

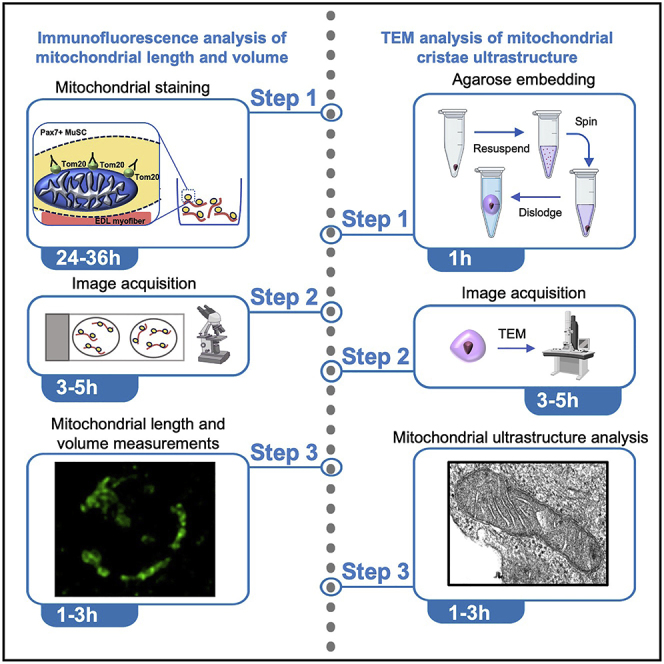
Summary
Since changes in mitochondrial morphology regulate key functions of stem cells, it is important to assess their structure under physiological and pathophysiological conditions. Here, we present techniques optimized in rare adult muscle stem cells (MuSCs). For evaluating mitochondrial length and volume within a compact cytoplasmic area in MuSCs on intact myofibers, we describe steps for mitochondrial staining, imaging, and quantification. For evaluating mitochondrial ultrastructure in small cell numbers, we describe steps for agarose embedding and quantification by TEM.
For complete details on generation and use of this protocol, please refer to Baker et al. (2022).1
Subject areas: Cell Biology, Metabolism, Microscopy, Stem Cells
Graphical abstract
Highlights
-
•
Staining, imaging, and 3D reconstruction of mitochondria in rare muscle stem cells
-
•
Manual mitochondrial length assessment in cells with a small cytoplasmic volume
-
•
Quantification of mitochondrial volume in muscle stem cells
-
•
Analysis of cristae architecture with transmission electron microscopy
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Since changes in mitochondrial morphology regulate key functions of stem cells, it is important to assess their structure under physiological and pathophysiological conditions. Here, we present techniques optimized in rare adult muscle stem cells (MuSCs). For evaluating mitochondrial length and volume within a compact cytoplasmic area in MuSCs on intact myofibers, we describe steps for mitochondrial staining, imaging, and quantification. For evaluating mitochondrial ultrastructure in small cell numbers, we describe steps for agarose embedding and quantification by transmission electron microscopy.
Before you begin
Preparation for immunofluorescence staining
Timing: is cell isolation specific
-
1.
Obtain cells of interest.
Note: The present protocol is focused on assessing mitochondrial length and volume within rare adult muscle stem cells (MuSCs) residing on isolated single myofibers in order to maintain the niche environment. Single extensor digitorum longus (EDL) skeletal muscle fibers are isolated using well-established techniques2,3,4 and can be utilized immediately following isolation and fixation or cultured for the required times. This method of visualization and assessment of mitochondria can also be generally applied to other stem cell populations or any cell types with a small cytoplasmic volume and clustered mitochondria, but will be subject to cell-type specific modifications.
-
2.Prepare 4% Paraformaldehyde (PFA).
 CRITICAL: Powdered PFA should always be used in a ventilated safety hood. Eye and skin exposure should be avoided. Follow safety data sheet and institutional guidelines when handling and disposing of PFA.
CRITICAL: Powdered PFA should always be used in a ventilated safety hood. Eye and skin exposure should be avoided. Follow safety data sheet and institutional guidelines when handling and disposing of PFA.-
a.Weigh 10 g of PFA powder and transfer to a 250 mL flask containing 200 mL of MilliQ H2O and a magnetic stirring rod.
-
b.Heat the mixture while stirring until it reaches a temperature of 60°C.
-
c.Add 50 μL of 5 N NaOH and heat PFA solution until it reaches 65°C and the solution becomes completely clear.
-
d.Turn off the heat and add 25 mL of 10× PBS and 25 mL of MilliQ H2O.
-
e.Filter the 4% PFA through Whatman Filter paper.
-
f.Adjust pH to 7.4 using 12 N HCl.
-
g.Immediately aliquot and freeze for long-term storage.Note: This solution can be made in advance and frozen at −20°C for up to three months.Note: Optimal results are achieved when 4% PFA is used immediately after thawing.CRITICAL: Cell fixation for assessment of mitochondrial morphology requires optimal PFA quality and concentration. If these requirements are not met, mitochondria may undergo rapid stress-induced fragmentation. To avoid adverse effects of the fixative on these organelles, users should adhere to the above guidelines.
-
a.
-
3.
Prepare Permeabilization Solution.
Note: This buffer can be made in advance and stored at room temperature for up to 1 month.
-
4.
Prepare Blocking Solution.
Note: This buffer must be made fresh and can be used for the two days of the staining protocol.
Preparation for transmission electron microscopy (TEM) of small cell number samples
-
5.
Isolate stem cells of interest.
Note: In the present protocol we focus on assessing mitochondrial ultrastructure in FACS isolated adult MuSCs from hindlimb muscles of mice using transmission electron microscopy (TEM), as previously described in Baker et al.1 FACS sorting was performed based on previously published methodologies, to isolate VCAM and α-Integrin7 positive cells.1,5,6 Other stem cell types with low population yields can be used and processed in a similar manner.
