

Abstract
Nitriles have broad applications in medicinal chemistry, with more than 60 small molecule drugs on the market containing the cyano functional group. In addition to the well-known noncovalent interactions that nitriles can perform with macromolecular targets, they are also known to improve drug candidates' pharmacokinetic profiles. Moreover, the cyano group can be used as an electrophilic warhead to covalently bind an inhibitor to a target of interest, forming a covalent adduct, a strategy that can present benefits over noncovalent inhibitors. This approach has gained much notoriety in recent years, mainly with diabetes and COVID-19-approved drugs. Nevertheless, the application of nitriles in covalent ligands is not restricted to it being the reactive center, as it can also be employed to convert irreversible inhibitors into reversible ones, a promising strategy for kinase inhibition and protein degradation. In this review, we introduce and discuss the roles of the cyano group in covalent inhibitors, how to tune its reactivity and the possibility of achieving selectivity only by replacing the warhead. Finally, we provide an overview of nitrile-based covalent compounds in approved drugs and inhibitors recently described in the literature.
This review highlights the roles of nitriles in covalent inhibitors, their reactivity, examples of pharmaceuticals containing the cyano group and recent developments of nitrile-based inhibitors.
Introduction
The nitrile (or cyano) group has several applications in diverse fields of chemistry, from superglues with methyl cyanoacrylate to drugs such as cimetidine. In the latter, nitriles have played a significant role, with over 70 approved drugs presenting this group in their chemical structure.1 Within these approved drugs, 61 are small organic molecules, with 55 containing only one nitrile group in their scaffold and six presenting this group twice.
Drugs containing the cyano group are used to treat various diseases, ranging from viral infections to different types of cancer.2 These drugs take advantage of the fact that nitriles can be an excellent group to improve the compounds' pharmacodynamic (PD) and pharmacokinetic (PK) profiles, as they can make different types of interactions with macromolecular targets and/or improve water solubility.2–4
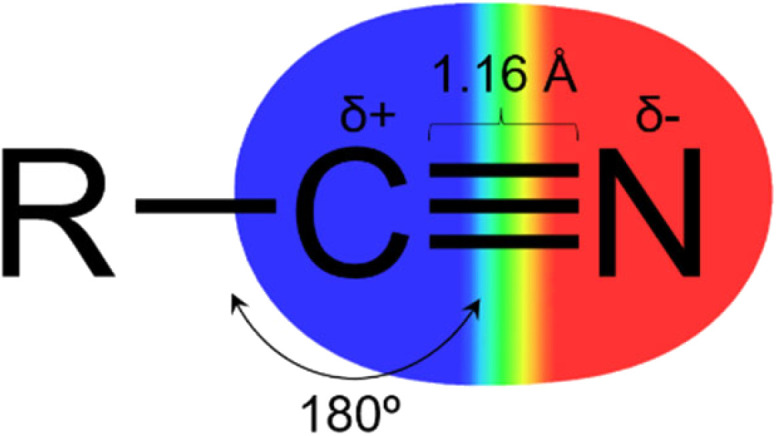
The nitrile group (Fig. 1) has a linear geometry with a nitrogen atom bonded to an sp hybridized carbon atom through a triple bond. The C sp atom can act as an electrophile due to its electron deficiency, promoted by the high electronegativity of the nitrogen and high dipole moment in the triple bond. And by the lone pair of the nitrogen atom, it is possible to interact with hydrogen bond donor groups.3–5 Owing to its linear shape and low molecular volume, it can fit properly in the subsites of target proteins and perform lipophilic interactions via the triple bond's pi system.3,4
Fig. 1. Representation of the nitrile (cyano) group.
In addition to the nonbonded interactions, nitriles are a remarkable group that can form covalent adducts with proteins, mainly linked to a reactive cysteine or serine side chain.6,7 The stability of the covalent bond between the ligand and the target can be modulated by different types of substituents in the vicinity of the nitrile, which also impacts its reactivity. The reactivity modulation is critical to the design of various types of inhibitors, from reversible to irreversible. In addition, the nitrile's electron-withdrawing property can be used in combination with other warheads, as is the case of cyano-acrylamides, in which the covalent bond is formed on the acrylamide β-carbon, while the cyano group increases the reactivity and promotes reversibility.8,9 All these characteristics make the nitrile an interesting and versatile moiety to be considered in drug design.
Apart from affecting the PD profile of drug molecules, nitriles can be incorporated to improve pharmacokinetic properties. There are examples in which the addition of a nitrile does not influence the potency of the inhibitor, but makes the compound 10-fold more soluble than its molecular pair by decreasing log P.10 In addition to this notable contribution to PK, the cyano functional group is considered a metabolically stable group, and may also reduce the susceptibility of hepatic oxidative metabolism.3 Nonetheless, cases relating to the toxicity of nitriles may happen due to a metabolically-induced release of cyanide into the body.11 Despite being unusual, this reaction can occur in a few cases, such as after ingesting fruit pits containing cyanogenic glycosides, although in a minimal amount. Overall, nitriles are considered a non-toxic group and are usually eliminated unchanged from the human body.
This review focuses on compounds in which the nitrile group is key to the ligand's reactivity, and is inspired by the successful development and approval examples of saxagliptin and nirmatrelvir (US approval in 2009 and 2021, respectively). Non-reactive nitrile-containing drugs and candidates were previously described in a set of high-quality, in-depth reviews in the literature.2–4,12
Herein, we will explain the function of nitriles in covalent inhibitors, followed by how to tune the reactivity of this group and discuss whether it is possible to achieve selectivity only by replacing the warhead. Ultimately, we review some successful cases of drugs and drug candidates that take advantage of the cyano group to covalently bind to macromolecules and recent applications of covalent nitrile-based inhibitors targeting cysteine proteases.
The role of nitriles in covalent inhibitors
Small molecule drugs can modulate a specific target, wherein the compound will interact with the receptor in an equilibrium process, represented by k1 and k−1 (Fig. 2). The inhibition constant (Ki) is determined using the initial equilibrium state.13 A compound that acts reversibly (without the formation of a covalent bond) is called a noncovalent inhibitor, and the main interactions with the receptor are noncovalent (e.g., hydrogen bonding, hydrophobic contact, van der Waals interactions), thus the Ki can be determined.
Fig. 2. General mechanism representing the ability of an inhibitor to form a noncovalent complex in equilibrium with an enzyme (E⋯I), followed by the formation of a covalently bound state (E–I).

On the other hand, some inhibitors can inactivate their biological targets by forming an irreversible covalent bond with a residue in the target of interest.13,14 This process is represented by the constant k2 or kinact (Fig. 2), and this class of compounds is dubbed irreversible covalent inhibitors. However, the covalent bond formed between the ligand and the target of interest can be reversible, depending on the stability of the formed adduct complex, as well as other possible mechanisms for the reverse reaction, and external factors, such as hydrolysis. The constant k−2 (Fig. 2) will guide the reversibility of the covalent complex, where k2 ≈ k−2 indicates a reversible system, but when k2 ≫ k−2, the complex turns out to be irreversible.14,15
In this scenario, the nitrile group can play a significant role in both cases. Regarding noncovalent inhibitors, nitriles can participate in several different interactions with the target of interest. Recently, Wang and colleagues reviewed the role of nitrile groups in protein–ligand interactions.4 They highlighted the hydrogen bond interactions that the nitrile group can perform with hydrogen bond donors from the protein backbone or side chains. In addition, the cyano group can interact with bridging water molecules and also participate in hydrophobic interactions.4
Nonetheless, the recent success of many covalent drugs like kinase inhibitors (ibrutinib, afatinib, and many others),16 nirmatrelvir to treat COVID-19,17 and sotorasib (the first KRAS-blocking drug)18 is catching the attention of the medicinal chemistry community, with an increasing number of reports describing new structures over time.19
Covalent drugs offer considerable advantages relative to noncovalent ones, the most notable potential benefits being the prolongation of therapeutic response, higher potency, improved selectivity, lower dosage and toxicity, and reduced probability of resistance mechanisms. Furthermore, the covalent inhibition approach prompted the targeting of enzymes that were considered intractable, such as KRAS G12C.19–21
The electrophilic characteristic of the nitrile's carbon atom makes it a functional reactive moiety (warhead) in the chemical scaffold of an inhibitor.6,7 In this way, the warhead can react with an active site nucleophilic residue of the target macromolecule to form a covalent bond. Cysteine and serine are the most common side-chain nucleophilic residues that react with nitrile to form a covalent adduct.6 Occasionally, the lysine side chain amino group may also react with the nitrile.6,22
Nevertheless, non-catalytic residues can also be targeted by covalent inhibitors, although reacting with residues other than an anionic thiolate from an active-site cysteine (CysS−) can be more challenging.23 Since the reactivity of the amino acid side chain is a function of its pKa, Cys residues have the advantage of presenting lower values of pKa in comparison with Ser and Lys. Moreover, Cys residues exposed to the surface may have an even lower pKa due to interactions that make them more polarized, such as hydrogen bonds with water or other polar amino acids.24 Nonetheless, buried non-catalytic Cys have also been extensively explored to discover new covalent inhibitors of kinases when a nucleophilic thiol is accessible in an allosteric pocket.9
Meanwhile, Ser is almost exclusively only nucleophilic at the active site of the protein, under a charge relay mechanism in a catalytic dyad or triad (e.g., protease catalytic mechanism) to activate the residue (SerO−).23 Finally, the Lys side chain residue is mostly protonated when exposed to the solvent, displaying a pKa ∼ 10.8 for the ε-amine. However, a buried Lys can have a pKa value of down to around 5.7 depending on the protein microenvironment, thus making it nucleophilic.25
The two-step process to form the covalent adduct when the warhead is a nitrile reacting with a cysteine protease is depicted in Fig. 3.26 Initially, the formation of the ionic pair between the catalytic dyad (Cys−/His+) takes place before the association of the ligand, a crucial step to the formation of the covalent adduct since the thiolate from Cys can act as a strong nucleophile.27 However, it is still an open question in the literature regarding the stage when the acid–base reaction between the cysteine and histidine residues occurs: (i) it may happen in the dissociated state; or (ii) in the presence of the ligand in the active site.27,28 In any case, the ligand is recognized by the enzyme's binding site in the association step (Fig. 3A), where the noncovalent state is formed in analogy to the Michaelis complex (therefore, enabling Ki determination represented in Fig. 2).
Fig. 3. Schematic reaction involving a reversible covalent inhibitor containing a nitrile warhead and a catalytic dyad Cys/His from a target enzyme. A) represents the dissociated state and the acid–base reaction; B) the noncovalent bound state; and C) the covalent state.

