

Abstract
A novel bacterial strain, designated GeG2T, was isolated from soils of the native Cerrado, a highly biodiverse savanna-like Brazilian biome. 16S rRNA gene analysis of GeG2T revealed high sequence identity (100%) to the alphaproteobacterium Novosphingobium rosa; however, comparisons with N. rosa DSM 7285T showed several distinctive features, prompting a full characterization of the new strain in terms of physiology, morphology, and, ultimately, its genome. GeG2T cells were Gram-stain-negative bacilli, facultatively anaerobic, motile, positive for catalase and oxidase activities, and starch hydrolysis. Strain GeG2T presented planktonic-sessile dimorphism and cell aggregates surrounded by extracellular matrix and nanometric spherical structures were observed, suggesting the production of exopolysaccharides (EPS) and outer membrane vesicles (OMVs). Despite high 16S rDNA identity, strain GeG2T showed 90.38% average nucleotide identity and 42.60% digital DNA–DNA hybridization identity with N. rosa, below species threshold. Whole-genome assembly revealed four circular replicons: a 4.1 Mb chromosome, a 2.7 Mb extrachromosomal megareplicon, and two plasmids (212.7 and 68.6 kb). The megareplicon contains a few core genes and plasmid-type replication/maintenance systems, consistent with its classification as a chromid. Genome annotation shows a vast repertoire of carbohydrate-active enzymes and genes involved in the degradation of aromatic compounds, highlighting the biotechnological potential of the new isolate. Chemotaxonomic features, including polar lipid and fatty acid profiles, as well as physiological, molecular, and whole-genome comparisons showed significant differences between strain GeG2T and N. rosa, indicating that it represents a novel species, for which the name Novosphingobium terrae is proposed. The type strain is GeG2T (= CBMAI 2313T = CBAS 753 T).
Supplementary information
The online version contains supplementary material available at 10.1007/s42770-022-00900-4.
Keywords: Novosphingobium, Cultivation, Sphingomonadales, Cerrado, Soils, Chromid
Introduction
Novosphingobium is a genus that belongs to the Alphaproteobacteria class, whose members possess diverse physiological profiles and colonize diverse niches such as soils and rhizospheres [1–3], groundwater [4], freshwater [5–7], deep-sea environments [8, 9], mangrove sediments [10], bioremediation systems [11], coolant lubricant emulsion [12], among many other natural and artificial habitats.
These organisms are Gram-negative non-sporulating small bacilli (1–4 µm × 0.3–1.0 µm), motile or non-motile, aerobic or facultative anaerobic [13]. Novosphingobium species are known for their production of different exopolysaccharides (EPS), commonly used in beverage and food industries [14], and their production is affected by environmental and in vitro culture conditions. Members of this genus also present the ability to degrade a wide variety of xenobiotics and aromatic compounds, such as polycyclic aromatic hydrocarbons (PAHs), lignin derivatives, heterocyclic compounds, steroid endocrine disruptors, and pesticides [11, 12, 15–17], qualifying them as candidates for different bioremediation processes.
At the time of writing, the List of Prokaryotic names with Standing in Nomenclature (LPSN) recognized 58 Novosphingobium species (https://lpsn.dsmz.de/genus/novosphingobium), accessed in August 2022). Despite the relatively high number of species described in this genus so far, to date, only 13 genomes are flagged as complete on GenBank (National Center for Biotechnology Information — NCBI) and some aspects regarding the biology of this important bacterial group remain elusive.
In this work, we describe the genomic and physiological characterization of a Novosphingobium strain (GeG2T) isolated from soils of a Brazilian savannah-like biome, known as Cerrado. A comprehensive morphological, chemotaxonomic, molecular, biochemical, and genome sequence analysis revealed that, despite sharing an identical 16S rRNA sequence with Novosphingobium rosa, GeG2T represents a new species, for which the name Novosphingobium terrae is proposed. Its environmental adaptation repertoire can be assessed by the existence of a varied set of genes related to polysaccharide catabolism, particularly xylan and hemicellulose, as well as genes conferring the ability to degrade recalcitrant aromatic compounds. Remarkably, in addition to the main chromosome, we could detect and annotate a secondary megabase-sized replicon, most likely a chromid [18], one of the largest reported to date.
Material and methods
Bacteria isolation and cultivation
The microorganism was isolated from soils sampled at a native area of Cerrado, a savannah-like Brazilian biome, located at the Reserva Ecológica do IBGE, Brasília, Brazil (15°55′ S, 47°51′ W), in January 2014. The isolate, denominated strain GeG2T, was initially grown in solid media, prepared homogenizing 5% (w/v) of the soil sample with distilled water, filtered in depth filters (pore size of 10 to 20 μm) to remove coarse particles, added with 1.5% (w/v) of agar, and sterilized in the autoclave. Media were supplemented with ampicillin (150 μg/mL), streptomycin (50 μg/mL), chloramphenicol (20 μg/mL), and itraconazole (0.25 mg/mL) before inoculation. Plates were incubated at 28 °C and cultures were transferred into fresh media approximately once a month. For strain GeG2T isolation, colonies were transferred to minimal medium (MM, per liter: 0.1 g KH2PO4; 0.2 g (NH4)2SO4; 0.1 g MgSO4.7H2O; 0.02 g CaCl2.2H2O; 0.2 g NaCl; 0.1 g yeast extract; 0.05% glucose; 15 g bacteriological agar, pH 5.5), using the standard streak plate technique to obtain pure cultures. Plates were incubated at 28 °C for 48 h and strain GeG2T was stored at − 80 °C in sterile MM containing 20% (v/v) glycerol.
16S rRNA gene analysis
16S rRNA gene was amplified from genomic DNA extracted from cultures using a standard phenol–chloroform method. Briefly, cells were harvested from culture plates and resuspended in a lysis buffer containing 25 mM Tris–HCl, 10 mM EDTA, 200 μg/mL proteinase K, 100 μg/mL RNAse A, and 0.6% (w/v) SDS. After incubation for 1 h at 37 °C, an equal volume of phenol:chloroform:isoamyl alcohol mixture (25:24:1) was added. The aqueous phase was then treated with chloroform:isoamyl alcohol (24:1) and DNA was precipitated with 0.3 M NaCl and cold ethanol. After washing with 70% ethanol (v/v), the extracted DNA was dried and re-suspended in ultra-pure H2O.
The 16S rRNA gene of strain GeG2T was amplified by polymerase chain reactions (PCR) using universal bacterial primers 27F (5′AGAGTTTGATCCTGGCTCAG3′)/1492R (5′ GGTTACCTTGTTACGACTT3′) [19]. PCR reactions were performed in a Bio-Rad PTC-100® (Peltier Thermal Cycler) employing the following cycle conditions: 5 min at 95 °C, followed by 30 cycles of 1 min at 95 °C, 1 min at 55 °C, 2 min at 72 °C, and a final extension of 10 min at 72 °C. PCR products were purified with Wizard® SV Gel and PCR Clean-Up System (Promega) and ligated to the pGEM-T easy® vector (Promega), according to the manufacturer’s instructions. Plasmid extraction was performed with phenol:chloroform:isoamyl alcohol at 25:24:1 (v:v:v) and Sanger sequencing was carried out by Macrogen Inc. (Seoul, Korea) using universal primers T7 e SP6. The EzTaxon-e server (available at https://www.ezbiocloud.net) [20] was used to calculate the similarity between 16S rRNA gene sequences of GeG2T and other strains (Accessed in August 2022).
Physiological and chemotaxonomic characterizations
A Novosphingobium rosa strain, DSM 7285T, was obtained from the Leibniz-Institut Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) and used as a reference strain for comparative phenotypic analyses against GeG2T. Growth of both strains in TSA (Trypticase Soy Agar—BD Difco™), LB (Luria–Bertani, per liter: 5 g yeast extract; 10 g NaCl; 10 g tryptone), NA (Nutrient Agar—Kasvi, Brazil), and MM media was assessed. Unless mentioned otherwise, for all further characterizations, both bacterial strains were cultured on MM containing glucose (0.05%—w/v) as a carbon source, at 28 °C.
Growth of GeG2T and reference strain DSM 7285T in different temperatures (4, 15, 20, 28, 33, 37, and 42 °C) and different pH (4.0 to 9.0 in 1.0-unit intervals, buffered with 50 mM MES—pH 4.0 to 6.0; MOPS—pH 7.0 or Tris—pH 8.0 and 9.0) was evaluated in agar plates incubated for 1 week. Salinity requirement and tolerance were evaluated on solid MM supplemented with 0, 0.1, 0.3, 0.5, 0.8, 1.0, 1.5, 2.0, and 3.0% (w/v) of NaCl. Hydrolysis of starch was analyzed in solid MM supplemented with soluble starch (0.5%—w/v) and revealed with iodine vapor after 7, 14, and 24 days [21]. Catalase activity was determined by assessing bubble production by cells in 3% (v/v) H2O2 [21]. Growth under anaerobic conditions was tested in agar medium supplemented with cysteine hydrochloride (0.05%—w/v), sodium sulfide (0.05%—w/v), and potassium nitrate (0.1%—w/v) [22]. Anaerobic glass jars were prepared under a nitrogen atmosphere, sealed with rubber stoppers and aluminum seals, and incubated at 28 °C for 35 days. Jars maintained under aerobic conditions were used as controls. Nitrate reduction, indole production, urease and gelatinase tests, assimilation and oxidation of various carbon compounds, and enzyme activities were carried out by the Identification Service of DSMZ Leibniz Institute, using the API 20NE and API ZYM kits (bioMérieux), according to the manufacturer’s instructions.
Antibiotic susceptibility profiles of strain GeG2T were evaluated by the agar diffusion method using antibiotic-impregnated discs [23, 24], with bacterial suspensions spread over MM plates, incubated at 28 °C for 48 h. The tested antibiotics were nalidixic acid (30 μg), amikacin (30 μg), amoxicillin + clavulanic acid (20 + 10 μg), ampicillin (10 μg), cephalothin (30 μg), cefepime (30 μg), cefoxitin (30 μg), ceftazidime (30 μg), cefuroxime (30 μg), ciprofloxacin (5 μg), clindamycin (2 μg), chloramphenicol (30 μg), erythromycin (15 μg), gentamicin (10 μg), levofloxacin (5 μg), meropenem (10 μg), nitrofurantoin (300 μg), norfloxacin (10 μg), oxacillin (1 μg), penicillin (6 μg), rifampicin (5 μg), tetracycline (30 μg), trimethoprim + sulfamethoxazole (1.25 + 23.75 μg), and vancomycin (30 μg).
Fatty acid compositions of strains GeG2T and DSM7285T grown in solid MM for 72 h were determined following the standard protocol of the Sherlock Microbial Identification System (version 6.2) [25]. Briefly, approximately 40 mg of bacterial biomass harvested from third quadrants was submitted to saponification in 1 mL methanol/sodium hydroxide solution (150 mL deionized water, 150 mL methanol, 45 g sodium hydroxide), followed by methylation in 2 mL of 6 mol/L methane in HCl and extraction with 1.25 mL hexane:tertbutyl ether (1:1). The fatty acid profiles were analyzed by gas chromatography (Agilent 7890A) using the RTSBA6 method/library. Analysis of strain GeG2T polar lipids and respiratory quinones was performed by the Identification Services of DSMZ Leibniz Institute, following standard protocols [21, 26, 27].
