect on RNA degradation that can result from a conformational change induced by seemingly small changes in RNA sequence.
The ability of nucleic-acid binding proteins to search their RNA or DNA targets for specific sites by one-dimensional diffusion has been debated for some time (37–39). However, the potential regulatory effect of obstacles to linear diffusion of this kind had received scant attention prior to the discovery of their impact on scanning by RNase E. A growing body of evidence now indicates that the protection afforded by obstacles to scanning by this endonuclease is a key mechanism for regulating RNA lifetimes in bacteria and that it plays an important role in pathogenesis, stress responses, and riboswitch function (15–17). Understanding this novel phenomenon and its impact on gene expression will require the detailed elucidation of the molecular mechanisms by which this essential enzyme is able to diffuse linearly along RNA and bypass small obstacles. The constraints imposed by our present findings establish a conceptual framework critical for achieving this goal.
DATA AVAILABILITY
The data underlying this article are available in the article and in its online supplementary material.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Alexander Serganov, Dan Luciano, Tithi Banerjee and Deblina Basu for their helpful suggestions.
Author contributions: J.R. and J.G.B. designed the research, J.R. conducted the experiments, and J.R. and J.G.B. interpreted the data and wrote the paper.
Contributor Information
Jamie Richards, Skirball Institute of Biomolecular Medicine, New York University School of Medicine, 540 First Avenue, New York, NY 10016, USA; Department of Microbiology, New York University School of Medicine, 430 E. 29th Street, New York, NY 10016, USA.
Joel G Belasco, Skirball Institute of Biomolecular Medicine, New York University School of Medicine, 540 First Avenue, New York, NY 10016, USA; Department of Microbiology, New York University School of Medicine, 430 E. 29th Street, New York, NY 10016, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R01GM123124, R35GM145359 to J.G.B.]. Funding for open access charge: NIH [R01GM123124].
Conflict of interest statement. None declared.
REFERENCES
- 1. Mackie G.A. RNase E: at the interface of bacterial RNA processing and decay. Nat. Rev. Microbiol. 2013; 11:45–57. [DOI] [PubMed] [Google Scholar]
- 2. McDowall K.J., Lin-Chao S., Cohen S.N.. A+U content rather than a particular nucleotide order determines the specificity of RNase E cleavage. J. Biol. Chem. 1994; 269:10790–10796. [PubMed] [Google Scholar]
- 3. Lin-Chao S., Wong T.-T., McDowall K.J., Cohen S.N.. Effects of nucleotide sequence on the specificity of rne-dependent and RNase E-mediated cleavages of RNA I encoded by the pBR322 plasmid. J. Biol. Chem. 1994; 269:10797–10803. [PubMed] [Google Scholar]
- 4. Kaberdin V.R. Probing the substrate specificity of Escherichia coli RNase E using a novel oligonucleotide-based assay. Nucleic Acids Res. 2003; 31:4710–4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Del Campo C., Bartholomäus A., Fedyunin I., Ignatova Z.. Secondary structure across the bacterial transcriptome reveals versatile roles in mRNA regulation and function. PLoS Genet. 2015; 11:e1005613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chao Y., Li L., Girodat D., Förstner K.U., Said N., Corcoran C., Śmiga M., Papenfort K., Reinhardt R., Wieden H.J.et al.. In vivo cleavage map illuminates the central role of RNase E in coding and non-coding RNA pathways. Mol. Cell. 2017; 65:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee K., Bernstein J.A., Cohen S.N.. RNase G complementation of rne null mutation identifies functional interrelationships with RNase E in Escherichia coli. Mol. Microbiol. 2002; 43:1445–1456. [DOI] [PubMed] [Google Scholar]
- 8. Hui M.P., Foley P.L., Belasco J.G.. Messenger RNA degradation in bacterial cells. Annu. Rev. Genet. 2014; 48:537–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mackie G.A. Ribonuclease E is a 5'-end-dependent endonuclease. Nature. 1998; 395:720–723. [DOI] [PubMed] [Google Scholar]
- 10. Callaghan A.J., Marcaida M.J., Stead J.A., McDowall K.J., Scott W.G., Luisi B.F.. Structure of Escherichia coli RNase E catalytic domain and implications for RNA turnover. Nature. 2005; 437:1187–1191. [DOI] [PubMed] [Google Scholar]
- 11. Celesnik H., Deana A., Belasco J.G.. Initiation of RNA decay in Escherichia coli by 5' pyrophosphate removal. Mol. Cell. 2007; 27:79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deana A., Celesnik H., Belasco J.G.. The bacterial enzyme RppH triggers messenger RNA degradation by 5' pyrophosphate removal. Nature. 2008; 451:355–358. [DOI] [PubMed] [Google Scholar]
