

Abstract
Background and Objectives
Deposition of myelin-associated glycoprotein (MAG) immunoglobulin M (IgM) antibodies in the sural nerve is a key feature in anti-MAG neuropathy. Whether the blood-nerve barrier (BNB) is disrupted in anti-MAG neuropathy remains elusive.We aimed to evaluate the effect of sera from anti-MAG neuropathy at the molecular level using our in vitro human BNB model and observe the change of BNB endothelial cells in the sural nerve of anti-MAG neuropathy.
Methods
Diluted sera from patients with anti-MAG neuropathy (n = 16), monoclonal gammopathies of undetermined significance (MGUS) neuropathy (n = 7), amyotrophic lateral sclerosis (ALS, n = 10), and healthy controls (HCs, n = 10) incubated with human BNB endothelial cells to identify the key molecule of BNB activation using RNA-seq and a high-content imaging system, and exposed with a BNB coculture model to evaluate small molecule/IgG/IgM/anti-MAG antibody permeability.
Results
RNA-seq and the high-content imaging system showed the significant upregulation of tumor necrosis factor (TNF-α) and nuclear factor-kappa B (NF-κB) in BNB endothelial cells after exposure to sera from patients with anti-MAG neuropathy, whereas the serum TNF-α concentration was not changed among the MAG/MGUS/ALS/HC groups. Sera from patients with anti-MAG neuropathy did not increase 10-kDa dextran or IgG permeability but enhanced IgM and anti-MAG antibody permeability. Sural nerve biopsy specimens from patients with anti-MAG neuropathy showed higher TNF-α expression levels in BNB endothelial cells and preservation of the structural integrity of the tight junctions and the presence of more vesicles in BNB endothelial cells. Neutralization of TNF-α reduces IgM/anti-MAG antibody permeability.
Discussion
Sera from individuals with anti-MAG neuropathy increased transcellular IgM/anti-MAG antibody permeability via autocrine TNF-α secretion and NF-κB signaling in the BNB.
Polyneuropathy associated with immunoglobulin M (IgM) monoclonal gammopathy of uncertain significance (MGUS) is a paraproteinemic polyneuropathy. Approximately half of patients with MGUS are positive for anti–myelin-associated glycoprotein (MAG) IgM antibodies.1 Patients with anti-MAG neuropathy typically show a chronic, progressive, predominantly sensory distal demyelinating neuropathy with ataxia and sometimes postural tremor.2,3 In some patients, immunologic treatments, including plasma exchange, IV immunoglobulin, rituximab, steroids, or anticancer drugs, may help reduce the MAG antibody titer and slow or prevent the progression of the disease.4
MAG is a member of the sialic acid–binding Ig-like lectins and is located in the Schwann cells of the peripheral nervous system (PNS), including the paranodal loops and Schmidt-Lanterman incisures.5 Anti-MAG IgM autoantibodies recognize the human natural killer 1 epitope on MAG or coreact with an acidic glycolipid, identified as sulfoglucuronosyl para-globoside (SGPG), in the ganglioside fraction of the human peripheral nerve.6 Sural nerve biopsies from patients with anti-MAG neuropathy show demyelination, widening of the myelin lamellae, and the deposition of IgM and complement on the myelin sheath and myelin lamellae, suggesting that MAG IgM antibodies cause demyelination with complement.7-9 Furthermore, systemic injection of purified IgM from individuals with anti-MAG neuropathy into susceptible host animals results in ataxic neuropathy and reproduces the human pathology (segmental peripheral nerve demyelination with deposition of IgM on the outer myelin lamellae and splitting of the myelin sheath), suggesting the pathogenicity of this antibody.10-12
It is not clear how serum MAG IgM antibody reaches the peripheral nerve. Nerve conduction studies show a uniformly slow conduction velocity with prolonged distal motor and sensory latencies, suggesting distal demyelination.13 MAG IgM antibody possibly accesses the dorsal root ganglia and peripheral nerve terminal, which lack a blood-CSF barrier and blood-nerve barrier (BNB).1,14,15 A recent study showed that the albumin quotient, which is an accurate marker of blood–spinal nerve root barrier (B-SNR-B) permeability, was increased in MAG neuropathy, indicating the disrupted B-SNR-B.16 Pathologic observation of sural biopsy specimens demonstrated gaps between adjacent endothelial cells of small endoneurial vessels in the patients with Waldenström macroglobulinemia, suggesting disruption of the BNB.17 Breakdown of the BNB may cause the entry of anti-MAG IgM antibodies into the PNS space and the binding of this antibody to the Schwann cells.
In the present study, we evaluated the contribution of sera from patients with anti-MAG neuropathy to BNB disruption using our human in vitro BNB model. We found that IgM and MAG IgM antibody permeability was increased after exposure to the sera of patients with anti-MAG neuropathy without an increase in small molecule permeability. RNA-Seq and a high-content imaging system showed the upregulation of autocrine/paracrine tumor necrosis factor (TNF)-α secreted by BNB endothelial cells and the induction of nuclear factor-kappa B (NF-κB) signaling after exposure to patient sera, suggesting the contribution of TNF-α/NF-κB signaling to increased IgM permeability.
Methods
Patient Serum Samples
Sera were collected from 16 patients with anti-MAG neuropathy with anti-MAG antibodies (males, n = 13; females, n = 3; mean age, 71.3 years) before immunomodulatory treatment, 7 patients with IgM monoclonal gammopathy–associated neuropathy (IgM-MG) who had IgM monoclonal gammopathies of undetermined significance (MGUS) or Waldenström macroglobulinemia without anti-MAG antibodies (males, n = 4; females, n = 3; mean age, 71.8 years), 10 patients with amyotrophic lateral sclerosis (ALS; male, n = 7; female, n = 3; mean age, 74.2 years) as disease controls and healthy controls (HCs; male, n = 6; female, n = 4; mean age, 32 years). Table 1 shows clinical information of patients with anti-MAG neuropathy and IgM-MG neuropathy. The anti-MAG antibody titer was assayed using an EK-MAG anti-MAG Autoantibodies ELISA (BÜHLMANN Laboratories AG, Table 1). The cutoff value was taken as +3SD of the anti-MAG antibody titer in HCs (eFigure 1, links.lww.com/NXI/A810).