Protein profiles analyses by MALDI-TOF
Protein profiles of strain GeG2T and the reference strain DSM7285T were determined by MALDI-TOF mass spectrometry. Proteins were extracted using the protocol described by [28] and then spotted (1 μL of protein extraction) in technical sextuplicates on a 96-well target steel plate, covered by 1 μL of 10 mg/mL α-cyano-4-hydroxycinnamic acid (HCCA) matrix (50% (v/v) acetonitrile, 0.3% (v/v) trifluoroacetic acid) and let dry at room temperature. Spectra were obtained in an Autoflex Speed II MALDI-TOF/TOF (Bruker Daltonics) in positive linear mode, 2000–20,000 m/z range, acquiring 2000 successful shots per spot using FlexControl 3.0 software. In total, 10,000 laser shots were accumulated for each spectrum. Spectra were further analyzed in MALDI Biotyper 3.0 software [29] which compares each sample mass spectrum to reference mass spectra in the Biotyper database and calculates a score value between 0 and 3 reflecting their similarity. GeG2T and DSM 7285T main spectra profiles (MSP) were compared to each other and Biotyper database. Scores of > 2.0 were accepted as reliable for identification at the species level, > 1.7 but < 2.0 at the genus level, and scores < 1.7 were considered unreliable, as specified by the manufacturer. All experiments were performed in a biological sextuplicate. A dendrogram was constructed using MSP information from each replicate on MALDI Biotyper 3.0 software.
Microscopy
GeG2T morphological characterizations were performed by light and electron microscopy analyses. Fresh or Gram-stained cells grown on solid or liquid media were observed by phase-contrast or bright field microscopy, respectively, in an Axio Scope.A1 (Zeiss, Germany) microscope. Scanning electronic microscopy (SEM) was performed in cells grown in liquid media for 72 h or 14 days and fixed on Karnovsky’s fixative solution [30]. Images were generated with a JEOL JSM-7001F microscope (JEOL Ltd., Tokyo, Japan). Transmission electron microscopy (TEM) analyses were conducted in the Center of Microscopy at the Universidade Federal de Minas Gerais using bacterial cells grown for 7 days in liquid or solid media and posteriorly cryofixed by high-pressure freezing (HPF) or cells grown for 14 days in liquid media and fixed on Karnovsky’s fixative solution.
Genome sequencing and assembly
Genomic DNA was extracted from cultures using the same phenol–chloroform method used for 16S rRNA amplification, mentioned above. Genomic DNA integrity and quality were assessed by agarose gel electrophoresis and spectrophotometry (NanoDrop™, Thermo Fisher Scientific Inc.). DNA quantification was carried out by fluorometric assay for double-stranded DNA (Qubit™, Thermo Fisher Scientific Inc.). Genome sequencing was performed at Macrogen Inc., Korea, using a combination of Illumina and PacBio technologies. Illumina libraries were constructed with TrueSeq DNA shotgun PCR-Free (350 bp) kits and PacBio 20 kb bluepippin systems. Sequencing was performed on Illumina HiSeq 2000 (paired-end sequencing) and PacBio RS (2 SMRT cells) platforms, respectively.
Quality control on Illumina short reads was performed with FastQC v0.11.5 [31] and TrimGalore v0.6.0 (https://github.com/FelixKrueger/TrimGalore) was used, with default parameters, to filter reads and trim bases based on quality threshold. PacBio reads were extracted using pbh5tools v0.8.0 (https://github.com/PacificBiosciences/pbh5tools), applying the ngs-preprocess pipeline (https://github.com/fmalmeida/ngs-preprocess), with default parameters. Additionally, Illumina paired-end reads were merged with PEAR v0.9.8 [32] to increase overall read length and help in the assembly step. Preprocessed Illumina and PacBio reads were assembled with the reads merged by PEAR using the program Unicycler v0.4.7 [33], with default parameters, in hybrid mode, and assembly statistics were assessed with QUAST v5.0.1 [34], both part of the MpGAP pipeline (https://github.com/fmalmeida/MpGAP). Genome completeness was assessed using BUSCO v3.1.0 [35] and CheckM v1.0.13 [36], using sphingomonadales_odb10 and o_Sphingomonadales (UID3310) reference datasets, respectively, with both tools executed with default parameters. Circular representations for the different genome replicons were generated with DNAPlotter [37].
Whole-genome-based taxonomic analyses
Genome-based taxonomic identification was performed by OGRI (overall genome relatedness index) estimations and phylogenomic analyses performed through the Type Strain Genome Server (TYGS) [38] and TrueBac ID [39] services. Average nucleotide identity (ANI) between the GeG2T genome and 198 Novosphingobium genomes downloaded from GenBank—NCBI (Accessed in September 2022) was calculated with FastANI v1.33–1 [40], using default parameters. The phylogenetic tree based on concatenated core orthologous genes from genomic sequences of 40 Novosphingobium strains, including strain GeG2T and ANI closely related strains, as well as reference species, was reconstructed by using the M1CR0B1AL1Z3R web server (https://microbializer.tau.ac.il/) [41]. Sphingomonas paucimobilis NCTC11030 (NCBI accession number GCA_900457515.1) was used as an outgroup.
Whole-genome alignment between strain GeG2T and N. rosa NBRC 15208T (GCF_001598555.1), the only genome available for the species in NCBI, was performed with MUMmer toolkit v3.1 [42]. Alignments with at least 1 kb length and 90% identity were used to draw a circular visualization of alignments with ggbio [43].
Genome functional annotation
Genome annotation was performed with Prokka v1.14.0 [44] and rRNA sequences were predicted with Barrnap v0.9 (https://github.com/tseemann/barrnap). Gene functions based on KEGG Orthology were predicted with KofamScan v1.3.0 [45], all orchestrated with the bacannot pipeline (https://github.com/fmalmeida/bacannot). Plasmid sequences were predicted using Plasflow v1.1 [46] and sequence similarity against known plasmids was calculated with the NCBI’s Microbial Nucleotide Blast. Dinucleotide relative abundance distance between genome replicons was determined as described in [47]. Clusters of Orthologous Genes (COG) assignments were performed by eggNOG-mapper v2 [48] and the two-proportions Z-test was used with the prop.test, R language built-in function, to calculate significance between the proportions of COG categories annotated in the replicons. Metabolic pathways and oxygenases were determined with the KEGG Orthology database [49]. Predictions of carbohydrate-active enzymes (CAZymes) were made with the dbCAN2 meta server [50], from which only CAZymes predicted by at least 2 tools were considered. All the tools were executed with default parameters.
Results and discussion
Isolation, growth, and 16S rDNA-based phylogenetic analysis
A bacterial strain, denominated GeG2T, was isolated from microbial enrichments obtained from the Cerrado soils, an extremely biodiverse environment with a vast microbial genetic repertoire and biotechnological potential [51, 52]. Growth in solid minimal medium (MM) resulted in white, circular (2–3 mm diameter), convex colonies, with regular edges and a shiny appearance. Light microscopy analysis revealed GeG2T cells to be Gram-negative, motile, and rod-shaped, with dimensions ranging from 1.3 to 2.3 μm in length and 0.3–0.6 μm in width.
Nearly complete 16S rRNA gene fragments of strain GeG2T were obtained by PCR amplification and sequenced. All 22 sampled amplicon sequences (1450 bp) were identical and comparisons with sequences from the EzBioCloud database [20] revealed the highest similarity with Novosphingobium rosa NBRC 15208T (100% rDNA sequence identity, with 95% of sequence coverage), followed by Novosphingobium lotistagni THG-DN6.20T (97.59%), Novosphingobium oryzae ZYY112T (97.02%), and Novosphingobium barchaimii LL02T (96.95%).
Although highly useful for the initial taxonomic classification of microorganisms, resolution limitations in the phylogenetic analysis based solely on 16S rRNA gene sequences are often reported, especially at the species level [53–55], indicating that additional approaches must be employed for the delimitation of bacterial species [56, 57]. For instance, despite a 99.9% similarity observed between Novosphingobium pentaromativorans US6-1T and Novosphingobium sp. PP1Y 16S rRNA genes, comparisons based on whole-genome sequences and chemotaxonomic traits suggest that these strains are different species [58, 59]. For this reason, physiological, chemical, morphological, and whole genome-based analyses of strain GeG2T were carried out, as described in the following sections.
Physiological and chemotaxonomic characterizations
Considering the highest 16S rRNA gene identity observed between strain GeG2T and Novosphingobium rosa, a type strain of this species, DSM 7285T, was obtained from DSMZ microorganism collection (Leibniz Institute, Germany) and used as the reference strain for comparative phenotypic analyses. Differently from N. rosa DSM 7285T, strain GeG2T was unable to grow in several rich media, such as Trypticase Soy Agar, Nutrient Agar, and Luria–Bertani medium (Table 1). Some Sphingomonadaceae members isolated from soils are known to exhibit better growth in low nutrient culture media [13] and the characteristic low nutrient availability of Cerrado soils [60] could probably have driven GeG2T strain adaptation to oligotrophic growth conditions.
Table 1.
Differential characteristics of strain GeG2T and type strains of related Novosphingobium species. Strains: 1. GeG2T; 2. N. rosa DSM 7285T (data from this study); 3. N. lotistagni THG-DN6.20T (data from [6]. All strains are positive for hydrolysis of aesculin; catalase; oxidase; assimilation of glucose, arabinose, and maltose; alkaline phosphatase, acid phosphatase, naphthol-AS-BI-phosphohydrolase, β-galactosidase, α-glucosidase, and β-glucosidase activities. All strains are negative for hydrolysis of gelatin and urea; indol production; glucose fermentation; assimilation of mannitol, caprate, adipate, malate, citrate, phenylacetate; arginine dihydrolase, lipase (C14), and α-mannosidase activities. + , positive; − , negative; w, weakly positive; -* no growth after first transfer; ND, no data
| Characteristic | 1 | 2 | 3 |
|---|---|---|---|
| Isolation source | Cerrado soil | Rose rhizosphere | Lotus pond |
| Colony color | White | Yellow | Yellow |
| Cell size (μm): | |||
| Width | 0.3–0.6 | 0.3–0.5 | 0.6–0.8 |
| Length | 1.3–2.3 | 1.0–1.9 | 0.9–4.2 |
| Motility | + | + | - |
| Growth on TSA | - | + | w |
| Growth on NA | -* | + | + |
| Growth on LB | -* | + | - |
| Growth conditions: | |||
| pH | 4–7 | 4–7 | ND |
| NaCl (%) | 0–1 | 0–1.5 | 0–1 |
| Temperature (°C) | 15–33 | 15–33 | 4–42 |
| Facultatively anaerobic growth | + | - | - |
| Hydrolysis of starch | + | - | - |
| Nitrate reduction | w | - | - |
| Assimilation of: | |||
| Mannose | + | + | - |
| N-Acetylglucosamin | + | w | w |
| Gluconate | + | w | - |
| Enzyme activities of: | |||
| Esterase (C4) | w | + | w |
| Esterase lipase (C8) | w | w | w |
| Leucin-arylamidase | + | + | w |
| Valin-arylamidase | + | + | w |
| Cystin-arylamidase | w | w | w |
| Trypsin | - | - | + |
| Chymotrypsin | w | - | + |
| α-Galactosidase | - | - | + |
| β-Glucuronidase | w | - | + |
| N-Acetyl-β-glucosaminidase | - | w | - |
| α-Fucosidase | - | - | w |
| DNA G + C content (mol%) | 63.57 | 64.50 | 63.10 |
Morphological differences were observed between colonies of strain GeG2T and N. rosa DSM 7285T grown in MM for 48 h, with colonies of strain GeG2T presenting a whitish and viscous aspect, while N. rosa DSM 7285T colonies were smaller, drier, and yellow-pigmented (Supplementary Fig. S1). Both bacterial strains grow in temperatures between 15 and 33 °C, with optimal growth at 28 °C, and pH 4.0 to 7.0, though while N. rosa DSM 7285T grows in NaCl concentrations ranging from 0 to 1.5%, strain GeG2T only grows up to 1% NaCl (Table 1). Starch hydrolysis was not observed for N. rosa DSM 7285T, whereas GeG2T was positive after 14 days of incubation in MM containing both glucose and starch, or only starch (Table 1, Supplementary Fig. S2). Furthermore, even though Novosphingobium members were initially described as strict aerobes [61], some species have recently been identified as facultative anaerobes [62, 63]. As reported for N. pentaromativorans US6-1T [62], the slow growth of strain GeG2T under anaerobic conditions was detected (after 30 days), which was never observed for N. rosa DSM 7285T (Table 1).
