

Abstract
Rapid detection of bloodstream pathogens would greatly facilitate clinicians to make precise antimicrobial treatment in patients with bacteremia. In this study, 114 plasma samples were collected from patients with identified or suspected bacteremia, and pathogens were detected by the conventional blood culture (BC) and cell-free DNA metagenomics next-generation sequencing (cfDNA mNGS). The present study indicated that 76% (38/50) of positive conventional blood culture (BC+ group) patients were positively detected by cfDNA mNGS, and only 4% were mismatched between cfDNA mNGS and conventional bacteria culture. Pathogens in 32.8% of suspected bacteremia patients with negative conventional blood culture (BC– group) were determined accurately by cfDNA mNGS combined with analyzing the patients’ clinical manifestations. Escherichia coli and Klebsiella pneumoniae were the most detected pathogens in identified bacteremia patients by cfDNA mNGS. 76.2% (16/21) of E. coli and 92.3% (12/13) of K. pneumoniae in bacteremia patients were identified by conventional blood cultures that were also detected by cfDNA mNGS. This study demonstrated that genomic coverage of E. coli and K. pneumoniae were more often detected in BC+ group patients and genomic coverage of Acinetobacter johnsonii and Paucibacter sp. KCTC 42545 was more often detected in BC– group patients. In conclusion, cfDNA mNGS could rapidly and precisely provide an alternative detection method for the diagnosis of bacteremia.
Keywords: cell-free DNA, next-generation sequencing, bacteremia, blood culture, pathogens
Introduction
Bacteremia can result in many serious diseases, such as sepsis, septic shock, and multi-organ failure (Laupland et al. 2004). Rapid detection of causative pathogens will be clinically helpful in optimizing accurate antimicrobial therapy in patients with suspected bacteremia. Conventional culture-based diagnostic procedures (e.g., blood cultures) currently remain the most reliable and clinically commonly used method for pathogens detection in patients with suspected bacteremia. However, the limitations of the conventional culture-based strategies for microbial tests in clinical practice have been widely discussed (e.g., it takes over 24 h to get results). Thus, several culture-independent molecular diagnostic procedures (such as polymerase chain reaction (PCR)-based techniques) have been introduced in clinics to identify the causative pathogens in patients with infectious diseases (Rutanga and Nyirahabimana 2016). Mounting evidence has demonstrated the diagnostic values of PCR-based techniques as additional tools to the conventional culture-based methods for identifying some known pathogens of patients with bacteremia and other infectious diseases (Blauwkamp et al. 2019).
Several recent studies have demonstrated the diagnostic values of cell-free DNA metagenomics next-generation sequencing (cfDNA mNGS) as a rapid and accurate method for identifying pathogens in patients with infectious diseases (Simner et al. 2018; Chiu and Miller 2019). The performance of the cfDNA mNGS for the identifying infectious microorganisms is based on the unbiased sequence analyses of the microbiome from the patient’s plasma (human DNA is removed) (Grumaz et al. 2016; Long et al. 2016). The conventional bacterial culture methods are commonly used as a gold standard for identifying bloodstream pathogens in patients with suspected bacteremia. Comparing diagnostic values of cfDNA mNGS and the conventional culture-based methods is seldomly reported in suspected bacteremia patients. In this study, the clinical values of the cfDNA mNGS were compared with the conventional culture-based methods. Moreover, the utility of cfDNA mNGS in suspected bacteremia patients was further evaluated as an alternative detection method.
Experimental
Materials and Methods
Patients and diagnostic standards. One hundred fourteen patients hospitalized at the Huazhong University of Science and Technology Union Shenzhen Hospital (Shenzhen, China) between August 2017 and May 2020 were enrolled in this study. The pathogen detection was performed synchronically in the sera of all 114 patients by both the conventional blood culture and cfDNA mNGS. The clinical diagnosis of these patients was shown in Table SI, and they were divided into two groups with identified or suspected bacteremia. Identified bacteremia patients were defined as those with the clinical symptoms of bloodstream infection and positive conventional bacteria culture from the blood samples (BC+ group). Suspected bacteremia patients were defined as those with the clinical symptoms of bloodstream infection and negative microbial detection in the conventional bacteria culture from the blood samples and other clinical specimens. The clinical symptoms of bloodstream infection mainly included as the following: 1) fevers with body temperatures > 38.0°C and recurrent chills; 2) clinical evidence of bacterial infection in some organs or systems; 3) no evidences of fever caused by non-infectious diseases. Suspected bacteremia patients were described as the BC– group in this study.