Table 1.
Clinical Information of Patients

All sera were stored at −80°C until use. Sera were inactivated at 56°C for 30 minutes before the experiments.
Standard Protocol Approvals, Registrations, and Patient Consents
We obtained written informed consent from each participant. This research was approved by the ethics committees of the Medical Faculties of Yamaguchi Universities (IRB#: H26-031-2).
Immunocytochemistry of NF-κB and TNF-α Using a High-Content Imaging Assay
Human BNB–comprising endothelial cells, immortalized with temperature-sensitive SV40 large T antigen (tsA58) and telomerase, named FH-BNB cells, were previously described.18 All experiments were performed 2 days after the shift in temperature from 33 to 37°C to inactivate immortalization. The high-content imaging system protocol was previously described.19 Briefly, 5,000 cells per well were plated and maintained on CELLSTAR® 96-well plates (Greiner) and then cultured in MCDB 131 medium containing 10% sera from patients with anti-MAG neuropathy, IgM-MG neuropathy, and ALS or HCs after substitution for MCDB 131 medium for 1 hour (NF-κB p65 immunostaining) and 24 hours (TNF-α immunostaining). For immunostaining, FH-BNB cells were (1) fixed with 4% paraformaldehyde, (2) incubated with 0.3% Triton X-100, (3) blocked for 12 hours in 5% fetal bovine serum/0.3% Triton X-100 in phosphate-buffered saline, (4) incubated with primary Abs (NF-κB p65 monoclonal antibody or TNF-α monoclonal antibody [Novus]), and (5) incubated with secondary Abs (Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 488 goat anti-mouse IgG [Thermo Fisher Scientific]).
Images in the plate were captured using an In Cell Analyzer 2000 (GE Healthcare) at ×20 magnification with 4 fields of view per well (equivalent to 800–1,000 cells). The images were then analyzed with the IN Carta image analysis software program (Cytiva) or the In Cell Analyzer software program (Cytiva). The data represent the mean values of 3 experiments.
Whole Transcriptome Analyses With RNA-Seq
We incubated MCDB 131 medium containing 10% sera from 4 patients with anti-MAG neuropathy, 4 patients with IgM-MG neuropathy, or 4 HCs with FH-BNB cells for 12 hours at 37°C. We used FH-BNB cells without incubation with 10% sera as controls. The method of the whole transcriptome analysis with RNA-seq was previously described.20 In brief, total RNA was extracted from FH-BNB cells using the RNeasy Mini Kit (Qiagen), and mRNA was purified. Complementary DNA libraries were produced using a NEBNext Ultra II RNA Library Prep kit (New England Biolabs, NEB) and NEBNextplex Oligos for Illumina, as described previously. In this approach, mRNA was fragmented in NEBNext First Strand Synthesis Reaction Buffer at 94°C for 15 minutes in the presence of NEBNext Random Primers and was reverse transcribed with NEBNext Strand Synthesis Enzyme Mix. The library fragments were then concentrated, and index sequences were inserted during PCR amplification. The products were purified using AMPure XP beads (Beckman Coulter), and the quality of the library was confirmed with an Agilent 2200 TapeStation (D1000, Agilent Thermo Fisher). The libraries mixed to equal molecular amounts were sequenced on an Illumina NextSeq DNA sequencer with a 75-bp pair-end cycle sequencing kit (Illumina). The data were then trimmed and mapped to the mouse reference genome GRCm38 release 92 using the CLC Genomics Workbench software program (ver.8.01; Qiagen) as previously described. The mapped read counts were normalized to transcripts per million, which were converted to log2 values after the addition of 1. For pathway analysis, factor loadings were calculated with principal component analysis (PCA) of JMP Pro ver.15.0.0 software, the 178 genes contributed to negative direction of principal component (PC)2 were selected, and the pathway analysis for the detected genes was examined using Ingenuity Pathway Analysis (Qiagen).
10-kDa Dextran/FITC-Labeled IgG/IgM/Anti-MAG Antibody Permeability
FH-BNB cells on the luminal side and human peripheral nerve pericytes on the abluminal side were cultured on 24-well collagen-coated Transwell culture inserts (pore size: 0.4 mm; Corning) for 5 days. MCDB 131 medium containing individual 10% sera from patients with anti-MAG neuropathy, IgM-MG neuropathy, and ALS and HCs was incubated with luminal FH-BNB cells for 24 hours at 37°C. After the cells were washed, FITC 10k-Da dextran fluorescence (Sigma-Aldrich) or fluorescein isothiocyanate (FITC)-IgG (Sigma-Aldrich) was added to the luminal insert (concentration, 1 mg/mL for 10-kDa dextran; mg/mL for IgG). A total of 100 μL of medium was then transferred from the abluminal chamber into 96-well black plates over 40 minutes. Fluorescence signals were calculated at 490/520 nm (absorption/emission) using a FlexStation 3 Multi-Mode Microplate Reader (Molecular Devices).
To evaluate IgM or anti-MAG antibody permeability, each MCDB 131 medium containing 10% sera from patients with anti-MAG neuropathy, IgM-MG neuropathy, and ALS and HCs was incubated in the luminal chamber for 24 hours (150 mL of conditioned medium including sera was incubated in the luminal chamber, and 300 mL of MCBD131 was added to the abluminal chamber). Then, MCBD131 in both the luminal and abluminal chambers was collected, and the IgM concentration/anti-MAG antibodies within both chambers were measured using an EK-MAG anti-MAG Autoantibodies ELISA (BÜHLMANN Laboratories AG) or Human IgM ELISA Kit (Abcam). The absorbance of the blank was used as the background, and data were normalized to the absorbance value of the blank.
