

Abstract
Extensive injury of endothelial cells in blood vasculature, especially in the microcirculatory system, frequently occurs in hosts suffering from sepsis and the accompanied systemic inflammation. Pathological factors, including toxic components derived from invading microbes, oxidative stress associated with tissue ischemia/reperfusion, and vessel active mediators generated during the inflammatory response, are known to play important roles in mediating endothelial injury. Collapse of microcirculation and tissue edema developed from the failure of endothelial barrier function in vital organ systems, including the lung, brain, and kidney, are detrimental, which often predict fatal outcomes. The host body possesses a substantial capacity for maintaining vascular homeostasis and repairing endothelial damage. Bone marrow and vascular wall niches house endothelial progenitor cells (EPCs). In response to septic challenges, EPCs in their niche environment are rapidly activated for proliferation and angiogenic differentiation. In the meantime, release of EPCs from their niches into the blood stream and homing of these vascular precursors to tissue sites of injury are markedly increased. The recruited EPCs actively participate in host defense against endothelial injury and repair of damage in blood vasculature via direct differentiation into endothelial cells for re-endothelialization as well as production of vessel active mediators to exert paracrine and autocrine effects on angiogenesis/vasculogenesis. In recent years, investigations on significance of EPCs in host defense and molecular signaling mechanisms underlying regulation of the EPC response have achieved substantial progress, which promotes exploration of vascular precursor cell-based approaches for effective prevention and treatment of sepsis-induced vascular injury as well as vital organ system failure.
Keywords: Endothelial progenitor cells, Host defense, Sepsis, Septic infection, Endothelial injury, Systemic inflammation, Microcirculation, Blood vasculature, Niche, Injury repair
1. Introduction
Sepsis is a life-threatening syndrome often accompanied with deterioration of microcirculation and a widespread endothelial injury/dysfunction (Hawiger, 2018; Ince et al., 2016; Jacobi, 2022; Vassiliou, Kotanidou, Dimopoulou, & Orfanos, 2020). Structural damage and functional collapse of microvasculature in vital organ systems is detrimental. In the lung, for example, the dedicated architecture of thin epithelium laying over the extensive capillary network in alveoli secures effective gas exchange. Loss of capillary integrity and the resulted edema in inter-stitial tissue of the terminal airways hamper gas exchange, which is the pathological hallmark of adult respiratory distress syndrome (ARDS) (Matthay, Ware, & Zimmerman, 2012). In hospitalized patients with sepsis, acute lung injury is the most frequent complication, often triggering and/or accelerating the development of multiple organ dysfunction syndrome (MODS). In addition, tissue edema due to the increase in capillary permeability in the brain can induce and aggravate encephalopathy in patients with sepsis, which predicts a worse prognosis (Gofton & Young, 2012). Since endothelial dysfunction represents a prominent feature of sepsis (Crouser & Matthay, 2017; Hawiger & Musser, 2011; Hawiger, Veach, & Zienkiewicz, 2015) and impairment of microcirculation evidently plays a critical role in the pathogenesis of MODS, the sequelae impacting the integrity of blood vasculature has been identified as the major cause of fatal outcome in patients with sepsis (Parikh, 2013).
Mounting investigations have been conducted focusing on etiology and pathogenesis of microvascular injury during sepsis and the systemic inflammatory response. In recent years, host defense mechanisms against endothelial injury in effort for maintaining microvascular homeostasis and repairing endothelial damage have caught increasing attention. The body processes vascular precursor cells, particularly endothelial progenitor cells (EPCs), which can be activated to participate in protecting endothelial cells from injury and/or repairing damage in the vasculature (Shi et al., 2017). This review discusses recent progress in studying the EPC response to sepsis and/or septic infection with an emphasis on its significance in host defense against endothelial injury. Effort in exploring the potential of EPC-based approaches for treatment of microvascular damage caused by sepsis will also be addressed.
2. Injury to microvascular endothelium
Microcirculation provides perfusion of blood to tissues. It dynamically interplays with tissue cells as well as their surrounding environments. Histologically, microvasculature is composed of arterioles, capillaries, and venules. Connecting arterioles and venules, capillaries are the smallest blood vessels with the wall consisting of only a single layer of endothelial cells. The structure of capillaries varies, which can be continuous, fenestrated, and sinusoidal (or discontinuous) depending on the type of tissue where they are located. The dedicated endothelial structures and organizations in capillaries are vital for carrying out normal function of transporting and exchanging materials through microcirculation in the body.
Injury to the microvascular endothelium consists of different forms varying from reversibly functional abnormality to irreversible death of cells. Multiple mechanisms are involved in causing endothelial damage during sepsis. Insults eliciting the inflammatory response and factors generated from host origins, can induce structural and/or functional abnormalities of endothelial cells in the microvasculature. Several recently published reviews have extended comprehensive discussions on mechanisms underlying endothelial injury during the host response to various pathogenic challenges (Dumnicka et al., 2017; Evans & Zhao, 2017; Hawiger et al., 2015; Matsuda, 2016; Vassiliou et al., 2020; Venkatesulu et al., 2018). Here, we will focus on the recent progress in studies on mechanisms by which biological agents/mediators induce structural and functional derangements of endothelial cells, particularly by those that are also involved in signaling vascular precursor cell activation for host defense against endothelial injury. Understanding the complex actions of these agents/mediators in mediating both the injury to endothelial cells and the host defense for maintaining and/or repairing the integrity of endothelium in microvasculature during the host defense response will shed light on identifying therapeutic targets for prevention and treatment of endothelial injury in hosts suffering from sepsis or septic infection.
2.1. Substances/toxins derived from pathogenic microbes
Numerous molecules derived from invading pathogens of viral, bacterial, and fungal origins can cause endothelial damage. Severe viral infections, from Ebola infection to novel coronavirus disease 2019 (COVID-19), are accompanied with massive damage of endothelial cells (Amraei & Rahimi, 2020; Eisa-Beygi & Wen, 2015; Nägele, Haubner, Tanner, Ruschitzka, & Flammer, 2020; Nicosia, Ligresti, Caporarello, Akilesh, & Ribatti, 2021). Septic infections caused by bacteria of either gram-positive or gram-negative strains result in endothelial damage and/or functional derangement in the microvasculature (Berube & Bubeck Wardenburg, 2013; Hawiger et al., 2015; Khakpour, Wilhelmsen, & Hellman, 2015; Remy, Qiu, Li, Cui, & Eichacker, 2013; Suffredini, Cui, Xu, Li, & Eichacker, 2017). Similarly, in life-threatening fungal infections, such as systemic candidiasis, substantial damage to endothelial cells may occur in multiple organ systems (Grubb et al., 2008; Sun et al., 2010). Mechanistically, microbial agents can cause endothelial damage by direct cytotoxicity and/or altering cell metabolic/functional activities. In addition, they can incite host immune response and activate coagulant system to injure endothelium through actions of activated immune cells, inflammatory mediators, and disseminated emboli clotting the microvasculature (Hawiger et al., 2015).
Lipopolysaccharides (LPS), also known as endotoxins, are cell wall components of gram-negative bacteria which cause two-thirds of septic infection in patients either alone or in combination with other pathogens (Vincent et al., 2009). LPS binds to its receptor, toll-like receptor 4 (TLR4, 95 KDa), and activates signaling pathways, including nuclear factor kappa-light-chain-enhancer of activated B cells, Rho/Rho-associated protein kinase (ROCK), mitogen-activated protein kinase (MAPK), and phosphatidylinositol 3-kinase (PI3K)/serine/threonine protein kinase B (PKB, also known as AKT) pathways, in endothelial cells (Aepfelbacher & Essler, 2001; Bannerman & Goldblum, 2003; Dauphinee & Karsan, 2006; Khakpour et al., 2015; Wójciak-Stothard & Ridley, 2002). The mediated downstream derangements include a). phosphorylation of myosin light chain (MLC) to promote interaction between myosin and actin, allowing myosin to slide along actin filaments to cause contractility of endothelial cells, which consequently leads to leakage of the endothelium; b). expression of coagulation pathway intermediaries resulting in microvascular thrombi and coagulopathy; c). up-regulation of adhesion molecule expression enhancing attachment of leukocytes; d). generation of inflammatory cytokines and lipid mediators; as well as e). activation of caspases mediating cleavage of junctional proteins responsible for maintaining the barrier function of endothelial cells and apoptosis of endothelial cells. Endothelial cells also express TLR2 which serves as the receptor for certain types of bacterial (i.e. lipoproteins)- and fungal (i.e. zymosan)-derived substances. Engagement of these pathogen-derived ligands with TLR2 activates the associated signaling pathways to mediate damage in endothelial cells similarly to that mediated by LPS/TLR4 signaling. In addition, microbial-derived ligands can bind to TLR4 and TLR2 receptors expressed by immune effector cells to activate their activity and production of cytotoxic substances, vessel active agents, and inflammatory mediators for inducing the secondary injury to endothelial cells.
2.2. Tumor necrosis factor-α (TNF-α)
TNF-α is a proximal pro-inflammatory cytokine produced by host immune effector cells and many other cell types. Initially, TNF-α is expressed as a 26-kDa membrane-bound precursor protein that is then cleaved by TNF-α converting enzyme (TACE) to generate the 17-kDa soluble TNF-α. Vascular endothelial cells express TNF receptors (TNFR1, 55 KDa and TNFR2, 75 KDa) and respond to TNF stimulation. Engagement of TNF-α with its receptors activates signal transduction pathways, including fas-associated death domain protein (FADD)-caspase, mitogen-activated protein kinase kinase kinase 3 (MEKK3)-apoptosis-inducing factor (AIF), Cdc42/Rac/Rho/ROCK, protein kinase C (PKC), MAPKs [including p38 MAPK, c-Jun N-terminal kinase (JNK)], in endothelial cells (Pober & Min, 2006; Siflinger-Birnboim & Johnson, 2003; Wójciak-Stothard & Ridley, 2002; Yoshizumi et al., 2004). TNF-α binds to TNFR1 [also called death receptor-1] activates FADD-caspase and MEKK3-AIF signaling which mediate apoptotic and non-apoptotic death (oncosis) of endothelial cells, respectively (Pober & Min, 2006). TNF-α activates the Cdc42/Rac/Rho /ROCK pathway leading to formation of stress fibers and contraction of endothelial cells via inhibiting dephosphorylation of MLC to strengthen phosphorylation of MLC (Petrache, Crow, Neuss, & Garcia, 2003). Activation of Rho/ROCK mediates TNF-α-induced progressive increase in endothelial permeability by disrupting tight junctions with removal of occludin and junctional adhesion molecule-A from tight junctions as well as redistribution of membrane-associated protein zonula occludens-1 (McKenzie & Ridley, 2007). TNF-α-induced activation of Rho/ROCK promotes apoptosis by mediating assembly of the death-inducing signaling complex leading to activation of caspase 7, 3, and 8 in endothelial cells (Petrache et al., 2003). TNF-α stimulation activates PKC-α and PKC-β in endothelial cells (Ferro, Neumann, Gertzberg, Clements, & Johnson, 2000). Binding of TNF-α to TNFR activates phospholipase C (PLC) via the activity of the TNFR-associated death domain, which subsequently activates PKC (Siflinger-Birnboim & Johnson, 2003). The interaction of PKC signaling with its adherens junction and cytoskeletal targets results in reorganization of cytoskeleton, alteration of adherence/junction, formation of intercellular gaps, and impairment of barrier function in endothelial cells (Siflinger-Birnboim & Johnson, 2003; Vandenbroucke, Mehta, Minshall, & Malik, 2008). TNF-α-induced activation of TNF receptor-associated factor 2-apoptosis signal-regulated kinase 1-JNK signaling mediates expression of adhesion molecules, such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), by endothelial cells (Yoshizumi et al., 2004), which promotes leukocyte adhesion. Activated leukocytes including granulocytes and monocytes may subsequently injure the endothelium by generation of reactive oxygen species and proteolytic enzymes. TNF-induced activation of JNK and p38 MAPK signaling also promotes apoptosis in endothelial cells (Pober & Min, 2006).
2.3. Interferon-gamma (IFN-γ)
IFN-γ is another proximal pro-inflammatory cytokine with a molecular weight of 17-kDa. Activated natural killer cells (NK cells), T lymphocytes, macrophages, and mucosal epithelial cells are major cell types producing IFN-γ. IFN-γ receptor is a heterodimeric glycoprotein consisting of two polypeptide chains, i.e. IFN-γ receptor 1 and 2 (IFNGR1, 90 kDa and IFNGR2, 60–67 kDa). IFNGR1 is the major ligand-binding subunit, while IFGR2 serves to increase the affinity of IFNGR1 for its ligand binding and transduce signaling. Janus family of protein tyrosine kinase 1 and 2 (JAK1 and 2) are constitutively associated with the cytoplasmic domains of IFNGR1 and 2, respectively. Engagement of IFN-γ to IFNGR leads to receptor oligomerization, with two IFNGR1 chains bound to one IFN-γ for recruitment of two IFNGR2 chains to form the receptor complex, which allows activation of the associated JAKs through sequential auto- and transphosphorylation. Activated JAKs catalyze phosphorylation of tyrosine residues on IFNGRs leading to recruitment and phosphorylation of signal transducers and activators of transcription 1 (STAT1). Activated STAT1 undergoes nuclear translocation to promote transcription of IFN-γ responsive gene products. Endothelial cells express IFNGR and respond to IFN-γ stimulation. Observations have shown that during the systemic inflammatory response, IFN-γ functions as a crucial co-factor potentiating the injurious effect of TNF on vascular endothelial cells (Pober & Min, 2006). In vivo administration of anti-IFN- γ has been reported to prevent lethality in mice caused by recombinant human TNF-α at doses four- to five-fold greater than the LD100. IFN- γ exposure causes apoptosis of endothelial cells, which involves the signal transduction pathway dependent on activation of tyrosine protein kinase (Wang, Redmond, Watson, Condron, & Bouchier-Hayes, 1999). IFN-γ stimulation increases permeability and reduces barrier properties of endothelial cells (Bonney et al., 2019). Following exposure to IFN-γ, cultured endothelial cells have been reported to show a marked increase in STAT1 phosphorylation and up-regulation of Rho-GTPases. In addition, ROCK activity in endothelial cells is elevated following IFN-γ treatment. This ROCK signaling mediates cytoskeletal contractions, resulting in junctional disorganization and cell-cell separation in endothelial cells. As described previously, TNF-α stimulates surface expression of adhesion molecule VCAM-1 by endothelial cells. IFN-γ promotes/amplifies TNF-α-induced gene transcription and protein expression of VCAM-1 by endothelial cells through the interferon-related factor-1 pathway (Lechleitner, Gille, Johnson, & Petzelbauer, 1998). Up-regulation of adhesion molecule VCAM-1 expression increases binding of leukocytes to the surface of endothelial cells, which in turn facilitates leukocyte-mediated endothelial injury during the inflammatory response.
2.4. Interleukin-6 (IL-6)
IL-6 is a soluble 21-kDa cytokine with a pleiotropic nature in biological activity. Immune cells and stromal cell types produce IL-6 during the inflammatory response (Jordan et al., 2017). IL-6 binds to its receptor [occurring in two forms: 80-kDa transmembrane receptor (mIL-6R) and 50–55-kDa soluble receptor (sIL-6R)] to form the IL-6/IL-6 receptor complex. Soluble IL-6 receptor is produced from mIL-6R through proteolytic cleavage by metalloproteinases or via translation of a differentially spliced IL-6 receptor mRNA (Kaur, Bansal, Kumar, & Bansal, 2020). Dependent on different forms of receptors engaged, two types of IL-6 signaling, i.e. classic signaling and trans-signaling, can be activated downstream of the IL-6/IL-6 receptor complex. Engagement of IL-6 with mIL-6R initiates classic signaling, whereas binding of IL-6 with sIL-6R starts trans-signaling (Mahmud-Al-Rafat et al., 2019). The IL-6/IL-6 receptor complex interacts with the signal-transducing component glycoprotein 130 kDa (gp130) on cells to mediate dimerization of gp130, which leads to activation of cell signaling pathways, including JAK/STAT, PI3K/AKT, and rat sarcoma protooncogene (Ras)/MAPK signaling cascades (Peppler, Townsend, & Wright, 2020; Rose-John, 2020; Tanaka, Narazaki, & Kishimoto, 2014). Endothelial cells express both IL-6 receptor and gp130 on their surface (Montgomery et al., 2021; Zegeye et al., 2018). It has been speculated that IL-6 elicits pro-inflammatory responses through its trans-signaling, while mediates anti-inflammatory activities via the classic signaling. Recent studies suggest that IL-6 classic signaling and trans-signaling have overlapping but distinct properties in regulating endothelial cell survival and inflammatory activations (Montgomery et al., 2021). Exposure of cultured endothelial cells to IL-6 has been shown to cause a significant increase in cell permeability with reduction of transendothelial electrical resistance (Couto et al., 2019). IL-6 down-regulates expression of endothelial intercellular junction proteins β-catenin and ZO-1 through JAK/STAT3 signaling. Suppressor of cytokine signaling-3 (SOCS3) is a negative regulator of JAK-STAT signaling. SOCS3 inhibits the tyrosine kinase activity of JAK by its kinase inhibitory region at the N-terminal adjacent to SH2 domain (Babon et al., 2012). SOCS3 also binds to IL-6 receptor associated gp130 to repress its pro-inflammatory signaling (Schmitz, Weissenbach, Haan, Heinrich, & Schaper, 2000). In addition, SOCS3 interacts with microtubule plus-end binding proteins CLIP-170 and CLASP2 via its N-terminal domain (Karki et al., 2021). The resulting SOCS3–CLIP-170/CLASP2 complex contributes to SOCS3-mediated protection against endothelial dysfunction (Karki et al., 2021). IL-6 promotes microtubule disassembly and disrupts interaction of SOCS3 with CLIP-170 and CLASP2, which may potentiate dysfunction of endothelium in microvasculature during the inflammatory response. Currently, observations about IL-6 classic signaling and trans-signaling in mediating alterations of endothelial cell activities remain inconsistent (Montgomery et al., 2021; Zegeye et al., 2018). Studies on human vascular endothelial cells have reported that IL-6 trans-signaling activates the PI3K/AKT and mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) pathways, while IL-6 classic signaling induces activation of the JAK/STAT3 pathway (Zegeye et al., 2018). In contrast to IL-6 classic signaling, IL-6 trans-signaling induces the release of the pro-inflammatory chemokine monocyte chemoattractant protein-1 (MCP-1) from endothelial cells. This IL-6-induced production of MCP-1 requires simultaneous activation of the JAK/STAT3 and PI3K/AKT pathways in endothelial cells. Other investigations on human endothelial cells have documented that in the inflammatory response, IL-6 trans-signaling mediates ICAM expression and secretion of C–C motif chemokine ligand 2, whereas IL-6 classic signaling induces secretion of C-X-C motif chemokine ligand 8 or interleukin-8 (IL-8)] and transmigration of neutrophil like cells (Montgomery et al., 2021). IL-6 classic signaling also protects endothelial cells against immune-mediated injury and stress-induced death. In this set of studies, endothelial cells respond to IL-6 classic signaling by activating the AKT and ERK1/2 pathways, whereas IL-6 trans-signaling activates the STAT3, AKT, and ERK1/2 pathways. Further investigations appear to be warranted for clarifying the existing discrepancies.