The conventional blood culture method. The conventional blood culture method was performed according to standard operations and incubated in the BD Bactec™ FX400 system (Becton Dickinson, USA). The bacterial species and antimicrobial susceptibility of blood culture positive samples were identified by the BD Phoenix™ M50 automated microbiology system (Becton Dickinson, USA).
Plasma preparation and cfDNA mNGS performance. Plasma samples were separated from the whole blood samples (before antibiotics were used) in EDTA anticoagulation blood collection tubes. The serum samples were collected from the whole blood samples in the tube without an anticoagulant. Plasma samples from five healthy volunteers were used as negative controls to calibrate and set detection thresholds. The cfDNA was extracted from 300 μl of plasma or serum using the TIANamp Micro DNA Kit (DP316, TIANGEN Biotech(Beijing)Co.,Ltd., China) according to the manufacturer’s protocol. The nucleic acid concentration was measured using the Qubit dsDNA HS Assay Kit (Life Technologies, Invitrogen, China). Library construction and mNGS were performed according to the sequencing protocol (BGI, China). DNA library quality was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, USA) combined with quantitative PCR to measure the adapters before sequencing. Qualified DNA libraries were prepared in a OneTouch system by emulsion PCR and then sequenced on the BGISEQ-100 or BGISEQ-50 sequencing platform (BGI, China).
NGS data processing. Raw data were preprocessed by removing short reads, low-quality reads, and sequencing adapters to generate clean reads. Raw data generated from the BGISEQ-50 sequencing platform were filtered using SOAPnuke software (version:1.5.6, BGI, China, https://github.com/BGI-flexlab/SOAP-nuke), which included removing reads in which at least 30% of the total reads had quality scores < 20, a proportion of unknown nucleotides > 2%, and less than 50 bp, and trimming adapters sequences. For raw data generated from the BGISEQ-100 sequencing platform, reads with less than 50 bp were discarded, and those with average quality scores of < 10 per 15 window lengths and sequencing adapters were trimmed. The alignment and analysis of the cfDNA reads by mNGS were performed according to the previous reports (Altschul et al. 1990; Wood and Salzberg 2014). Reads were normalized per 20 million mapped reads according to the clean reads as the relative abundance. Species with detection rates > 0.8 and sample standard deviations > 0.6 were retained. The ratio of the relative bacterial abundance in each sample was calculated and placed in descending order. Criteria for a positive cfDNA mNGS result were as per Miao and coworkers (Miao et al. 2018). Consistency between the microbial species identified by cfDNA mNGS and the BC results combined with clinical manifestations was evaluated. For mNGS+/BC– samples, whether the pathogen results were reliable after the combined analysis with clinical symptoms was assessed by three clinicians.
Statistical analysis. Data were analyzed using R software, version 3.5.0. A chi-square test was used to compare the prevalence of each species and their resistance and virulence genes between positive and negative BC samples. p < 0.05 was considered statistically significant. The Wilcoxon rank-sum test was used to calculate differences in the relative abundances of the resistance and virulence genes between positive and negative BC samples, with p < 0.05.
Results
Clinical information and cfDNA quantification from bacteremia patients’ plasma or serum samples. The microbes-derivative read numbers of the cfDNA extracted from bacteremia patients’ plasma and serum samples were compared. It suggested that more microbes-derivative read numbers could be detected in plasma compared to serum ones. It demonstrated that the plasma was more suitable for detecting cfDNA mNGS (Fig. 1a). Subsequently, 114 patients were divided into two groups according to the blood culture results and the criteria described above. Fifty patients were diagnosed with bacteremia (BC+ group) and 64 with suspected bacteremia (BC– group), and the diagnostic value of cfDNA mNGS in these groups was evaluated. The total cfDNA read numbers/concentrations (copies/ml) by cfDNA mNGS were significantly higher in the plasma samples of the BC+ group compared with the BC– group (Fig. 1b). The microbes-derivative cfDNA reads/total cfDNA reads showed no significant differences between these two groups (Fig. 1c).
Fig. 1.
