

Abstract

Recently, we constructed a hybrid thymine DNA glycosylase (hyTDG) by linking a 29-amino acid sequence from the human thymine DNA glycosylase with the catalytic domain of DNA mismatch glycosylase (MIG) from M. thermoautotrophicum, increasing the overall activity of the glycosylase. Previously, it was shown that a tyrosine to lysine (Y126K) mutation in the catalytic site of MIG could convert the glycosylase activity to a lyase activity. We made the corresponding mutation to our hyTDG to create a hyTDG-lyase (Y163K). Here, we report that the hybrid mutant has robust lyase activity, has activity over a broad temperature range, and is active under multiple buffer conditions. The hyTDG-lyase cleaves an abasic site similar to endonuclease III (Endo III). In the presence of β-mercaptoethanol (β-ME), the abasic site unsaturated aldehyde forms a β-ME adduct. The hyTDG-lyase maintains its preference for cleaving opposite G, as with the hyTDG glycosylase, and the hyTDG-lyase and hyTDG glycosylase can function in tandem to cleave T:G mismatches. The hyTDG-lyase described here should be a valuable tool in studies examining DNA damage and repair. Future studies will utilize these enzymes to quantify T:G mispairs in cells, tissues, and genomic DNA using next-generation sequencing.
Introduction
Substantial research efforts are currently focused on DNA repair enzymes because of the importance of DNA damage and repair to human diseases. Most endogenous DNA damage is repaired by the base excision repair (BER) pathway.1−5 The BER pathway is initiated by a series of lesion-specific glycosylases that recognize and remove a damaged base from DNA. The resulting abasic site is then cleaved by a lyase domain connected to the glycosylase, in the case of bifunctional glycosylases, or a separate lyase or endonuclease, in the case of monofunctional glycosylases. The repair cycle is then completed by inserting one or more nucleotides by a DNA polymerase, and a DNA ligase restores the phosphodiester backbone.
Current studies on DNA repair enzymes fall into at least three categories. First is understanding the types of DNA damage and how they are repaired. The DNA of all organisms is persistently damaged by endogenous reactions, including deamination and oxidation of the DNA bases.6−9 The repair of this damage is essential to prevent cell death and disease-causing mutations. An understanding of the repair efficiency and substrate selectivity of different DNA repair enzymes can reveal vulnerabilities in the genome. Second, DNA repair enzymes are potential pharmacological targets where DNA repair pathway inhibition, coupled with other metabolic deficiencies, could result in selective toxicity.10−14 Third, DNA repair enzymes can serve as valuable reagents for characterizing and quantifying specific types of DNA damage,15−18 identifying the location of specific types of DNA damage at nucleotide resolution,19−21 and for preparing input DNA for next-generation sequencing.22−26
There is an interest in developing specific DNA glycosylase inhibitors for cancer therapy.10 However, glycosylase assays are frequently based upon the cleavage of DNA strands following glycosylase excision of a target base.10,14 DNA strands, labeled with either 32P or fluorescent tags, can be separated by either gel electrophoresis or HPLC and quantified. While many DNA repair enzymes are bifunctional with the capacity to remove a base from DNA, generate an abasic site, and cleave the phosphodiester backbone, many are monofunctional and require a separate step to cleave the DNA or oligonucleotide backbone.10−13
Following the generation of an abasic site in a glycosylase activity assay, the DNA cleavage step can be accomplished by the addition of hydroxide or other strand cleavage catalysts. However, such reagents would be added at the end of the glycosylase assay, which would prevent continuous monitoring of glycosylase excision using FRET-based assays.10−13 Alternatively, a glycosylase could be coupled with a bifunctional glycosylase or an apurinic endonuclease such as apurinic/apyrimidinic endonuclease I (APE1). There are two challenges with the latter approach. First, the pairing of a glycosylase with an abasic (AP) endonuclease or lyase requires common reaction conditions for both the glycosylase and the endonuclease/lyase. For example, most glycosylase assays are conducted in the presence of EDTA and the absence of Mg++, as Mg++ has been reported to inhibit some glycosylases.27−29 A commonly used enzyme to cleave abasic sites is APE1. However, APE1 requires Mg++.30 Therefore, additional enzymes that cleave DNA at abasic sites under a wider range of experimental conditions could serve as useful reagents for glycosylase assays. Second, bifunctional glycosylases themselves remove some bases from DNA, potentially confounding the interpretation of studies with a target glycosylase.
In the third category, DNA repair enzymes can be used in studies to quantify and locate specific damage in DNA isolated from biological sources. One key example includes the deamination products derived from 5mC. In higher organisms, replacing C with 5mC is important in the epigenetic control of gene transcription and chromatin structure.31−33 Enzymatic DNA methylation usually occurs in the CpG dinucleotide, and C to T transition mutations at CpG dinucleotides represent the most frequent single-base change found in human tumors.34−37 The deamination of 5mC to T generates a T:G mismatch. After DNA replication, this results in a C to T mutation. The T:G mismatch is persistent in human DNA because members of the uracil-DNA glycosylase superfamily presumed to remove T from a T:G mispair have much weaker activity for T:G than for U:G, the product of cytosine deamination.38,39 Despite its prominent role in mutagenesis, methods to measure T:G mismatches have only recently been developed.18
Our work presented here with the development of a hybrid thymine DNA lyase (hyTDG-lyase) is part of our efforts in contributing to the second and third categories mentioned above. The development of this hybrid enzyme is based upon a thymine DNA glycosylase (MIG) first identified by Horst and Fritz in 1996 in the thermophile Methanobacterium thermoaquaticus.40 The value of this enzyme, in our hands, was its relatively good excision capability on a wide array of 5-substituted uracil analogues, including thymine, and the extremely high selectivity for uracil analogues mispaired with guanine.41 We attached a 29-amino acid peptide found in human TDG (hTDG) that increased the overall activity of hTDG to the catalytic domain of MIG to create a hybrid enzyme, hyTDG.18 MIG and hyTDG are monofunctional glycosylases that bind strongly to abasic sites, the product of the glycosylase activity. Pairing a monofunctional glycosylase with an AP endonuclease or bifunctional glycosylase has been shown to increase overall glycosylase activity, presumably by hydrolyzing the abasic site and allowing for turnover.42,43 Begley and Cunningham demonstrated that the substitution of tyrosine 126 with lysine in MIG converted the glycosylase to an AP lyase.43 We therefore introduced the corresponding mutation into our hyTDG (Y163K), creating hyTDG-lyase. We report here the characterization of hyTDG-lyase, and we demonstrate that the enzyme pair hyTDG and hyTDG-lyase can be a valuable tool in investigations of DNA damage and repair.
Results
Confirmation of the hyTDG-Lyase Primary Amino Acid Sequence
A Y163K mutant of our hybrid thymine DNA glycosylase (hyTDG) was constructed and is referred to as hyTDG-lyase. Our mutant protein has an apparent molecular weight of 26.5 kDa (Figure S1). Confirmation of the amino acid sequence of hyTDG-lyase was performed by analysis of tryptic peptides using nano-liquid chromatography-mass spectrometry (nLC-MS/MS). The amino acid sequence of the hyTDG-lyase is shown in Figure 1A. The Y163K substitution is indicated by the highlighted K (Figure 1A). One peptide, NRKAILDLPGVGKK, containing the Y163K substitution is underlined in Figure 1A. The corresponding mass spectrum of this peptide is shown in Figure 1B. Several other peptides derived from hyTDG-lyase were observed and are listed in Table S1.
Figure 1.

Confirmation of the primary amino acid sequence of hyTDG-lyase. (A) Amino acid sequence of hyTDG-lyase. Underlined and in bold is the peptide that contains the Y163K amino acid substitution (highlighted). The protein has a his-tag followed by a 29 amino acid sequence from human thymine DNA glycosylase on the N-terminus (blue). (B) Mass spectrum of the NRKAILDLPGVGKK peptide containing the Y163K substitution obtained by nLC-MS/MS. The fragmentation pattern confirms the predicted sequence. To obtain high sequence coverage and high-quality peptides, lysines were acetylated to prevent trypsin over digestion. Table S1 contains additional peptides that were detected.
Identification of hyTDG-Lyase Cleavage Products
AP lyases typically cleave abasic sites on the 3′-side and AP endonucleases cleave on the 5′-side of an abasic site.44 These different ends can result in different gel mobilities. We compared the gel mobilities of a 5′-FAM-labeled 18-base oligonucleotide containing an abasic site treated chemically or enzymatically with APE1, TTH endonuclease IV (TTH), formamidopyrimidine DNA glycosylase (FPG), NaOH, 8-oxoguanine DNA glycosylase (hOGG1), endonuclease III (Endo III), hyTDG-lyase, or N,N-dimethylethylenediamine (DMDA) (Figure S2). The 5′-FAM-labeled oligonucleotides containing a 3′-terminal 3′-OH or 3′-phospho-α,β-unsaturated aldehyde (PUA) were not separable from one another, but were distinguishable from a 3′-phosphate. We also confirmed the expected 3′-terminal ends by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (Table S2). The spectra are shown in Figures S3–S10.
To investigate the mechanism of cleavage by hyTDG-lyase, a 5′-FAM-labeled 18-base oligonucleotide duplex containing a U:G mispair was incubated with hyTDG and hyTDG-lyase at 65 °C for 2 h. The resulting oligonucleotide cleavage products were examined by MALDI-TOF-MS (Figure 2).45,46 The hyTDG-lyase cleavage resulted in two oligonucleotide fragments. The 11 base 3′-fragment of the abasic site contained a 5′-end with an observed m/z of 3446.58 Da, consistent with a 5′-phosphate (Figure 2B). The other oligonucleotide fragment, a 5′-FAM containing 6-base fragment with a 3′-terminus, had an observed m/z of 2601.2450 Da (Figure 2A). This was 78 mass units higher than the PUA. We propose that β-mercaptoethanol present in the hyTDG-lyase purification buffer reacted with the PUA to generate the product shown in Figure 2A.
Figure 2.

hyTDG-lyase generates a β-elimination product that undergoes a Michael addition with β-mercaptoethanol. An 18-base oligonucleotide duplex with a 5′-FAM label and a U:G mispair (100 pmol, 4 μM) was incubated with hyTDG (25 pmol, 1 μM) and hyTDG-lyase (12.5 pmol, 0.5 μM) for 2 h at 65 °C. The resulting fragments were examined by MALDI-TOF-MS. (A) The fragment that contains the 5′-FAM label and a 3′-terminus had a measured m/z of 2601.24. The observed mass is consistent with the formation of a β-elimination product, a PUA, that forms an adduct with β-ME (PUA-βME) (theoretical m/z 2601.48 Da). (B) Corresponding 11-base fragment formed from the cleavage of the 3′-end of the abasic site had a measured m/z of 3446.33 (theoretical m/z 3446.58 Da) consistent with a 5′-phosphate. The schematic demonstrates the possible structure of the proposed fragment consistent with the measured mass.