-
6.
Prepare TEM Fixation Buffer and adjust pH to 7.4 using NaOH.
Note: This buffer can be made in advance and stored at 4°C for up to 1 week.
-
7.Prepare 1× Tris-Acetate-EDTA Buffer (TAE) Buffer.
-
a.Dilute 10× TAE buffer by mixing 100 μL of 10× TAE with 900 μL of ddH2O.
-
a.
Note: 10× TAE can be made and stored at room temperature for up to 1 year.
Note: 1× TAE should be made and used fresh.
-
8.Prepare 4% low-melt agarose:
-
a.Add 0.04 g of low-melt agarose to 1 mL of 1×TAE.Note: 30 μL is needed per sample, however, for accuracy of weighing, it is advisable to make 1 mL at a time.
-
b.Dissolve by mixing with a pipette.
-
c.Heat in a water bath at 67°C until the agarose is completely clear.Note: This is made fresh prior to the EM-fixation steps detailed below.Note: Low-melt agarose solidifies at room temperature. To keep in liquid form, leave at 67°C until needed.
-
a.
Institutional permissions
Animal protocols were approved by the University of Ottawa’s Animal Care Ethics Committee and adhered to the guidelines of the Canadian Council on Animal Care. All mice were housed and maintained at the Animal Care and Veterinary Service (ACVS) of the University of Ottawa. Please ensure that these permissions are acquired based on the relevant institution and national guidelines.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse Pax7 (monoclonal) | DHSB | Cat# Pax7-S; 1:13 |
| Anti-rabbit Tom20 (polyclonal) | ProteinTech | Cat# 11802-AP; 1:500 RRID: AB_2207530 |
| Cy3 AffiniPure anti mouse IgG (H+L) | Jackson | Cat# 715-165-150; 1:1000 RRID:AB_2340813 |
| Alexa-Fluoro 488 anti mouse IgG (H+L) | Thermo Fisher Scientific | Cat# A11001; 1:1000 RRID:AB_2534069 |
| DAPI | Millipore Sigma | Cat# D9542 1:1500 |
| Chemicals, peptides, and recombinant proteins | ||
| 1× PBS | Wisent | Cat# 311-010-CL |
| Paraformaldehyde (PFA) powder | Sigma | Cat# P6148 |
| Horse serum | Wisent | Cat# 065150 |
| BSA | Sigma | Cat# A4503 |
| Low melting agarose | Invitrogen | Cat# 16250-050 |
| Triton x100 | Sigma | Cat# 1002560820 |
| Glycine | Promega | Cat# H5073 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | In-House | 8–10 weeks old male and female mice |
| Mouse: OPA1-floxed-Pax7CreERT2 | (Baker et al.1) | |
| Mouse: OPA1tg (OPA1-OE) | (Cogliati et al.7; Varanita et al.8) | |
| Software and algorithms | ||
| FIJI | NIH | https://fiji.sc |
| Imaris | Imaris Microscopy Imaging Software | https://imaris.oxinst.com |
| Other | ||
| Immu-Mount Mounting Media | Epredia | Cat# 9990402 |
| Pap Pen | Abcam | Cat# ab2601 |
| Charged Glass Slides | Fisher Scientific | Cat# 12-550-15 |
| Cover Slip (24 × 50 mm) | Fisher Scientific | Cat# 12544E |
| Whatman Filter Paper | GE Healthcare Life Sciences | Cat# 1001-240 |
Materials and equipment
4% Paraformaldehyde
| Reagent | Final concentration | Amount |
|---|---|---|
| PFA Powder | 4% (m/v) | 10 g |
| 5 N NaOH | 0.02% | 50 μL |
| MilliQ H2O | N/A | 225 mL |
| 10×PBS | N/A | 25 mL |
| Total | N/A | 250 mL |
Make in advance, aliquot and store at −20°C. Thaw on day of experiment.
Permeabilization Solution
| Reagent | Final concentration | Amount |
|---|---|---|
| 10% Triton x100 in 1×PBS | 0.1% (v/v) | 500 μL |
| Glycine | 0.1 M (m/v) | 0.37 g |
| 1×PBS | N/A | 49.5 mL |
| Total | N/A | 50 mL |
Make in advance and store at room temperature for up to a month.
Immunofluorescence Blocking Solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Horse Serum | 5% (v/v) | 500 μL |
| BSA | 2% (m/v) | 0.2 g |
| 10% Triton x100 in 1×PBS | 0.1% (v/v) | 100 μL |
| 1× PBS | N/A | 9.4 mL |
| Total | N/A | 10 mL |
Make fresh and store at 4°C for up to a week.
Primary Antibody Staining Solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Pax7 Anti Rabbit | 1:13 | 76.9 μL |
| Tom20 Anti Mouse | 1:500 | 2 μL |
| Blocking Solution | N/A | 920.1 μL |
| Total | N/A | 1 mL |
Make immediately prior to staining.
Secondary Antibody Staining Solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Cy3-Rabbit | 1:1000 | 1 μL |
| 488-Mouse | 1:1000 | 1 μL |
| DAPI | 1:1500 | N/A |
| Blocking Solution | N/A | 1,000 μL |
| Total | N/A | 1 mL |
Make immediately prior to staining.
Note: See below for details on DAPI addition to stain conditions.
TEM Fixative (pH 7.4)
| Reagent | Final concentration | Amount |
|---|---|---|
| Glutaraldehyde | 2.5% (v/v) | 5 mL |
| Sodium Cacodylate Buffer | 0.2 M | 25 mL |
| CaCl2 | 9.01 mM | 0.05 g |
| MilliQ H20 | N/A | 20 mL |
| Total | N/A | 50 mL |
Store at 4°C, can be used for up to one week.