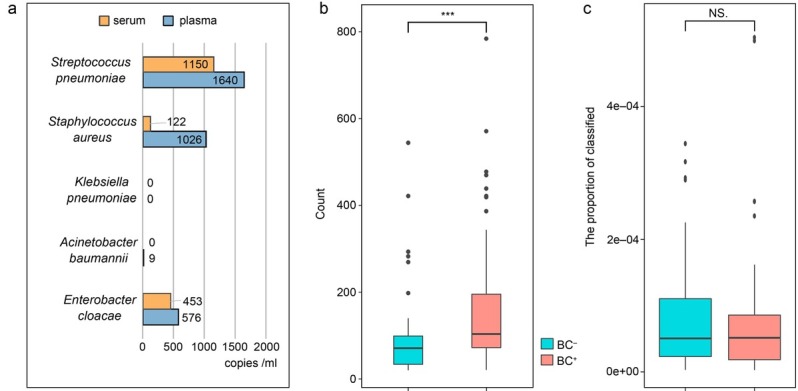
a) Comparison of cfDNA reads number/concentrations (copies/ml) of pathogens by cfDNA mNGS between plasma and serum; b) comparison of cfDNA read numbers/concentrations (copies/ml) between BC+ and BC–; c) comparison of the microbes-derivative cfDNA reads/total cfDNA reads between BC+ and BC– patients. *** – p < 0.001, NS – not significant, BC – blood culture, cfDNA mNGS – cell-free DNA metagenomics next-generation sequencing, + – positive, – – negative
Comparison of the diagnostic value between the conventional blood culture and cfDNA mNGS. The diagnostic values of pathogen detection by cfDNA mNGS and conventional blood culture were compared and are presented in Table I. In this study, 76% of the BC+ group (38/50) were positively recognized by cfDNA mNGS, indicating that 72% (36/50) were consistent with the BC results, and only two cases were mismatched between these two methods. Moreover, among the 64 cfDNA mNGS+ samples, 26 were BC–, and 80.7% (21/26) of cfDNA mNGS+/BC– samples were consistent with the clinical manifestation. This observation indicated that the pathogens in BC– samples were reliable determined by cfDNA mNGS. Thus, to promise the accuracy of cfDNA mNGS results in BC– groups, performing analysis combined with the clinical manifestation is necessary.
Table I.
Comparison of the consistency of the pathogen identification between cfDNA mNGS and BC.
| cfDNA mNGS+ | cfDNA mNGS– | Consistency | |
|---|---|---|---|
| BC+ | 38 | 12 | 36a |
| BC– | 26 | 38 | 21b |
| Total | 64 | 50 |
BC – blood culture, cfDNA mNGS – cell-free DNA metagenomics next-generation sequencing, a – consistency between the microbial species identified by cfDNA mNGS and the BC results, b – for NGS+/BC– samples, whether the pathogen results were reliable after the combined analysis with clinical symptoms were assessed by three clinicians
In this study, Escherichia coli and Klebsiella pneumoniae were the most detected pathogens in identified bacteremia patients by cfDNA mNGS (Fig. 2). cfDNA mNGS detected 76.2% (16/21) of E. coli and 92.3% (12/13) of K. pneumoniae in the patients in whom the bacteriemia was detected by conventional blood culture (Table SII).
Fig. 2.
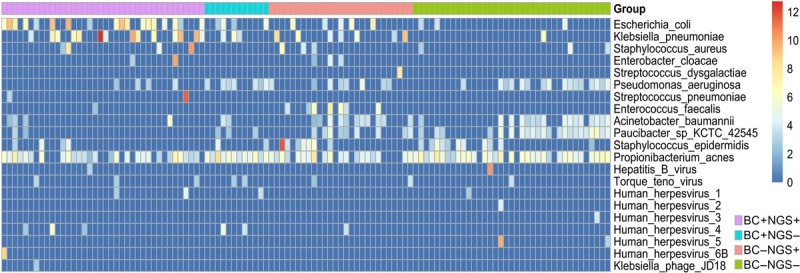
The relative abundance of cfDNA mNGS-detected bacteria and viruses in the 114 samples were shown by Heatmap. The relative abundance data used in the heatmap were log2‑transformed to compare among species. Staphylococcus epidermidis and Propionibacterium acnes were discarded as contaminants in the downstream analysis.