Immunohistochemistry and Electronic Microscopy of Sural Nerve Specimens From Patients With Anti-MAG Neuropathy
Immunohistochemistry was performed to analyze 4-μm-thick sections from paraffin-embedded sural nerve specimens from 4 patients with anti-MAG neuropathy, 2 patients with ALS (controls), and 1 patient with chronic inflammatory demyelinating polyneuropathy (CIDP). All paraffin sections were deparaffinized, and antigen (TNF-α) was activated by heating at 98°C for 10 minutes in citrate buffer. Indirect immunofluorescence was performed with anti-TNF-α antibodies (dilution 1:50, Novus) as primary antibodies and anti-rabbit secondary fluorescent antibodies (Alexa Fluor 488 goat anti-rabbit IgG, Invitrogen, dilution 1:200) as secondary antibodies. Electron microscopy was performed using 80-nm-thick sections from epoxy resin–embedded specimens stained with uranyl acetate and lead acetate. We collected the CSF data from 4 patients with anti-MAG neuropathy who received sural nerve biopsy (eTable 1, links.lww.com/NXI/A810). Q Alb LIM was calculated as ([age/15] + 4) as the cutoff value. The BNB damage was classified into 4 patterns ([1] absent, serum albumin quotient (QALB) < QAlbLIM; [2] mild BNB damage, QALB/QAlbLIM 1.0–2.0; [3] moderate BNB damage, QALB/QAlbLIM 2.0–5.0; and [4] severe BNB damage, QALB/QAlbLIM >5.0).16
Treatment With TNF-α–Neutralizing Antibody
To evaluate the effect of TNF-α on IgM or anti-MAG antibody permeability, cocultured in vitro BNB cells (luminal: FH-BNB cells/abluminal: peripheral nerve pericytes) were incubated with the MCBD media containing 10% sera from 2 patients with anti-MAG neuropathy, 1 patient with IgM-MG neuropathy, or 1 HC with/without 200 ng/mL of TNF-α–neutralizing antibody (R&D) in the luminal chamber for 24 hours. Then, MCBD131 in both the luminal and abluminal chambers was collected, and the IgM concentration and anti-MAG antibodies within both chambers were measured using an EK-MAG anti-MAG Autoantibodies ELISA (BÜHLMANN Laboratories AG) or Human IgM ELISA Kit (Abcam).
Serum TNF-α Concentration
The concentrations of serum TNF-α were measured using an ELISA (Abcam) according to the manufacturer's protocol.
Statistical Analyses
All statistical analyses were performed using the Prism 7 software program (Graph Pad). A paired Student t test (2 sided) was used for single comparisons. For multiple comparisons, a one-way analysis of variance (ANOVA) was used with Tukey multiple comparison test when the data were normally distributed. p Values of *p < 0.05, **p < 0.01, and ***p < 0.001 were considered to indicate statistical significance.
Data Availability
Data not provided within this article are available in anonymized form and can be shared by request from any qualified investigator. Sharing requires approval of a data transfer agreement by Yamaguchi University.
Results
Profiling of the Altered Gene Expression in FH-BNB After Exposure to Sera From Patients With MAG-Ab Neuropathy
FH-BNB cells were incubated with 10% sera from each patient with anti-MAG neuropathy (MAG group: n = 4) and neuropathy with IgM monoclonal gammopathy without anti-MAG neuropathy (IgM-MG group: n = 4) or HC (n = 4) for whole transcriptome analysis using RNA-seq. Over 32,000 genes of FH-BNB cells were detected from approximately 30 million reads in each sample. PCA revealed the separation on PC2 between the patients with MAG-Ab–associated disorder (n = 4) and HC/control groups (n = 4) (eFigure 2, links.lww.com/NXI/A810). Ingenuity Pathway Analysis was performed using 100 genes in IgM-MG or 178 genes in HC negatively contributed to the PC2 separation (factor loadings < −0.5) as shown in the heatmap to determine the signaling pathway of FH-BNB after exposure to IgGs from the MAG group in comparison to the HCs (eFigure 3, links.lww.com/NXI/A810) or IgM-MG group (eFigure 4, links.lww.com/NXI/A810). In the network analysis of the upregulated genes, TNF-α and NF-κB were detected in the center of the network analysis in the MAG group in comparison to in the HC (Figure 1, A and B) and IgM-MG group (Figure 1, C and D). Interleukin (IL)-6, IL-12, C-X-C motif chemokine ligand (CXCL)2, and CXCL3 were observed as downstream molecules of NF-κB signaling in the MAG group in comparison to the IgM-MG group (eFigure 6, links.lww.com/NXI/A810). CCL20, IL1A, CXCL1, CXCL3, and CXCL5 as downstream molecules of NF-κB signaling were observed in the MAG group in comparison to the HC group (eFigure 5, links.lww.com/NXI/A810).
Figure 1. Whole Transcriptome Analysis (Using RNA-Seq) of PnMECs After Exposure to Sera From Patients With Anti-MAG Neuropathy.

Human PnMECs were incubated with 10% sera from patients with anti-MAG neuropathy (MAG, n = 4), HCs (n = 4), and patients with IgM-MG (n = 4). PnMECs without exposure to IgGs were also used as controls. In the network analysis of upregulated genes, TNF-α and NF-κB were detected in the center of the network analysis in anti-MAG neuropathy in comparison to in HCs (A and B) or IgM-MG neuropathy (C and D). The red nodes show the upregulated genes in the RNA-seq analysis (FC > 1.5; p < 0.05). Anti-MAG = anti-MAG neuropathy; HC = healthy control; IgM-MG = IgM–monoclonal gammopathy neuropathy; MAG = myelin-associated glycoprotein; NF-κB = nuclear factor-kappa B; PnMECs = peripheral nerve microvascular endothelial cells; TNF = tumor necrosis factor.