10× TAE Buffer (pH 8.0):
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris | 0.4 M | 48.5 g |
| Glacial Acetic Acid (17.4 M) | 0.198 M | 11.4 mL |
| 0.5 M EDTA | 0.01 M | 20 mL |
| ddH2O | N/A | ∼950 mL |
| Total | N/A | 1 L |
Can be made and stored for up to a year in advance.
Note: When making 10× TAE, use 800 mL of ddH2O to dissolve and mix products and to pH. Subsequently, volume up to 1L with ddH2O.
4% Low-Melt Agarose
| Reagent | Final concentration | Amount |
|---|---|---|
| Agarose | 4% (w/v) | 0.04 g |
| 1× TAE | N/A | 1 mL |
| Total | N/A | 1 mL |
Make fresh and keep warmed at 67°C to prevent it from solidifying.
Step-by-step method details
Part 1: Stem cell fixation for immunofluorescence
As noted above, this protocol will outline the methods used to fix, stain, and visualize mitochondria in MuSCs residing on isolated single myofibers from the EDL muscles of mice; the model system utilized previously.1 Following isolation, EDL myofibers are collected in one well of a 24-well plate. The volumes discussed below are based on this experimental set up. This protocol can be adapted to other cell types that are either abundant or scarce in number.
-
1.
Once EDL myofibers are collected in a well(s) of a 24-well plate, all excess media/buffer should be removed carefully using a pipette to avoid loss of myofibers.
-
2.
Wash myofibers with 1 mL of pre-warmed sterile 1× PBS (37°C).
-
3.
Remove 500 μL of the 1× PBS using a pipette.
-
4.
Add an equal volume (i.e., 500 μL) of pre-warmed 4% PFA (37°C) to the remaining 1× PBS from the step above.
Note: This dilution will lead to the working concentration of 2% PFA, which has been optimized for the visualization and quantification of mitochondrial length and volume in the EDL-MuSC model while minimizing excessive background autofluorescence from the myofiber itself. The working concentration of PFA may differ in other cell-types and preparations. Users must optimize accordingly.
-
5.
Incubate with gentle rocking for 10 min at room temperature.
-
6.
Wash myofibers 3 × 5 min with 1 mL of 1× PBS.
Pause point: If not staining cells immediately, store myofibers at 4°C in 1× PBS until further use. To prevent evaporation of the PBS, myofibers should be stored in a sealed container (i.e., 1.5 mL tube, or a parafilm sealed plate). Additionally, optimal staining results are observed when myofibers are stained prior to 1 week storage.
Part 2: Immunofluorescence staining of muscle fibers to assess mitochondrial length and volume in resident muscle stem cells
The goal of this portion of the protocol is to stain the MuSCs and mount the EDL myofibers collected in Part 1. Utilizing a mitochondrial protein marker (i.e., Tom20) and a MuSC marker (i.e., Pax7) allows for visualization of these organelles in the MuSC population ex-vivo. If other cell types are being used, confirmation of cell lineage should be done by staining for a relevant cell-specific marker.
-
7.
Remove PBS from the samples collected in Part 1, leaving no more than ∼100 μL.
-
8.
Incubate each sample with 1 mL permeabilization solution for 10 min at room temperature with gentle rocking on a plate rocker.
-
9.
Remove all but ∼100 μL of permeabilization solution.
-
10.
Add 1 mL of blocking solution per sample.
-
11.
Incubate for 5 h at room temperature on a plate rocker.
Note: Less incubation time for blocking may lead to poor quality of mitochondrial images, thus it is advised that at least 5 h of incubation is followed.
-
12.
Remove all but ∼100 μL of the blocking solution.
-
13.
Add 100 μL of primary antibody staining solution. See “materials and equipment” section for suggested antibody concentrations.
-
14.
Incubate on a plate rocker overnight at 4°C.
-
15.
Wash 3 × 5 min in 1 mL of 1× PBS at room temperature on a plate rocker with gentle rocking.
-
16.
Add 200 μL secondary antibody staining solution. See “materials and equipment” section for suggested secondary antibody concentrations.
-
17.
Incubate with gentle rocking for 1 h at room temperature.
Note: Ensure samples are kept in the dark by wrapping in foil or covering with an opaque container.
-
18.
Add 200 μL of diluted DAPI for the final 5 min of this incubation. The working concentration of DAPI should be 1:1500.
Note: The DAPI is added to the existing secondary antibody staining solution by first generating a dilution from the 2 mg/mL stock DAPI of 1:750 and then adding an equal volume of this dilution (200 μL) to the sample to get a final concentration of 1:1500.
-
19.
Wash 3 × 5 min in 1 mL of 1× PBS at room temperature on a plate rocker while gently rocking in dark conditions.
-
20.Mount cells/fibers using charged glass slides and Mounting Media.
-
a.Create a circle on a charged glass slide using a Pap Pen.
-
b.Remove the 1× PBS from the sample until about 100–200 μL are left.
-
c.Using a p1000 pipette, gently pipette the EDL myofibers onto the glass slide.
-
d.Allow 2 min for the EDL myofibers to settle to the slide.
-
e.Gently remove the excess 1× PBS using a p200 pipette.
-
f.Allow samples to dry, until damp (this normally takes 1–2 min).Note: If the PBS dries completely, crystals will begin to form and will interfere with image quality. Thus, it is important to mount the samples in a timely manner.
-
g.Add a thin line of Mounting Media along the center of a rectangular (24 mm × 50 mm) cover slip.