Subsequently, during the acid–base reaction, and with the warhead group positioned in a proper orientation to form the covalent bond, the thiolate from Cys can perform a nucleophilic attack on the carbon of the nitrile (Fig. 3B). Furthermore, the proton from the protonated histidine will be transferred to neutralize and stabilize the nitrogen atom from the emergent thioimidate. This reaction mechanism can occur in a stepwise or concerted form, although computational studies made by our group with a nitrile-based cruzain inhibitor indicate that the concerted mechanism is most likely to happen.29 Therefore, the attack from the thiolate co-occurs with the proton transfer from His to the inhibitor.
Finally, the covalently bound adduct is formed (Fig. 3C), and the energy barrier of the reverse step will define the reversibility of the reaction through the rate constant k−2.13,14 Since the nitrile-bearing compound acts as a reversible covalent inhibitor, the barrier of the reverse reaction is not likely to exceed the value of around 23.5 kcal mol−1, corresponding to a residence time of 10 hours, typical for reversible covalent inhibitors.9,30 However, cases of reversible covalent inhibitors with a residence time over 150 hours are known, and factors other than the stability of the covalent bond may also influence the rate of the reverse reaction, such as the conformation of the covalent complex, the acidity of the α-proton in the adduct (in the case of cyanoacrylamides; vide infra), and the noncovalent interactions in the bound state.31
In addition to the reversible reaction, hydrolysis is another possible pathway to cleave the thioimidate product (Scheme 1). This would be similar to the deacylation step in the catalytic mechanism for cruzain or other papain-like cysteine proteases.32 However, studies have shown that the thioimidate is not usually hydrolyzed by papain, due to conformation changes that make the adduct inaccessible to the solvent.33,34 Interestingly, the addition of an external reactive thiol such as β-mercaptoethanol (βME) has been shown to result in a 100% yield for the hydrolysis of the nitrile to carboxylic acid by papain.35 Nevertheless, serine proteases can perform the hydrolysis of the nitrile adduct without the presence of an external thiol, as will be discussed later.35
Scheme 1. General hydrolysis reaction for the cleavage of the thioimidate adduct.

The versatile use of nitriles also led to exciting novel reactive moieties when combined with acrylamides, obtaining α-cyanoacrylamide warheads. Even though the nitrile is not the group attacked by the nucleophilic residue, it is a compelling approach because an irreversible warhead is converted into a reversible one.36
It is known that acrylamide-based inhibitors react irreversibly with their targets, and many approved kinase inhibitors have been designed to take advantage of this mechanism.8,9 However, the permanent inactivation of enzymes by covalent inhibition still raises concerns, mainly due to toxicity issues. Thus, replacing an irreversible warhead with a reversible one is, in most cases, a desirable approach.31,36
In this context, Taunton and coworkers36 pioneered the application of α-cyanoacrylamide warheads for kinase inhibition (Fig. 4). They observed that their reaction with β-mercaptoethanol (used to mimic the Cys residue) produced a reversible adduct. The reversibility of this reaction involved a β-elimination via an E1cB mechanism. Further studies37 showed that the rate of the elimination had an inverse correlation with the calculated proton affinity of the corresponding carbanions, that is, increasing the acidity of the α-C–H in the adduct resulted in faster thiol elimination. Notably, the acidity of the proton at the α-position in the adduct can be modulated by diverse electron-withdrawing groups (EWGs) attached to the acrylonitrile.37
Fig. 4. Reactions using the acrylamide warhead result in an irreversible adduct, but a reversible product is formed for the α-cyanoacrylamide reagent. The α-C–H in the adduct form is indicated with a green arrow.

Overall, the presence of the electron-withdrawing cyano group increases the reactivity of the acrylamide, and with it, the α-C–H acidity of the β-thioether adduct. Even though the cyanoacrylamide has a higher reactivity, it is also a reversible warhead, since the α-proton can be easily removed, which promotes the exit of the thiolate and thus the reverse reaction via an E1cB mechanism.36,37
Reactivity and selectivity of nitrile-based warheads
Nitriles are weaker electrophiles than most employed warheads, such as aldehydes and azanitriles, which are almost four and ten times more reactive, respectively.38 Despite that, nitriles are an excellent option for developing new covalent inhibitors, with the most recent successful case being the FDA-approved nirmatrelvir, as will be discussed further.
The reactivity of the nitrile group can be modulated by adding an adjacent electron-withdrawing group.3 Heteroaromatic nitriles, aminoacetonitriles, and cyanamides are examples of nitrile-derived warheads with improved reactivity.39–42 Among these, aminoacetonitriles are the weakest electrophilic groups because the electron-withdrawing amide is far from the reactive moiety. On the other hand, heteroaromatic nitriles (pyrimidine and triazine nitriles) and cyanamides are the most reactive warheads since they have electron-withdrawing atoms (e.g., nitrogen) directly bonded to the CN group, resulting in a more electrophilic carbon atom.
Modulation of reactivity can also be explored in prodrugs, like the masking of the nitrile warhead in diacylfuroxan inhibitors. Under aqueous conditions, diacylfuroxans form a masked nitrile oxide, a very reactive organic functional group. These compounds are being investigated as GPX4 inhibitors, a therapeutic target for drug-resistant cancers.43,44
The reactivity of nitriles can be investigated by computational methods. For example, the Fukui function and other DFT descriptors can be used for obtaining information about the reactivity site within a molecule.45 QM/MM calculations can also be utilized to study the reaction mechanism of an inhibitor; hence, it is possible to extract information about the reactivity of the warhead in the transition state.14 Two interesting studies discussing the reactivity and how to tune the electrophilicity of nitrile-based inhibitors were done by Oballa et al.46 and by Ehmke et al.,47 in which they proposed how to model them. Nevertheless, due to putative off-target effects, highly reactive inhibitors must be used carefully.48
Experimentally, HPLC-based kinetic assays are commonly used to assess nitriles' intrinsic reactivity. These assays can be performed in the presence of a cysteine surrogate (e.g., glutathione, βME, and proteins) to quantify their half-life (t1/2) for a nucleophilic attack from a soft thiol group.7 Correspondingly, the reactivity of nitrile-based warheads can be explored against N-α-acetyl-l-lysine (at pH 10.2) to mimic the attack from a deprotonated Lys residue.25
In Fig. 5, the reactivity order for a wide range of different types of nitrile warheads with GSH is shown. Noteworthily, depending on the GSH assay, the values may vary sharply. Still, the order of reactivity generally remains amidst studies.38,49–51
Fig. 5. HPLC-based kinetic assay using GSH to determine nitrile reactivity (decreasing from left to right) for different nitrile-based warheads. Values were retrieved from various academic publications based on comparable experimental assay conditions.38,49–51.