2.5. Vascular endothelial growth factor (VEGF)
VEGF is a secreted protein factor that plays an essential role in vasculogenesis and angiogenesis (Distler et al., 2003; Ferrara, 2009). The active forms of VEGF are mostly homodimers with a molecular weight of 45 kDa. Three types of VEGF receptors i.e., vascular endothelial growth factor receptor 1 (VEGFR1), VEGFR2 (Flk1/KDR, Flk1 or fms-like tyrosine kinase-1 is VEGFR2 in mice; KDR or kinase insert domain receptor is the human homolog of VEGFR2), and VEGFR3 have been identified (Distler et al., 2003). In adults, VEGFR1 and VEGFR2 are mainly expressed by endothelial cells in the blood vasculature while expression of VEGFR3 is generally restricted to endothelium in the lymphatic system. Due to differences in binding affinity for VEGF ligand and the receptor-associated kinase activity, VEGFR1 and 2 can exert contrast effects on vasculogenesis and angiogenesis. VEGFR1 has much stronger binding affinity for VEGF (approximately 10-fold) than that of VEGFR2, but the kinase activity of VEGFR1 is very weak (about one-tenth) in comparison to that of VEGFR2 (Shibuya, 2013). In addition, the VEGFR1 gene produces both membrane-bound VEGFR1 and soluble VEGFR1. The soluble VEGFR1 consists of only the extracellular domain for binding of VEGF without ability of transmitting signals across the cell membrane. Therefore, VEGFR2 primarily serves as a positive signal transducer, whereas VEGFR1, particularly soluble VEGFR1, commonly functions as an attenuator in the process of vasculogenesis and angiogenesis (Shibuya, 2006). Binding of a VEGF homodimer to its receptor induces dimerization of 2 receptors, triggering receptor auto-phosphorylation by the activity of receptor-associated tyrosine kinases. Subsequently, downstream signaling cascades, including PLC-γ/PKC, protein kinase D, PI3K, Src, Rho, Ras pathway members, MAPK, AKT, PKC, and focal adhesion kinase (FAK) are activated to mediate cell proliferation, differentiation, migration, and contraction (Bates, 2010; Gavard, Patel, & Gutkind, 2008; Schatteman & Awad, 2004; Sun, Breslin, Zhu, Yuan, & Wu, 2006).
In addition to robustly mitogenic and angiogenic activities for cells in the endothelial line, VEGF is a potent mediator regulating permeability of blood vasculature (Bates, 2010). Initially, VEGF was characterized as a vascular permeability factor secreted by tumor cells. Studies have shown that VEGF exposure rapidly alters endothelial morphology via inducing formation of fenestrations in cultured endothelial cells in vitro as well as in endothelia of small venules and capillaries in vivo (Esser et al., 1998; Roberts & Palade, 1995). Vascular endothelial cadherin (VE-cadherin) is specifically responsible for endothelial adherens junction assembly and barrier architecture (Gavard, 2013). Engagement of VEGF with VEGFR2 activates the small GTPase Rac (Gavard & Gutkind, 2006). Rac activation, in turn, promotes phosphorylation of VE-cadherin. The subsequent recruitment of beta-arrestin2 to serine-phosphorylated VE-cadherin mediates its internalization into clathrin-coated vesicles and disassembly of intercellular junctions. Rho signaling mediates MLC phosphorylation via either inhibition of MLC phosphatase or direct phosphorylation of MLC by ROCK. Activation of VEGFR2 by VEGF ligand binding activates Rho/ROCK signaling which mediates MLC phosphorylation, actin stress fiber formation, increase in endothelial permeability, and microvascular barrier dysfunction (Sun et al., 2006). FAK mediates endothelial cell-matrix interaction, adhesion, and migration in response to VEGF. VEGF promotes FAK phosphorylation and causes a significant morphological change of FAK from a punctate pattern to an elongated, dash-like distribution aligned with the longitudinal axis of the cells. This activation of FAK signaling participates in the regulation of VEGF-elicited hyperpermeability in endothelial cells (Wu, Guo, Yuan, & Granger, 2003). Blocking FAK signaling attenuates VEGF-induced endothelial hyperpermeability. Studies have shown that activation of other related signaling pathways, including PLC, PKC, MEK1/2, ERK1/2, PI3K/AKT, and endothelial nitric oxide synthase (eNOS), may also be involved in mediating VEGF-induced hyperpermeability of the endothelium (Aramoto, Breslin, Pappas, Hobson 2nd, & Durán, 2004; Bates, 2010; Becker et al., 2001; Fukumura et al., 2001; Lal, Varma, Pappas, Hobson 2nd, & Durán, 2001; Pocock & Bates, 2001).
Septic infection significantly elevates VEGF level in the systemic circulation in experimental animals (Tomita et al., 2020). VEGF gene expression by heart tissue increases in sheep with septic infection (Jonkam et al., 2009). In mice challenged with aerosol LPS, the level of tissue-associated VEGF increases along with the development of capillary leakage and edema in the lung (Karmpaliotis et al., 2002). LPS stimulates VEGF expression by vascular endothelial cells. Overexpression of VEGF gene in the lung directly induces pulmonary edema in mice (Bhandari et al., 2006; Kaner et al., 2000). Administration of VEGF to rats in the acute phase of focal cerebral ischemia causes a significant increase in blood–brain barrier leakage, hemorrhagic transformation, and ischemic lesions (Zhang et al., 2000). Intravenous administration of VEGF-neutralizing monoclonal antibody attenuates the increase in pulmonary vascular permeability in mice with LPS-induced lung inflammation (Tomita et al., 2020). VEGF exposure significantly increases protein permeability across cultured bovine microvascular endothelial cell monolayers (Godzich et al., 2006). This VEGF-mediated increase in endothelial permeability is accompanied with phosphorylation of the adherens junction protein E-cadherin and the formation of actin stress fibers. Pretreatment with an adenovirus encoding the soluble VEGF receptor, prevents increase in lung vascular permeability caused by ischemia/reperfusion injury in rats. In hospital settings, patients with septic infection exhibit an increase in VEGF level in the systemic circulation (Alves et al., 2011; Pickkers et al., 2005; Shapiro et al., 2008; van der Flier et al., 2005). Individuals with acute lung injury and pulmonary edema also show an elevated level of VEGF in the blood stream (Azamfirei et al., 2010; Thickett, Armstrong, Christie, & Millar, 2001).
At the present time, nevertheless, recognition about the role of VEGF in the regulation of microvascular integrity during the host defense response remains paradoxical (Mura, dos Santos, Stewart, & Liu, 2004). In normal adults, cells (particularly type II alveolar epithelial cells) in the lung express a high level of VEGF (Barratt, Medford, & Millar, 2014). Normal human respiratory epithelial lining fluid contains 500-fold greater concentration of VEGF than that in the plasma (Kaner & Crystal, 2001). The abundancy of VEGF in the lung appears necessary for securing homeostasis of pulmonary cells. VEGF is an alveolar epithelial mitogen and stimulant (Medford & Millar, 2006). It stimulates growth of airway epithelial cells and production of surfactant by alveolar type II cells (Mura et al., 2004). However, tissue edema and increase in capillary permeability are not seen in the healthy lung even in the presence of high VEGF level in the alveolar surface. The lung possesses ability for compartmentalizing VEGF (Kaner & Crystal, 2001). It has been postulated that this compartmentalization prevents exposure of endothelial cells in the pulmonary microvasculature to the high level of VEGF in the alveolar surface (Barratt et al., 2014; Medford & Millar, 2006). Pathological events cause decompartmentalization in the terminal airways, which will allow VEGF spillover to the microvascular side and induce endothelial hyperpermeability with pulmonary edema. Currently, direct evidence supporting this postulation has yet to be identified. Clinical investigations have repeatedly shown reduction of VEGF expression in the lung along with an elevation of VEGF level in the systemic circulation in patients with septic infection and ARDS (Abadie et al., 2005; Azamfirei et al., 2010; Thickett, Armstrong, & Millar, 2002; Tsokos, Pufe, Paulsen, Anders, & Mentlein, 2003). In these patients, lung injury score is inversely correlated with VEGF concentration in lung epithelial lining fluid (Koh et al., 2008; Thickett et al., 2002). The VEGF levels in lung epithelial lining fluid are significantly greater in survivors than in non-survivors (Koh et al., 2008). The altered VEGF levels in the lung and plasma return to normal in patients during their recovery from acute lung injury/ARDS (Azamfirei et al., 2010; Thickett et al., 2002). Analysis of gene polymorphisms in a cohort of 1253 patients with risk factors for ARDS treated in intensive care unit (ICU) has shown that patients with the +936TT and +936CT+TT genotypes are significantly associated with an increased mortality from ARDS. Whereas plasma VEGF levels in patients with ARDS are significantly lower in subjects with the +936CT+TT genotype than in those with the +936CC genotype (Zhai et al., 2007). Further studies on delineating the complex activities of VEGF in the regulation of endothelial function will provide a deeper insight into its effects.
2.6. Angiopoietin-2 (Ang-2)
Ang-2 is a member of the angiopoietin family which includes Ang-1, Ang-2, and Ang-3 (mouse)/Ang-4 (human) (Fagiani & Christofori, 2013). As a secreted glycoprotein, Ang-2 consists of 496 amino acids with the molecular weight around 70 kDa. The amino acid sequence of human Ang-2 is 85% identical to that of mouse. In the angiopoietin family, Ang-1 and Ang-2 are the best characterized. Both Ang-1 and Ang-2 are natural ligands of the Tie-2 receptor tyrosine kinase, which is expressed primarily on endothelial cells and early hematopoietic cells (Hsu et al., 2000; Parikh, 2013; Singh, Tahir, Alawo, Issa, & Brindle, 2011). Tie is the abbreviation for tyrosine kinase with immunoglobulin and EGF homology domains (Fagiani & Christofori, 2013). Unlike Ang-1 that is produced by perivascular cells (pericytes, vascular smooth muscle cells, and fibroblasts) and platelets to exert paracrine activities on the endothelium, Ang-2 is synthesized by endothelial cells largely for the purpose of autocrine actions (Akwii, Sajib, Zahra, & Mikelis, 2019; Fagiani & Christofori, 2013; Parikh, 2013; Thurston & Daly, 2012). Macrophages and smooth muscle cells also express Ang-2 (Parikh, 2013). In normal conditions, the expression of Ang-2 is low in quiescently mature vasculature. Newly synthesized Ang-2 protein is stored in cytoplasmic storage granules called Weibel–Palade bodies, that can be rapidly secreted from cells upon inflammatory and angiogenic stimulations (Fiedler et al., 2004; Thurston & Daly, 2012). Mediators, including TNF-α, phorbol 12-myristate 13-acetate, thrombin, histamine, VEGF, basic fibroblast growth factor, and hypoxia, stimulate Ang-2 production. Whereas Ang-1 and transforming growth factor-β1 inhibit Ang-2 expression. Clinical investigations have reported that the plasma level of Ang-2 increases substantially (up to 50 times) during the systemic inflammatory response and septic infection (David, Kümpers, van Slyke, & Parikh, 2013; Higgins et al., 2018; Parikh, 2013; van der Heijden, van Nieuw Amerongen, Koolwijk, van Hinsbergh, & Groeneveld, 2008). The elevated plasma level of Ang-2 is accompanied with the increase in vascular permeability, endothelial inflammation, edema, disseminated intravascular coagulation, tissue hypoperfusion, and worse clinical outcome (Ganter et al., 2008; Higgins et al., 2018; van der Heijden et al., 2008).
Both Ang-1 and Ang-2 bind to Tie-2 with nanomolar affinity via their similar carboxy-terminal fibrinogen-like domain modules (Parikh, 2013; Zhao et al., 2021). However, their actions for initiating the downstream signaling are substantially different. Under the physiological condition, Ang-1is constitutively expressed, that is not altered dramatically by most inflammatory and angiogenic stimuli (Thurston & Daly, 2012). As a potent agonist of Tie-2, Ang-1 engagement promotes auto-phosphorylation of Tie-2 and activates signaling cascades including PI3K, PKB/AKT, eNOS, MAPK, downstream of tyrosine kinase-2/non-catalytic region of tyrosine kinase adaptor protein/ p21-activated kinase, A20-binding inhibitor of NFjB-2, Wnt/b-catenin and delta-like 4/Notch pathways (David et al., 2013; Fagiani & Christofori, 2013; Master et al., 2001; Nicolini, Forini, Kusmic, Iervasi, & Balzan, 2019; Thurston & Daly, 2012). These signals promote endothelial cell survival, anti-apoptosis, integrity of barrier function, maintenance of quiescent status and vascular stabilization, extracellular deposition of type IV collagen, suppression of pro-inflammatory gene expression, and inhibition of adhesion molecule expression. Ang-2 primarily acts as an antagonist of Tie-2 to inhibit Ang-1-induced Tie-2 phosphorylation and the downstream signals responsible for promoting functional integrity of endothelium in the established vasculature (Akwii et al., 2019). It has been suggested that the different actions of Ang-1 and Ang-2 for initiating Tie-2 signaling are dependent on their N-terminal structures (Parikh, 2013). Activation of Tie-2 receptor needs the cluster of ligands at the tetramer or higher levels (Fagiani & Christofori, 2013). A unique region in the N-terminal of Ang-1 favors ligand multimerization into large aggregates which mediates intense Tie-2 clustering and cross-phosphorylation (Kim et al., 2005). Inability of Ang-2 to form large multimer aggregates limits activation of Tie-2 signaling following its binding to the receptor. Therefore, the competitive binding of Ang-2 against Ang-1 to Tie-2 receptor essentially exerts an antagonist action for blocking Tie-2 signaling, leading to destabilization of endothelium in the microvasculature. Further investigations suggest that Tie-2 is phosphorylated on multiple sites (Zhao et al., 2021). The relative degree of Tie-2 phosphorylation for different branches of signaling pathway (AKT, ERK, and FAK) varies. Unique structures of Ang-1 versus Ang-2 in the C-terminal fibrinogen-like domain also influence signaling transduction following Tie-2 engagement with these ligands. Vascular endothelial protein tyrosine phosphatase (VEPTP) mediates dephosphorylation of Tie-2 in endothelial cells of blood vasculature. The activity of VEPTP, therefore, sets a high threshold limiting Ang-2-mediated Tie-2 phosphorylation and the downstream signaling, but allows activation of Tie-2 by the strong agonist Ang-1 (Souma et al., 2018). In addition, Ang-2 has been reported to initiates proangiogenic/vascular-destabilizing signals via interplaying with integrin, particularly α5β1, mediated pathways (Felcht et al., 2012; Hakanpaa et al., 2015; Hakanpaa et al., 2018). In preclinical studies, reducing Ang-2 gene expression with genetic manipulation, blocking Ang-2 with neutralizing antibody, and promoting Ang-2 multimerization with clustering antibody have been shown to reduce vascular leak and organ injury as well as to improve survival following septic infection (David et al., 2012; Han et al., 2016; Ziegler et al., 2013).
Table 1 summarizes actions of related biological agents/mediators in inducing structural and functional derangements of endothelial cells.
Table 1.
Actions of related biological agents/mediators in inducing structural and functional derangements of endothelial cells.
| Soluble Factor | Endothelial Injury | Mechanism |
|---|---|---|
| LPS |
|
|
| TNF-α |
|
|
| IFN-γ |
|
|
| IL-6 |
|
|
| VEGF |
|
|
| Ang-2 |
|
|
3. EPCs
EPCs are upstream precursor cells that have substantial capacity of self-renewal, proliferation, and differentiation into endothelial cells. During the past two decades, efforts have been continually devoted to characterization of EPCs. It is now generally accepted that endothelial colony-forming cells (ECFCs) represent the true EPC population responsible for giving rise to endothelial cells and participating in neovascularization in vivo (Medina et al., 2017; Paschalaki & Randi, 2018; Shi et al., 2017). The bone marrow accommodates a major reserve of EPCs in the body. In addition, the vascular wall system also provides niches for housing vascular precursor cells including EPCs (Shi et al., 2017). These tissue resident EPCs may enter the circulation to become circulating EPCs. Specific surface markers have been selected for identifying, collecting, and expanding EPCs, although most of these strategies remain imperfect and require further refining. In humans, EPCs exhibit a phenotype of CD45−CD34+VEGFR2+ or CD45−CD34+CD133+VEGFR2+ (Peichev et al., 2000; Shi et al., 2017; Timmermans et al., 2007). For identifying ECFCs, it has been summarized that they express markers of CD31, CD146, VEGFR2, VE-cadherin, and CD34, but are negative for hematopoietic differentiation signatures of CD45 and CD14 (Dight, Zhao, Styke, Khosrotehrani, & Patel, 2022; Go & Yoder, 2021). In mice, a subset of lineage(lin)−stem cell factor receptor(c-kit)+stem cell antigen-1(Sca-1)+ cells expressing VEGFR2 have been identified to contain enriched EPCs (Rafii & Lyden, 2003). Initially, murine lin−c-kit+Sca1+ cell population was characterized to contain enriched hematopoietic stem cells (HSCs). The subsequent studies have demonstrated that transplantation of marrow lin−c-kit+Sca1+ cells can not only repopulate blood cells, but also give rise to endothelial cells that are integrated into blood vessels in various organ tissues in the recipients (Bailey et al., 2004). These investigations confirm that the marrow lin−c-kit+Sca1+ pool contains a heterogenous cell population encompassing different subsets of stem/progenitors in mice. In rats, a subset of lineage negative cells in the blood stream bearing lin−Hoechst+CD36+ markers have been isolated and validated to contain putative EPCs (Thomas et al., 2009). EPCs derived from rabbit peripheral blood have been shown to express CD34+VEGFR2+Ulex europaeus agglutinin-1+ markers (Gao, Chen, Liang, & Chen, 2011). These cells also uptake acetylated low-density lipoprotein. In chickens, bone marrow EPCs have been characterized, which are positive for expression of CD34, CD133, and VEGFR2 (Bai et al., 2012). In adult horse, ECFCs derived from peripheral blood have been reported positive for von Willebrand factor, VEGFR2, CD34, and CD105 (Salter et al., 2015).
Despite the established concept for EPCs or ECFCs, the cell composition in the pool of these vascular precursors within the body remains considerably heterogeneous (Aquino, Sierra, & Montaldo, 2021; Chopra, Hung, Kwong, Zhang, & Pow, 2018; Dight et al., 2022). Differences in their developmental stage, functional status, and phenotypic characteristics, as well as the niche environment where they reside warrant the heterogeneity of EPCs in the body. In a study for characterizing subsets of blood-derived EPCs from healthy volunteers, two phenotypes with markers of CD34+CD133+ and CD34−CD133+ have been analyzed (Bachelier, Bergholz, & Friedrich, 2020). Expression of CD31 and VEGFR2 is present on cells belonging to both subsets, but to a higher extent on those in the CD34+CD133+ subset. Compared with CD34+CD133+ cells, CD34−CD133+ cells exhibit stronger activities of adhesion, migration, and proliferation. Further, CD34−CD133+ cells show a significantly higher potential for multipotent differentiation, suggesting the upstream position of CD34−CD133+ cells in comparison to that of CD34+CD133+ cells. Studies on EPCs in murine models have reported even more diverse in phenotypes and biological characteristics. As described above, precursors with surface markers of lin−c-kit+Sca1+-VEGFR2+ have been identified as EPCs residing in different niche environments in mice (Rafii & Lyden, 2003). However, investigators have reported that subsets of marrow and circulating EPCs bearing surface makers of c-Kit+/CD31+, Sca-1+/Flk1+, CD34+/CD133+, and CD34+/Flk1+ show different patterns of response to limb ischemia injury in mice (Cui et al., 2020). In addition, studies on precursors from vascular origins in mice have revealed that cell populations with phenotypic signatures including lin−c-kit+Sca1+CD31+CD105+, CD45−CD31+VE-cadherin+Hoechstlow, CD45−CD31+VE-cadherin+CD157+CD200+, and lin−CD34+VE-cadherin+CD31lowVEGFR2low exhibit biological characteristics of EPCs (Dight et al., 2022). Further efforts in identification, characterization, and verification of EPC subsets will improve understanding the cell compositions in the EPC pool as well as the functional interplays among these vascular precursors.
