

Abstract
Immunosurveillance of cancer requires the presentation of peptide antigens on major histocompatibility complex class I (MHC-I)1,2,3,4,5. Current approaches to profile MHC-I associated peptides, collectively known as the “immunopeptidome”, are limited to in vitro investigation or bulk tumor lysates, limiting our understanding of cancer-specific patterns of antigen presentation in vivo6. To overcome these limitations, we engineered an inducible affinity tag into the mouse MHC-I gene (H2-K1) and targeted this allele to the KrasLSL-G12D/+; p53fl/fl (KP) mouse model (KP/KbStrep)7. This approach allowed us to precisely isolate MHC-I peptides from autochthonous pancreatic ductal adenocarcinoma (PDAC) and lung adenocarcinoma (LUAD) in vivo. In addition, we profiled the LUAD immunopeptidome from the alveolar type 2 cell-of-origin through late-stage disease. Differential peptide presentation in LUAD was not predictable by mRNA expression or translation efficiency and is likely driven by post-translational mechanisms. Vaccination with peptides presented by LUAD in vivo provoked CD8+ T cell responses in naïve and tumor-bearing mice. Many peptides unique to LUAD, including immunogenic peptides, exhibited minimal expression of the cognate mRNA, provoking reconsideration of antigen prediction pipelines that triage peptides according to transcript abundance8. Beyond cancer, the KbStrep allele is compatible with other Cre-driver lines to explore antigen presentation in vivo in the pursuit of understanding basic immunology, infectious disease, and autoimmunity.
The success of cancer immunotherapy has led to an explosion of interest in understanding immune recognition of cancer1,4. Studies in preclinical models and patient samples has revealed that responses to immunotherapy are dependent on the presentation of peptide antigens on major histocompatibility complex class I (MHC-I)4,5. MHC-I is a heterotrimeric complex consisting of a heavy chain (H2-K and H2-D in C57BL/6 mice, HLA-A, B, C in humans), a light chain (beta-2-microglobulin, B2M), and a peptide, generally 8-11 amino acids in length. Peptides presented by MHC-I are derived from proteolysis of intracellular proteins, giving rise to a diverse array of peptide MHC-I complexes (pMHC), known collectively as the “immunopeptidome”6. Expression of non-self proteins, such as those present in cancer or virally infected cells, results in the presentation of foreign peptides or neoantigens that are recognized by CD8+ T cells to drive anti-tumor immune responses9.
Numerous groups have contributed to the understanding of MHC-I trafficking, peptide loading onto MHC-I, and the biochemical features of immunopeptidome1,8,10–13. Notably, mass spectrometry analyses have characterized endogenously presented peptides in mouse and human cells and tissues, resulting in improved prediction algorithms for peptide-MHC binding and design of personalized, neo-antigen vaccines10,11,14–16. While these advances have undoubtedly improved the design of antigen-specific therapies, our understanding of the dynamic, context-dependent immunopeptidome is still lacking.
Proteomic studies interrogating the immunopeptidome have been carried out almost universally using antibody or affinity immunoprecipitation of MHC-I from cells grown in culture, which lack the microenvironmental and/or tissue-specific stimuli that are sure to impact the repertoire of peptides presented by cancer cells10,14,15. Significantly less focus has been paid to understand the immunopeptidome in vivo, where existing studies generally profile bulk tissue or tumor lysates, which are obscured by heterogenous cell mixtures.
A more precise comparison of tumor and normal immunopeptidomes is likely to uncover new cancer-specific epitopes. Unfortunately, the field currently lacks specialized tools to precisely isolate MHC-I peptides from cells of interest in vivo. Genetically engineered mouse models (GEMMs)—like the KrasLSL-G12D/+; Trp53fl/fl (KP) model—recapitulate the histopathological features of human cancer and are tractable systems to study tumor progression in the native tissue microenvironment7. Therefore, GEMMs represent an underappreciated tool to interrogate the tumor immunopeptidome at distinct stages of tumor progression, and uncover features of tumor antigen presentation in vivo that have thus far remained elusive17.
Here we report a novel GEMM that enables specific purification of pMHC-I complexes from cells of interest in vivo. Using this unique tool, we find that the cancer immunopeptidome reflects a loss of cell identity through tumor evolution and provide compelling evidence that patterns of cancer-specific antigen presentation are not driven by changes in RNA abundance or translational efficiency. Finally, we identify immunogenic epitopes that are presented on cancer cells in vivo and provide evidence that the universe of targetable antigens in cancer is potentially broader than currently appreciated.
Inducible affinity tagged MHC-I GEMM
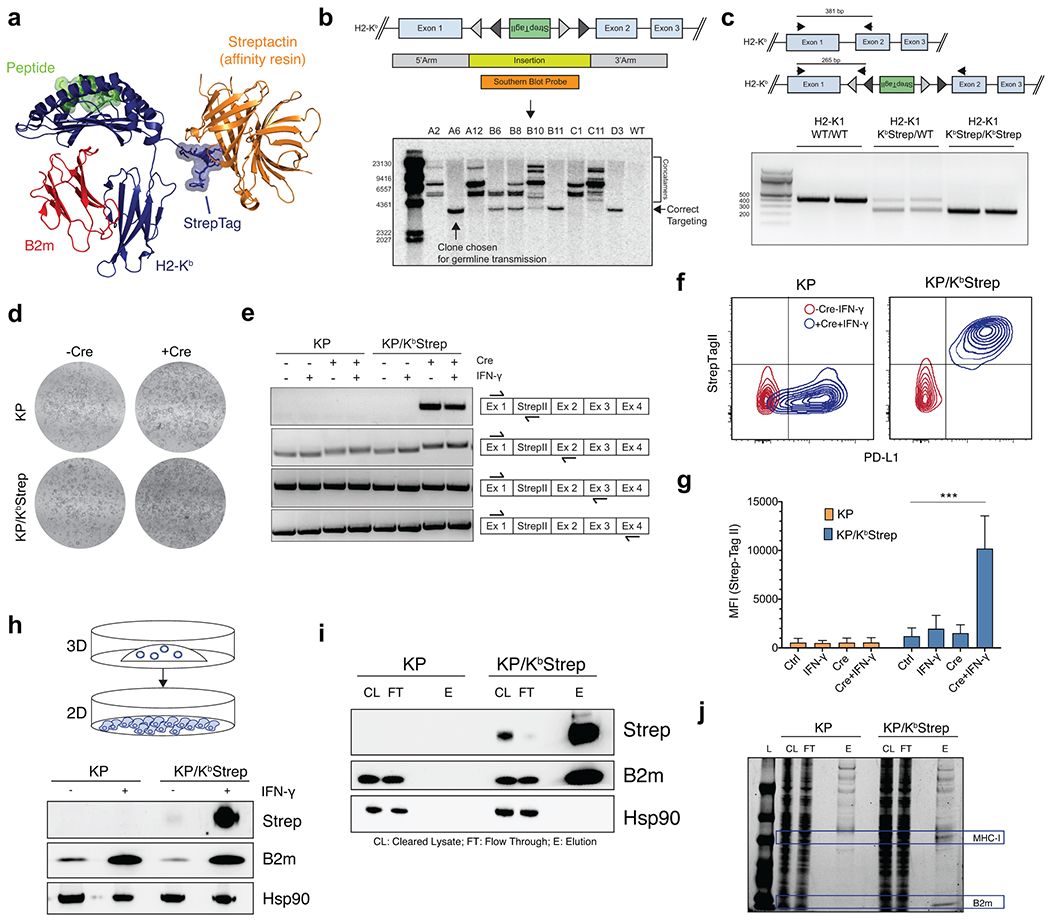
We engineered a Cre-inducible exon encoding the highly specific affinity tag, StrepTagII, into intron 1 of the H2-K1 locus (encoding the H2-Kb alloantigen of MHC-I) (Fig. 1a, KbStrep). This design results in Cre-dependent incorporation of the StrepTagII onto the mature H2-Kb protein. We targeted the KbStrep allele to embryonic stem cells derived from mice harboring the KrasLSL-G12D/+; p53fl/fl (KP) genotype. Thus, Cre recombinase activates oncogenic KrasG12D with simultaneous biallelic deletion of p53 and activation of the StrepTagII enabling specific purification of MHC-I complexes from autochthonous tumors in vivo (Fig. 1b, Extended Data Fig. 1a–c).
Figure 1. Design and Validation of the KP/KbStrep Mouse Model.
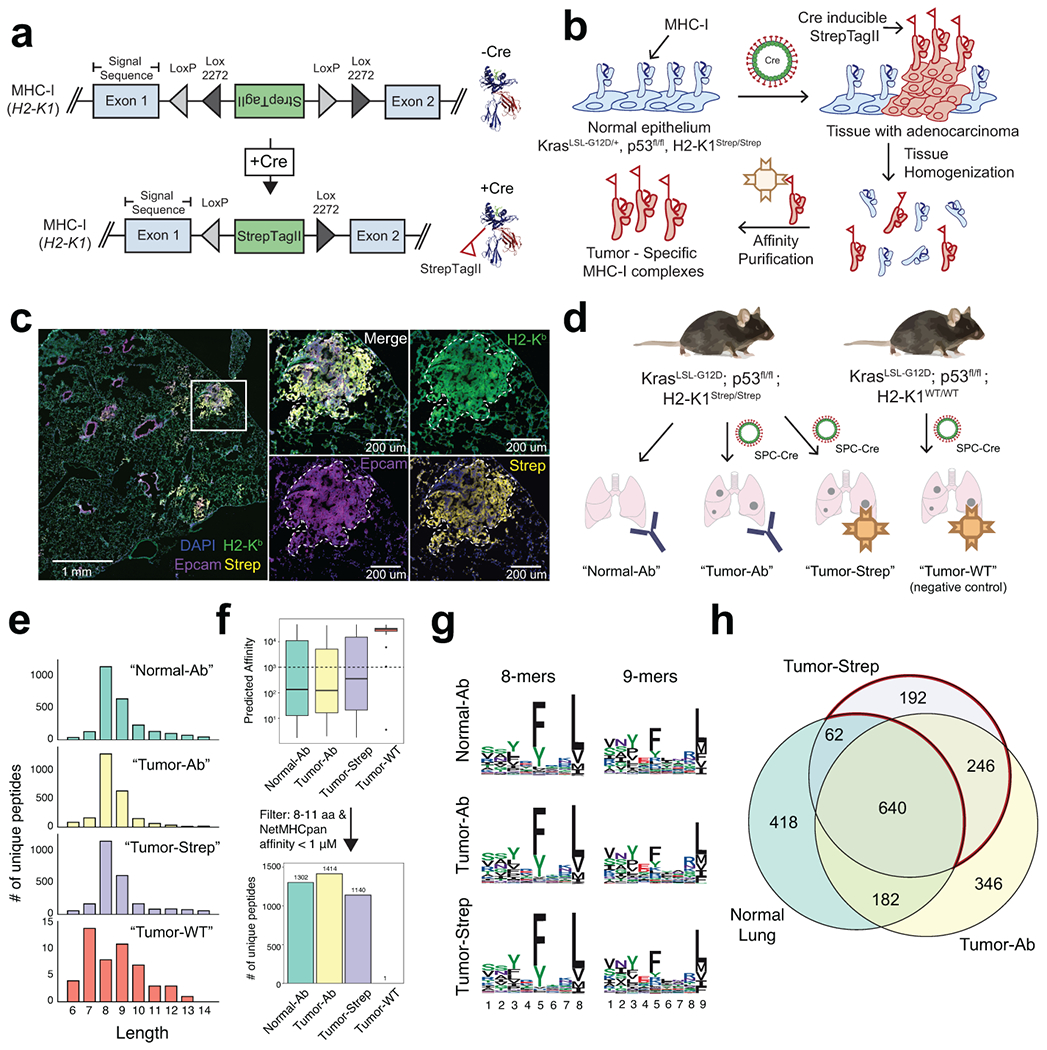
a) A Cre invertible exon encoding for the StrepTagII epitope was inserted into intron 1 of the H2-K1 gene (top). Cre-recombination induces incorporation of the StrepTagII onto the amino-terminus of MHC-I (bottom). b) Schematic illustration depicting how Cre activation of KbStrep enables tumor specific isolation of MHC-I in autochthonous tumors. c) Multiplex immunofluorescence of a representative KP/ KbStrep lung 8 weeks post tumor initiation. White box indicates the zoomed region on the right. d) Experimental schematic depicting the different in vivo sample types to be compared in downstream analyses. e) Length distribution of peptides isolated from healthy lung (Normal-Ab), KP/KbStrep tumor bearing lung with anti-H2-Kb antibody (Tumor-Ab), KP/KbStrep tumor bearing lung with Streptactin affinity purification (Tumor-Strep), or “wild type” KP tumor bearing lung with Streptactin affinity purification (Tumor-WT, negative control). f) Number of unique peptides identified in each sample type after filtering for length (8-11 amino acids) and NetMHCPan predicted affinity (<1000 nM). g) Peptide motifs of 8- and 9-mers isolated from Normal-Ab, Tumor-Ab, and Tumor-Strep samples. i) Venn diagram showing the relationship between peptides identified in Healthy Lung, tumor-bearing lung with antibody (Tumor-Ab), and tumor-specific MHC-I purification with Streptactin (Tumor-Strep). Peptides unique to tumors are outlined in red.
To validate this system, we derived pancreatic organoids from KP or KP/KbStrep mice and confirmed that the KbStrep allele was Cre-inducible, presented on the cell surface, and enabled purification of intact MHC-I complexes (Extended Data Fig. 1d–j). We next validated our system in vivo and confirmed that the StrepTagII was specifically detected on cancer cell nests in the tumor microenvironment (Extended Data Fig. 2a–c).
For immunopeptidome profiling, we isolated peptides from cells grown in a monolayer (2D), from orthotopically transplanted KP/KbStrep organoids (Ortho) or autochthonous tumors initiated through retrograde pancreatic ductal instillation of adenovirus expressing Cre recombinase into KP/KbStrep mice (Auto) or KP/KbWT mice (Auto-WT)18. We extracted peptides with the expected characteristics of H2-Kb binders in length, predicted affinity, and amino acid content from all samples except for the negative control Auto-WT samples (Extended Data Fig. 2d–j)12.
We next compared peptides isolated from orthotopic and autochthonous PDAC using our KbStrep system to those identified in normal pancreas tissue from a previous study using antibody-based immunoprecipitation of H2-Kb, revealing numerous peptides specific to PDAC tissue (Extended Data Fig. 2k)19. Analysis of source protein gene expression in scRNAseq data indicated that normal pancreas MHC-I peptides were non-specifically sampled across all cell types in the pancreas, whereas peptides identified in orthotopic and autochthonous samples were enriched for ductal cell features, reflecting the expected cellular phenotype of malignant cells in PDAC (Tabula Muris, Extended Data Fig. 2l–m). Collectively, these results validate that the KP/KbStrep system enables high resolution interrogation of cell type-specific immunopeptidomes in vivo.
Profiling the LUAD immunopeptidome in vivo
We next applied the KP/KbStrep model to autochthonous LUAD (Fig. 1c–h, Extended Data Fig. 3). Using antibody-based immunoprecipitation, we isolated H2-Kb peptides from healthy lung (Normal-Ab), 16-week tumor-bearing lung (Tumor-Ab) and compared the peptides to those captured with cancer-cell specific Streptactin affinity purification from 16 week, KP/KbStrep tumors (Tumor-Strep) or negative control KP/KbWT tumors (Tumor-WT) (Fig. 1d). Peptides isolated from all samples yielded length distributions, predicted affinities, and amino acid motifs that reflect Kb binding, except for negative control Tumor-WT samples (Fig. 1e–g, Extended Data Fig. 3j,k). In vitro biochemical experiments also demonstrated that StrepTagII incorporation does not impact MHC-I stability, trafficking, or recognition by T cells (Extended Data Fig. 4).
Comparison of the identity of peptides derived from the Normal-Ab, Tumor-Ab, and Tumor-Strep samples uncovered 438 peptides that were specific to tumors (Fig. 1h, red outline). While 246 of these peptides were also identified with the traditional antibody approach in tumor-bearing lungs, 192 were specific to Tumor-Strep samples, suggesting that cell type-specific isolation of MHC-I can provide deeper coverage of the immunopeptidome for cells of interest. 346 peptides were unique to the Tumor-Ab samples, but given the lack of specificity with this method, we could not conclude whether these peptides were derived from neoplastic cancer cells.
The immunopeptidome reflects cell identity
We next evaluated whether in vivo immunopeptidomes capture specific cellular identities or cell states within native tissue microenvironments. We applied gene signatures derived from peptides identified from healthy lung (Normal), antibody-purification from tumor-bearing lung (Ab), or affinity purification of MHC-I specifically from cancer cells (Strep) to scRNAseq data from healthy mouse lung (Tabula Muris, Figure 2a, Extended Data Fig. 5a, b)20. Direct comparison of the Strep and Normal signatures across all cell types revealed a highly significant enrichment for an AT2 phenotype in the Strep signature, consistent with tumor initiation driven by Cre recombinase expressed from an SPC promotor specific for AT2 cells (Fig. 1d, Fig. 2b, left). AT2 enrichment which was not as pronounced when comparing Ab to Normal (Fig. 2b, middle). Remarkably, comparison of the Strep and Ab signatures also resulted in robust enrichment for an AT2 phenotype in Strep samples, despite both datasets being derived from identical tumor bearing lung tissues (Fig. 2b, right). This suggests that peptides isolated with non-specific, antibody-mediated isolation of MHC-I from bulk tumor lysates are contaminated by peptides from tumor-infiltrating immune cells and other stromal cells in the microenvironment.
Figure 2. The LUAD immunopeptidome is dynamic and heterogenous throughout tumor evolution.