The reactivity of different warheads (electrophiles) with thiol-based compounds (nucleophiles) can be rationalized in terms of their intrinsic electrophilicity, as well as the nucleophilicity and polarizability of reacting chemical species. Thus, the principle of hard and soft acids and bases (HSAB) defined by Pearson52 is of great value, since the anionic cysteine sulfhydryl is a highly polarizable group (soft base), due to the high energy of the 3sp3 orbitals and large ionic radius. This means that softer electrophiles should react more synergistically with cysteine.
As shown in Fig. 5, it is clear that being bonded to an electron-withdrawing heteroaromatic ring and the presence of electron-withdrawing groups in the vicinity increases the reactivity of the nitrile group. This is because these EWGs promote polarization via inductive and resonance effects that decrease the electron density at the carbon atom of the cyano moiety.
Outstandingly, azanitriles are among the most reactive warheads in the series, even being reported as irreversible inhibitors.53 Several factors contribute to this, such as the inductive effect from the electronegative nitrogen, an extended electron density over the N–C N moiety, and a resonance contribution from the lone pair of the nitrogen atom to the sp2 carbon atom in the isothiosemicarbazide adduct, making it more stable.53,54
Even though a warhead replacement can be an excellent strategy to improve an inhibitor's potency, the effect on selectivity might not be so straightforward,47 since the new warhead will also affect potency over undesired targets if they present the same nucleophilic group. Nonetheless, this can be an interesting option when the target and its off-targets have different nucleophilic residues or a different chemical environment of the latter.14,55
Keserű and coworkers49 provided a protocol for prioritizing and designing warheads targeting the Cys residue. It was stated that the warhead chemistry could impact selectivity over undesirable targets. They noted that the selectivity of the electrophilic warhead group depended on the targeted cysteine nucleophilicity. Therefore, they could efficiently propose selective warheads for target enzymes from a high-homology family (e.g., human cysteine cathepsins) as long as these enzymes presented different cysteine nucleophilicity indexes. Thus, they concluded that modifications in the covalent fragment might be specific and conditional to the chemistry of the warhead, the cysteine reactivity, and the steric clashes that can happen between the ligand and the receptor.49
Still, probably the best way to design selective covalent inhibitors over undesired targets involves exploring the noncovalent interactions in combination with a proper choice of the warhead.
Nitrile-containing pharmaceuticals in the reactive group
Vildagliptin and saxagliptin
Dipeptidyl peptidase 4 is a serine protease that inactivates incretin hormones and is a widely exploited target for treating type 2 diabetes mellitus (T2DM). Sandoz discovered one of the first hits for DPP-4: valine pyrrolidide (18), Fig. 6. However, it was observed that noncovalent compounds were less selective toward enzymes DPP-8 and DPP-9.56 Therefore, further optimization of these noncovalent compounds involved the inclusion of warheads in their chemical scaffold. Phosphate diphenyl esters yielded less potent and irreversible inhibitors, and boronic acids were deemed too unstable. Nonetheless, it was noted that the nitrile warhead group could lead to inhibitors with nanomolar potency and adequate chemical stability to be administered orally. These studies eventually led to the development of the nitrile-based inhibitor DPP-728 (19), with improved glycemic control in patients with T2DM under clinical trials, providing the first proof-of-concept DPP-4 inhibitor.57,58
Fig. 6. Chemical structures of the pioneer DPP-4 inhibitor valine pyrrolidide and its optimized nitrile-containing analog DPP-728.

It was determined that the nitrile warhead was responsible for forming a reversible covalent bond to the Ser630 residue. However, kinetic studies showed that DPP-728 was, in fact, a substrate for DPP-4 with a slow dissociation rate and not a true competitive inhibitor, with the nitrile being hydrolyzed to a carboxylic acid. Consequently, further optimization of this chemical series was necessary to improve the pharmacokinetic profile. These optimizations led to the development and approval of vildagliptin (20) and saxagliptin (21) for the treatment of T2DM (Fig. 7).57,59
Fig. 7. Binding modes of DPP-4 inhibitors vildagliptin (PDB code: 6B1E)60 and saxagliptin (PDB code: 3BJM).61.

Vildagliptin and saxagliptin can form a reversible covalent bond with Ser630. Their binding modes were characterized by the crystal structure of these inhibitors bound to the enzyme in the covalent state (Fig. 7).60,61
Even though the chemical structures and their binding modes are similar, QM/MM studies have suggested that only saxagliptin can be considered a true reversible covalent inhibitor, as it mainly dissociates intact from the active site. On the other hand, vildagliptin is more likely to be hydrolyzed by DPP-4 at the nitrile end, forming a carboxylic acid (also observed for DPP-728). These compounds have been termed “pseudo-irreversible inhibitors”, even though this terminology may not appropriately reflect the observed mechanism.62 Moreover, such findings are corroborated by the residence times of both drugs: vildagliptin has a relatively short residence time of 6.6 min, while saxagliptin shows an extended duration of 5.1 h.63
The successful stories of vildagliptin and saxagliptin triggered many other research efforts in the field over the years, and many other DPP-4 nitrile-based inhibitors have been approved worldwide (Fig. 8). Anagliptin (22) and trelagliptin (23) were approved in Japan in 2012 and 2015, respectively. Alogliptin (24) was approved in 2013 in the United States and European Union. Additionally, melogliptin (25) and bisegliptin (26) presented successful phase II clinical trials.
Fig. 8. Chemical representation of drugs and candidates that inhibit DPP-4 to treat T2DM.

Nirmatrelvir
Researchers at Pfizer developed inhibitors for the SARS-CoV-1 main protease (Mpro/3CLpro) during the SARS outbreak in 2003. Among them, PF-00835231 (27) was identified as a promising lead with an α-hydroxymethyl ketone as a warhead. The compound did not go through clinical trials due to the containment of the SARS-CoV-1 episode. However, with the start of the SARS-CoV-2 pandemic at the end of 2019, Pfizer's researchers looked back to SARS-CoV-1 inhibitors to transform an old drug candidate into a new one.
They decided to use PF-00835231 (Fig. 9) as a starting point for developing novel drug candidates to treat SARS-CoV-2 since the main proteases of SARS-CoV-1 and SARS-CoV-2 share a very similar structure and sequence.64 As expected, PF-00835231 was also a potent inhibitor of the SARS-CoV-2 main protease (Mpro), with a Ki of 0.27 ± 0.1 nM.65
Fig. 9. Structure of SARS-CoV-1 and SARS-CoV-2 main protease inhibitor PF-00835231 emphasizing the P1–P3 substituents and the warhead moiety.

The major obstacle to the further development of PF-00835231 came from low gastrointestinal absorption. Therefore, the oral administration had to be replaced by the intravenous (IV) one. Since the goal of the pharmaceutical company was to develop a treatment to prevent the need for hospitalization, the chemical structure had to be optimized for oral bioavailability. One of the first strategies was the replacement of the α-hydroxymethyl ketone warhead with a chemical group that did not have hydrogen bond donors. Two series of compounds were produced, containing the nitrile (28) or the benzothiazol-2-yl ketone (29) warhead, with examples shown in Fig. 10.66,67
Fig. 10. Nitrile and benzothiazol-2-yl ketone derivatives of PF-00835231.

Both series led to promising structures, but the team decided to proceed with the nitrile derivatives for three main reasons: (i) the nitriles were more soluble, (ii) less prone to epimerization, and (iii) the synthesis was easier to scale up. Besides, another study showed that the new lead compound was selective over human cathepsins (CatB, CatL, CatS),68 reducing the probability of off-target effects.
Additional optimization efforts were made with the replacement of leucine by the cyclic P2 group of boceprevir to remove another hydrogen bond donor, also considered a positive point toward an oral drug. Finally, the chemical scaffold was modified to improve the potency and permeability by changing the P3 moiety and adding a P4 group.66,67
The outcome of these optimization efforts was the inhibitor PF-07321332, or nirmatrelvir (30), represented in Fig. 11. Nirmatrelvir, in combination with the HIV antiviral ritonavir (used to decrease the CYP metabolism), was given emergency use authorization in December 2021 by the FDA (followed by other regulatory agencies worldwide) for the treatment of COVID-19 under the trade name Paxlovid.
Fig. 11. Nirmatrelvir structure and the binding mode of its covalent form against SARS-CoV-2 Mpro. PDB code: 7RFW.66.