4. The EPC response
4.1. Change in circulating EPCs during the host defense response
Direct evidence of the EPC response to septic challenge is the alteration of EPC numbers and/or activities in the systemic circulation. Normally, EPCs are very rare in the systemic circulation. In humans, it has been reported that peripheral blood samples contain EPCs at 0.41 ± 0.47 colony-forming units (CFUs)/4 ml blood as analyzed by cell culture (Schlichting et al., 2011) and 2.70 ± 0.73 CD34+KDR+ cells/104 peripheral blood mononuclear cells as determined with flow cytometry (Zhu et al., 2021). In both adult and pediatric patients with septic infection, the level of EPCs in the circulation has been repeatedly reported to increase (Becchi et al., 2008; Kung et al., 2016; Patschan et al., 2011; Rafat et al., 2007a, 2007b; Siavashi et al., 2017; Zahran, Elsayh, Mohamad, Hassan, & Abdou, 2016). Commonly, the EPC response to acute vascular insult is very sensitive and effective. Investigations on patients with burn injuries or coronary artery bypass grafting have shown that EPCs in the peripheral circulation can increase 50-fold within the first 6–12 h (Gill et al., 2001). Similarly, the rapid change in circulating EPC counts represents a prominent feature in the EPC response to septic infection. A clinical observation has reported that patients with sepsis show a four-fold increase in the number of circulating EPCs in comparison to healthy controls (Becchi et al., 2008). This increase is evident at 6 h from their diagnosis, which reaches its maximum level at 72 h. Another report of 40 patients with sepsis admitted to ICU has documented that the level of circulating EPCs is significantly higher in all patients within 12 h after the diagnosis of sepsis (Patschan et al., 2011). An evaluation of 101 adult patients with severe sepsis and septic shock has shown that levels of EPCs in the blood are significantly higher in the patient group than in the control group within 24 h after admission (Kung et al., 2016). Likewise, a prospective study on 33 patients with septic shock treated in ICU has reported that the number of circulating EPCs is significantly greater in septic patients on day 1 of diagnosis compared to that in healthy controls (Skirecki et al., 2019). Further, analyzing blood samples 1 h post-birth of 40 preterm infants with sepsis has shown that the percentage of EPCs in the CD34+ cell population is significantly higher in comparison to that in preterm infants without sepsis (Siavashi et al., 2017). In experimental studies on a mouse model of LPS-induced acute lung injury, significant increase in circulating EPCs occurs 4 h following the LPS challenge (Yamada et al., 2004). In mice with sepsis induced by cecal ligation and puncture (CLP), a significant increase in circulating EPCs has been reported at 24 h and 48 h of sepsis (Pai, Shih, Shih, & Yeh, 2016). The active release of EPCs from their niches into the blood stream at the early stage of host response supports a speculation that these vascular precursors may participate in defense for maintaining the integrity of vascular endothelium or early repairing endothelial damage caused by sepsis and/or septic infection.
In addition to changes in EPC quantity in the blood stream, functional activity of circulating EPCs also alters during the host response to septic challenges. In a cohort of 45 patients with acute lung injury treated in ICU, 20 of them had septic shock (Burnham et al., 2005). Assessment of EPC colony-forming activity in blood samples by cell culture has demonstrated that patients with acute lung injury have significantly greater EPC CFU counts in comparison to healthy controls. Serum from patients with systemic inflammatory response syndrome has been shown to possess a significantly higher activity in enhancing EPC proliferation and migration (Pang et al., 2013). At the present time, nevertheless, available information about changing in functional activities of circulating EPCs during the host response to sepsis and/or septic infection remains limited and inconsistent. One clinical study comparing EPC colony-forming activity by culturing blood samples from ICU patients with and without severe sepsis has reported that no significant difference in circulating EPC colony counts exists in both cohorts at any time point [baseline (within 24 h of ICU admission) as well as 24–48 h and 120–144 h post-baseline] (Schlichting et al., 2011). Another investigation on a cohort of 86 patients with sepsis, 37 ICU controls, and 51 healthy controls has documented that blood samples from patients with sepsis have significantly lower potency of forming EPC colony in comparison to those from ICU controls and healthy controls (Cribbs et al., 2012). Similarly, characterization of circulating EPCs from preterm infants with sepsis has shown lower proliferative activity (as reflected by cell Ki-67 expression) (Siavashi et al., 2017). Further analysis of dynamic changes in functional activities of EPCs in the systemic circulation as well as in homing sites of vascular injury appears necessary for providing comprehensive information to completely understand the significance of the EPC response.
Many clinical and experimental studies have analyzed changes in circulating EPCs in correlation with disease progression of sepsis and/or septic infection. Majority of reported clinical investigations have shown an increase in circulating EPC level in patients with sepsis (Becchi et al., 2008; Patschan et al., 2011; Rafat et al., 2007a, 2007b; Siavashi et al., 2017; Zahran et al., 2016). Drop of EPC number in the circulation and impairment of EPC function during sepsis usually occur with worsening of the disease, development of organ failure, and increase in mortality (Cribbs et al., 2012; Kung et al., 2016; Liu et al., 2018; Rafat et al., 2007a, 2007b; van Ierssel et al., 2013; van Ierssel, Jorens, van Craenenbroeck, & Conraads, 2014; Zahran et al., 2016). Similarly, the level of circulating EPCs increases in patients with acute lung injury (Burnham & Moss, 2006). A greater number of circulating EPCs is usually associated with improved outcomes in patients with sepsis and acute lung injury (Burnham et al., 2005; Burnham, Mealer, Gaydos, Majka, & Moss, 2010; Burnham & Moss, 2006; Rafat, Tönshoff, Bierhaus, & Beck, 2013; Siavashi et al., 2017). Studies on both adult and pediatric patients with sepsis have shown that the increase in the level of circulating EPCs is correlated with the survival of patients (Rafat et al., 2013; Zahran et al., 2016). At the present time, however. This correlation in clinical patients remains to be further validated. Other investigators have reported inconsistent observations regarding this direct correlation (Cribbs et al., 2012; Patschan et al., 2011). In experimental studies on a pig model of MODS (mortality rate of 85%) induced by combined insults of hemorrhagic shock, resuscitation, and endotoxemia, the level of circulating EPCs has been observed to increase significantly following the combined challenges but drop sharply prior to the occurrence of MODS (Luo et al., 2009). The migratory and adhesive capacities of circulating EPCs exhibit a similarly biphasic alteration during the development of MODS.
4.2. Alteration of EPCs in their niche environment
In normal adults, the bone marrow houses a major population of EPCs (Shi et al., 2017). The vascular wall also provides niche environment for various types of vascular precursor cells. Currently, however, information regarding the alteration of EPCs in their niches in patients with sepsis and/or septic infection remains scant. In the experimental study on the pig model of MODS mentioned above, early significant increase in the number of EPCs has also been observed in the bone marrow following the combined insults (Luo et al., 2009). The drop of EPC quantity occurs in the bone marrow prior to that in the systemic circulation during the development of MODS. Further, defective activities of migration and adhesion also become apparent earlier in EPCs in the bone marrow than in those in the systemic circulation during the progression of MODS. In our experimental studies, significant increases in the marrow pool of EPCs have been observed in mice at 24 and 48 h following septic infection (Shi, Simms, Ewing, & Zhang, 2018). In addition to increase in number, the proliferative and angiogenic activities of EPCs in the bone marrow are also significantly enhanced during the host response to septic infection. These experimental studies have revealed that the increase in circulating EPCs during the host response to septic challenge is not a simple reflection of releasing these vascular precursor cells from their regular niches into the blood stream. Activation of proliferation as well as programing and/or reprograming functional capacity in EPCs in their niche environments also occur in parallel with the release of them into the systemic circulation.
4.3. EPC activities at homing sites of tissue injury
Circulating EPCs can home to tissue sites of injury where they actively participate in maintaining the integrity of vascular endothelium and/or repairing vascular injury (Cribbs, Martin, & Rojas, 2008; Shi et al., 2017; Sun, Huang, & Sun, 2020). Fully differentiated endothelial cells possess capacity of proliferation. At the site of endothelial injury, adjacent healthy endothelial cells contribute to injury repair via migration and proliferation (Critser, Voytik-Harbin, & Yoder, 2011; Mao, Ye, Liu, Song, & Liu, 2015). In case vascular endothelial injury is in a large scale and/or the damage is severe, the EPC response is evoked to join the effort of host defense. In experimental studies on endotoxemia-induced lung injury, it has been demonstrated that endothelial barrier restoration involves increase in proliferation of both resident endothelial cells and bone marrow derived EPCs (Mao et al., 2015). Resident endothelial cells proliferate in situ on endothelial layer, while bone marrow derived EPCs are engrafted into endothelial layer of lung microvasculature at active phase of barrier repair. During a period of eight weeks, the number of endothelial cells from resident endothelial cell and bone marrow-derived cell origins increase by 22- and 121-fold, respectively, in the lung. Recruited EPCs evidently contribute to re-endothelialization of the damaged vasculature and neovascularization of injured tissue through two major functions: structural incorporation by converting to functional vascular wall cells and signaling regulation via generation of mediators exerting paracrine as well as autocrine effects. For structural incorporation, direct evidence in humans for conversion of bone marrow-derived precursor cells to vascular endothelial cells has been observed in individuals receiving male-to-female hematopoietic stem cell transplantation (Suratt et al., 2003). Tissue specimens from recipients exhibit 35.7–42.3% endothelial chimerism in alveolar microvasculature during 200 to 462 days post transplantation. In chemic mice with reconstituted bone marrow cells expressing green fluorescent protein (GFP), a significant population of bone marrow-derived GFP positive cells have been observed homing to the lung and being integrated into alveolar capillary wall one week following LPS-induced lung injury (Yamada et al., 2004). Immunohistology analysis indicates that these recruited marrow precursor cells differentiate toward pulmonary capillary endothelial cells. Active proliferation and differentiation of recruited EPCs at the homing sites of vascular injury have been repeatedly observed in experimental studies. One estimation suggests that EPCs contribute anywhere between 2 and 25% of endothelial cells in newly formed vessels (Balaji, King, Crombleholme, & Keswani, 2013). Structural incorporation of recruited EPCs to blood vasculature at tissue sites of injury likely involves different patterns and/or processes, including direct repair of damaged endothelium in the existing vascular lumen via proliferation and differentiation into endothelial cells, as well as revascularization in ischemic tissue through vasculogenesis and angiogenesis (Fig. 1).
Fig. 1.

Participation of EPCs in maintaining homeostasis of endothelium and repair of microvascular damage.
EPCs produce angiogenic mediators and release extracellular vesicles (EVs), particularly exosomes, to exert paracrine and autocrine effects. Activated EPCs release angiogenic growth factors and cytokines, including VEGF, hepatocyte growth factor (HGF), insulin-like growth factor-1, platelet-derived growth factor, granulocyte colony–stimulating factor (G-CSF), granulocyte-macrophage colony–stimulating factor (GM-CSF), stromal cell-derived factor-1 (SDF-1, also called CXCL12), TNF-α, interleukin-1β (IL-1β), IL-3, IL-6, and IL-8 (He, Peterson, & Katusic, 2005; Rehman, Li, Orschell, & March, 2003; Rosell et al., 2013; Smadja et al., 2009; Urbich et al., 2005; Vašíček et al., 2021; Yan, Liu, Ding, & Zhang, 2022; Zhang et al., 2011). These soluble factors are well known as key players for promoting angiogenesis, regulating inflammatory reactions, and recruiting vascular precursor cells as well as immune cells to their homing sites for supporting homeostasis of local microvasculature and repairing endothelial damage. Production of EVs by EPCs, serves as an important mechanism for intercellular signaling. EVs is a generic term for lipid bilayer particles released from the cell, which cannot replicate (Salybekov, Kunikeyev, Kobayashi, & Asahara, 2021). These cell-released membrane vesicles carry diverse cargoes including proteins, nucleic acids (RNA species and DNA molecules), and lipids for cell-to-cell communication (Raposo & Stoorvogel, 2013; Xu, Greening, Zhu, Takahashi, & Simpson, 2016). Two major EV subtypes, i.e., exosomes and microvesicles, are well recognized based on their membrane origins. Exosomes are relatively small and homogenous in size (50–120 nm) from the endosomal origin (Xu et al., 2016). Biogenesis of exosomes is via inward budding in endosomes to generate intraluminal vesicles in multivesicular bodies, which subsequently fuse with the plasma membrane to be released into the extracellular environment. Microvesicles are formed/shed by cells through directly budding from the plasma membrane, with a large-scale of variation in sizes (50 to ~1500 nm). Release of EVs by EPCs is regulated by external stimuli, such as inflammatory environment and hypoxia (Zeng, Xu, Liu, & Lu, 2021). Piling investigations have shown that EPC-derived EVs promote endothelial cell survival, migration, proliferation, and tube formation, accelerate re-endothelialization of damaged blood vessels, as well as enhance angiogenesis (Salybekov et al., 2021; Terriaca et al., 2021; Yan et al., 2022; Zeng et al., 2021). An early interesting study has revealed that cultured human EPCs produce microvesicles, which activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA (Deregibus et al., 2007). The subsequent investigations have further explored the mechanistic significance of horizontal transferring genetic materials, such as microRNA (miRNA), mRNA, and proteins, for activation of the angiogenic program in endothelial cells. Exosomes carry a distinctive repertoire of miRNAs. Selective sorting of miRNA into exosomes by cells involves various modes including miRNA motif, sumoylated heterogeneous nuclear ribonucleoprotein-dependent pathway, neural sphingomyelinase 2-dependent pathway, and miRNA induced silencing complex-related pathway (Zeng et al., 2021). Exosomes contain higher proportion of miRNA in comparison to their parent cells and exosome-associated miRNAs are more stable than free miRNAs. Studies have shown that EPC-derived exosomes increase the expression of proangiogenic molecules in recipient endothelial cells, which include eNOS, hypoxia inducible factor 1 alpha, VEGF, VEGFR2, Ang-1, HGF, and platelet-derived growth factor subunit A (Li et al., 2016; Medica et al., 2021). This proangiogenic activity relies on RNA transfer from EVs to recipient endothelial cells (Medica et al., 2021). EPC-derived exosomes express high level of miRNA-221–3p which enhances cell expression of angiogenesis-related factors, including VEGF, CD31 and cell proliferation marker Ki67 (Xu et al., 2020). MiRNA-221–3p may also be involved in the advanced glycation end products-receptor for advanced glycation end products (RAGE) signaling pathway (Xu et al., 2020). EPC-derived EVs has been reported to inhibit endothelial cell reactive oxygen species production, lipid peroxidation, GSH consumption, and cell death via transferring miRNA-199a-3p to bind and inhibit specificity protein 1 activity (Li et al., 2021). Angiogenic miRNA-126 functions to repress negative regulators of the VEGF pathway, including the sprouty-related EVH1 domain-containing protein 1 and phosphoinositol-3 kinase regulatory subunit 2 (Fish et al., 2008). SDF-1α stimulates EPC expression of miRNA-126 and miRNA125b (Fan et al., 2014). Administration of EPCs protects vascular barrier in experimental animals with sepsis. This protective effect of EPCs on maintaining the integrity of microvasculature during sepsis occurs via exosome-mediated transfer of miRNAs, particularly miRNA-126–5p and 3p (Zhou et al., 2018). Exosome miRNA-126–5p and 3p suppress LPS-induced expression of high mobility group box 1 (HMGB1) and VCAM-1 by microvascular endothelial cells. Similarly, EPC-derived exosomes have been shown beneficial for mitigating LPS-induced acute lung injury, which appears partially through miRNA-126–5p and 3p mediated increase in expression of tight junction proteins (Zhou et al., 2018). Integrative analyses of miRNA-mRNA interactions have revealed that EPC-derived exosomes contain highly enriched miRNA-21–5p which specifically suppresses the expression of an angiogenesis inhibitor thrombospondin-1 in the recipient endothelial cells (Hu, Wang, Jiang, Li, & Zhao, 2019). These EPC-exosomes promote re-endothelialization in the early phase after endothelial damage in vivo and enhance proliferation, migration, as well as tube-forming ability of cultured endothelial cells in vitro. Except for delivering nuclear acid molecules, EPC-derived EVs carry proteins for intercellular communication. For example, exosome-mediated transfer of angiotensin-converting enzyme 2 from EPCs has been reported to enhance survival and function of endothelial cells (Wang et al., 1999). In addition to signaling between EPCs and the resident endothelial cells, EV-mediated intercellular communication may serve as an important mechanism underlying interactions of EPCs with other cell types, including fibroblasts and macrophages, for their survival, proliferation, as well as phenotypic transition in the integrated effort of maintaining microvascular homeostasis and injury repair (Ke et al., 2017; Lin et al., 2019; Ma et al., 2021).
4.4. Signaling regulation of the EPC response
Signaling regulation of the EPC response is complex. A typical process of the EPC response encompasses multiple steps/components, including EPC activation/reprograming in their niche environment, release of EPCs from their niches into the circulation, homing of circulating EPCs to tissue sites of vascular injury, EPC participation in maintaining homeostasis of vascular endothelium and repair of injury, as well as resolution of the response. Fig. 2 outlines steps/components in process of the EPC response to sepsis. Numerous mediators with the associated signaling pathways are known being involved in signaling EPC functional activities at each step of their response.
Fig. 2.