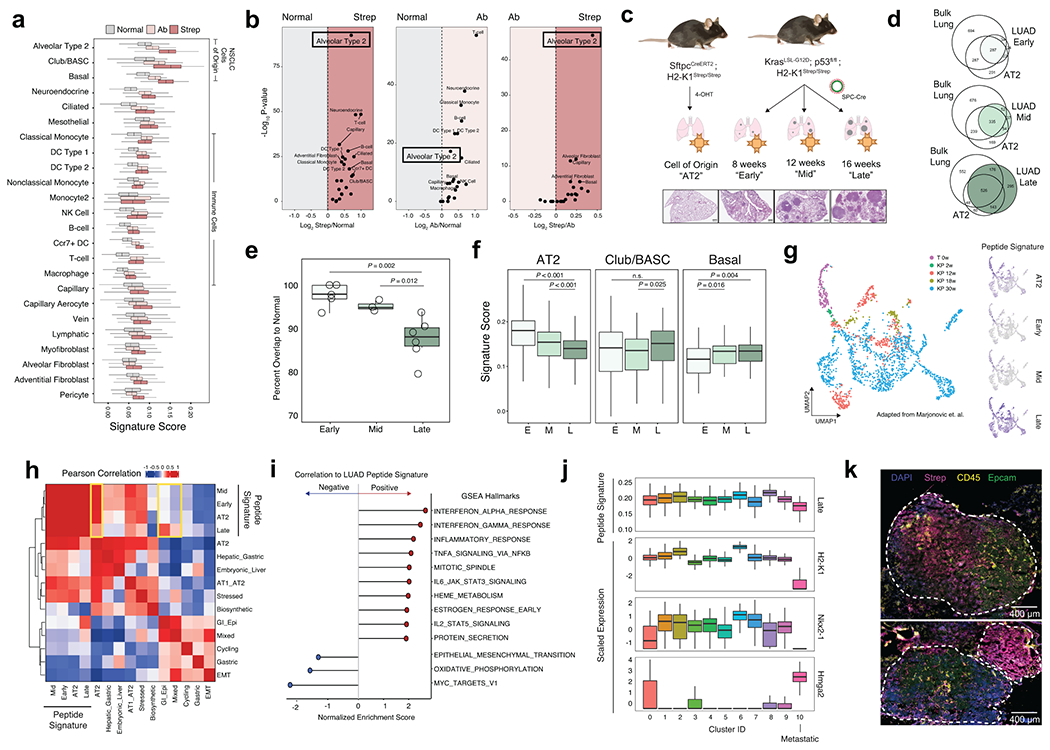
a) Comparison of the relative expression of gene signatures derived from Normal (gray), Tumor-Ab (pink), or Tumor-Strep (red) peptides across all cell types detected by scRNA-seq from healthy lung tissue (Adapted from Tabula Muris). b) Volcano plots for pairwise comparisons of peptide signatures for each cell type are shown on the right. P-value calculated with two-sided Student’s t-test with Bonferroni adjustment. c) Experimental schematic showing the types of samples for comparison of the immunopeptidome from the AT2 cell-of-origin through tumor progression. d) Overlap between peptides identified in Early, Mid, and Late-stage tumors versus bulk lung tissue and AT2 cells. e) Quantification of the percent overlap from Early (n=5), Mid (n=3), and Late (n=6) stage tumor peptides with Normal peptides (bulk lung + AT2). P calculated with Two-sided Student’s t-test. f) Relative expression of signatures derived from peptides identified in Early (E), Mid (M), and Late (L) stage tumors in alveolar type 2 (AT2), Club/BASC, and Basal cells in the healthy lung. P calculated with Mann-Whitney U Test. g) UMAP embedding of re-analyzed scRNAseq data from Marjonovic et. al. showing all AT2 (T 0w) and KP cells (KP 2-30w) throughout tumor progression (left) and expression of the AT2, Early-, Mid-, and Late-stage peptide signatures (right). h) Pearson correlation of AT2, Early, Mid, and Late signatures versus all gene modules described in Marjonovic et. al. i) Gene set enrichment analysis (GSEA) of genes ranked according to their correlation to the Late tumor peptide signature. Gene sets positively correlated to the Late signature are shown in red, and negatively correlated are shown in blue. j) Relative expression of the Late signature, Nxk2-1, Hmga2, or H2-K1 across clusters found in KP scRNAseq data. k) Multiplexed immunofluorescence depicting tumor-specific MHC-I expression (Strep, magenta) of a single late-stage KP/KbStrep tumor (top) or two adjacent tumors (bottom).
Recent data demonstrate that KP LUAD tumors progressively lose AT2 identity, prompting us to directly compare LUAD and AT2 immunopeptidomes21,22,23. To accomplish this, we crossed mice harboring the KbStrep allele to SftpcCreERT2 mice24, enabling tamoxifen-inducible incorporation of the StrepTagII specifically on AT2 cells in healthy lung tissue (Fig. 2c, Extended Data Fig. 5c–e). MHC-I peptides isolated from AT2 cells in vivo strongly reflect an AT2 cell identity (Extended Data Fig. 5f, g). We next evaluated the LUAD immunopeptidome through tumor progression at 8 weeks (Early), 12 weeks (Mid), and 16 weeks (Late) and compared these data with bulk lung and AT2 peptides (Fig. 2c, Extended Data Fig. 5c–e). Comparison of MHC-I peptides isolated from Early-, Mid-, and Late-stage tumors to those found in normal AT2 cells or lung tissue revealed that the tumor immunopeptidome progressively diverges from normal (Fig. 2d, e). We next derived signatures from genes encoding peptides identified in Early-, Mid-, and Late-stage tumors and applied them to AT2, club/BASC, and basal cell subsets from the healthy lung scRNAseq data. The immunopeptidome signatures exhibited progressive decline in signal within the AT2 compartment throughout tumor progression (Fig. 2f). In contrast, club/BASC cells and basal cells exhibited slightly increased association with Mid- and Late-stage signatures, indicating that as tumor cells lose AT2 identity and adopt alternative cellular phenotypes, a parallel alteration is observed in the tumor immunopeptidome (Fig. 2f)25.
Tumor evolution alters the immunopeptidome
To further understand the LUAD immunopeptidome in the context of tumor evolution, we applied AT2, Early, Mid, and Late peptide-derived gene signatures to published scRNAseq data from normal AT2 cells (T 0w) and KP tumor cells throughout progression (KP 2w, 12w, 18w, and 30w) (Fig. 2g)22. We observed dynamic changes in the correlation of peptide signatures with previously described expression modules in the KP model22 (Fig. 2g). Interestingly, the Late-stage signature exhibited decreased correlation to the AT2 module and increased correlation to Gastric-Epithelial and Highly Mixed transcriptional modules, previously found to be associated with phenotypic plasticity in late-stage KP tumor evolution (Fig. 2h, Extended Data Fig. 5j–l). Moreover, gene set enrichment analysis revealed that inflammatory cytokine signaling was highly correlated with the Late peptide signature, while Myc signaling, metabolic processes, and epithelial to mesenchymal transition (EMT) were anti-correlated with the Late signature (Fig. 2i). Additionally, the metastatic cluster of KP tumor cells (cluster 10, low Nkx2-1, high Hmga2), exhibits very low expression of H2-K1 and was the cluster with the lowest expression of the Late signature (Fig. 2j). Heterogeneity of antigen presentation across cancer cell states is further supported by multiplexed immunofluorescence of late-stage tumors revealing significant intra- and inter-tumor heterogeneity in MHC-I presentation (Fig. 2k). Consistent with this notion, dampening tumor inflammation with CD8a+ immune cell depletion decreased the number of unique MHC-I peptides we recovered, while provoking CD8+ T cell infiltration with agonistic CD40 antibody and Flt-3 ligand (CD40/Flt3L) increased unique peptide identification, highlighting that antigen presentation is responsive to inflammatory cues in the tumor microenvironment (Extended Data Fig. 5m, n)26. Collectively, these results demonstrate that antigen presentation by cancer cells is a highly integrative and complex process, subject to the evolution of cell identity and to microenvironmental cues in vivo. Importantly, our system captures each of these important features.
Features of LUAD-unique peptides
While we noted that gene signatures derived from the totality of the immunopeptidome reflect broad cellular identities as measured by RNA sequencing, we next sought to understand the features of peptides that were uniquely presented in KP tumors as compared to healthy bulk lung or AT2 cells (Fig. 3a, LUAD-Unique). To understand the relationship between transcription and presentation of LUAD-unique peptides, we used published bulk RNAseq data from sorted normal AT2 cells, or cells sorted from Early-, Mid-, or Late-stage KP tumors (Fig. 3b)27. Genes encoding LUAD-unique peptides exhibited varied patterns of RNA expression across timepoints, which did not correlate with either mean mRNA expression or predicted peptide affinity, suggesting that changes in mRNA expression cannot exclusively explain LUAD-unique presentation of individual peptides. To corroborate this finding, we identified genes that were upregulated at any stage of tumor progression and predicted the affinity of all possible 8- and 9-mer peptides from this list (Fig. 3c). This identified 17,130 peptides predicted to be enriched in tumor presentation based on mRNA expression and predicted affinity alone. However, this pipeline only identified 39 out of 312 total 8- and 9-mers in our LUAD-unique peptide list, reinforcing the importance of evaluating cell type/tissue-specific MHC-I presentation with empirical, mass spectrometry analysis (Fig. 3c).
Figure 3. Transcription and translation of LUAD-unique peptides.
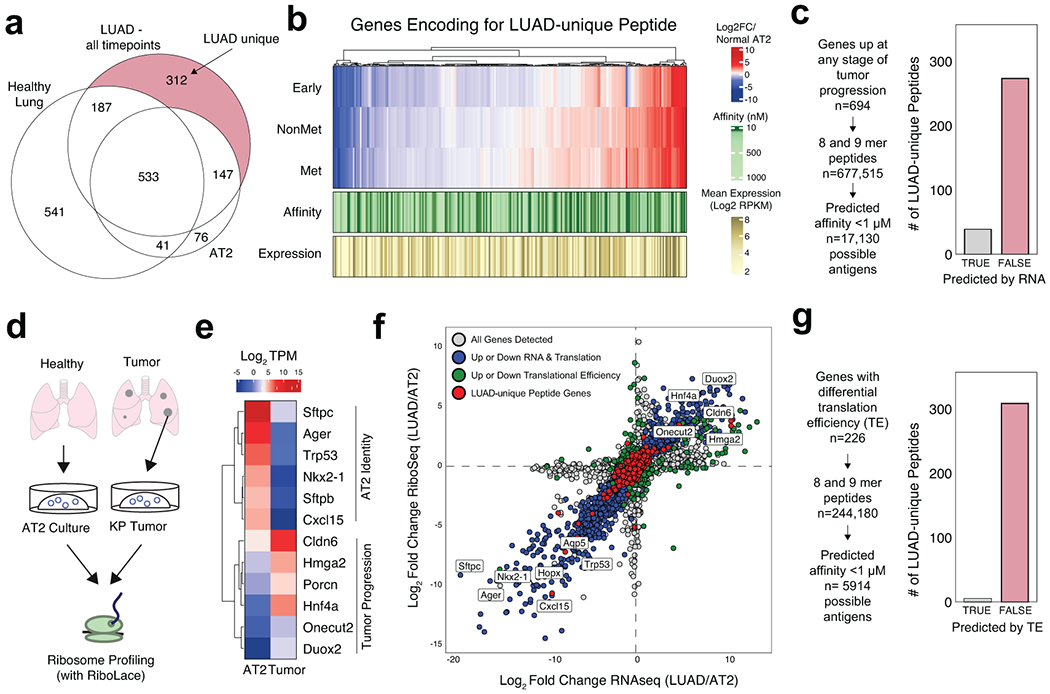
a) Venn diagram depicting the identification of LUAD-unique MHC-I Peptides. b) Heatmap showing the relative mRNA expression (red, white, blue), mean mRNA expression (gold), and predicted affinity (green) of genes encoding for LUAD-Unique peptides throughout tumor progression compared to normal AT2 cells (adapted from Chuang et. al.). c) Workflow depicting an in silico approach to predicting tumor-specific peptides based on RNA expression and predicted affinity. Histograms show the number of LUAD-unique peptides according to whether they were predicted (grey) or not predicted (red) by RNA/affinity analysis. d) Experiment outline for ribosome sequencing by RiboLace. e) Heatmap showing the relative translation intensity (TPM) for AT2 identity genes and genes associated with KP Tumor Progression. f) Comparison of RNAseq abundance (x-axis) and RiboSeq abundance (y-axis) in tumor organoids versus AT2 organoids. Genes coordinately up or down in both RNAseq & RiboSeq are shown in blue, genes exhibiting differential translational efficiency are shown in green, and genes encoding for LUAD-unique peptides are shown in red. g) Workflow depicting an approach to predict LUAD-unique peptides based on differential translation efficiency in LUAD versus AT2 (TE) and predicted affinity. Histograms show the number of LUAD-unique peptides according to whether they were predicted (grey) or not predicted (red) by differential TE.
We next assessed whether protein synthesis in tumor or normal cells could better predict the presentation of LUAD-unique peptides. To this end, we derived organoid cultures of normal AT2 cells or KP LUAD cells and performed ribosome profiling (RiboSeq) and RNAseq ex vivo (Fig. 3d, Extended Data Fig. 6a–d)28,29,30. Genes associated with AT2 identity (Sftpc, Sftpb, Ager, Nkx2-1, Cxcl15) exhibited higher translation rates in the AT2 samples, while genes associated with KP LUAD progression (Onecut2, Duox2, Hmga2, Porcn, Cldn6, Hnf4a) were translated at higher rates in LUAD samples (Fig. 3e). Integrated analysis of RNAseq and RiboSeq revealed numerous genes that were coordinately up- or downregulated at the mRNA and ribosome protected fragment (RPF) levels (Fig. 3f, blue) or exhibited differential translation efficiency (TE) (Fig. 3f, green, Extended Data Fig. 6 e–g). However, genes encoding LUAD-unique peptides generally exhibited no difference in mRNA or RPF abundance (Fig. 3f, red) and prediction of peptides based on differential TE yielded only 4 out of 312 empirically identified, LUAD-unique peptides (Fig. 3g).
The inability of mRNA abundance or translation efficiency to fully describe the LUAD-unique immunopeptidome prompted us to consider post-translational features of tumor-specific presentation31. While source protein length and thermal stability were similar in normal and LUAD-unique immunopeptidomes, we observed differences in source protein localization and a trend toward decreased protein half-life in source proteins giving rise to LUAD-unique peptides (Extended Data Fig. 7). We next sought to perturb post-translational processes through inhibition of the molecular chaperone heat shock protein 90 kDa (Hsp90), which we previously found to reshape the tumor immunopeptidome and stimulate anti-tumor immune responses32. Treatment with Hsp90i increased the number of unique MHC-I peptides we identified, including those derived from Hsp90 client proteins with lower thermal stability (Extended Data Fig 8a–k). Taken together, these data demonstrate that the immunopeptidome can be shaped through post-translational mechanisms and, more broadly, underscore the potential of the KP/KbStrep model to discover treatment-induced changes in the tumor immunopeptidome in vivo (Extended Data Fig. 8l).
Discovery of Tumor Antigens in KP LUAD
Given our ability to discern the LUAD immunopeptidome from that of normal tissues in vivo, we evaluated novel tumor epitopes in the KP/KbStrep model. We found 135 peptides that were recurrently presented on LUAD in vivo but not observed in healthy tissues from our study or published data19, which we termed putative non-mutated “Tumor Specific Antigens” (TSA) (Fig. 4a, Extended Data Fig. 9a). In addition, we nominated an additional 147 peptides that were only found on one tissue (not lung) and determined these peptides to be putative “Tumor-Associated Antigens” (TAA) (Fig. 4a, Extended Data Fig. 9b). Seeking to evaluate the immunogenicity of both classes of antigens, we vaccinated naïve mice with a pooled peptide vaccine (3 TSA, 5 TAA) using bone marrow derived dendritic cells (Fig. 4b)33. IFNγ ELISPOT of splenocytes from control and vaccinated mice, revealed 3 immunogenic peptides (Fig. 4c, Extended Data Fig. 10). Remarkably, 2/3 peptides were not presented by KP/KbStrep cells in vitro, and comparative RNA or RPF expression in LUAD versus AT2 cells would not have led to their prioritization (Fig. 4d, Extended Data Fig. 9c–f). Collectively, these data reinforce the importance of examining the immunopeptidome in vivo and challenge the notion that differential expression is required for tumor-specific antigen presentation.
Figure 4. Discovery of novel tumor antigens in LUAD.