-
h.Gently place coverslip onto to the glass slide and press down to create a seal.CRITICAL: If too much mounting media is used and/or the cover slip is placed too aggressively, fibers will be pushed off the glass slide and lost. Ensure careful application of the cover slip.
-
a.
-
21.
Allow slides to dry overnight in the dark at room temperature.
-
22.
Slides can subsequently be stored at 4°C in a slide box until ready for imaging.
Part 3: Generating 3D reconstructed mitochondria and assessing mitochondrial structure in adult muscle stem cells
Imaging and processing were completed on a LSM880 Airyscan Confocal Microscope (Zeiss) at the uOttawa Cell Biology Image Acquisition Core. The airyscan capability allows for enhanced signal-to-noise ratio and obtains high resolution information. Other microscopes and softwares with similar functionality may be sufficient to assess mitochondrial length and volume in these cell populations; however, users must optimize microscope settings and imaging parameters accordingly.
-
23.Image mitochondria in adult MuSCs by confocal microscopy.
-
a.Place the slide, with a drop of immersion oil onto a 63× oil immersion lens.
-
b.Locate a cell of interest and center it in the field of view.Note: For MuSCs on EDL myofibers, Pax7 immunofluorescence is used to locate cells of interest. A cell lineage marker should be used to identify specific cell-types, along with DAPI to confirm the staining of a cell.
-
c.Adjust Image settings to optimize laser intensity, pinhole diameter, gain, offset and ensure optimal signal: noise.Note: For this protocol, image settings were adjusted and optimized using the Zeiss Zen Black 2.3 software (Figure 1).
-
d.In order to create 3D reconstructed images, use the “Z-Stack” capability of the microscope to capture image slices spanning from the upper to the lower limits of the cells (yellow boxes in Figure 1).Note: When setting the upper and lower limits for the Z-stack, ensure that these limits are set to the presence of mitochondria. The purpose of this is two-fold, it ensures that 1) all mitochondria in the cell are being captured and that 2) the stack limits are not overestimated such that it would lead to excessively large files. To do this, the focus knob is rotated in one direction all the way until there are no mitochondria visible and this is set as the upper limit. Then the knob is turned in the opposite direction, again until there are no mitochondria visible, and this is set as the lower limit. This then establishes the upper and lower limits of the Z-stacks.Note: At this stage, it is most efficient to scan the upper and lower limits of the cell using only the channel with the mitochondrial stain (Tom20 immunofluorescence).Note: Each slice should be at 0.16 μm intervals. Using these settings, it may require anywhere between 20–45 slices to capture all mitochondria within MuSCs residing on EDL myofibers.
-
e.Acquire optimal Airyscan signal. Using the Zeiss Zen Black 2.3 software, ensure Airyscan processing is enabled to acquire images that are of sufficient quality for 3D reconstruction. This can be done by selecting the “Continuous” scanning option while in the Tom20 channel (pink box in Figure 1). The Airyscan window will specify a “Good” (in green) or “Bad” (in red) signal. Microscope settings can be adjusted until a “good” signal is reached. If “Good” is present, the user can proceed with imaging. If “Bad” is present, the user can adjust the Z-plane of the cell until “Good” is reached.
- f.
-
g.Once the z-stack is complete, process the image using the Airyscan Processing Window. This is performed by selecting the “Processing” window and clicking on “Airyscan Processing” (blue boxes in Figure 1). Press “Select” in the “Method Parameters”, which will then preview the captured image following Airyscan. Select “Apply” and wait for the Airyscan feature to complete.
-
h.Save image in the .czi format that retains all the metadata required for subsequent analysis. Proceed with analysis.
-
a.
-
24.Measure and quantify mitochondrial length using the FIJI Software.
-
a.Open image in Fiji.13 When prompted with the “Bio-Formats Import Options” window, ensure that “View Stack with” is set to “hyperstack”, and “autoscale” is selected.
- b.
-
c.It may be necessary to adjust the brightness and contrast of the image to better visualize mitochondrial staining (Figure 2Bi). To do this, go to: “Image” > “Adjust” > “Brightness/Contrast” and adjust the histogram of brightness and contrast (Figure 2Bii). Click on “Apply” in the “Brightness/Contrast” window to ensure that these modifications are saved for all Z-stacks. Repeat this for all channels if needed.Note: If it is difficult to distinguish individual mitochondria the user can adjust the image using built-in image processing in Fiji. Adjustments include 1) adjusting the brightness and contrast features to resolve mitochondria (Figure 2Bii), 2) sharpening the image (Figure 2Biii), and 3) rotating the image using the toolbar below the image.
-
d.Select the Line Function in the toolbar (Figure 2C). Selecting “Freehand” allows for more flexibility when tracing individual mitochondria for subsequent measurement (Figure 2C).Note: When quantifying mitochondrial length, users can turn off the alternate channels to allow for visualization of all mitochondria within the cell. This can be done by following the path “Image” -> “Color” -> “Channels Tool”.
-
e.Draw a line across an individual mitochondrion and click “M” on the keyboard to measure and record its length. This will be repeated for each mitochondrion in the image. All discernable mitochondria within a cell are to be quantified (Figure 2C).Note:Figure 2C depicts a zoomed region of mitochondria within the cell to provide a representative view of mitochondrial measurements. It is important to note that all discernable mitochondria should be measured within a cell to prevent any bias in measurements. As such, data for mitochondrial length, presented as scatter plots (Figures 3B, 3F, and 3J), mean mitochondrial length (Figures 3C, 3G, and 3K) or percent mitochondrial length distribution (Figures 3D, 3H, and 3L) shown in Figure 3 was generated by quantification of all mitochondria within the cell.