Recently, the Gates and Guengerich laboratories have independently described thiol adducts formed at abasic sites and abasic site-derived cleavage products.47−49 In Figure 2, we present a structure consistent with the observed mass, although other structural isomers are possible. Interestingly, we find that the PUA is reactive with a broad range of nucleophiles. We observed different adducts such as the addition of a water molecule with Endo III, β-ME with hyTDG-lyase, and an adduct with DMDA—a secondary amine reported to promote β-elimination (Figure S2 and Table S2).50
Kinetics of hyTDG and hyTDG-Lyase
To examine the kinetics of hyTDG-lyase (Figure 3), an 18-base oligonucleotide duplex containing a U:G mispair (0.6 pmol, 12.5 μL reaction volume, 0.05 μM oligo) was incubated with UDG (0.84 pmol, 0.07 μM) at 37 °C for 1 h (Table S3). Alkaline hydrolysis of the abasic site, followed by gel electrophoresis analysis, revealed that the U from the U:G duplex had been completely excised, generating an abasic site. Preliminary experiments revealed that the hyTDG-lyase did not turn over, and therefore, kinetic data were obtained under predominantly single-turnover conditions. Increasing concentrations of hyTDG-lyase (0.02, 0.33, 0.67, 1.00, and 1.34 μM) were added to an abasic site containing oligonucleotide (0.05 μM) followed by incubation at 65 °C for up to 80 min (Figure 3A). Oligonucleotide substrates and products were resolved by electrophoresis and visualized with a STORM imager (Figure S11). One predominant band was seen corresponding to the β-elimination product in the gel assay. Each time course yielded an observed initial rate (kobs) that was subsequently plotted against increasing hyTDG-lyase to obtain kmax and Kd (Figure 3). The apparent Kd was found to be 0.22 ± 0.12 μM, and the kmax was found to be 4.20 ± 0.56 min–1. For comparison purposes, the kinetic properties of the hyTDG glycosylase for removal of U on a U:G substrate were measured under single-turnover conditions, as it is product inhibited by the resulting abasic site (Table S4 and Figure S12). The apparent Kd was found to be 0.16 ± 0.05 μM, and the kmax was 0.97 ± 0.06 min–1 (Figure S13). Fitted rate constants, their amplitude, and y-intercept are shown in Table S5.
Figure 3.

hyTDG-lyase kinetics. (A) To estimate the kmax and Kd of hyTDG-lyase, we incubated various concentrations of hyTDG-lyase: 0.02, 0.33, 0.67, 0.96, and 1.34 μM, at 65 °C with a FAM-labeled duplex oligonucleotide (0.05 μM) containing an AP:G site. This was done by first incubating a U:G oligonucleotide with UDG for 1 h at 37 °C to generate an abasic site. Product formation was monitored as a function of time using a gel and fit a single exponential (smooth curves) to individual data points (dots) to obtain the kobs for each concentration of hyTDG-lyase. The arrow represents increasing hyTDG-lyase concentration [E]. (B) We then fit our kobs data as a function of hyTDG-lyase concentration to determine kmax and Kd using a nonlinear hyperbolic fit (solid curve). Errors for kmax and Kd are reported as the standard error of the mean associated with the fit of the curve.
hyTDG-Lyase Is Active over a Broad Range of Temperatures
The activity of the hyTDG-lyase as a function of temperature was then compared with APE1 and the FPG (Figure 4). To test the AP lyase activity of the mutant protein hyTDG-lyase, an 18-base oligonucleotide duplex was constructed containing a U:G mispair and a 5′-FAM label (Figure S2). This duplex was incubated with UDG at 37 °C for 1 h to generate an abasic site. The hyTDG-lyase was then added, and the reaction mixture was incubated at defined temperatures from 25 to 95 °C. Substrate and product oligonucleotides were resolved by gel electrophoresis and imaged with a STORM imager. The hyTDG-lyase effectively cleaved the abasic site containing oligonucleotide from 25 to 95 °C (Figure 4A).
Figure 4.

hyTDG-lyase is thermostable. A FAM-labeled 18-base oligonucleotide containing a U:G mispair (2.5 pmol, 0.2 μM) was incubated with UDG (2.5 U, 0.84 pmol, 0.07 μM) at 37 °C for 1 h in TDG buffer. Then hyTDG-lyase (16.8 pmol, 1.34 μM), APE1 (5 U, 0.18 pmol, 0.01 μM), FPG (4 U, 6.44 pmol, 0.52 μM), or a no lyase control was added at the indicated temperatures for 1 h. (A) hyTDG-lyase cleaves oligonucleotides (S) at an abasic site generated by UDG at all temperatures tested and produces a PUA-βME adduct. (B) APE1 cleaves an abasic site from 25 to 45 °C to form a free 3′-OH but was inactive at higher temperatures (55–95 °C). Spontaneous β and β,δ-elimination occurred at the abasic site at higher temperatures (65–95 °C), resulting in both formation of a PUA and 3′-OPO3–, respectively. (C) FPG was highly active from 25 to 55 °C and produces a 3′-OPO3–. Its activity was greatly reduced at higher temperatures (65–95 °C). (D) As a control, we heated the abasic site containing oligo for the same duration at the indicated temperatures. Heating an abasic site for 1 h at 65–75 °C produced a β-elimination product. At higher temperatures, all the abasic sites were cleaved and we observed a mixture of both β and β,δ-elimination, upper and lower product bands respectively, seen in panels B–D.
To compare the activity of our hyTDG-lyase with other AP endonucleases and lyases, an abasic site containing duplex was also incubated with APE1 (Figure 4B) and FPG (Figure 4C). APE1 was active from 25 to 45 °C. It cleaves on the 5′-side of the abasic site to generate a 5′-FAM-labeled cleavage product with a 3′-OH terminus (Figure S3). At temperatures above 45 °C, cleavage is observed due to nonenzymatic β-elimination. FPG cleaves the abasic site-containing oligonucleotide from 25 to 55 °C. Spontaneous β-elimination is seen at higher temperatures as with APE1. Spontaneous β-elimination (higher band) and β,δ-elimination (lower band, 3′-phosphate) of abasic site-containing oligonucleotides were observed at temperatures above 55 °C (Figure 4D) in the absence of any AP lyase or endonuclease. Intact oligonucleotides containing no abasic sites did not spontaneously cleave under these conditions.
hyTDG-Lyase and Other Lyases Do Not Require Mg++
Next, we examined the activity of our hyTDG-lyase in various buffer systems (Figure 5). Using an 18-base oligonucleotide duplex containing a U:G mispair, the U was cleaved with UDG in UDG buffer to generate an abasic site. Either EDTA (2 mM) or Mg-acetate (10 mM) was then added to test the effect of Mg++ on the subsequent cleavage reaction. The abasic site was cleaved by hyTDG-lyase and the lyase activity of FPG and endonuclease III in the presence of EDTA. In contrast, the abasic site-containing oligonucleotide was not cleaved by APE1 in the presence of EDTA, but only when Mg++ was added. Our data indicate that hyTDG-lyase functions in the presence of either EDTA or Mg++.
Figure 5.

AP endonucleases but not AP lyases require magnesium. APE1 requires the presence of Mg2+ for strand cleavage, while FPG, Endo III, hOGG1, and hyTDG-lyase do not. This suggests that these bifunctional glycosylases and our hyTDG-lyase are AP lyases and not endonucleases. An abasic site was generated by treating an 18-base oligonucleotide containing uracil with UDG and subsequently adding the indicated enzyme. 2.5 units of UDG (0.84 pmol, 0.07 μM) for 1 h at 37 °C. This substrate was then treated with 5 units of APE1 (0.18 pmol, 0.01 μM), 4 units of FPG (6.44 pmol, 0.52 μM), 5 units of Endo III (0.13 pmol, 0.01 μM), 0.25 μg of hOGG1 (6.2 pmol, 0.5 μM), or 0.5 μg of hyTDG-lyase (16.8 pmol, 1.34 μM) for an addition 1 h. The reaction with hyTDG-lyase was incubated at 65 °C, while the others were held at 37 °C. Reactions were prepared in TDG buffer (10 mM K2HPO4, 30 mM NaCl, 40 mM KCl, pH 7.9) supplemented with either 2 mM EDTA or 10 mM Mg-Ac, as indicated. Each reaction had 2.5 pmol (0.2 μM) of oligo.
hyTDG-Lyase Maintains Some of the Base-Pairing Preference of hyTDG/MIG
The hyTDG glycosylase is specific for uracil analogues mispaired with G.41 The Y163K mutation converts the enzyme from a glycosylase to an AP lyase, but this mutation would not be expected to have a substantial impact on the preference of the AP lyase for an abasic site opposite G. To test the opposite-base preferences of hyTDG-lyase, a single-stranded oligonucleotide containing a uracil base, as well as U:G, U:A, U:C and U:T duplexes were incubated with UDG (Figure 6) to generate abasic sites. The abasic site containing oligonucleotides were then incubated with either NaOH or hyTDG-lyase. A NaOH control showed that UDG had completely removed uracil from all the oligonucleotides under these conditions (data not shown). The hyTDG-lyase was able to cleave 77% of the abasic site opposite to G substrates within 30 s, but only 14–20% of the other substrates. The original gel images are shown in Figure S14.
Figure 6.

hyTDG-lyase prefers cleaving an abasic site opposite G. hyTDG-lyase retains a preference for activity opposite G but can cleave abasic sites in single-stranded DNA and all other base-pairing contexts. (A) Representative gel of a 30 s reaction of hyTDG-lyase with an abasic site containing oligonucleotides in different base-pairing contexts. (B) Quantification of gel pictures (n = 3). Error bars represent the standard deviation. A 5′-FAM-labeled single-stranded oligonucleotide containing U was used or was annealed to a complementary strand containing either a G, A, C, or T opposite U. DNA substrates (2.5 pmol, 0.2 μM), were incubated with 2.5 units of UDG (0.84 pmol, 0.07 μM) in buffer at 37 °C for 1 h to generate an abasic site. hyTDG-lyase was then added, and cleavage of the abasic site was quantified after incubating at 65 °C for 30 s.
hyTDG-Lyase 3′-End Is Not Extendable by BER
The 3′-terminus of an oligonucleotide generated by hyTDG-lyase would be a β-ME-substituted deoxyribose-3′-phosphate (Figure 2) which, presumably, could not be extended by Pol β. To verify this expectation, we created a fluorescently tagged 79/77 base pair oligonucleotide duplex containing a central U:G mispair as described previously (Figure S15).51 The intact 79/77 base duplex is shown in Figure 7 lane 1. When this duplex was incubated with UDG for 1 h followed by APE1 for 30 min at 37 °C, the upper FAM-labeled strand (green) was cleaved (lane 2). When UDG, APE1, Pol β, dCTP, E. coli ligase, and NAD+ were added, the abasic site was repaired and filled with dCTP and the resulting DNA nick was ligated resulting in 93% full-length oligo (lane 3).
Figure 7.

3′-terminus produced by hyTDG-lyase cannot be extended by DNA Pol β but is resolved by APE1. In lane 1, A 5′-FAM-labeled 79 base oligonucleotide containing a U in a U:G mispair (2.5 pmol, 0.2 μM). In lane 2, the oligonucleotide was incubated with UDG (2.5 U, 0.84 pmol, 0.07 μM) for 1 h at 37 °C in CutSmart buffer, and subsequently, the abasic site was cleaved by incubating with APE1 (5 U, 0.18 pmol, 0.01 μM) for an additional 30 min. In lane 3, Pol β (6.2 pmol, 0.5 μM) and E. coli ligase (5 U, 4 pmol, 0.32 μM), dCTP (250 pmol, 20 μM), and NAD+ (325 pmol, 26 μM) were added for an additional 1 h to simulate short-patch BER. Lane 4 was otherwise identical to lane 2, but hyTDG-lyase was used instead of APE1 at 37 °C for 30 min. Lane 5 was otherwise identical to lane 3, except hyTDG-lyase (26.9 pmol, 2.15 μM) was used instead of APE1 at 37 °C. In lane 6, we similarly generated an abasic site that was then cleaved by hyTDG-lyase for 30 min at 37 °C. Then APE1, Pol β, dCTP, and E. coli ligase were added and incubated for an additional 1 h. Lane 6 demonstrates that the AP endonuclease activity of APE1 can clean up the PUA-βME 3′-end produced by the hyTDG-lyase. The complementary strand is labeled with a 5′-Cy5 fluorophore (red). The overlap between the full-length 5′-FAM-labeled oligo and its 5′-Cy5 complement is depicted as yellow.