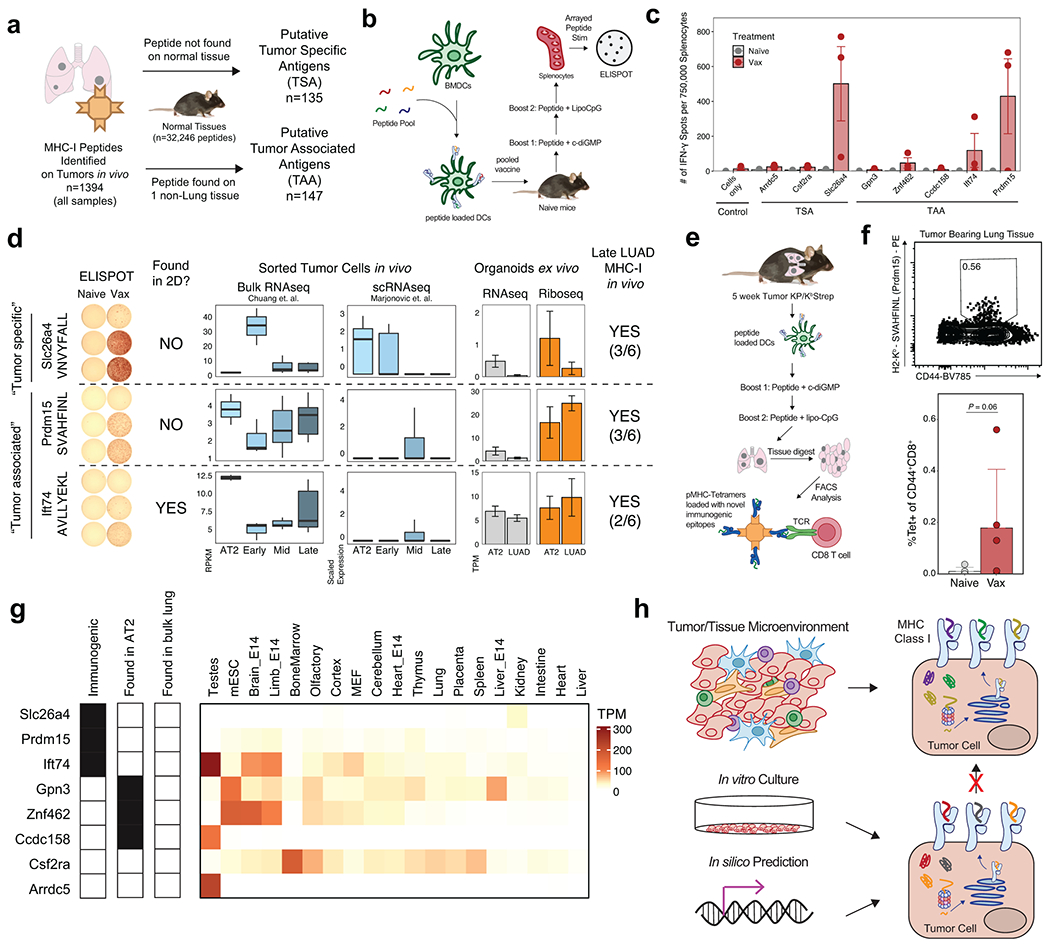
a) Workflow for identifying putative non-mutated Tumor Specific Antigens (TSA) or Tumor Associated Antigens (TAA). b) Experimental schematic for a pooled peptide vaccine strategy in naïve mice. c) Quantification of interferon-gamma (IFN-γ) ELISPOT data of splenocytes from naïve mice or mice vaccinated with pooled peptides. Each peptide in the pool was used individually to stimulate splenocytes prior to ELISPOT. n=3 mice per group. d) Comparison of in vivo bulk RNAseq, single cell RNAseq and ex vivo RNAseq and RiboSeq for genes encoding immunogenic peptides. Identification of peptides by in vitro and in vivo immunopeptidomics is also indicated. For in vivo peptide identification, the fraction of late-stage tumor samples where the peptide was identified is indicated. e) Pooled vaccine strategy for KP tumor-bearing animals. f) Flow cytometry plots depicting pMHC-I Tetramer staining for a representative TAA (SVAHFINL, Prdm15) in the lung tissue of naïve and vaccinated, tumor-bearing mice. P calculated with one-sided Mann-Whitney test. g) Transcript abundance across healthy mouse tissues for peptides included in the pooled vaccine. Detection of the peptide on AT2 cells or bulk lung tissue is also indicated. h) Model depicting the incongruence between the immunopeptidome derived from in silico prediction methods, in vitro mass spectrometry, and tumor-specific in vivo immunopeptidomics.
To further explore the immunogenicity of these epitopes, we vaccinated 5-week KP tumor-bearing animals and measured CD8+ T cell reactivity with custom peptide:MHC-I Tetramers (Fig. 4e)34. We detected CD8+ T cells that recognize a peptide derived from the TAA Prdm15 (SVAHFINL) in tumor-bearing lung tissue in vaccinated mice, but not control mice (Fig. 4f, Extended Data Fig. 9h). Seeking to understand the lack of immunogenicity of the remaining 5/8 vaccine peptides, we cross-referenced our AT2 immunopeptidome data and found that 3/5 were presented in AT2 but not bulk lung tissue (Fig. 4g). RNA expression patterns across healthy mouse tissue (Mouse Encode Project) for genes encoding peptides presented by AT2 cells would incorrectly predict potential cancer testis antigens (Ccdc158), or oncofetal antigens (Gpn3, Znf462). These analyses demonstrate that in vivo, cell type specific immunopeptidomics provides an opportunity to empirically evaluate cell and tissue specific presentation patterns and more accurately classify potential antigens compared to in vitro or in silico approaches (Fig. 4h, Extended Data Fig. 9j, k).
Discussion
We developed a novel mouse model that enables specific isolation of MHC-I complexes from discrete cell populations of interest in vivo. Application of this system to AT2 cells and LUAD revealed that the immunopeptidome is highly dynamic through tumor progression and evolves with cellular states adopted by tumor cells. This raises the intriguing possibility that distinct stages of tumor development are associated with an ever-changing landscape of targets for antigen-specific T cells through tumor progression.
Notably, our results suggest that neither mRNA expression nor translation efficiency fully explain LUAD-specific antigen presentation in vivo. While many neo-antigen prediction pipelines include mRNA abundance as a method to triage potential neoantigens, our data suggest that many peptides are recurrently presented in vivo despite apparently low transcript levels, highlighting the importance of empirical mass spectrometry evidence to evaluate MHC-I presentation. Further study is needed to elucidate post-translational mechanisms that shape tumor-specific antigen presentation in vivo, which may ultimately improve neoantigen prediction.
TSAs and TAAs are known to elicit antigen-specific responses against tumors35,36,37,38. Consistent with recent work from our laboratory26,39–41, we found that vaccination was required to provoke responses against antigens identified in vivo, likely due to suboptimal priming of CD8+ T cells in our model. Systematic interrogation of epitopes identified with this mouse model will likely uncover additional immunogenic epitopes. These strategies will ultimately expand our understanding of the “altered-self” presented by cancer cells, which can be used to design and evaluate new cancer immunotherapies in preclinical models42.
We also envision that this model system can identify peptides derived from cryptic translation events29,43,44,45, post-translational modifications46, transposable elements47, and the microbiome48 as many of these events will be influenced by physiological cues from the in vivo microenvironment.
Given the prevalence of cell/tissue specific Cre-driver mouse strains, the KbStrep allele presents a unique opportunity to map a high resolution in vivo Immunopeptidome Atlas19, as we demonstrated with AT2 cells in this study. Cross referencing these data to other “omics” based tissue atlases can elucidate the relationship between cellular phenotype and the immunopeptidome, which cannot be accomplished using bulk tissue measurements.
In summary, we have uncovered important aspects of the tumor immunopeptidome in vivo that should provoke further investigation into context-specific antigen presentation and will ultimately improve our understanding of tumor-immune interactions. In turn, we describe a versatile tool for the broader research community to interrogate mechanisms of antigen presentation with unprecedented resolution in health and disease.
Materials and Methods
Mice
All animal studies described in this study were approved by the MIT Institutional Animal Care and Use Committee. All animals were maintained on a pure C57BL/6J genetic background, except for ES cell chimeras which were a mix of C57BL/6J and albino C57BL/6J. Generation of KrasLSL-G12D/+ and Trp53flox/flox (KP) mice has previously been described49,50. SftpcCreERT2/CreER/T2 mice on a C57BL/6J background were purchased from Jackson Labs (Strain# 028054)24 and bred to H2-K1Strep/Strep mice to produce the SftpcCreERT2/CreERT2 ; H2-K1Strep/Strep strain.
mESC culture and CRISPR-assisted gene targeting
Targeted insertion of the Cre-invertible StrepTag allele (KbStrep) into the endogenous H2-K1 locus was performed in KP*1 embryonic stem cells, which were generated by crossing a hormone-primed C57BL/6J Trp53fl/fl female with a C57BL/6J KrasLSL-G12D/+; Trp53fl/fl male. At 3.5 days post-coitum, blastocysts were flushed from the uterus, isolated, and cultured on a mouse embryonic fibroblast (MEF) feeder layer in ‘ESCM+LIF+2i’ [Knockout DMEM (Gibco), 15% FBS (Hyclone), 1% NEAA (Sigma), 2 mM Glutamine (Gibco), 0.1 mM β-mercaptoethanol (Sigma-Aldrich) 50 IU Penicillin, 50 IU Streptomycin, 1000 U/ml LIF (Amsbio), 3 μM CHIR99021 (AbMole), 1 μM PD0325901(AbMole)]. After 5-7 days in culture the outgrown inner cell mass was isolated, trypsinized and re-plated on a fresh MEF layer. ES cell lines were genotyped for KrasLSL-G12D/+;Trp53fl/fl, and Zfy (Y-chromosome specific). Primer sequences available upon request. ES cell lines were tested for pluripotency by injection into host blastocysts from albino mice to generate chimeric mice.
DNA mixes containing 3:1 mixes of H2-K1-Strep targeting vector:U6-sgH2-K1-eCas9v1.1-T2A-BlastR were ethanol precipitated prior to Lipofectamine 2000 (Thermo Fisher) transfection of approximately 3 x 105 KP*1 mESCs according to manufacturer’s instructions. Transfected mESCs were plated on MEF feeder cells and selected with 4 ug/mL blasticidin for 48 hours. Cells were then trypsinized and plated at low density onto a fresh plate with MEF feeder cells. After 5-6 days, large colonies were picked using a dissecting microscope and replated into a 96 well plate containing MEF feeder cells. After 4-5 days, each well of the 96 well plate was trypsinized and 1:2 of the material was frozen in ESCM + 10% DMSO and the remainder of the cells were transferred to a 96 well PCR plate for PCR based integration screening. Clones containing homozygous targeting events were then thawed and expanded into 24 well dishes, which were then subjected to genomic DNA extraction and overnight restriction digest with SapI. Digestions were then electrophoresed on a 0.7% agarose gel prior to transfer onto an Amersham Hybond XL nylon membrane (GE Healthcare). Blots were then probed with P32-labeled DNA probes comprised of an internal sequence homologous to the genomic insertion containing the StrepTag exon.
Correctly targeted clones were injected into albino C57BL/6 blastocysts. Coat color was used as a surrogate marker for chimerism. Low degree chimeras were chosen for pancreatic organoid generation and high degree chimeras were bred to KP* mice for germline transmission.
Pancreatic Organoid Isolation
Low degree KP; KbStrep chimeras were chosen for organoid isolation using previously described methods41. Briefly, the pancreas was manually dissected, transferred to a petri dish and thoroughly minced with a razor blade. Minced tissue was then transferred to a 1.5 mL microcentrifuge tube with 1 mL of PBS supplemented with 125 U/mL collagenase IV (Worthington) and incubated rotating at 37°C for 20-30 min. Cell suspensions were diluted with 9 mL of PBS, and centrifuged at 2000 r.p.m. for 2 min. Cell pellets were then washed with 10 mL of PBS and centrifuged at 2000 r.p.m. for 2 min. The resulting cell pellet was then resuspended in 100% Matrigel (Corning) and plated as 50 μL domes and solidified at 37°C. Organoids were then cultured in organoid complete media. Purification of cells derived from the targeted ES cells was accomplished with puromycin selection at 6 μg/mL (a puromycin resistance gene is encoded within the LSL cassette upstream of the KrasG12D allele).
Media for pancreatic organoids was formulated based on L-WRN cell conditioned media (L-WRN CM)51. Briefly, L-WRN CM was generated by collecting 8 days of supernatant from L-WRN cells, grown in Advanced DMEM/F12 (Gibco) supplemented with 20% fetal bovine serum (Hyclone), 2 mM GlutaMAX, 100 U/mL of penicillin, 100 μg/mL of streptomycin, and 0.25 μg/mL amphotericin. L-WRN CM was diluted 1:1 in Advanced DMEM/F12 (Gibco) and supplemented with additional RSPO-1 conditioned media (10% v/v), generated using Cultrex HA-R-Spondin1-Fc 293T Cells. The following molecules were also added to the growth media: B27 (Gibco), 1 μM N-acetylcysteine (Sigma-Aldrich), 10 μM nicotinamide (Sigma-Aldrich), 50 ng/mL EGF (Novus Biologicals), 500 nM A83-01 (Cayman Chemical), 10 μM SB202190 (Cayman Chemical), and 500 nM PGE2 (Cayman Chemical).
Pancreatic organoids were serially passaged with TrypLE Express (Life Technologies). After 4 passages, KP or KP; KbStrep organoids were then subjected to ex vivo transformation by dissociation, mixing with adenoviral Cre (Ad-CMV-Cre; MOI=500), and re-embedding in Matrigel. After 72 hours, transformants were selected with Nutlin-3a (10 μM, Sigma Aldrich) to select for loss of p53.
Orthotopic transplantation of pancreatic organoids
Orthotopic transplantation of organoids was performed as previously described41. Briefly, animals were anesthetized using Isoflurane, the left subcostal region was depilated (using clippers or Nair) and the surgical area was disinfected with alternating Betadine/Isopropyl alcohol. A small (~2 cm) skin incision was made in the left subcostal area and the spleen was visualized through the peritoneum. A small incision (~2 cm) was made through the peritoneum overlying the spleen and the spleen and pancreas were exteriorized using ring forceps. A 30-gauge needle was inserted into the pancreatic parenchyma parallel to the main pancreatic artery and 100 μL (containing 1.25*105 organoid cells in 50% PBS + 50% Matrigel) was injected into the pancreatic parenchyma. Successful injection was visualized by formation of a fluid-filled region within the pancreatic parenchyma without leakage. The pancreas/spleen were gently internalized and the peritoneal and skin layers were sutured independently using 5-0 vicryl sutures. All mice received pre-operative analgesia with Bup-SR and were followed post-operatively for any signs of discomfort or distress. Organoid/Matrigel mixes were kept on ice throughout the entirety of the procedure to prevent solidification prior to injection. For orthotopic transplantation, syngeneic C57BL/6J mice (aged 4-12 weeks) were transplanted. Male pancreatic organoids were only transplanted back into male recipients.
Retrograde pancreatic duct delivery
Retrograde pancreatic duct instillation of lentivirus has been previously described 18,41. Briefly, the ventral abdomen was depilated (using clippers or Nair) 1-2 days prior to surgery. Animals were anesthetized with Isoflurane and the surgical area was disinfected with alternating Betadine/Isopropyl alcohol. A small skin incision was made in the anterior abdomen (~2-3 cm midline incision extending caudally from the xiphoid process). A subsequent incision was made through the linea alba and incision edges were secured in place with a Colibri retractor. The remainder of the procedure was conducted under a Nikon stereomicroscope. A moistened (with sterile 0.9% saline) sterile cotton swab was used to gently move the left lobe of the liver cranially towards the diaphragm. A second moistened sterile cotton swab was used to gently reposition the colon/small intestine into the right lower abdominal quadrant, until the duodenum was visualized. The duodenum was gently repositioned (still in the abdominal cavity) using moistened cotton swabs until the pancreas, common bile duct and sphincter of Oddi were well visualized. The common bile duct and cystic duct were gently separated from the portal vein and hepatic artery using blunt dissection with Moria forceps. A microclip was placed over the common bile duct (cranial to pancreatic duct branching) to prevent influx of the viral particles into the liver or gallbladder, forcing the viral vector retrograde through the pancreatic duct. To infuse the viral vector, the common bile duct was cannulated with a 30-gauge needle at the level of the sphincter of Oddi and 150 μL of virus was injected over the course of 30 seconds. Gentle pressure was applied at the sphincter of Oddi upon needle exit to prevent leakage into the abdominal cavity. Subsequently, the microclip and Colibri retractor were removed. The peritoneum was closed using running 5-0 Vicryl sutures. The cutis and fascia were closed using simple interrupted 5-0 Vicryl sutures. The entire procedure was conducted on a circulating warm water heating blanket to prevent intra-operative hypothermia. All mice received pre-operative analgesia with sustained-release Buprenorphine (Bup-SR) and were followed post-operatively for any signs of discomfort or distress. For retrograde pancreatic ductal installation, male mice (aged 3-6 weeks) and female mice (aged 3-8 weeks) were transduced with 7e8 PFU of Ad5-Pft1a-Cre (University of Iowa Viral Core) in serum-free media (Opti-MEM; Gibco).
Intratracheal Administration of Adenovirus
Adenovirus expressing Cre recombinase from an SPC promoter (Ad5-SPC-Cre, Univ. of Iowa) was prepared by diluting virus stocks to 400 p.f.u./μL into OptiMEM (Gibco) followed by addition of CaCl2 to a final concentration of 10 μM7. Viral suspensions were then mixed and incubated at room temperature for 20 min before placing on ice. Mice were anesthetized with Isoflurane prior to setting on a custom wire platform to open the mouth. The trachea was then canulated with 22G catheters (Exel) and 50 μL of viral suspension was added to the catheters. Once the mouse aspirated the viral suspension, they were transferred to a prewarmed cage to recover from anesthesia and monitored daily for 3 days to ensure recovery from the procedure. Dilutions of virus were used within 1 hour of preparation.
Flow cytometry of cultured cells and lung tissue
For staining pancreatic organoids, cells were plated into 4 x 20 uL domes per well in a 12 well plate. Cells were either left untreated or incubated with 20 ng/mL IFN-γ for 48 hours. At the time of harvest, organoid media was aspirated, and the Matrigel domes were mechanically disrupted with vigorous pipetting in PBS. Cell suspensions were then transferred to 15 mL conical tubes passivated with 0.1% BSA and centrifuged at 2000 rpm for 2 min. The resulting cell pellet was then incubated with 1 mL TrypLE Express for 10-15 minutes. The dissociation reaction was quenched with 10 mL PBS followed by centrifugation at 2000 rpm for 2 min. Cell pellets were then resuspended in 200 uL PBS and transferred to a 96 well U-bottom plate for staining. Cells were then incubated with Zombie Aqua fixable viability stain (1:1000, Biolegend) for 15 min prior to staining with primary antibodies diluted in PBS+2% Heat Inactivated FBS (FACS buffer) for 30 min on ice (see Antibody table). Cells were then washed twice with FACS buffer prior to analysis on a LSR II analytical flow cytometer.