Following the publication of the chemical structure of nirmatrelvir, Vankadara and coworkers performed a warhead substitution study on this scaffold.69 They found that four warheads (aldehyde, hydroxymethylketone, ketoamide and ketobenzothiazole) led to equally or more potent compounds towards Mpro. Two of these compounds (27 and 29) had already been explored by Pfizer, and 27 has been evaluated in clinical trials through IV administration. In line with the success of nirmatrelvir, novel nitrile-based Mpro inhibitors were developed.68,70 Interestingly, Breidenbach and coworkers employed the azanitrile warhead (31) and extended the chemical scaffold to interact with the S5 subsite, thereby improving the potency against Mpro (Ki = 24.0 nM), Fig. 12. In addition, they noted that the compound bound irreversibly with the target with a kinact/Ki of 37 500 m−1 s−1.53
Fig. 12. Azanitrile-based inhibitor of the SARS-CoV-2 Mpro target.

Rilzabrutinib and compounds targeting kinases
Bruton's tyrosine kinase (BTK), an enzyme involved in intracellular signaling and immune pathways, is a target of interest for cancer and autoimmune diseases.71 Targeting a cysteine residue (Cys481) proximal to the adenosine triphosphate (ATP) binding pocket has been a commonly explored mechanism for BTK inhibition. Notably, this residue is present in only ten kinases, thereby achieving excellent selectivity profiles.72,73 Using this strategy, several irreversible inhibitors of BTK have been discovered, such as the approved drugs ibrutinib and acalabrutinib.9,16
Irreversible covalent inhibitors still raise safety concerns despite numerous successful clinical trials, especially when searching for a novel and selective autoimmune disease treatment. Therefore, reversible covalent inhibitors of BTK have been the subject of many research projects.73
Acrylamide-based inhibitors were explored, based on the previous knowledge that acrylate/acrylamide-based kinase inhibitors would show an improved reactivity profile with the addition of a cyano group to the electrophilic β-carbon, while avoiding the formation of irreversible adducts.36,74 However, previous studies with other kinases connected the β-carbon directly to a kinase-recognition scaffold.74 Since the targeted cysteine group in BTK is distant from the ATP binding site, the authors postulated that an “inverted” cyanoacrylamide warhead, with the electrophilic β-carbon pointing away from the site, would have a more appropriate orientation for the formation of the covalent bond.31 It is worth mentioning that this “inversion” was relative to previously published cyanoacrylamide-based inhibitors, but it is in fact the “normal” orientation in classical acrylamide-based kinase inhibitors. Both warhead orientations are depicted in Fig. 13 to illustrate this design approach.
Fig. 13. The design orientation of the cyanoacrylamide warhead is based on (a) the traditional positioning of the investigated kinase inhibitors and (b) the “inverted” approach. The electrophilic carbon is marked in blue, while the warhead is shown in red.

The “inverted” warhead positioning, combined with optimization via addition of a branched-alkyl capping group to the electrophilic carbon provided excellent results, such as a residence time of 167 ± 21 h for one of the compounds in a kinetic-competition assay against BTK.31 The improvement in residence time was also observed in in vivo assays. Hence, further optimization efforts led to the development of PRN1008, or rilzabrutinib (32), a very potent covalent reversible inhibitor of BTK (IC50 = 1.3 ± 0.5 nM) with long BTK occupancy and increased efficacy in rat models. Still, the compound is highly selective against more than 200 kinases (Fig. 14).73
Fig. 14. Structure of the BTK inhibitor rilzabrutinib.

Despite failing on a phase 3 clinical trial for pemphigus,75 rilzabrutinib was granted FDA Fast Track Designation in 2020 for immune thrombocytopenia. In addition, a phase 2 study is being conducted for the autoimmune condition known as IgG4-related disease.76
Cyanamides have also been explored in this context of targeting noncatalytic cysteines in kinases. This electrophile has the cyano group bonded to a nitrogen atom and is more reactive than its carbon-bonded counterparts.
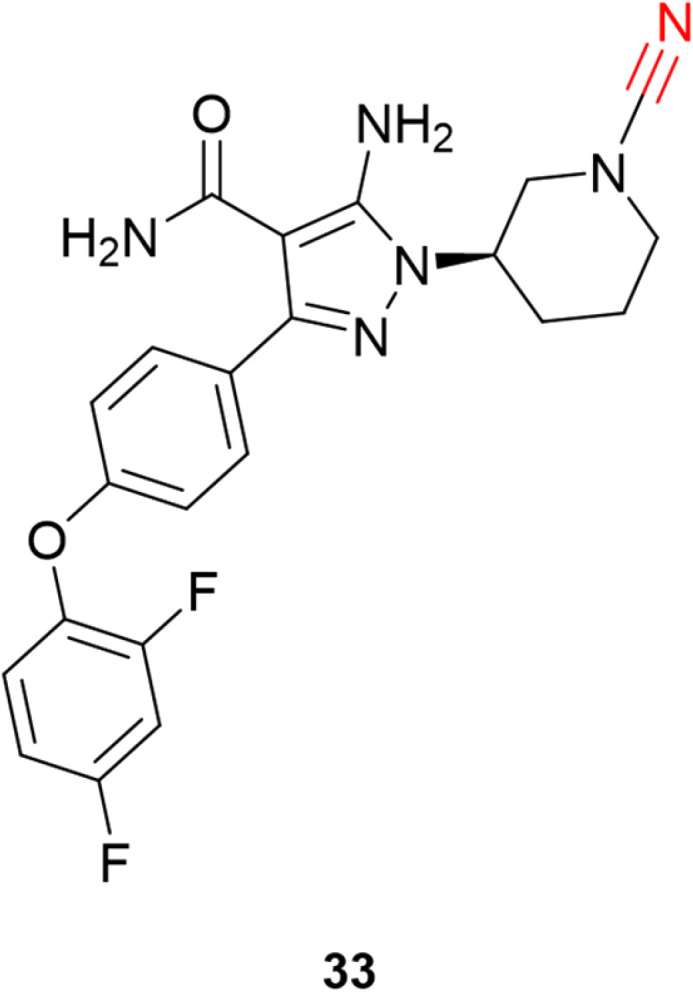
Schnute and colleagues77 synthesized a series of compounds containing cyanamide as a warhead to replace the acrylamide-based irreversible BTK inhibitors. The authors managed to obtain reversible inhibitors with improved selectivity against a panel of 51 kinases and excellent rat pharmacokinetic profiles by targeting Cys481. Compound 33 exhibited excellent results for inhibiting wild-type BTK (IC50 = 1.5 nM) with high oral bioavailability in rats, Fig. 15.
Fig. 15. Potent and selective inhibitor of BTK by targeting noncatalytic Cys481.
Janus kinase 3 (JAK3) is another kinase that has a noncatalytic cysteine (Cys909) to be targeted. It has been the subject of studies involving the development of covalent reversible inhibitors based on both cyanoacrylamide78 and cyanamide.79 Both warheads showed promising results for JAK3 inhibition, and selectivity over the kinome, also having good PK profiles.
Recently, one more interesting option for targeting kinases has been the use of proteolysis targeting chimeras (PROTACs).80 In a prominent study, Gabizon et al. employed the cyanoacrylamide as the electrophilic moiety (34) targeting BTK, wherein it achieved high degradation potency (DC50 = 6 nM) and maximal degradation of the protein (Dmax = 85%).81 The chemical structure of the compound was based on ibrutinib with the replacement of the acrylamide warhead by cyanoacrylamide, thereby creating a reversible covalent PROTAC (Fig. 16). A reversible warhead is preferable, as irreversible PROTACs could not take advantage of the catalytic mechanism of reversible PROTACs. However, mixed results were achieved for PROTACs that exploit the covalent mechanism of action.81–83
Fig. 16. Structure of the PROTAC containing the cyanoacrylamide warhead with the ibrutinib scaffold highlighted by the dashed line.

The recent development of nitrile-based inhibitors against cysteine proteases
Parasitic cysteine proteases
Cysteine protease inhibitors containing a nitrile warhead have been investigated for many years, especially for human cathepsins and related enzymes from viruses and parasites,41,84–88 yet no human or parasitic cysteine protease inhibitor has ever been approved to treat any disease. Nevertheless, this area of research is still prolific and will likely gain popularity after the approval of nirmatrelvir. Therefore, we will review recent developments for parasitic targets involved in several neglected diseases and advances in targeting human cathepsins.
Malaria is an infectious disease caused by protozoa of the genus Plasmodium. Despite the existing drugs available for its treatment, more than 600 000 people die each year from the disease.89 The parasite variability among the species, infection stage, drug resistance and many other factors hinder the identification of efficacious drugs.90
Nitrile-containing inhibitors of the parasitic cysteine protease falcipain-2 have been developed and evaluated as potential new treatments for malaria, such as pyrimidine nitrile-pyrazolines (35, IC50 = 1.63 μM),91 and peptide-like compounds (36, IC50 = 2.7 nM), Fig. 17.92 Nevertheless, the diversity and complexity of Plasmodium parasites make drug resistance a significant problem, with single target inhibitors being especially prone to the emergence of resistant strains. To overcome this issue, researchers modified endoperoxide-containing compounds by appending to them nitrile moieties from known falcipain-2 inhibitors, thereby creating hybrid structures with two mechanisms of action: oxidative stress inducer coupled to falcipain-2 inhibition. The resulting compound (37, Fig. 17) showed excellent enzyme inhibition values (IC50 = 3.4 nM) while also presenting activity against three strains of P. falciparum resistant to commonly used drugs.93
Fig. 17. Examples of nitrile-based inhibitors of cysteine protease falcipain-2.