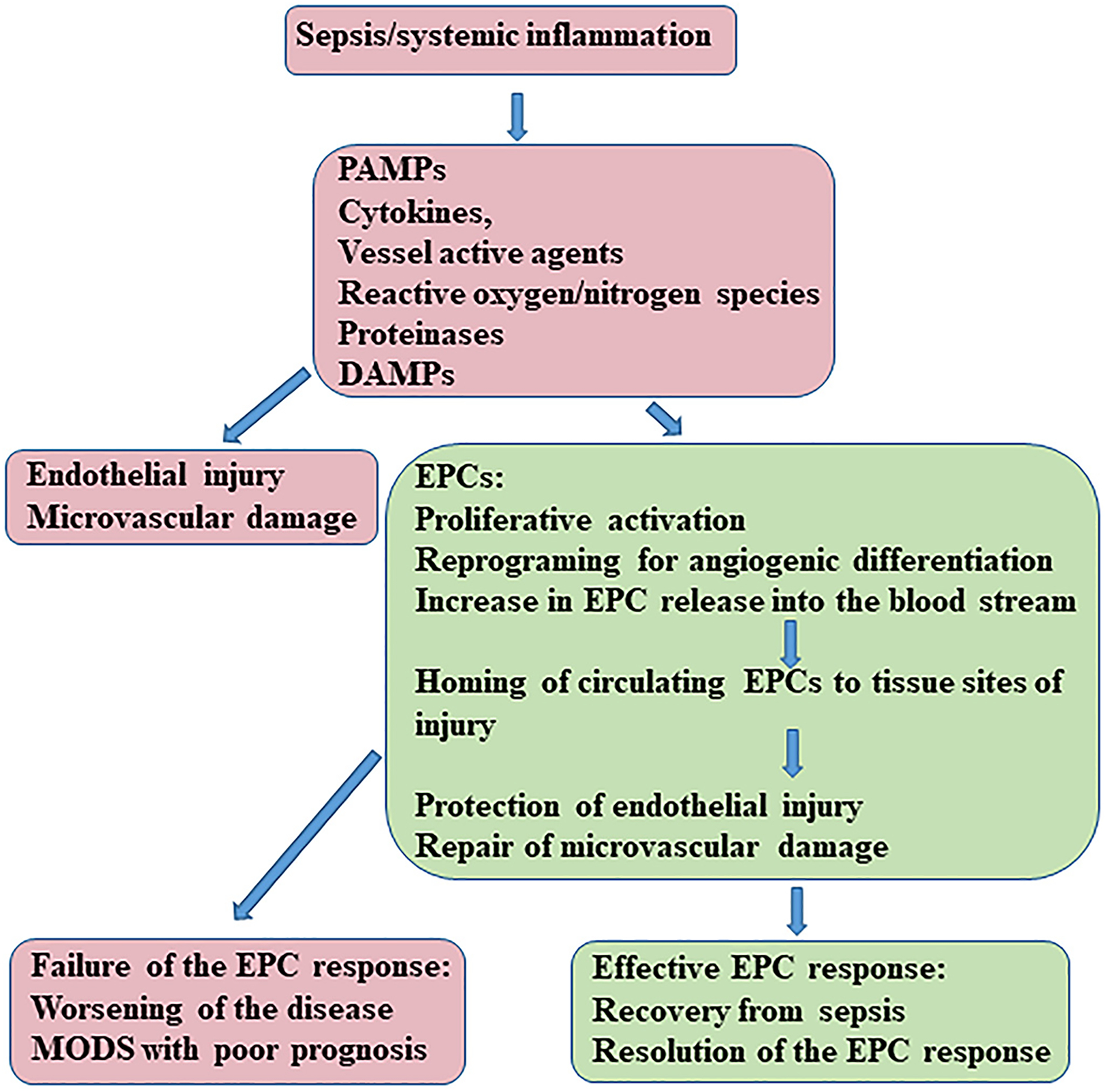
Process of the EPC response to sepsis.
EPC proliferative activation along with reprograming for enhancement of angiogenic activity commonly occurs from the initial stage of the EPC response. EPCs express a profile of pattern recognition receptors, such as TLRs and RAGE (Heidarzadeh et al., 2020; Zhang et al., 2019), as well as cytokine receptors, including VEGFR2, Tie-2, TNFR2, IFNGR, IL-1β Receptor-I (IL-1βRI), and IL-6R among others (Barkestani et al., 2021; Fan et al., 2008; Lee et al., 2018; Rosell et al., 2009; Sasi et al., 2014; Shaw, Basch, & Shamamian, 2004). During septic infection, the cognate ligand mediators including LPS, HMGB1, VEGF, Ang-2, TNF-α, IFN-γ, IL-1, and IL-6 are frequently increased in the systemic circulation. Ligand engagement to their receptors activate EPCs in their niche environment to promote proliferation and angiogenic activity of these vascular precursors (Shi et al., 2017). Since changes in the circulating level of soluble mediators are dynamic with various patterns, it remains to be defined how the initiated signaling pathways are coordinated for the integrated regulation of EPC activation. Further investigations are also required to delineate if signaling mechanisms exist for activation of selective EPC subsets in the niche network in different phases of the EPC response.
Mobilization of EPCs involves cell detachment from the niche stroma and egress into the circulation. As described previously, EPCs and HSCs have a close relationship and these two types of precursors may share similar mechanisms for mobilization, particularly from the bone marrow. Niche structures contain different stromal cell types (Anthony & Link, 2014; Silberstein & Lin, 2013). Several pairs of ligand/receptor molecules, including kit ligand [kitL, also called stem cell factor (SCF)]/c-kit (CD117), SDF-1/CXCR4, and VCAM-1/integrins (a4b1, a4b7, and a9b1), are known to play important roles in bonding (or sticking) stem/progenitor cells to the stroma for their niche retention. (Anthony & Link, 2014; Bonig & Papayannopoulou, 2013; Kunisaki et al., 2013; Lévesque, Hendy, Winkler, Takamatsu, & Simmons, 2003; Morrison & Scadden, 2014; Silberstein & Lin, 2013). Both proteolytic degradation of expressed niche ligand/receptor molecules on cell membrane and down regulation of niche ligand expression by stromal cells facilitate detachment of stem/progenitor cells from the niche network. Matrixmetalloproteinase-9 (MMP-9) secreted by niche stromal cells and hematopoietic cells catalyzes proteolytic cleavage of membrane-bound kitL, c-kit, and SDF-1 (Anthony & Link, 2014; Christopher, Liu, Hilton, Long, & Link, 2009; Hattori, Heissig, & Rafii, 2003; Jin et al., 2008; Petit et al., 2002). Many soluble mediators produced during the inflammatory response, including SDF-1, VEGF, SCF, G-CSF, GM-CSF, macrophage colony stimulating factor, TNF-a, IL-3, IL-6, IL-8, and pattern recognition receptor ligands stimulate cell secretion of MMP-9 (Anthony & Link, 2014; Ratajczak, 2015; Tanaka, Yamane, & Matsuda, 2001). The resulted increase in MMP-9 activity in the niche environment facilitates uncoupling of kitL/c-kit and SDF-1/CXCR4 for EPC mobilization. Neutrophil-derived proteolytic enzymes catalyze cleavage of SDF-1 (by elastase) and VCAM-1 (by elastase and cathepsin G) expressed by stromal cells (Jin et al., 2008; Lévesque, Helwani, & Winkler, 2010). G-CSF stimulates marrow neutrophil release of elastase and cathepsin G for proteolytic degradation of membrane bound SDF-1 and VCAM-1 (Jin et al., 2008; Lévesque et al., 2010). Except for mediating proteolytic degradation, certain soluble mediators, such as G-CSF, Flt3 ligand, and SCF induce down-regulation of SDF-1 expression by marrow niche cells, which facilitate stem/progenitor cell mobilization (Chang, Liou, & Sun, 2021; Mohty & Ho, 2011; Semerad et al., 2005).
Egress of unleashed EPCs from their niche environment into the circulation is guided by chemotactic forces. Soluble chemoattractant molecules, including SDF-1, growth-related oncogene protein (Gro)/IL-8, and sphingosine-1-phosphate (S1P), have been shown to exert chemotactic effects on EPCs during their mobilization. In contrast to what occurs in the niche environment, the concentration of SDF-1 in the systemic circulation is increased during septic infection. Patients with septic shock have shown an elevated blood level of SDF-1, which is accompanied with mobilization of EPCs into the blood stream (Patry et al., 2018; Patschan et al., 2011; Skirecki et al., 2019). With the reduced retentive force of SDF-1 in the niche network, increase in pulling force of SDF-1 in the circulation promotes egress of EPC from their niche compartment. Administration of AMD3100 (a selective CXCR4 antagonist) to experimental animals causes a rapid elevation of SDF-1 in the peripheral circulation (Dar et al., 2011). The resulted reverse of SDF-1 gradient across the bone marrow sinusoidal endothelium facilitates stem/progenitor cell mobilization. Gro and IL-8 are ELR+ CXC chemokines, which are increased in the blood circulation during sepsis and/or septic infection (Harris et al., 2005; Lin, Lin, Hanasawa, Tani, & Kodama, 2000; Manoura et al., 2010; Orman, Ierapetritou, Berthiaume, & Androulakis, 2012; Steinbach et al., 2007). Studies have shown that Gro and IL-8 alone or in combination with other agents facilitate the mobilization of stem/progenitor cells including EPCs from the bone marrow niche (Fukuda, Bian, King, & Pelus, 2007; Hristov, Zernecke, Liehn, & Weber, 2007; Pelus & Fukuda, 2006; Ratajczak & Kim, 2012). Lipid mediator S1P is a strong chemoattractant for migration of EPCs (Wang et al., 2019; Wang, Cai, Li, & Huang, 2015). Many types of tissue cells produce S1P via phosphorylation of sphingosine by sphingosine-kinases (Bode & Gräler, 2012). In normal status, the level of S1P in peripheral blood is much higher than in bone marrow microenvironment (Ishii & Kikuta, 2013). The low level of S1P in tissue space results from rapid degradation and dephosphorylation of S1P molecules by intracellular S1P lyase and S1P phosphatases, respectively. In the circulatory system, vascular endothelial cells, erythrocytes, and platelets continuously secrete large amounts of S1P to maintain a high concentration of S1P in the blood stream (Ratajczak, Borkowska, & Ratajczak, 2013). In the plasma, albumin and high-density lipoprotein are major molecules for carrying S1P, which allow access of S1P to S1P receptors expressed by cells (Bode & Gräler, 2012). The resulted stiff gradient of S1P concentration across the niche barrier provides a pulling force for release of unleashed EPCs from their niche environment into the systemic circulation. At the present time, however, the role of S1P in EPC mobilization during the host response to sepsis remains to be further clarified. Studies have reported that septic infection causes significant decrease in the level of circulating S1P and plasma S1P levels are inversely correlated with the severity of disease (Frej et al., 2016;Winkler et al., 2015; Winkler et al., 2019). S1P receptors bind S1P with high affinity (at low nanomolar range) (Chun, Hla, Lynch, Spiegel, & Moolenaar, 2010). Even if plasma levels of S1P in patients with sepsis reduce to 40–50% of the normal control levels as reported, they remain in the range of 260–580 nM (Winkler et al., 2015; Winkler et al., 2019). Further investigations need to verify if this range of reduced S1P concentration in the blood stream would still be sufficient to provide chemotactic force for mobilizing unlashed EPCs from their niche environment during the host response to sepsis.
Actions of other cellular and molecular mechanisms that have been known to contribute to mobilizing stem/progenitor cells may also be involved in mobilization of EPCs during the host response to septic challenges. These include changes in cell membrane lipid rafts, activation of the complement cascade and fibrinolytic system, interaction of extracellular ATP with the inflammasome system, as well as regulation through the sympathetic nervous system (Alvarez et al., 2013; Lapid et al., 2012; Ratajczak et al., 2019).
EPC homing from the blood circulation to tissue sites of injury involves interplays among multiple sets of chemoattractant molecules with their receptors and adhesion ligands with adhesion receptors. Further, proteases released by EPCs facilitate invasion of these vascular precursors via matrix degradation. Angiogenic activation of the recruited EPCs for participation in maintaining integrity of vascular endothelium as well as in repairing vascular injury via angiogenesis and vasculogenesis encompasses activities of various angiogenic mediators generated during the inflammatory response and interactions of EPCs with other recruited vascular precursor cells, resident tissue cells, as well as extracellular matrix. Signaling mechanisms underlying the regulation of these processes have been extensively discussed (Balaji et al., 2013; Evans, Iruela-Arispe, & Zhao, 2021; Shi et al., 2017). Here, details are referred to reading those recent articles.
5. Therapeutic approaches
Existing efforts in exploring EPC-targeted therapeutic approach for treatment of organ tissue injury during the inflammatory response to septic challenges are largely in the preclinical stage. Three lines of investigations, including transplanting exogenous EPCs, delivering EPC-derived EVs/exosomes, and pharmacological intervention via administering bioactive agents/molecules have been explored.
5.1. Exogenous EPCs
As described previously, the level of EPCs in the systemic circulation increases in response to septic challenges and a higher level of circulating EPCs is usually associated with better outcomes in patients with sepsis and acute lung injury. Studies have been conducted to test if administration of EPCs would be beneficial for hosts with sepsis and/or septic infection. In a murine model of sepsis induced by CLP, intravenous injection of exogenous EPCs has been reported to improve survival of experimental animals (Fan et al., 2014). This improvement is associated with alleviation of vascular leakage in the lung as well as reduction of tissue injury in the liver and kidney. Similarly, intravenous administration of in vitro expanded human EPCs to mice with CLP sepsis has been shown to reduce tissue edema and improve lung mechanics (Güldner et al., 2015). Attenuation of the inflammatory response to sepsis has also been observed in these animals following administration of the expanded EPCs. In a rat model of endotoxemia, intravenous administration of GFP expressing EPCs has been documented that these exogenous EPCs home to peripheral tissue in the body with alleviation of tissue edema (Xu et al., 2015). Systemic administration of gene transfected EPCs with enhanced expression of VEGFA and heme oxygenase-1 to rats has been reported to synergistically promote angiogenesis in tissues with ischemic damage (Long et al., 2013). Although treatment with EPCs may potentially generate some beneficial effects, there are concerns on the clinical application of this approach. These concerns include the consistent cell source with stable phenotype and activity, cellular rejection, infusion toxicity, ectopic tissue formation, as well as possible tumorigenicity (Terriaca et al., 2021).
5.2. EPC-derived EVs/exosomes
In recent years, EVs including exosomes have been demonstrated to play important roles in intercellular signaling. EPC-derived EVs mediate paracrine effects on attenuating the inflammatory response and promoting angiogenesis as well as vasculogenesis. In mice with CLP sepsis, intravenous administration of EPC-derived exosomes has been reported to attenuate the systemic inflammatory response, improve survival, alleviate vascular leakage in the lung and kidney, as well as reduce tissue injury in the liver and kidney (Zhou et al., 2018). These beneficial effects appear involving exosomal miRNA-126 mediated repression of HMGB1 and VCAM-1 expression. Similarly, in mice with CLP sepsis, intratracheal administration of EPC-derived exosomes has been shown to promote VEGF expression, reduce pulmonary vascular leakage, attenuate the inflammatory response, and improve pathological changes in the lung via miRNA-125b-5p-mediated inhibition of topoisomerase II alpha axis (Jiang et al., 2021). Additionally, EPC-derived exosomal miRNA-21–5p has been identified for alleviating acute kidney injury by inhibiting expression of runt-related transcription factor 1 signaling in rats with CLP sepsis (Zhang et al., 2021). Extended investigations in the future will further characterize the targets of treatment with EPC-derived EVs/exosomes and the underlying molecular mechanisms.
5.3. Pharmacological agents
Since certain angiogenic factors are known to exert adverse effects on the integrity of endothelial barrier function, application of these factors directly for treatment of endothelial injury and repair microvascular damage during sepsis and/or septic infection remains facing challenges and restrictions. Regardless, SDF-1 in the blood circulation and tissue sites of injury has been known to promote EPC mobilization and homing (Dar et al., 2011; Shen, Gao, Qian, Sun, & Ge, 2011). SDF-1 also activates EPC activity for angiogenesis and vasculogenesis (Cun et al., 2021; Jiang et al., 2020). Enhancing local expression of SDF-1 promotes EPC engraftment and neovascularization in tissue sites with ischemic injury (Kuliszewski, Kobulnik, Lindner, Stewart, & Leong-Poi, 2011). Systemic administration of SDF-1α -loaded pH-sensitive polymeric micelles in a rat model has been reported to increase expression of SDF-1α in tissues with ischemic injury (Kim et al., 2015). This targeted delivery of SDF-1α enhances angiogenesis without influencing local cell survival or inflammation.
Erythropoietin (EPO) stimulates erythropoiesis. In addition, it promotes EPC mobilization into the systemic circulation with a lower pro-inflammatory profile (Aicher, Zeiher, & Dimmeler, 2005). Patients with sepsis exhibit an elevated serum level of EPO along with the increase in the number of circulating EPCs (Rafat et al., 2007a, 2007b). Administration of EPO has been reported to expend EPC pool in the bone marrow and promote mobilization of EPCs into the blood stream in experimental animals (Heeschen et al., 2003). Similarly, administration of EPO causes an increase in the number of EPCs in peripheral blood in humans (Bahlmann et al., 2004). Endothelial line cells express EPO receptor and respond to stimulation of EPO for enhancing survival, proliferation, differentiation, and migration (Beleslin-Cokic et al., 2004; Urao et al., 2006). Signaling involved in the regulation of these cell activities include activation of JAK2-STAT5 and AKT-eNOS pathways, as well as increase in nitric oxide (NO) production (Schröder et al., 2009; Urao et al., 2006). Further, recent investigations have revealed that the multiple, non-erythropoietic effects of EPO are mediated by a heteromeric receptor (termed tissue protective receptor or TPR) distinctive from the homodimeric receptor responsible for erythropoiesis (Brines, 2010; Hand & Brines, 2011). Due to the lower affinity of TPR for EPO, activation of TPR requires a higher concentration of EPO. In a rat model of hemorrhagic shock, it has been shown that pretreatment with EPO significantly enhances EPC mobilization along with attenuation of tissue injury and organ dysfunction in the kidney, liver, and neuromuscular system following hemorrhagic shock (Nandra et al., 2013). Similarly, in a murine model of endotoxemia-induced acute kidney injury, treatment with EPO has been reported to ameliorate microvascular injury in the kidney (Stoyanoff et al., 2018). Based on the potentially beneficial effect of EPO in promoting EPC mobilization and protecting microvascular endothelium, investigators have postulated that EPC application may be considered as an adjuvant therapeutic strategy for management of acute lung injury/ARDS in patients with COVID-19 (Sahebnasagh et al., 2020). Regardless, EPO at higher doses has adverse consequences including stimulation of hematopoietic and pro-coagulant pathways. To overcome these obstacles, efforts have been devoted to modifying the EPO molecule for it to optimally interact with TPR. Studies have shown that these compounds appear safe and effective for protecting organ tissues from injury (Brines, 2010; Erbayraktar et al., 2009; Hand & Brines, 2011; Patel et al., 2011).
Angiotensin-converting enzyme inhibitors (ACEI) inhibit biosynthesis of angiotensin II, which helps relaxing blood vessels and reducing blood pressure. Both experimental studies and clinical observations have demonstrated that ACEI promotes mobilization of EPCs into the peripheral circulation and exerts pleiotropic effects on the blood vasculature (Lee & Poh, 2014; Sun et al., 2020). In a murine model of transaortic constriction, ramipril administration has been reported to promote EPC mobilization, enhance neoangiogenesis, and increase the quantity of bone marrow derived endothelial cells in the myocardium (Müller et al., 2009). In addition, this ramipril treatment prevents apoptosis of endothelial cells. Similarly, treatment with enalapril has been shown to significantly increase the level of circulating EPCs and substantially enhance the contribution of bone marrow derived EPCs to post-ischemia neovascularization in experimental animals (Wang et al., 2006). A prospective, multicenter, randomized, double-blind, placebo-controlled trial of 96 recipients for heart transplantation has reported that ramipril treatment stabilizes levels of EPCs in the circulation and improves microvascular function, which is associated with improved long-term survival (Fearon et al., 2017). Mechanisms underlying beneficial effects of ACEI on EPC-targeted treatment appear involving multiple lines of signaling regulation. Angiotensin II induces EPC senescence and dysfunction via gp91phox-mediated increase in oxidative stress (Imanishi, Hano, & Nishio, 2005). Inhibition of angiotensin II generation by ACEI contributes to offsetting the negative effect on EPCs exerted by angiotensin II. Further, administration of enalapril has been reported to increase SDF-1 concentration in the blood stream while reduce SDF-1 level in the bone marrow via regulating the CD26/dipeptidylpeptidase IV system, which facilitates EPC mobilization into the circulation (Wang et al., 2006). Perindopril treatment has also been shown to increase the expression of SDF-1/CXCR4 in myocardium with ischemic injury as well as to activate VEGF/AKT/eNOS signaling and upregulate expression of MMP-9 in the bone marrow (Sun et al., 2016).