For FACS analysis of tumor bearing lung tissue, KP or KP/KbStrep mice (n=3 each) were euthanized 12 weeks post tumor initiation. 2 minutes prior to sacrifice, intravascular staining antibody (anti-CD45-APC-eFluor786, Biolegend) was injected retro-orbitally to stain circulating immune cells. Mice were then euthanized with cervical dislocation, and the lungs were removed and placed on ice. 100-200 mg of tumor bearing lung tissue was then thoroughly minced with Noyes scissors, prior to incubation with digestion buffer (HBSS supplemented with 5% HI FBS, 125 U/mL collagenase IV (Worthington) and DNase (Roche) with rotation for 30 min at 37 °C. Cell suspensions were then macerated through 70 μM cell strainers (Corning) and centrifuged at 500 x g for 5 min. Cell pellets were resuspended in 1 mL ACK Lysis Buffer (Gibco) and incubated at room temperature for 5 min before quenching with 10 mL RPMI + 10% HI FBS and centrifugation at 500 x g for 5 min. Cell pellets were then resuspended in 200 uL FACS buffer and transferred to 96 well U bottom plates for staining with Fc block (BD Biosciences) for 20 minutes, zombie fixable viability stain for 20 min, and primary antibody cocktail for 30 min on ice. Cells were then washed twice times with FACS buffer before fixation overnight with FoxP3/Transcription Factor Staining Buffer Set (eBioscience) according to manufacturer’s instructions prior analysis on a Fortessa flow cytometer. All flow cytometry data was analyzed in FlowJo v10.
Affinity Purification of H2-Kb with Streptactin
Whenever possible, great care was taken to keep samples ice cold at all times to maintain MHC-I complex stability. For cultured cells, ~6 x 107 cells were used for each replicate (4 x 15 cm dishes). In the culture dish, cells were washed twice with PBS, prior to lysis with 2 mL MHC Extraction Buffer (MEB: 20 mM Tris pH 8.0, 100 mM NaCl, 1 mM EDTA, 1% Triton-X100, 60 mM octyl-glucopyranoside, 6 mM MgCl2, and 1X HALT protease inhibitors (Pierce)10. Cell lysates were then transferred to 2 mL microcentrifuge tubes and supplemented with 20 U benzonase and 10 U avidin (to block endogenously biotinylated protein) prior to incubating with rotation at 4 °C. Lysates were then cleared by centrifugation at 16,000 x g for 15 min prior to incubation with MagStrep Type 3 Streptactin XT beads (IBA Biosciences).
For pancreatic and lung tissues, tumor bearing tissue was dissected and immediately lysed or flash frozen in liquid N2 for later processing. For lysis, fresh or frozen tissue was quickly minced with Noyes scissors and transferred to a 7 mL glass Dounce homogenizer (Sigma), precooled on ice. 4 mL MEB was then added, and the tissue was thoroughly homogenized with 10-20 passes of a loose-fitting pestle followed by 5-10 passes of a tight-fitting pestle. Tissue homogenates were transferred to 5 mL centrifuge tubes (Eppendorf) and supplemented with 20 U benzonase and 10 U avidin prior to incubating with rotation at 4 °C for 20 min and subsequent removal of debris by centrifugation at 16,000 x g for 15 min.
Prior to incubating with cleared lysate, Streptactin beads were equilibrated by magnetizing and washing 1x with MEB. For each cell culture sample or tissue sample, 1 mL of bead suspension (50 uL bed volume) was used. Equilibrated beads were then added to cleared lysates and incubated with rotation at 4 °C for 1-3 hrs. After incubation, beads were washed 2 x with MEB, 2 x with TBS, and 2x with 20 mM Tris. On the last wash, suspended beads were transferred to a new Lo-Bind microcentrifuge tube prior to elution. Strep tagged H2-Kb was then eluted from the Streptactin resin by adding 400 uL of 0.5X Buffer BXT (IBA Biosciences) and incubating on ice for 20-30 min with occasional flicking of the tube to maintain the beads in suspension. Beads were then magnetized, and the supernatant was transferred to a new Lo-Bind microcentrifuge tube. Biotin, H2-Kb heavy chain, and B2m light chain were then precipitated by adding 1% TFA slowly while gently vortexing the elution. The fluffy white precipitate was then pelleted by centrifugation at 20,000 x g for 10 min. Supernatants containing liberated MHC-I peptides were then directly aspirated into 8 ug binding capacity C18 solid phase extraction pipette tips (Pierce). Tips were preequilibrated with 50% Acetonitrile (ACN) and 0.1 % formic acid according to manufacturer’s instructions. The 400 uL of eluted material was loaded, 20 uL at a time until the entire volume of the elution was passed over the C18 sorbent. Tips were then washed twice with 5% ACN/0.1% FA prior to elution in 10 uL of 30% ACN/0.1% FA. Desalted peptides were then dried down prior to reconstitution in 3% ACN/0.1% FA in an autosampler vial for LC-MS/MS analysis.
Antibody Immunoprecipitation of MHC-I
Peptide MHC isolation was performed as described previously33. Healthy lung tissue or tumor bearing lung tissue was homogenized and cleared as described for Streptactin purification. Per sample, 1 mg of Anti-H2-Kb (clone Y3, BioXCell) was bound to 20 μL (bed volume) FastFlow Protein A Sepharose beads (GE Healthcare) by incubating for 1 hour at 4°C. Beads were then washed with lysis buffer and samples were incubated for 2-4 hours rotating at 4°C. Beads were then centrifuged at 1,000 rpm, washed twice with MEB, twice with 1× TBS, and eluted with 10% acetic acid at room temperature. Eluate was then filtered using 10 kDa MWCO spin filters (PALL Life Science), which were passivated with 0.1% BSA and acidified with 10% acetic acid prior to filtration. Filtered peptides were then further purified with 8 μg binding capacity C18 tips (Pierce) prior to LC/MS-MS analysis. . For multiplexing, lyophilized pMHC were resuspended in 33 μl of labeling buffer (50% ethanol, 150 mM TEAB) and mixed with 40 μg of pre-aliquoted TMT 6plex (Thermo Scientific) resuspended in 10ul of anhydrous acetonitrile. Labeling reaction was carried out on a shaker for 1 hour at room temperature and quenched with 0.3% hydroxylamine. Samples were combined and dried in SpeedVac prior to being cleaned up with SP3 protocol as previously described (Stopfer et. al. 2020).
Liquid Chromatography Tandem Mass Spectrometry
pMHC samples were analyzed using an Exploris 480 Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Scientific) coupled with an UltiMate 3000 RSLC Nano liquid chromatography (LC) system (Dionex), Nanospray Flex ion source (Thermo Scientific), and column oven heater (Sonation). Samples were resuspended in 0.1% formic acid and directly loaded onto an analytical capillary chromatography column with an integrated electrospray tip (~1 μm orifice), prepared and packed in-house (50 μm ID × 15 cm & 1.9 μm C18 beads, ReproSil-Pur). All or half the pMHC elutions was analyzed in a given LC-MS/MS analysis. Peptides were eluted using a linear gradient with 6-25% buffer B (70% Acetonitrile, 0.1% formic acid) for 53 minutes, 25-45% for 12 minutes, 45-97% for 3 minutes, hold for 1 minute, and 97% to 3% for 1 minute.
Standard mass spectrometry parameters were as follows: spray voltage, 2.0-2.5 kV; no sheath or auxiliary gas flow; heated capillary temperature, 275 °C. The Exploris was operated in data dependent acquisition (DDA) mode. Full scan mass spectra (350-1200 m/z, 60,000 resolution) were detected in the orbitrap analyzer after accumulation of 3E6 ions (normalized automatic gain control (AGC) target of 300%), automatic maximum injection time (IT). For every full scan, MS2 spectrum were collected during a 3 second cycle time. Ions were isolated (0.4 m/z isolation width) for a maximum IT of 150/250 ms or 1E5/7.5E4 ions (100%/75% normalized AGC target) and fragmented by higher energy disassociation collision (HCD) with 30% collision energy (CI) at a resolution of 60,000. Charge states < 2 and > 4 were excluded, and precursors were excluded from selection for 30 seconds if fragmented n=2 times within 20 second window.
Synthetic peptides were ordered from Genscript at >= 85% purity, with no TFA removal, and stock solutions were dissolved in DMSO at a peptide concentration of 10 mM. Synthetic peptides analysis was also performed on the Exploris 480 coupled to an Agilent 1260 LC system using a custom analytical chromatography column, prepared as described above. Peptides were eluted using a 105-minute gradient (3.4% for 10 min, 3.4-6.9% for 2 min, 6.9-29% for 53 min, 51-100% for 3 min, hold for 1 min, 100% to 3.4% for 1 min) with 70% acetonitrile in 0.2 M acetic acid at the flow rate of 0.2 ml/min and a pre-column split of 2,000:1. The Exploris was operated with a precursor scan range of 440-544 m/z and a targeted inclusion list for synthetic peptides (Supplementary Table 10). Additional acquisition parameters are as follows: full scan spectra were collected at 60,000 resolution, 300% normalized AGC, automatic IT, MS2 spectra were collected at 60,000 resolution, isolation width 0.4 m/z, maximum IT of 250 ms, 100% normalized AGC target fragmented by HCD with 30% CI with a 3 second cycle time.
TMT labeled samples were analyzed on a Q Exactive Plus mass spectrometer coupled to Agilent 1260 LC. Samples were resuspended in 3% acetonitrile, 0.1% formic acid and one third of the labeled mixture was loaded onto a precolumn (100 um ID x 10 cm) packed in-house with 10 μm C18 beads (YMC gel, ODS-A, AA12S11) connected in tandem to an in-house packed analytical column (50 μM ID x 15 cm & 5 μm C18 beads). Peptides were separated by a 105-minute LC gradient with a flow rate of 0.2 ml/min and a similar pre-column split ratio. MS1 scans were performed with the following settings: 350-1200 m/z range, resolution of 70,000, AGC target of 3E6, and maximum IT of 50 ms. The top 15 abundance ions were isolated and fragmented with HCD (31% CI) with the following parameters: resolution of 70,000, AGC target of 1E5, maximum IT of 350 ms and isolation window of 0.4 m/z. Charge states < 2 and > 4 were excluded.
Mass Spectrometry data analysis
All mass spectra were analyzed with Proteome Discoverer (PD, version 2.5) and searched using Mascot (version 2.4) against the mouse SwissProt database (2021_03, 2021 02 for label free quantification). Peptides were searched with no enzyme and variable methionine oxidation. Peptide spectrum matches (PSMs) were further filtered according to the following criteria: ions score ≥ 15, search engine rank = 1 and results from technical replicates of each sample analysis were combined. Median retention time (RT) was calculated using the RT values of filtered PSMs from all replicates of a given sample. Label free quantitation was done using the Minora Feature Detector (precursor abundance values measured based on area under the curve) in PD with match between runs enabled and filtered for peptides with ions score ≥ 15 and search engine rank = 1. Abundances were averaged across technical and biological replicates. All data were processed in R studio (R version 4.1.0) and Microsoft Excel (version 16.57)
Endogenous and synthetic peptide spectra were compared by head-to-tail plots using MSnbase R package52. Pearson correlations and dot products were calculated using all the detected fragment ions in both spectra. Spectra that had the highest ions score in tumor samples (endogenous) or synthetic peptide sample were used (details listed in Supplementary Table 10) for head-to-tail plots and visualization by Interactive Peptide Spectral Annotator (IPSA)53. For IPSA, all fragment ions were plotted.
Immunofluorescence of Lung and Pancreas Tissue
Tumor bearing lung or pancreas tissue was manually dissected and embedded in optimal cutting temperature (OCT) compound and slowly frozen on dry ice. Frozen tissue sections were stored at −80 °C until sectioning. On the day of sectioning, frozen tissue was allowed to equilibrate to −20 °C in the cryostat for at least 1 hour. 8 μM sections were then cut and transferred to microscope slides (Fisher) prior to fixation in 100% acetone at −20 °C for 10 min and dried for 15 minutes at room temperature. Slides were then stored at −20 °C until staining.
Tissue sections were circled with a hydrophobic pen and then rehydrated with PBS for 5-10 mins and then blocked in PBS supplemented with 5% BSA for 45 minutes. Primary antibodies were then added at the indicated dilutions (see Antibody Table) for 1 hour at 25 °C. Slides were then washed 4 times with PBS + BSA, and incubated with fluorescently labeled secondary antibodies where indicated for 1 hour at 25 °C. Stained sections were then washed 3 times with PBS, incubated with DAPI for 5 min, washed once with PBS, and then mounted with Prolong Diamond AntiFade Mountant (Thermo). Slides were scanned on a VERSA 8 slide scanner (Leica) prior to analysis in ImageScope v64 and ImageJ.
Immunoblotting
For organoids, cells were dissociated with TrypLE, washed once with PBS, and then lysed in cell lysis buffer (CLB, 50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton-X100, 0.1% SDS, 1X HALT protease inhibitors). For monolayer cultures, media was aspirated cells were washed twice with PBS prior to lysis with CLB. Protein concentration was quantified with BCA (Pierce) and samples were loaded onto 4-12% Bis-Tris SDS-PAGE gels (Invitrogen) and electrophoresed at 150V until the loading dye reached the bottom of the gel. Gels were then transferred onto 0.45 uM nitrocellulose membranes overnight at 20V in a cold room. Blots were blocked with PBST (PBS + 0.5% Tween20) + 5% milk for 30 min at room temperature, incubated with primary antibody for 1 hour at room temperature, washed 4 times in PBST, incubated with HRP-conjugated secondary antibody for 1 hour at room temperature, and then washed 4 times in PBST. Blots were developed with Clarity or Clarity-Max ECL substrate (BioRad) and imaged on a ChemiDoc Gel Imaging System (BioRad).
RT-PCR
RNA was isolated from organoid cultures by aspirating media, and then resuspending the Matrigel domes in 1 mL Trizol (Ambion). RNA was then extracted from the homogenate using the PureLink RNA Mini Kit (Ambion) according to manufacturer’s instructions with with the final elution step in 40 μL of nuclease free water. Isolated RNA was then quantified on a NanoDrop 2000 (Thermo). 1 μg of total RNA was then added to a cDNA synthesis reaction using a High Capacity cDNA Reverse Transcription Kit (Applied Biosciences) according to manufacturer’s instructions with random hexamer’s as primers and including RNase inhibitor in the reaction. cDNA was then diluted 1:10 with nuclease free water prior to adding 1 μL to a PCR reaction containing 500 uM forward and reverse primer and 1X Q5 Master Mix (NEB). Reactions were cycled 35 times with annealing temperatures calculated by NEB Tm calculator prior to loading on 2% agarose gels. DNA bands were imaged by staining with ethidium bromide and UV transillumination.
Ribosome Protected Fragment Isolation with RiboLace
Ribosome profiling was performed using a previously described method using Puromycin conjugated magnetic beads to isolated actively translating ribosomes28. To obtain cellular material for Ribosome sequencing, organoid cultures of normal alveolar type 2 cells or organoids derived from 12 week KP tumors were transiently adapted to monolayer culture by washing tissue cultures dishes with a 10% Matrigel solution prior to plating cells in either complete lung organoid medium for AT2 cells or lung organoid base medium for tumor organoids 30. After 48 hours, cells were treated with 100 μM cycloheximide for 5 min and immediately transfer to ice. Plates were then washed once with 10 mL PBS + 100 μM cycloheximide followed by direct lysis and thorough mechanical disruption in the plate according to the RiboLace Protocol (Immagina Biotech). Lysates were then cleared with centrifugation at 20,000 x g for 15 min. Nucleic acid content was quantified with a NanoDrop and each lysate was normalized to 0.3 a.u.260nm in 150 μL of W-buffer. 0.3 μL of SS solution and diluted RNAse Nux solution (5 μL of 1:66.7 dilution). RNase digestion was then stopped with 0.5 μL SUPERaseIn for 10 min on ice.
RiboLace beads were prepared according to RiboLace kit instructions. Briefly, per sample, 90 μL of beads were magnetized, washed once with buffer OH, once with nuclease free water, twice with B-buffer, and then resuspended in 30 μL RiboLace probe followed by incubation shaking for 1 hour at room temperature. Beads were then passivated with 3 μL PEG for 15 min at room temperature and washed twice with 500 μL of buffer W.
Prepared beads were then resuspended in RNAse digested lysates, and incubated with slow rotation for 70 min at 4 °C. Beads were then washed twice with 500 μL of Buffer-W before protease digestion by addition of 20 μL of SDS and 5 μL of proteinase K and incubating for 75 min at 37 °C. RNA was then extracted with Phenol-Chloroform, precipitated overnight with isopropanol, and then electrophoresed on denaturing 15% TBE-Urea gels. Gel pieces were excised from the region according for 20-40 nucleotides to enrich for ribosome protected fragments (RPFs). Gel pieces were then crushed by centrifugation through a punctured 0.5 mL tube placed in a 1.5 mL microcentrifuge tube. Gel debris was then incubated overnight rotating in 400 μL of gel elution buffer (20 mM Tris pH 7.5, 250 mM sodium acetate, 1 mM EDTA, 0.25% SDS). RNA was precipitated from the elutions by adding 700 μL of isopropanol and 1.5 μL of GlycoBlue (Thermo) followed by overnight incubation at −80°C. RNA was then pelleted by centrifugation at 20,000 x g for 30 min at 4 °C. The RNA pellet was then washed with 70% EtOH, air dried for 2-3 min and resuspended in 10 μL of nuclease free water.