-
f.Calculations and presentation of mitochondrial length measurements can be performed in several ways. The raw individual mitochondrial length measurements can be presented in a scatter plot to showcase value distributions. The average mitochondrial length can also be calculated and presented as a bar graph of “mean mitochondrial length”. Although this method can be informative, subtle differences may be lost when quantifying in this manner. Thus, investigators can also present the data by binning mitochondria based on their length distribution.1 Data in Figure 3 demonstrate mitochondrial length in MuSCs under conditions that lead to increased or decreased mitochondrial length by genetic or pharmacological means (Figure 3).
-
a.
-
25.Measure mitochondrial volume in Imaris Software (version 9.7.2).
-
a.Open the image on Imaris.
-
b.To create a surface rendering of the image, first click on the “add new surfaces” button.Note: Leave the settings as is.
-
c.Select the Tom20 channel to create a surface over mitochondria.Note: Do not smooth the image, as this may reduce the resolution of individual mitochondria.
-
d.Enable background subtraction in the thresholding option.
-
e.For the background subtraction, the value inputted is equivalent to the smallest diameter of the object for which the surface is being created. In this case, that value would represent the width of a mitochondrion. Measure the width of a single mitochondrion and copy that distance into the surface creator.
-
f.In the next step, adjust the thresholding on the histogram to create an appropriate surface rendering that is representative of the mitochondrial staining, while at the same time minimizing the background noise effectively.Note: Depending on the quality and resolution of the staining, it may be useful to enable the option for “split touching objects”, where it is evident that individual mitochondria are being merged into one surface.
-
g.Once the surface is generated, adjustments can be made in the number of voxels histogram to remove background or noise.Note: If the rendering is not properly detecting all mitochondria, repeat previous steps adjusting the background subtraction and thresholding.CRITICAL: Imaris will automate a surface rendering of the inputted image based on the user’s adjustments in previous steps. It is strongly advised that this rendering be validated for accuracy by the investigator. If individual mitochondria are not resolved properly this may lead to an over or under estimation of individual organelle volume.
-
h.Once the above steps are completed, and the user clicks “Done”, the output measures will be automatically generated.
-
i.To view values, users should click on “Detailed” in the analysis tab of the software. Values for mitochondrial volume (both individual and total) will appear, amongst other measures.CRITICAL: If the investigator notices extreme values (i.e., very large or very small), they can be validated as individual mitochondria. To do so, click on the value in question, which will highlight the product in the rendered surface. Upon validation, if these values are not accurate (i.e., they pertain to debris or background staining) they can be excluded and/or the investigator can go back and produce a more appropriate surface of the image.
-
a.
Figure 1.

Confocal and z-stack imaging of mitochondria
Screenshot view of the Zen software control panel with parameters used to image mitochondria. White Box: “Live” Button allows for live viewing of staining in real time. While using this feature, users can set the upper and lower limits of the Z-stack as addressed below. Pink Box: “Continuous” Button allows for live viewing in continuous scanning to adjust for the Airyscan signal. Red Box: “Channels” parameters is used to modify pinhole, channel gain, laser selection, and other imaging parameters for each individual channel. Selecting different tracks allows the visualization and image capture of the selected channel(s). Yellow Box: “Z-Stack” window is used to set the parameters for the upper and lower limits of the cell of interest. Green Box: “Start Experiment” Button is used to capture the Z-stack image once all the parameters for the experimented have been selected. Teal boxes: Outlines the steps to follow to implement the Airyscan processing on the acquired image.
Figure 2.

Generation of 3D projection confocal images and quantification of mitochondrial length in Fiji
(Ai) Once the .czi image file is opened in Fiji, a 3D Projection can be generated by selecting the “Image” tool in the toolbar and “3D Project” in the “Stacks” column.
(Aii) The “3D Projection” window will appear. The user should ensure the settings are correct and then select “Ok”.
(Bi) A 3D projection of the z-stack image will appear, revealing the mitochondrial (Tom20) and MuSC (Pax7) staining (inset shows merged view).
(Bii) To enhance the quality of Tom20 staining (i.e., mitochondria), the brightness/contrast can be adjusted.
(Biii) Application of the “Sharpen” feature will define individual mitochondria for accurate measurements to be made. Dotted box shows zoomed views of mitochondria before and after application of the “sharpen” feature. Scale = 2 μm.
(C) The “Freehand Line” tool, selected from the Line tool in the toolbar, is used to trace the individual length of a mitochondrion to acquire mitochondrial length measurements. Zoomed view shows examples of mitochondrial tracings (yellow lines) used to acquire length measurements.
Figure 3.

Changes in mitochondrial length observed in MuSCs upon genetic or pharmacological manipulation of mitochondrial dynamics proteins
(A, E, and I) Representative confocal 3D reconstructed images of mitochondria (Tom20) in muscle stem cells (Pax7+). (A) Freshly isolate MuSCs from OPA1 WT and OPA1 KO mice, showing decreased mitochondrial length upon loss of the mitochondrial fusion protein OPA1.1 (E) Freshly isolated MuSCs from WT and OPA1 OE mice, showing increased mitochondrial length upon over-expression of OPA1.1,7,8 (I) MuSCs isolated from wild-type mice and cultured for 4 h with or without 2 μm Mdivi-1, showing increased mitochondrial length upon inhibition of the mitochondrial fission protein DRP1. Tom20 inset shows zoomed view of mitochondria in dotted box. Scale = 2 μm.
(B–D, F–H, J–L) Three individual cells were quantified for each condition and the data are presented in three ways. (B, F, and J) Scatter plots of the compiled raw length values of individual mitochondria from three cells; n > 90 mitochondria. (C, G, and K) Bar graphs of mean mitochondrial length within each independent cell; n = 3 cells. (D, H, and L) Graphs of the percentage distribution of mitochondria based on each length binning; n = 3 cells. Data presented as mean ± SEM. ∗∗p < 0.01, ∗∗∗p < 0.001 student’s unpaired t-test.