Antibiotic susceptibility profiles based on disc-diffusion tests (see Methods) revealed strain GeG2T to be resistant to several antibiotics, with susceptibility observed only to tetracycline and rifampicin. A similar resistance pattern was observed for N. rosa DSM 7285T, which, in addition to tetracycline and rifampicin, was also susceptible to trimethoprim/sulfamethoxazole. Although information regarding resistance mechanisms in Sphingomonadaceae is still scarce, soil and rhizosphere isolates belonging to this family are generally resistant to various kinds of antimicrobial agents [64] and have been identified to constitute an environmental reservoir of the antibiotic resistome [65].
Additional biochemical characterizations were performed with API 20NE and API Zym kits and revealed similar overall biochemical profiles for strain GeG2T and N. rosa DSM 7285T (Table 1). However, although weak positive activities of chymotrypsin, β-glucuronidase, and nitrate reduction were detected for strain GeG2T, N. rosa DSM 7285T presented negative results for these traits. Moreover, while GeG2T was negative for N-acetyl-β-glucosaminidase, a weak positive activity was observed for N. rosa DSM 7285T (Table 1).
As expected for members of the Sphingomonadaceae family [13, 61], the major respiratory quinone identified in GeG2T cells was Q10 (> 95%), with Q9 also detected in smaller amounts. Major fatty acids were C16:0 (24.62%), C18:0 (21.84%) and C18:1ω7ϲ/C18:1ω6ϲ (18.18%) (Table 2). Despite presenting similar composition, proportions of different fatty acids varied considerably between GeG2T and N. rosa DSM 7285T, for which the major fatty acids detected were C18:1ω7ϲ/C18:1ω6ϲ (38.71%), followed by C14:0 2-OH (19.65%) and C16:0 (14.84%) (Table 2). Notably, while the unsaturated fatty acid C18:1ω9ϲ represented a considerable fraction of fatty acids (8.29%) in GeG2T cells, it was only detected in trace amounts (< 1%) in N. rosa DSM 7285T cells grown under the same conditions (Table 2).
Table 2.
Whole-cell fatty acid contents (%) of strain GeG2T and the closest related species Novosphingobium rosa DSM 7285T. Both strains were grown on MM agar at 28 °C for 72 h. TR, trace (< 1%); -, not detected
| Fatty acid | GeG2T | N. rosa DSM 7285T |
|---|---|---|
| C12:0 | TR | - |
| C13:0anteiso | TR | TR |
| C14:0 | 1.85 | 1.30 |
| C14:0 2-OH | 7.22 | 19.65 |
| C16:0 | 24.62 | 14.84 |
| C17:0 anteiso | TR | - |
| C17:0 cyclo | TR | - |
| C17:0 | TR | - |
| C18:1ω9ϲ | 8.29 | TR |
| C18:0 | 21.84 | 7.82 |
| C19:0 iso | TR | - |
| C19:0 cyclo ω8ϲ | 1.92 | 2.18 |
| C20:0 | TR | - |
| C16:1ω7ϲ/C16:1ω6ϲ | 11.97 | 13.88 |
| C18:1ω7ϲ/C18:1ω6ϲ | 18.18 | 38.71 |
The polar lipids of strain GeG2T included phosphatidylglycerol (PG), phosphatidylethanolamine (PE), sphingoglycolipid (SGL), diphosphatidylglycerol (DPG), besides an aminolipid (AL) and unidentified lipids (Supplementary Fig. S3). Interestingly, even though phosphatidylmonomethylethanolamine (PME) and phosphatidyldimethylethanolamine (PDE) are commonly identified in Novosphingobium members [66], including N. rosa [67], these polar lipids were not detected in strain GeG2T. Differences in polar lipids and fatty acids profiles between strain GeG2T and the N. rosa strain suggest that these bacteria do not belong to the same species, as sphingomonads species can be distinguished from each other due to qualitative and/or quantitative variations in these chemotaxonomic components [68].
Protein profile analyses by MALDI-TOF
Comparative analysis of protein profiles by MALDI-TOF mass spectrometry was performed to further demonstrate the distinctive phenotype of strain GeG2T. While main spectra profiles (MSP) obtained for strain DSM 7285T matched those from the species Novosphingobium rosa (scores > 2.0), MSP generated from GeG2T protein extracts showed identification scores lower than 2.0 when compared to DSM 7285T MSP and MALDI Biotyper® database, suggesting that this strain represents a new species, not yet represented in the database containing over 8000 strains. Furthermore, dendrogram analysis of generated MSP shows that GeG2T and N. rosa DSM 7285T are grouped in different clades, even when external groups are added (Supplementary Fig. S4), further indicating that they belong to different species.
Morphological characterization of GeG2T cells by electronic microscopy
Transmission electron micrographs of strain GeG2T cells revealed one or two intracytoplasmic electron-dense granules (70–150 μm in diameter) per cell, mostly located in the central portion of the cytoplasm (Fig. 1A and B). The observed granules exhibit a characteristic aspect of polyphosphate (poly-P) granules or acidocalcisomes, subcellular structures enriched in phosphorus compounds, and different cations [69]. While acidocalcisomes are membrane-encapsulated, poly-P granules lack surrounding membranes and, even though recent studies suggested that these are different subcellular structures that can be simultaneously found in alphaproteobacterial species [70], the presence of surrounding membranes in GeG2T granules could not be determined by electron microscopy. Further microscopic and chemical analyses are required to elucidate the nature and function of the electron-dense granules observed in strain GeG2T. However, as poly-P molecules are involved in diverse physiological and regulatory mechanisms in bacteria such as response to nutrient starvation and oxidative, acid, osmotic, or UV stresses [69], it is tempting to speculate that the granules observed in GeG2T cytoplasm could represent an adaptative strategy to thrive in nutrient deprived conditions encountered in the minimal medium as well as native Cerrado soils [60].
Fig. 1.

Transmission electron micrograph of strain GeG2T cells showing intracytoplasmic electron-dense granules (A and B) and scanning electron micrographs of strain GeG2T cells surrounded by amorphous polymeric matrix, with the characteristic aspect of exopolysaccharides (EPS) (C) and presenting smooth or granular surfaces (D). Magnifications and scale bars are indicated under each micrograph. IG, intracytoplasmic granule. CM, cytoplasmic membrane. OM, outer membrane. P, peptidoglycan layer
Scanning electron micrographs of GeG2T cells grown in liquid MM revealed aggregated cells surrounded by an amorphous polymeric matrix, with the characteristic aspect of exopolysaccharides (EPS) (Fig. 1C). Sphingomonadaceae members are known for their ability to produce diverse EPS, generically denominated sphingans, presenting important applications in bioremediation, pharmaceutical, and food industries [14]. Some of the functions already described for bacterial EPS in soils are drought protection, nutrient trapping, aggregation, and biofilm structure, providing important benefits in stress responses [71]. A recent study revealed that Enterobacter EPS can alter the soil microbiome, as well as nutrients availability, to allow the microorganisms to better cope with heavy metal soil contamination [72].
Moreover, heterogeneities in cell surfaces among different GeG2T cells have also been identified: some showed a smooth uniform aspect, while others were granular and rough, a characteristic aspect of outer membrane vesicle (OMV) producers (Fig. 1D) [73, 74]. Although initially characterized in pathogenic Gram-negative bacteria [75], OMVs produced by environmental species have been increasingly studied and their varied composition under different conditions suggests diverse biological functions, including nutrient acquisition, stress responses, biodegradation of aromatic compounds, surfactant actions, and biofilm formation [73, 76–79].
Planktonic-sessile dimorphism of strain GeG2T
After sequential transfers in liquid MM, isolate GeG2T was found to grow as either planktonic motile cells or sessile-aggregated non-motile cells that form macroscopic flocks of different sizes (Supplementary Fig. S5). This phenomenon, also observed in other sphingomonads, is known as planktonic sessile dimorphism [80–82]. As reported for other species [80], flock formation by strain GeG2T seemed to be influenced by different growth parameters. While culture agitation increased flock formation, cultures incubated without agitation or in flasks containing greater volumes of media showed fewer macroscopic flocks and more homogeneous turbidity, indicating that greater oxygen levels may favor cell aggregation and extracellular matrix production. Increased flock formation was also observed in non-agitated cultures grown for extended incubation periods (over 4 days).
Scanning electron micrographs from agitated cultures grown for 14 days and presenting several flocks revealed long extracellular fibers forming a network connecting the cells, as well as an extracellular matrix enclosing them (Fig. 2A). An increased number of cells with granular surfaces were also identified, suggesting that OMV production could be favored in these cultivation conditions (Fig. 2B). Moreover, spherical structures resembling large vesicles, with diameters ranging from 150 to 250 nm, were identified among cell aggregates (Fig. 2B). Similar extracellular structures were reported for environmental bacteria grown in minimal media containing different hydrocarbons, in which an emulsifying activity and optimization of substrate assimilation roles were proposed [83, 84]. Furthermore, the formation of macroscopic flocks composed of aggregated cells embedded in an extracellular matrix rich in size-varying vesicle structures has recently been described for Novosphingobium sp. PP1Y grown in minimal media containing glutamate as the sole carbon source, suggesting a potential role of vesicles in nutrient acquisition under limiting conditions [79]. Likewise, it is possible that the vesicles associated with GeG2T cell aggregates observed after long incubation periods may be related to better nutrient assimilation under oligotrophic conditions.
Fig. 2.
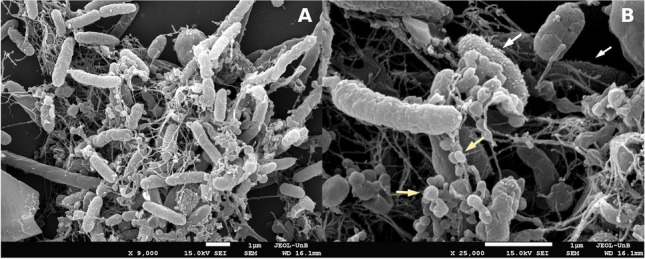
Scanning electron micrographs of strain GeG2T cultures presenting macroscopic flocks grown in MM for 14 days. Cells presenting granular surfaces are indicated by white arrows and spherical structures resembling large vesicles are indicated by yellow arrows. Magnifications and scale bars are indicated under each micrograph
Genome assembly and genome-based taxonomic analyses
To further improve its characterization, the complete genome sequence of strain GeG2T was assembled using a combination of long and short-read sequencing approaches, resulting in a high-quality and contiguous assembly totalizing 7,162,928 bp, distributed in 6 contigs, with a total GC content of 63.57% (Table 3). Four circular replicons were identified: a 4,164,843 bp chromosome (GC content: 64.23%), a 2,710,928 bp extrachromosomal megareplicon (GC content: 62.93%), and two plasmids – pGeG2a and pGeG2b—with 212,687 and 68,553 bp (GC content of 61.41 and 55.61%, respectively) (Fig. 3). Two additional contigs containing the entire rRNA operon and tRNA genes (with 5,558 bp and 359 bp) were assembled separately from the chromosome by Unicycler (Supplementary Fig. S6) possibly due to difficulties associated with the resolution of highly repetitive regions [33].