Dephosphorylation of RPFs was performed by adding 23 μL nuclease free water, 5 μL T4 PNK buffer (NEB), 1 μL SUPER In (Invitrogen), and 1 μL T4 PNK (NEB) followed by incubation for 60 min at 37 °C, then 10 min at 70 °C, and cooled to room temperature. RNA was then precipitated by adding 39 μL nuclease free water, 10 μL 3M sodium acetate, 150 μL isopropanol, and 1 μL GlycoBlue and incubated at −80 °C overnight. RNA was pelleted by centrifugation at 20,000 x g for 30 min, washed with 70% EtOH and then resuspended in 7 μL of nuclease free water.
RPF Sequencing Library Preparation
Illumina sequencing libraries of isolated RPFs was performed using a LACEseq kit (Immagina Biotechnology). Library preparation was performed according to the manufacturer’s protocol with the following parameters. 5 ng RPFs were 5’ phosphorylated in a 50 μL reaction containing 5 μL Buffer BPK, 5 μL ATP, 1 μL PK, and 34 μL nuclease free water. The reaction was incubated for 1 hour at 37 °C. RNA was then purified with an RNA Clean and Concentrator - 5 Kit (Zymo) and eluted in 6 μL. A linker was then ligated with 6 μL of RNA from the previous step, 1 μL Buffer BA, 0.5 μL GTP, 0.6 μL MnCl2, 1 μL Enzyme Mix A, 0.25 μL Linker MC, and 0.75 μL nuclease free water. The reaction was then incubated for 1 hour at 37 °C and purified with an RNA Clean and Concentrator Kit with 8 μL elution in nuclease free water. Circularization was then performed with 8 μL of the eluted RNA, 2 μL Buffer BLB, 1 μL ATP, 8 μL PEG8000, and 1 μL Enzyme Mix B. The resulting reaction was then incubated for 2 hours at 25 °C. The reaction was then purified with an RNA Clean and Concentrator Kit with 10 μL elution in nuclease free water. The 10 μL of circularized RNAs were then primed by adding 1 μL of dNTPs, 1 μL of RT_T Primer, and 2 μL of nuclease free water and incubating at 70 °C for 5 minutes. Next, a mastermix containing 4 μL Buffer BRT, 1 μL DTT, and 1 μL RT enzyme was added followed by an incubation at 50 °C for 40 minutes and heat inactivation for 5 min at 80 °C. cDNA was then amplified b adding 50 μL Amplification Mix, 0.8 μL forward primer, 0.8 μL reverse primer, and 28.4 μL nuclease free water. The reactions were then cycled 7 times according to the manufacturer’s PCR parameters. PCR reactions were cleaned up using AMPURE XP beads at a 1.6x ratio and elution in 40 μL of nuclease free water. A second PCR amplification was performed using 20 μL from the previous PCR reaction, 50 μL Amplification Mix, 1 μL LACEseq UDIs, and 29 μL nuclease free water. Reactions were cycles 6 times according for manufacturer’s parameters. The final libraries were size selected using PAGE purification, and cleaned up using the DNA Clean and Concentrator (Zymo) prior to Illumina sequencing.
RNAseq Library Preparation
RNA was extracted from the remaining AT2 or Tumor lysates that were used for RiboSeq using Trizol according to manufacturer’s instructions. Aqueous phase containing RNA was precipitated with 2 volumes isopropanol and washed once with 70% ethanol and air dried. Pellets were resuspended in nuclease-free water prior to polyA+ selective mRNA library preparation at the MIT BioMicroCenter.
RiboSeq Data Analysis
Raw fastq files from each RiboSeq library were trimmed according to recommendations in the LACE-seq protocol (Immagina Biosciences). Fastq files from each sample type were then merged prior to submission to the RiboToolKit server (http://rnabioinfor.tch.harvard.edu/RiboToolkit/) using a 26-38 nt size filter and a predicted ORF cutoff of p < 0.05. Data were then downloaded and visualized in R. For analysis of translational efficiency (TE), raw RNAseq and Riboseq data were aligned to the transcriptome with kallisto 0.46.0. Aligned reads were then imported into R for DEseq2 analysis using the tximport package. Differential expression at the RNA and RPF levels were calculated using standard DEseq2 workflows. For TE analysis, the DEseq2 results object was generated incorporating both sequence type (Ribo vs RNA) and sample (LUAD vs AT2) as independent variables to identify genes that were differentially translated in LUAD vs AT2.
Hsp90 Inhibitor Treatment
Hsp90 inhibitor (NVP-HSP990, Selleck Chemical) was administered in the drinking water32. The average water consumption of the mice was calculated by measuring water bottle weight before and after 72 hours of housing to determine the average consumption per mouse, per day. Across all experiments, C57Cl/6 mice consumed approximately 4 mL every day. Using these water consumption values, and mouse weight, a 4 mg/mL stock solution of NVP-HSP990 (in 100% PEG400) was diluted directly into the drinking water to achieve a target dose of 0.5 mg/kg/day. Hsp90i treatment began 8 weeks post tumor induction and water was replaced twice per week for 4 weeks prior to sacrifice.
CD8 Depletion and CD40/Flt3 Ligand Treatment
KP/KbStrep tumor-bearing mice at 2 (chronic depletion) or 14 weeks (acute depletion) post tumor initiation were treated with 100 μg of anti-CD8a depleting antibody (BioXCell, BE0061) every three or four days until animals reached 16 weeks post tumor initiation. At necropsy, a small portion of tumor-bearing lung tissue (~100 mg) was taken for flow cytometry confirmation of CD8a+ immune cell depletion. The remaining tissue was flash frozen and processed for pMHC isolation.
KP/KbStrep tumor-bearing mice at 15 weeks post tumor initiation were treated for 7 days with 10 μg Flt-3L-Ig (BioXCell, BE0342) with intraperitoneal injection 26. On day three of Flt-3L-Ig treatment, mice were also dosed with 100 μg of agonistic CD40 antibody (BioXCell, BP0016-2) through intraperitoneal injection. On day 7 of treatment (16 weeks post tumor initiation), mice were euthanized and a small portion of tumor-bearing lung tissue (~100 mg) was taken for flow cytometry analysis of tumor infiltrating CD8+ T cells and the remaining tissue was flash frozen for pMHC isolation.
OT-I Killing Assay
OT-I transgenic mice on a Rag2−/− background were euthanized by CO2 inhalation and spleens were dissected and macerated through a 70 μm cell strainer. Splenocytes were then pelleted by centrifugation at 500 x g for 5 min, followed by red blood cell lysis using ACK lysis buffer for 2 min at room temperature. ACK was quenched with RPMI 1640 + 10% HI FBS, and splenocytes were pelleted by centrifugation at 500 x g for 5 min. The resulting cell pellet was then resuspended in 10 mL of T cell media (RPMI 1640 supplemented with 10% HI FBS, 1X penicillin/streptomycin (Gibco), 1X non-essential amino acids (Gibco), 1X L-glutamine (Gibco), 1X HEPES (Gibco), and recombinant IL-2 (Peprotech)). At the time of plating, 1 μM SIINFEKL peptide was added to stimulate T cell activation and differentiation. 18 hours after plating, cells SIINFEKL peptide was removed and cells were replated in T cell media. 72 hours after addition of SIINFEKL cells were prepped for target killing assays.
Target cells were KP or KP/KbStrep cell lines transduced with either lentivirus encoding mScarlet or mScarlet-SIINFEKL. On the day of the assay, target cells were trypsinized, washed, and counted. 1 x 106 KP-mScarlet cells were incubated with 100 nM CFSE (KP CFSE lo) and 1 x 106 KP-mScarlet-SIINFEKL cells were incubated with 5 μM CFSE (KP CFSE hi) for 20 min at room temperature in PBS54. In parallel, 1 x 106 KP/KbStrep-mScarlet cells were incubated with 100 nM CFSE (KP/KbStrep CFSE lo) and 1 x 106 KP/KbStrep mScarlet-SIINFEKL cells were incubated with 5 μM CFSE (KP/KbStrep CFSE hi) for 20 min at room temperature in PBS. Labeling reactions were quenched with DMEM + 10% FBS and cells pelleted at 500 x g for 5 min. KP CFSE hi/lo and KP/KbStrep CFSE hi/lo were mixed in a 1:1 ratio and plated into round bottom 96 well plates at 20,000 cells per well (10,000 hi and 10,000 lo). Transgenic OT-I T cells were then counted, washed, and plated into wells containing target cells at 0:1, 5:1, and 1:1 effector: target ratios. After 4 hours, cells were processed for flow cytometry. Cells were gated on live/dead viability stain, and CD45+ cells were excluded. Specific killing was calculated with the following formula: 100 − ((CFSEhi/CFSElo − with effector cells)/(CFSEhi/CFSElo without effector cells) * 100.
Bone Marrow Derived Dendritic Cell (BMDC) Vaccination
Peptides were prioritized for vaccination as described in Figure 4a. Candidates were then further prioritized through manual inspection of RNA expression patterns across healthy mouse tissues. We selected peptides that have very low RNA expression in healthy tissue (Slc26a4, Prdm15), peptides that resemble cancer testis antigens (Ift74, Ccdc158, Arrdc5), oncofetal antigens (Gpn3, Znf462), and widespread expression despite tumor specific presentation (Csf2ra). This effort was not comprehensive of all potential antigens and continued interrogation of peptides from our model is warranted.
Isolation of BMDCs was carried out by euthanizing mice and dissecting the femur and tibia into PBS33. Ends of the bones were then clipped and the open end of each bone was placed facing down into a 0.5 mL tube with a hole in the bottom and placed into a 1.5 mL tube. The tube assembly was then quickly centrifuged on a tabletop microcentrifuge. Red blood cells were removed from the pelleted cell mass with ACK lysis buffer (Gibco) and the resulting cell pellet was resuspended at 1.5 x 106 cells/mL in BMDC media (RPMI 1640 supplemented with 10% FBS, penicillin/streptomycin, 50 μM beta-mercaptoethanol, 600 ng/mL Flt3L (BioXCell) and 5 ng/mL GM-CSF (R&D Systems)) and cultured in non-tissue culture treated dishes. After 5 days, cells were washed, recounted and replated in BMDC media. On Day 10, cells were activated with 10 μg CpG ODN 1826 (Abeomics) for 18-24 hours.
On Day 11, activated BMDCs were gently washed with BMDC media and pulsed with 10 ug of each peptide in a 15 mL conical tube with the lid not fully tightened for 2 hours in a 37 °C tissue culture incubator. Pulsed cells were then washed once with BMDC media, and two times with PBS. For injection, 1.5 x 106 peptide loaded BMDCs were injected subcutaneously into the flank. As a negative control, mice received BMDCs that were not pulsed with peptide.
10 days after BMDC injection, mice received a booster of 6 μg of each peptide with 25 ug of c-di-GMP as adjuvant. 14 days after the first boost (24 days after BMDC injection), mice received a booster of 6 ug of each peptide with 1 nmol of lipoCpG as adjuvant (kindly provided from the Irvine Laboratory at MIT). 7 days after the second boost, splenocytes were harvested for ELISPOT analysis or tetramer staining for tumor bearing animals.
Custom pMHC Tetramer Generation
H2-Kb tetramers were generated in-house using previously described protocols 34. Specifically, we used disulfide bridged H2-Kb (Y84C, A139C). Briefly, recombinant MHC-I heavy chain (DS-Kb) and light chain (H. sapiens B2m) were expressed in E. coli using the lac-operon regulated pET-15b plasmid. Inclusion bodies were prepped as previously described 55. Inclusion bodies were solubilized in 8M urea, 100 mM Tris-HCl pH 8.0 and added to a refolding buffer (100 mM Tris-HCl pH 8.0, 400 mM L-arginine (Sigma), 5 mM reduced glutathione (Sigma), 0.5 mM oxidized glutathione (Sigma), 2 mM EDTA (Gibco), 1x HALT protease inhibitor (Roche)). At the time of refolding, 1 mM Gly-Leu dipeptide was added to refolding buffer, followed by 2 μM B2m, 1 μM DS-Kb, and 1 mM PMSF for three consecutive days. After 72 hours, the reaction was concentrated to ~ 2 mL by applying the reaction to a 10,000 MWCO filter by nitrogen flow. The resulting concentrated protein solution was then purified by gel-filtration on an s200 Sephadex column in HBS (100 mM HEPES pH 8.0, 150 mM NaCl). Relevant fractions were collected and concentrated to ~8 mL. Purified monomers were then biotinylated using BirA Ligase according to manufacturer’s instruction (Avidity). Biotinylated monomers were then further purified with another round of gel-filtration on an s200 Sephadex in HBS and the relevant fractions were collected and concentrated to 2 mg/mL and 50 μL aliquots were flash frozen in liquid nitrogen until use.
To generate tetramers, 15.9 μL of Streptavidin-PE (SA-PE, Molecular Probes) was added to 50 μL DS-Kb monomers and incubated for 10 min at room temperature, protected from light. Addition of SA-PE was repeated 9 times for a total of 10 times resulting in a final volume of 209 μL tetramerized DS-Kb. At the time of staining, 20 μL of tetramer was incubated with 50 μM peptide of interest for 1 hour on ice. The resulting solution was used at a 1:200 dilution in flow cytometry staining solutions.
Enzyme-linked immune adsorbent spot (ELISPOT) Assay
Prior to plating cells, wells of a precoated ELISPOT plate were loaded with 100 L of DMEM + 10% FBS + Pen/Strep containing 10 μM of individual peptides, media only, or PMA/ionomycin as a positive control. Spleens from control or vaccinated mice were dissected and macerated through a 70 μM filter. Cells were then centrifuged for 5 min at 1500 rpm and pellets were then lysed with ACK lysis buffer and the remaining splenocytes were counted with a hemocytometer. Cells were then plated across control and peptide stimulation conditions at a concentration of 750,000 cells/well and gently mixed prior to incubation for 20 hours. After overnight incubation, cells were decanted and the plates were washed and IFN-γ spots detected using the procedure outlined in the Mouse IFN-γ ELISPOT kit (ImmunoSpot). After development, plates were dried overnight prior to automated spot counting on an ImmunoSpot scanner.
Data Analysis, Statistics, and Visualization
All quantitative data was processed and visualized in R (R version 4.0.2, RStudio version 1.3.959). Peptides from PDAC or LUAD samples were concatenated and organized in Microsoft Excel with predicted affinity values calculated by NetMHCPan 4.1. Following import into R, peptide lists were filtered for length (8-11 aa) and predicted affinity (<1000 nM) before further analysis. Non-metric multidimensional scaling (nmds) analysis was performed using the published R script from Sarkizova et. al. and applied to 8- and 9-mer peptides separately. Peptide clusters were empirically chosen through hierarchical clustering of the calculated nmds matrix. Peptide motifs were generated with the ggseqlogo package in R. All boxplots are in the Tukey style with the horizontal mark at the median, the box extending from quartile 1 to quartile 3, and whiskers extending 1.5 x the interquartile range from the box.
Bulk RNA sequencing was adapted from Chuang et. al. by taking the mean expression of each gene across all normal, early, nonmet, and metastatic primary tumor samples prior to cross comparison to peptide data.
Single cell RNA sequencing data (scRNAseq) was analyzed with Seurat (version 4.0.1). Cells by genes matrices were downloaded from the Tabula Muris (healthy lung and pancreas) or from the Broad single cell RNA seq Portal (KP scRNAseq data). Data were filtered with a gene count cutoff of 1100 prior to normalization, scaling, and dimensionality reduction with UMAP according to standard Seurat pipelines (https://satijalab.org/seurat/). Custom modules were added with the AddModuleScore function according to gene lists from peptide data or published reports. Volcano plots for comparing signatures across cell types in the healthy lung data were calculated with two-tailed Student’s t-test for all pairwise comparisons of Normal/Ab/Strep signatures within each cell type with a Bonferroni multiple comparisons adjustment. The median score for each signature was then used to calculate the Log2 fold change and P values were transformed to −Log10(P) for plotting. Pearson correlations between gene signatures were calculated by AddModuleScore in Seurat, extraction of metadata, conversion of the data to a matrix containing cells x signatures, and calculation of Pearson correlation for signatures across cells. For correlation of peptide signatures to all genes, the scaled data from Seurat was extracted to a matrix, and the Late/Tumor-Unique Peptide signature was appended onto the matrix. Pearson correlation was then calculated between the peptide signature versus each individual gene. Genes were then ranked by correlation and analyzed with preranked gene set enrichment in GSEA (Broad Institute).
Gene Ontology analysis was performed on Gene Lists in StringDb (www.stringdb.org) with high confidence interaction cutoffs. Gene Set enrichment analysis (Figure 2) was performed with the GSEA software package using a preranked gene list according to correlation to the Late or Tumor-Unique peptide signature.
Subcellular compartment analysis was performed by extracting compartment locations from UniProt for source proteins which had available localization data. Comparison of subcellular distributions was performed with Fisher’s Exact test with Monto Carlo simulation. Protein Length information was also extracted from Uniprot for each source protein. Thermal stability and protein half-life were obtained from published data sets30.
Statistical analyses were performed in R. To compare median fluorescence intensity from flow cytometry data, two-tailed Student’s t-tests were performed for all pairwise comparisons. For comparisons of gene expression or signature scores an unpaired two sample Wilcoxon Test (Mann-Whitney U) was performed. For analysis of peptide abundance distributions in Veh and Hsp90i treated samples, Kolmogorov-Smirnov Tests were used.