Another parasitic illness, Chagas disease, caused by the protozoa Trypanosoma cruzi, has only two nitro-heterocyclic approved drugs (nifurtimox and benznidazole). However, the use of these drugs is related to toxic effects, bringing the need to find potential new drugs to treat this disease.94 Accordingly, inhibitors of the main protease of T. cruzi, cruzain, have been explored.
Dipeptidyl nitrile derivatives (38, Ki = 0.5 μM) were investigated as cruzain inhibitors by our research group, Fig. 18.95 The results showed that increasing the electrophilicity of the warhead by replacing the nitrile with an aldehyde (39, Ki = 0.005 μM, Fig. 18) or oxime (Ki = 0.1 μM) increased the potency against the enzyme, however, at the cost of increasing the toxicity according to the follow-up cell assays. Of particular interest, compounds bearing the azanitrile (40, Fig. 18)54 were the most potent against cruzain, with two orders of magnitude higher than the corresponding nitrile counterparts. Presumably, the higher potency of the azanitrile warhead comes from its intrinsic reactivity (Fig. 5).95
Fig. 18. The potency of T. cruzi cysteine protease cruzain inhibitors depicted here increases from nitrile to aldehyde and to azanitrile, following the reactivity profile of the warhead.

Further studies with azanitrile derivatives reinforced the higher potency against cruzain than other warheads.38 Computer simulations and experimental assays were coupled to analyze the compounds' half-life after the incubation with GSH or cysteine. These results linked the intrinsic reactivity of the warhead to the inhibition against cruzain.38
The use of nitrile as a warhead for reversible covalent inhibitors of cysteine proteases is not limited to dipeptidyl scaffolds. A series of bioisosteric replacements of the P2–P3 amide bond by a trifluoromethylamine moiety (41) has been explored, yielding compounds with excellent potency (Ki = 1.58 nM) and selectivity over human cathepsins. Moreover, it is less likely to be hydrolyzed, improving metabolic stability, a known weak spot of peptide-like structures.96,97 Another interesting approach involved the preparation of peptoids (42) based on the peptide-like scaffold. Using peptoid-based compounds led to an inhibitor with a Ki of 0.16 μM against cruzain. Even though this is a reasonably good affinity for a hit compound, the inhibition was lower than for the corresponding peptide. This difference can be rationalized according to the displacement of the P2 position in the S2 recognition site of the protein, as suggested by docking studies (Fig. 19).98
Fig. 19. Further expansion of the chemical diversity of cruzain inhibitors was made for nitrile-based dipeptidyl (41) and peptoid (42) derivatives.

Azanitrile-based inhibitors were also explored for Leishmania mexicana cysteine protease B (LmCPB), another parasitic cysteine protease target found in the causative agent of leishmaniasis. A wide potency range was achieved with nitrile-based inhibitors (Ki ranging from 50 μM to 5 nM) coming from the modulation of noncovalent interactions with the amino acid residues in the active site.99 Azanitrile-based compounds were more potent than their nitrile counterparts. Compound (43) is the most potent LmCPB inhibitor reported to date (Ki = 0.2 nM) and it was used to obtain the first X-ray crystal structure of LmCPB (Fig. 20).99 Although azanitriles present promising results in terms of potency for several targets containing catalytic cysteine residues, more studies will be required to verify if these compounds can be appropriately modulated for selectivity and toxicity, given the high reactivity of the warhead and its irreversible mechanism of inhibition.
Fig. 20. Azanitrile-based inhibitor of LmCPB and its cocrystalized structure (PDB code: 6P4E).99.

Another disease tackled by targeting a cysteine protease is African human trypanosomiasis (HAT, sleeping sickness), an endemic disease in sub-Saharan Africa caused by Trypanosoma brucei.100,101 Currently, four drugs are approved to treat HAT (suramin, pentamidine, melarsoprol, and eflornithine). However, these drugs have severe toxicity issues that limit their uses, as for Chagas disease treatments. Hence, developing new therapies against HAT is crucial, with the cysteine protease rhodesain being a widely explored target for developing new inhibitors since it is vital for the parasite's life cycle.102
Recently, di Chio and coworkers developed a series of dipeptidyl nitriles as rhodesain inhibitors,103 once again highlighting the value of peptidomimetic nitriles targeting cysteine proteases. The structure-based drug design was used to design compounds that achieved nanomolar inhibition potency against rhodesain and micromolar potency in in vitro assays against T. brucei brucei. Fig. 21 depicts the most potent rhodesain inhibitor (44, Ki = 14.1 μM) and the best antiparasitic compound (45, EC50 = 8.8 μM) developed in this work.
Fig. 21. Promising compounds to treat HAT by the inhibition of rhodesain.

Another impressive potential candidate for treating HAT is compound 46, with a Ki = 7.4 nM against rhodesain and IC50 = 18.8 nM against T. brucei rhodesiense, also displaying a good PK profile and selectivity over other human cysteine proteases. In addition, derivatives of this compound exhibited promising in vivo results when administrated orally, reducing almost 50% parasitemia compared to untreated mice.104
Human cysteine proteases
Eleven human cysteine proteases constitute the family of enzymes called cathepsins (B, C, F, H, K, L, O, S, V, W, and X). These enzymes are essential regulators of physiological processes.105,106 However, many pathological conditions observed in humans are related to the dysfunction of these enzymes, making them attractive targets for developing new drugs.106,107 There is still no approved drug targeting human cathepsins, due to side effects observed in clinical trials.
The most studied cathepsins are CatB, CatK, and CatL, since these three enzymes have an increased expression in cancer cells, indicating that they may be involved with neoplastic progression.108,109 CatB is also related to neurodegenerative diseases such as Parkinson's and Alzheimer's, where the cells responsible for the defense of the central nervous system secrete this enzyme due to upregulation, causing apoptosis.110,111 CatK is widely known to be an interesting target in bone-related diseases, as it is highly expressed in osteoclasts. When secreted, it is responsible for degrading the bone matrix.112,113 CatL has become an interesting target to impair the viral replication of SARS-CoV-2 since it is a crucial enzyme related to the virus entry and replication in the host cells.114,115
Schmitz and colleagues employed click chemistry to synthesize a series of CatB inhibitors containing the nitrile group as the warhead, achieving nanomolar potency.116 Compound 47 (Fig. 22) and its analogs were designed to interact with the occluding loop of the CatB in the S1′. This improved the selectivity more than 10-fold over CatK, CatL, and CatS.
Fig. 22. The nitrile-based inhibitor of CatB targeting the occluding loop region of the enzyme.

Hardegger and coworkers described for the first time the importance of the halogen bond in the context of CatL inhibition to achieve selectivity over other cathepsins and increase potency.117 The series of nitrile-containing compounds presented outstanding CatL inhibition values, and the potency increased with the use of heavier halogen atoms (Fig. 23). The sigma-hole, which is a region of positive electrostatic potential in the halogen's surface due to the anisotropic distribution of the charges, is responsible for the halogen bond interaction with Gly61. The electrostatic potential in the sigma-hole will be more positive in the order I > Br > Cl, with Fig. 23 illustrating the halogen bond for compound 50 in the enzyme active site. Further studies have been done exploiting the halogen bond to model new CatL inhibitors.26,118
Fig. 23. Series of CatL inhibitors developed emphasizes the importance of the halogen bond to enzyme recognition. The cocrystalized structure of 50 with CatL (PDB code: 2XU1)117 is also represented. Cl and the dashed lines representing the halogen bond between the Cl atom and the main chain of the Gly61 oxygen atom are shown in orange.