The experiment was then repeated, but APE1 was replaced by hyTDG-lyase. Under these conditions, the U was removed by UDG (37 °C for 1 h) and the abasic site was cleaved by hyTDG-lyase by incubating at 37 °C for an additional 30 min (Figure 7, lane 4). However, after treating the U:G mispair in this manner, incubation with Pol β, dCTP, ligase, and NAD+ did not result in dCTP incorporation and ligation (lane 5). This contrasts with incubation with APE1 (lane 3) and is consistent with the observed 3′-end obtained with hyTDG-lyase (Figure 2).
In addition to its AP endonuclease activity, APE1 can remove a deoxyribose-5′-phosphate generated from an abasic site, generating a 3′-OH end which can be extended by Pol β. To test if APE1 could repair the 3′-end generated by hyTDG-lyase, the U:G mispaired oligonucleotide was first incubated with UDG at 37 °C for 1 h, then hyTDG-lyase at 37 °C for an additional 30 min, and finally incubated with APE1, Pol β, dCTP, and NAD+ for 1 h (Figure 7, lane 6). Both dCTP incorporation and ligation are observed. This experiment verified that the 3′-end generated by hyTDG-lyase cleavage could not be extended by Pol β (lane 5). However, if the end was subsequently treated with APE1, it could be repaired and extended with a DNA polymerase. These findings are similar to the Gates group that recently demonstrated that a glutathione adduct with an abasic site could be repaired by APE1.49 The original gel image is found in Figure S16.
Competition Between hyTDG and hyTDG-Lyase
We then performed a series of experiments to determine if hyTDG glycosylase and hyTDG-lyase could be used simultaneously (Figure 8). A 5′-FAM-labeled 18-base oligonucleotide duplex containing a U:G mispair (lane 1) was incubated with varying concentrations of hyTDG-lyase. Subsequent incubation with NaOH and examination of the product oligonucleotide by gel electrophoresis revealed 94% abasic site formation (lane 2). The experiment was then repeated with a fixed amount of hyTDG [0.67 μM] and increasing amounts of hyTDG-lyase (lanes 3–8). Increased oligonucleotide cleavage was observed up to the point where the concentrations of the hyTDG glycosylase and AP lyase were similar: 71% cleavage at equimolar hyTDG and hyTDG-lyase and 72% when there was a twofold excess of the hyTDG-lyase. However, when the amount of the hyTDG-lyase exceeded the glycosylase by a factor of four, oligonucleotide cleavage was reduced to 54% (Figure 8A). In addition, when the U:G mispair was incubated with hyTDG-lyase alone at the highest concentration [2.68 μM], no cleavage was shown, demonstrating no functional glycosylase activity (lane 9). These results show that the hyTDG-lyase has no apparent glycosylase activity. Furthermore, when hyTDG glycosylase and hyTDG-lyase are incubated together, they can compete with one another. At high enough concentrations of hyTDG-lyase, this can diminish overall cleavage (Figure 8B). The original gel images are found in Figure S17.
Figure 8.

hyTDG-lyase competitively inhibits hyTDG glycosylase. (A) 5′-FAM-labeled 18-baseoligonucleotides containing a U:G mispair (0.6 pmol, 0.05 μM) was treated with hyTDG (8.4 pmol, 0.67 μM) alone or simultaneously incubated with increasing concentrations of hyTDG-lyase (1.05, 2.1, 4.2, 8.4, 16.8, or 33.6 pmol) (0.08–2.68 μM) or hyTDG-lyase alone (33.6 pmol, 2.68 μM) in TDG buffer. To confirm removal of U following incubation with hyTDG, one sample was treated with NaOH. To confirm that hyTDG-lyase has no glycosylase activity, we had a hyTDG-lyase only control lane. All samples were incubated for 1 h at 65 °C. (B) Quantification of three independent experiments. Error bars represent the standard deviation.
Removing DNA Damage Products Prior to PCR and Next-Generation Sequencing
In the final experiment (Figure 9), a 79/77 base pair fluorescently labeled oligonucleotide was used to construct a duplex with a normal C:G base pair within a HpaII restriction enzyme recognition sequence (CCGG) and a corresponding duplex with a T:G mispair (CTGG), as shown in Figure S15. This experiment simulates the potential contamination of a normal C:G duplex with T:G mispairs that could arise from the deamination of 5mC.
Figure 9.
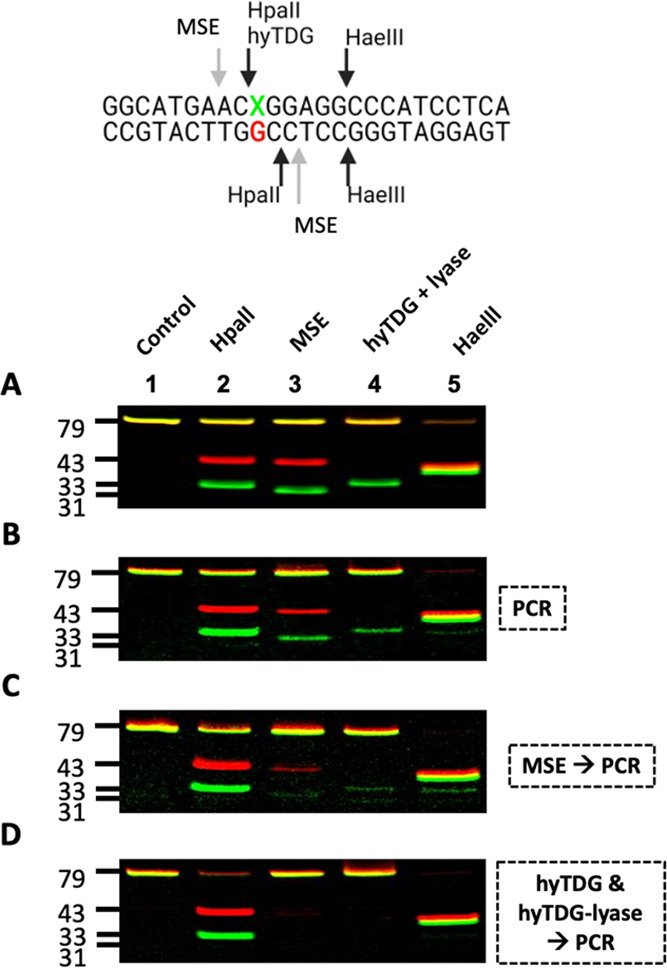
hyTDG and hyTDG-lyase are more thorough than MSE at removing T:G mismatches in DNA prior to PCR amplification for NGS applications. (A) 79/77 base oligonucleotide duplexes containing a C:G or T:G mismatch were prepared by ligation and mixed in a 0.9:1 ratio, respectively (X = C or T). This mixture was then treated with HpaII, MSE, hyTDG and hyTDG-lyase, or HaeIII (lanes 1–5). HpaII cleaves both strands of only a C:G oligo. MSE cleaves both strands of a T:G mispaired oligo with a two-nucleotide overhang 5′ to the T:G mismatch. hyTDG and hyTDG-lyase remove the T and cleave the T-containing strand. HaeIII cleaves only duplex DNA. (B) PCR amplification: Unlabeled C:G and T:G oligos were mixed in a 1:1 ratio with a 10% excess of the complementary G strand and then used as PCR template to amplify the DNA and attach fluorescent labels for visualization purposes. PCR products were column purified and treated with enzymes, as in A. (C) MSE then PCR: The same oligo mixture in B was first treated with MSE and then used as a PCR template. MSE does not remove the T and leaves overhangs, which can provide a template during PCR, resulting in amplification and T:A mutations. In lane 2, HpaII digestion shows a significant amount of full-length T:A oligo. To confirm this, in lanes 3 and 4, we see only minor cleavage with MSE or hyTDG and hyTDG-lyase. (D) hyTDG and hyTDG-lyase then PCR: Same as C except DNA was first treated with hyTDG for 1 h followed by the addition of hyTDG-lyase for an additional 1 h, at 65 °C. Quantification is found in Table S6.
Panel A represents the control characterization of the duplex. A 0.9:1 mixture of the C:G and T:G containing duplex oligonucleotides were treated with enzymes (Figure 9, panel A). The C:G oligonucleotide in the mixture was cut with HpaII (lane 2), and the T:G mispaired oligonucleotide was cleaved with the mismatch-specific endonuclease (MSE) (lane 3).52,53 The T-containing oligonucleotide (green gel band) of the T:G mismatch was removed by hyTDG, and the backbone was cleaved by the subsequent addition of hyTDG-lyase (sequential reaction). All of the oligonucleotides were cleaved by HaeIII, which has a GGCC recognition sequence common to all duplexes. A 0.9:1 mixture of ligated oligos was used to equalize the fluorescence between the batches of oligos and account for variance in the substrate concentration. Quantification of gels is shown in Figure 9 and is presented in Table S6.
An unlabeled mixture (1:1) of a C:G and T:G duplex was then PCR-amplified for 10 cycles with fluorescent primers (Figure 9). PCR amplification would amplify the C:G duplex, but the T:G duplex would be converted to a T:A duplex and a C:G duplex. The C:G and T:A duplexes would be expected in a 3:1 ratio. Examination of the products (Figure 9B) reveals increased cleavage of the mixture by HpaII as expected. The T:A duplex is not cleaved at the HpaII site. Surprisingly, the duplex mixture is also cleaved by MSE and hyTDG/hyTDG-lyase, indicating the presence of some remaining T:G duplex. The T:G duplex could arise during the heat denaturation/renaturation PCR step where a G-containing oligonucleotide would reanneal with a T-containing complementary strand.
The mixture of the C:G and T:G duplexes was first incubated with MSE under conditions that would lead to complete cleavage of the T:G duplex in both strands, prior to PCR amplification (Figure 9C). Following PCR amplification, the products were examined. As expected, the mixture was predominantly cleaved with HpaII, indicating that most, but not all of the product oligonucleotides contained C:G. However, a fraction of oligonucleotides after PCR were cleaved again by MSE (lane 3) and hyTDG/hyTDG-lyase (lane 4). This indicates that the T:G duplex reappeared following PCR amplification.
MSE cleavage generates fragments with 3′-OH and 5′-phosphate ends which could potentially serve as a primer and ligateable end, respectively. The MSE-cleaved T-containing oligonucleotide could serve as a template and generate a short T:A duplex. The A-containing strand can then serve as a primer and participate in PCR amplification and produce full-length T:A duplex. As in panel B, the C:G and T:A duplexes could swap complementary strands during heat denaturation and renaturation during PCR, resulting in a T:G mispaired duplex.
In Figure 9D, the mixture of the C:G and T:G duplexes was incubated with hyTDG followed by hyTDG-lyase under conditions that result in complete cleavage of the T-containing strand. As the T is removed from the oligonucleotide, and the abasic site is subsequently cleaved, the incubation product can no longer participate in strand elongation or PCR amplification. Following PCR amplification as mentioned above, the product duplex mixture is mostly cleaved by HpaII, indicating selective amplification of the C:G duplex. No cleavage products were observed following incubation by MSE or hyTDG/hyTDG-lyase, unlike pretreatment with MSE. This indicates that the original T:G duplex, simulating a deamination artifact, was completely removed from the starting C:G and T:G mixture. The original gel images are found in Figure S18.
Discussion
Our work presented here with the development of a hybrid thymine DNA AP lyase (hyTDG-lyase) reflects our efforts in developing useful genomic tools. These tools include identifying potential drugs that can selectively block selected DNA repair pathways, creating reagents useful for quantifying and identifying the sequence location of specific types of DNA damage and preparing biological DNA samples for NGS sequencing.