Table 3.
Statistics of strain GeG2T genome assembly performed with a hybrid approach using both short (Illumina) and long reads (PacBio)
| Feature | Value |
|---|---|
| Number of contigs | 6 |
| Largest contig (bp) | 4,164,843 |
| Total size (bp) | 7,162,928 |
| N50 | 4,164,843 |
| L50 | 1 |
| GC (%) | 63.57 |
Fig. 3.

Circular maps and genetic features of the chromosome, chromid, and plasmids of strain GeG2T. From outside to center: forward CDS (green), reverse CDS (light blue), genomic islands (brown), transposases (pink), tRNAs (purple), GC content, and GC skew (red and dark blue). Replicons are not shown to scale
The complete genome sequence of strain GeG2T offers a new perspective regarding its relationship with N. rosa, given the context of 100% sequence identity of their 16S rRNA genes. Initially, we could ascertain that the GeG2T 16S rRNA gene sequence predicted from the genome assembly (1,486 bp) was identical to the sequences obtained by direct colony PCR amplification followed by Sanger sequencing (1450 bp). As recently proposed [56], overall genome-related indexes (OGRI) were calculated to better evaluate the taxonomic classification of strain GeG2T. As shown in Table 4, average nucleotide identity (ANI) and digital DNA:DNA hybridization (dDDH) values obtained for these isolates (90.38% and 42.60%) are markedly divergent, considering the generally accepted species boundary of 95–96% and 70% for ANI and dDDH, respectively [56]. Moreover, genome-based taxonomic analyses performed in both Type Strain Genome Server (TYGS) [38] and TrueBac ID [39] servers identified that strain GeG2T does not belong to any species currently found in their databases, further indicating that it represents a new species within the genus Novosphingobium (Supplementary Fig. S7, Supplementary File S1). Finally, we carried out a whole-genome alignment between strain GeG2T and N. rosa NRBC 15208T. Setting a threshold of 90% identity in 1 kb blocks, it is possible to observe in Fig. 4 a sizeable number of unaligned regions in the chromosome (24% unaligned blocks) and even more in the megareplicon (65%), indicating that the genomes are divergent from each other.
Table 4.
Statistics of genome similarity between strain GeG2T and closest Novosphingobium genomes obtained from the TYGS analysis
| Genome | ANI | dDDH (%) | 16S rRNA (%) |
|---|---|---|---|
| N. rosa NBRC 15208T | 90.339 | 42.6 | 100.000 |
| N. naphthalenivorans NBRC 102051T | 78.857 | 14.0 | 96.774 |
| N. subarcticum KF1T | 78.836 | 14.1 | 96.445 |
| N. aromaticivorans DSM 12444T | 78.582 | 13.8 | 96.306 |
| N. lindaniclasticum DSM 25409T | 78.469 | 14.1 | 96.577 |
| N. guangzhouense DSM 32207T | 78.449 | 14.0 | 96.774 |
Fig. 4.
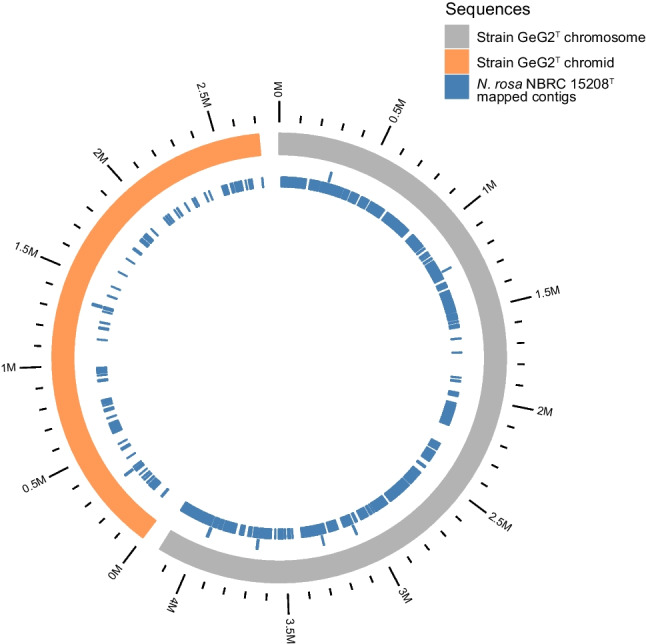
Circular visualization of whole-genome alignment between strain GeG2T (chromosome in grey and chromid in orange) and N. rosa NBRC 15208T reference genome (in blue). Only genome alignment blocks with at least 1,000 nucleotides and 90% identity are shown
Expanding the comparative genomics analysis to investigate the phylogenetic relationships of this genus, we used the genome sequences from 40 Novosphingobium species (32 from different species, 6 from indeterminate species, and the two strains above) to extract orthologous gene sets. Using the M1CR0B1AL1Z3R web server, a total of 178 core genes (subset of genes shared among the 40 species) were recovered and used to construct a maximum-likelihood phylogenetic tree, which was compared to a tree built using only the 16S rRNA gene sequences from the same species above. This comparison is shown in Fig. 5 and reveals a close, but divergent, evolutionary link between strain GeG2T and N. rosa NRBC 15208T. Moreover, despite their 16S rRNA gene identity, which was the only case among the 40 genomes analyzed, it is evident the lower resolution of 16S rRNA trees (in terms of branch lengths) and branch divergence compared to the core genes tree.
Fig. 5.

Comparison of phylogenetic trees of 40 Novosphingobium species based on core genes (left) and 16S rRNA genes (right). The trees show phylogenetic positions of strain GeG2T and the 39 Novosphingobium genomes selected based on the highest ANI values to GeG2T. A total of 178 core genes were identified and used to construct a maximum-likelihood phylogenetic tree using the M1CR0B1AL1Z3R web server [41]. The 16S rRNA gene-based tree was inferred from Global Blast Distance Phylogeny (GBDP) distances by the TYGS server [38]. Branch lengths are displayed in the bottom scales and only the bootstrap support values below 100% are indicated in the core genes tree. Sphingomonas paucimobilis NCTC 11030T was used as an outgroup
The genome is bipartite and contains a chromid
Besides the chromosome and two plasmids, an interesting feature identified in the genome of strain GeG2T was the presence of an extrachromosomal megareplicon (2.7 Mb) (Fig. 3). Gene content and genomic signature analyses of this replicon revealed both chromosomal and plasmid features, commonly associated with chromids: replicons that are normally larger than the accompanying plasmids but smaller than the chromosome, presenting nucleotide composition close to that of the chromosome, but plasmid-type replication and maintenance systems [18].
While plasmid replication and segregation genes belonging to the alphaproteobacterial RepABC family have been identified in the megareplicon of strain GeG2T, close GC content (∆= 1.2%) and dinucleotide relative abundance distance from the chromosome (< 0.4) were observed, supporting its classification as a putative chromid [47]. Furthermore, the presence of one or more core genes that can be found in the chromosome of related species is considered a distinctive characteristic of chromids [18]. Indeed, genes involved in important cellular functions, such as DNA polymerase III ε and α subunits (genes dnaQ and dnaE), pentose phosphate, and pyruvate oxidation pathway genes were detected both in the chromosome and in the putative chromid of strain GeG2T, based on KEGG annotations. Moreover, the alignment of sequences with coding potential predicted in the chromosome and the megareplicon has allowed the identification of 29 genes with high nucleotide and amino acid sequence similarities (> 85%) (Supplementary File S2).
Secondary megareplicons (> 350 kb) were identified in all six fully resolved Novosphingobium genomes evaluated by [17], suggesting that multipartite genomes may be a common characteristic of this genus. In this context, strain GeG2T stands out since its 2.7 Mb secondary replicon is the largest reported to date for Novosphingobium species, followed by a 2.2 Mb replicon identified in Novosphingobium sp. P6W [85], a 1.7 Mb replicon from N. resinivorum SA1T [17] up to a 487.3 kb putative chromid in the genome of N. aromaticivorans DSM 12444 T [47]. Interestingly, the N. rosa NBRC 15208T genome assembly (GCF_001598555.1), used for comparative purposes with GeG2T, does not report any plasmids or chromids. However, the pairwise genome alignment, shown in Fig. 4, reveals several NBRC 15208T contigs mapping to GeG2T chromid, which may be an indicator of additional replicons yet unresolved and underscores the benefits of hybrid sequencing strategy, using both short and long reads, resulting in the high-contiguity assembly obtained for GeG2T.
Next, we set out to explore if the chromosome and chromid of GeG2T share sequence identity to probe their evolutionary paths. DNA sequence alignments of both replicons were performed using NCBI’s blastn and results were filtered to keep only hits with 100% similarity and longer than 1000 bp. Using these thresholds, only six hits of approximately 1300 bp have been identified. Interestingly, we found a single region of the chromid (between the positions 1,657,174 and 1,655,854) matching three different locations in the chromosome. Inside this genomic region, two IS3 family transposases, namely IS868 and ISRtr2, have been found in tandem. Altogether, these results indicate distinct evolutionary routes between the chromosome and chromid, and that transposable elements may mediate DNA exchange between these megareplicons in GeG2T, even though on a very limited scale.
Gene annotation
In total, 6,124 genes (3,697 in the chromosome) were predicted in the genome of strain GeG2T, including 3 rRNAs (collapsed in the rRNA operon in contig 5), 57 tRNAs, and 6,063 protein-coding sequences (CDS). A putative function was assigned to 3,706 of the protein-coding genes, while 2,357 were annotated as hypothetical proteins.
Functional analysis based on clusters of orthologous groups of proteins (COGs) assigned 5,746 CDS predicted from strain GeG2T genome to one or more COG functional classes, which were then sorted into 21 groups, as shown in Fig. 6A. As frequently observed in bacterial multipartite genomes [86–88], functional biases were detected between the genomic replicons of strain GeG2T. While proteins involved in essential processes such as DNA replication and repair (L), cell division (D), translation machinery (J), and cell wall/ membrane biogenesis (M) are mostly present in the chromosome, enrichment of genes involved in inorganic ion transport and metabolism (P), transcription (K) and carbohydrate transport and metabolism (G) are observed in the secondary megareplicon (Fig. 6A). These categories were shown to be primarily enriched in chromids rather than in megaplasmids [47], further corroborating the classification of GeG2T megareplicon as a chromid and indicating that it is likely related to adaptative roles and responsive behaviors to environmental changes [88]. However, unlike other bacterial species in which cellular motility (N) and signal transduction mechanism (T) categories are overrepresented in chromids [88, 89], most genes associated with these functions are chromosomal in strain GeG2T (Fig. 6A). Also, the majority of putative proteins assigned to the unknown function COG category (S) are present in the chromosome, differently from other sphingomonads, in which hypothetical proteins were mainly found in secondary replicons [90].