Sera From Patients With Anti-MAG Neuropathy Induced the Increase of TNF-α and NF-κB
To confirm the data suggested by RNA-seq and the pathway analysis, the change in the protein levels of TNF-α and NF-κB after exposure to 10% sera from patients with MAG, IgM-MG, and ALS or HCs was evaluated using a high-content imaging system (MAG, n = 16; IgM-MG, n = 7; ALS, n = 10; HC, n = 10). The amount of TNF-α in the FH-BNB was significantly increased in patients with MAG neuropathy in comparison to patients with IgM-MG, patients with ALS, and HCs, and the proportion of NF-κB p65 nuclear-positive cells was also significantly increased in patients with MAG neuropathy in comparison to patients with ALS and HCs (Figure 2, A–C). The serum TNF-α concentration was not changed among the groups (Figure 2D).
Figure 2. Changes in the TNF-α and NF-κb of PnMECs After Exposure to Sera From Patients With MAG Neuropathy.

(A) Immunostaining for TNF-α and NF-κB p65 (green) in PnMECs after exposure to 10% sera from patients with MAG neuropathy. Images were captured by an In cell analyzer 2000. Scale bar, 50 μm. Arrows indicate representative cells with NF-kB p65-positive nuclei. (B and C) Scatter plots of the intensity of TNF-α (B) or the number of nuclear NF-κB p65-positive PnMECs (C), as determined by high-content imaging after exposure to sera from patients with MAG neuropathy (MAG, n = 16), patients with polyneuropathy associated with IgM-MG neuropathy (n = 8), patients with ALS (n = 10), and HCs (n = 10). (D) The concentration of serum TNF-α was assayed among patients with MAG neuropathy (MAG: n = 16), patients with IgM-MG neuropathy (n = 8), patients with ALS (n = 10), and HCs (n = 10). The data were normalized to cultures that had not been exposed to sera and represent values obtained from 3 independent experiments. Data are shown as the mean ± standard error of the mean (SEM). p Values were determined by a one-way ANOVA followed by the Tukey multiple comparison test (*p < 0.05 and **p < 0.01 vs IgM-MG, ALS, or HC followed by the Tukey multiple comparison test). ALS = amyotrophic lateral sclerosis; ANOVA = analysis of variance; HC = healthy control; IgM-MG = IgM–monoclonal gammopathy neuropathy; MAG = myelin-associated glycoprotein; NF-κB = nuclear factor-kappa B; PnMECs = peripheral nerve microvascular endothelial cells; TNF = tumor necrosis factor.
IgM and Anti-MAG Antibody Permeability Across the BNB Was Increased After Exposure to Sera From Patients With MAG Neuropathy
Using an in vitro coculture BNB model with FH-BNB and peripheral nerve pericytes, 10-kDa dextran, IgG, IgM, and MAG IgM antibody permeability was measured after exposure to 10% sera from patients with MAG, IgM-MG, and ALS or HCs (eFigure 7, links.lww.com/NXI/A810). The 10-kDa dextran and IgG permeability was not changed (Figure 3, A and B); however, IgM permeability and MAG IgM antibody concentration in the lower chamber were found to be significantly increased after incubation with sera from patients with anti-MAG neuropathy, in comparison to after incubation with sera from patients with ALS and HCs (Figure 3, C and D). The anti-MAG antibody permeability index could not be determined because the anti-MAG antibody titer was below the limit of detection in both the upper and lower chambers in many of the samples from patients with IgM-MG neuropathy and ALS and HCs (eTable 2, links.lww.com/NXI/A810).
Figure 3. Changes of the 10-kDa Dextran, IgG, IgM, and Anti-MAG Antibody Permeability Evaluated Using an In Vitro Human BNB Coculture Model Exposed to Sera From Individuals With MAG Neuropathy.

PnMECs on the luminal side and human peripheral nerve pericytes on the abluminal side were cultured on a 24-well collagen-coated Transwell culture insert (pore size: 0.4 mm). MCDB 131 medium containing individual 10% sera from patients with anti-MAG neuropathy, patients with IgM-MG neuropathy, patients with ALS, and HCs were incubated with luminal side. The change in the 10-kDa dextran permeability coefficient (A), IgG permeability index (B), IgM permeability index (C), and MAG antibody (MAG-Ab) permeability (D) was calculated. Data are shown as the mean ± SEM. p Values were determined by a one-way ANOVA followed by the Tukey multiple comparison test (*p < 0.05, **p < 0.01, and ***p < 0.001 vs ALS or HC group followed by the Tukey multiple comparison test). ALS = amyotrophic lateral sclerosis; ANOVA = analysis of variance; BNB = blood-nerve barrier; IgM-MG = IgM–monoclonal gammopathy neuropathy; MAG = myelin-associated glycoprotein; PnMECs = peripheral nerve microvascular endothelial cells.
Upregulation of TNF-α Was Observed in BNB Endothelial Cells of Sural Nerve Biopsy Specimens From Patients With Anti-MAG Neuropathy
We observed sural nerve biopsy specimens from 4 patients with anti-MAG neuropathy, 2 patients with ALS as noninflammatory control samples, and 1 patient with CIDP as inflammatory neuropathy control samples. TNF-α immunostaining showed the expression of TNF-α in BNB endothelial cells from patients with anti-MAG neuropathy but not in noninflammatory and inflammatory controls (Figure 4A). In addition, electronic microscopy demonstrated the hypertrophy of endothelial cells and the preservation of the tight junctions between adjacent endothelial cells of BNB endoneurial vessels (Figure 4B) and more vesicles in the BNB endothelial cells from patients with anti-MAG neuropathy in comparison to BNB endothelial cells from noninflammatory and inflammatory controls (Figure 4C).
Figure 4. Sural Nerve Biopsy Specimens From Patients With Anti-MAG Neuropathy.