In addition to bacterial pathogens, cfDNA mNGS indicated that 54.39% of the blood samples (62/114) came from patients with coexisting viral infections (Table SIII). The Torque Teno virus (TTV) and human herpesvirus 4 (EBV) were the top two species detected in this study. EBV had a significantly higher detection rate in the BC+ samples than in the BC– group. Notably, most viral read numbers were < 20, and the relative abundance of the hepatitis B virus (HBV) in the BC– group was over 1,000, which nearly equaled the viral copy number determined by quantitative fluorescent PCR (Fig. 2).
The genomic mapping of the microbes detected by cfDNA mNGS in BC+ and BC– groups. Furthermore, the whole genome of individual microbial species, such as E. coli and K. pneumoniae were mapped from the cfDNA reads detected by mNGS, and compared between the BC+ and BC– groups. This study demonstrated that genomic coverage of E. coli and K. pneumoniae was most often detected in BC+ group patients, and genomic coverage of Acinetobacter johnsonii and Paucibacter sp. KCTC 42545 was more often detected in BC– patients (Fig. 3).
Fig. 3.
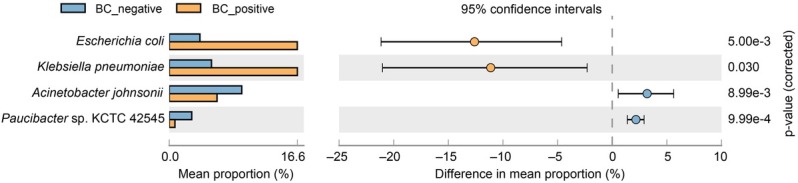
Relative abundances of read numbers of microbial species between the BC+ and BC– samples. Confidence intervals and p-values are indicated for each species, and the differences in proportions were calculated as the mean proportion of BC– minus BC+ samples with 95% confidence intervals.
Proteobacteria was the most common phylum in both BC+ and BC– patients (Fig. 4a). Gammaproteobacteria had the highest relative abundance ratio at the class level, and was more abundant in the BC+ patients (p = 0.03), whereas Alphaproteobacteria (p < 0.001) and Betaproteobacteria (p < 0.001) were more abundant in the BC– samples (Fig. 4b). At the order level, Enterobacterales (p < 0.001) was significantly abundant in the BC+ samples, whereas Rhizobiales (p = 0.001) and Pseudomonadales (p = 0.01) were more abundant in the BC– samples. At the family level, Enterobacteriaceae (p < 0.001) was significantly abundant in the BC+ samples, whereas Moraxellaceae (p = 0.01) and Caulobacteraceae (p < 0.001) were more abundant in the BC– group. At the genus level, Escherichia (p < 0.001) was relatively higher in the BC+ samples, whereas Acinetobacter (p = 0.01) was relatively higher in the BC– samples (Fig. 5).
Fig. 4.
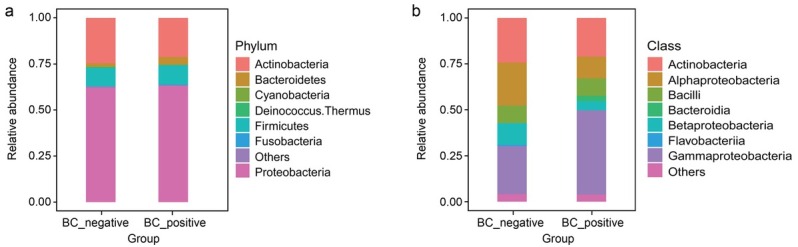
Microbial compositions of the BC+ and BC– samples.
a) At phylum level; b) at class level.
Fig. 5.