Extended Data
Extended Data Figure 1. (Related to Figure 1). In vitro validation of the KbStrep allele.
a) Structural model depicting the topology of the KbStrep protein during affinity purification. The StrepTagII engagement with Streptactin affinity resin does not interfere with peptide or B2m binding. b) Southern blot analysis of KbStrep targeted KP* ES cells. c) Representative genotyping for WT/WT, KbStrep/WT heterozygotes, and KbStrep/KbStrep homozygotes. d) Brightfield images of KP and KP/ KbStrep pancreatic organoids pre- and post- Ad-CMV-Cre mediated transformation ex vivo. e) RT-PCR analysis of KP or KP/KbStrep pancreatic organoids with or without Cre recombination and with or without IFN-γ treatment. Each row represents a distinct primer set showing no discernable alterations in mRNA splicing with or without StrepTagII activation. f) Representative flow cytometry plots detecting cell surface expression of PD-L1 and StrepTagII at baseline (red) and following Cre activation and IFN-γ treatment (blue). g) Quantification of the median fluorescence intensity (MFI) of StrepTagII staining in KP (orange) and KP/KbStrep (blue) organoids in control, IFN-γ treated, Cre transformed, and Cre+IFN-γ treated samples. Data are mean ± sem (n=3). Two-sided Student’s t-test. h) Immunoblot analysis of whole cell lysate from KP or KP/KbStrep PDAC cells after adaptation to 2D following treatment with IFN-γ. i) Immunoblot depicting affinity purification of intact MHC-I with Streptactin resin as evidenced by the co-precipitation of B2m. j) Coomassie staining of samples taken from KP or KP/KbStrep lysates at various stages of purification. In this experiment, the elution was taken by incubating washed Streptactin resin with SDS-PAGE loading buffer.
Extended Data Figure 2. (related to Figure 1). Isolation of MHC-I complexes from PDAC in vivo.
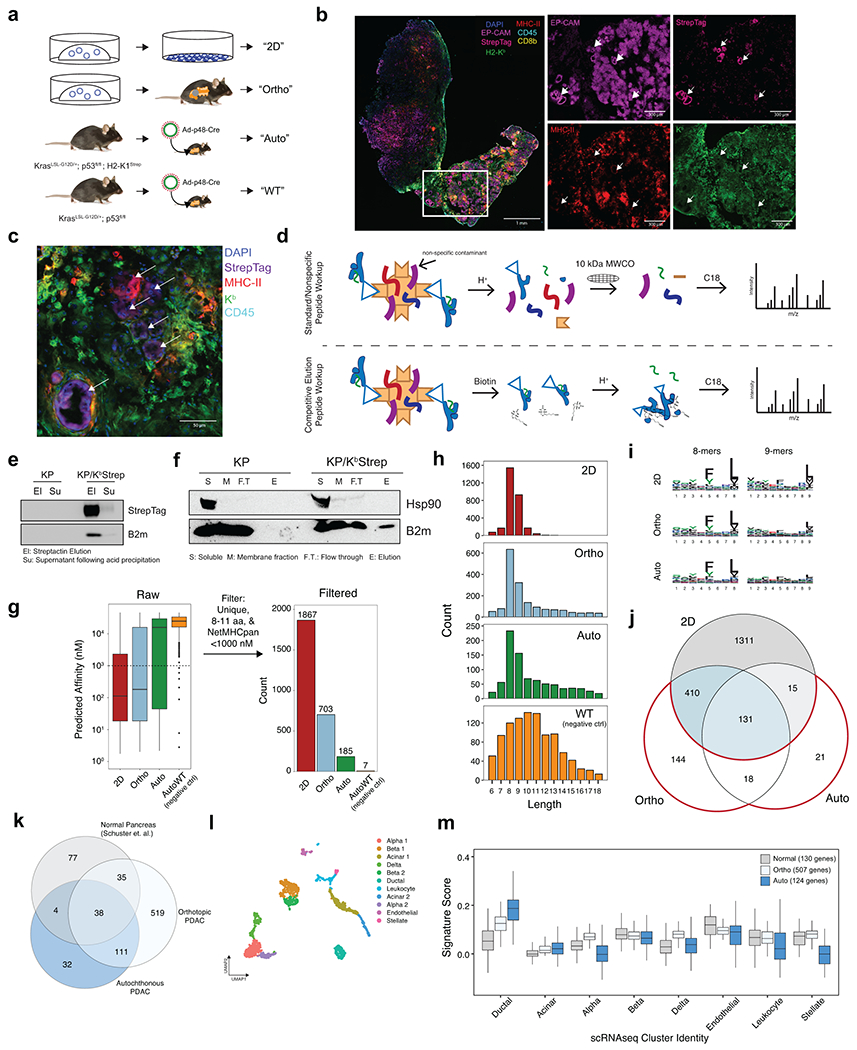
a) Experimental illustration of samples used for immunopeptidome comparison in PDAC. b) Multiplexed immunofluorescence of a representative autochthonous PDAC tumor. White arrows indicate cancer cell nests. c) High magnification multiplexed immunofluorescence image depicting the specificity of StrepTagII staining on cancer cells. d) Schematic illustration of the traditional method of peptide extraction (top) and the method used for competitive elution of MHC-I complexes with biotin used in this study (bottom). e) Immunoblot demonstrating efficient precipitation of the MHC-I heavy and light chain with acid (1% TFA) prior to peptide clean-up with C18 ziptip. f) Representative immunoblot demonstrating purification of MHC-I specifically from KP/KbStrep tumor bearing mice. g) Number of unique peptides identified in each sample type after filtering for length (8-11 amino acids) and NetMHCPan predicted affinity (<1000 nM). h) Amino acid length distribution of all peptides identified from 2D, Ortho, Auto, or WT samples. i) Peptide motifs of 8- and 9-mers isolated from 2D, orthotopic, and autochthonous tumors. j) Venn diagram comparison of peptides found in 2D, Ortho, or Auto samples. Peptides only found in vivo are outlined in red. k) Venn diagram comparing MHC-I peptides derived from normal pancreas in Schuster et. al. (gray) versus orthotopic transplant (light blue) or autochthonous PDAC (dark blue) in this study. l) UMAP embedding of reanalyzed pancreatic scRNAseq data from the Tabula Muris. For clarity of presentation in Extended Data Figure 2m, cells from a specific lineage were collapsed into a single cluster if they originally separated into multiple clusters (i.e. Alpha 1 and Alpha 2 → Alpha). m) Expression of gene signatures derived from genes encoding for normal pancreas peptides (gray), orthotopic PDAC peptides (light blue), and autochthonous PDAC peptides (dark blue) in all cell types of the normal pancreas as measured by scRNAseq from Tabula Muris.
Extended Data Figure 3. (related to Figure 1). In vivo validation of KbStrep allele in KP LUAD.
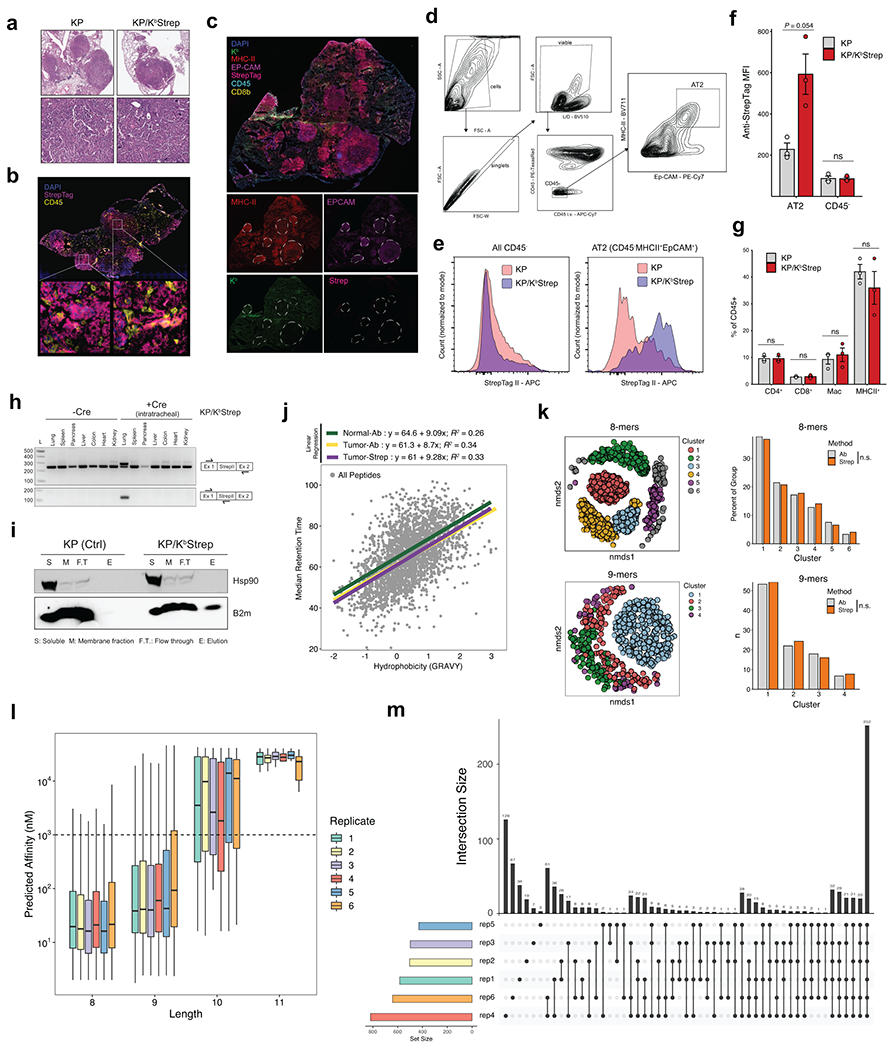
a) Representative H&E images demonstrating adenocarcinoma in KP and KP/KbStrep tumors. b) Multiplexed immunofluorescence of 16-week KP/KbStrep tumors demonstrating specific StrepTagII detection on tumor cells within the tumor microenvironment. c) Multiplexed immunofluorescence of a WT KP tumor demonstrating no detection of the StrepTagII in tumors outlined in white dotted lines. d) Gating strategy for isolating cells positive for an alveolar type 2 (AT2) phenotype from KP tumor-bearing lung tissue. e) Histograms depicting StrepTagII staining intensity across all CD45− cells (left) or after gating for AT2 cells (right) in KP (pink) or KP; KbStrep (purple) tumors. f) Quantification of StrepTagII MFI on AT2 or CD45 cells in the tumor microenvironment from KP or KP/KbStrep tumors. Data are mean ± sem. Two-sided Student’s t-test. g) Relative abundance of CD4+ T cells, CD8+ T cells, Macrophages, and CD45+MHCII+ immune cells in the tumor microenvironment of KP and KP/KbStrep tumors. Data are mean ± sem. Two-sided Student’s t-test. h) RT-PCR analysis of diverse tissues in KP/KbStrep mice with and without intratracheal Adeno-SPC-Cre administration. Expression of the Strep tagged Kb allele is only present in the lung after Cre induction. i) Immunoblot depicting isolation of intact MHC-I complexes specifically from KP/KbStrep tissue as evidenced by co-purification of B2m. j) Comparison of predicted peptide hydrophobicity (GRAVY) versus median peptide retention time in MS analysis for all peptides presented in Figure 1i. Linear regression is shown for peptides identified in Normal-Ab, Tumor-Ab, and Tumor-Strep datasets indicating no detectable differences in the biochemical features of identified peptides across methods. k) (left) Non-metric multidimensional scaling (nmds) plots depicting clusters of 8- and 9-mer peptides identified from Normal-Ab, Tumor-Ab, and Tumor-Strep samples. (right) Histogram showing the distribution of unique peptides from antibody (Ab) and Streptactin affinity purification (Strep) methods across peptide clusters identified with nmds analysis (n.s. – not significant, Fisher’s exact test). (l) Distribution of predicted peptide affinity for MHC-I peptides identified in 6 KP/KbStrep replicates. m) Upset plot depicting the peptide identification overlap between 6 KP/KbStrep replicates after length and affinity filtering. >77% of all identified peptides were found in at least 2/6 replicates.
Extended Data Figure 4. (related to Figure 1). Biochemical comparison of WT and Strep tagged H2-Kb.
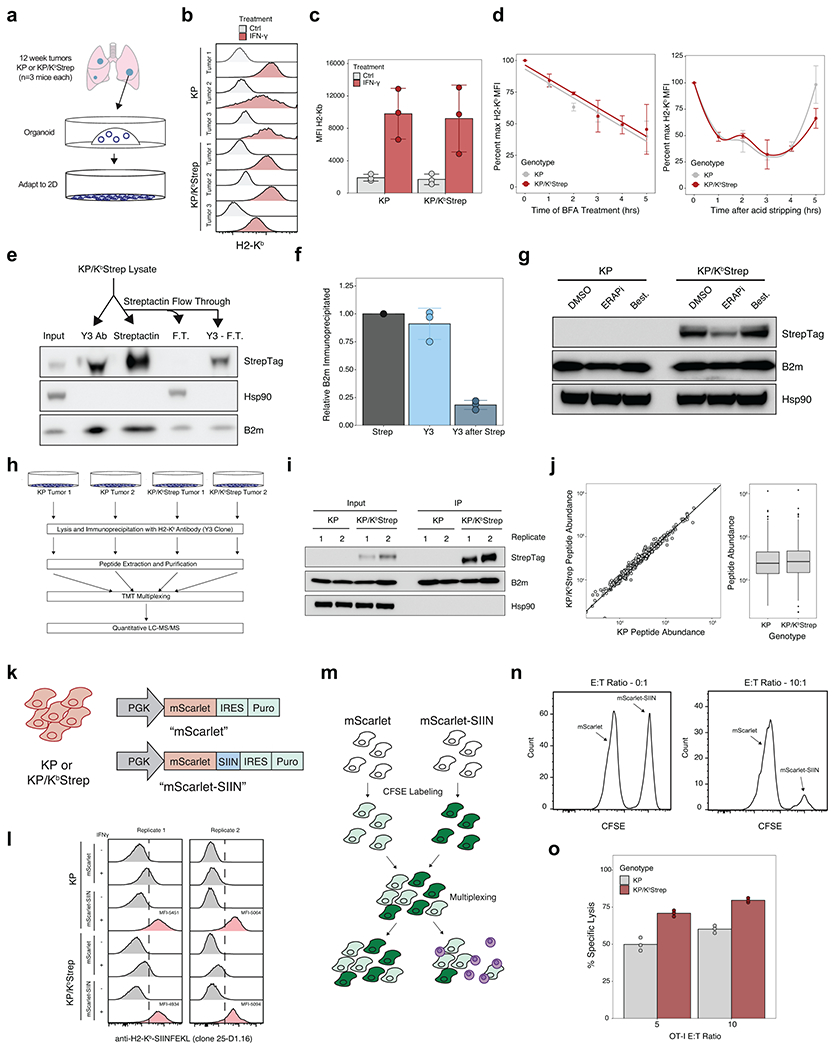
a) Isolation strategy for KP and KP/KbStrep cell lines. b) Histograms depicting fluorescence staining intensity of H2-Kb across KP and KP/KbStrep cell lines at baseline (gray) or following treatment with IFN-γ (red). c) Median fluorescence intensity quantification of H2-Kb staining across KP or KP/KbStrep cell lines at baseline (gray) or following treatment with IFN-γ (red). d) Relative H2-Kb staining intensity on KP (gray) or KP/KbStrep (red) cell lines following incubation with brefeldin A (BFA, left) or acute stripping with acid (300 mM glycine, pH 3.0, right) for the indicated times. e) Representative immunoblot depicting relative amounts of H2-Kb immunoprecipitation with antibody (Y3-Ab), Streptactin, or antibody following Streptactin (Y3-F.T.). f) Densitometric quantification of immunoprecipitated B2m intensity following antibody (Y3), streptactin, or antibody following streptactin (Y3 after Strep) purification schemes. g) Representative immunoblot depicting Strep-tagged H2-Kb expression in KP or KP/KbStrep cell lines following incubation with the aminopeptidase inhibitors ERAP1-in-1 or Bestatin. h) Experimental schematic for comparison of KP and KP/KbStrep immunopeptidomes from cultured cells using a quantitative, tandem mass tag (TMT) mass spectrometry strategy. i) Immunoblot depicting the abundance of immunoprecipitated MHC-I from samples described in h). j) Quantitative abundance comparison between all peptides identified in KP and KP/KbStrep samples. k) Illustration of the lentiviral constructs used for stable expression of SIINFEKL in KP and KP/KbStrep cell lines. l) Flow cytometric analysis of SIINKFEKL-H2-Kb complex surface expression using 25-D1.16 antibody. m) Experimental schematic used to evaluate specific T cell killing mediated by OT-I TCR transgenic T cells. n) Representative flow cytometry histograms depicting raw data used for calculating % specific lysis. o) Quantification of OT-I T cell killing in KP and KP/KbStrep cells.
Extended Data Figure 5. (Related to Figure 2). Analysis of the LUAD immunopeptidome throughout tumor evolution.