Recently, an article was published showing strategies to achieve selectivity for CatB over CatL (or vice versa) for nitrile-based inhibitors.41 In addition, a review discussing patents of CatB and CatL inhibitors119 highlighted that some of the most promising compounds presented nitrile as the reactive group.
Ultimately, the most notable inhibitors of CatK are the nitrile-containing compounds odanacatib (53) and balicatib (54), Fig. 24. Both reached clinical trials (phases III and II, respectively); however, the studies were discontinued due to undesirable side effects. Nevertheless, these compounds were crucial for developing the next generation of nitrile-based inhibitors for CatK. Like CatB and CatL inhibitors, the most attractive reversible covalent inhibitors patented for CatK present the nitrile group as their reactive center.120
Fig. 24. Structures of CatK inhibitors odanacatib and balicatib that evolved to clinical trials.

Compounds targeting CatK may feature a variety of groups in their chemical scaffold.121 For instance, Benýšek and coworkers developed azanitriles to reach picomolar potency against CatK (55, Ki = 13 pM), Fig. 25.122 In the same work, the authors developed a structurally distinct azanitrile-based compound (56) to inhibit CatK with a Ki of 0.91 nM. Rankovic and colleagues employed a 2-cyanopyrimidine-based compound to obtain highly selective and potent compounds for CatK. In addition, compound 57 (Fig. 25) showed an excellent pharmacokinetic profile with the possibility to be administrated orally.123 Researchers from Hanlim Pharmaceutical Co., LTD. designed a series of compounds employing 2-cyanopyrimidine as the reactive group in urea-based compounds (58), reaching a nanomolar range of inhibition; the company patented the resulting series containing more than 190 compounds.124
Fig. 25. Highly potent nitrile-containing CatK inhibitors described in the literature.