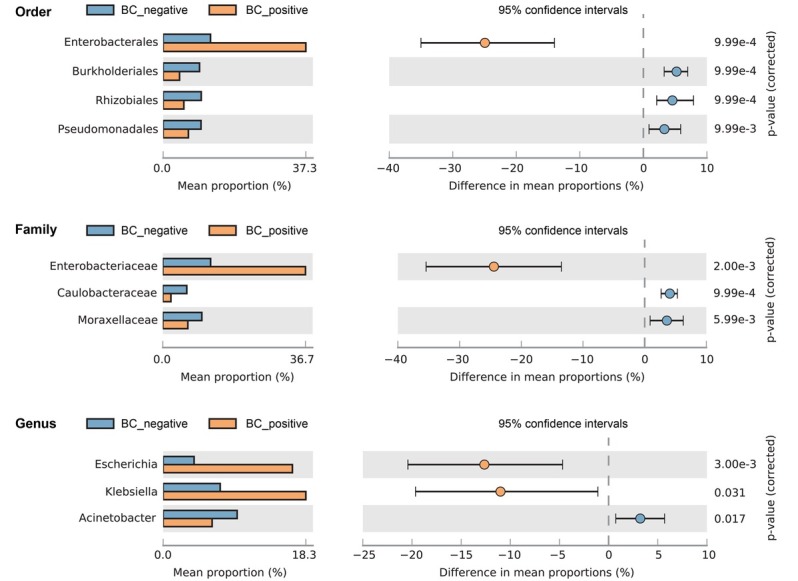
Comparison of community compositions at the order, family and genus levels. Confidence intervals and p-values are indicated in each case, and the difference in proportions was calculated by the mean proportion of BC– minus BC+ samples with 95% confidence intervals.
BC – blood culture, + – positive, – – negative
Dynamics of microbial derivative cfDNA read numbers determined by mNGS during antibiotic treatment. The read numbers of cfDNA from the plasma samples collected from three patients at different time points were analyzed to investigate the dynamic changes of the microbial derivative cfDNA read numbers during antimicrobial treatment (Fig. 6). In patients of A47 and A45, the cfDNA read numbers of K. pneumoniae and E. coli significantly decreased after of the effective antimicrobial treatment. It showed that the dynamics of the bacterial read numbers in these two cases reflected the efficacy of the antibiotic treatment. However, the cfDNA read numbers of Staphylococcus aureus in patient A48 remained positive with a transient increase of cfDNA read number after eight days of antibiotic treatment with remission of clinical symptoms. Subsequently, with persistent antibiotic treatment, the S. aureus cfDNA read numbers decreased and became negative (Fig. 6). With the decrease of cfDNA read numbers, the fever symptoms gradually improved, and while blood cells, C-reactive protein, and procalcitonin were gradually reduced (Fig. 7).
Fig. 6.
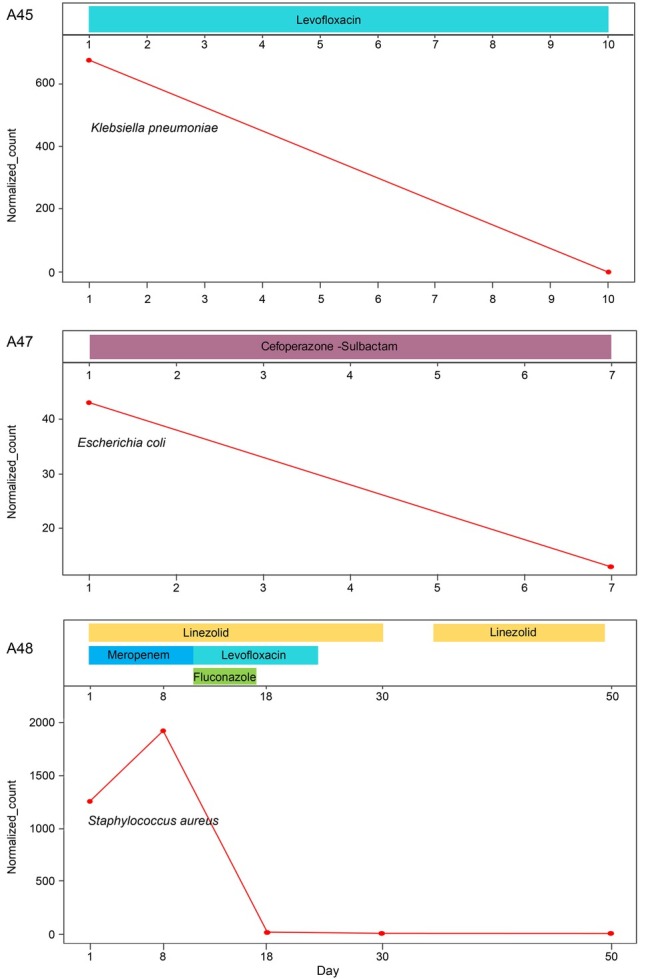
Antibiotic treatments and relative pathogen abundances at different times after disease onset.