Certain angiotensin receptor blockers (ARB) used for treatment of hypertension have been reported to promote EPC mobilization and modulate EPC activities. Losartan administration in rats increases the number of EPCs in the circulation (Yao et al., 2007). Mobilized EPCs in rats with losartan treatment exhibit enhanced activities of colony formation and migration. Similarly, irbesartan administration has been observed to increase EPC mobilization and inhibit endothelial injury in a hamster model (Georgescu, Alexandru, Nemecz, Titorencu, & Popov, 2013). In vitro culture studies have revealed that telmisartan increases colony forming activity of EPCs from healthy human volunteers (Honda et al., 2009). Knowledge about mechanisms underlying ARB-induced EPC mobilization and enhancement of EPC function remains incomplete. Evidence suggests that signaling regulations may involve inhibition of NAD(P)H oxidases and activation of the PPAR-γ-I3K/AKT pathway (Honda et al., 2009; Lee & Poh, 2014).
Statins [also known as 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors) promote EPC mobilization and regulate EPC functional activities in addition to its therapeutic effects on reducing blood cholesterol level via inhibiting biosynthesis of cholesterol. Administration of simvastatin in mice has been shown to promote mobilization of EPCs from the bone marrow into the blood stream and recruitment of EPCs in neovascularization (Llevadot et al., 2001). Simvastatin rapidly activates AKT protein kinase in EPCs, enhancing EPC survival, proliferation, and migration. Overexpression of dominant negative AKT blocks these functional activities of EPC. In hypercholesterolemic patients, rosuvastatin therapy has been reported to significantly increase the number of circulating EPCs (Pirro et al., 2009). A prospective clinical study has reported that in patients treated for acute myocardial infarction, those discharged on a high-intensity statin therapy maintain circulating levels of EPCs and present higher EPC levels than patients not on an intensive statin therapy at 3 months of follow-up (António et al., 2014). Similarly, a randomized, controlled trial has shown that preoperative short-term use of atorvastatin increases the level of circulating EPCs with less myocardial damage and high-sensitivity C-reactive protein levels post-operatively (Baran et al., 2012). A systematic review of randomized trials confirms that statin therapy mobilizes EPCs into the circulation in humans (Hibbert et al., 2013). Mechanistically, statins activate the PI3K/AKT pathway, upregulate expression of VEGF and VEGFR, as well as stimulate NO biosynthesis (Cantoni et al., 2012; Mangialardi et al., 2011), which facilitate mobilization of EPCs and re-endothelization of injured microvasculature. Studies have also shown that statins upregulate CXCR4 expression by EPCs to promote EPC homing to tissue sites of neovascularization (António et al., 2014; Chiang et al., 2015). Gamma-tocotrienol (GT3), a vitamin E isoform, is a potent inhibitor of HMG-CoA reductase (Berbée et al., 2009). In a murine model, administration of GT3 has been shown to significantly increase EPCs and hematopoietic progenitors in the blood stream (Ray et al., 2013). Administration of GT3 ameliorates radiation-induced vascular injury and improves survival in mice (Berbée et al., 2009; Berbée et al., 2011). Mechanisms underlying GT3-induced mobilization EPCs remain incompletely understood. GT3 stimulates G-CSF production in vivo (Kulkarni et al., 2012; Kulkarni, Singh, Ghosh, Posarac, & Singh, 2013), which may facilitate EPC mobilization (Kulkarni et al., 2013; Singh et al., 2014).
In supporting the beneficial effect of using anti-hypertension and anti-hyperlipidemia drugs for EPC-targeted treatment of sepsis and/or septic infection, several meta-analyses of septic patients treated in different hospital settings have demonstrated that pre-admission use of ACEI, ARB, or statins is associated with a significantly reduced risk of in-hospital mortality (Hasegawa et al., 2022; Hsieh, How, Hsieh, & Chen, 2020; Lee, Suh, Jang, & Lee, 2021; Mortensen et al., 2007)
6. Prospective remarks
Sepsis with systemic inflammation causes a widespread injury to endothelium in the blood vasculature, which evokes the EPC response. Proliferative activation and angiogenic reprograming of EPCs occur rapidly in their niche environment. Increase in EPC release into the systemic circulation and homing of these released vascular precursor cells to the site of tissue injury contribute to protection of endothelial homeostasis and repair of endothelial damage in microvasculature. Therefore, the EPC response constitutes an important component of host defense against septic challenges. Future investigations on the heterogenicity of EPCs, the dynamic process of the EPC response, and the underlying signaling regulations will provide deeper insights into the functional significance of EPCs in the host defense system. Identifying molecular targets in the EPC response will facilitate developing new therapeutic approaches for effective prevention and treatment of vascular injury as well as vital organ system failure caused by sepsis and the accompanied systemic inflammation.
Acknowledgments
This work is supported by NIH grant R01GM132449 and Watanakunakorn Chair Endowment Fund. The funding sources have not been involved in study design, in the collection, analysis, and interpretation of data, in the writing, and in the decision to submit the paper for publication.
Abbreviations:
- ACEI
angiotensin-converting enzyme inhibitors
- Ang-1
angiopoietin-1
- Ang-2
angiopoietin-2
- Ang-3
angiopoietin-3
- Ang-4
angiopoietin-4
- AIF
apoptosis-inducing factor
- ARB
angiotensin receptor blocker
- ARDS
adult respiratory distress syndrome
- CFUs
colony-forming units
- c-kit
stem cell factor receptor
- CLP
cecal ligation and puncture
- COVID-19
novel coronavirus disease 2019
- CXCR4
CXC receptor 4
- ECFCs
endothelial colony-forming cells
- eNOS
endothelial nitric oxide synthase
- EPCs
endothelial progenitor cells
- ERK
extracellular signal-regulated kinases
- EVs
extracellular vesicles
- FADD
fas-associated death domain protein
- FAK
focal adhesion kinase
- Flk1
fms-like tyrosine kinase-1
- G-CSF
granulocyte colony–stimulating factor
- GFP
green fluorescent protein
- GM-CSF
granulocyte-macrophage colony–stimulating factor
- gp130
glycoprotein 130 kDa
- Gro
growth-related oncogene protein
- HGF
hepatocyte growth factor
- HMGB1
high mobility group box 1
- HMG-CoA
3-hydroxy-3-methylglutaryl-coenzyme A
- HSCs
hematopoietic stem cells
- ICAM-1
intercellular adhesion molecule-1
- ICU
intensive care unit
- IFN-γ
interferon-gamma
- IFNGR1
interferon-gamma receptor 1
- IFNGR2
interferon-gamma receptor 2
- IL-1β
interleukin-1β
- IL-3
interleukin-3
- IL-6
interleukin-6
- IL-8
interleukin-8
- JAK1
Janus family of protein tyrosine kinase 1
- JAK2
Janus family of protein tyrosine kinase 2
- JNK
c-Jun N-terminal kinase
- KDR
kinase insert domain receptor
- kitL
kit ligand
- Lin
lineage
- LPS
lipopolysaccharides
- MAPK
mitogen-activated protein kinase
- MCP-1
monocyte chemoattractant protein-1
- MEKK3
mitogen-activated protein kinase kinase kinase 3
- mIL-6R
interleukin-6 transmembrane receptor
- miRNA
microRNA
- MLC
myosin light chain
- MMP-9
matrixmetalloproteinase-9
- MODS
multiple organ dysfunction syndrome
- NO
nitric oxide
- PI3K
phosphatidylinositol 3-kinase
- PKB or AKT
serine/threonine protein kinase B
- PKC
protein kinase C
- PLC
phospholipase C
- RAGE
advanced glycation end products
- Ras
rat sarcoma protooncogene
- ROCK
Rho-associated protein kinase
- S1P
sphingosine-1-phosphate
- Sca-1
stem cell antigen-1
- SCF
stem cell factor
- SDF-1
stromal cell-derived factor-1
- sIL-6R
interleukin-6 soluble receptor
- SOCS3
suppressor of cytokine signaling-3
- STAT1
signal transducers and activators of transcription 1
- STAT5
signal transducers and activators of transcription 5
- TACE
TNF-α converting enzyme
- Tie
tyrosine kinase with immunoglobulin and EGF homology domains
- TLR2
toll-like receptor 2
- TLR4
toll-like receptor 4
- TNF-α
tumor necrosis factor-α
- TNFR1
TNF receptor 1
- TNFR2
TNF receptor 2
- TPR
tissue protective receptor
- VCAM-1
vascular cell adhesion molecule-1
- VE-cadherin
vascular endothelial cadherin
- VEGF
vascular endothelial growth factor
- VEGFR1
vascular endothelial growth factor receptor 1
- VEGFR2
vascular endothelial growth factor receptor 2
- VEGFR3
vascular endothelial growth factor receptor 3
- VEPTP
Vascular endothelial protein tyrosine phosphatase
Footnotes
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Data availability
The authors are unable or have chosen not to specify which data has been used.
References
- Abadie Y, Bregeon F, Papazian L, Lange F, Chailley-Heu B, Thomas P, et al. (2005). Decreased VEGF concentration in lung tissue and vascular injury during ARDS. The European Respiratory Journal 25, 139–146. [DOI] [PubMed] [Google Scholar]
- Aepfelbacher M, & Essler M (2001). Disturbance of endothelial barrier function by bacterial toxins and atherogenic mediators: A role for Rho/Rho kinase. Cellular Microbiology 3, 649–658. [DOI] [PubMed] [Google Scholar]
- Aicher A, Zeiher AM, & Dimmeler S (2005). Mobilizing endothelial progenitor cells. Hypertension 45, 321–325. [DOI] [PubMed] [Google Scholar]
- Akwii RG, Sajib MS, Zahra FT, & Mikelis CM (2019). Role of Angiopoietin-2 in vascular physiology and pathophysiology. Cells 8, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez P, Carrillo E, Vélez C, Hita-Contreras F, Martínez-Amat A, Rodríguez-Serrano F, et al. (2013). Regulatory systems in bone marrow for hematopoietic stem/progenitor cells mobilization and homing. BioMed Research International 2013, Article 312656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves BE, Montalvao SA, Aranha FJ, Lorand-Metze I, De Souza CA, Annichino Bizzacchi JM, et al. (2011). Time-course of sFlt-1 and VEGF-A release in neutropenic patients with sepsis and septic shock: A prospective study. Journal of Translational Medicine 9, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amraei R, & Rahimi N (2020). COVID-19, Renin-angiotensin system and endothelial dysfunction. Cells 9, 1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony BA, & Link DC (2014). Regulation of hematopoietic stem cells by bone marrow stromal cells. Trends in Immunology 35, 32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- António N, Fernandes R, Soares A, Soares F, Lopes A, Carvalheiro T, et al. (2014). Impact of prior chronic statin therapy and high-intensity statin therapy at discharge on circulating endothelial progenitor cell levels in patients with acute myocardial infarction: A prospective observational study. European Journal of Clinical Pharmacology 70, 1181–1193. [DOI] [PubMed] [Google Scholar]
- Aquino JB, Sierra R, & Montaldo LA (2021). Diverse cellular origins of adult blood vascular endothelial cells. Developmental Biology 477, 117–132. [DOI] [PubMed] [Google Scholar]
- Aramoto H, Breslin JW, Pappas PJ, Hobson RW 2nd, & Durán WN (2004). Vascular endothelial growth factor stimulates differential signaling pathways in in vivo microcirculation. American Journal of Physiology. Heart and Circulatory Physiology 287, H1590–H1598. [DOI] [PubMed] [Google Scholar]
- Azamfirei L, Gurzu S, Solomon R, Copotoiu R, Copotoiu S, Jung I, et al. (2010). Vascular endothelial growth factor: A possible mediator of endothelial activation in acute respiratory distress syndrome. Minerva Anestesiologica 76, 609–616. [PubMed] [Google Scholar]
- Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN, et al. (2012). Suppression of cytokine signaling by SOCS3: Characterization of the mode of inhibition and the basis of its specificity. Immunity 36, 239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachelier K, Bergholz C, & Friedrich EB (2020). Differentiation potential and functional properties of a CD34-CD133+ subpopulation of endothelial progenitor cells. Molecular Medicine Reports 21, 501–507. [DOI] [PubMed] [Google Scholar]
- Bahlmann GH, De Groot K, Spandau J, Landry AL, Hertel B, Duckert T, et al. (2004). Erythropoietin regulates endothelial progenitor cells. Blood 103, 921–926. [DOI] [PubMed] [Google Scholar]
- Bai C, Hou L, Zhang M, Pu Y, Guan W, & Ma Y (2012). Characterization of vascular endothelial progenitor cells from chicken bone marrow. BMC Veterinary Research 8, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey AS, Jiang S, Afentoulis M, Baumann CI, Schroeder DA, Olson SB, et al. (2004). Transplanted adult hematopoietic stems cells differentiate into functional endothelial cells. Blood 103, 13–19. [DOI] [PubMed] [Google Scholar]
- Balaji S, King A, Crombleholme TM, & Keswani SG (2013). The role of endothelial progenitor cells in postnatal vasculogenesis: Implications for therapeutic neovascularization and wound healing. Advances in Wound Care 2, 283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannerman DD, & Goldblum SE (2003). Mechanisms of bacterial lipopolysaccharide induced endothelial apoptosis. American Journal of Physiology. Lung Cellular and Molecular Physiology 284, L899–L914. [DOI] [PubMed] [Google Scholar]
- Baran C, Durdu S, Dalva K, Zaim C, Dogan A, Ocakoglu G, et al. (2012). Effects of preoperative short term use of atorvastatin on endothelial progenitor cells after coronary surgery: A randomized, controlled trial. Stem Cell Reviews and Reports 8, 963–971. [DOI] [PubMed] [Google Scholar]
- Barkestani MN, Shamdani S, Afshar Bakshloo M, Arouche N, Bambai B, Uzan G, et al. (2021). TNFα priming through its interaction with TNFR2 enhances endothelial progenitor cell immunosuppressive effect: New hope for their widespread clinical application. Cell Communication and Signaling: CCS 19, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barratt S, Medford AR, & Millar AB (2014). Vascular endothelial growth factor in acute lung injury and acute respiratory distress syndrome. Respiration; International Review of Thoracic Diseases 87, 329–342. [DOI] [PubMed] [Google Scholar]
- Bates DO (2010). Vascular endothelial growth factors and vascular permeability. Cardiovascular Research 87, 262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becchi C, Pillozzi S, Fabbri LP, Al Malyan M, Cacciapuoti C, Della Bella C, et al. (2008). The increase of endothelial progenitor cells in the peripheral blood: A new parameter for detecting onset and severity of sepsis. International Journal of Immunopathology and Pharmacology 21, 697–705. [DOI] [PubMed] [Google Scholar]
- Becker PM, Verin AD, Booth MA, Liu F, Birukova A, & Garcia JG (2001). Differential regulation of diverse physiological responses to VEGF in pulmonary endothelial cells. American Journal of Physiology. Lung Cellular and Molecular Physiology 281, L1500–L1511. [DOI] [PubMed] [Google Scholar]
- Beleslin-Cokic BB, Cokic VP, Yu X, Weksler BB, Schechter AN, & Noguchi CT (2004). Erythropoietin and hypoxia stimulate erythropoietin receptor and nitric oxide production by endothelial cells. Blood 104, 2073–2080. [DOI] [PubMed] [Google Scholar]
- Berbée M, Fu Q, Boerma M, Wang J, Kumar KS, & Hauer-Jensen M (2009). gamma-Tocotrienol ameliorates intestinal radiation injury and reduces vascular oxidative stress after total-body irradiation by an HMG-CoA reductase-dependent mechanism. Radiation Research 171, 596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbée M, Fu Q, Garg S, Kulkarni S, Kumar KS, & Hauer-Jensen M (2011). Pentoxifylline enhances the radioprotective properties of γ-tocotrienol: Differential effects on the hematopoietic, gastrointestinal and vascular systems. Radiation Research 175, 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berube BJ, & Bubeck Wardenburg J (2013). Staphylococcus aureus α-toxin: Nearly a century of intrigue. Toxins 5, 1140–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari V, Choo-Wing R, Chapoval SP, Lee CG, Tang C, Kim YK, et al. (2006). Essential role of nitric oxide in VEGF-induced, asthma-like angiogenic, inflammatory, mucus, and physiologic responses in the lung. Proceedings of the National Academy of Sciences of the United States of America 103, 11021–11026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode C, & Gräler MH (2012). Immune regulation by sphingosine 1-phosphate and its receptors. Archivum Immunologiae et Therapiae Experimentalis 60, 3–12. [DOI] [PubMed] [Google Scholar]
- Bonig H, & Papayannopoulou T (2013). Hematopoietic stem cell mobilization: updated conceptual renditions. Leukemia 27, 24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonney S, Seitz S, Ryan CA, Jones KL, Clarke P, Tyler KL, et al. (2019). Gamma interferon alters junctional integrity via rho kinase, resulting in blood-brain barrier leakage in experimental viral encephalitis. mBio 10 e01675–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brines M (2010). The therapeutic potential of erythropoiesis-stimulating agents for tissue protection: A tale of two receptors. Blood Purification 29, 86–92. [DOI] [PubMed] [Google Scholar]
- Burnham E, & Moss M (2006). Progenitor cells in acute lung injury. Minerva Anestesiologica 72, 369–374. [PubMed] [Google Scholar]
- Burnham EL, Mealer M, Gaydos J, Majka S, & Moss M (2010). Acute lung injury but not sepsis is associated with increased colony formation by peripheral blood mono-nuclear cells. American Journal of Respiratory Cell and Molecular Biology 43, 326–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnham EL, Taylor WR, Quyyumi AA, Rojas M, Brigham KL, & Moss M (2005). Increased circulating endothelial progenitor cells are associated with survival in acute lung injury. American Journal of Respiratory and Critical Care Medicine 172, 854–860. [DOI] [PubMed] [Google Scholar]
- Cantoni S, Cavallini C, Bianchi F, Bonavita F, Vaccari V, Olivi E, et al. (2012). Rosuvastatin elicits KDR-dependent vasculogenic response of human placental stem cells through PI3K/AKT pathway. Pharmacological Research 65, 275–284. [DOI] [PubMed] [Google Scholar]
- Chang HH, Liou YS, & Sun DS (2021). Hematopoietic stem cell mobilization. Tzu Chi Medical Journal 34, 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang K, Cheng W, Shih C, Lin Y, Tsao N, Kao Y, et al. (2015). Statins, HMG-CoA reductase inhibitors, improve neovascularization by increasing the expression density of CXCR4 in endothelial progenitor cells. PLoS One 10, Article e0136405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra H, Hung MK, Kwong DL, Zhang CF, & Pow E (2018). Insights into endothelial progenitor cells: Origin, classification, potentials, and prospects. Stem Cells International 2018, 9847015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopher MJ, Liu F, Hilton MJ, Long F, & Link DC (2009). Suppression of CXCL12 production by bone marrow osteoblasts is a common and critical pathway for cytokine-induced mobilization. Blood 114, 1331–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J, Hla T, Lynch KR, Spiegel S, & Moolenaar WH (2010). International union of basic and clinical pharmacology. LXXVIII. Lysophospholipid receptor nomenclature. Pharmacological Reviews 62, 579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couto M, Coelho-Santos V, Santos L, Fontes-Ribeiro C, Silva AP, & Gomes C (2019). The interplay between glioblastoma and microglia cells leads to endothelial cell monolayer dysfunction via the interleukin-6-induced JAK2/STAT3 pathway. Journal of Cellular Physiology 234, 19750–19760. [DOI] [PubMed] [Google Scholar]
- Cribbs SK, Martin GS, & Rojas M (2008). Monitoring of endothelial dysfunction in critically ill patients: The role of endothelial progenitor cells. Current Opinion in Critical Care 14, 354–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribbs SK, Sutcliffe DJ, Taylor WR, Rojas M, Easley KA, Tang L, et al. (2012). Circulating endothelial progenitor cells inversely associate with organ dysfunction in sepsis. Intensive Care Medicine 38, 429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critser PJ, Voytik-Harbin SL, & Yoder MC (2011). Isolating and defining cells to engineer human blood vessels. Cell Proliferation 44(Suppl. 1), 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouser ED, & Matthay MA (2017). Endothelial damage during septic shock: Significance and implications for future therapies. Chest 152, 1–3. [DOI] [PubMed] [Google Scholar]
- Cui Y, Liu L, Xiao Y, Li X, Zhang J, Xie X, et al. (2020). N-acetylcysteine differentially regulates the populations of bone marrow and circulating endothelial progenitor cells in mice with limb ischemia. European Journal of Pharmacology 881, Article 173233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cun Y, Diao B, Zhang Z, Wang G, Yu J, Ma L, et al. (2021). Role of the stromal cell derived factor-1 in the biological functions of endothelial progenitor cells and its underlying mechanisms. Experimental and Therapeutic Medicine 21, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar A, Schajnovitz A, Lapid K, Kalinkovich A, Itkin T, Ludin A, et al. (2011). Rapid mobilization of hematopoietic progenitors by AMD3100 and catecholamines is mediated by CXCR4-dependent SDF-1 release from bone marrow stromal cells. Leukemia 25, 1286–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauphinee SM, & Karsan A (2006). Lipopolysaccharide signaling in endothelial cells. Laboratory Investigation; A Journal of Technical Methods and Pathology 86, 9–22. [DOI] [PubMed] [Google Scholar]
- David S, Kümpers P, van Slyke P, & Parikh SM (2013). Mending leaky blood vessels: The angiopoietin-Tie2 pathway in sepsis. The Journal of Pharmacology and Experimental Therapeutics 345, 2–6. [DOI] [PubMed] [Google Scholar]
- David S, Mukherjee A, Ghosh CC, Yano M, Khankin EV, Wenger JB, et al. (2012). Angiopoietin-2 may contribute to multiple organ dysfunction and death in sepsis*. Critical Care Medicine 40, 3034–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deregibus MC, Cantaluppi V, Calogero R, Lo Iacono M, Tetta C, Biancone L, et al. (2007). Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood 110, 2440–2448. [DOI] [PubMed] [Google Scholar]
- Dight J, Zhao J, Styke C, Khosrotehrani K, & Patel J (2022). Resident vascular endothelial progenitor definition and function: The age of reckoning. Angiogenesis 25, 15–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distler JH, Hirth A, Kurowska-Stolarska M, Gay RE, Gay S, & Distler O (2003). Angiogenic and angiostatic factors in the molecular control of angiogenesis. The Quarterly Journal of Nuclear Medicine 47, 149–161. [PubMed] [Google Scholar]
- Dumnicka P, Maduzia D, Ceranowicz P, Olszanecki R, Drożdż R, & Kuśnierz-Cabala B (2017). The interplay between inflammation, coagulation and endothelial injury in the early phase of acute pancreatitis: Clinical implications. International Journal of Molecular Sciences 18, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisa-Beygi S, & Wen XY (2015). Could pharmacological curtailment of the RhoA/Rho kinase pathway reverse the endothelial barrier dysfunction associated with Ebola virus infection? Antiviral Research 114, 53–56. [DOI] [PubMed] [Google Scholar]
- Erbayraktar Z, Erbayraktar S, Yilmaz O, Cerami A, Coleman T, & Brines M (2009). Nonerythropoietic tissue protective compounds are highly effective facilitators of wound healing. Molecular Medicine 15, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser S, Wolburg K, Wolburg H, Breier G, Kurzchalia T, & Risau W (1998). Vascular endothelial growth factor induces endothelial fenestrations in vitro. The Journal of Cell Biology 140, 947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CE, Iruela-Arispe ML, & Zhao YY (2021). Mechanisms of endothelial regeneration and vascular repair and their application to regenerative medicine. The American Journal of Pathology 191, 52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CE, & Zhao YY (2017). Impact of thrombosis on pulmonary endothelial injury and repair following sepsis. American Journal of Physiology. Lung Cellular and Molecular Physiology 312, L441–L451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagiani E, & Christofori G (2013). Angiopoietins in angiogenesis. Cancer Letters 328, 18–26. [DOI] [PubMed] [Google Scholar]
- Fan H, Goodwin AJ, Chang E, Zingarelli B, Borg K, Guan S, et al. (2014). Endothelial progenitor cells and a stromal cell-derived factor-1α analogue synergistically improve survival in sepsis. American Journal of Respiratory and Critical Care Medicine 189, 1509–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Ye J, Shen F, Zhu Y, Yeghiazarians Y, Zhu W, et al. (2008). Interleukin-6 stimulates circulating blood-derived endothelial progenitor cell angiogenesis in vitro. Journal of Cerebral Blood Flow and Metabolism 28, 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon EF, Okada K, Kobashigawa JA, Kobayashi Y, Luikart H, Sean Sana S, et al. (2017). Angiotensin-converting enzyme inhibition early after heart transplantation. Journal of the American College of Cardiology 69, 2832–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felcht M, Luck R, Schering A, Seidel P, Srivastava K, Hu J, et al. (2012). Angiopoietin-2 differentially regulates angiogenesis through TIE2 and integrin signaling. The Journal of Clinical Investigation 122, 1991–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N (2009). Vascular endothelial growth factor. Arteriosclerosis, Thrombosis, and Vascular Biology 29, 789–791. [DOI] [PubMed] [Google Scholar]
- Ferro T, Neumann P, Gertzberg N, Clements R, & Johnson A (2000). Protein kinase C alpha mediates endothelial barrier dysfunction induced by TNF-alpha. American Journal of Physiology. Lung Cellular and Molecular Physiology 278, L1107–L1117. [DOI] [PubMed] [Google Scholar]
- Fiedler U, Scharpfenecker M, Koidl S, Hegen A, Grunow V, Schmidt JM, et al. (2004). The Tie-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood 103, 4150–4156. [DOI] [PubMed] [Google Scholar]
- Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD, et al. (2008). miR 126 regulates angiogenic signaling and vascular integrity. Developmental Cell 15, 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier M, van Leeuwen HJ, van Kessel KP, Kimpen JL, Hoepelman AI, & Geelen SP (2005). Plasma vascular endothelial growth factor in severe sepsis. Shock 23, 35–38. [DOI] [PubMed] [Google Scholar]
- Frej C, Linder A, Happonen KE, Taylor FB, Lupu F, & Dahlbäck B (2016). Sphingo-sine 1-phosphate and its carrier apolipoprotein M in human sepsis and in Escherichia coli sepsis in baboons. Journal of Cellular and Molecular Medicine 20, 1170–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda S, Bian H, King AG, & Pelus LM (2007). The chemokine GRObeta mobilizes early hematopoietic stem cells characterized by enhanced homing and engraftment. Blood 110, 860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, et al. (2001). Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proceedings of the National Academy of Sciences of the United States of America 98, 2604–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganter MT, Cohen MJ, Brohi K, Chesebro BB, Staudenmayer KL, Rahn P, et al. (2008). Angiopoietin-2, marker and mediator of endothelial activation with prognostic significance early after trauma? Annals of Surgery 247, 320–326. [DOI] [PubMed] [Google Scholar]
- Gao X, Chen W, Liang Z, & Chen L (2011). Autotransplantation of circulating endothelial progenitor cells protects against lipopolysaccharide-induced acute lung injury in rabbit. International Immunopharmacology 11, 1584–1590. [DOI] [PubMed] [Google Scholar]
- Gavard J (2013). Endothelial permeability and VE-cadherin: A wacky comradeship. Cell Adhesion & Migration 7, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavard J, & Gutkind JS (2006). VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nature Cell Biology 8, 1223–1234. [DOI] [PubMed] [Google Scholar]
- Gavard J, Patel V, & Gutkind JS (2008). Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Developmental Cell 14, 25–36. [DOI] [PubMed] [Google Scholar]
- Georgescu A, Alexandru N, Nemecz M, Titorencu I, & Popov D (2013). Irbesartan administration therapeutically influences circulating endothelial progenitor cell and microparticle mobilization by involvement of pro-inflammatory cytokines. European Journal of Pharmacology 711, 27–35. [DOI] [PubMed] [Google Scholar]
- Gill M, Dias S, Hattori K, Rivera ML, Hicklin D, Witte L, et al. (2001). Vascular trauma induces rapid but transient mobilization of VEGFR2(+)AC133(+) endothelial precursor cells. Circulation Research 88, 167–174. [DOI] [PubMed] [Google Scholar]
- Go E, & Yoder MC (2021). Identification of endothelial cells and their progenitors. Methods in Molecular Biology 2206, 27–37. [DOI] [PubMed] [Google Scholar]
- Godzich M, Hodnett M, Frank JA, Su G, Pespeni M, Angel A, et al. (2006). Activation of the stress protein response prevents the development of pulmonary edema by inhibiting VEGF cell signaling in a model of lung ischemia-reperfusion injury in rats. FASEB Journal 20, 1519–1521. [DOI] [PubMed] [Google Scholar]
- Gofton TE, & Young GB (2012). Sepsis-associated encephalopathy. Nature reviews. Neurology 8, 557–566. [DOI] [PubMed] [Google Scholar]
- Grubb SE, Murdoch C, Sudbery PE, Saville SP, Lopez-Ribot JL, & Thornhill MH (2008). Candida albicans-endothelial cell interactions: A key step in the pathogenesis of systemic candidiasis. Infection and Immunity 76, 4370–4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güldner A, Maron-Gutierrez T, Abreu SC, Xisto DG, Senegaglia AC, Barcelos PR, et al. (2015). Expanded endothelial progenitor cells mitigate lung injury in septic mice. Stem Cell Research & Therapy 6, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakanpaa L, Kiss EA, Jacquemet G, Miinalainen I, Lerche M, Guzmán C, et al. (2018). Targeting β1-integrin inhibits vascular leakage in endotoxemia. Proceedings of the National Academy of Sciences of the United States of America 115, E6467–E6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakanpaa L, Sipila T, Leppanen VM, Gautam P, Nurmi H, Jacquemet G, et al. (2015). Endothelial destabilization by angiopoietin-2 via integrin β1 activation. Nature Communications 6, 5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Lee SJ, Kim KE, Lee HS, Oh N, Park I, et al. (2016). Amelioration of sepsis by TIE2 activation-induced vascular protection. Science Translational Medicine 8, 335ra55. [DOI] [PubMed] [Google Scholar]
- Hand CC, & Brines M (2011). Promises and pitfalls in erythopoietin-mediated tissue protection: Are nonerythropoietic derivatives a way forward? Journal of Investigative Medicine 59, 1073–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris MC, D’Angio CT, Gallagher PR, Kaufman D, Evans J, & Kilpatrick L (2005). Cytokine elaboration in critically ill infants with bacterial sepsis, necrotizing entercolitis, or sepsis syndrome: Correlation with clinical parameters of inflammation and mortality. The Journal of Pediatrics 147, 462–468. [DOI] [PubMed] [Google Scholar]
- Hasegawa D, Lee YI, Prasitlumkum N, Chopra L, Nishida K, Smith NK, et al. (2022). Premorbid angiotensin converting enzyme inhibitors or angiotensin II receptor blockers in patients with sepsis. The American Journal of Emergency Medicine 62, 69–77. [DOI] [PubMed] [Google Scholar]
- Hattori K, Heissig B, & Rafii S (2003). The regulation of hematopoietic stem cell and progenitor mobilization by chemokine SDF-1. Leukemia & Lymphoma 44, 575–582. [DOI] [PubMed] [Google Scholar]
- Hawiger J (2018). Heartfelt sepsis: Microvascular injury due to genomic storm. Kardiologia Polska 76, 1203–1216. [DOI] [PubMed] [Google Scholar]
- Hawiger J, & Musser JM (2011). How to approach genome wars in sepsis? Critical Care 15, 1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawiger J, Veach RA, & Zienkiewicz J (2015). New paradigms in sepsis: From prevention to protection of failing microcirculation. Journal of Thrombosis and Haemostasis: JTH 13, 1743–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He T, Peterson TE, & Katusic ZS (2005). Paracrine mitogenic effect of human endothelial progenitor cells: Role of interleukin-8. American Journal of Physiology. Heart and Circulatory Physiology 289, H968–H972. [DOI] [PubMed] [Google Scholar]
- Heeschen C, Aicher A, Lehmann R, Fichtlscherer S, Vasa M, Urbich C, et al. (2003). Erythropoietin is a potent physiologic stimulus for endothelial progenitor cell mobilization. Blood 102, 1340–1346. [DOI] [PubMed] [Google Scholar]
- Heidarzadeh M, Roodbari F, Hassanpour M, Ahmadi M, Saberianpour S, & Rahbarghazi R (2020). Toll-like receptor bioactivity in endothelial progenitor cells. Cell and Tissue Research 379, 223–230. [DOI] [PubMed] [Google Scholar]
- van der Heijden M, van Nieuw Amerongen GP, Koolwijk P, van Hinsbergh VW, & Groeneveld AB (2008). Angiopoietin-2, permeability oedema, occurrence and severity of ALI/ARDS in septic and non-septic critically ill patients. Thorax 63, 903–909. [DOI] [PubMed] [Google Scholar]
- Hibbert B, Simard T, Ramirez FD, Pourdjabbar A, Raizman JE, Maze R, et al. (2013). The effect of statins on circulating endothelial progenitor cells in humans: A systematic review. Journal of Cardiovascular Pharmacology 62, 491–496. [DOI] [PubMed] [Google Scholar]
- Higgins SJ, De Ceunynck K, Kellum JA, Chen X, Gu X, Chaudhry SA, et al. (2018). Tie2 protects the vasculature against thrombus formation in systemic inflammation. The Journal of Clinical Investigation 128, 1471–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda A, Matsuura K, Fukushima N, Tsurumi Y, Kasanuki H, & Hagiwara N (2009). Telmisartan induces proliferation of human endothelial progenitor cells via PPARgamma-dependent PI3K/Akt pathway. Atherosclerosis 205, 376–384. [DOI] [PubMed] [Google Scholar]
- Hristov M, Zernecke A, Liehn EA, & Weber C (2007). Regulation of endothelial progenitor cell homing after arterial injury. Thrombosis and Haemostasis 98, 274–277. [PubMed] [Google Scholar]
- Hsieh M, How C, Hsieh VC, & Chen P (2020). Preadmission antihypertensive drug use and sepsis outcome: Impact of angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs). Shock 53, 407–415. [DOI] [PubMed] [Google Scholar]
- Hsu HC, Ema H, Osawa M, Nakamura Y, Suda T, & Nakauchi H (2000). Hematopoietic stem cells express Tie-2 receptor in the murine fetal liver. Blood 96, 3757–3762. [PubMed] [Google Scholar]
- Hu H, Wang B, Jiang C, Li R, & Zhao J (2019). Endothelial progenitor cell-derived exosomes facilitate vascular endothelial cell repair through shuttling miR-21–5p to modulate Thrombospondin-1 expression. Clinical Science 133, 1629–1644. [DOI] [PubMed] [Google Scholar]
- van Ierssel SH, Jorens PG, van Craenenbroeck EM, & Conraads VM (2014). The endothelium, a protagonist in the pathophysiology of critical illness: Focus on cellular markers. BioMed Research International 2014, Article 985813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ierssel SH, van Craenenbroeck EM, Hoymans VY, Vrints CJ, Conraads VM, & Jorens PG (2013). Endothelium dependent vasomotion and in vitro markers of endothelial repair in patients with severe sepsis: An observational study. PLoS One 8, Article e69499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imanishi T, Hano T, & Nishio I (2005). Angiotensin II accelerates endothelial progenitor cell senescence through induction of oxidative stress. Journal of Hypertension 23, 97–104. [DOI] [PubMed] [Google Scholar]
- Ince C, Mayeux PR, Nguyen T, Gomez H, Kellum JA, Ospina-Tascón GA, et al. (2016). The endothelium in sepsis. Shock 45, 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii M, & Kikuta J (2013). Sphingosine-1-phosphate signaling controlling osteoclasts and bone homeostasis. Biochimica et Biophysica Acta 1831, 223–227. [DOI] [PubMed] [Google Scholar]
- Jacobi J (2022). The pathophysiology of sepsis-2021 update: Part 1, immunology and coagulopathy leading to endothelial injury. American Journal of Health-System Pharmacy 79, 329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Li R, Ma X, Hu H, Guo J, & Zhao J (2020). AMD3100 and SDF-1 regulate cellular functions of endothelial progenitor cells and accelerate endothelial regeneration in a rat carotid artery injury model. Molecular Medicine Reports 22, 3201–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Ni J, Shen G, Xia Z, Zhang L, Xia S, et al. (2021). Upregulation of endothelial cell-derived exosomal microRNA-125b-5p protects from sepsisi-nduced acute lung injury by inhibiting topoisomerase II alpha. Inflammation Research 70, 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin F, Zhai Q, Qiu L, Meng H, Zou D, Wang Y, et al. (2008). Degradation of BM SDF-1 by MMP-9: The role in G-CSF-induced hematopoietic stem/progenitor cell mobilization. Bone Marrow Transplantation 42, 581–588. [DOI] [PubMed] [Google Scholar]
- Jonkam CC, Lange M, Traber DL, Maybauer DM, Maybauer MO, Bansal K, et al. (2009). Cardiovascular collapse and vascular permeability changes in an ovine model of methicillin-resistant Staphylococcus aureus sepsis. Shock 32, 621–625. [DOI] [PubMed] [Google Scholar]
- Jordan SC, Choi J, Kim I, Wu G, Toyoda M, Shin B, et al. (2017). Interleukin-6, a cytokine critical to mediation of inflammation, autoimmunity and allograft rejection: Therapeutic implications of IL-6 receptor blockade. Transplantation 101, 32–44. [DOI] [PubMed] [Google Scholar]
- Kaner RJ, & Crystal RG (2001). Compartmentalization of vascular endothelial growth factor to the epithelial surface of the human lung. Molecular Medicine 7, 240–246. [PMC free article] [PubMed] [Google Scholar]
- Kaner RJ, Ladetto JV, Singh R, Fukuda N, Matthay MA, & Crystal RG (2000). Lung overexpression of the vascular endothelial growth factor gene induces pulmonary edema. American Journal of Respiratory Cell and Molecular Biology 22, 657–664. [DOI] [PubMed] [Google Scholar]
- Karki P, Ke Y, Zhang CO, Li Y, Tian Y, Son S, et al. (2021). SOCS3-microtubule interaction via CLIP-170 and CLASP2 is critical for modulation of endothelial inflammation and lung injury. The Journal of Biological Chemistry 296, Article 100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmpaliotis D, Kosmidou I, Ingenito EP, Hong K, Malhotra A, Sunday ME, et al. (2002). Angiogenic growth factors in the pathophysiology of a murine model of acute lung injury. American Journal of Physiology. Lung Cellular and Molecular Physiology 283, L585–L595. [DOI] [PubMed] [Google Scholar]
- Kaur S, Bansal Y, Kumar R, & Bansal G (2020). A panoramic review of IL-6: Structure, pathophysiological roles and inhibitors. Bioorganic & Medicinal Chemistry 28, Article 115327. [DOI] [PubMed] [Google Scholar]
- Ke X, Yang D, Liang J, Wang X, Wu S, Wang X, et al. (2017). Human endothelial progenitor cell-derived exosomes increase proliferation and angiogenesis in cardiac fibroblasts by promoting the mesenchymal-endothelial transition and reducing high mobility group box 1 protein B1 expression. DNA and Cell Biology 36, 1018–1028. [DOI] [PubMed] [Google Scholar]
- Khakpour S, Wilhelmsen K, & Hellman J (2015). Vascular endothelial cell toll-like receptor pathways in sepsis. Innate Immunity 21, 827–846. [DOI] [PubMed] [Google Scholar]
- Kim DH, Seo YK, Thambi T, Moon GJ, Son JP, Li G, et al. (2015). Enhancing neurogenesis and angiogenesis with target delivery of stromal cell derived factor-1α using a dual ionic pH-sensitive copolymer. Biomaterials 61, 115–125. [DOI] [PubMed] [Google Scholar]
- Kim KT, Choi HH, Steinmetz MO, Maco B, Kammerer RA, Ahn SY, et al. (2005). Oligomerization and multimerization are critical for angiopoietin-1 to bind and phosphorylate Tie2. The Journal of Biological Chemistry 280, 20126–20131. [DOI] [PubMed] [Google Scholar]
- Koh H, Tasaka S, Hasegawa N, Asano K, Kotani T, Morisaki H, et al. (2008). Vascular endothelial growth factor in epithelial lining fluid of patients with acute respiratory distress syndrome. Respirology 13, 281–284. [DOI] [PubMed] [Google Scholar]
- Kuliszewski MA, Kobulnik J, Lindner JR, Stewart DJ, & Leong-Poi H (2011). Vascular gene transfer of SDF-1 promotes endothelial progenitor cell engraftment and enhances angiogenesis in ischemic muscle. Molecular Therapy: The Journal of the American Society of Gene Therapy 19, 895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni S, Singh PK, Ghosh SP, Posarac A, & Singh VK (2013). Granulocyte colony-stimulating factor antibody abrogates radioprotective efficacy of gamma-tocotrienol, a promising radiation countermeasure. Cytokine 62, 278–285. [DOI] [PubMed] [Google Scholar]
- Kulkarni SS, Cary LH, Gambles K, Hauer-Jensen, Kumar MKS, & Ghosh SP (2012). Gamma-tocotrienol, a radiation prophylaxis agent, induces high levels of granulocyte colony-stimulating factor. International Immunopharmacology 14, 495–503. [DOI] [PubMed] [Google Scholar]
- Kung CT, Su CM, Chen CT, Cheng HH, Chang MW, Hung CW, et al. (2016). Circulating endothelial progenitor cells may predict outcomes in adult patients with severe sepsis in the emergency department. Clinica Chimica Acta; International Journal of Clinical Chemistry 455, 1–6. [DOI] [PubMed] [Google Scholar]
- Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, et al. (2013). Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal BK, Varma S, Pappas PJ, Hobson RW 2nd, & Durán WN (2001). VEGF increases permeability of the endothelial cell monolayer by activation of PKB/akt, endothelial nitric-oxide synthase, and MAP kinase pathways. Microvascular Research 62, 252–262. [DOI] [PubMed] [Google Scholar]
- Lapid K, Glait-Santar C, Gur-Cohen S, Canaani J, Kollet O, & Lapidot T (2012). Egress and mobilization of hematopoietic stem and progenitor cells: A dynamic multi-facet process. StemBook. Cambridge, MA: Harvard Stem Cell Institute. [PubMed] [Google Scholar]
- Lechleitner S, Gille J, Johnson DR, & Petzelbauer P (1998). Interferon enhances tumor necrosis factor-induced vascular cell adhesion molecule 1 (CD106) expression in human endothelial cells by an interferon-related factor 1-dependent pathway. The Journal of Experimental Medicine 187, 2023–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Chang HK, Son YS, Lee D, Kwon SM, Kim PH, et al. (2018). IFN-γ enhances the wound healing effect of late EPCs (LEPCs) via BST2-mediated adhesion to endothelial cells. FEBS Letters 592, 1705–1715. [DOI] [PubMed] [Google Scholar]
- Lee HW, Suh JK, Jang E, & Lee S (2021). Effect of angiotensin converting enzyme inhibitor and angiotensin II receptor blocker on the patients with sepsis. The Korean Journal of Internal Medicine 36, 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PSS, & Poh KK (2014). Endothelial progenitor cells in cardiovascular diseases. World Journal of Stem Cells 6, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévesque JP, Helwani FM, & Winkler IG (2010). The endosteal “osteoblastic” niche and its role in hematopoietic stem cell homing and mobilization. Leukemia 24, 1979–1992. [DOI] [PubMed] [Google Scholar]
- Lévesque JP, Hendy J, Winkler IG, Takamatsu Y, & Simmons PJ (2003). Granulocyte colony-stimulating factor induces the release in the bone marrow of proteases that cleave c-KIT receptor (CD117) from the surface of hematopoietic progenitor cells. Experimental Hematology 31, 109–117. [DOI] [PubMed] [Google Scholar]
- Li L, Wang H, Zhang J, Chen X, Zhang Z, & Li Q (2021). Effect of endothelial progenitor cell-derived extracellular vesicles on endothelial cell ferroptosis and atherosclerotic vascular endothelial injury. Cell Death Discovery 7, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Chen C, Wei L, Li Q, Niu X, Xu Y, Wang Y, & Zhao J (2016). Exosomes derived from endothelial progenitor cells attenuate vascular repair and accelerate reendothelialization by enhancing endothelial function. Cytotherapy 18, 253–262. [DOI] [PubMed] [Google Scholar]
- Lin F, Zeng Z, Song Y, Li L, Wu Z, Zhang X, Li Z, Ke X, & Hu X (2019). YBX-1 mediated sorting of miR-133 into hypoxia/reoxygenation-induced EPC-derived exosomes to increase fibroblast angiogenesis and MEndoT. Stem Cell Research & Therapy 10, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KJ, Lin J, Hanasawa K, Tani T, & Kodama M (2000). Interleukin-8 as a predictor of the severity of bacteremia and infectious disease. Shock 14, 95–100. [DOI] [PubMed] [Google Scholar]
- Liu J, Zou GJ, Yang L, Rong S, Li BQ, Tong ZH, et al. (2018). Early prediction of persistent organ failure by circulating endothelial progenitor cells in patients with acute pancreatitis. Shock 50, 265–272. [DOI] [PubMed] [Google Scholar]
- Llevadot J, Murasawa S, Kureishi Y, Uchida S, Masuda H, Kawamoto H, et al. (2001). HMG-CoA reductase inhibitor mobilizes bone marrow–derived endothelial progenitor cells. The Journal of Clinical Investigation 108, 399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J, Wang S, Zhang Y, Liu X, Zhang H, & Wang S (2013). The therapeutic effect of vascular endothelial growth factor gene- or heme oxygenase-1 gene-modified endothelial progenitor cells on neovascularization of rat hindlimb ischemia model. Journal of Vascular Surgery 58, 756–765 e2. [DOI] [PubMed] [Google Scholar]
- Luo TH, Wang Y, Lu ZM, Zhou H, Xue XC, Bi JW, et al. (2009). The change and effect of endothelial progenitor cells in pig with multiple organ dysfunction syndromes. Critical Care 13, R118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Zhang W, Cui B, Gao J, Liu Q, Yao M, et al. (2021). Functional delivery of lncRNA TUG1 by endothelial progenitor cells derived extracellular vesicles confers anti-inflammatory macrophage polarization in sepsis via impairing miR-9–5p-targeted SIRT1 inhibition. Cell Death & Disease 12, 1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmud-Al-Rafat A, Majumder A, Taufiqur Rahman KM, Mahedi Hasan AM, Didarul Islam KM, Taylor-Robinson AW, et al. (2019). Decoding the enigma of antiviral crisis: Does one target molecule regulate all? Cytokine 115, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangialardi G, Monopoli A, Ongini E, Spinetti G, Fortunato O, Emanueli C, et al. (2011). Nitric oxide-donating statin improves multiple functions of circulating angiogenic cells. British Journal of Pharmacology 164, 570–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoura A, Gourgiotis D, Galanakis E, Matalliotakis E, Hatzidaki E, Korakaki E, et al. (2010). Circulating concentrations of α- and β-chemokines in neonatal sepsis. International Journal of Infectious Diseases 14, e806–e809. [DOI] [PubMed] [Google Scholar]
- Mao SZ, Ye X, Liu G, Song D, & Liu SF (2015). Resident endothelial cells and endothelial progenitor cells restore endothelial barrier function after inflammatory lung injury. Arteriosclerosis, Thrombosis, and Vascular Biology 35, 1635–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Master Z, Jones N, Tran J, Jones J, Kerbel RS, & Dumont DJ (2001). Dok-R plays a pivotal role in angiopoietin-1-dependent cell migration through recruitment and activation of Pak. The EMBO Journal 20, 5919–5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N (2016). Alert cell strategy in SIRS-induced vasculitis: Sepsis and endothelial cells. Journal of Intensive Care 4, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthay MA, Ware LB, & Zimmerman GA (2012). The acute respiratory distress syndrome. The Journal of Clinical Investigation 122, 2731–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie JA, & Ridley AJ (2007). Roles of Rho/ROCK and MLCK in TNF-alpha-induced changes in endothelial morphology and permeability. Journal of Cellular Physiology 213, 221–228. [DOI] [PubMed] [Google Scholar]
- Medford AR, & Millar AB (2006). Vascular endothelial growth factor (VEGF) in acute lung injury (ALI) and acute respiratory distress syndrome (ARDS): Paradox or paradigm? Thorax 61, 621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medica D, Franzin R, Stasi A, Castellano G, Migliori M, Panichi V, et al. (2021). Extracellular vesicles derived from endothelial progenitor cells protect human glomerular endothelial cells and podocytes from complement- and cytokine-mediated injury. Cells 10, 1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina RJ, Barber CL, Sabatier F, Dignat-George F, Melero-Martin JM, Khosrotehrani K, et al. (2017). Endothelial progenitors: A consensus statement on nomenclature. Stem Cells Translational Medicine 6, 1316–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohty M, & Ho AD (2011). In and out of the niche: Perspectives in mobilization of hematopoietic stem cells. Experimental Hematology 39, 723–729. [DOI] [PubMed] [Google Scholar]
- Montgomery A, Tam F, Gursche C, Cheneval C, Besler K, Enns W, et al. (2021). Overlapping and distinct biological effects of IL-6 classic and trans-signaling in vascular endothelial cells. American Journal of Physiology. Cell Physiology 320, C554–C565. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, & Scadden DT (2014). The bone marrow niche for haematopoietic stem cells. Nature 505, 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen WM, Restrepo MI, Copeland LA, Augh JA, Anzueto A, Cornell JE, et al. (2007). Impact of previous statin and angiotensin II receptor blocker use on mortality in patients hospitalized with sepsis. Pharmacotherapy 27 1619–1026. [DOI] [PubMed] [Google Scholar]
- Müller P, Kazakov A, Jagoda P, Semenov A, Böhm M, & Laufs U (2009). ACE inhibition promotes upregulation of endothelial progenitor cells and neoangiogenesis in cardiac pressure overload. Cardiovascular Research 83, 106–114. [DOI] [PubMed] [Google Scholar]
- Mura M, dos Santos CC, Stewart D, & Liu M (2004). Vascular endothelial growth factor and related molecules in acute lung injury. Journal of Applied Physiology 97, 1605–1617. [DOI] [PubMed] [Google Scholar]
- Nägele MP, Haubner B, Tanner FC, Ruschitzka F, & Flammer AJ (2020). Endothelial dysfunction in COVID-19: Current findings and therapeutic implications. Atherosclerosis 314, 58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandra KK, Collino M, Rogazzo M, Fantozzi R, Patel NSA, & Thiemermann C (2013). Pharmacological preconditioning with erythropoietin attenuates the organ injury and dysfunction induced in a rat model of hemorrhagic shock. Disease Models & Mechanisms 6, 701–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolini G, Forini F, Kusmic C, Iervasi G, & Balzan S (2019). Angiopoietin 2 signal complexity in cardiovascular disease and cancer. Life Sciences 239, Article 117080. [DOI] [PubMed] [Google Scholar]
- Nicosia RF, Ligresti G, Caporarello N, Akilesh S, & Ribatti D (2021). COVID-19 vasculopathy: Mounting evidence for an indirect mechanism of endothelial injury. The American Journal of Pathology 191, 1374–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orman MA, Ierapetritou MG, Berthiaume F, & Androulakis IP (2012). Long-term dynamic profiling of inflammatory mediators in double-hit burn and sepsis animal models. Cytokine 58, 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai MH, Shih YM, Shih JM, & Yeh CL (2016). Glutamine administration modulates endothelial progenitor cell and lung injury in septic mice. Shock 46, 587–592. [DOI] [PubMed] [Google Scholar]
- Pang T, Chen W, Lu ZM, Luo TH, Zhou H, Xue XC, et al. (2013). Endothelial progenitor cells are influenced by serum of patients with systemic inflammatory response syndrome or multiple organ dysfunction. European Review for Medical and Pharmacological Sciences 17, 3169–3177. [PubMed] [Google Scholar]
- Parikh SM (2013). Dysregulation of the angiopoietin-Tie-2 axis in sepsis and ARDS. Virulence 4, 517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschalaki KE, & Randi AM (2018). Recent advances in endothelial colony forming cells toward their use in clinical translation. Frontiers in Medicine 5, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel NSA, Nandra KK, Brines M, Collino M, Wong WF, Kapoor A, et al. (2011). A nonerythropoietic peptide that mimics the 3D structure of erythropoietin reduces organ injury/dysfunction and inflammation in experimental hemorrhagic shock. Molecular Medicine 17, 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patry C, Stamm D, Betzen C, Tönshoff B, Yard BA, Beck GC, et al. (2018). CXCR-4 expression by circulating endothelial progenitor cells and SDF-1 serum levels are elevated in septic patients. Journal of Inflammation 15, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patschan SA, Patschan D, Temme J, Korsten P, Wessels JT, Koziolek M, et al. (2011). Endothelial progenitor cells (EPC) in sepsis with acute renal dysfunction (ARD). Critical Care 15, R94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peichev M, Naiyer AJ, Pereira D, Zhu Z, Lane WJ, Williams M, et al. (2000). Expression of VEGFR-2 and AC133 by circulating human CD34(+) cells identifies a population of functional endothelial precursors. Blood 95, 952–958. [PubMed] [Google Scholar]
- Pelus LM, & Fukuda S (2006). Peripheral blood stem cell mobilization: The CXCR2 ligand GRObeta rapidly mobilizes hematopoietic stem cells with enhanced engraftment properties. Experimental Hematology 34, 1010–1020. [DOI] [PubMed] [Google Scholar]
- Peppler WT, Townsend LK, & Wright DC (2020). Recent advances in the role of interleukin-6 in health and disease. Current Opinion in Pharmacology 52, 47–51. [DOI] [PubMed] [Google Scholar]
- Petit I, Szyper-Kravitz M, Nagler A, Lahav M, Peled A, Habler L, et al. (2002). G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nature Immunology 3, 687–694. [DOI] [PubMed] [Google Scholar]
- Petrache I, Crow MT, Neuss M, & Garcia JG (2003). Central involvement of rho family GTPases in TNF-alpha-mediated bovine pulmonary endothelial cell apoptosis. Biochemical and Biophysical Research Communications 306, 244–249. [DOI] [PubMed] [Google Scholar]
- Pickkers P, Sprong T, Eijk LV, Hoeven HV, Smits P, & Deuren MV (2005). Vascular endothelial growth factor is increased during the first 48 hours of human septic shock and correlates with vascular permeability. Shock 24, 508–512. [DOI] [PubMed] [Google Scholar]
- Pirro M, Schillaci G, Romagno PF, Mannarino MR, Bagaglia F, Razzi R, et al. (2009). Influence of short-term rosuvastatin therapy on endothelial progenitor cells and endothelial function. Journal of Cardiovascular Pharmacology and Therapeutics 14, 14–21. [DOI] [PubMed] [Google Scholar]
- Pober JS, & Min W (2006). Endothelial cell dysfunction, injury and death. Handbook of Experimental Pharmacology 176(Pt 2), 135–156. [DOI] [PubMed] [Google Scholar]
- Pocock TM, & Bates DO (2001). In vivo mechanisms of vascular endothelial growth factor mediated increased hydraulic conductivity of Rana capillaries. The Journal of Physiology 534, 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafat N, Hanusch C, Brinkkoetter PT, Schulte J, Brade J, Zijlstra JG, et al. (2007a). Increased circulating endothelial progenitor cells in septic patients: Correlation with survival. Critical Care Medicine 35, 1677–1684. [DOI] [PubMed] [Google Scholar]
- Rafat N, Hanusch C, Brinkkoetter PT, Schulte J, Brade J, Zijlstra JG, et al. (2007b). Increased circulating endothelial progenitor cells in septic patients: Correlation with survival. Critical Care Medicine 35, 1677–1684. [DOI] [PubMed] [Google Scholar]
- Rafat N, Tönshoff B, Bierhaus A, & Beck GC (2013). Endothelial progenitor cells in regeneration after acute lung injury: Do they play a role? American Journal of Respiratory Cell and Molecular Biology 48, 399–405. [DOI] [PubMed] [Google Scholar]
- Rafii S, & Lyden D (2003). Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nature Medicine 9, 702–712. [DOI] [PubMed] [Google Scholar]
- Raposo G, & Stoorvogel W (2013). Extracellular vesicles: Exosomes, microvesicles, and friends. The Journal of Cell Biology 200, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajczak MZ (2015). A novel view of the adult bone marrow stem cell hierarchy and stem cell trafficking. Leukemia 29, 776–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajczak MZ, Adamiak M, Thapa A, Bujko K, Brzezniakiewicz-Janus K, & Lenkiewicz AM (2019). NLRP3 inflammasome couples purinergic signaling with activation of the complement cascade for the optimal release of cells from bone marrow. Leukemia 33, 815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajczak MZ, Borkowska S, & Ratajczak J (2013). An emerging link in stem cell mobilization between activation of the complement cascade and the chemotactic gradient of sphingosine-1-phosphate. Prostaglandins & Other Lipid Mediators 104–105, 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajczak MZ, & Kim C (2012). The use of chemokine receptor agonists in stem cell mobilization. Expert Opinion on Biological Therapy 12, 287–297. [DOI] [PubMed] [Google Scholar]
- Ray S, Kulkarni SS, Chakraborty K, Pessu R, Hauer-Jensen M, Kumar MKS, et al. (2013). Mobilization of progenitor cells into peripheral blood by gamma-tocotrienol: A promising radiation countermeasure. International Immunopharmacology 15, 557–564. [DOI] [PubMed] [Google Scholar]
- Rehman J, Li J, Orschell CM, & March KL (2003). Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation 107, 1164–1169. [DOI] [PubMed] [Google Scholar]
- Remy KE, Qiu P, Li Y, Cui X, & Eichacker PQ (2013). B. anthracis associated cardiovascular dysfunction and shock: The potential contribution of both non-toxin and toxin components. BMC Medicine 11, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts WG, & Palade GE (1995). Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. Journal of Cell Science 108, 2369–2379. [DOI] [PubMed] [Google Scholar]
- Rose-John S (2020). Interleukin-6 signalling in health and disease. F1000Research 9 F1000 Faculty Rev-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosell A, Arai K, Lok J, He T, Guo S, Navarro M, et al. (2009). Interleukin-1beta augments angiogenic responses of murine endothelial progenitor cells in vitro. Journal of Cerebral Blood Flow and Metabolism 29, 933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosell A, Morancho A, Navarro-Sobrino M, Martínez-Saez E, Hernández-Guillamon M, Lope-Piedrafita S, et al. (2013). Factors secreted by endothelial progenitor cells enhance neurorepair responses after cerebral ischemia in mice. PLoS One 8, Article e73244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahebnasagh A, Mojtahedzadeh M, Najmeddin F, Najafi A, Safdari M, Ghaleno HR, et al. (2020). A perspective on erythropoietin as a potential adjuvant therapy for acute lung injury/acute respiratory distress syndrome in patients with COVID-19. Archives of Medical Research 51, 631–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MM, Seeto WJ, DeWitt BB, Hashimi SA, Schwartz DD, Lipke EA, et al. (2015). Characterization of endothelial colony-forming cells from peripheral blood samples of adult horses. American Journal of Veterinary Research 76, 174–187. [DOI] [PubMed] [Google Scholar]
- Salybekov AA, Kunikeyev AD, Kobayashi S, & Asahara T (2021). Latest advances in endothelial progenitor cell-derived extracellular vesicles translation to the clinic. Frontiers in Cardiovascular Medicine 8, Article 734562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasi SP, Song J, Park D, Enderling H, McDonald JT, Gee H, et al. (2014). TNF TNFR2/p75 signaling inhibits early and increases delayed nontargeted effects in bone marrow-derived endothelial progenitor cells. The Journal of Biological Chemistry 289, 14178–14193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatteman GC, & Awad O (2004). Hemangioblasts, angioblasts, and adult endothelial cell progenitors. The Anatomical Record. Part A, Discoveries in Molecular, Cellular, and Evolutionary Biology 276, 13–21. [DOI] [PubMed] [Google Scholar]
- Schlichting DE, Waxman AB, O’Brien LA, Wang T, Naum CC, Rubeiz GJ, et al. (2011). Circulating endothelial and endothelial progenitor cells in patients with severe sepsis. Microvascular Research 81, 216–221. [DOI] [PubMed] [Google Scholar]
- Schmitz J, Weissenbach M, Haan S, Heinrich PC, & Schaper F (2000). SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP2 recruitment site of gp130. The Journal of Biological Chemistry 275, 12848–12856. [DOI] [PubMed] [Google Scholar]
- Schröder K, Kohnen A, Aicher A, Liehn EA, Büchse T, Stein S, et al. (2009). NADPH oxidase Nox2 is required for hypoxia-induced mobilization of endothelial progenitor cells. Circulation Research 105, 537–544. [DOI] [PubMed] [Google Scholar]
- Semerad CL, Christopher MJ, Liu F, Short B, Simmons PJ, Winkler I, et al. (2005). G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood 106, 3020–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro NI, Yano K, Okada H, Fischer C, Howell M, Spokes KC, et al. (2008). A prospective, observational study of soluble FLT-1 and vascular endothelial growth factor in sepsis. Shock 29, 452–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw JP, Basch R, & Shamamian P (2004). Hematopoietic stem cells and endothelial cell precursors express Tie-2, CD31 and CD45. Blood Cells, Molecules & Diseases 32, 168–175. [DOI] [PubMed] [Google Scholar]
- Shen L, Gao Y, Qian J, Sun A, & Ge J (2011). A novel mechanism for endothelial progenitor cells homing: The SDF-1/CXCR4-Rac pathway may regulate endothelial progenitor cells homing through cellular polarization. Medical Hypotheses 76, 256–258. [DOI] [PubMed] [Google Scholar]
- Shi X, Simms KJ, Ewing TJ, & Zhang P (2018). Sca-1 signaling in the marrow endothelial progenitor cell response to septic infection. Shock 49(supplement 1), 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Zhang W, Yin L, Chilian WM, Krieger J, & Zhang P (2017). Vascular precursor cells in tissue injury repair. Translational Research: The Journal of Laboratory and Clinical Medicine 184, 77–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M (2006). Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis. Journal of Biochemistry and Molecular Biology 39, 469–478. [DOI] [PubMed] [Google Scholar]
- Shibuya M (2013). Vascular endothelial growth factor and its receptor system: Physiological functions in angiogenesis and pathological roles in various diseases. Journal of Biochemistry 153, 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siavashi V, Asadian S, Taheri-Asl M, Keshavarz S, Zamani-Ahmadmahmudi M, & Nassiri SM (2017). Endothelial progenitor cell mobilization in preterm infants with sepsis is associated with improved survival. Journal of Cellular Biochemistry 118, 3299–3307. [DOI] [PubMed] [Google Scholar]
- Siflinger-Birnboim A, & Johnson A (2003). Protein kinase C modulates pulmonary endothelial permeability: A paradigm for acute lung injury. American Journal of Physiology. Lung Cellular and Molecular Physiology 284, L435–L451. [DOI] [PubMed] [Google Scholar]
- Silberstein LE, & Lin CP (2013). A new image of the hematopoietic stem cell vascular niche. Cell Stem Cell 13, 514–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H, Tahir TA, Alawo DO, Issa E, & Brindle NP (2011). Molecular control of angiopoietin signalling. Biochemical Society Transactions 39, 1592–1596. [DOI] [PubMed] [Google Scholar]
- Singh VK, Wise SY, Fatanmi OO, Scott J, Romaine PLP, Newman VL, et al. (2014). Progenitors mobilized by gamma-tocotrienol as an effective radiation countermeasure. PLoS One 9, Article e114078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skirecki T, Mikaszewska-Sokolewicz M, Godlewska M, Dołęgowska B, Czubak J, Hoser G, et al. (2019). Mobilization of stem and progenitor cells in septic shock patients. Scientific Reports 9, 3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smadja DM, Bièche I, Susen S, Mauge L, Laurendeau I, d’Audigier C, et al. (2009). Interleukin 8 is differently expressed and modulated by PAR-1 activation in early and late endothelial progenitor cells. Journal of Cellular and Molecular Medicine 13, 2534–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souma T, Thomson BR, Heinen S, Carota IA, Yamaguchi S, Onay T, et al. (2018). Context-dependent functions of angiopoietin 2 are determined by the endothelial phosphatase VEPTP. Proceedings of the National Academy of Sciences of the United States of America 115, 1298–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach G, Bölke E, Schulte am Esch J, Peiper M, Zant R, Schwarz A, et al. (2007). Comparison of whole blood interleukin-8 and plasma interleukin-8 as a predictor for sepsis in postoperative patients. Clinica Chimica Acta; International Journal of Clinical Chemistry 378, 117–121. [DOI] [PubMed] [Google Scholar]
- Stoyanoff TR, Rodríguez JP, Todaro JS, Colavita JPM, Torres AM, & Aguirre MV (2018). Erythropoietin attenuates LPS-induced microvascular damage in a murine model of septic acute kidney injury. Biomedicine & Pharmacotherapy 107, 1046–1055. [DOI] [PubMed] [Google Scholar]
- Suffredini DA, Cui X, Xu W, Li Y, & Eichacker PQ (2017). The potential pathogenic contributions of endothelial barrier and arterial contractile dysfunction to shock due to B. anthracis lethal and edema toxins. Toxins 9, 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Breslin JW, Zhu J, Yuan SY, & Wu MH (2006). Rho and ROCK signaling in VEGF-induced microvascular endothelial hyperpermeability. Microcirculation 13, 237–247. [DOI] [PubMed] [Google Scholar]
- Sun J, Xie J, Kang L, Ferro A, Dong L, Biao X, & B. (2016). Amlodipine ameliorates ischemia-induced neovascularization in diabetic rats through endothelial progenitor cell mobilization. BioMed Research International 2016, 3182764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JN, Solis NV, Phan QT, Bajwa JS, Kashleva H, Thompson A, et al. (2010). Host cell invasion and virulence mediated by Candida albicans Ssa1. PLoS Pathogens 6, Article e1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R, Huang J, & Sun B (2020). Mobilization of endothelial progenitor cells in sepsis. Inflammation Research 69, 1–9. [DOI] [PubMed] [Google Scholar]
- Suratt BT, Cool CD, Serls AE, Chen L, Varella-Garcia M, Shpall EJ, et al. (2003). Human pulmonary chimerism after hematopoietic stem cell transplantation. American Journal of Respiratory and Critical Care Medicine 168, 318–322. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Yamane Y, & Matsuda H (2001). Mast cell MMP-9 production enhanced by bacterial lipopolysaccharide. The Journal of Veterinary Medical Science 63, 811–813. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Narazaki M, & Kishimoto T (2014). IL-6 in inflammation, immunity, and disease. Cold Spring Harbor Perspectives in Biology 6, Article a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terriaca S, Fiorelli E, Scioli MG, Fabbri G, Storti G, Cervelli V, & Orlandi A (2021). Endothelial progenitor cell-derived extracellular vesicles: Potential therapeutic application in tissue repair and regeneration. International Journal of Molecular Sciences 22, 6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thickett DR, Armstrong L, Christie SJ, & Millar AB (2001). Vascular endothelial growth factor may contribute to increased vascular permeability in acute respiratory distress syndrome. American Journal of Respiratory and Critical Care Medicine 164, 1601–1605. [DOI] [PubMed] [Google Scholar]
- Thickett DR, Armstrong L, & Millar AB (2002). A role for vascular endothelial growth factor in acute and resolving lung injury. American Journal of Respiratory and Critical Care Medicine 166, 1332–1337. [DOI] [PubMed] [Google Scholar]
- Thomas RA, Pietrzak DC, Scicchitano MS, Thomas HC, McFarland DC, & Frazier KS (2009). Detection and characterization of circulating endothelial progenitor cells in normal rat blood. Journal of Pharmacological and Toxicological Methods 60, 263–274. [DOI] [PubMed] [Google Scholar]
- Thurston G, & Daly C (2012). The complex role of angiopoietin-2 in the angiopoietin-tie signaling pathway. Cold Spring Harbor Perspectives in Medicine 2, Article a006550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermans F, Van Hauwermeiren F, De Smedt M, Raedt R, Plasschaert F, De Buyzere ML, et al. (2007). Endothelial outgrowth cells are not derived from CD133+ cells or CD45+ hematopoietic precursors. Arteriosclerosis, Thrombosis, and Vascular Biology 27, 1572–1579. [DOI] [PubMed] [Google Scholar]
- Tomita K, Saito Y, Suzuki T, Imbaby S, Hattori K, Matsuda N, et al. (2020). Vascular endothelial growth factor contributes to lung vascular hyperpermeability in sepsis associated acute lung injury. Naunyn-Schmiedeberg’s Archives of Pharmacology 393, 2365–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokos M, Pufe T, Paulsen F, Anders S, & Mentlein R (2003). Pulmonary expression of vascular endothelial growth factor in sepsis. Archives of Pathology & Laboratory Medicine 127, 331–335. [DOI] [PubMed] [Google Scholar]
- Urao N, Okigaki M, Yamada H, Aadachi Y, Matsuno K, Matsui A, et al. (2006). Erythropoietin-mobilized endothelial progenitors enhance reendothelialization via Akt-endothelial nitric oxide synthase activation and prevent neointimal hyperplasia. Circulation Research 98, 1405–1413. [DOI] [PubMed] [Google Scholar]
- Urbich C, Aicher A, Heeschen C, Dernbach E, Hofmann WK, Zeiher AM, et al. (2005). Soluble factors released by endothelial progenitor cells promote migration of endothelial cells and cardiac resident progenitor cells. Journal of Molecular and Cellular Cardiology 39, 733–742. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke E, Mehta D, Minshall R, & Malik AB (2008). Regulation of endothelial junctional permeability. Annals of the New York Academy of Sciences 1123, 134–145. [DOI] [PubMed] [Google Scholar]
- Vašíček J, Baláži A, Tirpáková M, Svoradová A, Ondruška Ľ, Parkányi V, et al. (2021). Secretome analysis of rabbit and human mesenchymal stem and endothelial progenitor cells: A comparative study. International Journal of Molecular Sciences 22, 12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassiliou AG, Kotanidou A, Dimopoulou I, & Orfanos SE (2020). Endothelial damage in acute respiratory distress syndrome. International Journal of Molecular Sciences 21, 8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesulu BP, Mahadevan LS, Aliru ML, Yang X, Bodd MH, Singh PK, et al. (2018). Radiation-induced endothelial vascular injury: A review of possible mechanisms. JACC. Basic to Translational Science 3, 563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent JL, Rello J, Marshall J, Silva E, Anzueto A, Martin CD, et al. (2009). International study of the prevalence and outcomes of infection in intensive care units. JAMA 302, 2323–2329. [DOI] [PubMed] [Google Scholar]
- Wang C, Verma S, Hsieh I, Chen Y, Kuo L, Yang N, et al. (2006). Enalapril increases ischemia-induced endothelial progenitor cell mobilization through manipulation of the CD26 system. Journal of Molecular and Cellular Cardiology 41, 34–43. [DOI] [PubMed] [Google Scholar]
- Wang H, Cai KY, Li W, & Huang H (2015). Sphingosine-1-phosphate induces the migration and angiogenesis of epcs through the Akt signaling pathway via sphingosine 1-phosphate receptor 3/platelet-derived growth factor receptor-β. Cellular & Molecular Biology Letters 20, 597–611. [DOI] [PubMed] [Google Scholar]
- Wang JH, Redmond HP, Watson RW, Condron C, & Bouchier-Hayes D (1999). Involvement of tyrosine protein kinase in IFN-gamma-induced human endothelial cell apoptosis. Shock 11, 311–318. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhan E, Lu G, Mu Q, Zhang T, & Yang N (2019). Sphingosine-1-phosphate improves the biological features of mouse bone marrow-derived EPCs partially through PI3K/AKT/eNOS/NO pathway. Molecules 24, 2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler MS, Märtz KB, Nierhaus A, Daum G, Schwedhelm E, Kluge S, et al. (2019). Loss of sphingosine 1-phosphate (S1P) in septic shock is predominantly caused by decreased levels of high-density lipoproteins (HDL). Journal of Intensive Care 7, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler MS, Nierhaus A, Holzmann M, Mudersbach E, Bauer A, Robbe L, et al. (2015). Decreased serum concentrations of sphingosine-1-phosphate in sepsis. Critical Care 19, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wójciak-Stothard B, & Ridley AJ (2002). Rho GTPases and the regulation of endothelial permeability. Vascular Pharmacology 39, 187–199. [DOI] [PubMed] [Google Scholar]
- Wu MH, Guo M, Yuan SY, & Granger HJ (2003). Focal adhesion kinase mediates porcine venular hyperpermeability elicited by vascular endothelial growth factor. The Journal of Physiology 552, 691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Bai S, Cao Y, Liu L, Fang Y, Du J, et al. (2020). miRNA-221–3p in endothelial progenitor cell-derived exosomes accelerates skin wound healing in diabetic mice. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy 13, 1259–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Greening DW, Zhu HJ, Takahashi N, & Simpson RJ (2016). Extracellular vesicle isolation and characterization: Toward clinical application. The Journal of Clinical Investigation 126, 1152–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Yang J, Li N, Wu R, Tian H, Song H, et al. (2015). Role of endothelial progenitor cell transplantation in rats with sepsis. Transplantation Proceedings 47, 2991–3001. [DOI] [PubMed] [Google Scholar]
- Yamada M, Kubo H, Kobayashi S, Ishizawa K, Numasaki M, Ueda S, et al. (2004). Bone marrow-derived progenitor cells are important for lung repair after lipopolysaccharide induced lung injury. The Journal of Immunology 172, 1266–1272. [DOI] [PubMed] [Google Scholar]
- Yan F, Liu X, Ding H, & Zhang W (2022). Paracrine mechanisms of endothelial progenitor cells in vascular repair. Acta Histochemica 124, Article 151833. [DOI] [PubMed] [Google Scholar]
- Yao E, Fukuda N, Matsumoto T, Kobayashi N, Katakawa M, Yamamoto C, et al. (2007). Losartan improves the impaired function of endothelial progenitor cells in hypertension via an antioxidant effect. Hypertension Research 30, 1119–1128. [DOI] [PubMed] [Google Scholar]
- Yoshizumi M, Fujita Y, Izawa Y, Suzaki Y, Kyaw M, & Ali N (2004). Ebselen inhibits tumor necrosis factor-alpha-induced c-Jun N-terminal kinase activation and adhesion molecule expression in endothelial cells. Experimental Cell Research 292, 1–10. [DOI] [PubMed] [Google Scholar]
- Zahran AM, Elsayh KI, Mohamad IL, Hassan GM, & Abdou MA (2016). Circulating endothelial cells and endothelial progenitor cells in pediatric sepsis. Pediatric Emergency Care 32, 163–167. [DOI] [PubMed] [Google Scholar]
- Zegeye MM, Lindkvist M, Fälker K, Kumawat AK, Paramel G, Grenegård M, et al. (2018). Activation of the JAK/STAT3 and PI3K/AKT pathways are crucial for IL-trans-signaling-mediated pro-inflammatory response in human vascular endothelial cells. Cell Communication and Signaling: CCS 16, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng CY, Xu J, Liu X, & Lu YQ (2021). Cardioprotective roles of endothelial progenitor cell-derived exosomes. Frontiers in Cardiovascular Medicine 8, Article 717536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai R, Gong MN, Zhou W, Thompson TB, Kraft P, Su L, et al. (2007). Genotypes and haplotypes of the VEGF gene are associated with higher mortality and lower VEGF plasma levels in patients with ARDS. Thorax 62, 718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Huang H, Liu W, Liu S, Wang XY, Diao ZL, et al. (2021). Endothelial progenitor cells-derived exosomal microRNA-21–5p alleviates sepsis-induced acute kidney injury by inhibiting RUNX1 expression. Cell Death & Disease 12, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, You B, Liu X, Chen J, Peng Y, & Yuan Z (2019). High-mobility group box 1 (HMGB1) induces migration of endothelial progenitor cell via receptor for advanced glycation end-products (RAGE)-dependent PI3K/Akt/eNOS signaling pathway. Medical Science Monitor: International Medical Journal of Experimental and Clinical Research 25, 6462–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Ito WD, Hopfner U, Böhmert B, Kremer M, Reckhenrich AK, et al. (2011). The role of single cell derived vascular resident endothelial progenitor cells in the enhancement of vascularization in scaffold-based skin regeneration. Biomaterials 32, 4109–4117. [DOI] [PubMed] [Google Scholar]
- Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, et al. (2000). VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. The Journal of Clinical Investigation 106, 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YT, Fallas JA, Saini S, Ueda G, Somasundaram L, Zhou Z, et al. (2021). F-domain valency determines outcome of signaling through the angiopoietin pathway. EMBO Reports 22, Article e53471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Li P, Goodwin AJ, Cook JA, Halushka PV, Chang E, & Fan H (2018). Exosomes from endothelial progenitor cells improve the outcome of a murine model of sepsis. Molecular Therapy: The Journal of the American Society of Gene Therapy 26, 1375–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Zhu J, Tian X, Zhang F, Zhao L, Zhu J, et al. (2021). Circulating endothelial progenitor cells from septic patients are associated with different infectious organisms. Annals of Palliative Medicine 10, 549–559. [DOI] [PubMed] [Google Scholar]
- Ziegler T, Horstkotte J, Schwab C, Pfetsch V, Weinmann K, Dietzel S, et al. (2013). Angiopoietin 2 mediates microvascular and hemodynamic alterations in sepsis. The Journal of Clinical Investigation 123, 3436–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors are unable or have chosen not to specify which data has been used.