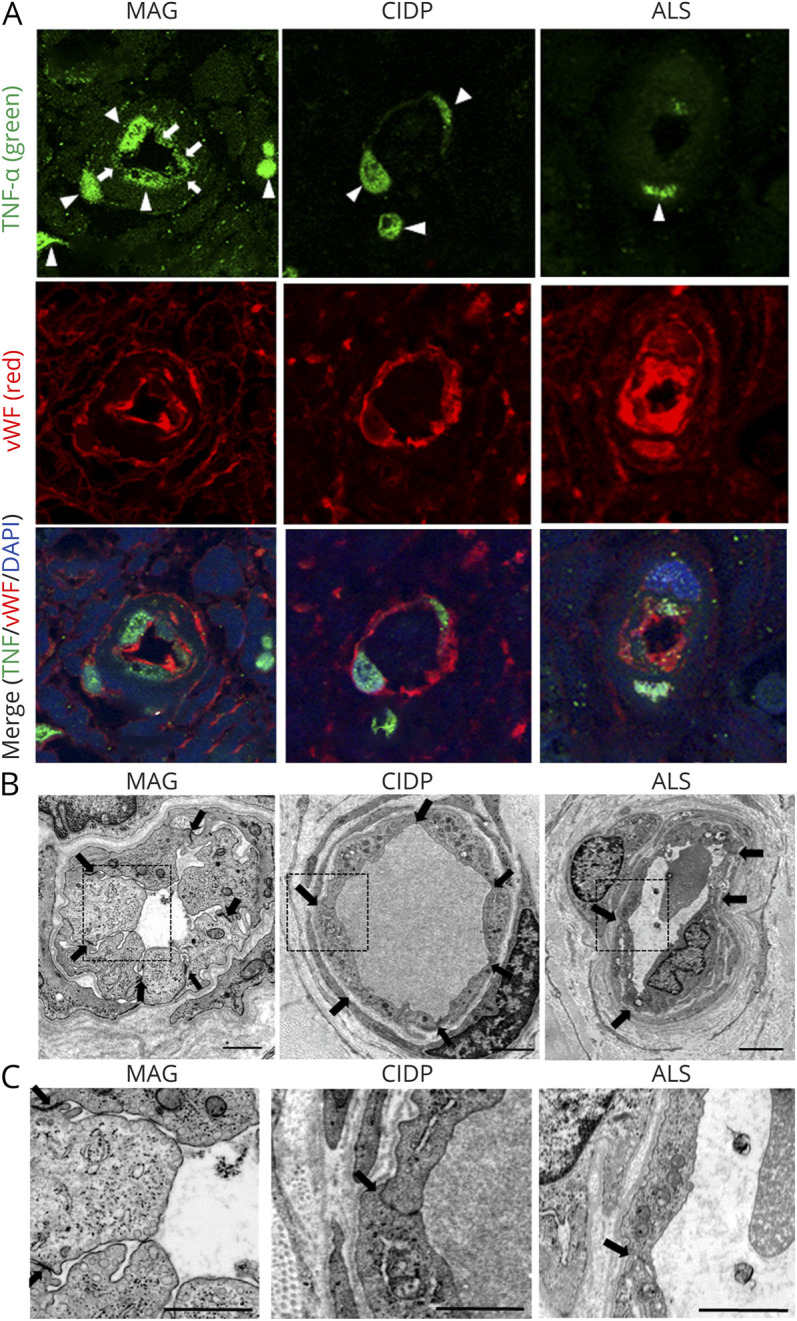
Sural nerve biopsy specimens obtained from 4 patients with MAG IgM neuropathy, 2 patients with ALS as noninflammatory control samples and 1 patient with CIDP as inflammatory control were analyzed. (A) TNF-α immunostaining showed the expression of TNF-α in BNB endothelial cells of biopsy specimens from patients with anti-MAG neuropathy (arrows), but not those from noninflammatory or inflammatory controls. Nuclear staining of TNF-α observed in all samples of MAG neuropathy, CIDP, and ALS was nonspecific (arrowheads). Upper panel: TNF-α (green); middle panel: Von Willebrand Factor (vWF) (red), an endothelial cell marker; lower panel: merge TNF-α (green)/vWF (red)/DAPI (blue). (B and C) Electronic microscopy showed the hypertrophy of endothelial cells and the preservation of the tight junctions between adjacent endothelial cells of BNB endoneurial vessels in biopsy specimens from patients with anti-MAG neuropathy (B) and more abundant vesicles in BNB endothelial cells in biopsy specimens from patients with anti-MAG neuropathy, in comparison to those from patients with ALS and CIDP (C). Scale bar, 1 μm. ALS = amyotrophic lateral sclerosis; BNB = blood-nerve barrier; CIDP = chronic inflammatory demyelinating polyneuropathy; IgM-MG = IgM–monoclonal gammopathy neuropathy; MAG = myelin-associated glycoprotein; TNF = tumor necrosis factor.
Treatment With TNF-α–Neutralizing Antibody Attenuated the IgM and MAG IgM Antibody Permeability Across the BNB Induced by Sera
To analyze the contribution of TNF-α to the penetration of MAG IgM antibody across the BNB, TNF-α activity was inhibited using TNF-α–neutralizing antibody. IgM and MAG IgM antibody permeability across the BNB was significantly attenuated after exposure to sera from 2 patients with anti-MAG neuropathy incubated with TNF-α–neutralizing antibody (Figure 5, A and B). Sera from 1 patient with IgM-MG and 1 HC as controls did not have the same effect on the BNB coculture model with/without TNF-α–neutralizing antibodies (Figure 5, A and B).
Figure 5. Effect of the Inhibition of TNF-α From Sera of Patients With MAG Neuropathy on IgM and MAG Antibody Permeability in the BNB Coculture Model.

TNF-α inhibition using TNF-α–neutralizing antibody from 2 10% sera with MAG neuropathy (MAG1 and MAG2) significantly decreased the IgM (A) and MAG antibody (B) permeability by approximately 50% in an in vitro human BNB coculture model consisting of both PnMECs on the luminal side and peripheral nerve pericytes on the abluminal side. After exposure to 10% sera from patients with IgM-MG (IgM-MG) or HCs, TNF-α–neutralizing antibody did not change the IgM/MAG antibody permeability. Data are shown as the mean ± SEM of 4 independent experiments. Statistical significance was determined by a paired 2-tailed t test. +TNF-α–neutralizing antibody, sera with TNF-α–neutralizing antibody. BNB = blood-nerve barrier; HC = healthy control; IgM-MG = IgM–monoclonal gammopathy neuropathy; MAG = myelin-associated glycoprotein; PnMECs = peripheral nerve microvascular endothelial cells; TNF = tumor necrosis factor.
Discussion
MAG IgM antibodies are considered pathogenic because IgM and complement are deposited on the myelin sheath in sural nerve biopsy specimens from patients with anti-MAG neuropathy.1 However, it is still elusive how this IgM antibody, which is a high-molecular-weight protein, passes through the intact BNB. We therefore hypothesized that the breakdown of the BNB may induce the entry of IgM into the peripheral nerves in anti-MAG neuropathy. In the present study, we first found that the genes associated with TNF-α, NF-κB, and downstream pathway molecules of NF-κB (IL-6, IL-12, CXCL2, and CXCL3) were significantly upregulated after exposure to sera from patients with anti-MAG neuropathy, in comparison to sera from those with IgM-MG neuropathy and HCs, using whole RNA-seq and a pathway analysis. Next, a high-content imaging system showed the induction of TNF-α upregulation and NF-κB nuclear translocation in the BNB endothelial cells after exposure to sera from patients with anti-MAG neuropathy, in comparison to sera from patients with IgM-MG neuropathy and ALS and HCs. Serum TNF-α concentration did not differ among patients with anti-MAG neuropathy, IgM-MG neuropathy, and ALS or HCs, suggesting that autocrine/paracrine TNF-α secretion induced by the sera from patients from anti-MAG neuropathy can activate BNB endothelial cells. The 10-kDa dextran and IgG permeability was not changed, although IgM and MAG IgM antibody permeability was significantly increased after incubation with sera from the MAG group, in comparison to that from ALS/HC groups. This result suggests that the penetration of IgM/MAG IgM antibodies across the BNB occurs without disruption of the tight junction and increased small molecule permeability. Pathologic analyses revealed the expression of TNF-α in BNB endothelial cells, preservation of the structural integrity of the tight junctions, and greater numbers of vesicles in BNB endothelial cells from patients with anti-MAG neuropathy. Finally, the neutralization of TNF-α after exposure to sera from patients with anti-MAG neuropathy resulted in the reduction of IgM and MAG IgM antibody permeability, suggesting that autocrine/paracrine TNF-α–induced activation of BNB endothelial cells may lead to increased MAG IgM antibody permeability, possibly through transcytosis (eFigure 8, links.lww.com/NXI/A810).
A previous study revealed increased serum IL-6 and IL-10, but not serum TNF-α, in patients with anti-MAG neuropathy.21 Our study also demonstrated that the serum TNF-α concentration was not increased in patients with anti-MAG neuropathy. We demonstrated that sera from patients with anti-MAG neuropathy induced the upregulation of TNF-α in the BNB endothelial cells, which influences the BNB endothelial cells via NF-κB signaling in an autocrine/paracrine manner. In this study, we could not clarify the precise factors, including anti-MAG IgM antibodies or other soluble factors, in the sera, which had this effect. TNF-α is known to be a major proinflammatory cytokine and is one of the most potent physiologic inducers of the NF-κB.22 Several reports have demonstrated that the activation of NF-κB stimulates the expression of additional inflammatory cytokines and adhesion molecules and increases the receptor-mediated transcytosis of low-density lipoprotein (LDL) via the LDL receptor in endothelial cells.23-27 In addition, TNF-α/NF-κB signaling induced the upregulation of SGPG in endothelial cells,28 suggesting that expression of receptor against MAG IgM antibodies become more abundant after the stimulation of TNF-α. In our study, electron microscopy revealed more vesicles in BNB endothelial cells in sural nerve biopsy specimens from patients with anti-MAG neuropathy. We speculate that the autocrine TNF-α/NF-κB signal may stimulate the expression of SGPG and activate the receptor-mediated transcytosis of MAG IgM antibodies through SGPG across the BNB (eFigure 5, links.lww.com/NXI/A810). Further analysis regarding the upregulation of endosomal vesicles in in vitro BNB endothelial cells after exposure to sera from anti-MAG neuropathy will be required to support this hypothesis.
It is still unclear whether the BNB disruption was observed in patients with anti-MAG neuropathy. Our in vitro study and pathologic observation revealed that the tight junction in the BNB was preserved in anti-MAG neuropathy. This suggests that there is little paracellular entry of MAG IgM antibodies into the peripheral nerve. These observations support the findings that the deposition of IgM is abundant in the dorsal root ganglia and peripheral nerve terminal without the BNB.1 We previously showed a decrease of the tight junction protein, claudin-5, after exposure to sera from patients with CIDP using our in vitro BNB model and in the sural nerve of patients with CIDP, unlike anti-MAG neuropathy.29
In conclusion, the present study shows increased transcellular IgM/MAG IgM antibody permeability via TNF-α/NF-κB signaling in anti-MAG neuropathy. Several TNF-α inhibitors have been administered, but the effect of these antibodies against anti-MAG neuropathy has not been established. The present study shows that TNF-α inhibitors may have the potential to reduce the deposition of anti-MAG antibodies on myelin. Some studies showed the efficacy of rituximab in anti-MAG neuropathy, although rituximab leads to neurologic improvement in only 30%–50% of these patients.1,4,30 Rituximab possibly has no effect on decreasing the penetration of anti-MAG antibodies across the BNB but reduces anti–MAG antibody–producing B cells in sera because this drug cannot inhibit TNF-α/NF-κB signaling in the BNB endothelial cells induced by sera from patients with anti-MAG neuropathy. The combination of rituximab plus chemotherapy, including B-cell lymphoma 2 (BCL2) inhibitor treatment, has recently been proven to improve the level of treatment response in anti-MAG neuropathy.31 Our findings suggest the possibility that combination treatment with rituximab plus an anti–TNF-α inhibitor may reduce the amount of anti-MAG antibodies in both sera and the peripheral nerves and prevent the accumulation of deposition of anti-MAG antibodies on myelin, thereby leading to neurologic improvement in anti-MAG neuropathy. Further in vivo analyses and clinical trials to evaluate the efficacy of the combination treatment with rituximab plus anti–TNF-α inhibitor in patients with anti-MAG neuropathy are required to clarify the pathophysiology of anti-MAG neuropathy.
Glossary
- ALS
amyotrophic lateral sclerosis
- ANOVA
analysis of variance
- BCL-2
B-cell lymphoma 2
- BNB
blood-nerve barrier
- B-SNR-B
blood–spinal nerve root barrier
- CIDP
chronic inflammatory demyelinating polyneuropathy
- CXCL
C-X-C motif chemokine ligand
- FITC
fluorescein isothiocyanate
- HC
healthy control
- IgM
immunoglobulin M
- IL
interleukin-6
- LDL
low-density lipoprotein
- MAG
myelin-associated glycoprotein
- MGUS
monoclonal gammopathies of undetermined significance
- MGUS
monoclonal gammopathy of uncertain significance
- NF-κB
nuclear factor-kappa B
- PCA
principal component analysis
- PNS
peripheral nervous system
- QALB
serum albumin quotient
- SGPG
sulfoglucuronosyl para-globoside
- TNF
tumor necrosis factor
- vWF
von Willebrand Factor
Appendix. Authors
Study Funding
This work was funded by (1) research grants (Nos. 18K07526 and 20H00529) from the Japan Society for the Promotion of Science, Tokyo, Japan, (2) Takeda Research Foundation, and (3) research grant from Health and Labour Sciences Research Grants for research on intractable diseases (Neuroimmunological Disease Research Committee) from the Ministry of Health, Labour and Welfare of Japan.
Disclosure
All authors report no financial disclosure relevant to the submitted manuscripts. Dr. Kanda received speaking fees outside this work from Teijin Pharma Limited, Nihon Pharmaceutical Co., Ltd., Kaketsuken, Mitsubishi Tanabe Pharma, Takeda Pharmaceutical Company, Novartis, and Biogen. Go to Neurology.org/NN for full disclosures.
References
- 1.Dalakas MC. Advances in the diagnosis, immunopathogenesis and therapies of IgM-anti-MAG antibody-mediated neuropathies. Ther Adv Neurol Disord. 2018;11:175628561774664. doi. 10.1177/1756285617746640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steck AJ, Stalder AK, Renaud S. Anti-myelin-associated glycoprotein neuropathy. Curr Opin Neurol. 2006;19(5):458-463. doi. 10.1097/01.wco.0000245368.36576.0d. [DOI] [PubMed] [Google Scholar]
- 3.Svahn J, Petiot P, Antoine JC, et al. . Anti-MAG antibodies in 202 patients: clinicopathological and therapeutic features. J Neurol Neurosurg Psychiatry. 2018;89(5):499-505. doi. 10.1136/jnnp-2017-316715. [DOI] [PubMed] [Google Scholar]
- 4.Lunn MP, Nobile-Orazio E. Immunotherapy for IgM anti-myelin-associated glycoprotein paraprotein-associated peripheral neuropathies. Cochrane Database Syst Rev. 2016;10:CD002827. doi. 10.1002/14651858.CD002827.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalakas MC. Pathogenesis and treatment of anti-MAG neuropathy. Curr Treat Options Neurol. 2010;12(2):71-83. doi. 10.1007/s11940-010-0065-x. [DOI] [PubMed] [Google Scholar]
- 6.Quarles RH. Myelin-associated glycoprotein (MAG): past, present and beyond. J Neurochem. 2007;0(0):1431-1448. doi. 10.1111/j.1471-4159.2006.04319.x. [DOI] [PubMed] [Google Scholar]
- 7.Ritz MF, Erne B, Ferracin F, Vital A, Vital C, Steck AJ. Anti-MAG IgM penetration into myelinated fibers correlates with the extent of myelin widening. Muscle Nerve. 1999;22(8):1030-1037. doi. . [DOI] [PubMed] [Google Scholar]
- 8.Gabriel JM, Erne B, Bernasconi L, et al. . Confocal microscopic localization of anti-myelin-associated glycoprotein autoantibodies in a patient with peripheral neuropathy initially lacking a detectable IgM gammopathy. Acta Neuropathol. 1998;95(5):540-546. doi. 10.1007/s004010050835. [DOI] [PubMed] [Google Scholar]
- 9.Monaco S, Bonetti B, Ferrari S, et al. Complement-mediated demyelination in patients with IgM monoclonal gammopathy and polyneuropathy. N Engl J Med. 1990;322(10):649-652. doi. 10.1056/nejm199003083221002. [DOI] [PubMed] [Google Scholar]
- 10.Willison HJ, Trapp BD, Bacher JD, Dalakas MC, Griffin JW, Quarles RH. Demyelination induced by intraneural injection of human antimyelin-associated glycoprotein antibodies. Muscle Nerve. 1988;11:1169-1176. doi. 10.1002/mus.880111111. [DOI] [PubMed] [Google Scholar]
- 11.Tatum AH. Experimental paraprotein neuropathy, demyelination by passive transfer of human IgM anti-myelin-associated glycoprotein. Ann Neurol. 1993;33(5):502-506. doi. 10.1002/ana.410330514. [DOI] [PubMed] [Google Scholar]
- 12.Ilyas AA, Gu Y, Dalakas MC, Quarles RH, Bhatt S. Induction of experimental ataxic sensory neuronopathy in cats by immunization with purified SGPG. J Neuroimmunol. 2008;193(1-2):87-93. doi. 10.1016/j.jneuroim.2007.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaku DA, England JD, Sumner AJ. Distal accentuation of conduction slowing in polyneuropathy associated with antibodies to myelin-associated glycoprotein and sulphated glucuronyl paragloboside. Brain. 1994;117(5):941-947. doi. 10.1093/brain/117.5.941. [DOI] [PubMed] [Google Scholar]