Part 4: Preparing primary muscle stem cells for electron microscopy and analysis of mitochondrial ultrastructure
Below is a generalized protocol that is optimized and utilized for assessing mitochondrial ultrastructure via electron microscopy on a small number of primary MuSCs. Although this methodology was utilized on MuSCs, this protocol can be adapted and used for other rare cell populations.
Note: Our FACS-based protocol for isolating muscle stem cells yields ∼100,000 cells, which is sufficient for transmission electron microscopy (TEM) analysis only when embedded in low-melt agarose.1 However, if the cell yield is less than this number, samples may need to be combined after fixation. See below for details.
-
26.
Following isolation of cells, pellet cells via centrifugation.
Note: FACS isolated MuSCs are spun in a 1.5 mL tube at 4,200 rpm for 2 min.
-
27.
Remove the supernatant using a pipette, carefully avoiding the cell pellet.
-
28.
Gently add 1 mL of sterile 1× PBS.
Note: Be very gentle when adding the PBS so that the pellet is not disturbed.
-
29.
Centrifuge at 1,200 rpm for 5 min at room temperature.
-
30.
Remove liquid using a pipette and gently add 500 μL of cold EM Fixative.
Note: Be very gentle when adding the fixative so that the pellet is not disturbed/resuspended.
-
31.
Incubate for 15 min at 4°C.
-
32.
Centrifuge at 5,000 rpm for 8 min.
-
33.
Remove liquid using a pipette.
-
34.
Add 500 μL of cold EM Fixative.
-
35.
Combine cell pellets from multiple sorts if required. To do so, gently resuspend a pellet in one tube, using the EM Fixative buffer within the tube, and add it to the tube containing the second pellet.
-
36.
Whether combining pellets or not, spin the sample at 5,000 rpm for 8 min and then remove the supernatant.
-
37.Agarose embed the samples as per the instructions below.CRITICAL: Low-melt agarose solidifies at room temperature. To keep in liquid form, leave at 67°C until needed.
-
a.Prewarm pellets to 37°C in a heating block for a few minutes.Note: This can be done while the agarose is melting (see instructions above for agarose preparation).
-
b.Quickly add 30 μL of agarose to the cell pellet and gently resuspend.Note: It is important to work quickly to ensure that the agarose remains liquid while spinning for the MuSC pellet to be encased.
-
c.Spin at 2,500 rpm for 3 min.
-
d.Incubate the agarose embedded pellet at 4°C for 10 min.
-
e.Gently loosen the agarose encased pellet from the side of the tube using a sterile, stainless steel micro-spatula.CRITICAL: Proceed with caution. Gently liberate the agarose for the wall of the tube, ensuring that the agarose is not pierced in the process.
-
f.Add 500 μL of EM fixative so that the agarose encased pellet is free floating.
-
g.Store at 4°C until needed.
-
a.
-
38.
Process samples utilizing standard EM-sectioning protocols.
Note: Agarose-embedded samples for this protocol were processed and sectioned by the Facility for Electron Microscopy Research at McGill University.
-
39.
Visualize mitochondria in MuSCs via TEM.
Note: Electron micrograph images for this protocol were captured at the uOttawa TEM core facility using an JEOL JEM-1400Flash 120 kv transmission electron microscope equipped with a GATAN 4K CMOS OneView digital camera and a Lab6 filament as emission source for enhanced contrast. We recommend first taking an image at lower magnification (i.e., 1500×) to visualize the whole cell of interest (Figure 4A). Subsequently, investigators should take an image at a high enough magnification (i.e., 20,000×) to capture mitochondria (Figure 4B).
Figure 4.

Quantification of mitochondrial ultrastructure in muscle stem cells by transmission electron microscopy (TEM) in muscle stem cells
(A) TEM image of mitochondria within a freshly FACS isolated and agarose embedded muscle stem cell.
(B) Zoomed view of an individual mitochondrion from TEM image in (A) with visible organellar components.
(C) Representative line drawn for mitochondrial length measurement.
(D) View of an individual cristae showing lines drawn at three locations representing the opposing edges (#1 and #3) and the center (#2) of the cristae that are used to calculate the mean cristae width.
(E) Inset shows zoomed view (white dotted square) of a cristae junction with a representative line drawn for measurement of cristae junction width. IMM; inner mitochondrial membrane, OMM; outer mitochondrial membrane.
-
40.
Save TEM images.
Note: For this protocol, images were saved in .dm4 format, which contain embedded metadata and retain information for image calibration (i.e., scaling of pixel size).
-
41.Quantify mitochondrial ultrastructure (Figures 4C–4E).
-
a.Open the .dm4 image file on Fiji software.
-
b.Measure the length of a mitochondrion using the Freehand Line tool in Fiji (Figure 4C).
-
c.Count the number of cristae in the mitochondrion.
-
d.Measure the width of each cristae using the Freehand Line tool in Fiji (Figure 4D).Note: To ensure a representative quantification of cristae width, the average of three measurements should be used (Figure 4D). One measure should be at the top region of the cristae, another in the middle portion, and finally one at the bottom of the cristae (designated as #1, 2 and 3 in Figure 4D).
-
e.Measure the width of the cristae junction (Figure 4E).Note: This is the distance between the two edges at the base of a cristae.
-
f.Perform the following calculations:
-
i.Cristae # per mitochondria or Cristae # per mitochondrial length.
-
ii.Cristae width.
-
iii.Cristae junction width.