- 13. Richards J., Belasco J.G.. Distinct requirements for 5'-monophosphate-assisted RNA cleavage by Escherichia coli RNase E and RNase G. J. Biol. Chem. 2016; 291:5038–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luciano D.J., Vasilyev N., Richards J., Serganov A., Belasco J.G.. A novel RNA phosphorylation state enables 5′ end-dependent degradation in Escherichia coli. Mol. Cell. 2017; 67:44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Richards J., Belasco J.G.. Obstacles to scanning by RNase E govern bacterial mRNA lifetimes by hindering access to distal cleavage sites. Mol. Cell. 2019; 74:284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Richards J., Belasco J.G.RNase E.. Widespread protection of RNA cleavage sites by a riboswitch aptamer that folds as a compact obstacle to scanning by. Mol. Cell. 2021; 81:127–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lodato P.B., Hsieh P.K., Belasco J.G., Kaper J.B.RNase E.. The ribosome binding site of a mini-ORF protects a T3SS mRNA from degradation by. Mol. Microbiol. 2012; 86:1167–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Datsenko K.A., Wanner B.L.. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Neidhardt F.C., Bloch P.L., Smith D.F.. Culture medium for enterobacteria. J. Bacteriol. 1974; 119:736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Santoro S.W., Joyce G.F.. A general purpose RNA-cleaving DNA enzyme. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Richards J., Luciano D.J., Belasco J.G.. Influence of translation on RppH-dependent mRNA degradation in Escherichia coli. Mol. Microbiol. 2012; 86:1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gollnick P., Babitzke P., Antson A., Yanofsky C.. Complexity in regulation of tryptophan biosynthesis in Bacillus subtilis. Annu. Rev. Genet. 2005; 39:47–68. [DOI] [PubMed] [Google Scholar]
- 23. Antson A.A., Dodson E.J., Dodson G., Greaves R.B., Chen X., Gollnick P.. Structure of the trp RNA-binding attenuation protein, TRAP, bound to RNA. Nature. 1999; 401:235–242. [DOI] [PubMed] [Google Scholar]
- 24. Babitzke P., Yealy J., Campanelli D. Interaction of the trp RNA-Binding attenuation protein (TRAP) of Bacillus subtilis with RNA: effects of the number of GAG repeats, the nucleotides separating adjacent repeats, and RNA secondary structure. J. Bacteriol. 1996; 178:5159–5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shen L.X., Tinoco I.. The structure of an RNA pseudoknot that causes efficient frameshifting in mouse mammary tumor virus. J. Mol. Biol. 1995; 247:963–978. [DOI] [PubMed] [Google Scholar]
- 26. Su L., Chen L., Egli M., Berger J.M., Rich A.. Minor groove RNA triplex in the crystal structure of a ribosomal frameshifting viral pseudoknot. Nat. Struct. Biol. 1999; 6:285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Theimer C.A., Blois C.A., Feigon J.. Structure of the human telomerase RNA pseudoknot reveals conserved tertiary interactions essential for function. Mol. Cell. 2005; 17:671–682. [DOI] [PubMed] [Google Scholar]
- 28. Plant E.P., Pérez-Alvarado G.C., Jacobs J.L., Mukhopadhyay B., Hennig M., Dinman J.D.. A three-stemmed mRNA pseudoknot in the SARS coronavirus frameshift signal. PLoS Biol. 2005; 3:e172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huston N.C., Wan H., Strine M.S., de Cesaris Araujo Tavares R., Wilen C.B., Pyle A.M. Comprehensive in vivo secondary structure of the SARS-CoV-2 genome reveals novel regulatory motifs and mechanisms. Mol. Cell. 2021; 81:584–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Joachimi A., Benz A., Hartig J.S.. A comparison of DNA and RNA quadruplex structures and stabilities. Bioorg. Med. Chem. 2009; 17:6811–6815. [DOI] [PubMed] [Google Scholar]
- 31. Do N.Q., Phan A.T.. Monomer-dimer equilibrium for the 5'-5' stacking of propeller-type parallel-stranded G-quadruplexes: NMR structural study. Chemistry. 2012; 18:14752–14759. [DOI] [PubMed] [Google Scholar]
- 32. Roman C., Lewicka A., Koirala D., Li N.S., Piccirilli J.A.. The SARS-CoV-2 programmed -1 ribosomal frameshifting element crystal structure solved to 2.09 Å using chaperone-assisted RNA crystallography. ACS Chem. Biol. 2021; 16:1469–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Woolstenhulme C.J., Guydosh N.R., Green R., Buskirk A.R.. High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep. 2015; 11:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Meydan S., Marks J., Klepacki D., Sharma V., Baranov P.V., Firth A.E., Margus T., Kefi A., Vázquez-Laslop N., Mankin A.S.. Retapamulin-assisted ribosome profiling reveals the alternative bacterial proteome. Mol. Cell. 2019; 74:481–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xiao C.D., Shibata T., Yamamoto Y., Xu Y.. An intramolecular antiparallel G-quadruplex formed by human telomere RNA. Chem. Commun. (Camb.). 2018; 54:3944–3946. [DOI] [PubMed] [Google Scholar]
- 36. Shao X., Zhang W., Umar M.I., Wong H.Y., Seng Z., Xie Y., Zhang Y., Yang L., Kwok C.K., Deng X.. RNA G-quadruplex structures mediate gene regulation in bacteria. Mbio. 2020; 11:e02926-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Berg O.G., Winter R.B., Hippel P.H.. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry. 1981; 20:6929–6948. [DOI] [PubMed] [Google Scholar]
- 38. Halford S.E. An end to 40 years of mistakes in DNA-protein association kinetics?. Biochem. Soc. Trans. 2009; 37:343–348. [DOI] [PubMed] [Google Scholar]
- 39. Mechetin G.V., Zharkov D.O.. Mechanisms of diffusional search for specific targets by DNA-dependent proteins. Biochemistry (Mosc.). 2014; 79:496–505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.