- 14.Dalakas MC, Papadopoulos NM. Paraproteins in the spinal fluid of patients with paraproteinemic polyneuropathies. Ann Neurol. 1984;15(6):590-593. doi. 10.1002/ana.410150612. [DOI] [PubMed] [Google Scholar]
- 15.Yu RK, Ariga T, Kohriyama T, Kusunoki S, Maeda Y, Miyatani N. Autoimmune mechanisms in peripheral neuropathies. Ann Neurol. 1990;27(S1):S30-S35. doi. 10.1002/ana.410270709. [DOI] [PubMed] [Google Scholar]
- 16.Ruiz M, Puthenparampil M, Campagnolo M, et al. Oligoclonal IgG bands in chronic inflammatory polyradiculoneuropathies. J Neurol Neurosurg Psychiatry. 2021;92(9):969-974. doi. 10.1136/jnnp-2020-325868. [DOI] [PubMed] [Google Scholar]
- 17.Kanda T, Usui S, Beppu H, Miyamoto K, Yamawaki M, Oda M. Blood-nerve barrier in IgM paraproteinemic neuropathy: a clinicopathologic assessment. Acta Neuropathol. 1998;95(2):184-192. doi. 10.1007/s004010050785. [DOI] [PubMed] [Google Scholar]
- 18.Abe M, Sano Y, Maeda T, et al. . Establishment and characterization of human peripheral nerve microvascular endothelial cell lines: a new in vitro blood-nerve barrier (BNB) model. Cell Struct Funct. 2012;37(2):89-100. doi. 10.1247/csf.11042. [DOI] [PubMed] [Google Scholar]
- 19.Shimizu F, Takeshita Y, Sano Y, et al. . GRP78 antibodies damage the blood-brain barrier and relate to cerebellar degeneration in Lambert-Eaton myasthenic syndrome. Brain. 2019;142(8):2253-2264. doi. 10.1093/brain/awz168. [DOI] [PubMed] [Google Scholar]
- 20.Kohno M, Kobayashi S, Yamamoto T, et al. . Enhancing calmodulin binding to cardiac ryanodine receptor completely inhibits pressure-overload induced hypertrophic signaling. Commun Biol. 2020;3(1):714. doi. 10.1038/s42003-020-01443-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stork ACJ, Rijkers GT, Vlam L, et al. . Serum cytokine patterns in immunoglobulin m monoclonal gammopathy-associated polyneuropathy. Muscle Nerve. 2019;59(6):694-698. doi. 10.1002/mus.26462. [DOI] [PubMed] [Google Scholar]
- 22.Liu ZG. Molecular mechanism of TNF signaling and beyond. Cell Res. 2005;15(1):24-27. doi. 10.1038/sj.cr.7290259. [DOI] [PubMed] [Google Scholar]
- 23.Pan W, Kastin AJ. Tumor necrosis factor and stroke: role of the blood-brain barrier. Prog Neurobiol. 2007;83(6):363-374. doi. 10.1016/j.pneurobio.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2(1):17023. doi. 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhong L, Simard MJ, Huot J. Endothelial microRNAs regulating the NF-κB pathway and cell adhesion molecules during inflammation. FASEB J. 2018;32(8):4070-4084. doi. 10.1096/fj.201701536r. [DOI] [PubMed] [Google Scholar]
- 26.Mussbacher M, Salzmann M, Brostjan C, et al. . Cell type-specific roles of NF-κB linking inflammation and thrombosis. Front Immunol. 2019;10:85. doi. 10.3389/fimmu.2019.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Yang X, Bian F, et al. . TNF-α promotes early atherosclerosis by increasing transcytosis of LDL across endothelial cells: crosstalk between NF-κB and PPAR-γ. J Mol Cel Cardiol. 2014;72:85-94. doi. 10.1016/j.yjmcc.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 28.Dasgupta S, Silva J, Wang G, Yu RK. Sulfoglucuronosyl paragloboside is a ligand for T cell adhesion: regulation of sulfoglucuronosyl paragloboside expression via nuclear factor κB signaling. J Neurosci Res. 2009;87(16):3591-3599. doi. 10.1002/jnr.22153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimizu F, Sawai S, Sano Y, et al. . Severity and patterns of blood-nerve barrier breakdown in patients with chronic inflammatory demyelinating polyradiculoneuropathy: correlations with clinical subtypes. PLoS One. 2014;9(8):e104205. doi. 10.1371/journal.pone.0104205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dalakas MC, Rakocevic G, Salajegheh M, et al. . Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein antibody demyelinating neuropathy. Ann Neurol. 2009;65(3):286-293. doi. 10.1002/ana.21577. [DOI] [PubMed] [Google Scholar]
- 31.Briani C, Visentin A, Castellani F, Cacciavillani M, Trentin L. The BCL2 inhibitor venetoclax plus rituximab is active in MYD88 wild-type polyneuropathy with anti-MAG antibodies. Neurol Neuroimmunol Neuroinflamm. 2022;9(4):e1181. doi. 10.1212/nxi.0000000000001181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data not provided within this article are available in anonymized form and can be shared by request from any qualified investigator. Sharing requires approval of a data transfer agreement by Yamaguchi University.