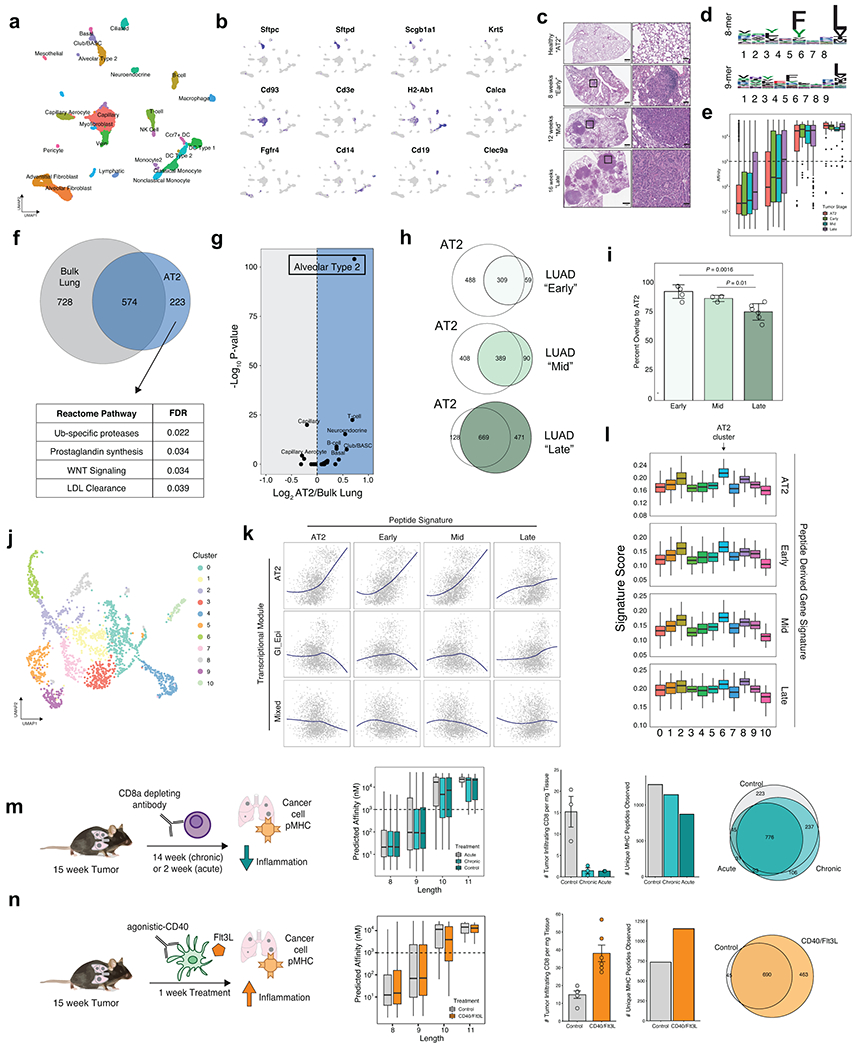
a) UMAP embedding of clusters used for signature expression analysis in Figure 2a. b) Gene expression profiles of cell type marker genes indicating robust clustering of known cell types in the healthy lung. c) Representative H&E stains for healthy lung (AT2), Early-, Mid- and Late-stage tumor samples. d) Peptide motifs of 8- and 9-mer peptides identified from healthy AT2 cells. e) Length and affinity characteristics of peptides identified in AT2, Early-, Mid-, and Late-stage tumors. f) Venn diagram comparison of peptides identified in bulk, healthy lung versus those identified specifically presented by normal AT2 cells. Pathways enriched by gene ontology depicted on bottom. g) Comparative analysis of gene signatures derived from peptides detected on AT2 cells versus bulk lung applied to the Tabula Muris data. Volcano plot shows the strong enrichment for an AT2 phenotype in the AT2 immunopeptidome versus bulk lung immunopeptidome. h) Comparison of AT2 and Early-, Mid-, and Late-Stage immunopeptidomes. i) Quantification of percent overlap from h). j) UMAP embedding of clusters from reanalyzing scRNAseq data from Marjonovic et. al. and used for analysis of Figure 2j. k) Loess regression analysis across all cells scored for AT2, Early, Mid, and Late peptide signatures versus the AT2, GI-Epi, and Mixed transcriptional modules. l) Signature distribution for AT2-, Early-, Mid-, and Late-Stage peptide signatures across all KP scRNAseq clusters. m) Comparison of the immunopeptidome in control tumors (gray), tumors chronically depleted of CD8 T-cells (light teal), and tumors acutely depleted of CD8 T-cells (dark teal). n) Comparison of the immunopeptidome from control tumors (gray) or tumors treated with agonistic-CD40 antibody and Flt3-L (orange). CD8 depletion experiments and CD40/Flt3L experiments were carried out independently and analyzed on different MS runs separated by ~2.5 months.
Extended Data Figure 6. (Related to Figure 3). Identification of differentially translated genes in LUAD versus AT2 cells.
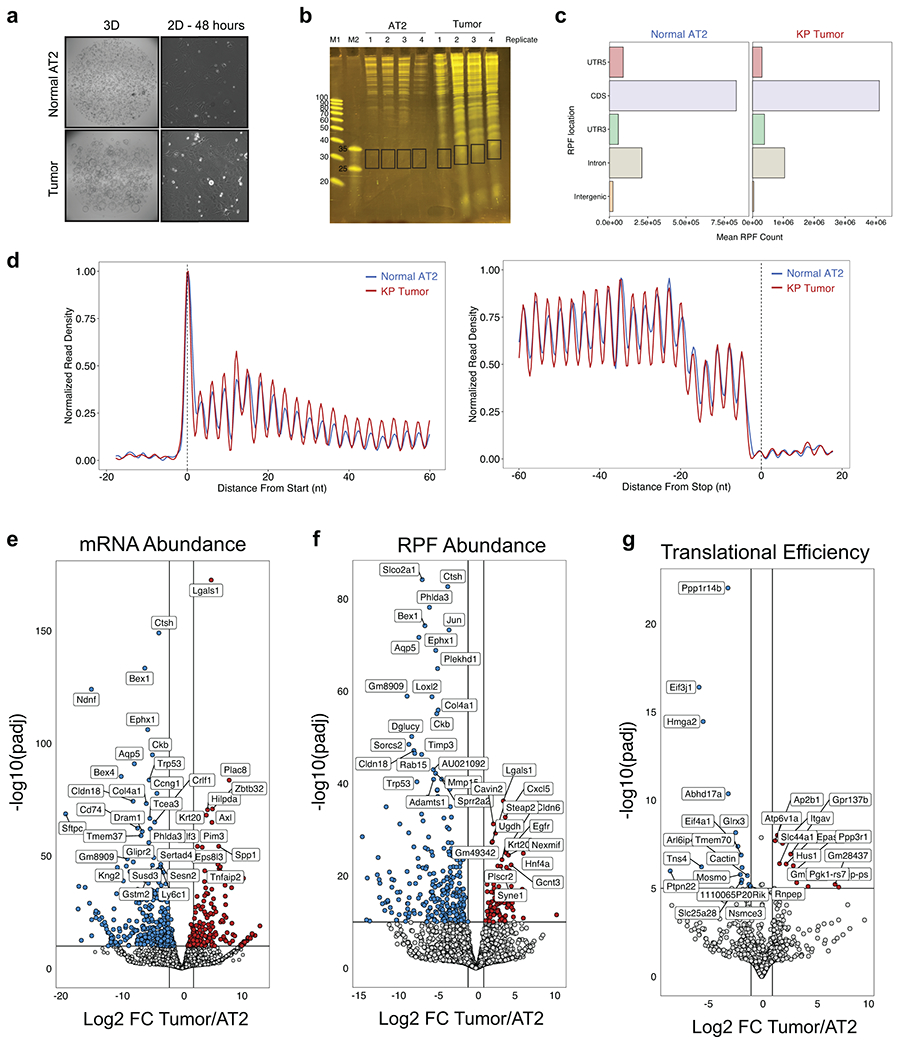
a) Representative images of normal AT2 and tumor organoid cultures in 3D organotypic culture or after transient adaptation to 2D monolayer culture prior to RiboSeq and RNAseq processing. b) Denaturing PAGE gels of RNA purified from RiboLace purification. Excised bands used for RiboSeq are indicated on the right and were selected for RNA that was ~ 30 bp in length. c) Localization of RPF alignments within transcripts depicting enrichment for annotated coding sequences (CDS). d) Normalized metagene density profiles of reads from normal and tumor cells at translation initiation (left) and termination (right). Both normal and tumor metaprofiles exhibit 3 nucleotide periodicity, indicative of active translation. e) Volcano plot depicting differential mRNA expression in tumor versus normal AT2 cells. f) Volcano plot depicting differential RPF abundance in tumor versus normal AT2 cells. g) Volcano plot depicting differential translation efficiency in tumor versus normal AT2 cells.
Extended Data Figure 7. (Related to Figure 3). Transcriptomic and proteomic data associated with LUAD-unique peptides.
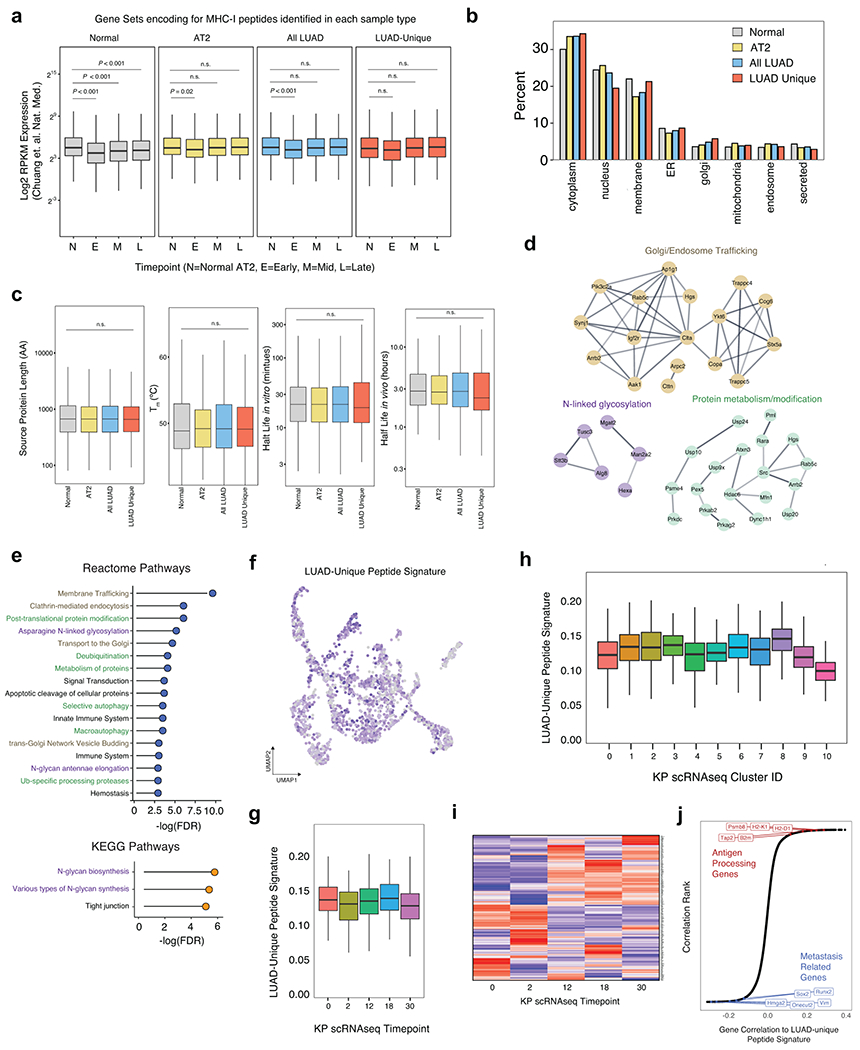
a) mRNA expression of genes encoding for Normal (bulk-lung), AT2, All-LUAD, or LUAD-unique peptides across normal AT2 cells, early-, mid-, and late-stage sorted tumor cells. (Adapted from Chuang et. al.). b) Subcellular compartment distribution of source proteins for peptides found in bulk Normal Lung, AT2 cells, all tumor peptides, and LUAD-unique peptides. c) Distribution of Protein length, thermal stability, or protein half-life for source proteins of peptides found in Normal Tissue, All Tumor peptides, or LUAD-unique peptides. d) StringDb analysis of source proteins for LUAD-unique peptides indicated in Figure 3a. Clusters of enriched protein families are depicted. e) Gene ontology analysis of LUAD-unique peptides from KEGG and Reactome databases. f) Expression of the LUAD-unique signature across all cells in the KP scRNAseq dataset (Marjonovic et. al.) g) Expression of the LUAD-unique signature across tumor progression in KP scRNAseq. h) Expression of LUAD-unique peptide signature across clusters in KP scRNAseq. i) Expression of individual genes encoding LUAD-unique peptides across KP timepoints. j) Correlation of the LUAD-unique peptide signature to all genes detected in scRNAseq. Genes related to antigen presentation are highlighted in red and genes related to metastasis are highlighted in blue.
Extended Data Figure 8. (Related to Figure 3). Modulation of protein folding through Hsp90 inhibition alters the immunopeptidome in vivo.
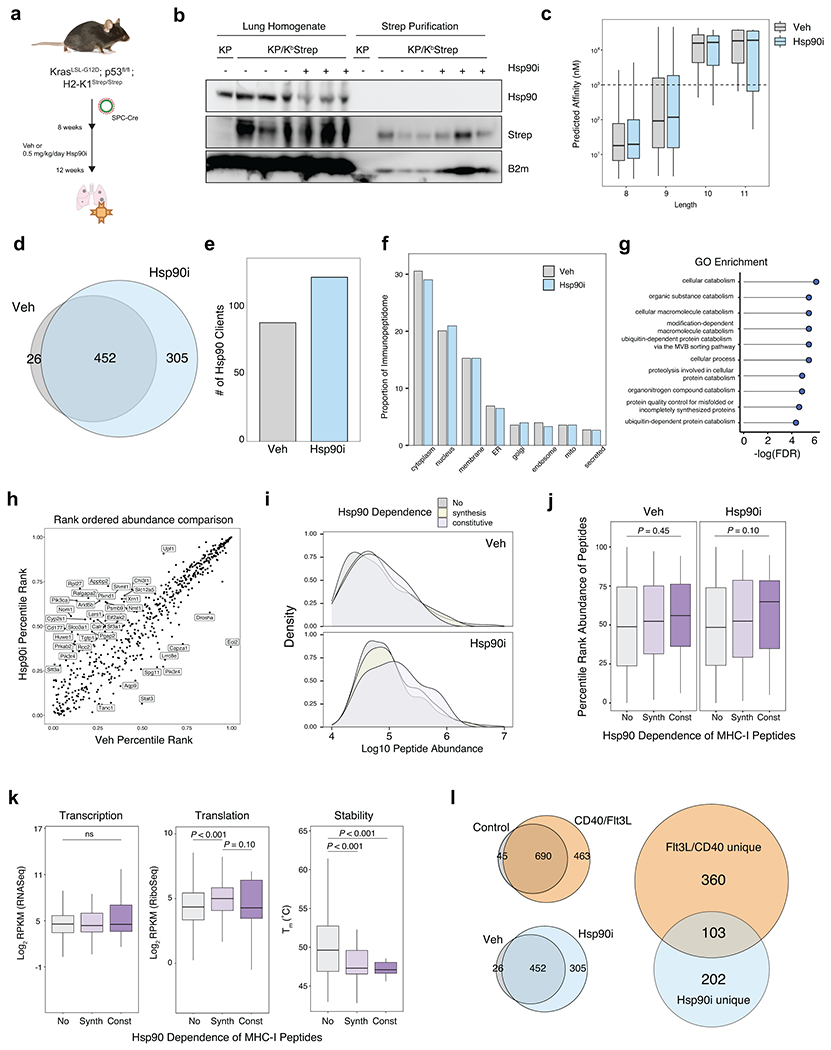
a) Experimental schematic of KP/KbStrep tumor treatment with either vehicle control or 0.5 mg/kg/day NVP-HSP990 prior to tumor specific MHC-I isolation. b) Immunoblot analysis of purified MHC-I from Vehicle (Veh) and Hsp90 inhibitor (Hsp90i) treated tumor samples or KP control tumors. c) Length and affinity distribution of peptides found in Veh (grey) and Hsp90i (blue) treated samples. d) Venn diagram of peptides found in 12-week control tumors or Hsp90i treated samples. e) Number of Hsp90 clients giving rise to peptide in either Veh (grey) or Hsp90i (blue) samples. f) Distribution of peptides identified in Veh or Hsp90i treated samples across subcellular compartments. g) Gene ontology analysis of source proteins for peptides that were only found in Hsp90i treated samples ranked according to FDR enrichment significance. h) Comparing the rank ordered abundance of all common peptides between Hsp90i treatment and control. i) Density plots of raw peptide abundance for non-clients, synthesis clients or constitutive clients in Veh (top) or Hsp90i (bottom) treated samples. j) Rank ordered abundance of peptides derived from Non-clients (No, gray), synthesis clients (Synth, light purple) or constitutive clients (Const, dark purple) in Veh and Hsp90i treated samples. P calculated with the Kologorov-Smirnov Test. k) RNA abundance, translation rate, and melting temperatures across non-clients (No), synthesis clients (Synth) and constitutive clients (Const) that are source proteins for MHC-I presentation. P calculated with Mann-Whitney U Test. l) Comparison of peptides unique to Flt3L/aCD40 treatment and those unique to Hsp90i.
Extended Data Figure 9. (Related to Figure 4). Expression and presentation of putative tumor specific and tumor associated antigens.