Conclusions
Nitriles are present in many approved drugs, most of which take advantage of their ability to perform different types of noncovalent interactions with their respective targets. In addition, incorporating a cyano group into a drug molecule candidate can also improve pharmacokinetic properties.
Besides these characteristics, introducing the nitrile group in a compound can also result in covalent interactions with the macromolecule. This is an attractive approach for designing new molecules since forming a covalent bond between an inhibitor and its target presents remarkable benefits. In particular, using nitriles as the reactive center within a compound may result in the formation of a reversible covalent adduct, which is more compelling than the irreversible counterpart in terms of toxicity and selectivity.
Another great use of nitriles comes from the exploration of its electron-withdrawing property, for instance, by attaching it to an acrylamide, converting an irreversible warhead into a reversible covalent reactive center. This is a valuable strategy for developing reversible covalent inhibitors for kinases. Likewise, the use of α-cyanoacrylamides in PROTACs seems to have a bright future for treatments exploring protein degradation.
The reactivity of the nitrile warhead can be modulated by changing the atoms or groups close to the reactive center, which allows for the synthesis of a wide range of groups with increasing reactivity that can be explored to achieve a desired potency and/or selectivity. Furthermore, even less reactive warheads, such as aminoacetonitriles, have been observed to make covalent bonds with less reactive groups, such as lysines,125 showing that, with an appropriate orientation of the inhibitor on the target enzyme, covalent inhibition can be achieved while avoiding the use of more reactive warheads.
We have presented several examples of covalent inhibitors and approved drugs that contain nitriles and derivatives as their electrophilic moiety. We have shown that including nitriles in inhibitors is a versatile strategy that can be used for a wide range of biological targets and in different contexts, such as the conversion of non-covalent to covalent inhibitors, from irreversible to reversible inhibitors, or by modulating the reactivity profile of other warheads. Therefore, we expect to observe more nitrile-based or nitrile-modified drug candidates being disclosed in the coming years, given the wide interest of the medicinal chemistry community in kinase enzymes and also in the recent interest in cysteine proteases, especially after the successful development of nirmatrelvir.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We acknowledge the Fundação de Amparo à Pesquisa do Estado de São Paulo – FAPESP grants #2021/01633-3, #2020/04653-2, and 2018/15904-6. The authors also thank Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001, and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) #306708/2020-5, #304030/2018-0 and #305182/2021-8.
References
- DrugBank, https://go.drugbank.com/, (accessed May 2022)
- Wang X. Wang Y. Li X. Yu Z. Song C. Du Y. RSC Med. Chem. 2021;12:1650–1671. doi: 10.1039/D1MD00131K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming F. F. Yao L. Ravikumar P. C. Funk L. Shook B. C. J. Med. Chem. 2010;53:7902–7917. doi: 10.1021/jm100762r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. Du Y. Huang N. Future Med. Chem. 2018;10:2713–2728. doi: 10.4155/fmc-2018-0252. [DOI] [PubMed] [Google Scholar]
- le Questel J.-Y. Berthelot M. Laurence C. J. Phys. Org. Chem. 2000;13:347–358. doi: 10.1002/1099-1395(200006)13:6<347::AID-POC251>3.0.CO;2-E. [DOI] [Google Scholar]
- Ábrányi-Balogh P. and Keserű G. M., in Advances in Chemical Proteomics, ed. X. Yao, Elsevier, 2022, pp. 47–73 [Google Scholar]
- Gehringer M. Laufer S. A. J. Med. Chem. 2019;62:5673–5724. doi: 10.1021/acs.jmedchem.8b01153. [DOI] [PubMed] [Google Scholar]
- Ferguson F. M. Gray N. S. Nat. Rev. Drug Discovery. 2018;17:353–377. doi: 10.1038/nrd.2018.21. [DOI] [PubMed] [Google Scholar]
- Abdeldayem A. Raouf Y. S. Constantinescu S. N. Moriggl R. Gunning P. T. Chem. Soc. Rev. 2020;49:2617–2687. doi: 10.1039/C9CS00720B. [DOI] [PubMed] [Google Scholar]
- Hunt J. T. Ding C. Z. Batorsky R. Bednarz M. Bhide R. Cho Y. Chong S. Chao S. Gullo-Brown J. Guo P. Kim S. H. Lee F. Y. F. Leftheris K. Miller A. Mitt T. Patel M. Penhallow B. A. Ricca C. Rose W. C. Schmidt R. Slusarchyk W. A. Vite G. Manne V. J. Med. Chem. 2000;43:3587–3595. doi: 10.1021/jm000248z. [DOI] [PubMed] [Google Scholar]
- Singh P. D. A. Jackson J. R. James S. P. Biochem. Pharmacol. 1985;34:2207–2209. doi: 10.1016/0006-2952(85)90420-4. [DOI] [PubMed] [Google Scholar]
- Brogi S. Ibba R. Rossi S. Butini S. Calderone V. Gemma S. Campiani G. Molecules. 2022;27:2561. doi: 10.3390/molecules27082561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhirter C., in Annual Reports in Medicinal Chemistry, ed. R. A. Ward and N. P. Grimster, Academic Press, 2021, vol. 56, pp. 1–31 [Google Scholar]
- Awoonor-Williams E. Walsh A. G. Rowley C. N. Biochim. Biophys. Acta, Proteins Proteomics. 2017;1865:1664–1675. doi: 10.1016/j.bbapap.2017.05.009. [DOI] [PubMed] [Google Scholar]
- Singh J. Petter R. C. Baillie T. A. Whitty A. Nat. Rev. Drug Discovery. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Cohen P. Cross D. Jänne P. A. Nat. Rev. Drug Discovery. 2021;20:551–569. doi: 10.1038/s41573-021-00195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb Y. N. Drugs. 2022;82:585–591. doi: 10.1007/s40265-022-01692-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair H. A. Drugs. 2021;81:1573–1579. doi: 10.1007/s40265-021-01574-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutanto F. Konstantinidou M. Dömling A. RSC Med. Chem. 2020;11:876–884. doi: 10.1039/D0MD00154F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Cesco S. Kurian J. Dufresne C. Mittermaier A. K. Moitessier N. Eur. J. Med. Chem. 2017;138:96–114. doi: 10.1016/j.ejmech.2017.06.019. [DOI] [PubMed] [Google Scholar]
- Singh J. J. Med. Chem. 2022;65:5886–5901. doi: 10.1021/acs.jmedchem.1c02134. [DOI] [PubMed] [Google Scholar]
- Pettinger J. Jones K. Cheeseman M. D. Angew. Chem., Int. Ed. 2017;56:15200–15209. doi: 10.1002/anie.201707630. [DOI] [PubMed] [Google Scholar]
- Jones L. H., in Annual Reports in Medicinal Chemistry, ed. R. A. Ward and N. P. Grimster, Academic Press, 2021, vol. 56, pp. 95–134 [Google Scholar]
- Marino S. M. Gladyshev V. N. J. Biol. Chem. 2012;287:4419–4425. doi: 10.1074/jbc.R111.275578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahal U. P. Gilbert A. M. Obach R. S. Flanagan M. E. Chen J. M. Garcia-Irizarry C. Starr J. T. Schuff B. Uccello D. P. Young J. A. MedChemComm. 2016;7:864–872. doi: 10.1039/C6MD00017G. [DOI] [Google Scholar]
- Bonatto V. Shamim A. Rocho F. R. Leitão A. Luque F. J. Lameira J. Montanari C. A. J. Chem. Inf. Model. 2021;61:4733–4744. doi: 10.1021/acs.jcim.1c00515. [DOI] [PubMed] [Google Scholar]
- Oanca G. Asadi M. Saha A. Ramachandran B. Warshel A. J. Phys. Chem. B. 2020;124:11349–11356. doi: 10.1021/acs.jpcb.0c08192. [DOI] [PubMed] [Google Scholar]
- Kneller D. W. Phillips G. Weiss K. L. Zhang Q. Coates L. Kovalevsky A. J. Med. Chem. 2021;64:4991–5000. doi: 10.1021/acs.jmedchem.1c00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos A. M. Cianni L. Vita D. Rosini F. Leitão A. Laughton C. A. Lameira J. Montanari C. A. Phys. Chem. Chem. Phys. 2018;20:24317–24328. doi: 10.1039/C8CP03320J. [DOI] [PubMed] [Google Scholar]
- Mondal D. Warshel A. Biochemistry. 2020;59:4601–4608. doi: 10.1021/acs.biochem.0c00782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw J. M. McFarland J. M. Paavilainen V. O. Bisconte A. Tam D. Phan V. T. Romanov S. Finkle D. Shu J. Patel V. Ton T. Li X. Loughhead D. G. Nunn P. A. Karr D. E. Gerritsen M. E. Funk J. O. Owens T. D. Verner E. Brameld K. A. Hill R. J. Goldstein D. M. Taunton J. Nat. Chem. Biol. 2015;11:525–531. doi: 10.1038/nchembio.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai X. Meek T. D. Biochemistry. 2018;57:3176–3190. doi: 10.1021/acs.biochem.7b01250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon J. B. Coleman R. S. Hanzlik R. P. J. Am. Chem. Soc. 1986;108:1350–1351. doi: 10.1021/ja00266a066. [DOI] [Google Scholar]
- Liang T.-C. Abeles R. H. Arch. Biochem. Biophys. 1987;252:626–634. doi: 10.1016/0003-9861(87)90068-3. [DOI] [PubMed] [Google Scholar]
- Gour-Salin B. J. Storer A. C. Castelhano A. Krantz A. Robinson V. Enzyme Microb. Technol. 1991;13:408–411. doi: 10.1016/0141-0229(91)90203-M. [DOI] [Google Scholar]
- Serafimova I. M. Pufall M. A. Krishnan S. Duda K. Cohen M. S. Maglathlin R. L. McFarland J. M. Miller R. M. Frödin M. Taunton J. Nat. Chem. Biol. 2012;8:471–476. doi: 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan S. Miller R. M. Tian B. Mullins R. D. Jacobson M. P. Taunton J. J. Am. Chem. Soc. 2014;136:12624–12630. doi: 10.1021/ja505194w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonatto V. Batista P. H. J. Cianni L. Vita D. Silva D. G. Cedron R. Tezuka D. Y. Albuquerque S. Moraes C. B. Franco C. H. Lameira J. Leitão A. Montanari C. A. RSC Med. Chem. 2020;11:1275–1284. doi: 10.1039/D0MD00097C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podgorski I. Future Med. Chem. 2009;1:21–34. doi: 10.4155/fmc.09.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L. H. RSC Chem. Biol. 2020;1:298–304. doi: 10.1039/D0CB00174K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianni L. Rocho F. R. Bonatto V. Martins F. C. P. Lameira J. Leitão A. Montanari C. A. Shamim A. Bioorg. Med. Chem. 2021;29:115827. doi: 10.1016/j.bmc.2020.115827. [DOI] [PubMed] [Google Scholar]
- Cianni L. Rocho F. R. Rosini F. Bonatto V. Ribeiro J. F. R. Lameira J. Leitão A. Shamim A. Montanari C. A. Bioorg. Chem. 2020;101:104039. doi: 10.1016/j.bioorg.2020.104039. [DOI] [PubMed] [Google Scholar]