Fig. 7.
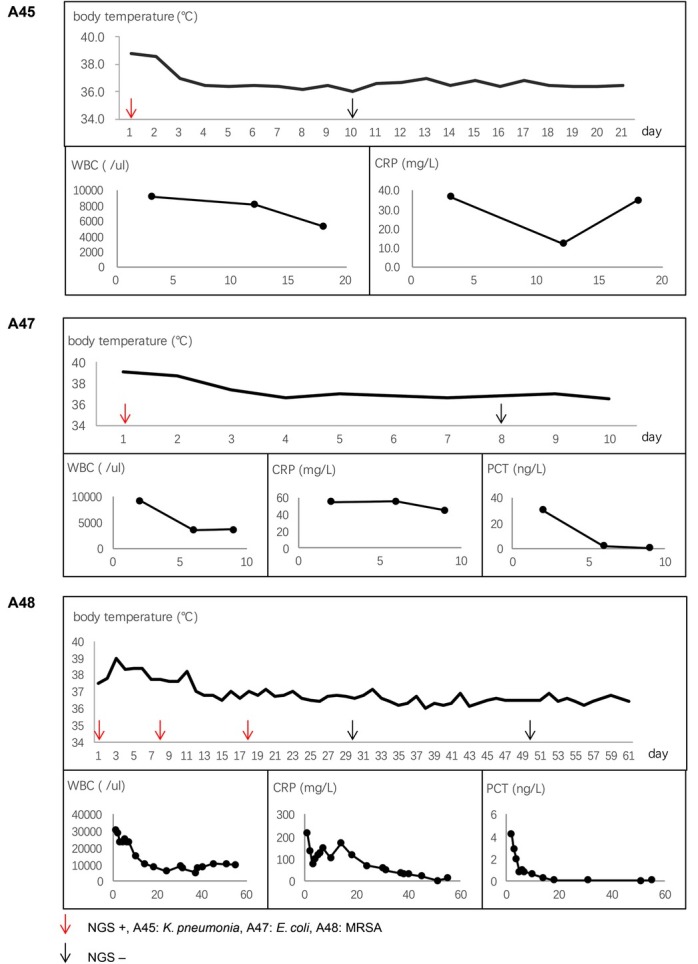
Patient symptoms with antibiotic treatment.
WBC – white blood cells, CRP – C-reactive protein, PCT – procalcitonin, MRSA – methicillin-resistant S. aureus, NGS – cell-free DNA metagenomics next-generation sequencing
Discussion
An ideal diagnostic test would enable the simultaneous detection and identification of pathogens from various clinical specimens, including rare or unculturable ones. cfDNA-based mNGS has been widely reported as a promising diagnostic tool for detecting microorganism from various clinical samples, such as bronchoalveolar lavage fluid and cerebrospinal fluid (Horiba et al. 2018; Miao et al. 2018; Miller et al. 2019). However, the evaluation of the diagnostic values of cfDNA mNGS for microbial testing of bacteremia patients’ blood samples has rarely been reported. Here, our data showed that cfDNA mNGS could detect pathogens in plasma samples independently of BC tests, demonstrating that 72% of the mNGS-positive samples were consistent with the gold standard approach of BC.
One of the most urgent needs in clinics is to evaluate the clinical significance of cfDNA mNGS for determining microbial species in the BC– group. i.e., patients with suspected bacteremia (Horiba et al. 2018). Our data indicated that cfDNA mNGS yielded accurate microbial results in 32.8% of the BC– group of patients with suspected bacteremia, supporting the future application of this approach in clinics. Although the cost of cfDNA NGS is five to six times that of conventional blood culture method, it takes only 24 hours from blood sample collection to NGS detection, bioinformatics analysis, and final results. Especially for those patients with severe bloodstream infections, it might be helpful to obtain a clinical prognosis in the shortest time and start timely anti-infection treatment.
Previous studies have indicated that herpes viruses (cytomegalovirus, Epstein-Barr virus, herpes simplex virus types 1, and human herpesviruse types 6), polyomaviruses (JC and BK), and an anellovirus (Torque Teno virus) are significant and commonly found in plasma samples of septic patients (Béland et al. 2014; Walton et al. 2014). Whether the increased propensity for infections with relatively weakly pathogenic organisms results from viral-mediated effects to impair immunity and whether viral reactivation occurs more readily in more profoundly immunosuppressed septic patients remains unknown. Applying cfDNA-based NGS may facilitate monitoring serum viral titers and identifying viral reactivation or flare in serum samples of patients with identified or suspected bacteremia. Therefore, discovering occult DNA viruses in host plasma samples may be an advantage in the future.
The antimicrobial susceptibility of traditional microbial culture depends on the microbial isolation and identification. Some molecular diagnostic methods, such as PCR, can both analyze the microbial species and determine the antimicrobial-resistance genes to provide the additional guidance for the antibiotic treatments (Gu et al. 2019). Previous researches have indicated that mNGS detection of clinical samples can reveal information about both the microbial species and the antimicrobial gene distribution, which can facilitate undertaking treatment choices for bacteremia patients (Grumaz et al. 2016; Horiba et al. 2018; Charalampous et al. 2019). In this study, blood culture results showed that these bacteria were not resistant to commonly used antibiotics, so we did not conduct in-depth analysis of resistance genes in cfDNA mNGS sequencing results. This study was probably limited by its single-center nature, and small number of patients. Thus, to some extent, it may affect the accuracy and reliability of the results.
Conclusion
In conclusion, cfDNA mNGS may be an alternative detection method for pathogen identification in the patients with bacteremia. Applying cfDNA mNGS, especially combined with BC and clinical manifestation, can significantly enhance the detection rate and accuracy of cfDNA microbial detection in patients with bacteremia. High read numbers of microbial cfDNA will facilitate using cfDNA mNGS to determine the antimicrobial susceptibility and dynamical detection of the cfDNA-derivative microbial read numbers. The achieve the accurate diagnosis by cfDNA mNGS one should avoid the operational contamination and exclude the interference from the extra or occult microbial species. Additional clinical trials are needed to determine the sensitivity and specificity of this NGS method.
Acknowledgments
The authors would like to thank these organizations for financial supports and the anonymous reviewers are acknowledged for their critical comments and suggestions which have considerably improved the manuscript.
Funding Statement
This work was supported by the Technology and Innovation Commission of Shenzhen Municipality of Key Funds (JCYJ20180302144721183; JCYJ20170412143551332) and Basic Research Funds (JCYJ20180302144345028; JCYJ20180302144403714) and National Key R&D Program of China O. 2016YFE0205800, Science, Technology and Shenzhen Key Medical Discipline Construction Fund (No. SZXK06162).
Footnotes
Availability of data and material
The raw whole-genome sequencing data was posted in the Sequence Read Archive (SRA) database under accession number PRJNA837746 (https://dataview.ncbi.nlm.nih.gov/object/ PRJNA837746).
Ethical statement
All procedures involving human participants were performed following the ethical standards of Huazhong University of Science and Technology Union Shenzhen Hospital and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. As this study only involves the detection of patients’ blood samples and does not carry out drug treatment, patients do not need to sign informed consent according to the requirements of the Ethics Committee of Huazhong University of Science and Technology Union Shenzhen Hospital.
Author contributions
CZ participated in the design of this study, collected strains, extracted cfDNA from plasma or serum, and drafted the manuscript. HC analyze the cfDNA mNGS data. YZ participated in the collection of strains and cfDNA mNGS test. JC participated in the collection of strains and extraction of cfDNA from plasma or serum. ML, ZY and XS participated in the design of this study, collected strains and analyze the cfDNA mNGS data. PL and YS collected clinical information of patients and participated in the cfDNA mNGS test. JM and JZ designed the study, conducted the data analysis, and provided critical revisions of the manuscript for valuable intellectual content.
Conflict of interest
The authors do not report any financial or personal connections with other persons or organizations, which might negatively affect the contents of this publication and/ or claim authorship rights to this publication.
Supplementary Material.