-
i.
-
a.
Expected outcomes
The expected outcomes below are based on previous findings1 in freshly isolated MuSCs (i.e., near quiescent). These values change as MuSCs undergo activation, commitment, and differentiation. Furthermore, these measurements are a guideline in MuSCs, but are likely different in other cell populations.
Mitochondrial length measurements
Mitochondria within MuSCs are variable in length. We typically find that in MuSCs on EDL myofibers isolated from wild-type C57BL-6 mice, mitochondrial length ranges between ∼0.1–1.0 μm, with an average of 0.4–0.75 μm. However, it should be noted that this will vary depending on the mouse strain and background. Data in Figure 3 demonstrates mitochondrial length in MuSCs under conditions that lead to increased or decreased mitochondrial length by genetic or pharmacological means (Figure 3). These conditions include 1) OPA1 WT and OPA1 KO MuSCs (genetic fragmentation model) (Figures 3A–3D) 2) WT and OPA1 OE MuSCs (genetic elongation model) (Figures 3E–3H), and 3) WT MuSCs treated in culture (t = 4 h) with vehicle or 2 μm of Mdivi-1 (pharmacologic elongation model) (Figures 3I–3L); whereby data was collected and analyzed from 3 independent MuSCs.
Mitochondrial volume measurements
The volume of individual mitochondria within MuSCs display the same variability as length. We find that the average volume of mitochondria within MuSCs is ∼0.045 μm3.
Mitochondrial ultrastructure measurements
Like mitochondrial length and volume, mitochondrial ultrastructure characteristics display variability. In FACS-isolated MuSCs we have found that mean mitochondrial length is ∼0.4 μm, mean cristae width is 0.009 μm, mean # of cristae/mitochondria are ∼3, and there are approximately 7 cristae per μm of mitochondrial length.
Limitations
This protocol was designed and optimized to assess mitochondrial morphology in muscle stem cells, which have a small cytoplasmic volume and represent a population of rare cells. This protocol serves as a suitable starting point for fixing, staining and visualizing mitochondria, however the protocol as presented requires validation in other cell populations prior to experimental applications.
In this protocol, mitochondrial measurements are performed manually, which could potentially present the injection of bias. Although the use of well validated automated pipelines would alleviate such bias, these approaches were not found to be suitable for measuring mitochondrial length in MuSCs, whereby the cytoplasmic volume of these cells, particularly during quiescence and the early stages of commitment and differentiation, is dramatically less compared to other cell types. Due to this intrinsic characteristic, mitochondria within MuSCs are often clustered together and condensed into a small area. This makes it difficult for automated pipelines to identify individual mitochondria and thus assessing mitochondrial length using these programs leads to an overestimation of the mitochondrial length. It is therefore important for the investigator to understand their cell type and make an informed decision to use manual or automated mitochondrial quantifications methods.
The protocols described have been optimized in freshly isolated stem cell populations. However, removal from their native environment activates these cells, and thus does not reflect a truly deep quiescent state. Thus, the morphology of mitochondria observed with this protocol may not necessarily reflect the mitochondrial state of cells in deep quiescence. Visualization of mitochondrial morphology in their native, deep quiescent, state may be achieved using techniques such as in situ fixation. However, concentrations of fixative and rate of PFA perfusion would need optimization to prevent stress-induced damage to mitochondria within these rare cell populations.
Troubleshooting
Problem 1
Loss of EDL myofibers when washing and mounting onto glass slide and coverslip (Part 2, step 20).
Potential solution
-
•
When removing buffer from the myofibers (i.e., wash steps) it is integral that the investigator carefully pipette excess liquid off without touching the fibers. To do so, it is recommended that users pipette liquid from the top of the well, without placing the tip near the bottom of the well. In addition, tilting the plate will aid in confining the myofibers towards the bottom of the well and will allow for easier removal of the liquid.
-
•
Ensure there is very little PBS on the slide prior to adding the coverslip. This will allow for the EDL myofibers to stick to the charged glass slide. Failure to remove an adequate volume of liquid and/or not allowing enough time to dry will cause the EDL myofibers to be expelled off the slide together with the excess liquid and mounting media when the coverslip is mounted.
Problem 2
Poor resolution of individual mitochondrial morphology by immunofluorescence (Part 3, step 24).
Potential solution
-
•
Use the “Sharpen” tool on Fiji Software. Sharpening the image will further distinguish the spacing between adjacent mitochondria, allowing for more accurate measurements of single mitochondrion (depicted in Figure 2Biii).
-
•
Increase the blocking time. This may help remove background fluorescence and make organelles more distinguishable.
-
•
Eliminate excess antibody by adding more wash steps after incubation with primary and/or secondary antibodies.
-
•
Decrease scanning speed per Z-stack while imaging.
Note: This will increase the resolution collected per stack. Although this will aid in distinguishing individual mitochondria, it will also increase file size and imaging time.
Problem 3
Difficulty measuring mitochondrial length due to organelle clustering (Part 3, step 24).
Note: In muscle stem cells, where cytoplasmic volume is small, mitochondria often appear clustered. This renders it difficult to distinguish individual mitochondria.
Potential solution
-
•
Rotate the 3D reconstructed image to view the mitochondria from multiple angles. Rotating the image will allow for the investigator to properly distinguish individual mitochondria that are clustered together.
-
•
Use the “Sharpen” feature in the Fiji software to further distinguish mitochondria.
Problem 4
Inability to see a pellet following FACS isolation and during EM-preparation (Part 4, steps 26–37).
Note: This is a common issue that occurs when working with rare cell populations or low-yield cell isolations. Following fixation with EM fixative, the subsequent pellets may appear as a smear on the bottom of the tube.