Fig. 6.

COG functional categories (A) and carbohydrate-active enzymes (CAZymes) (B) predicted in each replicon of strain GeG2T genome. COG categories with significantly different proportions as shown by the two-proportions Z-test are indicated with *. D: cell cycle control, cell division, chromosome partitioning; M: cell wall/membrane/envelop biogenesis; N: cell motility; O: post-translational modification, protein turnover, chaperone functions; T: signal transduction mechanisms; U: intracellular trafficking, secretion, and vesicular transport; V: defense mechanisms; Z: cytoskeleton; A: RNA processing and modification; B: chromatin structure and dynamics; J: translation, ribosomal structure, and biogenesis; K: transcription; L: replication and repair; C: energy production and conversion; E: amino acid metabolism and transport; F: nucleotide metabolism and transport; G: carbohydrate metabolism and transport; H: coenzyme metabolism and transport; I: lipid metabolism and transport; P: inorganic ion transport and metabolism; Q: secondary metabolites biosynthesis, transport, and catabolism; S: function unknown. GT: glycosyl transferases; GH: glycosyl hydrolases; CE: carbohydrate esterases; CBM: carbohydrate-binding module; AA: enzymes for the auxiliary activities; PL: polysaccharide lyases
Regarding the plasmids detected in strain GeG2T (pGeG2a and pGeG2b), sequence similarity searches against the NCBI database did not result in any significant alignment, indicating they are newly identified plasmids with still unknown functional properties, as depicted by the fact that most CDS detected in these replicons were annotated as hypothetical proteins. Genes related to cation efflux (cusC) and drug efflux systems (emrB, emrK, stp) have been predicted in pGeG2a, suggesting a potential role of this plasmid in drug resistance [91]. Moreover, functional analyses based on COGs revealed that while transcription (K), amino acid transport/metabolism (E), and carbohydrates transport/metabolism (G) were the main functional categories detected in pGeG2a, proteins predicted in pGeG2b were predominantly (almost 40%) attributed to trafficking, secretion and vesicular transport category (U) (Supplementary Table S1), frequently associated with resistance to toxic compounds [47, 92, 93]. Considering the indication of vesicle production by strain GeG2T detected in SEM micrographs (Fig. 2B), it is tempting to speculate that the numerous genes from this functional category identified in its plasmid pGeG2b, as well as in its chromosome and chromid (Fig. 6A), could be related to this phenotype. Furthermore, a considerable fraction of predicted proteins in both plasmids were also attributed to replication, recombination, and repair functions (category L) (Supplementary Table S1). Gene enrichments from categories L and K have been reported for plasmids of several bacterial species and are possibly associated with replication and conjugation processes [47, 92], and the identification of tra and trb genes in both plasmids from strain GeG2T indicates their conjugative nature.
Biochemical pathways inferred from gene annotation
Strain GeG2T genome encodes an extensive repertoire of enzymes involved in carbohydrate metabolism, comprising 41 glycosyl transferases (GTs), 131 glycosyl hydrolases (GHs), 11 carbohydrate esterases (CEs), two polysaccharide lyases (PLs), and seven enzymes for auxiliary activities (AAs), totalizing 192 carbohydrate-active enzymes (CAZymes; Fig. 6B, Supplementary File S3), a higher number than previously reported for other Sphingomonadaceae [90, 94]. As shown by COG analysis, many genes encoding CAZymes are found in the chromid (89; 47%), especially GHs and AAs (Fig. 6B), indicating an important role of this replicon in polysaccharide catabolism [95].
Among GHs encoded in the genome, 40 (23 in the chromid and 17 in the chromosome) belonging to families related to the degradation of xylan and other hemicellulose components (GH3, GH5, GH8, GH10, GH16, GH30, GH43, GH51, GH67, GH115) were identified, as well as CEs involved in the removal of the side chains from substituted xylose units (CE1 and CE4), revealing a large potential of strain GeG2T for plant biomass degradation [95]. Moreover, five GHs classified in families GH106 and GH78 of α-L-rhamnosidases, enzymes involved in many essential microbial functions and biotechnological applications [96], were detected in the GeG2T chromid (three GH106 genes) and chromosome (one GH78 and one GH106). The recent isolation and characterization of an α-L-rhamnosidase produced by Novosphingobium sp. PP1Y hydrolyzing various glycosylated flavonoids reveal interesting biotechnological potential associated with these enzymes [97]. Furthermore, CAZymes associated with the biosynthesis of oligosaccharides, polysaccharides, and glycoconjugates (GTs) are mainly encoded in the chromosome (Fig. 6B). As reported for different EPS producers [98, 99], most GTs were classified in families GT2 and GT4, which include enzymes with diverse origins and functions (e.g., cellulose synthases, chitin synthases, glycosyltransferases) [100].
Metabolic profiles predicted from KEGG revealed that, as reported for other 22 Novosphingobium genomes [17], strain GeG2T encodes the complete glycolysis, pentose phosphate, and tricarboxylic acid cycle pathways. While many genes involved in carbon metabolism are found both in the chromosome and the chromid (Supplementary Fig. S8), genes involved in nitrogen metabolism are encoded exclusively in the chromosome, including extracellular nitrite and nitrate transporter, nitrite reductases (nirB/nirD), and nitronate monooxygenase (ncd2), as well as genes encoding the Amt family of ammonium transporters.
Interestingly, member genes were interspersed in the chromosome and the chromid for some pathways. While alkane sulfonate assimilation genes (ssuABCD), the only extracellular sulfur assimilation pathway detected in the GeG2T genome, were found exclusively in the chromid, genes involved in sulfate, sulfite, and sulfide metabolism are encoded solely in the chromosome (Fig. 7). Moreover, except for hisC, all genes related to histidine biosynthesis (hisG, hisE, hisJ, hisA, hisF, hisB, hisN, hisD) are exclusively found in the chromosome, whereas genes encoding enzymes responsible for the conversion of histidine into glutamate (hutH, hutU, hutI, hutF, hutG) were detected in the chromid. Similarly, genes involved in de novo purine synthesis are encoded in the chromosome and many genes for purine degradation and salvage pathways were identified exclusively in the chromid of strain GeG2T. Curiously, even though taurine dioxygenase (tauD) was identified in all 27 Novosphingobium genomes from diverse habitats analyzed by [101], none of the genes associated with the taurine assimilation pathway could be detected in the GeG2T genome (Fig. 7).
Fig. 7.

Schematic representation of sulfur metabolism genes identified in strain GeG2T chromosome (in blue) and chromid (in orange). Genes associated with the assimilation of environmental sulfur compounds that were not detected in the genome of strain GeG2T but can be found in other Novosphingobium species [101] are represented in grey. APS, adenosine phosphosulfate; PAPS, phosphoadenosine phosphosulfate
Genes for three secretion systems as well as the twin-arginine translocation (Tat) and secretion (Sec) pathways were detected in the genome of strain GeG2T. Interestingly, all genes associated with type IV secretion system (T4SS), Tat, and Sec pathways are located in the chromosome, while complete type I and type VI secretion systems (T1SS and T6SS) are coded in the chromid. T4SS has been identified in the chromosome of many bacterial species [102, 103], including Sphingomonas [14], and has been associated with horizontal DNA transfer and secretion of bacterial interaction mediators [104]. T1SS is widely distributed among Gram-negative bacteria and is frequently associated with efflux mechanisms that lead to resistance to several compounds in Sphingomonadaceae [14]. Although initially identified as a typical virulence factor [105], T6SS is now recognized as a versatile secretion system, commonly found in soil bacteria, with roles in the assimilation of scarce ions, inter-bacterial competition, and different interactions with the environment [106]. The detection of T1SS and T6SS secretion systems in the chromid of strain GeG2T can further suggest an important role of this replicon in adaptative responses to environmental changes and interactions with other soil microorganisms.
Aromatic compound degradation
Members of Novosphingobium and related genera are recognized for their ability to degrade several xenobiotics and aromatic compounds [17]. A large number of monooxygenases and dioxygenases were detected in the strain GeG2T genome (Supplementary Table S2). These enzymes catalyze the ring cleavage step critical to the aerobic degradation of aromatic compounds and are essential for the metabolism of a wide variety of recalcitrant substances [107]. Although additional experimental confirmations are still necessary, taking into consideration the number and classes of monooxygenases and dioxygenases identified in the genome of strain GeG2T (Supplementary Table S2) that share the same annotation with the ones previously reported for Novosphingobium species known to be able to degrade compounds such as hexachlorocyclohexane, pentachlorophenol, biphenyl, phenanthrene, pyrene, benzo(a)pyrene, naphthalene, fluorene, among others [11, 17, 62, 82, 100, 108], strain GeG2T is probably able to metabolize different aromatic compounds.
Interestingly, contrary to the observed for N. aromaticivorans DSM 12444T, Novosphingobium sp. P6W, Novosphingobium sp. PP1Y, N. pentaromativorans US6-1T, and Novosphingobium sp. THN1, in which mono and dioxygenases are primarily encoded in chromosomes instead of their secondary megareplicons [17], most monooxygenases from strain GeG2T are found in the chromid. On the other hand, dioxygenases are detected in the chromosome and chromid (Supplementary Table S2). Analysis of known pathways for aromatic compound degradation revealed that, although monooxygenases usually associated with the first steps of toluene and xylene degradation (tmoA and xylM) could not be annotated in strain GeG2T genome, all other genes for enzymes associated with the biodegradation of these compounds could be identified (Fig. 8). Furthermore, many genes from the upper pathway of aromatic compounds degradation producing catechol intermediates were identified both in the chromosome and the chromid, while all genes related to catechol orthoclivage to tricarboxylic acids are encoded in the chromosome (Fig. 8).
Fig. 8.

Pathways associated with the degradation of aromatic compounds, obtained with the KEGG Mapper tool, indicating genes detected in the chromosome (in blue), the chromid (in orange), or in both replicons (in pink) of strain GeG2T genome. 1.1.1.90: aryl-alcohol dehydrogenase; 1.2.1.28: benzaldehyde dehydrogenase (xylC); 1.14.12.10: benzoate/toluate 1,2-dioxygenase subunit alpha (benA-xylX); 1.3.1.25: dihydroxycyclohexadiene carboxylate dehydrogenase (benD-xylL); 1.13.11.1: catechol 1,2-dioxygenase (catA); 5.5.1.1: muconate cycloisomerase (catB); 5.3.3.4: muconolactone D-isomerase (catC); 3.1.1.24: 3-oxoadipate enol-lactonase (pcaD); 1.13.11.2: catechol 2,3-dioxygenase (dmpB-xylE); 5.3.2.6: 4-oxalocrotonate tautomerase (praC-xylH); 4.2.1.80: 2-keto-4-pentenoate hydratase (mhpD)
Conclusion
In this study, we isolated a novel bacterial strain (GeG2T) from soils of a native area of Cerrado, a highly biodiverse biome located in Central Brazil. Based on the 16S rRNA gene sequence, strain GeG2T belongs to the alphaproteobacterial genus Novosphingobium, presenting 100% nucleotide identity (95% coverage) with previously characterized species Novosphingobium rosa. Albeit this fact, we conducted thorough morphological, biochemical, and genomic analyses to describe GeG2T. This strain presented planktonic-sessile dimorphism and microscopical analyses indicated the production of exopolysaccharide, variable-sized extracellular vesicles as well as intracytoplasmic electron-dense granules with the characteristic aspect of polyphosphate granules or acidocalcisomes. The complete genome of GeG2T was resolved through a combination of long and short-read sequencing approaches revealing a multipartite architecture consisting of a chromosome (4.2 Mb), an extrachromosomal megareplicon (2.7 Mb), and two plasmids (212 and 68 kb). Although experimental demonstrations would be ideally necessary for a definitive classification of the secondary megareplicon identified in GeG2T, the identification of chromosomal signatures and plasmid-type replication and maintenance systems indicates that it is a chromid [47]. Genome-based taxonomic identification, including overall genome relatedness index (OGRI) estimations and phylogenomic analysis, indicated a clear distinction between strain GeG2T and other Novosphingobium representatives, even N. rosa, despite their 100% 16S rRNA gene similarity. Furthermore, whole-genome sequence alignment between strain GeG2T and N. rosa NBRC 15208T further demonstrated their dissimilarity. In terms of gene space, a broad spectrum of carbohydrate metabolism enzymes and the large number of monooxygenases and dioxygenases identified in the genome of strain GeG2T reveal its great potential for plant biomass degradation, polysaccharide production, and degradation of diverse aromatic compounds. In summary, a polyphasic characterization, including physiologic, chemotaxonomic, MALDI-TOF protein profile, and whole genome-based analyses, indicates that strain GeG2T represents a new species within the Novosphingobium genus for which the name Novosphingobium terrae sp. nov. is proposed.