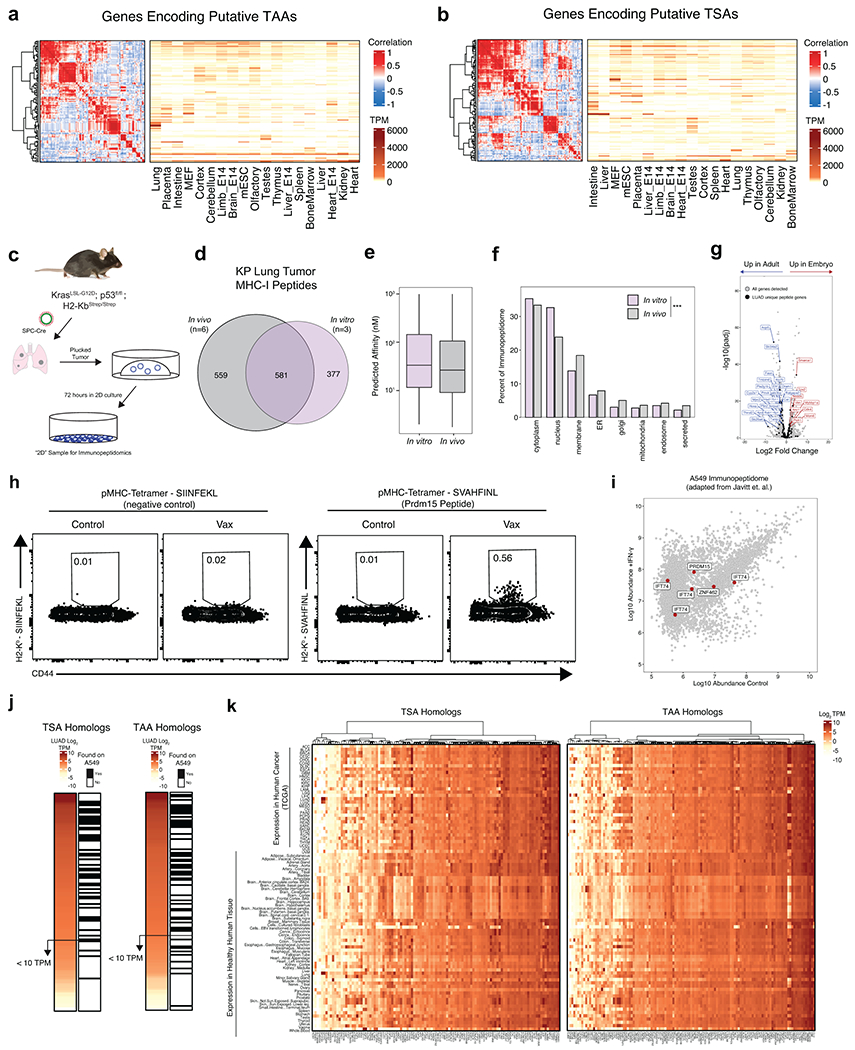
a) Correlelogram and heatmap depicting transcript abundance (transcripts per million, TPM) of putative TSA genes across mouse tissues. b) Correlelogram and heatmap depicting TPM abundance of putative TAA genes across mouse tissues. c) Experimental schematic showing the derivation of samples for 2D immunopeptidomics. d) Venn diagram depicting the relationship between peptides identified by KP tumors in vivo and those identified in vitro. e) Boxplot showing the predicted affinity distributions of peptides isolated in vivo and in vitro. f) Distribution of source protein subcellular compartments for peptides identified in vivo (gray) and in vitro (green). P calculated with Fisher’s Exact test with Monte Carlo simulation. g) Volcano plot indicating differentially expressed genes between EPCAM+ cells from embryonic day 16.5 and post-natal day 28 mouse lung (Adapted from Lung Map Project). Data analyzed and P calculated with DEseq2. All genes detected are shown in grey and genes encoding for LUAD-unique peptides are indicated with black dots. h) Flow cytometry analysis of tumor-bearing lung tissue from naïve and vaccinated mice stained with control pMHC-I tetramer (SIINFEKL) or TAA tetramer (SVAHFINL). i) Peptides identified in A549 cells (Javitt et. al.) with and without treatment of IFN-γ/TNFα. Peptides derived from source proteins homologous to those using in the pooled vaccine are indicated in red. j) Heatmap depicting expression of the human homologs of putative TSAs and TAAs from this study and whether or not peptides derived from those genes were found to be presented on A549 cells from Javitt et. al. k) Heatmap depicting the RNA Expression of homologs of potential TSA and TAA genes as found in Figure 4a across all individual human tissues and 33 cancer types within TCGA.
Extended Data Figure 10. (Related to Figure 4). Mass spectrometry validation of immunogenic epitopes with synthetic peptides.
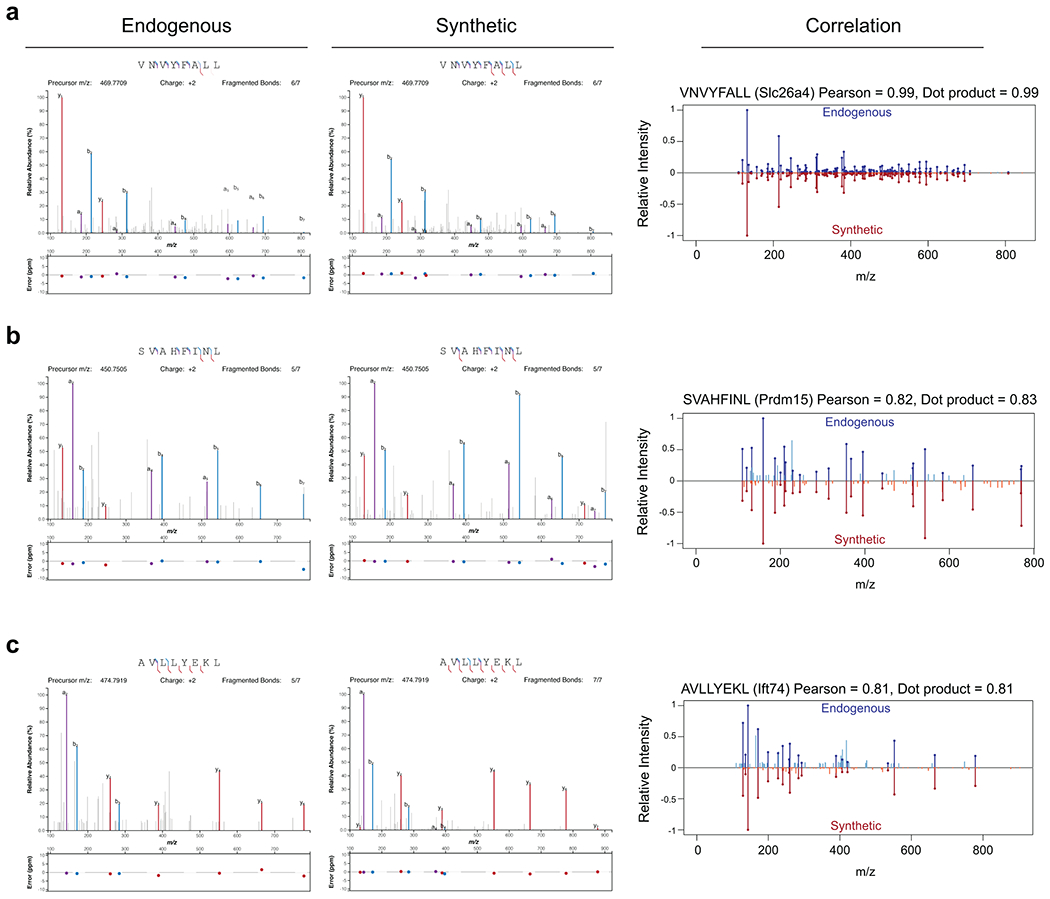
a) Mass spectrometry comparison of spectra from endogenously identified VNVYFALL peptide (Slc26a4) and a synthetic standard. b) Mass spectrometry comparison of spectra from endogenously identified SVAHFINL peptide (Prdm15) and a synthetic standard. c) Mass spectrometry comparison of spectra from endogenously identified AVLLYEKL peptide (Ift74) and a synthetic standard. In the left panel, y-, b-, and a-ions are colored in bold. In the right panel, common peaks are drawn in darker color.
Supplementary Material
Acknowledgments
We would like to thank members of the Jacks Lab for critical comments on the work presented here, especially Zack Ely and Nimisha Pattada for conceptual and technical support. We would like to thank George Eng for support with organoid work. In addition, we thank Toni Koller and Richard Schiavoni of the Koch Institute Proteomics core for technical insight on proteomic workflows. Data accessibility from the Tabula Muris was essential for multiple analyses in this manuscript. This work was supported by the Damon Runyon Cancer Research Foundation (A.M.J), an NCI K99 Pathway to Independence Award (A.M.J.), the Howard Hughes Medical Institute, the Johnson and Johnson Lung Cancer Initiative, NCI Cancer Center Support Grant P30-CA1405, the Lustgarten Foundation Pancreatic Cancer Research Laboratory at MIT, the Stand Up To Cancer-Lustgarten Foundation Pancreatic Cancer Interception Translational Cancer Research Grant (Grant Number: SU2C-AACR-DT25-17, W.F.P, T.J.), the MIT Center for Precision Cancer Medicine (L.E.S., R.A., F.M.W.), the Margaret A. Cunningham Immune Mechanisms of Cancer Research Fellowship (L.E.S.), the Melanoma Research Alliance (L.E.S.), NIH Training Grant (T32-ES007020), the Ludwig Center at MIT (R.A.), the Pew-Stewart Scholars Program for Cancer Research (A.K.S.), and the Cancer Research Institute (T.F., S.S.). In addition, we thank the Koch Institute Swanson Biotechnology Center for technical support, specifically the Flow Cytometry, Histology, Preclinical Modeling, Imaging and Testing, and Integrative Genomics and Bioinformatics core facilities.
Declaration of Interests
T.J. is a member of the Board of Directors of Amgen and Thermo Fisher Scientific, and a co-Founder of Dragonfly Therapeutics and T2 Biosystems. T.J. serves on the Scientific Advisory Board of Dragonfly Therapeutics, SQZ Biotech, and Skyhawk Therapeutics. T.J. is also President of Break Through Cancer. His laboratory currently receives funding from Johnson & Johnson and The Lustgarten Foundation, and funds from the Lustgarten Foundation supported the research described in this manuscript. S.S. is a co-founder of Danger Bio and serves on the Scientific Advisory Board of Related Sciences/DanderBio, Ankyra Therapeutics, Arcus Biosciences, TAKEDA, Ribon, and serves as an advisor for Dragonfly Therapeutics and Merck. S.S. lab currently receives funding from Leap Therapeutics. A.K.S. reports compensation for consulting and/or SAB membership from Merck, Honeycomb Biotechnologies, Cellarity, Repertoire Immune Medicines, Ochre Bio, Third Rock Ventures, Hovione, Relation Therapeutics, FL82, Empress Therapeutics, and Dahlia Biosciences. None of these affiliations influenced the work conducted or analysis of data presented in this manuscript.
Data Availability
All mass spectrometry data have been deposited to the Proteomics Identifications Database (PRIDE) repository with the dataset identifier PXD033232. Raw ribosome sequencing data has been submitted to the Gene Expression Omnibus with dataset identifier [GEO178944].
References
- 1.Jhunjhunwala S, Hammer C & Delamarre L Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer 1–15 (2021) doi: 10.1038/s41568-021-00339-z. [DOI] [PubMed] [Google Scholar]
- 2.Donia M et al. Acquired Immune Resistance Follows Complete Tumor Regression without Loss of Target Antigens or IFNγ Signaling. Cancer Res 77, 4562–4566 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Yamamoto K et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581, 100–105 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schumacher TN & Schreiber RD Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Keenan TE, Burke KP & Allen EMV Genomic correlates of response to immune checkpoint blockade. Nat Med 25, 389–402 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dersh D, Hollý J & Yewdell JW A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat Rev Immunol 1–13 (2020) doi: 10.1038/s41577-020-0390-6. [DOI] [PubMed] [Google Scholar]
- 7.DuPage M, Dooley AL & Jacks T Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc 4, 1064–1072 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wells DK et al. Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell 183, 818–834.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghorani E et al. The T cell differentiation landscape is shaped by tumour mutations in lung cancer. Nat Cancer 1, 546–561 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abelin JG et al. Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-allelic Cells Enables More Accurate Epitope Prediction. Immunity 46, 315–326 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chong C et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat Commun 11, 1293 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yadav M et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Granados DP et al. Impact of genomic polymorphisms on the repertoire of human MHC class I-associated peptides. Nat Commun 5, 3600 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarkizova S et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat Biotechnol 38, 199–209 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stopfer LE, Mesfin JM, Joughin BA, Lauffenburger DA & White FM Multiplexed relative and absolute quantitative immunopeptidomics reveals MHC I repertoire alterations induced by CDK4/6 inhibition. Nat Commun 11, 2760 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khodadoust MS et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature 543, 723–727 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jamal-Hanjani M et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. New Engl J Medicine 376, 2109–2121 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Chiou S-H et al. Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Gene Dev 29, 1576–1585 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schuster H et al. A tissue-based draft map of the murine MHC class I immunopeptidome. Sci Data 5, 180157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schaum N et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.LaFave LM et al. Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma. Cancer Cell 38, 212–228.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marjanovic ND et al. Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell 38, 229–246.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Concepcion CP et al. SMARCA4 inactivation promotes lineage-specific transformation and early metastatic features in the lung. Cancer Discov 12, candisc.0248.2021 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rock JR et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc National Acad Sci 108, E1475–E1483 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han X et al. Transdifferentiation of lung adenocarcinoma in mice with Lkb1 deficiency to squamous cell carcinoma. Nat Commun 5, 3261 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schenkel JM et al. Conventional type I dendritic cells maintain a reservoir of proliferative tumor-antigen specific TCF-1+ CD8+ T cells in tumor-draining lymph nodes. Immunity 54, 2338–2353.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chuang C-H et al. Molecular definition of a metastatic lung cancer state reveals a targetable CD109–Janus kinase–Stat axis. Nat Med 23, 291–300 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clamer M et al. Active Ribosome Profiling with RiboLace. Cell Reports 25, 1097–1108.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Chen J et al. Pervasive functional translation of noncanonical human open reading frames. Science 367, 1140–1146 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naranjo S et al. Modeling diverse genetic subtypes of lung adenocarcinoma with a next-generation alveolar type 2 organoid platform. Biorxiv 2021.12.07.471632 (2021) doi: 10.1101/2021.12.07.471632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perrin J et al. Identifying drug targets in tissues and whole blood with thermal-shift profiling. Nat Biotechnol 38, 303–308 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Jaeger AM et al. Rebalancing Protein Homeostasis Enhances Tumor Antigen Presentation. Clin Cancer Res 25, 6392–6405 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perez CR & Palma MD Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat Commun 10, 5408 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saini SK et al. Empty peptide-receptive MHC class I molecules for efficient detection of antigen-specific T cells. Sci Immunol 4, eaau9039 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Zitvogel L, Perreault C, Finn OJ & Kroemer G Beneficial autoimmunity improves cancer prognosis. Nat Rev Clin Oncol 1–12 (2021) doi: 10.1038/s41571-021-00508-x. [DOI] [PubMed] [Google Scholar]
- 36.Sahin U et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 585, 107–112 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Nelson CE et al. Robust Iterative Stimulation with Self-Antigens Overcomes CD8+ T Cell Tolerance to Self- and Tumor Antigens. Cell Reports 28, 3092–3104.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vonderheide RH CD40 Agonist Antibodies in Cancer Immunotherapy. Annu Rev Med 71, 1–12 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Burger ML et al. Antigen dominance hierarchies shape TCF1+ progenitor CD8 T cell phenotypes in tumors. Cell 184, 4996–5014.e26 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Westcott PMK et al. Low neoantigen expression and poor T-cell priming underlie early immune escape in colorectal cancer. Nat Cancer 2, 1071–1085 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Freed-Pastor WA et al. The CD155/TIGIT axis promotes and maintains immune evasion in neoantigen-expressing pancreatic cancer. Cancer Cell (2021) doi: 10.1016/j.ccell.2021.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yarmarkovich M et al. Cross-HLA targeting of intracellular oncoproteins with peptide-centric CARs. Nature 599, 477–484 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Cuevas MVR et al. Most non-canonical proteins uniquely populate the proteome or immunopeptidome. Cell Reports 34, 108815 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erhard F et al. Improved Ribo-seq enables identification of cryptic translation events. Nat Methods 15, 363–366 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ouspenskaia T et al. Thousands of novel unannotated proteins expand the MHC I immunopeptidome in cancer. Biorxiv 2020.02.12.945840 (2020) doi: 10.1101/2020.02.12.945840. [DOI] [Google Scholar]
- 46.Raposo B et al. T cells specific for post-translational modifications escape intrathymic tolerance induction. Nat Commun 9, 353 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Griffin GK et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 1–6 (2021) doi: 10.1038/s41586-021-03520-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kalaora S et al. Identification of bacteria-derived HLA-bound peptides in melanoma. Nature 592, 138–143 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marino S, Vooijs M, Gulden H. van der, Jonkers J & Berns A Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Gene Dev 14, 994–1004 (2000). [PMC free article] [PubMed] [Google Scholar]
- 50.Jackson EL et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Gene Dev 15, 3243–3248 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.VanDussen KL, Sonnek NM & Stappenbeck TS L-WRN conditioned medium for gastrointestinal epithelial stem cell culture shows replicable batch-to-batch activity levels across multiple research teams. Stem Cell Res 37, 101430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gatto L, Gibb S & Rainer J MSnbase, Efficient and Elegant R-Based Processing and Visualization of Raw Mass Spectrometry Data. J Proteome Res 20, 1063–1069 (2021). [DOI] [PubMed] [Google Scholar]
- 53.Brademan DR, Riley NM, Kwiecien NW & Coon JJ Interactive Peptide Spectral Annotator: A Versatile Web-based Tool for Proteomic Applications*. Mol Cell Proteomics 18, S193–S201 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Noto A, Ngauv P & Trautmann L Cell-based Flow Cytometry Assay to Measure Cytotoxic Activity. J Vis Exp e51105 (2013) doi: 10.3791/51105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grotenbreg GM et al. Discovery of CD8+ T cell epitopes in Chlamydia trachomatis infection through use of caged class I MHC tetramers. Proc National Acad Sci 105, 3831–3836 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All mass spectrometry data have been deposited to the Proteomics Identifications Database (PRIDE) repository with the dataset identifier PXD033232. Raw ribosome sequencing data has been submitted to the Gene Expression Omnibus with dataset identifier [GEO178944].