Literature
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. Oct 5. [DOI] [PubMed] [Google Scholar]
- Béland K, Dore-Nguyen M, Gagné MJ, Patey N, Brassard J, Alvarez F, Halac U. Torque Teno virus in children who underwent orthotopic liver transplantation: New insights about a common pathogen. J Infect Dis. 2014;209(2):247–254. doi: 10.1093/infdis/jit423. Jan 15. [DOI] [PubMed] [Google Scholar]
- Blauwkamp TA, Thair S, Rosen MJ, Blair L, Lindner MS, Vilfan ID, Kawli T, Christians FC, Venkatasubrahmanyam S, Wall GD. Analytical and clinical validation of a microbial cell-free DNA sequencing test for infectious disease. Nat Microbiol. 2019;4(4):663–674. doi: 10.1038/s41564-018-0349-6. et al. Apr. [DOI] [PubMed] [Google Scholar]
- Charalampous T, Kay GL, Richardson H, Aydin A, Baldan R, Jeanes C, Rae D, Grundy S, Turner DJ, Wain J. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat Biotechnol. 2019;37(7):783–792. doi: 10.1038/s41587-019-0156-5. et al. Jul. [DOI] [PubMed] [Google Scholar]
- Chiu CY, Miller SA. Clinical metagenomics. Nat Rev Genet. 2019;20(6):341–355. doi: 10.1038/s41576-019-0113-7. Jun. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumaz S, Stevens P, Grumaz C, Decker SO, Weigand MA, Hofer S, Brenner T, von Haeseler A, Sohn K. Next-generation sequencing diagnostics of bacteremia in septic patients. Genome Med. 2016;8(1):73. doi: 10.1186/s13073-016-0326-8. Jul 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Miller S, Chiu CY. Clinical metagenomic next-generation sequencing for pathogen detection. Annu Rev Pathol. 2019;14:319–338. doi: 10.1146/annurev-pathmechdis-012418-012751. Jan 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiba K, Kawada JI, Okuno Y, Tetsuka N, Suzuki T, Ando S, Kamiya Y, Torii Y, Yagi T, Takahashi Y. Comprehensive detection of pathogens in immunocompromised children with bloodstream infections by next-generation sequencing. Sci Rep. 2018;8(1):3784. doi: 10.1038/s41598-018-22133-y. et al. Feb 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laupland KB, Gregson DB, Zygun DA, Doig CJ, Mortis G, Church DL. Severe bloodstream infections: A population-based assessment. Crit Care Med. 2004;32(4):992–997. doi: 10.1097/01.ccm.0000119424.31648.1e. Apr. [DOI] [PubMed] [Google Scholar]
- Long Y, Zhang Y, Gong Y, Sun R, Su L, Lin X, Shen A, Zhou J, Caiji Z, Wang X. Diagnosis of sepsis with cell-free DNA by next-generation sequencing technology in ICU patients. Arch Med Res. 2016;47(5):365–371. doi: 10.1016/j.arcmed.2016.08.004. et al. Jul. [DOI] [PubMed] [Google Scholar]
- Miao Q, Ma Y, Wang Q, Pan J, Zhang Y, Jin W, Yao Y, Su Y, Huang Y, Wang M. Microbiological diagnostic performance of metagenomic next-generation sequencing when applied to clinical practice. Clin Infect Dis. 2018;67(suppl_2):S231–S240. doi: 10.1093/cid/ciy693. et al. Nov 13. [DOI] [PubMed] [Google Scholar]
- Miller S, Naccache SN, Samayoa E, Messacar K, Arevalo S, Federman S, Stryke D, Pham E, Fung B, Bolosky WJ. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. 2019;29(5):831–842. doi: 10.1101/gr.238170.118. et al. May. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutanga JP, Nyirahabimana T. Clinical significance of molecular diagnostic tools for bacterial bloodstream infections: A systematic review. Interdiscip Perspect Infect Dis. 2016;2016:6412085. doi: 10.1155/2016/6412085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simner PJ, Miller S, Carroll KC. Understanding the promises and hurdles of metagenomic next-generation sequencing as a diagnostic tool for infectious diseases. Clin Infect Dis. 2018;66(5):778–788. doi: 10.1093/cid/cix881. Feb 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton AH, Muenzer JT, Rasche D, Boomer JS, Sato B, Brownstein BH, Pachot A, Brooks TL, Deych E, Shannon WD. Reactivation of multiple viruses in patients with sepsis. PLoS One. 2014;9(2):e98819. doi: 10.1371/journal.pone.0098819. et al. Jun 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014;15(3):R46. doi: 10.1186/gb-2014-15-3-r46. Mar 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.