Description of Novosphingobium terrae sp. nov.
Novosphingobium terrae (ter’rae. L. gen. fem. n. terrae, of soil, referring to the isolation source of the type strain). Cells are Gram-negative bacilli, with 0.3–0.6 µm in width and 1.3–2.3 µm in length. Colonies grown in minimal media (MM) were white, circular, convex, with regular edges, shiny appearance, and about 2–3 mm in diameter. Growth occurs at 15–33 °C, pH 4–7, and NaCl concentrations from 0 to 1% (w/v). The cells are positive for aesculin hydrolysis, catalase and oxidase activity, assimilation of glucose, arabinose, mannose, N-acetyl-glucosamine, maltose, gluconate, and activities of alkaline phosphatase, leucine-arylamidase, valine-arylamidase, acid phosphatase, naphthol-AS-BI-phosphohydrolase, β-galactosidase, α-glucosidase, and β-glucosidase weakly positive for nitrate reduction, esterase C4 and esterase lipase C8. negative for hydrolysis of gelatin and urea, indol production, glucose fermentation, assimilation of mannitol, caprate, adipate, malate, citrate, phenylacetate, arginine dihydrolase, lipase C14, and α-mannosidase activities.
The type strain, GeG2T (= CBMAI 2313 T = CBAS 753 T) was isolated from Cerrado soils collected in Brasilia, Brazil (15°55′ S, 47°51′ W). The DNA GC content of the type strain is 63.57 mol %. The 16S rRNA gene sequence (MT325926.1) and the complete genome sequence of Novosphingobium terrae GeG2T (GCA_017163935.1) have been deposited in GenBank.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors would like to thank Dr. Heloisa Sinatora Miranda for her assistance in soil collections. We acknowledge the Center of Microscopy at Federal University of Minas Gerais (http://www.microscopia.ufmg.br) and the Laboratory of Microscopy at the University of Brasilia for providing the equipment and technical support for experiments involving transmission electron microscopy and scanning electron microscopy, respectively. We would also like to thank the Laboratory of Mass Spectrometry at Embrapa for providing the equipment and support for the MALDI Biotyper analysis.
Author contribution
AB cultivated and isolated the bacterial strain. AB and CV performed the microbiological characterizations. FA, RR, AB, RK, and GP analyzed the sequence data. AB and MR performed MALDI-TOF analyses. MT performed fatty acid analysis. CK and RK acquired funding for this study. AB, FA, CK, and GP wrote the manuscript. All authors revised and approved the manuscript.
Funding
This research was supported by grants from the National Council for Scientific and Technological Development (CNPq), Brazilian Federal Agency for Support and Evaluation of Graduate Education (CAPES), and Fundação de Apoio à Pesquisa do Distrito Federal (FAPDF-Grant: 00193–00000129/2019–80). AB and FA acknowledge fellowships from CNPq.
Data availability
The whole-genome sequencing dataset is available under the NCBI Bioproject PRJNA624997.
Code availability
The genome assembly and annotation pipelines developed by our group are available at the following links: https://github.com/fmalmeida/ngs-preprocess, https://github.com/fmalmeida/MpGAP, and https://github.com/fmalmeida/bacannot.
Declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
All authors consent to participate in the paper’s publication.
Conflict of interest
The authors declare no competing interests.
Footnotes
Cynthia Maria Kyaw and Georgios J. Pappas share senior authorship
Responsible Editor: Luiz Henrique Rosa
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Aline Belmok, Email: abelmokadias@gmail.com.
Cynthia Maria Kyaw, Email: malta@unb.br.
Georgios J. Pappas, Jr, Email: gpappas@unb.br.
References
- 1.Takeuchi M, Sakane T, Yanagi M, Yamasato K, Hamana K, Yokota A. Taxonomic study of bacteria isolated from plants: proposal of Sphingomonas rosa sp. nov., Sphingomonas pruni sp. nov., Sphingomonas asaccharolytica sp. nov., and Sphingomonas mali sp. nov. Int J Syst Bacteriol. 1995;45(2):334–341. doi: 10.1099/00207713-45-2-334. [DOI] [PubMed] [Google Scholar]
- 2.Kämpfer P, Young CC, Busse H, Lin SY, Rekha PD, Arun AB, et al. Novosphingobium soli sp. Nov., isolated from soil. Int J Syst Evol Microbiol. 2011;61(Pt 2):259–263. doi: 10.1099/ijs.0.022178-0. [DOI] [PubMed] [Google Scholar]
- 3.Kämpfer P, Martin K, McInroy JA, Glaeser SP. Proposal of Novosphingobium rhizosphaerae sp. nov., isolated from the rhizosphere. Int J Syst Evol Microbiol. 2015;65(Pt 1):195–200. doi: 10.1099/ijs.0.070375-0. [DOI] [PubMed] [Google Scholar]
- 4.Lee JC, Kim SG, Whang KS. Novosphingobium aquiterrae sp. nov., isolated from ground water. Int J Syst Evol Microbiol. 2014;64(Pt 9):3282–3287. doi: 10.1099/ijs.0.060749-0. [DOI] [PubMed] [Google Scholar]
- 5.Baek SH, Lim JH, Jin L, Lee HG, Lee ST. Novosphingobium sediminicola sp. nov. isolated from freshwater sediment. Int J Syst Evol Microbiol. 2011;61(Pt 10):2464–2468. doi: 10.1099/ijs.0.024307-0. [DOI] [PubMed] [Google Scholar]
- 6.Ngo HT, Trinh H, Kim JH, Yang JE, Won KH, Kim JH, et al. Novosphingobium lotistagni sp. nov., isolated from a lotus pond. Int J Syst Evol Microbiol. 2016;66(11):4729–4734. doi: 10.1099/ijsem.0.001418. [DOI] [PubMed] [Google Scholar]
- 7.Sheu SY, Chen ZH, Chen WM. Novosphingobium piscinae sp. nov., isolated from a fish culture pond. Int J Syst Evol Microbiol. 2016;66(3):1539–1545. doi: 10.1099/ijsem.0.000914. [DOI] [PubMed] [Google Scholar]
- 8.Yuan J, Lai Q, Zheng T, Shao Z. Novosphingobium indicum sp. nov., a polycyclic aromatic hydrocarbon-degrading bacterium isolated from a deep-sea environment. Int J Syst Evol Microbiol. 2009;59(Pt 8):2084–2088. doi: 10.1099/ijs.0.002873-0. [DOI] [PubMed] [Google Scholar]
- 9.Huo YY, You H, Li ZY, Wang CS, Xu XW. Novosphingobium marinum sp. nov., isolated from seawater. Int J Syst Evol Microbiol. 2015;65(Pt 2):676–80. doi: 10.1099/ijs.0.070433-0. [DOI] [PubMed] [Google Scholar]
- 10.Lee LH, Azman AS, Zainal N, Eng SK, Fang CM, Hong K, Chan KG. Novosphingobium malaysiense sp. nov. isolated from mangrove sediment. Int J Syst Evol Microbiol. 2014;64(pt4):1194–1201. doi: 10.1099/ijs.0.059014-0. [DOI] [PubMed] [Google Scholar]
- 11.Tiirola MA, Busse HJ, Kämpfer P, Männistö MK. Novosphingobium lentum sp. nov., a psychrotolerant bacterium from a polychlorophenol bioremediation process. Int J Syst Evol Microbiol. 2005;55(Pt 2):583–588. doi: 10.1099/ijs.0.63386-0. [DOI] [PubMed] [Google Scholar]
- 12.Kämpfer P, Busse HJ, Glaeser SP. Novosphingobium lubricantis sp. nov., isolated from a coolant lubricant emulsion. Int J Syst Evol Microbiol. 2018;68(5):1560–1564. doi: 10.1099/ijsem.0.002702. [DOI] [PubMed] [Google Scholar]
- 13.Glaeser SP, and Kämpfer P (2014) The family Sphingomonadaceae. In: The Prokaryotes. Springer, Berlin, Heidelberg. 10.1007/978-3-642-30197-1_302
- 14.Wu M, Huang H, Li G, Ren Y, Shi Z, Xiaoyan L, et al. The evolutionary life cycle of the polysaccharide biosynthetic gene cluster based on the Sphingomonadaceae. Sci Rep. 2017;7:46484. doi: 10.1038/srep46484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hegedűs B, Kós PB, Bálint B, Maróti G, Gan HM, et al. Complete genome sequence of Novosphingobium resinovorum SA1, a versatile xenobiotic-degrading bacterium capable of utilizing sulfanilic acid. J Biotechnol. 2017;241:76–80. doi: 10.1016/j.jbiotec.2016.11.013. [DOI] [PubMed] [Google Scholar]
- 16.Sheu SY, Huang CW, Chen JC, Chen ZH, Chen WM. Novosphingobium arvoryzae sp. nov., isolated from a flooded rice field. Int J Syst Evol Microbiol. 2018;68:2151–2157. doi: 10.1099/ijsem.0.002756. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Wang C, Li J, Bai P, Li Q, Shen M, et al. Comparative genomics of degradative Novosphingobium strains with special reference to microcystin-degrading Novosphingobium sp. THN1. Front Microbiol. 2018;9:2238. doi: 10.3389/fmicb.2018.02238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harrison PW, Lower RP, Kim NK, Young JP. Introducing the bacterial ‘chromid’: not a chromosome, not a plasmid. Trends Microbiol. 2010;18(4):141–148. doi: 10.1016/j.tim.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 19.Lane DJ. 16S/23S rRNA sequencing in nucleic acid techniques in bacterial systematics. New York: John Wiley and Sons; 1991. pp. 115–175. [Google Scholar]
- 20.Yoon SH, Ha SM, Kwon S, Lim J, Kim Y, Seo H, Chun J. Introducing EzBioCloud: a taxonomically united database of 16S rRNA and whole genome assemblies. Int J Syst Evol Microbiol. 2017;67:1613–1617. doi: 10.1099/ijsem.0.001755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tindall BJ, Sikorski J, Smibert RA, Krieg NR. Methods for General and Molecular Bacteriology. 3. Washington DC: American Society for Microbiology; 2007. Phenotypic characterization and the principles of comparative systematics; pp. 330–393. [Google Scholar]
- 22.Blume LR, Noronha EF, Leite J, et al. Characterization of Clostridium thermocellum isolates grown on cellulose and sugarcane bagasse. Bioenerg Res. 2013;6:763–775. doi: 10.1007/s12155-013-9295-6. [DOI] [Google Scholar]
- 23.Bauer AW, Kirby WMM, Sherris JC, Turck M. Antibiotic susceptibility testing by a standardized single disk method. Am J Clin Pathol. 1966;45(4):493–496. doi: 10.1093/ajcp/45.4_ts.493. [DOI] [PubMed] [Google Scholar]
- 24.Sha S, Zhong J, Chen B, Lin L, Luan T. Novosphingobium guangzhouense sp. nov., with the ability to degrade 1-methylphenanthrene. Int J Syst Evol Microbiol. 2017;67(2):489–497. doi: 10.1099/ijsem.0.001669. [DOI] [PubMed] [Google Scholar]
- 25.Sasser M. Identification of bacteria by gas chromatography of cellular fatty acids. MIDI Tech Note (MIDI Newark Delaware) 1990;101:1–7. [Google Scholar]