Rationale for Developing Tools to Measure Deaminated Cytosine and 5-Methylcytosine DNA Damage
One key example of the use of DNA repair enzymes for DNA damage studies includes the measurement of deamination products derived from 5mC.18 In higher organisms, the replacement of C with 5mC is important in the epigenetic control of gene expression and also for chromatin structure.31−33 Enzymatic DNA methylation usually occurs in the CpG dinucleotide, and C to T transition mutations at CpG dinucleotides represent the most frequent single-base change found in human tumors.34−37 The deamination of 5mC to T generates a T:G mismatch and, upon DNA replication, results in a C to T mutation. The T:G mismatch is persistent in human DNA because members of the uracil-DNA glycosylase superfamily that remove T from a T:G mispair have much weaker activity for T:G than for U:G, the product of cytosine deamination.38,39
The measurement of a T:G mispair presents some unique challenges. Traditional methods for the measurement of DNA adducts require the enzymatic or acid hydrolysis of DNA prior to the separation of the DNA nucleosides or bases and analysis by mass spectrometry-based methods. As T resulting from 5mC deamination is indistinguishable from T in a normal T:A base pair by such methods, hydrolysis would result in the loss of base-pairing context and elimination of the possibility of distinguishing T arising from a T:G mispair.
Although the T:G mispair is the intermediate in the formation of a disproportionate number of point mutations in human tumors,34−37 few glycosylases remove the mispaired T from DNA with reasonable efficiency. In 1996, Horst and Fritz identified a mismatch thymine DNA glycosylase (MIG) encoded by Methanobacterium thermoautotrophicum that removed both T and U when mispaired with G.40 The thermophile M. thermoautotrophicum has a restriction-modification system in which specific C residues are enzymatically converted to 5mC, although the specific sequence motif modified is yet to be identified.54M. tthermoautotrophicum has an optimum growth temperature of 65 °C, and it is presumed that the reason for the existence of MIG is to repair T:G mispairs derived from 5mC deamination.
Our previous studies examined the base selectivity of MIG.41 We observed that the activity of MIG for T:G was close to that for U:G, and further, several other 5-substituted uracil analogues were substrates as well. A series of purine analogues were placed opposite the target pyrimidine to determine if MIG-purine interactions dictated the selectivity for T:G over T:A. We observed that modification of any of the potential G base contact points, including the O6 carbonyl, the N3 imino proton, and the 2-amino group, diminished selectivity. The observed selectivity for T:G over T:A was approximately a million to one, a level of selectivity required to prevent inadvertent damage to normal T:A base pairs in DNA in vivo. Selectivity for MIG excision can be attributed in part to the reduced thermal stability of a mispair over a normal base pair and enzyme interactions with the widowed or orphaned guanine, a mechanism previously attributed to the interaction of the mispaired uracil glycosylase.55
The strong selectivity of MIG for T:G over T:A suggested to us that MIG might be useful for measuring T in T:G mispairs. However, to increase the activity of MIG, we explored making a hybrid enzyme inspired by the work of the Drohat group.56 Coey et al. demonstrated that a 29-amino acid peptide from the human TDG (hTDG) increased the overall glycosylase activity. The 29-amino acid sequence is unstructured, even when bound to DNA. It has an abundance of positively charged amino acids and is suggested to facilitate both specific and nonspecific DNA interactions. Although MIG does not turnover, we envisioned that the 29-amino acid arm would facilitate scanning of the genome, and so we created hyTDG. We previously demonstrated that in the presence of excess calf thymus DNA, hyTDG was faster than its MIG counterpart.18 We used hyTDG to remove T and U mispaired with G in calf thymus DNA, which was separated by spin filtration and measured by GC–MS/MS, thus allowing for the first measurement of T:G mispairs in a DNA sample.18
Single-turnover kinetic studies on MIG reported by Begley and Cunningham revealed a kst (kmax) of 0.83 ± 0.10 min–1, similar to the value of 0.68 min–1 measured by Mol and Tainer.57,58 The maximal rate constant for our hyTDG (Figure S13) is similar, 0.97 ± 0.15 min–1 at 65 °C using our 18-base nucleotide containing a U:G mispair. The 29-amino acid peptide from hTDG that we appended to hyTDG would not be expected to change the maximal rate of cleavage when the enzyme was saturating. Instead, we envisioned that the appended peptide might facilitate DNA scanning and target location.
In 2003, Begley and Cunningham reported additional enzymatic properties of MIG.57 MIG is a member of the helix-hairpin-helix (HhH) family of glycosylases which includes endonuclease III and as with many other monofunctional glycosylases, MIG was shown to bind tightly to its enzymatic product, an abasic site. Examination of the crystal structure of the MIG protein, by analogy with endonuclease III bound to a DNA substrate, suggested mechanisms for the target selectivity of MIG.58 Whereas UDG excludes T from binding to the pyrimidine binding pocket with a tyrosine residue that acts as a steric gate,59 the Tyr126 in MIG is essential for T recognition and assisting with twisting the T by ∼90°, facilitating water attack on the C1′ position and base excision.58
Characterization of a Compatible AP Lyase to Cleave the Resulting Abasic Site Following hyTDG Glycosylase
Having identified an enzyme that could selectively excise T and T analogues from mispairs in DNA, we sought to identify proteins that could cleave the resulting abasic site. Begley and Cunningham explored the impact of selected mutations on MIG based on homology with Endo III.43 The lysine at position 120 acts as a nucleophile, attacking the C1′ of the target pyrimidine and displacing the base. Lys120 can then form a Schiff base, catalyzing β-elimination and cleavage of the phosphodiester backbone, accounting for the dual glycosylase/lyase activities. The analogous position in MIG is Tyr126, which forms part of the extrahelical base recognition pocket. Upon the basis of the structural similarity between MIG and endonuclease III, these investigators proposed that the replacement of MIG Tyr126 with Lys (Y126K) might convert the glycosylase to a bifunctional glycosylase/lyase. The MIG Y126K mutant was found to have high-affinity binding to an oligonucleotide containing a U:G mismatch, but no U:G glycosylase activity. Instead, MIG Y126K had lyase activity on abasic sites.43
Motivated by this study, we prepared the corresponding mutant of our hyTDG, which we call hyTDG-lyase.43 The amino acid sequence of the Y163K mutant was verified by mass spectrometry analysis of tryptic peptides (Figure 1). The purity and approximate MW (theoretical 29.7 kDa) of the protein were demonstrated by gel electrophoresis (Figure S1). Examination of the products of the lyase reaction on an 18-base oligonucleotide duplex containing an abasic site opposite G revealed that hyTDG-lyase generated a 5′-phosphate 11-base product and a 5′-FAM-labeled 6-base product with a 3′-terminus containing an abasic deoxyribose β-ME adduct (Figure 2). We propose that the lyase activity generates an α,β-unsaturated aldehyde, which then reacts with β-ME in the enzyme buffer. Reports from other laboratories have described similar adducts with oligonucleotides containing 3′-α,β-unsaturated sugars.47−49
Previously, it was shown that both MIG and MIG-Y126K bound to the U:G mispair, as well as to an oligonucleotide containing an abasic site, with a similar affinity. Mol et al. measured the affinity of MIG (Kd 2.5–2.6 μM) and MIG-Y126K (Kd 2.9–3.1 μM) using a nonhydrolyzable tetrahydrofuran-containing oligonucleotide opposite G and a gel shift assay at room temperature.58 In our studies, we measured an apparent Kd by measuring the cleavage rate as a function of enzyme concentration at 65 °C (Figure 3). In our study, the Kd for hyTDG to a U:G duplex was measured to be 0.16 ± 0.05 μM and for hyTDG-lyase to the abasic-site oligo 0.22 ± 0.12 μM. When incubated with 0.5 molar equivalents of an abasic site substrate, approximately half of the substrate was cleaved in 80 min. We therefore measured the kinetics of cleavage under single-turnover conditions. The turnover number for hyTDG-lyase on an abasic site opposite G substrate was measured to be 4.20 ± 0.56 min–1. Although our kinetic results indicated that hyTDG-lyase bound tightly to its product, we saw no evidence of an irreversible oligonucleotide–protein complex in our gel assays. As with Endo III, hyTDG-lyase forms a Schiff base intermediate with the abasic site, which would be expected to increase its affinity to the abasic site containing oligonucleotide.
The binding of MIG to an abasic site is reported to be strongest when opposite G and weakest when opposite A.57,58 However, the relative magnitude changes by less than a factor of 4 upon changing the opposing base with hyTDG-lyase. Similarly, the binding of MIG (Y126K) to a series of oligonucleotides containing a nonhydrolyzable abasic site showed that binding was highest opposite G, and differences among the other bases varied by less than a factor of 4.58 In accord with prior studies with MIG, our results (Figure 6) show that hyTDG-lyase similarly will preferentially cleave an abasic site in a duplex when opposite G. However, it also cleaves an abasic site opposite C, T and A as well as an abasic site in a single-stranded oligonucleotide. Collectively, these data show that the very high substrate selectivity of MIG/hyTDG for T:G over T:A also depends upon the active site tyrosine in the glycosylase that shapes the active site pocket by binding with and orienting the target pyrimidine. In contrast, when the tyrosine is mutated to lysine to make hyTDG-lyase, the preference for abasic site cleavage opposite G diminishes substantially.
Begley and Cunningham originally mutated the active site tyrosine of MIG to a lysine to determine if a monofunctional glycosylase could be converted to a bifunctional glycosylase/lyase.43 The resulting MIG (Y126K) mutant, as with our hyTDG-lyase, lost glycosylase activity on undamaged pyrimidines but gained lyase activity on oligonucleotides containing an abasic site. Due to the wide buffer and temperature conditions compatible with both hyTDG and hyTDG-lyase, we considered the possibility that the pair of proteins could be used simultaneously to generate a bifunctional activity, even if not on the same protein. On the other hand, the glycosylases (MIG and hyTDG) show single-turnover kinetics as they bind tightly to the product abasic site. The tyrosine to lysine mutant lyases retains affinity for the original mismatched U:G or T:G mispairs. The similarity in the binding properties of the MIG/hyTDG glycosylases and lyases suggests that they could also interfere with one another.
In Figure 8, we show that abasic sites generated by hyTDG can be cleaved by hyTDG-lyase when both proteins are incubated simultaneously with a target oligonucleotide. However, under conditions where hyTDG results in the generation of ∼94% abasic sites (lane 2), the combination of hyTDG and hyTDG-lyase results in maximal cleavage of ∼72% (lane 8). At even higher hyTDG-lyase concentrations, oligonucleotide cleavage is reduced to ∼54%, suggesting the hyTDG-lyase might bind to the U:G mispair and block hyTDG excision.
Applications of hyTDG-Lyase: Next-Generation Sequencing
Having extensively characterized hyTDG-lyase, we examined other properties of hyTDG-lyase that are important in determining potential applications. We show that hyTDG-lyase maintains the thermal stability demonstrated for hyTDG (Figure 4). We also found that hyTDG-lyase can function in the presence or absence of Mg++ (Figure 5). Both properties extend the range of conditions in which hyTDG-lyase might be used.
Sequences obtained by next-generation DNA sequencing (NGS) are cluttered with minor sequence variants that could represent true underlying mutations or might have arisen due to DNA damage occurring during DNA isolation, storage, or PCR amplification.22−26 Several groups are currently examining the use of DNA repair enzymes to remove damaged DNA adducts from biological samples prior to NGS.23,25,26 During DNA extraction, processing, and thermal cycling, DNA bases can be deaminated, oxidized, and DNA strands can be depurinated. These damage events can result in sequencing errors, increased noise, and decreased sensitivity.
The removal of T arising from 5mC deamination remains a problem.25 C deamination results in a U:G mispair that can miscode and result in a C > T transition mutation artifact. Similarly, 5mC deamination results in a T:G mispair, which also causes the same artifact in sequencing. While a U:G mispair can be repaired by UDG, T:G mispairs are much more challenging to remove.23,25 5mC also deaminates at a similar rate to C60 but is much more poorly repaired by the human enzymes believed to be involved in their repair.38,39 Consequently, T:G mispairs are believed to be one of the most common DNA damage adducts. Until recently, enzymes that remove T:G efficiently were not readily available.18 In addition, emerging methods, based upon NGS, are creating new opportunities to quantify and locate specific damage or enzymatic modifications in DNA isolated from biological sources using DNA repair enzymes.61 Our hyTDG/hyTDG-lyase combination might assist with the reduction of 5mC deamination artifacts and aid in the detection of endogenous T:G mispairs.
Incubation of the DNA sample at various stages of NGS library preparation with repair enzymes could increase the quality of the NGS results. For example, abasic sites can be generated by both glycosylase removal of damaged bases as well as depurination. Abasic sites can block the progression of polymerases, but they can also miscode, generating artifact DNA sequencing data. The thermostable hyTDG-lyase described here could be included in the DNA processing protocol where it would continuously cleave abasic sites, including those resulting from spontaneous depurination. As shown in Figure 7, the 3′-ends generated by hyTDG-lyase, in contrast to those generated by APE1, would not be extendable. Therefore, damaged strands would be eliminated from subsequent PCR amplification. We then demonstrated for the first time how we could deplete T:G mispairs prior to PCR amplification, using hyTDG and hyTDG-lyase (Figure 9D).
In summary, we have generated a hyTDG-lyase by introducing point mutation to our hyTDG.18 The hyTDG-lyase binds tightly to its product and does not cycle in kinetic studies. However, the hyTDG and hyTDG-lyase can function sequentially under identical buffer conditions and temperature to eliminate T from T:G mispairs. We used this approach to demonstrate the elimination of a potential T:G contamination prior to PCR amplification. This approach provides a novel method to purge a DNA sample of T:G mispairs, arising from 5mC deamination, prior to NGS analysis. This approach could similarly be exploited to map the sequence location of T:G mispairs in a DNA sample, potentially allowing the rates of formation and repair of the important T:G mispair to be measured in biological samples.
Experimental Procedures
Enzymes
DNA repair enzymes uracil-DNA glycosylase (UDG, #M0280S), human apurinic/apyrimidinic endonuclease 1 (APE1, #M0282S), formamidopyrimidine DNA glycosylase (FPG, #M0240S) and E. coli DNA ligase (ligase, #M0205S), endonuclease III (Endo III, #M0268S), and TTH endonuclease IV (TTH, #M0294S) were obtained from New England Biolabs (NEB). Mismatch-specific endonuclease I (MSE, #M0678S) and restriction enzymes HpaII (#R0171S) and HaeIII (#R0108S) were also purchased from NEB. Human 8-oxoguanine DNA glycosylase (hOGG1, #NBP1-45318-0.1mg) and DNA polymerase β (Pol β, #NBP1-72434-0.5mg) were purchased from Novus Biologicals. Our hybrid thymine DNA glycosylase (hyTDG) was prepared as previously described.18
Buffers
The following buffers were used in this study: CutSmart buffer (NEB, #B6004): 50 mM potassium acetate, 20 mM Tris-acetate, 10 mM magnesium acetate, 100 mg/mL bovine serum albumin, pH 7.9; UDG buffer (NEB, #B0280SVIAL): 20 mM Tris–HCl, 1 mM dithiothreitol, 1 mM EDTA, pH 8.0; NEBuffer 2.1 (NEB, #B7002S): 50 mM NaCl, 10 mM Tris–HCl, 10 mM MgCl2, 100 μg/mL BSA, pH 7.9; NEBuffer 4 (NEB, #B7004S): 50 mM potassium acetate, 20 mM Tris-acetate, 10 mM magnesium acetate, 1 mM DTT, pH 7.9. TDG buffer: 10 mM K2HPO4, 30 mM NaCI, 40 mM KCI, pH 7.7.
Oligonucleotide Synthesis
All 18-base oligonucleotides were synthesized on an Expedite 8909 synthesizer using phosphoramidites from Glen Research (Sterling, VA). The 18-base oligonucleotides containing U, or the unlabeled complementary strand, were synthesized using standard phosphoramidites (Bz-dA, Bz-dC, iBu-dG, dT) and a 6-fluorescein (FAM) phosphoramidite without DMT. Oligonucleotides were deprotected in ammonium hydroxide at 60 °C for 15 h. The top strand sequence is 5′-FAM-CGT GGC UGG CCA CGA CGG-3′, and the bottom strand sequence is 5′-CCG TCG TGG CCX GCC ACG-3′, where X = G, A, C, and T. A 3′-BHQ1 CPG column was used for the synthesis of complementary G oligonucleotide used in the MALDI mass spectrometry assays.
HPLC purification of oligonucleotides was performed on a Hewlett Packard 1050 HPLC with a PDA detector. DMT-on oligonucleotides were purified using a Hamilton PRP-1 column (10 × 250 mm) and a gradient of acetonitrile in 10 mM potassium phosphate, pH 7.4. Detritylation of the complementary G oligonucleotide was performed using 2% trifluoroacetic acid. DMT-off oligonucleotides were purified using a Phenomenex Clarity-RP column (4.6 × 250 mm) and a gradient of acetonitrile in water.
Preparation of the Expression Vector and Site-Directed Mutagenesis to Generate hyTDG-Lyase
To introduce Y163K point mutation to hyTDG,18 site-directed mutagenesis PCR was performed using a Q5 Site-Directed Mutagenesis Kit (NEB, #E0554) and pET-28a(+)-his-hyTDG plasmid DNA as a template, and with forward primer 5′-TGTGGGCAAAAAAACCTGCGCGG-3′, where desired bases are underlined, and reverse primer 5′-CCCGGCAGATCCAGAATCG-3′ according to the manufacturer’s protocol for the kit, with an annealing temperature of 69 °C. A fraction of the PCR product was used for kinase/ligation/digestion reactions and further transformed into DH5α competent cells provided with the kit according to the manufacturer’s protocol. Antibiotic-resistant clones were selected on Luria broth (LB)-agar plates containing kanamycin (50 μg/mL) and inoculated in 5 mL LB (Fisher Scientific, #BP9723-500). After overnight culture, plasmid DNA was purified from the NEB 5-alpha competent cells, using a plasmid DNA mini prep kit (NEB, #T1010) following manufacturer’s instructions. The coding sequence was confirmed by Sanger sequencing for N-terminal 6× His tagged hyTDG-lyase.
Expression and Purification of hyTDG-Lyase
Plasmid DNA was transformed to E. coli strain BL21 (DE3) (NEB, #C2527). Transformants were selected on LB plates containing 1.4% agar and kanamycin (50 μg/mL). Expression of the target protein was confirmed by SDS-PAGE and Coomassie brilliant blue staining in a small-scale culture after induction with IPTG (1 mM). Selected clones were further cultured in 100 mL LB (Fisher Scientific, #BP9723-500) containing kanamycin (50 μg/mL) at 37 °C on a shaker (250 rpm) until the optical density reached to 0.4–0.8 at 600 nm.
The expression of His-tagged hyTDG-lyase was induced with IPTG (1 mM) at 250 rpm and 30 °C for 6 h. The cells were harvested by centrifugation at 4100 rpm for 5 min and stored −80 °C until use. The purification of the target protein was performed as previously described with slight modification.18 Briefly, the cell pellet was thawed and suspended in 4 mL of lysis buffer and sonicated on ice. After removal of cell debris by centrifugation, the supernatant was loaded on previously equilibrated HisPur Ni NTA Resin (Thermo Scientific, #88221) and incubated for 1.5 h at 4 °C on a see-saw shaker. The suspension of HisPur Ni NTA Resin beads and cell lysate was centrifuged using a centrifuge column (Pierce, #89896) at 1000 g, 4 °C for 5 min. The beads were washed with 3 mL of wash buffer A (2×), 3 mL of wash buffer B (2×), and 3 mL of wash buffer C (6×). Bound protein was eluted from the beads in 1.2 mL of elution buffer. Protein concentration was quantified with a Bradford protein assay (Bio-Rad, #5000006) using bovine serum albumin as a standard. Purified protein was resolved by gel electrophoresis [12% Tris-Glycine PAGE (Bio-Rad, #4561044) and Coomassie blue staining], and the purity of the target protein band was determined by densitometry using ImageJ software, using a picture obtained after separation of the protein.
Proteomic Verification of the Protein Sequence
Proteomics was performed as previously described.18 10 μg of hyTDG-lyase protein was separated by SDS-PAGE. The gel bands with molecular weight around 26.5 kDa were removed from the gel and destained with 50% methanol in water. Gel bands were dried under reduced pressure and suspended in 50 μL of acetic anhydride and 200 μL of acetic acid to acetylate protein lysine residues and incubated at 37 °C on a shaker for 1 h. Liquid was decanted, and the gel bands were washed three times with deionized water (1 mL). Washed gel bands were dried and ground into a fine powder with a tip-sealed 200 μL pipette tip. 100 μL of buffer (50 mM NH4HCO3) was added, and the pH of the resultant jelly was adjusted to be approximately 8 using NH3·H2O. 2 μg of trypsin was added to the sample and digested overnight at 37 °C. Digested peptides were extracted with acetonitrile, dried, and resuspended in 50 μL of 1% formic acid for nLC-MS/MS analysis.
Peptide mixtures were separated by reversed-phase liquid chromatography using an Easy-nLC 1200 equipped with an autosampler (Thermo Fisher Scientific, #LC140). A PicoFrit 25 cm length × 75-μm id, ProteoPep analytical column packed with a mixed (1:1) packing material (Waters XSelect HSS T3, 5 μm, and Waters YMC ODS-AQ, S-5, 100 Å) was used to separate peptides by reversed-phase liquid chromatography (solvent A, 0.1% formic acid in water; solvent B, 0.1% formic acid in acetonitrile), with a 100 min gradient from 2 to 45% of solvent B with a flow rate of 300 μL/min. The QExactive mass analyzer was set to acquire data at a resolution of 35,000 in full scan mode and 17,500 in MS/MS mode. The top 15 most intense ions in each MS survey scan were automatically selected for MS/MS.
Peptides were identified with PEAKS 8.5 (Bioinformatics Solutions Inc., On, Canada) to perform a de novo sequencing-assisted database search against the hyTDG-lyase protein sequence. Acetylation of lysine, serine, threonine, cysteine, tyrosine, and histidine (K, S, T, C, Y, and H), oxidation of methionine, and deamination of asparagine and glutamine were set as variable modifications. The false discovery rate was estimated by the ratio of decoy hits over target hits among peptide spectrum matches. The maximum allowed −10log(q-value) is greater than or equal to 15.
MALDI Mass Spectrometry
A 20 μM stock solution containing one equivalent of an 18-base U-containing oligonucleotide and two equivalents of the complementary oligo with a 3′-BHQ1 and a G directly opposite U in TDG buffer was prepared. From this stock solution, a 5 μL aliquot (100 pmol) was treated in a 25 μL reaction containing 25 pmol of hyTDG and 12.5 pmol hyTDG-lyase, or 20 U of TTH (12.4 pmol), at 65 °C for 2 h. In addition, U:G oligonucleotides were also incubated with 1 U of UDG (0.34 pmol) and 10 U of APE1 (0.36 pmol), 8 U of FPG (12.8 pmol), 10 U of Endo III (0.26 pmol), 1 μg of hOGG1 (24.5 pmol), or DMDA (Sigma, #D157805-5G) (100 mM) at 37 °C for 2 h to generate additional cleavage products. Reactions containing hOGG1 or TTH used 1× NEB4 Buffer (50 mM potassium acetate, 20 mM Tris-acetate, 10 mM Mg-acetate, 1 mM DTT, pH 7.9); otherwise, all other reactions used 1× TDG buffer. Chemical cleavage with NaOH was done after the 2 h incubation with UDG alone followed by addition of 1 M NaOH to a final concentration of 0.17 M and heated at 95 °C for 10 min.
Reaction samples were then desalted using Bio-Rad Micro Bio-Spin 6 columns (Hercules, CA), eluted, dried in vacuo, and resuspended in 5 μL distilled water with 2 μL of ammonium cation exchange resin for 40 min.45,46 Aliquots (1 μL) were then placed on a MALDI plate and spotted with 1 μL of 3-hydroxpicolinic acid matrix (70 mg/mL 3-HPA, 10 mg/mL diammonium citrate, in 50/50 ACN/distilled water and 0.1% trifluoroacetic acid).
Samples were analyzed with a high-resolution MALDI-ToF-ToF Ultraflextreme (Bruker, MA) to identify cleavage products following glycosylase and lyase reactions. The reflectron positive ion mode was used with the “ultra” laser beam parameter set, and laser fluency was manually optimized for oligonucleotide standards (∼70%). Pulsed ion extraction was set to 170 ns, IS2 voltage: 17.85 kV and lens: 7.50 kV. Mass accuracy was calibrated using Bruker’s low molecular weight oligonucleotide standard mixture prior to data acquisition using a cubic enhanced fit. A minimum of 1000 spectra were acquired per spot. Data were exported into Mmass, using the Bruker CompassXport software, and then baseline-corrected and Savitsky–Golay-smoothed. MALDI spectra are plotted using the PRISM software.
hyTDG-Lyase Thermostability and Preference Assay
To examine if hyTDG-lyase is thermostable, A 5′-FAM-labeled 18-base oligonucleotide duplex containing U:G (2.5 pmol, 0.2 μM) was incubated with 2.5 units of UDG (0.84 pmol, 0.07 μM) at 37 °C for 1 h. After confirmation of abasic site generation using NaOH, samples were treated with hyTDG-lyase (16.8 pmol, 1.34 μM), 5 units of APE1 (0.18 pmol, 0.01 μM), 4 units of FPG (6.44 pmol, 0.052 μM), or with no enzyme control samples for 1 h at indicated temperatures. To examine the preferences for opposite nucleotide to the abasic site, 18-base oligonucleotide containing U, or U:G, U:A, U:C and U:T (2.5 pmol, 0.2 μM) duplexes were pretreated with 2.5 units of UDG (0.84 pmol, 0.07 μM) for 1 h at 37 °C. Then abasic site containing oligonucleotides were treated with hyTDG-lyase (16.8 pmol, 1.34 μM) at 65 °C for 30 s. Finally, samples were mixed with an equal volume of formamide and resolved in denaturing 20% polyacrylamide gel.
hyTDG and hyTDG-Lyase Competition Assay
To test if hyTDG and hyTDG-lyase can be used simultaneously, a 5′-FAM-labeled 18-base oligonucleotide containing a U:G (0.6 pmol, 0.05 μM) was treated with hyTDG (8.4 pmol, 0.67 μM) alone or co-incubated with increasing concentrations of hyTDG-lyase (2.1, 4.2, 8.4, 16.8, or 33.6 pmol) (0.8–2.68 μM) or hyTDG-lyase alone (33.6 pmol, 2.68 μM) at 65 °C, for 1 h in TDG buffer. To confirm the removal of U following incubation with hyTDG, one sample was treated with NaOH. To confirm that hyTDG-lyase had no glycosylase activity, we included a hyTDG-lyase-treated sample (hyTDG-lyase only) as a control lane.
Gel Electrophoresis and Quantification
To separate 5′-FAM-labeled 18-base oligonucleotides after glycosylase excision and AP-site cleavage reactions, samples were mixed with an equal volume of formamide and loaded into a 20% polyacrylamide gel containing 6 M urea and run at 180 V for 35–45 min in 1× TBE buffer.
To separate the dual-labeled (FAM and Cy5) 79-base oligonucleotides after repair reactions, samples were mixed with an equal volume of formamide, heated to 95 °C for 1 min, and loaded onto a 15% polyacrylamide gel containing 8 M urea and run at 180 V for 50 min in 1× TBE buffer.
Gels were visualized using a STORM 860 gel imager. FAM and Cy5 scans were adjusted for brightness and contrast, pseudo-colored, and overlayed. Gel band peak areas were quantified in ImageJ (Version 1.53 k).
Kinetic Analysis
Single-turnover kinetics data were processed as described previously.62−64 For kinetic experiments with hyTDG-lyase, a 5′-FAM-labeled 18-base oligonucleotide duplex containing a U:G mispair (0.6 pmol, 0.05 μM) was treated with 2.5 U of UDG (0.84 pmol, 0.07 μM) at 37 °C for 1 h to generate an abasic site. The oligonucleotide containing an abasic site was then incubated with different concentrations of hyTDG-lyase (0.026–16.8 pmol, 0.02–1.34 μM) at 65 °C for indicated amounts of time (min). Reactions were quenched by adding an equal volume of formamide and storing on ice. The total reaction volume for all experiments was 12.5 μL and used 1× TDG buffer.
For kinetic experiments with our hyTDG glycosylase, the same FAM-labeled U:G oligonucleotide (0.6 pmol, 0.05 μM) was treated with different concentrations of hyTDG ranging from 0.16 to 2.68 μM (2–33.6 pmol) at 65 °C for the indicated time period (min). Reactions were quenched by adding 1 M NaOH to a final concentration of 0.16 μM. Samples were then heated at 96 °C for 8 min to cleave the product at the abasic site. Results were visualized by gel electrophoresis and quantified.
The observed product formation (% cleavage) was monitored as a function of time. We fit these data using a single exponential fit in PRISM 9.4.1 (eq 1).
| 1 |
Y represents the percent product, A represents the amplitude, t represents time (min), kobs represents the observed rate constant in units of min–1, and C is the y-intercept. The y-intercept was constrained to be greater than 0, and both A and kobs were fit (Table S5).
To determine the maximal enzyme rate, kmax, and the concentration of enzyme required to obtain half kmax, Kd, we titrated either hyTDG or hyTDG-lyase concentration with a constant amount of substrate. Each reaction was monitored over time and fit to a single exponential, as described, to obtain kobs. Then, kobs as a function of enzyme concentration was fit with a hyperbolic fit (eq 2) using PRISM 9.4.1.64kmax is reported in units of min–1 and Kd, in units of concentration (μM).
| 2 |
AP Endonuclease and Lyase Magnesium Dependency
An abasic site was generated by treating an 18-base oligonucleotide duplex 2.5 pmol (0.2 μM) containing uracil opposite G with 2.5 units of UDG (0.84 pmol, 0.07 μM) for 1 h at 37 °C. This substrate was then treated with 5 units of APE1 (0.18 pmol, 0.01 μM), 4 units of FPG (6.44 pmol, 0.52 μM), 5 units of Endo III (0.13 pmol, 0.01 μM), 0.25 μg of hOGG1 (6.2 pmol, 0.5 μM), or 0.5 μg of hyTDG-lyase (16.8 pmol, 1.34 μM) for an addition 1 h. The reaction with hyTDG-lyase was incubated at 65 °C, while the others were held at 37 °C. Reactions were prepared in TDG buffer supplemented with either 2 mM EDTA or 10 mM magnesium acetate, as indicated. Results were visualized by gel electrophoresis.
Construction of Dual Fluorescently Labeled Duplexes
The construction of dual 5′-FAM/Cy5-labeled 79/77-base oligonucleotide duplexes was described previously.51 Briefly, we ligated six oligonucleotides purchased from Integrated DNA Technologies to form a full-length duplex (Figure S15). The full-length product was purified on an 8 M urea denaturing gel.
Short-Patch Repair with a Fluorescent Oligonucleotide
A 79-base oligonucleotide duplex was prepared containing an upper strand that has a 5′-FAM label and contained U, while the complementary strand was 5′-Cy5-labeled and contained a G opposite the U to produce a U:G mispair. An enzymatic repair reaction was performed in three sequential steps: glycosylase treatment, cleavage, and repair. Each 12.5 μL reaction initially consisted of 79 base U:G-containing oligonucleotide (2.5 pmol, 0.2 μM), UDG (2.5 units, 0.84 pmol, 0.07 μM), dCTP (250 pmol, 20 μM), and NAD+ (325 pmol, 26 μM), as indicated in 1× CutSmart buffer. In the glycosylase (UDG) reaction step, samples were incubated for 1 h at 37 °C to allow for removal of U and creation of AP-site abasic sites. Next, cleavage was performed by adding APE1 (5 U, 0.18 pmol, 0.01 μM) or hyTDG-lyase (26.9 pmol, 2.15 μM) to the glycosylase reactions. Samples were incubated for an additional 30 min at 37 °C to allow for cleavage of the phosphodiester backbone. Repair reactions were completed by adding Pol β (6.2 pmol, 0.5 μM) and E. coli ligase (5 U, 4 pmol, 0.32 μM) to simulate short-patch BER and incubating for an additional 1 h at 37 °C.
To determine if the 3′-end left by hyTDG-lyase could be removed by APE1 and repaired, we first incubated the abasic site with hyTDG-lyase for 30 min at 37 °C. Subsequently, we added APE1 along with Pol β and ligase and incubated for an additional 1 h. Results were visualized by gel electrophoresis and quantified.
Removing T:G Mispairs before PCR Amplification
Cleavage of Ligated Control Oligos
Ligated 79/77 base duplexes containing a mixture of a correct C:G and mispaired T:G were prepared and mixed in a 0.9:1.0 ratio. Enzymatic reactions were prepared in 12.5 μL of appropriate buffer, 2.5 pmol of each oligo duplex (0.2 μM each), 10 units HpaII, 2 units MSE (44.9 pmol), 10 units HaeIII, or 16.8 pmol hyTDG followed by 16.8 pmol of hyTDG-lyase (1.34 μM each) as indicated. CutSmart buffer was used with HpaII and HaeIII. NEBuffer 2.1 was used with MSE, and TDG buffer was used with hyTDG. Reactions were incubated for 2 h at 37 °C, except for hyTDG containing reactions. hyTDG reactions were first incubated at 65 °C for 1 h to generate an abasic site, and then hyTDG-lyase was added and the incubation was continued for an additional 1 h. Results were visualized by gel electrophoresis and quantified (Table S6).
PCR Reactions
Unlabeled DNA was used for PCR reactions. Unlabeled full-length oligos were purchased from IDT and annealed with a 10% excess of the complementary G strand. Enzymatic reactions were prepared in 12.5 μL of appropriate buffer, 2.5 pmol of each oligo duplex (0.2 μM each), and 2 units MSE (44.9 pmol) or 16.8 pmol hyTDG, followed by 16.8 pmol of hyTDG-lyase (1.3 μM each) as indicated. Reactions were incubated for 2 h at 37 °C. CutSmart buffer was used with HpaII and HaeIII. NEBuffer 2.1 was used with MSE, and TDG buffer was used with hyTDG. hyTDG reactions instead were first incubated at 65 °C for 1 to generate an abasic site, then hyTDG-lyase was added and the incubation was continued for an additional 1 h. After incubation, 1 μL (0.2 pmol of each duplex) was used as the template for PCR. PCR reactions consisted of 2.5 μM of each fluorescent primer (oligos 1 and 6 from Figure S15), 300 μM dNTPs, 5 units LongAmp Taq (NEB, #E5200S), and 1 μL template DNA in 25 μL of LongAmp buffer. Reactions were placed into a thermocycler preheated to 94 °C for 30 s and then cycled 10× at 94 °C for 20 s and 60 °C for 15 s, with a final extension at 60 °C for 5 min.
DNA Cleanup
An excess of primers and polymerase remained in PCR reactions which interfere with visualization and cause band shifting. PCR product (25 μL) was purified using a Monarch PCR and DNA Cleanup Kit (NEB, #T1030S) to remove the primers, polymerase, and dNTPs. The following modifications to the protocol were made: after passing DNA through the column the first time, the flowthrough was collected and passed through a second time to increase total yield; DNA was eluted in 20 μL.
Post-PCR Enzymatic Reactions and Visualization
Post-PCR enzymatic reactions were prepared in 12.5 μL of appropriate buffer, 1 μL of PCR product (approximately 2.5 pmol), 10 units HpaII, 2 units MSE (44.9 pmol), 10 units HaeIII, or 16.8 pmol hyTDG followed by 16.8 pmol of hyTDG-lyase (1.34 μM each) as indicated. CutSmart buffer was used with HpaII and HaeIII. NEBuffer 2.1 was used with MSE and TDG buffer was used with hyTDG. Reactions were incubated for 2 h at 37 °C except for hyTDG containing reactions. hyTDG reactions were first incubated at 65 °C for 1 to generate abasic sites, then hyTDG-lyase was added, and the incubation was continued for an additional 1 h. Results were visualized by gel electrophoresis and quantified.
Acknowledgments
This work was funded in part by a grant from the NIH NCI R01CA228085, the John Sealy Distinguished chair in Cancer Biology, the Keating Endowment, and the NSF EFRI1933321. J.W.C. was funded in part by the Computational Cancer Biology Training Program fellowship (CCBTP) Grant No. RP170593. M.L.S. and B.C. were supported in part by the UTMB physician-scientist training program. We thank Ellie Cherryhomes for improving the visualization of the manuscript. We thank the editor and reviewers for their helpful suggestions that improved the quality of the manuscript.
Glossary
Abbreviations
- UDG
uracil-DNA glycosylase
- TDG
thymine DNA glycosylase
- hTDG
human TDG
- hyTDG
hybrid TDG
- MIG
mismatch DNA glycosylase from Methanobacterium thermoautotrophicum
- FPG
formamidopyrimidine DNA glycosylase
- Endo III
endonuclease III
- Pol β
DNA polymerase β
- BER
base excision repair
- MSE
mismatch-specific endonuclease
- BHQ1
black hole fluorescence quencher 1
- FAM
6-fluorescein
- MS
mass spectrometry
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemrestox.2c00172.
Associated gel images, tables for kinetic data, MALDI mass spectra, schemes, and other related data (PDF)
Author Contributions
T.B., M.L.S., and L.C.S.: conceptualization; T.B., M.L.S., B.C., J.W.C., and L.C.S.: methodology; T.B., M.L.S., and J.W.C.: validation; T.B., M.L.S., J.C., L.C.H., J.L.H., and K.Z.: formal analysis; T.B., M.L.S., J.C., J.W.C., B.C., and H.T.: investigation; T.B., M.L.S., J.W.C., K.Z., and L.C.S.: resources; T.B., and L.C.S.: writing–original draft; T.B., M.L.S., J.W.C. and L.C.S.: writing–review and editing; T.B., M.L.S., J.W.C., B.C., K.Z., and L.C.S.: visualization; T.B., and L.C.S.: supervision; L.C.S.: project administration; L.C.S.: funding acquisition.
The authors declare the following competing financial interest(s): The authors have submitted a patent application for hyTDG and hyTDG-lyase. Otherwise, the authors declare that they have no conflicts of interest with the contents of this article.
Notes
All data are contained within the article and Supporting Information.
Supplementary Material
References
- Friedberg E. C. A History of the DNA Repair and Mutagenesis Field. The Discovery of Base Excision Repair. DNA Repair 2016, 37, A35–A39. 10.1016/j.dnarep.2015.12.003. [DOI] [PubMed] [Google Scholar]
- Howard M. J.; Wilson S. H. DNA Scanning by Base Excision Repair Enzymes and Implications for Pathway Coordination. DNA Repair 2018, 71, 101–107. 10.1016/j.dnarep.2018.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins E. A.; Rodriguez A. A.; Bradley N. P.; Eichman B. F. Emerging Roles of DNA Glycosylases and the Base Excision Repair Pathway. Trends Biochem. Sci. 2019, 44, 765–781. 10.1016/j.tibs.2019.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S.; Tadesse S.; Kidane D.. Significance of Base Excision Repair to Human Health, 1st ed.; Elsevier Inc., 2021; Vol. 364. [DOI] [PubMed] [Google Scholar]
- Bordin D. L.; Lirussi L.; Nilsen H. Cellular Response to Endogenous DNA Damage: DNA Base Modifications in Gene Expression Regulation. DNA Repair 2021, 99, 103051 10.1016/j.dnarep.2021.103051. [DOI] [PubMed] [Google Scholar]
- Lindahl T.; Nyberg B. Heat-Induced Deamination of Cytosine Residues in DNA. Biochemistry 1974, 13, 3405–3410. 10.1021/bi00713a035. [DOI] [PubMed] [Google Scholar]
- Ames B. N. Mutagenesis and Carcinogenesis: Endogenous and Exogenous Factors. Environ. Mol. Mutagen. 1989, 14, 66–77. 10.1002/em.2850140614. [DOI] [PubMed] [Google Scholar]
- Mullaart E.; Lohman P. H. M.; Berends F.; Vijg J. DNA Damage Metabolism and Aging. Mutat. Res. 1990, 237, 189–210. 10.1016/0921-8734(90)90001-8. [DOI] [PubMed] [Google Scholar]
- Marnett L. J.; Plastaras J. P. Endogenous DNA Damage and Mutation. Trends Genet. 2001, 17, 214–221. 10.1016/S0168-9525(01)02239-9. [DOI] [PubMed] [Google Scholar]
- Li J.; Svilar D.; McClellan S.; Kim J. H.; Ahn E. Y. E.; Vens C.; Wilson D. M.; Sobol R. W. DNA Repair Molecular Beacon Assay: A Platform for Real-Time Functional Analysis of Cellular DNA Repair Capacity. Oncotarget 2018, 9, 31719–31743. 10.18632/oncotarget.25859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visnes T.; Grube M.; Hanna B. M. F.; Benitez-Buelga C.; Cázares-Körner A.; Helleday T. Targeting BER Enzymes in Cancer Therapy. DNA Repair 2018, 71, 118–126. 10.1016/j.dnarep.2018.08.015. [DOI] [PubMed] [Google Scholar]
- Kurthkoti K.; Kumar P.; Sang P. B.; Varshney U. Base Excision Repair Pathways of Bacteria: New Promise for an Old Problem. Future Med. Chem. 2020, 12, 339–355. 10.4155/fmc-2019-0267. [DOI] [PubMed] [Google Scholar]
- Hans F.; Senarisoy M.; Naidu C. B.; Timmins J. Focus on Dna Glycosylases—a Set of Tightly Regulated Enzymes with a High Potential as Anticancer Drug Targets. Int. J. Mol. Sci. 2020, 21, 9226. 10.3390/ijms21239226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visnes T.; Cázares-Körner A.; Hao W.; Wallner O.; Masuyer G.; Loseva O.; Mortusewicz O.; Wiita E.; Sarno A.; Manoilov A.; Astorga-Wells J.; Jemth A. S.; Pan L.; Sanjiv K.; Karsten S.; Gokturk C.; Grube M.; Homan E. J.; Hanna B. M. F.; Paulin C. B. J.; Pham T.; Rasti A.; Berglund U. W.; von Nicolai C.; Benitez-Buelga C.; Koolmeister T.; Ivanic D.; Iliev P.; Scobie M.; Krokan H. E.; Baranczewski P.; Artursson P.; Altun M.; Jensen A. J.; Kalderén C.; Ba X.; Zubarev R. A.; Stenmark P.; Boldogh I.; Helleday T. Small-Molecule Inhibitor of OGG1 Suppresses Proinflammatory Gene Expression and Inflammation. Science 2018, 362, 834–839. 10.1126/SCIENCE.AAR8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy G. J.; Parsons J. L. Base Excision Repair and Its Implications to Cancer Therapy. Essays Biochem. 2020, 64, 831–843. 10.1042/EBC20200013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount B. C.; Ames B. N. Analysis of Uracil in DNA by Gas Chromatography-Mass Spectrometry. Anal. Biochem. 1994, 219, 195–200. 10.1006/abio.1994.1257. [DOI] [PubMed] [Google Scholar]
- Minko I. G.; Vartanian V. L.; Tozaki N. N.; Coskun E.; Coskun S. H.; Jaruga P.; Yeo J.; David S. S.; Stone M. P.; Egli M.; Dizdaroglu M.; McCullough A. K.; Lloyd R. S. Recognition of DNA Adducts by Edited and Unedited Forms of DNA Glycosylase NEIL1. DNA Repair 2020, 85, 102741 10.1016/j.dnarep.2019.102741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu C. W.; Sowers M. L.; Baljinnyam T.; Herring J. L.; Hackfeld L. C.; Tang H.; Zhang K.; Sowers L. C. Measurement of Deaminated Cytosine Adducts in DNA Using a Novel Hybrid Thymine DNA Glycosylase. J. Biol. Chem. 2022, 298, 101638 10.1016/j.jbc.2022.101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holton N. W.; Ebenstein Y.; Gassman N. R. Broad Spectrum Detection of DNA Damage by Repair Assisted Damage Detection (RADD). DNA Repair 2018, 66–67, 42–49. 10.1016/j.dnarep.2018.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassman N. R.; Holton N. W. Targets for Repair: Detecting and Quantifying DNA Damage with Fluorescence-Based Methodologies. Curr. Opin. Biotechnol. 2019, 55, 30–35. 10.1016/j.copbio.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. P.; Angerer B.; Loeb L. A. Incorporation of Reporter-Labeled Nucleotides by DNA Polymerases. BioTechniques 2005, 38, 257–264. 10.2144/05382RR02. [DOI] [PubMed] [Google Scholar]
- Briggs A. W.; Heyn P.. Preparation of Next-Generation Sequencing Libraries from Damaged DNA. In Methods in Molecular Biology; Shapiro B., Hofreiter M., Eds.; 2012; Vol. 840, pp 143–154. [DOI] [PubMed]
- Do H.; Wong S. Q.; Li J.; Dobrovic A. Reducing Sequence Artifacts in Amplicon-Based Massively Parallel Sequencing of Formalin-Fixed Paraffin-Embedded DNA by Enzymatic Depletion of Uracil-Containing Templates. Clin. Chem. 2013, 59, 1376–1383. 10.1373/clinchem.2012.202390. [DOI] [PubMed] [Google Scholar]
- Costello M.; Pugh T. J.; Fennell T. J.; Stewart C.; Lichtenstein L.; Meldrim J. C.; Fostel J. L.; Friedrich D. C.; Perrin D.; Dionne D.; Kim S.; Gabriel S. B.; Lander E. S.; Fisher S.; Getz G. Discovery and Characterization of Artifactual Mutations in Deep Coverage Targeted Capture Sequencing Data Due to Oxidative DNA Damage during Sample Preparation. Nucleic Acids Res. 2013, 41, e67 10.1093/nar/gks1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbeithuber B.; Makova K. D.; Tiemann-Boege I. Artifactual Mutations Resulting from DNA Lesions Limit Detection Levels in Ultrasensitive Sequencing Applications. DNA Res. 2016, 23, 547–559. 10.1093/dnares/dsw038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Liu P.; Evans T. C. Jr.; Ettwiller L. M. DNA Damage Is a Pervasive Cause of Sequencing Errors, Directly Confounding Variant Identification. Science 2017, 355, 752–756. 10.1126/science.aai8690. [DOI] [PubMed] [Google Scholar]
- Eide L.; Luna L.; Gustad E. C.; Henderson P. T.; Essigmann J. M.; Demple B.; Seeberg E. Human Endonuclease III Acts Preferentially on DNA Damage Opposite Guanine Residues in DNA. Biochemistry 2001, 40, 6653–6659. 10.1021/bi0028901. [DOI] [PubMed] [Google Scholar]
- Adhikari S.; Toretsky J. A.; Yuan L.; Roy R. Magnesium, Essential for Base Excision Repair Enzymes, Inhibits Substrate Binding of N-Methylpurine-DNA Glycosylase. J. Biol. Chem. 2006, 281, 29525–29532. 10.1074/jbc.M602673200. [DOI] [PubMed] [Google Scholar]
- Sidorenko V. S.; Mechetin G. V.; Nevinsky G. A.; Zharkov D. O. Ionic Strength and Magnesium Affect the Specificity of Escherichia Coli and Human 8-Oxoguanine-DNA Glycosylases. FEBS J. 2008, 275, 3747–3760. 10.1111/j.1742-4658.2008.06521.x. [DOI] [PubMed] [Google Scholar]
- Manvilla B. A.; Pozharski E.; Toth E. A.; Drohat A. C. Structure of Human Apurinic/Apyrimidinic Endonuclease 1 with the Essential Mg2+ Cofactor. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 2555–2562. 10.1107/S0907444913027042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H. S.; Shin W. J.; Lee J. E.; Do J. T. CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function. Genes 2017, 8, 148. 10.3390/genes8060148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornaletti S.; Pfeifer G. P. Complete and Tissue-Independent Methylation of CpG Sites in the P53 Gene: Implications for Mutations in Human Cancers. Oncogene 1995, 10, 1493–1499. [PubMed] [Google Scholar]
- Riggs A. D.; Jones P. A.. 5-Methylcytosine, Gene Regulation, and Cancer. Adv. Cancer Res.1983, 40, 1–30, 10.1016/S0065-230X(08)60678-8. [DOI] [PubMed] [Google Scholar]
- Mancini D.; Singh S.; Ainsworth P.; Rodenhiser D. Constitutively Methylated CpG Dinucleotides as Mutation Hot Spots in the Retinoblastoma Gene (RB1). Am. J. Hum. Genet. 1997, 61, 80–87. 10.1086/513898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper D. N.; Mort M.; Stenson P. D.; Ball E. v.; Chuzhanova N. A. Methylation-Mediated Deamination of 5-Methylcytosine Appears to Give Rise to Mutations Causing Human Inherited Disease in CpNpG Trinucleotides, as Well as in CpG Dinucleotides. Hum. Genomics 2010, 4, 406–410. 10.1186/1479-7364-4-6-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulos R. C.; Olivier J.; Wong J. W. H. The Interaction between Cytosine Methylation and Processes of DNA Replication and Repair Shape the Mutational Landscape of Cancer Genomes. Nucleic Acids Res. 2017, 45, 7786–7795. 10.1093/nar/gkx463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iengar P. An Analysis of Substitution, Deletion and Insertion Mutations in Cancer Genes. Nucleic Acids Res. 2012, 40, 6401–6413. 10.1093/nar/gks290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutte C.; Yang A. S.; Beart R. W.; Jones P. A. Base Excision Repair of U:G Mismatches at a Mutational Hotspot in the P53 Gene Is More Efficient Than Base Excision Repair of T:G Mismatches in Extracts of Human Colon Tumors. Cancer Res. 1995, 55, 3742–3746. [PubMed] [Google Scholar]
- Baljinnyam T.; Sowers M. L.; Hsu C. W.; Conrad J. W.; Herring J. L.; Hackfeld L. C.; Sowers L. C. Chemical and Enzymatic Modifications of 5-Methylcytosine at the Intersection of DNA Damage, Repair, and Epigenetic Reprogramming. PLoS One 2022, 17, e0273509 10.1371/journal.pone.0273509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horst J. P.; Fritz H. J. Counteracting the Mutagenic Effect of Hydrolytic Deamination of DNA 5-Methylcytosine Residues at High Temperature: DNA Mismatch N-Glycosylase Mig.Mth of the Thermophilic Archaeon Methanobacterium Thermoautotrophicum THF. EMBO J. 1996, 15, 5459–5469. 10.1002/j.1460-2075.1996.tb00929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.; Burdzy A.; Sowers L. C. Substrate Recognition by a Family of Uracil-DNA Glycosylases: UNG, MUG, and TDG. Chem. Res. Toxicol. 2002, 15, 1001–1009. 10.1021/tx020030a. [DOI] [PubMed] [Google Scholar]
- Fitzgerald M. E.; Drohat A. C. Coordinating the Initial Steps of Base Excision Repair: Apurinic/Apyrimidinic Endonuclease 1 Actively Stimulates Thymine DNA Glycosylase by Disrupting the Product Complex. J. Biol. Chem. 2008, 283, 32680–32690. 10.1074/jbc.M805504200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley T. J.; Cunningham R. P. Methanobacterium Thermoformicicum Thymine DNA Mismatch Glycosylase: Conversion of an n-Glycosylase to an AP Lyase. Protein Eng. 1999, 12, 333–340. 10.1093/protein/12.4.333. [DOI] [PubMed] [Google Scholar]
- Bailly V.; Verly W. G. AP Endonucleases and AP Lyases. Nucleic Acids Res. 1989, 17, 3617–3618. 10.1093/NAR/17.9.3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwanto A.; Farrel A.; Rogstad D. K.; Sowers L. C. Characterization of DNA Glycosylase Activity by Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry. Anal. Biochem. 2009, 394, 13–23. 10.1016/j.ab.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z.; Theruvathu J. A.; Farrel A.; Burdzy A.; Sowers L. C. Characterization of Synthetic Oligonucleotides Containing Biologically Important Modified Bases by Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry. Anal. Biochem. 2008, 379, 196–207. 10.1016/j.ab.2008.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldar T.; Jha J. S.; Yang Z.; Nel C.; Housh K.; Cassidy O. J.; Gates K. S. Unexpected Complexity in the Products Arising from NaOH-, Heat-, Amine-, and Glycosylase-Induced Strand Cleavage at an Abasic Site in DNA. Chem. Res. Toxicol. 2022, 35, 218–232. 10.1021/acs.chemrestox.1c00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodke P. P.; Matse J. H.; Dawson S.; Guengerich F. P. Nucleophilic Thiol Proteins Bind Covalently to Abasic Sites in DNA. Chem. Res. Toxicol. 2022, 35, 1805–1808. 10.1021/acs.chemrestox.2c00068. [DOI] [PubMed] [Google Scholar]
- Jha J. S.; Yin J.; Haldar T.; Yang Z.; Wang Y.; Gates K. S. Reconsidering the Chemical Nature of Strand Breaks Derived from Abasic Sites in Cellular DNA: Evidence for 3′-Glutathionylation. J. Am. Chem. Soc. 2022, 144, 10471–10482. 10.1021/jacs.2c02703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mchugh P. J.; Knowland J. Novel Reagents for Chemical Cleavage at Abasic Sites and UV Photoproducts in DNA. Nucleic Acids Res. 1995, 23, 1664–1670. 10.1093/nar/23.10.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu C. W.; Conrad J. W.; Sowers M. L.; Baljinnyam T.; Herring J. L.; Hackfeld L. C.; Hatch S. S.; Sowers L. C. A Combinatorial System to Examine the Enzymatic Repair of Multiply Damaged DNA Substrates. Nucleic Acids Res. 2022, 50, 7406–7419. 10.1093/nar/gkac530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishino S.; Nishi Y.; Oda S.; Uemori T.; Sagara T.; Takatsu N.; Yamagami T.; Shirai T.; Ishino Y. Identification of a Mismatch-Specific Endonuclease in Hyperthermophilic Archaea. Nucleic Acids Res. 2016, 44, 2977–2986. 10.1093/nar/gkw153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae S.; Hijikata A.; Tsuji T.; Yonezawa K.; Kouyama K. i.; Mayanagi K.; Ishino S.; Ishino Y.; Shirai T. Structure of the EndoMS-DNA Complex as Mismatch Restriction Endonuclease. Structure 2016, 24, 1960–1971. 10.1016/j.str.2016.09.005. [DOI] [PubMed] [Google Scholar]
- Hayashi M.; Sugahara K.; Yamamura A.; Iida Y. Evaluation of the Properties of the DNA Methyltransferase from Aeropyrum Pernix K1. Microbiol. Spectrum 2021, 9, e0018621 10.1128/spectrum.00186-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T. E.; Savva R.; Panayotou G.; Barlow T.; Brown T.; Jiricny J.; Pearl L. H. Crystal Structure of a G:T/U Mismatch-Specific DNA Glycosylase: Mismatch Recognition by Complementary-Strand Interactions. Cell 1998, 92, 117–129. 10.1016/S0092-8674(00)80904-6. [DOI] [PubMed] [Google Scholar]
- Coey C. T.; Malik S. S.; Pidugu L. S.; Varney K. M.; Pozharski E.; Drohat A. C. Structural Basis of Damage Recognition by Thymine DNA Glycosylase: Key Roles for N-Terminal Residues. Nucleic Acids Res. 2016, 44, 10248–10258. 10.1093/nar/gkw768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley T. J.; Haas B. J.; Morales J. C.; Kool E. T.; Cunningham R. P. Kinetics and Binding of the Thymine-DNA Mismatch Glycosylase, Mig-Mth, with Mismatch-Containing DNA Substrates. DNA Repair 2003, 2, 107–120. 10.1016/S1568-7864(02)00190-8. [DOI] [PubMed] [Google Scholar]
- Mol C. D.; Arvai A. S.; Begley T. J.; Cunningham R. P.; Tainer J. A. Structure and Activity of a Thermostable Thymine-DNA Glycosylase: Evidence for Base Twisting to Remove Mismatched Normal DNA Bases. J. Mol. Biol. 2002, 315, 373–384. 10.1006/jmbi.2001.5264. [DOI] [PubMed] [Google Scholar]
- Savva R.; Mc Auley-Hecht K.; Brown T. The Structural Basis of Specific Base-Excision Repair by Uracil–DNA Glycosylase. Nature 1995, 373, 487–493. 10.1038/373487a0. [DOI] [PubMed] [Google Scholar]
- Shen J.-C.; Rideout W. M.; Jones P. A. The Rate of Hydrolytic Deamination of 5-Methylcytosine in Double-Stranded DNA. Nucleic Acids Res. 1994, 22, 972–976. 10.1093/nar/22.6.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingard C.; Wu J.; McKeague M.; Sturla S. J. Next-Generation DNA Damage Sequencing. Chem. Soc. Rev. 2020, 49, 7354–7377. 10.1039/d0cs00647e. [DOI] [PubMed] [Google Scholar]
- Leiros I.; Nabong M. P.; Grøsvik K.; Ringvoll J.; Haugland G. T.; Uldal L.; Reite K.; Olsbu I. K.; Knævelsrud I.; Moe E.; Andersen O. A.; Birkeland N. K.; Ruoff P.; Klungland A.; Bjelland S. Structural Basis for Enzymatic Excision of N1-Methyladenine and N3-Methylcytosine from DNA. EMBO J. 2007, 26, 2206–2217. 10.1038/sj.emboj.7601662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knvælsrud I.; Slupphaug G.; Leiros I.; Matsuda A.; Ruoff P.; Bjelland S. Opposite-Base Dependent Excision of 5-Formyluracil from DNA by HSMUG1. Int. J. Radiat. Biol. 2009, 85, 413–420. 10.1080/09553000902818915. [DOI] [PubMed] [Google Scholar]
- Coey C. T.; Drohat A. C. Kinetic Methods for Studying DNA Glycosylases Functioning in Base Excision Repair. Methods Enzymol. 2017, 592, 357–376. 10.1016/bs.mie.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.