Potential solution
-
•
Investigators must ensure they are cautious when performing EM fixation steps. The pellet from samples should not be re-suspended; as this will result in loss of cells. Rather, buffers should be gently removed from the pellet/smear with a pipette and new buffer should be pipetted gently.
-
•
Following spinning, a “smear” rather than a pellet may be observed. An acceptable cell pellet for agarose embedding will be circular with a clearly defined border located near the bottom of the tube. A smear appears as an elongated pellet, resembling a vertical line along the inside of the tube. If a “smear” is present after the spin, one solution is to follow with a second spin. Additionally, the experimenter can also adjust the number of washes with PBS following isolation of the cells. Buffers containing FBS cause the cells to be more “slippery” which may prevent the formation of a pellet and result in a smear. Repeat PBS wash steps until a compact pellet is observed. It should be noted that while this will aid in the formation of a pellet, it may result in some loss of cells.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Mireille Khacho (mkhacho@uottawa.ca).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This work was supported by funds from Canada Research Chair (CRC), CIHR, Stem Cell Network, and NSERC, J.P. Bickell to M.K., an NSERC-CGSM and OGSD scholarship to S.W., and a Muscular Dystrophy Canada and Neuromuscular Disease Network for Canada (MDC-NMD4C) Research Fellowship to M.T. We thank the University of Ottawa Cell Biology and Image Acquisition Core Facility (CBIA RRID [SCR_021845], Fernando Ortiz at the OHRI (Ottawa Hospital Research Institute) Flow Cytometry & Cell Sorting Core Facility, and the Facility for Electron Microscopy Research at McGill University.
Author contributions
M.K. conceptualized and led the study. M.K. and N.B. developed and optimized the protocols. M.K., N.B., M.T., and S.W. designed and performed the experiments, and M.T. quantified data. M.T. wrote the manuscript and contributed to designing the figures. S.W. contributed to generating the figures and writing the paper. M.K. contributed to writing the manuscript, generating the figures, and edited and finalized the paper and figures.
Declaration of interests
The authors declare no competing interests.
Data and code availability
This study did not generate any unique data sets or code.
References
- 1.Baker N., Wade S., Triolo M., Girgis J., Chwastek D., Larrigan S., Feige P., Fujita R., Crist C., Rudnicki M.A., et al. The mitochondrial protein OPA1 regulates the quiescent state of adult muscle stem cells. Cell Stem Cell. 2022;29:1315–1332.e9. doi: 10.1016/j.stem.2022.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brun C.E., Wang Y.X., Rudnicki M.A. Single EDL myofiber isolation for analyses of quiescent and activated muscle stem cells. Methods Mol. Biol. 2018;1686:149–159. doi: 10.1007/978-1-4939-7371-2_11. [DOI] [PubMed] [Google Scholar]
- 3.Hüttner S.S., Ahrens H.E., Schmidt M., Henze H., Jung M.J., Schüler S.C., von Maltzahn J. Isolation and culture of individual myofibers and their adjacent muscle stem cells from aged and adult skeletal muscle. Methods Mol. Biol. 2019;2045:25–36. doi: 10.1007/7651_2019_209. [DOI] [PubMed] [Google Scholar]
- 4.Pasut A., Jones A.E., Rudnicki M.A. Isolation and culture of individual myofibers and their satellite cells from adult skeletal muscle. J. Vis. Exp. 2013:e50074. doi: 10.3791/50074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu L., Cheung T.H., Charville G.W., Rando T.A. Isolation of skeletal muscle stem cells by fluorescence-activated cell sorting. Nat. Protoc. 2015;10:1612–1624. doi: 10.1038/nprot.2015.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maesner C.C., Almada A.E., Wagers A.J. Established cell surface markers efficiently isolate highly overlapping populations of skeletal muscle satellite cells by fluorescence-activated cell sorting. Skelet. Muscle. 2016;6:35. doi: 10.1186/s13395-016-0106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cogliati S., Frezza C., Soriano M.E., Varanita T., Quintana-Cabrera R., Corrado M., Cipolat S., Costa V., Casarin A., Gomes L.C., et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–171. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Varanita T., Soriano M.E., Romanello V., Zaglia T., Quintana-Cabrera R., Semenzato M., Menabò R., Costa V., Civiletto G., Pesce P., et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015;21:834–844. doi: 10.1016/j.cmet.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gambini E., Martinelli I., Stadiotti I., Vinci M.C., Scopece A., Eramo L., Sommariva E., Resta J., Benaouadi S., Cogliati E., et al. Differences in mitochondrial membrane potential identify distinct populations of human cardiac mesenchymal progenitor cells. Int. J. Mol. Sci. 2020;21:7467. doi: 10.3390/ijms21207467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang B.B., Wang D.G., Guo F.F., Xuan C. Mitochondrial membrane potential and reactive oxygen species in cancer stem cells. Fam. Cancer. 2015;14:19–23. doi: 10.1007/s10689-014-9757-9. [DOI] [PubMed] [Google Scholar]
- 11.Sukumar M., Liu J., Mehta G.U., Patel S.J., Roychoudhuri R., Crompton J.G., Klebanoff C.A., Ji Y., Li P., Yu Z., et al. Mitochondrial membrane potential identifies cells with enhanced stemness for cellular therapy. Cell Metab. 2016;23:63–76. doi: 10.1016/j.cmet.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rovini A., Heslop K., Hunt E.G., Morris M.E., Fang D., Gooz M., Gerencser A.A., Maldonado E.N. Quantitative analysis of mitochondrial membrane potential heterogeneity in unsynchronized and synchronized cancer cells. FASEB J. 2021;35:e21148. doi: 10.1096/fj.202001693R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate any unique data sets or code.