- 26.Tindall BJ. A comparative study of the lipid composition of Halobacterium saccharovorum from various sources. Syst Appl Microbiol. 1990;13:128–130. doi: 10.1016/S0723-2020(11)80158-X. [DOI] [Google Scholar]
- 27.Tindall BJ. Lipid composition of Halobacterium lacusprofundi. FEMS Microbiol Letts. 1990;66:199–202. doi: 10.1016/0378-1097(90)90282-U. [DOI] [Google Scholar]
- 28.Agustini BC, Silva LP, Bloch C, Jr, Bonfim TM, da Silva GA. Evaluation of MALDI-TOF mass spectrometry for identification of environmental yeasts and development of supplementary database. Appl Microbiol Biotechnol. 2014;98:5645–5654. doi: 10.1007/s00253-014-5686-7. [DOI] [PubMed] [Google Scholar]
- 29.Ramasamy D, Mishra AK, Lagier JC, Padhmanabhan R, Rossi M, Sentausa E, et al. A polyphasic strategy incorporating genomic data for the taxonomic description of novel bacterial species. Int J Syst Evol Microbiol. 2014;64:384–391. doi: 10.1099/ijs.0.057091-0. [DOI] [PubMed] [Google Scholar]
- 30.Souza W (2007) Técnicas de microscopia eletrônica aplicadas às ciências biológicas. 3a edição. Sociedade Brasileira de Microscopia, Rio de Janeiro
- 31.Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- 32.Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2014;30(5):614–620. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PloS Comput. Biol. 2017;13(6):e1005595. doi: 10.1371/journal.pcbi.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waterhouse RM, Seppey M, Simão FA, Manni M, Ioannidis P, Klioutchnikov G, et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol. 2018;35(3):543–548. doi: 10.1093/molbev/msx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2014;25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carver T, Thomson N, Bleasby A, Berriman M, Parkhill J. DNAPlotter: circular and linear interactive genome visualization. Bioinformatics. 2009;25(1):119–120. doi: 10.1093/bioinformatics/btn578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meier-Kolthoff JP, Göker M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat Commun. 2019;10:2182. doi: 10.1038/s41467-019-10210-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ha SM, Kim CK, Roh J, Byun JH, Yang SJ, Choi SB, et al. Application of the whole genome-based bacterial identification system, TrueBac ID, using clinical isolates that were not identified with three matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) systems. Ann Lab Med. 2019;39(6):530–536. doi: 10.3343/alm.2019.39.6.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9(1):5114. doi: 10.1038/s41467-018-07641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Avram O, Rapoport D, Portugez S, Pupko T. M1CR0B1AL1Z3R-a user-friendly web server for the analysis of large-scale microbial genomics data. Nucl Acids Res. 2019;47:W88–W92. doi: 10.1093/nar/gkz423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yin T, Cook D, Lawrence M. ggbio: an R package for extending the grammar of graphics for genomic data. Genome Biol. 2012;13(8):R77. doi: 10.1186/gb-2012-13-8-r77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 45.Aramaki T, Blanc-Mathieu R, Endo H, Ohkubo K, Kanehisa M, Goto S, Ogata H. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics. 2020;36(7):2251–2252. doi: 10.1093/bioinformatics/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krawczyk PS, Lipinski L, Dziembowski A. PlasFlow: predicting plasmid sequences in metagenomic data using genome signatures. Nucleic Acids Res. 2018;46(6):e35. doi: 10.1093/nar/gkx1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.diCenzo GC, Finan TM. The divided bacterial genome: structure, function, and evolution. Microbiol Mol Biol Rev. 2017;81(3):e00019–e117. doi: 10.1128/MMBR.00019-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, von Mering C, Bork P. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol Biol Evol. 2017;34(8):2115–2122. doi: 10.1093/molbev/msx148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang H, Yohe T, Huang L, et al. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018;46(W1):W95–W101. doi: 10.1093/nar/gky418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alves-Prado HF, Pavezzi FC, de Leite RS, Oliveira VM, Sette LD, Dasilva R. Screening and production study of microbial xylanase producers from Brazilian Cerrado. Appl Biochem Biotechnol. 2010;161(1–8):333–346. doi: 10.1007/s12010-009-8823-5. [DOI] [PubMed] [Google Scholar]
- 52.Peixoto J, Silva LP, Krüger RH. Brazilian Cerrado soil reveals an untapped microbial potential for unpretreated polyethylene biodegradation. J Hazard Mater. 2017;324(Pt B):634–644. doi: 10.1016/j.jhazmat.2016.11.037. [DOI] [PubMed] [Google Scholar]
- 53.Fox GE, Wisotzkey JD, Jurtshuk P., Jr How close is close: 16S rRNA sequence identity may not be sufficient to guarantee species identity. Int J Syst Bacteriol. 1992;42(1):166–170. doi: 10.1099/00207713-42-1-166S. [DOI] [PubMed] [Google Scholar]
- 54.Stackebrandt E, Goebel BM. Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Evol Microbiol. 1994;44(4):846–849. doi: 10.1099/00207713-44-4-846. [DOI] [Google Scholar]
- 55.Jaspers E, Overmann J. Ecological significance of microdiversity: identical 16S rRNA gene sequences can be found in bacteria with highly divergent genomes and ecophysiologies. Appl Environ Microbiol. 2004;70(8):4831–4839. doi: 10.1128/AEM.70.8.4831-4839.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chun J, Oren A, Ventosa A, Christensen H, Arahal DR, da Costa MS, et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int J Syst Evol Microbiol. 2018;68(1):461–466. doi: 10.1099/ijsem.0.002516. [DOI] [PubMed] [Google Scholar]
- 57.Raina V, Nayak T, Ray L, Kumari K, Suar M. Microbial diversity in the genomic era. India: Elsevier; 2019. Approach for designation and description of novel microbial species; pp. 137–152. [Google Scholar]
- 58.Choi DH, Kwon YM, Kwon KK, Kim SJ. Complete genome sequence of Novosphingobium pentaromativorans US6-1(T) Stand Genomic Sci. 2015;2015(10):107. doi: 10.1186/s40793-015-0102-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Troncone L (2011) A study of the biotechnological applications of Novosphingobium puteolanum PP1Y. PhD thesis. Università di Napoli Federico II, Naples, Italy
- 60.Haridasan M. Nutritional adaptations of native plants of the cerrado biome in acid soils. Braz J Plant Physiol. 2008;20(3):183–195. doi: 10.1590/S1677-04202008000300003. [DOI] [Google Scholar]
- 61.Takeuchi M, Hamana K, Hiraishi A. Proposal of the genus Sphingomonas sensu stricto and three new genera, Sphingobium, Novosphingobium and Sphingopyxis, on the basis of phylogenetic and chemotaxonomic analyses. Intern J System Bacteriol. 2001;51(Pt 4):1405–1417. doi: 10.1099/00207713-51-4-1405. [DOI] [PubMed] [Google Scholar]
- 62.Sohn JH, Kwon KK, Kang JH, Jung HB, Kim SJ. Novosphingobium pentaromativorans sp. nov., a high-molecular-mass polycyclic aromatic hydrocarbon-degrading bacterium isolated from estuarine sediment. Int J Syst Evol Microbiol. 2004;54(Pt 5):1483–1487. doi: 10.1099/ijs.0.02945-0. [DOI] [PubMed] [Google Scholar]
- 63.Sheu SY, Cai CY, Kwon SW, Chen WM. Novosphingobium umbonatum sp. nov., isolated from a freshwater mesocosm. Int J Syst Evol Microbiol. 2020;70(2):1122–1132. doi: 10.1099/ijsem.0.003889. [DOI] [PubMed] [Google Scholar]
- 64.Yabuuchi E, Kosako Y, Fujiwara N, Naka T, Matsunaga I, Ogura H, Kobayashi K. Emendation of the genus Sphingomonas Yabuuchi et al. 1990 and junior objective synonymy of the species of three genera, Sphingobium, Novosphingobium and Sphingopyxis, in conjunction with Blastomonas ursincola. Int J Syst Evol Microbiol. 2002;52(Pt 5):1485–1496. doi: 10.1099/00207713-52-5-1485. [DOI] [PubMed] [Google Scholar]
- 65.Dantas G, Sommer MO, Oluwasegun RD, Church GM. Bacteria subsisting on antibiotics. Science. 2008;320(5872):100–103. doi: 10.1126/science.1155157. [DOI] [PubMed] [Google Scholar]
- 66.Glaeser SP, Bolte K, Martin K, Busse HJ, Grossart HP, Kämpfer P, Glaeser J. Novosphingobium fuchskuhlense sp. nov., isolated from the north-east basin of Lake Grosse Fuchskuhle. Int J Syst Evol Microbiol. 2013;63(Pt 2):586–592. doi: 10.1099/ijs.0.043083-0. [DOI] [PubMed] [Google Scholar]
- 67.Kämpfer P, Denner EB, Meyer S, Moore ER, Busse HJ. Classification of “Pseudomonas azotocolligans” in the genus Sphingomonas as Sphingomonas trueperi sp. nov. Int J Syst Bacteriol. 1997;47:577–583. doi: 10.1099/00207713-47-2-577. [DOI] [PubMed] [Google Scholar]
- 68.Busse HJ, Kämpfer P, Denner EB. Chemotaxonomic characterisation of Sphingomonas. J Ind Microbiol Biotechnol. 1999;23(4–5):242–251. doi: 10.1038/sj.jim.2900745. [DOI] [PubMed] [Google Scholar]
- 69.Docampo R (2006) Acidocalcisomes and polyphosphate granules. In: Inclusions in Prokaryotes - Microbiology Monographs, vol 1. Springer, Berlin, Heidelberg. 10.1007/3-540-33774-1_3
- 70.Frank C, Jendrossek D. Acidocalcisomes and polyphosphate granules are different subcellular structures in Agrobacterium tumefaciens. Appl Environ Microbiol. 2020;86(8):e02759–e2819. doi: 10.1128/AEM.02759-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Costa OYA, Raaijmakers JM, Kuramae EE. Microbial extracellular polymeric substances: ecological function and impact on soil aggregation. Front Microbiol. 2018;23(9):1636. doi: 10.3389/fmicb.2018.01636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li Y, Shi X, Ling Q, Li S, Wei J, Xin M, Xie D, Chen X, Liu K, Yu F. Bacterial extracellular polymeric substances: impact on soil microbial community composition and their potential role in heavy metal-contaminated soil. Ecotoxicol Environ Saf. 2022;240:113701. doi: 10.1016/j.ecoenv.2022.113701. [DOI] [PubMed] [Google Scholar]