- Eaton J. K. Ruberto R. A. Kramm A. Viswanathan V. S. Schreiber S. L. J. Am. Chem. Soc. 2019;141:20407–20415. doi: 10.1021/jacs.9b10769. [DOI] [PubMed] [Google Scholar]
- Eaton J. K. Furst L. Ruberto R. A. Moosmayer D. Hilpmann A. Ryan M. J. Zimmermann K. Cai L. L. Niehues M. Badock V. Kramm A. Chen S. Hillig R. C. Clemons P. A. Gradl S. Montagnon C. Lazarski K. E. Christian S. Bajrami B. Neuhaus R. Eheim A. L. Viswanathan V. S. Schreiber S. L. Nat. Chem. Biol. 2020;16:497–506. doi: 10.1038/s41589-020-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattaraj P. K. Giri S. Duley S. Chem. Rev. 2011;111:PR43–PR75. doi: 10.1021/cr100149p. [DOI] [PubMed] [Google Scholar]
- Oballa R. M. Truchon J.-F. Bayly C. I. Chauret N. Day S. Crane S. Berthelette C. Bioorg. Med. Chem. Lett. 2007;17:998–1002. doi: 10.1016/j.bmcl.2006.11.044. [DOI] [PubMed] [Google Scholar]
- Ehmke V. Quinsaat J. E. Q. Rivera-Fuentes P. Heindl C. Freymond C. Rottmann M. Brun R. Schirmeister T. Diederich F. Org. Biomol. Chem. 2012;10:5764–5768. doi: 10.1039/C2OB00034B. [DOI] [PubMed] [Google Scholar]
- Ward R. A., in Reference Module in Biomedical Sciences, ed. O. Wolkenhauer, Academic Press, Oxford, 2021, pp. 174–189 [Google Scholar]
- Ábrányi-Balogh P. Petri L. Imre T. Szijj P. Scarpino A. Hrast M. Mitrović A. Fonovič U. P. Németh K. Barreteau H. Roper D. I. Horváti K. Ferenczy G. G. Kos J. Ilaš J. Gobec S. Keserű G. M. Eur. J. Med. Chem. 2018;160:94–107. doi: 10.1016/j.ejmech.2018.10.010. [DOI] [PubMed] [Google Scholar]
- Berteotti A. Vacondio F. Lodola A. Bassi M. Silva C. Mor M. Cavalli A. ACS Med. Chem. Lett. 2014;5:501–505. doi: 10.1021/ml400489b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeley A. Ábrányi-Balogh P. Keserű G. M. MedChemComm. 2019;10:263–267. doi: 10.1039/C8MD00327K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson R. G. J. Am. Chem. Soc. 1963;85:3533–3539. doi: 10.1021/ja00905a001. [DOI] [Google Scholar]
- Breidenbach J. Lemke C. Pillaiyar T. Schäkel L. al Hamwi G. Diett M. Gedschold R. Geiger N. Lopez V. Mirza S. Namasivayam V. Schiedel A. C. Sylvester K. Thimm D. Vielmuth C. Vu L. Phuong. Zyulina M. Bodem J. Gütschow M. Müller C. E. Angew. Chem., Int. Ed. 2021;60:10423–10429. doi: 10.1002/anie.202016961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löser R. Frizler M. Schilling K. Gütschow M. Angew. Chem., Int. Ed. 2008;47:4331–4334. doi: 10.1002/anie.200705858. [DOI] [PubMed] [Google Scholar]
- Martin J. S. MacKenzie C. J. Fletcher D. Gilbert I. H. Bioorg. Med. Chem. 2019;27:2066–2074. doi: 10.1016/j.bmc.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veken P. Haemers A. Augustyns K. Curr. Top. Med. Chem. 2007;7:621–635. doi: 10.2174/156802607780091046. [DOI] [PubMed] [Google Scholar]
- Foley J. E. Ahrén B. Eur. Endocrinol. 2017;13:56–61. doi: 10.17925/EE.2017.13.02.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes T. E. Mone M. D. Russell M. E. Weldon S. C. Villhauer E. B. Biochemistry. 1999;38:11597–11603. doi: 10.1021/bi990852f. [DOI] [PubMed] [Google Scholar]
- Villhauer E. B. Brinkman J. A. Naderi G. B. Burkey B. F. Dunning B. E. Prasad K. Mangold B. L. Russell M. E. Hughes T. E. J. Med. Chem. 2003;46:2774–2789. doi: 10.1021/jm030091l. [DOI] [PubMed] [Google Scholar]
- Berger J. P. SinhaRoy R. Pocai A. Kelly T. M. Scapin G. Gao Y.-D. Pryor K. A. D. Wu J. K. Eiermann G. J. Xu S. S. Zhang X. Tatosian D. A. Weber A. E. Thornberry N. A. Carr R. D. Endocrinol., Diabetes Metab. 2018;1:e00002. doi: 10.1002/edm2.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzler W. J. Yanchunas J. Weigelt C. Kish K. Klei H. E. Xie D. Zhang Y. Corbett M. Tamura J. K. He B. Hamann L. G. Kirby M. S. Marcinkeviciene J. Protein Sci. 2008;17:240–250. doi: 10.1110/ps.073253208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.-H. Zhang F. Diao H. Wu R. ACS Catal. 2019;9:2292–2302. doi: 10.1021/acscatal.8b05051. [DOI] [Google Scholar]
- Tummino P. J. Copeland R. A. Biochemistry. 2008;47:5481–5492. doi: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- Naqvi A. A. T. Fatima K. Mohammad T. Fatima U. Singh I. K. Singh A. Atif S. M. Hariprasad G. Hasan G. M. Hassan Md. I. Biochim. Biophys. Acta, Mol. Basis Dis. 2020;1866:165878. doi: 10.1016/j.bbadis.2020.165878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman R. L. Kania R. S. Brothers M. A. Davies J. F. Ferre R. A. Gajiwala K. S. He M. Hogan R. J. Kozminski K. Li L. Y. Lockner J. W. Lou J. Marra M. T. Mitchell L. J. Murray B. W. Nieman J. A. Noell S. Planken S. P. Rowe T. Ryan K. Smith G. J. Solowiej J. E. Steppan C. M. Taggart B. J. Med. Chem. 2020;63:12725–12747. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D. R. Allerton C. M. N. Anderson A. S. Aschenbrenner L. Avery M. Berritt S. Boras B. Cardin R. D. Carlo A. Coffman K. J. Dantonio A. Di L. Eng H. Ferre R. Gajiwala K. S. Gibson S. A. Greasley S. E. Hurst B. L. Kadar E. P. Kalgutkar A. t. S. Lee J. C. Lee J. Liu W. Mason S. W. Noell S. Novak J. J. Obach R. S. Ogilvie K. Patel N. C. Pettersson M. Rai D. K. Reese M. R. Sammons M. F. Sathish J. G. Singh R. S. P. Steppan C. M. Stewart A. E. Tuttle J. B. Updyke L. Verhoest P. R. Wei L. Yang Q. Zhu Y. Science. 2021;374:1586–1593. doi: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- Halford B. ACS Cent. Sci. 2022;8:405–407. doi: 10.1021/acscentsci.2c00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B. Arutyunova E. Khan M. B. Lu J. Joyce M. A. Saffran H. A. Shields J. A. Kandadai A. S. Belovodskiy A. Hena M. Vuong W. Lamer T. Young H. S. Vederas J. C. Tyrrell D. L. Lemieux M. J. Nieman J. A. RSC Med. Chem. 2021;12:1722–1730. doi: 10.1039/D1MD00247C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vankadara S. Dawson M. D. Fong J. Y. Oh Q. Y. Ang Q. A. Liu B. Chang H. Y. Koh J. Koh X. Tan Q. W. Joy J. Chia C. S. B. ACS Med. Chem. Lett. 2022;13:1345–1350. doi: 10.1021/acsmedchemlett.2c00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller D. W. Li H. Phillips G. Weiss K. L. Zhang Q. Arnould M. A. Jonsson C. B. Surendranathan S. Parvathareddy J. Blakeley M. P. Coates L. Louis J. M. Bonnesen P. Kovalevsky A. Nat. Commun. 2022;13:2268. doi: 10.1038/s41467-022-29915-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crofford L. J. Nyhoff L. E. Sheehan J. H. Kendall P. L. Expert Rev. Clin. Immunol. 2016;12:763–773. doi: 10.1586/1744666X.2016.1152888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung L., in Annual Reports in Medicinal Chemistry, ed. R. A. Ward and N. P. Grimster, Academic Press, 2021, vol. 56, pp. 33–74 [Google Scholar]
- Owens T. D. Brameld K. A. Verner E. J. Ton T. Li X. Zhu J. Masjedizadeh M. R. Bradshaw J. M. Hill R. J. Tam D. Bisconte A. Kim E. O. Francesco M. Xing Y. Shu J. Karr D. LaStant J. Finkle D. Loewenstein N. Haberstock-Debic H. Taylor M. J. Nunn P. Langrish C. L. Goldstein D. M. J. Med. Chem. 2022;65:5300–5316. doi: 10.1021/acs.jmedchem.1c01170. [DOI] [PubMed] [Google Scholar]
- Miller R. M. Paavilainen V. O. Krishnan S. Serafimova I. M. Taunton J. J. Am. Chem. Soc. 2013;135:5298–5301. doi: 10.1021/ja401221b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons L., Sanofi's oral BTK inhibitor rilzabrutinib fails Phase III trial, https://www.pharmatimes.com/news/sanofis_oral_btk_inhibitor_rilzabrutinib_fails_phase_iii_trial_1376448, (accessed May 2022)
- Sanofi, Rilzabrutinib granted FDA Fast Track Designation for treatment of immune thrombocytopenia, https://www.sanofi.com/en/media-room/press-releases/2020/2020-11-18-07-15-00, (accessed May 2022)
- Schnute M. E. Benoit S. E. Buchler I. P. Caspers N. Grapperhaus M. L. Han S. Hotchandani R. Huang N. Hughes R. O. Juba B. M. Kim K.-H. Liu E. McCarthy E. Messing D. Miyashiro J. S. Mohan S. O'Connell T. N. Ohren J. F. Parikh M. D. Schmidt M. Selness S. R. Springer J. R. Thanabal V. Trujillo J. I. Walker D. P. Wan Z.-K. Withka J. M. Wittwer A. J. Wood N. L. Xing L. Zapf C. W. Douhan J. ACS Med. Chem. Lett. 2019;10:80–85. doi: 10.1021/acsmedchemlett.8b00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster M. Chaikuad A. Bauer S. M. Holstein J. Robers M. B. Corona C. R. Gehringer M. Pfaffenrot E. Ghoreschi K. Knapp S. Laufer S. A. Cell Chem. Biol. 2016;23:1335–1340. doi: 10.1016/j.chembiol.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casimiro-Garcia A. Trujillo J. I. Vajdos F. Juba B. Banker M. E. Aulabaugh A. Balbo P. Bauman J. Chrencik J. Coe J. W. Czerwinski R. Dowty M. Knafels J. D. Kwon S. Leung L. Liang S. Robinson R. P. Telliez J.-B. Unwalla R. Yang X. Thorarensen A. J. Med. Chem. 2018;61:10665–10699. doi: 10.1021/acs.jmedchem.8b01308. [DOI] [PubMed] [Google Scholar]
- Békés M. Langley D. R. Crews C. M. Nat. Rev. Drug Discovery. 2022;21:181–200. doi: 10.1038/s41573-021-00371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabizon R. Shraga A. Gehrtz P. Livnah E. Shorer Y. Gurwicz N. Avram L. Unger T. Aharoni H. Albeck S. Brandis A. Shulman Z. Katz B.-Z. Herishanu Y. London N. J. Am. Chem. Soc. 2020;142:11734–11742. doi: 10.1021/jacs.9b13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue G. Chen J. Liu L. Zhou D. Zuo Y. Fu T. Pan Z. Chem. Commun. 2020;56:1521–1524. doi: 10.1039/C9CC08238G. [DOI] [PubMed] [Google Scholar]
- Tinworth C. P. Lithgow H. Dittus L. Bassi Z. I. Hughes S. E. Muelbaier M. Dai H. Smith I. E. D. Kerr W. J. Burley G. A. Bantscheff M. Harling J. D. ACS Chem. Biol. 2019;14:342–347. doi: 10.1021/acschembio.8b01094. [DOI] [PubMed] [Google Scholar]
- Otto H.-H. Schirmeister T. Chem. Rev. 1997;97:133–172. doi: 10.1021/cr950025u. [DOI] [PubMed] [Google Scholar]
- Greenspan P. D. Clark K. L. Tommasi R. A. Cowen S. D. McQuire L. W. Farley D. L. van Duzer J. H. Goldberg R. L. Zhou H. Du Z. Fitt J. J. Coppa D. E. Fang Z. Macchia W. Zhu L. Capparelli M. P. Goldstein R. Wigg A. M. