

Abstract
RNA regulation can be performed by a second targeting RNA molecule, such as in the microRNA regulation mechanism. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) probes the structure of RNA molecules and can resolve RNA:protein interactions, but RNA:RNA interactions have not yet been addressed with this technique. Here, we apply SHAPE to investigate RNA-mediated binding processes in RNA:RNA and RNA:RNA-RBP complexes. We use RNA:RNA binding by SHAPE (RABS) to investigate microRNA-34a (miR-34a) binding its mRNA target, the silent information regulator 1 (mSIRT1), both with and without the Argonaute protein, constituting the RNA-induced silencing complex (RISC). We show that the seed of the mRNA target must be bound to the microRNA loaded into RISC to enable further binding of the compensatory region by RISC, while the naked miR-34a is able to bind the compensatory region without seed interaction. The method presented here provides complementary structural evidence for the commonly performed luciferase-assay-based evaluation of microRNA binding-site efficiency and specificity on the mRNA target site and could therefore be used in conjunction with it. The method can be applied to any nucleic acid-mediated RNA- or RBP-binding process, such as splicing, antisense RNA binding, or regulation by RISC, providing important insight into the targeted RNA structure.
Keywords: RNA biophysics, MiR targeting/RISC, RNA secondary structure, SHAPE, RNA:RNA binding
INTRODUCTION
MicroRNAs (miRs) are small noncoding RNAs that regulate messenger RNA (mRNA) translation by either inhibiting protein translation or initiating mRNA degradation (Filipowicz et al. 2008; Frédérick and Simard 2021). After maturation, the miR is loaded into the Argonaute protein (AGO) and forms the RNA-inducing silencing complex (RISC). Each miR can target and regulate many mRNAs by interacting with approximately six complementary bases, called the seed, usually located at the 3′-untranslated region (3′-UTR) of the mRNA (Bartel 2009; Lau and MacRae 2009; Hombach and Kretz 2016; Gebert and MacRae 2019). The interaction between miR loaded into AGO (miR-AGO) and the target mRNA initiates from the 5′ seed region (Fig. 1), then continues toward the 3′-end of miR, the compensatory region. Generally, the number of continuous canonical Watson–Crick (WC) base pairs in the seed determines the efficiency of mRNA down-regulation (Filipowicz et al. 2008; Bartel 2018; Frédérick and Simard 2021), while non-WC base pairs for nucleotides 7 and 8 of the seed can reduce binding and down-regulation (Bartel 2009; Agarwal et al. 2015). In rare cases, a strong interaction in the 3′-compensatory region can overcome the detrimental effect of mismatches in the seed region (Brennecke et al. 2005).
FIGURE 1.

The RNA:RNA binding by SHAPE (RABS) approach. (A) The RABS workflow starting from the RNA scaffold design until the interpretation of the binding based on the data obtained. (B) Trans-scaffold design containing the mSIRT1 sequence, and (C) the cis-scaffold containing mSIRT1 and miR-34a sequences. In B and C, the seed region in mSIRT1 sequence and in mSIRT1:miR-34a, respectively, are between green dotted lines. (D) Overview of the three different experimental conditions that we used to evaluate miR:mRNA binding with the trans-scaffold (target sequence in light blue, miR in pink and AGO in light green).
X-ray structures of miR-AGO2 (human AGO2 loaded with miR) showed that AGO2 prearranges the miR seed into an A-form helix and that AGO2 uses its helix-7 to control for continued interaction after seed nucleotide 6 toward the 3′-compensatory region (Klum et al. 2018). A previous NMR study of hsa-miR-34a-5p (miR-34a) bound to the silent information regulator 1 mRNA (mSIRT1) targeting site revealed that structural changes can affect down-regulation efficiency (Baronti et al. 2020). Our group determined the low-resolution structure and dynamics of the mSIRT1:miR-34a complex using an NMR-informed MD approach (Steiner et al. 2016; Ebrahimi et al. 2019; Baronti et al. 2020). Still, little is known about whether the compensatory region interaction is defined by AGO or by the miR or to what degree targeting specificity is determined by the miR sequence outside the seed (Kilikevicius et al. 2021).
One approach to study RNA secondary structure is selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) (Merino et al. 2005). SHAPE reagents chemically modify nucleotides in flexible conformations by adding covalent 2′-O-adducts (Velema and Kool 2020). In the next step, complementary DNA (cDNA) is produced via reverse transcription (RT), which aborts at the modified nucleotide (SHAPE-Seq and SHAPE-CE) or incorporates a mutation (SHAPE-MaP). The resulting cDNA sequences are analyzed by capillary electrophoresis (SHAPE-CE), or Illumina sequencing (SHAPE-Seq or SHAPE-MaP) (Wilkinson et al. 2006; Smola et al. 2015b). Besides revealing secondary structure information within RNA molecules, SHAPE can be used to study the interaction between other nonnatural nucleic acids, such as LNA (locked nucleic acids [Dethoff et al. 2018]) RNA and RNA-binding proteins (RBPs) by analyzing the protein footprint (f-SHAPE or deltaSHAPE, Lucks et al. 2011; Smola et al. 2015a), or RNA conformation in an RNA:DNA hetero duplex (Kladwang and Das 2010; Palka et al. 2020). Different SHAPE approaches are available to study RNA structure in high throughput and its conformational space (Washietl et al. 2012; Cheng et al. 2015; Lee et al. 2015; Tomezsko et al. 2020; Morandi et al. 2021). Interestingly, evaluating RNA binding to a second RNA molecule (RNA:RNA) or an RNA-mediated protein binding (RNA:RNA-RBP), as in the mRNA:miR-AGO complex, has to our knowledge not yet been addressed using SHAPE, although SHAPE represents an ideal technique to study structural details of miR targeting.
Here, we applied SHAPE to study miR targeting in vitro, abbreviated to RNA:RNA binding by SHAPE (RABS), by implementing the mRNA target site into a trans-acting RNA scaffold (trans-scaffold, Fig. 1B). This setup allows the analysis of the mRNA target interaction with miR (RNA:RNA) or miR-loaded AGO2 (RNA:RNA-RBP) with single base-pair resolution (Fig. 1A). The proof-of-concept experiments confirmed the interaction pattern found by NMR (Baronti et al. 2020). We used two control sequences: “scrambled” which does not bind to miR-34a and controls for interaction specificity of the RNA-mediated protein binding (RNA:RNA-RBP), and “scrambled seed,” which has mismatches to the seed nucleotides 2–7 but is otherwise complementary to miR-34a. Using RABS, we show the “scrambled seed” target is bound by the miR-34a in the absence of AGO2 via the compensatory region, while this binding is reduced when interacting with miR-34a loaded in AGO2, because of the strongly reduced binding affinity, confirming current models (Bartel 2018) and demonstrating the specificity of the presented approach.
RESULTS
Here, we extended the SHAPE application to study RNA:RNA interaction in miR targeting, using the example of miR-34a and its target mRNA SIRT1 (mSIRT1). We generated two RNA scaffolds for this purpose: the trans- and cis-scaffold. The trans-scaffold involves intermolecular binding (Fig. 1B), by which one RNA sequence of interest (in this case mSIRT1) is inserted into the main loop of the scaffold and is subsequently bound by RNA or RNA-RBP (here miR-34a or miR-34a-AGO2, respectively). In the cis-scaffold (Fig. 1C), both RNA sequences are inserted in the same scaffold, resembling a typical SHAPE scaffold. The use of both scaffolds enables a better understanding of the impact of RNA:RNA binding on RNA structure, and a comparison of RNA structure between RNA:RNA and RNA:RNA-RBP complexes.
For the trans-scaffold, three different conditions were tested: (i) unbound; (ii) +RNA; leading to the RNA:RNA complex; and (iii) +RNA-RBP; leading to the RNA:RNA-RBP complex (Fig. 1D). The unbound trans-scaffold acts as a control for three factors: first, it quality controls the scaffold for unwanted stable secondary structures between the scaffold itself and the inserted target sequence (Fig. 1A); second, it probes for self-folding of the target sequence, which can compete with binding of the second component (miR-34a or miR-34a-AGO2); and third, it provides a baseline for sequence-dependent reactivity biases. The trans-scaffold provides the reactivity read-out for the inserted target RNA sequence only, while the cis-scaffold reads out secondary structure for both strands in the RNA:RNA duplex (here mSIRT1:miR-34a). However, because the 3′- and 5′-end of the microRNA must be anchored inside AGO2 to function (Sheu-Gruttadauria et al. 2019a,b), the trans-scaffold provides the unique advantage of monitoring the effect of miR-AGO2 binding on the mRNA target over traditional cis-type scaffolds.
Optimizing chemical probing for RNA:RNA binding
For probing of the target RNA, we designed two different trans-scaffolds (TS#1 and TS#2; Supplemental Fig. S1a), which differ in the buffer nucleotides in the main loop (Supplemental Table S1). The reactivity profiles from two independent experimental replicates (each with three technical replicates) were used to predict the secondary structure formation of the main loop containing the target RNA insert, using both RNAprobing and MC-Fold (Parisien and Major 2008; Washietl et al. 2012). No stable secondary structure of the main loops was observed for either TS#1 or TS#2 (Supplemental Fig. S1), although some nucleotides show reduced reactivity (Supplemental Fig. S1b), namely tC4 to tC6, tA14, and tC16, which could be caused by a transient fold of the target sequence (Kladwang and Das 2010; Bindewald et al. 2011; Mlýnský and Bussi 2018; Strobel et al. 2018), sequence-specific biases by the chemical modification (Weeks and Mauger 2011) or the reverse transcription step (Kladwang et al. 2020). Generally, base-paired nucleotides are supposed to show low (near zero) reactivity values (Weidmann et al. 2021). Most of the lower reactivity is observed for sequential cytosines, possibly caused by stacking of the single-stranded region (Tubbs et al. 2013), and confirmed by our nine cytosine-containing control; however, our NMR data indicates a flexible loop, shown by mixed sugar puckers (Supplemental Fig. S2). As neither of the scaffolds adopt stable secondary structures where the buffer nucleotides interact with the mSIRT1 sequence, we selected TS#1 for the rest of the experiments, as it showed lower propensity of structure formation in the target sequence (Supplemental Fig. S1b).
A binding interaction is dependent on the binding constant (KD), and the concentration of the components. Therefore, we optimized the concentration ratios between the trans-scaffold, containing the mSIRT1 binding site, and the binding components (miR-34a and miR-34a-AGO2) by titration (Supplemental Fig. S3). The concentration was varied from a ratio of 1:0.05 to 1:5 of mSIRT1:miR-34a (with [miR-34a] from 4 to 400 nM and [mSIRT1] kept constant and equal to 80 nM), considering the previously determined KD of 124.3 ± 21.7 nM by electrophoretic mobility shift assay (Baronti et al. 2020). For nucleotides that showed a change in reactivity, we calculated the KD and the probability to be bound at the 1:2 ratio for each mSIRT1 nucleotide (Supplemental Fig. S3). The KD ranged from 4 to 160 nM and the fraction bound at 1:2 ratio from 89% (first nucleotide, reduced by end-fraying) to 99%, indicating that the majority of molecules are bound at this condition. Furthermore, the reactivity pattern agrees with the binding pattern obtained from NMR data (Baronti et al. 2020) already at a ratio of 1:1. A ratio of 1:2 was chosen, as it provided reduced error of the reactivity signal in the experiment (Supplemental Table S2; Supplemental Fig. S3).
To study the mSIRT1:miR-34a-AGO2 interaction, we produced human Argonaute 2 (AGO2) and loaded miR-34a into the miR-34a-AGO2 complex by incubation (Elkayam et al. 2012) and confirmed loading efficiency by northern blot (Supplemental Fig. S4) at 5%. The 5% miR-34a-AGO2 was compensated by using a 20-fold excess of the whole miR-AGO2 heterogeneous sample to achieve a 1:1 ratio for mSIRT1:miR-34a-AGO2 binding. The presence of the high (AGO2) protein reduced the efficiency of the following RT step. Therefore, we added a proteinase K digestion step, after the chemical modification and before the cDNA preparation, to remove the protein and increase the signal intensity. Unfortunately, adding this digestion step reduces the overall signal intensities in the CE profiles, likely due to lower RNA recovery after deactivating of proteinase K, and a dilution effect of the scaffold (+165 µL to a 17µL sample). We confirmed this by adding the proteinase K digestion step to the unbound trans-scaffold (Supplemental Table S3; Supplemental Fig. S5), reproducing the reduction in signal intensity. Increasing the AGO2 concentration to a 1:2 ratio (meaning 40-fold excess of random miR loaded AGO2 heterogeneous sample), and thus mimicking the de facto ratio in the RNA:RNA experiments, reduces the final signal intensities to near noise-levels for the mSIRT1:miR experiment (Supplemental Fig. S6). We estimated KD and the probability to be bound at the 1:1 ratio mSIRT1:miR-34a-AGO2 complex by titration as for the mSIRT1:miR-34a (Supplemental Fig. S3), ranging from ratios 1:0.25 to 1:1.5 (with [miR-34a-AGO2] from 20 nM to 120 nM and [mSIRT1] equal to 80 nM), which was the highest concentration to achieve an interpretable signal (ratio 1:2 shows very little signal [Supplemental Fig. S6b]). The KD ranged from 320 to 20 nM, and the fraction bound at a 1:1 ratio from 60% (second nucleotide, possibly reduced by end-fraying) to 87%, indicating that the majority of molecules are bound in this condition, however, with a somewhat weaker KD and binding probability than observed for mSIRT1:miR-34a (Supplemental Fig. S7). We could also show that increasing the AGO2 concentration, for example, to a ratio of 1:1.5, shows little change (right lane, Supplemental Table S4), indicating that the use of a 1:1 ratio is reliable.
As a negative control, we designed a trans-scaffold containing a scrambled sequence (scrm), which did not bind either miR-34a or the miR-34a:AGO2 complex (Fig. 2A; Supplemental Table S1). We confirmed experimentally that no change in reactivity was observed upon addition of miR-34a or miR-34a-AGO2 compared to the unbound scrm trans-scaffold (Fig. 2B,C; Supplemental Table S5). Importantly, with this control we could exclude that the target site nonspecifically binds the differently loaded 20-fold excess AGO2.
FIGURE 2.
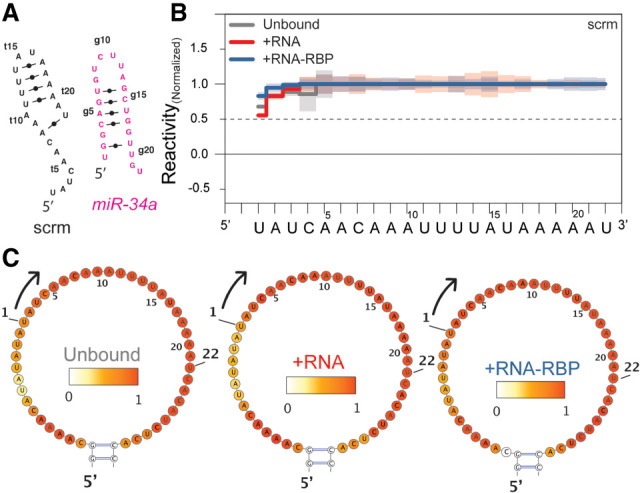
Scrambled sequence, the negative control for nonspecific binding. (A) Predicted secondary structures of unbound miR-34a (pink) and unbound scrambled control (scrm, black) using MC-Fold (23). (B) Normalized reactivity of the scrm control inserted in the unbound trans-scaffold (gray), bound by miR-34a (red) and miR-34a loaded AGO2 with respective heatmaps in C, indicating that no unspecific interactions with the RNA or the RNA-RBP complex occur.
Structural probing of RNA:RNA binding: the trans-scaffold
To simplify data interpretation, we limited the reactivity scale to 0–1 and introduced a threshold to distinguish between base-paired and unbound nucleotides (see Data analysis in Methods). Furthermore, to compare results from the same scaffold in different experimental conditions, we renormalized the reactivity values of each nucleotide in the main loop over the averaged reactivity values of the buffer nucleotides, nucleotides before and after the target sequence in the main loop (Fig. 1B; Supplemental Table S1). Limiting the reactivity scale to between 0 and 1 (Washietl et al. 2012) addresses the binary question of being base-paired while eliminating negative reactivity due to averaging artifacts or large positive values, representing different levels of dynamics, which are not relevant here. The threshold of 0.5 for base-pairing has been used before (Wilkinson et al. 2008; Zarringhalam et al. 2012) and was confirmed by the observable imino resonances in NMR spectra for the same complex (Baronti et al. 2020), which are also indicators of stable base-pairing (Fig. 3A,B).
FIGURE 3.

SIRT1 mRNA target binding measured by RABS using a trans-scaffold. (A) Observed base-pairing pattern of mSIRT1:miR-34a duplex as determined by NMR spectroscopy (Baronti et al. 2020). (B) Averaged reactivity values of three independent replicates for the unbound scaffold containing the mSIRT1 sequence (gray), RNA:RNA (red, +RNA) and RNA:RNA-RBP complexes (blue, +RNA-RBP), with one standard deviation indicated by shaded regions. (C) Reactivity values after normalization over buffer nucleotides of the main loop. Values >1 and <0 are set to 1 and 0, respectively. (D) Heatmaps of the same data presented in B. (E) ΔReactivity values in which the unbound scaffold reactivity is subtracted from the bound reactivity at each nucleotide. Nucleotides in the mSIRT1 target sequence interacting with the miR-34a seed region are indicated by green underlining and letters.
To compare different experiments and subtract background, we calculated ΔReactivity (equation 2, Smola et al. 2015a), which is negative when a nucleotide becomes base-paired upon binding to the second component. We calculated a Z-factor (Zhang et al. 1999) on the renormalized values to compare different experimental conditions (see tables in the Supplemental Material for the different conditions). We controlled for the potential of DMSO, solvent for 1M7, to destabilize RNA folding (Supplemental Table S6; Supplemental Fig. S8; Lee et al. 2013).
Typically, reactivity is displayed on a SHAPE-based predicted structure; however, here this leads to incorrect interpretation, as the information of the binding partner is missing because only one component is accessible for probing. Instead, we show heatmaps for the trans-scaffold without structural predictions (Fig. 3D) and display the reactivity along the sequence (Fig. 3B). For structural prediction we instead compared the unbound trans-scaffold reactivity to the reactivity of the RNA:RNA and RNA:RNA-RBP complexes.
There are minor differences in the reactivity patterns of the mSIRT1:miR-34a and the mSIRT1:miR-34a-AGO2 interaction (Fig. 3; Supplemental Table S7). In both complexes the seed and the 3′-compensatory region are base-paired, with a flexible 4-nt bulge in between, agreeing with previous NMR experiments (Baronti et al. 2020). By comparing ΔReactivity we observe a difference in bulge size for mSIRT1:miR-34a and mSIRT1:miR-34a-AGO2, especially tC16 and tA17 (Z-factor of 0.55 and 0.22, respectively, where Z-factor >0 indicates a statistically significant difference with P-value <0.05) (Fig. 3C; Supplemental Table S7). The larger bulge size in the RNA:RNA complex could be due to the larger flexibility of unpaired nucleotides, which could be constrained by AGO2 in the mSIRT1:miR-34a-AGO2 interaction. The data indicate that AGO2 increases the stability of the interaction between mSIRT1 and miR-34a by stabilizing the 3′-compensatory region in the dedicated “Supplemental chamber” and thereby reduces the end-fraying effect the bulge has on the neighboring nucleotides without the AGO2 protein, as previously reported with X-ray crystallography (Sheu-Gruttadauria et al. 2019b). The first and last nucleotides in the mSIRT1 sequence in both the mSIRT1:miR-34a and mSIRT1:miR-34a-AGO2 complexes, experience reactivity >0.5 (Fig. 3), which can be explained by the end-fraying effect, the increase of dynamic of closing base pairs, observed as well by NMR (Baronti et al. 2020).
While the unbound mSIRT1 targeting sequence shows no secondary structure propensity, probing the unbound miR-34a scaffold reveals secondary structure formation within the sequence (Supplemental Fig. S9), similar to the one observed by NMR (Baronti et al. 2020). Some reduced reactivity is also observed in the 5′ buffer region that is resolved in the mSIRT1 bound form, which could be due to residual interaction of miR-34a with the scaffold. The inverted scaffold probes miR-34a binding to added mSIRT1 in a 1:2 ratio, similar to mSIRT1:miR-34a, and showed that miR-34a is fully bound to mSIRT1 (Supplemental Table S8), confirming the mSIRT1 trans-scaffold data (Fig. 3). Nucleotides gU7 to gA13 display a somewhat higher reactivity, though still within the bound range (i.e., ≤0.5), and these also show higher reactivity in the unbound trans-scaffold, which could indicate that the mSIRT1:miR-34a complex might not be 100% formed, but a small fraction could be competing with self-folding of miR-34a.
The trans-scaffold only provides information on the mSIRT1 half of the complex, because the miR sequence is inserted by the 5′ and 3′ ends into AGO2 (Sheu-Gruttadauria et al. 2019b), which prevents it from being inserted into a scaffold. Furthermore, AGO2 prestructures miR and likely protects it from modification, therefore even isolating the miR after probing will not provide information about mRNA:miR interaction per se.
Structural probing of RNA:RNA interaction within the same construct: the cis-scaffold
As mentioned above, the main limitation of the trans-scaffold is that only the targeted sequence included in the scaffold can be probed. To provide insights into both interacting RNA molecules, we returned to classical SHAPE and designed the cis-scaffold (Fig. 1C), as “interacting on the same strand.” The cis-scaffold removes the concentration-dependence of the binding interactions, as both binding partners are now within the same construct and can therefore be considered as a reference for a fully bound trans-scaffold. We applied the cis-scaffold to study the interaction between mSIRT1:miR-34a and scrm-seed:miR-34a, with experimental conditions similar to the unbound trans-scaffold experiments.
Figure 4A shows the observed reactivity values for the mSIRT1:miR-34a complex in the cis-scaffold, which agree with the binding pattern from NMR spectroscopy (Fig. 3A), and are similar to the observed pattern of the mSIRT1:miR-34a complex in the trans-scaffold (Fig. 3; Supplemental Table S9). To compare the scaffolds quantitatively, we plotted the averaged reactivity values of both scaffolds for the mSIRT1 sequence against each other (Fig. 4C) and were able to fit the data linearly, further indicating similar structures. The slope of the linear fit deviates from 1, as the maximum reactivity observed depends on a number of variables, that is, sequence bias or different structural dynamics on top of base-pairing.
FIGURE 4.

Cis-scaffold comparison to trans-scaffold. (A, top) Reactivity values (three independent replicates) of mSIRT1 in the cis-scaffold (orange) and in trans-scaffold (red). (Bottom) miR-34a reactivity values in the cis-scaffold (light purple) and in the trans-scaffold (dark purple). The base-pairing indicated by lines between the sequences is based on SHAPE reactivity values (RNAprobing, Washietl et al. 2012). (B) Overlay of the reactivity values of the mSIRT1 sequence in the trans-scaffold bound to the miR-34a-AGO2 complex (+RNA-RBP, dark blue) and the mSIRT1 sequence in the cis-scaffold (orange). One standard deviation is indicated with shading. For charts in panels A and B, the observed reactivity values >1 and <0 are set to 1 and 0, respectively. (C) Correlating reactivity values of mSIRT1 sequence from RABS using the trans- (y-axis) versus cis-scaffold (x-axis). The linear fit indicates correlations for most nucleotides, with tC16, tU18, and tA26 as outliers likely due to overall stabilization of the construct and therefore reduced end-fraying. The trans-scaffold reactivity values presented in this figure are not additionally normalized (see Material and Methods).
The cis- and trans-scaffold have minor differences, such as diminished end-fraying in the cis-scaffold, as expected, even though bulges were introduced at the end of the sequence to provide a native-like environment. The 4-nt bulge (tC16, to tU19) shows maximum reactivity values for the mSIRT1 target sequence in both scaffolds (Fig. 4), and the 3′-compensatory region agrees well (Fig. 4A). The seed region (tA20 to tA26, green in Fig. 4) correlates less well in comparison with the 3′-compensatory region, which might be due to a less stable complex in the trans-scaffold than the cis-scaffold. The seed helix has seven base pairs, of which two are AU closing base pairs (tA20:gU7 and tA26:gU1) that are more prone to end fraying, which is reduced in the cis-scaffold. Interestingly, the stability of the seed in the cis-scaffold compared to the trans-scaffold looks similar to the RNA:RNA-RBP complex probed with the mSIRT1 trans-scaffold, which could be interpreted as another indicator for a stabilization of the seed region by AGO2 in the mRNA:miR-AGO2 complex (Fig. 4B), as also described by X-ray crystallography previously (Schirle et al. 2014). In the 3′-compensatory region, tC15, which is located right before the 4-nt flexible bulge (Fig. 3A), is the only nucleotide that appears almost unbound in both trans-(mSIRT1:miR-34a) and cis-scaffolds (tCcis-scaffold15 = 0.6 ± 0.2, tCtrans-scaffold15 = 0.4 ± 0.3) (Fig. 4A), while it is paired in the trans-scaffold probing the ternary complex, that is, mSIRT1:miR-34a-AGO2 (Fig. 4B). This shows that the flexible bulge can destabilize the tC15:gG8 closing base pair via end-fraying, even in the cis-scaffold despite representing a more stable conformation than the trans-scaffold.
Seed binding is the first threshold
The results of chemical probing for the mSIRT1 mRNA target sequence in the trans- and cis-scaffolds agree with NMR data. Nevertheless, to confirm the specificity of the interaction between mSIRT1 and miR-34a or miR-34a-AGO2, we used another control sequence: “scrambled seed” (scrm-seed) (Salzman et al. 2016). This sequence binds miR-34a strongly in the 3′-compensatory region and is designed not to interact with 6 nt of the seed region (tA16 through tG21 in scrm-seed, Fig. 5A). The results of probing the scrm-seed:miR-34a complex in the trans-scaffold show reactivity values ≤0.5, for nucleotide tC2 to tC15, indicating base-pairing of the entire 3′-compensatory region with miR-34a, while the seed region shows reactivity values >0.5 and is not base-paired, as expected (Fig. 5; Supplemental Fig. S13).
FIGURE 5.
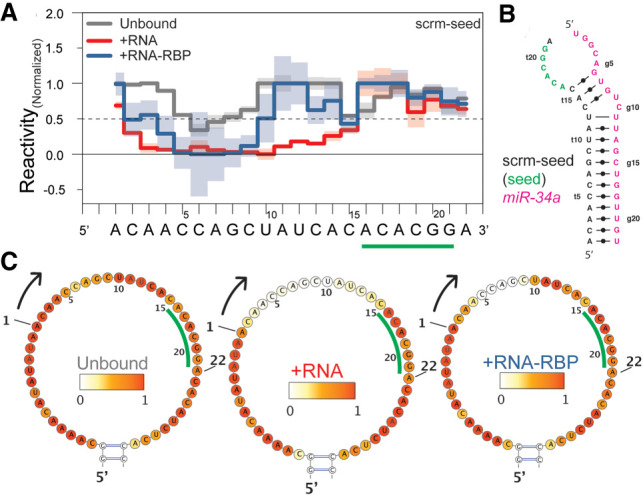
AGO2 controls for seed binding. (A) Normalized reactivity of unbound (gray), RNA:RNA (red), and RNA:RNA-RBP complexes (blue) for scrm-seed and the respective heatmaps (B), with the nucleotides opposing the miR-34a seed in green letters/underlined. (C) Secondary structure of the lowest energy conformation of scrm-seed binding to miR-34a predicted with RABS-restrained MC-Fold (Parisien and Major 2008). The normalized reactivity shows different binding patterns for RNA:RNA versus RNA:RNA-RBP, indicating that miR-34a binds the 3′-compensatory region of the scrm-seed, while miR-34a loaded AGO2 does not.
The scrm-seed:miR-34a complex in the cis-scaffold shows a similar reactivity pattern as the scrm-seed:miR-34a trans-scaffolds (Supplemental Fig. S10; Supplemental Table S10), especially in the 3′-compensatory region. Differences to the trans-scaffold are observed in the seed region, especially for tA16 and tC17 (Supplemental Fig. S10b,c; Supplemental Table S10). The structures predicted from the reactivities of trans- and cis-scaffolds by MC-Fold (Parisien and Major 2008), although energetically the same, present different secondary structures (Supplemental Fig. S10). This highlights a difference between the trans- and cis-scaffolds on secondary structure formation, which can lead to different structural interpretation, for example, by changes in the overall restraint of the ends of the interacting RNAs.
Interestingly, the reactivity profile of the scrm-seed:miR-34a-AGO2 interaction is different from scrm-seed:miR-34a, especially in the 3′-compensatory region. The difference is most pronounced for nucleotides tA11 through tC13, which are base-paired in scrm-seed:miR-34a (Fig. 5A; Supplemental Table 11). On the other hand, the reactivity profiles of the 3′-compensatory region of the scrm-seed:miR-34a-AGO2 complex and unbound are not completely similar either (Supplemental Table S11). This could indicate a transient scrm-seed:miR-34a-AGO2 interaction in the 3′-compensatory region, possibly because AGO2 permits interaction in the 3′-compensatory region only once seed-pairing has been established and helix-7 releases the binding constraint (Schirle and MacRae 2012; Chandradoss et al. 2015; Klum et al. 2018). Some of the lower reactivity, especially around the tC5 and tC6, can be attributed to artifacts/biases for sequential cytosines as well as lower overall reactivity in AGO2-containing experiments. Accordingly, the reactivity values of the scrm-seed:miR-34a-AGO2 complex could be interpreted as an average between transient bound/unbound conditions of the 5 to 6 nt in the 3′-compensatory region. The scrm-seed construct thus serves as an essential control for bona fide binding of miR-34a loaded AGO2, since only miR-34a alone completely binds the compensatory region in the absence of AGO2 (Fig. 5A). To our knowledge, this has never been observed directly for the targeted mRNA moiety, as the flexible nature of the mRNA part of the complex interferes with structure determination techniques.
DISCUSSION
In this report, we introduced a new approach to study RNA:RNA binding based on SHAPE (abbreviated RABS). We designed two RNA scaffolds: trans- and cis-scaffolds (Fig. 1). The trans-scaffold contains the mRNA target sequence that is probed upon binding the microRNA component, and thus allowed us to study binding of an mRNA target to a complex mediated via the RNA bound to the RNA-binding protein (RNA-RBP) (Fig. 3). RABS can be combined with different read-outs of the scaffold, such as SHAPE-Seq or -MaP (Wilkinson et al. 2006; Smola et al. 2015b).
We applied our method to study the interaction between hsa-miR-34a-5p (miR-34a) and one of its target mRNAs, the silent information regulator 1 mRNA (mSIRT1), where results agreed well with our recent NMR data (Baronti et al. 2020). Based on this NMR structure, we defined a range of reactivity values ≤0.5 representing base-pairing and provided a path for analysis that is reminiscent of RNAprobing (Washietl et al. 2012) or used by the Weeks Lab (Wilkinson et al. 2008). Furthermore, we designed a second RNA scaffold, a cis-scaffold, in which both mSIRT1 and miR-34a sequences can be probed together, analogous to the classic SHAPE experiments (Fig. 1C). Binding patterns obtained from the trans- and cis-scaffolds for the mSIRT1:miR-34a duplex are in agreement, for example, reactivity of tC15 indicates it is not base-paired in either (Fig. 4).
Probing of the mSIRT1 structure upon binding to miR-34a-loaded human Argonaute 2 (mSIRT1:miR-34a-AGO2, or RNA-RBP experiments) yielded similar results to the mSIRT1:miR-34a complex, indicating that indeed it is the RNA component that defines target specificity. Minor differences were observed between the mSIRT1:miR-34a and the mSIRT1:miR-34a-AGO2 complexes, for example, tC15 that locates at the 5′ of the flexible bulge, which is likely due to enhanced stability of RISC binding target mRNA (Fig. 3).
Controls were implemented to exclude nonspecific binding of the miR-34a-AGO2 RNA-RBP complex (Fig. 2). One such control, the scrambled seed (scrm-seed), demonstrates the strength of the method by allowing the investigation of RISC complex-specific binding of the miR-34a. In the absence of AGO-loading, miR-34a binds the compensatory region. In contrast, the miR-34a-AGO2 complex does not bind, or only transiently binds, the scrm-seed construct as the seed binding is absent. This checkpoint control performed by RISC seems to require seed binding before 3′-compensatory binding (Fig. 5) and is crucial in identifying correct targets, as has been previously shown (Schirle and MacRae 2012; Chandradoss et al. 2015; Klum et al. 2018).
In sum, the approach presented here is a reliable method to study any type of RNA:RNA and RNA:RNA-RBP interactions/binding, and is particularly well suited to reveal structural details of microRNA targeting. RABS could easily be adopted to investigate interactions with other nucleic acid-mediated binding complexes, for example, splicing machinery, RNA-mediated protein aggregation or RNA in liquid–liquid phase separation, and could thus serve as a new tool in the RNA structure toolkit.
MATERIALS AND METHODS
RNA scaffold design
We designed two different RNA scaffolds and named them based on whether both interacting RNAs are in the same scaffold (cis-scaffold) or the monitored RNA is alone in the scaffold (trans-scaffold). Sequences are in Supplemental Information, Supplemental Table S1. In both types of scaffolds, two small hairpin structures were designed as references near the terminal ends of the RNA scaffold; important for data analysis to correct for a decrease in reactivity over the length of the sequence (Fig. 1B,C; Lee et al. 2015). In the trans-scaffolds, the sequence of interest (i.e., mSIRT1 or miR-34a) was introduced into the large hairpin structure located in the middle of the scaffold (called main loop, Fig. 1B). Here, the sequence of the main loop was engineered to avoid stable secondary structures in the RNA scaffold itself or with the target sequences. For the cis-scaffolds, instead of the main loop, the mRNA target and miR-34a sequences were each introduced in a 5′ to 3′ orientation, with addition of symmetric 4-nt bulges at the non-seed end of the interacting RNAs (at 5′-end of mSIRT1 and 3′-end of the miR-34a sequence) and a 10-nt closing loop at the seed end (Fig. 1C). For both scaffolds, to stabilize the overall secondary structure, we used GC-rich sequences for the stems, and A-rich sequences for the buffer, single stranded regions, based on guidelines (Wilkinson et al. 2006). The probability of secondary structure formation in each RNA scaffold was predicted using the RNAstructure package (Reuter and Mathews 2010).
Control sequence design
We designed two control sequences (Supplemental Table S1). One “scrambled” (scrm) control was generated in which the sequence was designed to have the lowest base-pairing possibility with the miR-34a sequence, indicated by the highest minimum free energy (MFE) value (MC-Fold, Parisien and Major 2008), and that miR-34a prefers to bind to itself rather than binding to the scrambled sequence. No guanosine nucleotides were used in this sequence, in order to prevent the formation of GC WC base pairs, or GU/GG/GA mismatches, as all have a reasonably high base-pairing probability. The scrm sequence was validated against the miRBase database (Griffiths-Jones et al. 2008) to avoid off-target effects in luciferase assays. Only one hit was found within the miRBase human database, hsa-miR-384 with an E-value of 85, which is too high for any statistical significance. The “scrambled seed” (scrm-seed) control (Salzman et al. 2016) was designed to have no interaction with the miR-34a seed nucleotides (numbering from 5′-end, Supplemental Table S1).
Preparing RNA scaffolds
All DNA templates were produced by PCR assembly (Tian and Das 2017). The primers were designed using Primerize web server (Tian and Das 2017), so that every sense strand starts with a T7 polymerase promotor sequence (sequence overview is in Supplemental Table S1). All primers were purchased from Integrated DNA Technologies (IDT). Phusion High-Fidelity PCR Master Mix with HF buffer (Thermo Fisher) was used to amplify each DNA template. The size of all PCR products were controlled in agarose gels (1% agarose, 0.5× TBE buffer [45 mM Tris-Borate, 1 mM EDTA, pH 8.3]), 1 h at 150 V to confirm correct PCR assembly. Afterward, DNA templates were purified by NucleoSpin Gel and PCR Clean-up Kit (Macherey-Nagel), and concentrations were calculated based on A260 absorption.
Next, RNA scaffolds were produced by in vitro transcription (IVT) of the DNA templates (Karlsson et al. 2020). Each IVT reaction contains 100 mM Tris-HCl pH 8.0, 10 mM MgCl2, 10 mM dithiothreitol, 20 mM spermidine, 5 mM GMP, 3 mM NTPs, 0.3 mg/mL T7 polymerase (produced in-house) (Borkotoky and Murali 2018), 0.1 mg/mL inorganic pyrophosphatase (iPPase, produced in-house) (Kern and Davis 1997), and 3.6 ng/mL of dsDNA template in a total volume of 100 µL. All RNA scaffolds were purified with RNAClean XP beads (Beckman Coulter). Before and after purification, the size and purity of RNA scaffolds were checked by denaturing polyacrylamide gel (15% polyacrylamide, 8 M urea, 1× TBE buffer [90 mM Tris-Borate, 2 mM EDTA, pH 8.3], 350 V, 2 h). Finally, the concentration of RNA scaffolds was calculated based on A260 absorption.
Preparing miR-34a and mSIRT1 RNA binding sequences
To perform RABS, we prepared both miR-34a and mSIRT1 RNA sequences as the second component, using IVT. The miR-34a sample was prepared using a plasmid DNA template with tandem repeats followed by RNase H-derived cleavage of the transcript (Feyrer et al. 2020). The DNA plasmid containing 26 tandem repeats of miR-34a was linearized using 1 µg DNA plasmid, 5 µL BamHI buffer (NEB), 1 µL BamHI (NEB), in a total volume of 50 µL; incubated at 37°C for 1 h. Next, the linearized plasmid was used for large scale IVT (200 mM Tris-Cl pH 8.0, 10 mM MgCl2, 10 mM dithiothreitol [DTT], 25 mM spermidine, 5 mM GMP, 3 mM NTPs, 0.3 mg/mL T7 polymerase, 0.1 mg/mL iPPase, 100 ng/mL RNase H [in-house], 20 ng/mL of DNA template, 20 µM of cleavage guide [IDT], in a total volume of 10 mL; incubated overnight at 37°C). For the mSIRT1 RNA sequence, the template DNA was purchased from IDT and used for IVT (200 mM Tris-Cl pH 8.0, 10 mM MgCl2, 10 mM dithiothreitol [DTT], 25 mM spermidine, 5 mM GMP, 3 mM NTPs, 0.3 mg/mL T7 polymerase, 0.1 mg/mL iPPase, 3.6 ng/mL of DNA template, preannealed with T7 promoter [Karlsson et al. 2020], in a total volume of 10 mL; incubated overnight at 37°C). Next, the IVT products were purified with ion-exchange high performance liquid chromatography (IE-HPLC). The buffer system used was loading buffer: 20 mM NaOAc with 20 mM NaClO4; and eluting buffer: 20 mM NaOAc with 600 mM NaClO4 (for details, see Karlsson et al. 2020). The purity of the IE-HPLC fractions were checked with denaturing polyacrylamide gel electrophoresis (20% polyacrylamide, 8 M urea, 1× TBE buffer, 350 V, 1 h).
Preparing miR-34a-AGO2 complex
Cloning and recombinant baculovirus production
The full-length human Argonaute protein 2 (AGO2) was cloned into pFastBac dual expression vector (Thermo Fisher) and contained an amino-terminal His-tag followed by a TEV cleavage site. The construct was under the control of a polyhedrin promotor. In order to generate and transform recombinant Bacmid, the AGO2 construct was transformed into chemical competent DH10EmBacY cells (Geneva Biotech). Following the “Bac-to-Bac TOPO Expression System” user manual (Thermo Fisher, Version A,15 December 2008 A10606), phenotype verification by blue/white screening, bacmid isolation and PCR analysis was used to confirm the recombinant Bacmid. In brief, fresh 8 × 105 Sf9 cells/plate (1.5 × 106 cells/mL) were transfected with recombinant Bacmid (P0-AGO2), and amplification was achieved by two rounds of additional transfections (P1-AGO2 and P2-AGO2). Finally, 300 mL of P2-AGO2 baculovirus stock was filtered and FBS (2% final conc.) was added. Bacmid internal GFP expression was used for monitoring throughout baculovirus production. Overexpression of AGO2 was confirmed by western blot (Abcam, anti-6X His tag AB [ab18184]).
Expression, loading and purification of miR-34a-AGO2
A total of 10 mL P2-AGO2 baculovirus stock was used to transfect 1 L of Sf9 cells (1.5 × 106 cells/mL, passage 22) of total 6 L grown in Sf-900 II SFM media (Thermo Fisher). AGO2 was expressed for ∼72 h at 27°C in an orbital shaker and monitored by Bacmid internal GFP expression under the control of a polyhedrin promotor. After 72 h, cells were centrifuged and the pellet washed with PBS. The total wet cell mass was ∼45 g. The cell pellet was resuspended in IMAC buffer A (50 mM Tris-HCl, 300 mM NaCl, 10 mM Imidazole, 1mM TCEP, 5% glycerol v/v), and 25× EDTA-free protease inhibitor cocktail (Merck) was added. First, cells were lysed by three cycles of freeze/thawing on dry ice. Then, cells were sonicated for 10 min (30% amplitude, 10 sec ON, 10 sec OFF) to reduce the viscosity of the lysate. The lysate was centrifuged for 1 h at 50,000 RCF (4°C). Afterward, the supernatant was filtered through a 0.22 µm sterile filter before applying onto a preequilibrated (IMAC buffer A) HisTrap-Ni2+ column (Cytiva, HisTrap HP). A linear gradient was applied with buffer A (50 mM Tris-HCl, 300 mM NaCl, 10 mM Imidazole, 1 mM TCEP, 5% glycerol v/v) and buffer B (50 mM Tris-HCl, 300 mM NaCl, 300 mM Imidazole, 1 mM TCEP, 5% glycerol). All protein-containing fractions were pooled, with a total volume of ∼15 mL, and concentrated to ∼5 mL by using a 30 kDa cut-off Amicon centrifugal filter unit (Sigma Aldrich). Next, 3 mL of a crude miR-34a IVT was concentrated to ∼600 µL and added for loading of miR-34a into AGO2 in a water bath at 37°C for 4–5 h. A slight turbidity was observed at the end of the reaction. After loading, the loaded protein solution was dialyzed (Spectrum, 3000 MWCO), then 200 µL of TEV (in-house production) added and dialyzed against 2 L IMAC buffer A overnight at 8°C. The next day, significant protein precipitation in the dialysis bag was observed, which was confirmed to not contain AGO2. The precipitate was removed by centrifugation and the supernatant was loaded onto a preequilibrated HisTrap-Ni2+ column to remove TEV-protease and other impurities. The flow-through was collected and applied onto the preequilibrated (buffer C: 20 mM HEPES, 100 mM KCl, 1 mM TCEP, 5% glycerol) size exclusion chromatography (SEC) column (Cytiva, HiLoad 16/600 Superdex 200 pg). All fractions of both HisTrap-Ni2+ columns and SEC were analyzed by SDS-PAGE (Thermo Fisher, NuPAGE 4 to 12%, Bis-Tris, 1.0 mm). Protein concentration was determined by a Bradford-based assay (Thermo Fisher, Pierce Detergent Compatible Bradford Assay Kit) and BSA as standard (Thermo Fisher). All experimental steps were followed as described in the instructions for the microplate procedure. Absorption measurements at 595 nm were performed on a Varioskan LUX multimode microplate reader (Thermo Fisher). Northern blot was performed to detect and estimate the loading efficiency (Baronti et al. 2020).
RNA SHAPE modification
The structure-specific RNA modification process for SHAPE is based on Cordero et al. (2014) with adaptations as described for the specific RNA scaffolds. To fold the RNA scaffolds, 1.2 pmol of RNA in 74 mM Na-HEPES (pH 8) was incubated at 95°C for 3 min, then snap cooled on ice for 20 min. After adding MgCl2 (1.5 µL from 100 mM stock per reaction), samples were incubated at room temperature for 30 min (final concentration: 0.08 µM RNA, 67 mM Na-HEPES, 10 mM MgCl2). To reduce sampling error, every RNA scaffold was folded in a total volume of 150 µL, corresponding to a master mix for 10 reactions, which was then divided into eight reactions of 15 µL each for the experiment.
The eight reactions were treated as follows: (i) one reaction without modifier (control); (ii) three reactions plus 1-methyl-7-nitroisatoic anhydride (1M7, Mortimer and Weeks 2007) (technical replicates); (iii) four reactions without modifier as Sanger sequencing ladder. A total of 1.72 µL 1M7 (100 mM stock, dissolved in anhydrous DMSO [Sigma Aldrich]) was added as the chemical modifier for RNA modification per reaction, and incubated for 15 min at room temperature. In reactions without a modifier, 1.72 µL of ddH2O was added instead to reach the same total volume. A control for the influence of DMSO, the solvent of 1M7, was performed, where instead of 1.72 µL of ddH2O, 100% DMSO was added.
Next, 9.8 µL of Recovery mix (0.25 M Na-MES pH 6, 1.5 M NaCl, 0.006 µM 5′-poly-dA-FAM-labeled primers [IDT, Supplemental Table S1]), which are complementary to the 20 nt at the 3′-end, and 1.5 µL poly-dT Dynabeads (Thermo Fisher), which bind the poly(A) of the FAM-labeled primers, were added to each reaction. After mixing, the samples were incubated at room temperature for 5 min and then for another 5 min on a magnetic rack (Thermo Fisher). Next, supernatants were removed, and the magnetic beads were washed three times with 70% ethanol (100 µL each time). After the last ethanol wash, samples were air dried at room temperature (at least for 1 h, on the magnetic rack) until the remaining ethanol had completely evaporated.
For experiments containing either miR-34a (RNA:RNA) or AGO2 loaded with miR-34a (RNA:RNA-RBP), these additional steps were taken after folding of the RNA trans-scaffold: the miR-AGO2 in RNA:RNA-RBP reactions was prepared as previously described (Baronti et al. 2020). After folding the trans-scaffold, the second component (i.e., miR-34a or miR-AGO2 complex) was added to the trans-scaffold in an RNA:miR ratio of 1:1 and 1:2; or an RNA:miR-AGO2 ratio of 1:1, where 20-fold access of AGO2 was used, to provide a 1:1 ratio of miR-34a-AGO2 to the RNA scaffold with miR-34a-AGO2 being only loaded 5%. For the RNA:RNA-RBP experiments, proteinase K (PK) treatment was applied after the RNA SHAPE modification by adding 45 µL of proteinase K mix (20 µg proteinase K PCR grade [Sigma Aldrich], 50 mM Tris-Cl pH 7.5, 75 mM NaCl, 6.25 mM EDTA, 1% SDS [w/v]) per reaction and incubated at 65°C for 1 h. To inactivate proteinase K, 120 µL of 70% ethanol (per reaction) was added and samples were kept on ice for 5 min. To recover the RNA, after deactivating proteinase K, 19.6 µL of Recovery mix was added per reaction. The rest of the protocol for the RNA:RNA-RBP samples was the same as mentioned above from the Recovery mix step on.
Primer extension
Primer extension was performed following the protocol by Cordero et al. (2014). Specifically, after adding 2.5 µL of ddH2O and resuspending Dynabeads, 2.5 µL of reverse transcription (RT) mix (1 µL 5× First strand buffer [Thermo Fisher], 0.01 M DTT, 1.6 mM dNTPs, 0.1 µL SuperScript III Reverse Transcriptase [Thermo Fisher]) was added to the control and 1M7 reactions. The RT mix for four reactions that were used for Sanger sequencing were prepared separately. For each of the Sanger reactions, 2.5 µL of the RT mix including a 1:6 ratio of dNTP:ddNTP were used. All samples were incubated at 50°C for 30 min. Next, RNA hydrolysis was performed by adding 5 µL of NaOH (0.4 M) and incubating at 90°C for 3 min. After snap cooling on ice for 3 min, 5 µL of Acid mix (1 volume of 1.25 M NaCl, 1 volume of 0.5 M HCl, 2 volumes of 1 M NaOAc) was added per reaction to balance the pH of the samples. After three ethanol washes (70% ethanol, 100 µL per reaction/time) and the air drying as described above, a mix of Hi-Di Formamide (Thermo Fisher) and 350 ROX size standard (Thermo Fisher) was added per reaction (11 µL from a stock of 1000 µL Formamide mixed with 8 µL of 350 ROX) to elute cDNA from Dynabeads. Finally, the eluted cDNA was sent for capillary electrophoresis (CE) by an in-house core-facility in two different dilutions: saturated samples were prepared from 4 µL of sample, diluted with 8 µL of Formamide-ROX mix; and diluted samples were prepared from 1 µL of sample diluted with 14 µL of Formamide-ROX mix.
Data analysis
The CE profiles were analyzed with the high-throughput robust analysis for the capillary electrophoresis (HiTRACE) (Lee et al. 2015) pipeline. Next, HiTRACE calculated the normalized reactivity values, based on the observed reactivity of the reference loops (Fig. 1) and adjusted for the decrease in reactivity over the length of the construct. To check the quality of obtained results between technical replicates of each scaffold/experiment, the Spearman's rank correlation coefficient (RS) of the main loop nucleotides in the trans-scaffold was calculated. For cis-scaffolds, from 5′ to 3′, the reactivity values of the first nucleotide in the target sequence (mSIRT1 or scrm-seed) till the last nucleotide of miR-34a (including the 10-nt closing loop) were used to calculate RS. Experiments with RS ≥ 0.7 for three technical replicates were used for further data analysis (see Supplemental File at the GitHub repository, data availability). To account for differences in experimental conditions, an additional normalization step was applied. We renormalized the reactivity values of the nucleotides in the main loop to the averaged reactivity values of the buffer nucleotides, the nucleotides before and after the target sequence in the main loop. These values then were used to calculate the Z-factor (equation 1 in Zhang et al. 1999) for the nucleotides of the target sequence between unbound and experiment with ligand (miR-34a and miR-34a-AGO2), to identify significant changes in reactivity values. Nucleotides with Z-factor >0 are considered as showing significantly different reactivity values (P-value ≤0.05).
| (1) |
In this equation, σU and σL are one standard deviation of reactivity values (calculated between independent replicates of each scaffold/experiment) of unbound and with ligand experiments, respectively. We also calculate the ΔReactivity, which represents the difference between reactivity values of two different experimental conditions (equation 2, Smola et al. 2015a).
| (2) |
We also used ΔReactivity as a way to visualize the difference between different experimental conditions for trans-scaffolds. For this, the ΔReactivity value is calculated from renormalized values after setting reactivity values larger than 1 to 1, and smaller than 0 to 0. We used these reactivity values (after renormalization and between 1 to 0) to calculate the P-value (Wilcoxon paired test) of the whole target sequence, nucleotides of the seed region, or the 3′-compensatory region between different experimental conditions. For the seed region, the reactivity value of the last nucleotide (at the 3′-end of the target sequence) is excluded as they commonly show high reactivity values due to the end-fraying effect (Pinamonti et al. 2019). The significant difference between unbound and with ligand experiments (+RNA or +RNA:RNA-RBP) for each sequence was considered as a check-point of having successful interaction.
For all trans-scaffolds, the results from HiTRACE, without the added normalization steps, are presented in Supplemental Figures S9–S11. For each construct, the averaged values of at least two independent replicates, each consisting of three technical replicates, were used. The standard deviation was calculated from the independent replicates and data are presented as means plus/minus one standard deviation of the average value of the independent replicates, per nucleotide. The results of all scaffolds/sequences that we used in this research (raw data and processed data) are available on our lab GitHub page (see the Data deposition section).
To predict the secondary structure of the mSIRT1:miR-34a complex based on the reactivity values from cis-scaffolds, we used the RNAProbing webserver (Washietl et al. 2012). The statistical analysis was performed using GraphPad Prism 9.0 for mac OS (GraphPad Software). Graphs and figures were created using OriginPro 2017 (OriginLab Corporation) and Adobe Illustrator.
DATA DEPOSITION
Raw data are available on our GitHub page: https://github.com/PetzoldLab/RABS_data. The data can also be found on the RMDB.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
T7 polymerase, RNase H, iPPase and TEV that were used for IVT and AGO2 purification in this project were produced at the Protein Science Facility (PSF) at the Karolinska Institutet. Insect cells were used and grown at the Protein Expression and Characterization platform, SciLifeLab. Fragment length analysis of all the samples in this project were performed at KIGene Core Facility. We thank the Petzold Lab for discussion of the work. M.D.S acknowledges funding from NIGMS R01-GM095850. K.P. acknowledges funding from Cancerfonden (CAN 2018/715), the Karolinska Institute and the Department of Medical Biochemistry and Biophysics (2-481/2016, 2-2111/2019 and support for the purchase of a 600-MHz Bruker NMR spectrometer), the Ragnar Söderberg Stiftelse (M91/14), and the Stiftelse för Strageskic Forskning (FFL15-0178). K.P and E.R.A. acknowledge funding from the Knut och Alice Wallenberg foundation (project grant KAW 2016.0087).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079190.122.
Freely available online through the RNA Open Access option.
MEET THE FIRST AUTHOR
Elnaz Banijamali.

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Elnaz Banijamali is the first author of this paper, “RNA:RNA interaction in ternary complexes resolved by chemical probing.” Elnaz received a PhD from the University of Tehran, and then moved to Sweden to join Katja Petzold's lab at the Karolinska Institutet as a postdoctoral research fellow.
What are the major results described in your paper and how do they impact this branch of the field?
We introduced RNA:RNA binding by SHAPE (RABS) as a research approach, which is suitable to study the structural details of RNA:RNA interactions in a ternary complex. This could be helpful in a better understanding of complex cellular mechanisms, such as microRNA targeting.
What led you to study RNA or this aspect of RNA science?
As a biologist, I'm interested in understanding and studying biomolecules that help a living cell to function (e.g., RNAs and proteins), individually and together. Mostly, to get the function you need the structure—hence the reason that I chose this research field.
If you were able to give one piece of advice to your younger self, what would that be?
Never forget: “This too shall pass.”
Are there specific individuals or groups who have influenced your philosophy or approach to science?
Generally, everybody needs a mentor in life to show them “the” way. For sure it's the same in science. I think I'm very lucky because I have/had two incredible mentors instead of one—my PhD cosupervisors, Dr. Mehriar Amininasab and Dr. Alireza Mashaghi.
What are your subsequent near- or long-term career plans?
Certainly establishing my own research group!
REFERENCES
- Agarwal V, Bell GW, Nam J-W, Bartel DP. 2015. Predicting effective microRNA target sites in mammalian mRNAs. Elife 4: e05005. 10.7554/eLife.05005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baronti L, Guzzetti I, Ebrahimi P, Friebe Sandoz S, Steiner E, Schlagnitweit J, Fromm B, Silva L, Fontana C, Chen AA, et al. 2020. Base-pair conformational switch modulates miR-34a targeting of Sirt1 mRNA. Nature 583: 139–144. 10.1038/s41586-020-2336-3 [DOI] [PubMed] [Google Scholar]
- Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. 2018. Metazoan microRNAs. Cell 173: 20–51. 10.1016/j.cell.2018.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindewald E, Wendeler M, Legiewicz M, Bona MK, Wang Y, Pritt MJ, Le Grice SFJ, Shapiro BA. 2011. Correlating SHAPE signatures with three-dimensional RNA structures. RNA 17: 1688–1696. 10.1261/rna.2640111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkotoky S, Murali A. 2018. The highly efficient T7 RNA polymerase: a wonder macromolecule in biological realm. Int J Biol Macromol 118: 49–56. 10.1016/j.ijbiomac.2018.05.198 [DOI] [PubMed] [Google Scholar]
- Brennecke J, Stark A, Russell RB, Cohen SM. 2005. Principles of microRNA-target recognition. PLoS Biol 3: 0404–0418. 10.1371/journal.pbio.0030085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandradoss SD, Schirle NT, Szczepaniak M, MacRae IJ, Joo C. 2015. A dynamic search process underlies microRNA targeting. Cell 162: 96–107. 10.1016/j.cell.2015.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Chou F-C, Kladwang W, Tian S, Cordero P, Das R. 2015. Consistent global structures of complex RNA states through multidimensional chemical mapping. Elife 4: e07600. 10.7554/eLife.07600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero P, Kladwang W, VanLang CC, Das R. 2014. The mutate-and-map protocol for inferring base pairs in structured RNA. Methods Mol Biol 1086: 53–77. 10.1007/978-1-62703-667-2_4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethoff E, Boerneke MA, Gokhale NS, Muhire BM, Martin DP, Sacco MT, McFadden MJ, Weinstein JB, Messer WB, Horner SM, et al. 2018. Pervasive tertiary structure in the dengue virus RNA genome. Proc Natl Acad Sci 115: 11513–11518. 10.1073/pnas.1716689115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi P, Kaur S, Baronti L, Petzold K, Chen AA. 2019. A two-dimensional replica-exchange molecular dynamics method for simulating RNA folding using sparse experimental restraints. Methods 162–163: 96–107. 10.1016/j.ymeth.2019.05.001 [DOI] [PubMed] [Google Scholar]
- Elkayam E, Kuhn C-D, Tocilj A, Haase AD, Greene EM, Hannon GJ, Joshua-Tor L. 2012. The structure of human argonaute-2 in complex with miR-20a. Cell 150: 100–110. 10.1016/j.cell.2012.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyrer H, Munteanu R, Baronti L, Petzold K. 2020. One-pot production of RNA in high yield and purity through cleaving tandem transcripts. Molecules 25: 1142. 10.3390/molecules25051142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N. 2008. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 9: 102–114. 10.1038/nrg2290 [DOI] [PubMed] [Google Scholar]
- Frédérick P-M, Simard MJ. 2021. Regulation and different functions of the animal microRNA-induced silencing complex. Wiley Interdiscip Rev RNA 12: e1701. 10.1002/wrna.1701 [DOI] [PubMed] [Google Scholar]
- Gebert LFR, MacRae IJ. 2019. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol 20: 21–37. 10.1038/s41580-018-0045-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. 2008. miRBase: tools for microRNA genomics. Nucleic Acids Res 36: D154–D158. 10.1093/nar/gkm952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombach S, Kretz M. 2016. Non-coding RNAs: classification, biology and functioning. In Non-coding RNAs in colorectal cancer (ed. Slaby O, Calin GA), pp. 3–17. Springer International Publishing, Cham: [DOI] [PubMed] [Google Scholar]
- Karlsson H, Baronti L, Petzold K. 2020. A robust and versatile method for production and purification of large-scale RNA samples for structural biology. RNA 26: 1023–1037. 10.1261/rna.075697.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern JA, Davis RH. 1997. Application of solution equilibrium analysis to in vitro RNA transcription. Biotechnol Prog 13: 747–756. 10.1021/bp970094p [DOI] [PubMed] [Google Scholar]
- Kilikevicius A, Meister G, Corey DR. 2021. Reexamining assumptions about miRNA-guided gene silencing. Nucleic Acids Res 50: 617–634. 10.1093/nar/gkab1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kladwang W, Das R. 2010. A mutate-and-map strategy for inferring base pairs in structured nucleic acids: proof of concept on a DNA/RNA helix. Biochemistry 49: 7414–7416. 10.1021/bi101123g [DOI] [PubMed] [Google Scholar]
- Kladwang W, Topkar VV, Liu B, Rangan R, Hodges TL, Keane SC, Al-Hashimi H, Das R. 2020. Anomalous reverse transcription through chemical modifications in polyadenosine stretches. Biochemistry 59: 2154–2170. 10.1021/acs.biochem.0c00020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klum SM, Chandradoss SD, Schirle NT, Joo C, MacRae IJ. 2018. Helix-7 in Argonaute2 shapes the microRNA seed region for rapid target recognition. EMBO J 37: 75–88. 10.15252/embj.201796474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau P-W, MacRae IJ. 2009. The molecular machines that mediate microRNA maturation. J Cell Mol Med 13: 54–60. 10.1111/j.1582-4934.2008.00520.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Vogt CE, McBrairty M, Al-Hashimi HM. 2013. Influence of dimethylsulfoxide on RNA structure and ligand binding. Anal Chem 85: 9692–9698. 10.1021/ac402038t [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kim H, Tian S, Lee T, Yoon S, Das R. 2015. Automated band annotation for RNA structure probing experiments with numerous capillary electrophoresis profiles. Bioinformatics 31: 2808–2815. 10.1093/bioinformatics/btv282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucks JB, Mortimer SA, Trapnell C, Luo S, Aviran S, Schroth GP, Pachter L, Doudna JA, Arkin AP. 2011. Multiplexed RNA structure characterization with selective 2′-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq). Proc Natl Acad Sci 108: 11063–11068. 10.1073/pnas.1106501108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. 2005. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE). J Am Chem Soc 127: 4223–4231. 10.1021/ja043822v [DOI] [PubMed] [Google Scholar]
- Mlýnský V, Bussi G. 2018. Molecular dynamics simulations reveal an interplay between SHAPE reagent binding and RNA flexibility. J Phys Chem Lett 9: 313–318. 10.1021/acs.jpclett.7b02921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morandi E, Manfredonia I, Simon LM, Anselmi F, van Hemert MJ, Oliviero S, Incarnato D. 2021. Genome-scale deconvolution of RNA structure ensembles. Nat Methods 18: 249–252. 10.1038/s41592-021-01075-w [DOI] [PubMed] [Google Scholar]
- Mortimer SA, Weeks KM. 2007. A fast-acting reagent for accurate analysis of RNA secondary and tertiary structure by SHAPE chemistry. J Am Chem Soc 129: 4144–4145. 10.1021/ja0704028 [DOI] [PubMed] [Google Scholar]
- Palka C, Forino NM, Hentschel J, Das R, Stone MD. 2020. Folding heterogeneity in the essential human telomerase RNA three-way junction. RNA 26: 1787–1800. 10.1261/RNA.077255.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien M, Major F. 2008. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature 452: 51–55. 10.1038/nature06684 [DOI] [PubMed] [Google Scholar]
- Pinamonti G, Paul F, Noé F, Rodriguez A, Bussi G. 2019. The mechanism of RNA base fraying: molecular dynamics simulations analyzed with core-set Markov state models. J Chem Phys 150: 154123. 10.1063/1.5083227 [DOI] [PubMed] [Google Scholar]
- Reuter JS, Mathews DH. 2010. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11: 129. 10.1186/1471-2105-11-129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman DW, Nakamura K, Nallur S, Dookwah MT, Metheetrairut C, Slack FJ, Weidhaas JB. 2016. miR-34 activity is modulated through 5′-end phosphorylation in response to DNA damage. Nat Commun 7: 10954. 10.1038/ncomms10954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirle NT, MacRae IJ. 2012. The crystal structure of human Argonaute2. Science 336: 1037–1040. 10.1126/science.1221551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirle NT, Sheu-Gruttadauria J, MacRae IJ. 2014. Structural basis for microRNA targeting. Science 346: 608–613. 10.1126/science.1258040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu-Gruttadauria J, Pawlica P, Klum SM, Wang S, Yario TA, Schirle Oakdale NT, Steitz JA, MacRae IJ. 2019a. Structural basis for target-directed microRNA degradation. Mol Cell 75: 1243–1255.e7. 10.1016/j.molcel.2019.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu-Gruttadauria J, Xiao Y, Gebert LF, MacRae IJ. 2019b. Beyond the seed: structural basis for supplementary microRNA targeting by human Argonaute2. EMBO J 38: e101153. 10.15252/embj.2018101153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smola MJ, Calabrese JM, Weeks KM. 2015a. Detection of RNA-protein interactions in living cells with SHAPE. Biochemistry 54: 6867–6875. 10.1021/acs.biochem.5b00977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smola MJ, Rice GM, Busan S, Siegfried NA, Weeks KM. 2015b. Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nat Protoc 10: 1643–1669. 10.1038/nprot.2015.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner E, Schlagnitweit J, Lundström P, Petzold K. 2016. Capturing excited states in the fast-intermediate exchange limit in biological systems using (1) HNMR spectroscopy. Angew Chem Int Ed Engl 55: 15869–15872. 10.1002/anie.201609102 [DOI] [PubMed] [Google Scholar]
- Strobel EJ, Yu AM, Lucks JB. 2018. High-throughput determination of RNA structures. Nat Rev Genet 19: 615–634. 10.1038/s41576-018-0034-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian S, Das R. 2017. Primerize-2D: automated primer design for RNA multidimensional chemical mapping. Bioinformatics 33: 1405–1406. 10.1093/bioinformatics/btw814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomezsko PJ, Corbin VDA, Gupta P, Swaminathan H, Glasgow M, Persad S, Edwards MD, Mcintosh L, Papenfuss AT, Emery A, et al. 2020. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature 582: 438–442. 10.1038/s41586-020-2253-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs JD, Condon DE, Kennedy SD, Hauser M, Bevilacqua PC, Turner DH. 2013. The nuclear magnetic resonance of CCCC RNA reveals a right-handed helix, and revised parameters for AMBER force field torsions improve structural predictions from molecular dynamics. Biochemistry 52: 996–1010. 10.1021/bi400401j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velema WA, Kool ET. 2020. The chemistry and applications of RNA 2′-OH acylation. Nat Rev Chem 4: 22–37. 10.1038/s41570-019-0147-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S, Hofacker IL, Stadler PF, Kellis M. 2012. RNA folding with soft constraints: reconciliation of probing data and thermodynamic secondary structure prediction. Nucleic Acids Res 40: 4261–4272. 10.1093/nar/gks009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks KM, Mauger DM. 2011. Exploring RNA structural codes with SHAPE chemistry. Acc Chem Res 44: 1280–1291. 10.1021/ar200051h [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidmann CA, Mustoe AM, Jariwala PB, Calabrese JM, Weeks KM. 2021. Analysis of RNA–protein networks with RNP-MaP defines functional hubs on RNA. Nat Biotechnol 39: 347–356. 10.1038/s41587-020-0709-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson KA, Merino EJ, Weeks KM. 2006. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat Protoc 1: 1610–1616. 10.1038/nprot.2006.249 [DOI] [PubMed] [Google Scholar]
- Wilkinson KA, Gorelick RJ, Vasa SM, Guex N, Rein A, Mathews DH, Giddings MC, Weeks KM. 2008. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol 6: e96. 10.1371/journal.pbio.0060096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarringhalam K, Meyer MM, Dotu I, Chuang JH, Clote P. 2012. Integrating chemical footprinting data into RNA secondary structure prediction. PLoS ONE 7: e45160. 10.1371/journal.pone.0045160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4: 67–73. 10.1177/108705719900400206 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.