- 73.Gilewicz M, Ni’matuzahroh, Nadalig T, Budzinski H, Doumenq P, Michotey V, et al. Isolation and characterization of a marine bacterium capable of utilizing 2-methylphenanthrene. Appl Microbiol Biotechnol. 1997;48:528–533. doi: 10.1007/s002530051091. [DOI] [PubMed] [Google Scholar]
- 74.Coppotelli BM, Ibarrolaza A, Dias RL, Del Panno MT, Berthe-Corti L, Morelli IS. Study of the degradation activity and the strategies to promote the bioavailability of phenanthrene by Sphingomonas paucimobilis strain 20006FA. Microb Ecol. 2010;59(2):266–276. doi: 10.1007/s00248-009-9563-3. [DOI] [PubMed] [Google Scholar]
- 75.Toyofuku M, Nomura N, Eberl L. Types and origins of bacterial membrane vesicles. Nat Rev Microbiol. 2019;17:13–24. doi: 10.1038/s41579-018-0112-2. [DOI] [PubMed] [Google Scholar]
- 76.Choi CW, Park EC, Yun SH, Lee SY, Lee YG, Hong Y, et al. Proteomic characterization of the outer membrane vesicle of Pseudomonas putida KT2440. J Proteome Res. 2014;13(10):4298–4309. doi: 10.1021/pr500411d. [DOI] [PubMed] [Google Scholar]
- 77.Schwechheimer C, Kuehn MJ. Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat Rev Microbiol. 2015;13(10):605–619. doi: 10.1038/nrmicro3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yun SH, Lee SY, Choi CW, Lee H, Ro HJ, Jun S, et al. Proteomic characterization of the outer membrane vesicle of the halophilic marine bacterium Novosphingobium pentaromativorans US6-1. J Microbiol. 2017;55(1):56–62. doi: 10.1007/s12275-017-6581-6. [DOI] [PubMed] [Google Scholar]
- 79.De Lise F, Mensitieri F, Rusciano G, Dal Piaz F, Forte G, Di Lorenzo F, et al. Novosphingobium sp. PP1Y as a novel source of outer membrane vesicles. J Microbiol. 2019;57(6):498–508. doi: 10.1007/s12275-019-8483-2. [DOI] [PubMed] [Google Scholar]
- 80.Pollock T, Armentrout R. Planktonic/sessile dimorphism of polysaccharide-encapsulated sphingomonads. J Ind Microbiol Biotech. 1999;23:436–441. doi: 10.1038/sj.jim.2900710. [DOI] [PubMed] [Google Scholar]
- 81.Tiirola MA, Männistö MK, Puhakka JA, Kulomaa MS. Isolation and characterization of Novosphingobium sp. strain MT1, a dominant polychlorophenol-degrading strain in a groundwater bioremediation system. Appl Environ Microbiol. 2002;68(1):173–180. doi: 10.1128/aem.68.1.173-180.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Notomista E, Pennacchio F, Cafaro V, Smaldone G, Izzo V, Troncone L, et al. The marine isolate Novosphingobium sp. PP1Y shows specific adaptation to use the aromatic fraction of fuels as the sole carbon and energy source. Microb Ecol. 2011;61(3):582–594. doi: 10.1007/s00248-010-9786-3. [DOI] [PubMed] [Google Scholar]
- 83.Goutx M, Mutaftshiev S, Bertrand J. Lipid and exopolysaccharide production during hydrocarbon growth of a marine bacterium from the sea surface. Mar Ecol Prog Ser. 1987;40(3):259–265. doi: 10.3354/meps040259. [DOI] [Google Scholar]
- 84.Husain DR, Goutx M, Bezac C, Gilewicz M, Bertrand J-C. Morphological adaptation of Pseudomonas nautica strain 617 to growth on eicosane and modes of eicosane uptake. Letters Appl Microbiol. 1997;24:55–58. doi: 10.1046/j.1472-765X.1997.00345.x. [DOI] [Google Scholar]
- 85.Gogoleva NE, Nikolaichik YA, Ismailov TT, Gorshkov VY, Safronova VI, Belimov AA, Gogolev Y. Complete genome sequence of the abscisic acid-utilizing strain Novosphingobium sp. P6W. 3 Biotech. 2019;9(3):94. doi: 10.1007/s13205-019-1625-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mackenzie C, Choudhary M, Larimer FW, Predki PF, Stilwagen S, Armitage JP, et al. The home stretch, a first analysis of the nearly completed genome of Rhodobacter sphaeroides 2.4.1. Photosynth Res. 2001;70(1):19–41. doi: 10.1023/A:1013831823701. [DOI] [PubMed] [Google Scholar]
- 87.Chain PS, Denef VJ, Konstantinidis KT, Vergez LM, Agulló L, Reyes VL. Burkholderia xenovorans LB400 harbors a multi-replicon, 9.73-Mbp genome shaped for versatility. Proc Natl Acad Sci USA. 2006;103(42):15280–15287. doi: 10.1073/pnas.0606924103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Janssen PJ, Van Houdt R, Moors H, Monsieurs P, Morin N, Michaux A, et al. The complete genome sequence of Cupriavidus metallidurans strain CH34, a master survivalist in harsh and anthropogenic environments. PLoS One. 2010;5(5):e10433. doi: 10.1371/journal.pone.0010433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Frank O, Göker M, Pradella S, Petersen J. Ocean’s twelve: flagellar and biofilm chromids in the multipartite genome of Marinovum algicola DG898 exemplify functional compartmentalization. Environ Microbiol. 2015;17:4019–4034. doi: 10.1111/1462-2920.12947. [DOI] [PubMed] [Google Scholar]
- 90.Aylward FO, McDonald BR, Adams SM, Valenzuela A, Schmidt RA, Goodwin LA, et al. Comparison of 26 sphingomonad genomes reveals diverse environmental adaptations and biodegradative capabilities. Appl Environ Microbiol. 2013;79(12):3724–3733. doi: 10.1128/AEM.00518-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li XZ, Nikaido H. Efflux-mediated drug resistance in bacteria: an update. Drugs. 2009;69(12):1555–1623. doi: 10.2165/11317030-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zheng J, Guan Z, Cao S, Peng D, Ruan L, Jiang D, Sun M. Plasmids are vectors for redundant chromosomal genes in the Bacillus cereus group. BMC Genomics. 2015;16(1):6. doi: 10.1186/s12864-014-1206-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Suzuki Y, Nishijima S, Furuta Y, Yoshimura J, Suda W, Oshima K, et al. Long-read metagenomic exploration of extrachromosomal mobile genetic elements in the human gut. Microbiome. 2019;7(1):119. doi: 10.1186/s40168-019-0737-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.D'Argenio V, Notomista E, Petrillo M, Cantiello P, Cafaro V, Izzo V, et al. Complete sequencing of Novosphingobium sp. PP1Y reveals a biotechnologically meaningful metabolic pattern. BMC Genomics. 2014;15(1):384. doi: 10.1186/1471-2164-15-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nguyen STC, Freund HL, Kasanjian J, Berlemont R. Function, distribution, and annotation of characterized cellulases, xylanases, and chitinases from CAZy. Appl Microbiol Biotechnol. 2018;102(4):1629–1637. doi: 10.1007/s00253-018-8778-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Manzanares P, Vallés S, Ramòn D, Orejas M (2007) α-L-rhamnosidases: old and new insights. In: Industrial Enzymes. Springer, Dordrecht. 10.1007/1-4020-5377-0_8
- 97.De Lise F, Mensitieri F, Tarallo V, Ventimiglia N, Vinciguerra R, Tramice A, et al. RHA-P. Isolation, expression and characterization of a bacterial α-l-rhamnosidase from Novosphingobium sp. PP1Y. J Mol Catalysis B: Enzymatic. 2016;134:136–147. doi: 10.1016/j.molcatb.2016.10.002. [DOI] [Google Scholar]
- 98.Wang J, Salem DR, Sani RK. Extremophilic exopolysaccharides: a review and new perspectives on engineering strategies and applications. Carbohydr Polym. 2019;205:8–26. doi: 10.1016/j.carbpol.2018.10.011. [DOI] [PubMed] [Google Scholar]
- 99.Deo D, Davray D, Kulkarni R. A diverse repertoire of exopolysaccharide biosynthesis gene clusters in lactobacillus revealed by comparative analysis in 106 sequenced genomes. Microorganisms. 2019;7(10):444. doi: 10.3390/microorganisms7100444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Breton C, Snajdrová L, Jeanneau C, Koca J, Imberty A. Structures and mechanisms of glycosyltransferases. Glycobiology. 2006;16(2):29R–37R. doi: 10.1093/glycob/cwj016. [DOI] [PubMed] [Google Scholar]
- 101.Kumar R, Verma H, Heider S, Bajaj A, Sood U, Ponnusamy K, et al. Comparative genomic analysis reveals habitat-specific genes and regulatory hubs within the genus Novosphingobium. mSystems. 2017;2:e00020–17. doi: 10.1128/mSystems.00020-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wallden K, Rivera-Calzada A, Waksman G. Type IV secretion systems: versatility and diversity in function. Cell Microbiol. 2010;12(9):1203–1212. doi: 10.1111/j.1462-5822.2010.01499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fischer W, Tegtmeyer N, Stingl K, Backert S. Four chromosomal type IV secretion systems in Helicobacter pylori: composition, structure and function. Front Microbiol. 2020;11:1592. doi: 10.3389/fmicb.2020.01592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Alegria MC, Souza DP, Andrade MO, Docena C, Khater L, Ramos CH, et al. Identification of new protein-protein interactions involving the products of the chromosome- and plasmid-encoded type IV secretion loci of the phytopathogen Xanthomonas axonopodis pv. citri. J Bacteriol. 2005;187:2315–2325. doi: 10.1128/JB.187.7.2315-2325.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, et al. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci USA. 2006;103(5):1528–1533. doi: 10.1073/pnas.0510322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Coulthurst S. The type VI secretion system: a versatile bacterial weapon. Microbiology. 2019;165(5):503–515. doi: 10.1099/mic.0.000789. [DOI] [PubMed] [Google Scholar]
- 107.Ladino-Orjuela G, Gomes E, da Silva R, Salt C, Parsons JR (2016) Metabolic pathways for degradation of aromatic hydrocarbons by bacteria. In: Reviews of Environmental Contamination and Toxicology Volume 237, Springer, Cham. 10.1007/978-3-319-23573-8_5 [DOI] [PubMed]
- 108.Saxena A, Anand S, Dua A, Sangwan N, Khan F, Lal R. Novosphingobium lindaniclasticum sp. nov., a hexachlorocyclohexane (HCH)-degrading bacterium isolated from an HCH dumpsite. Int J Syst Evol Microbiol. 2013;63(Pt 6):2160–2167. doi: 10.1099/ijs.0.045443-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The whole-genome sequencing dataset is available under the NCBI Bioproject PRJNA624997.
The genome assembly and annotation pipelines developed by our group are available at the following links: https://github.com/fmalmeida/ngs-preprocess, https://github.com/fmalmeida/MpGAP, and https://github.com/fmalmeida/bacannot.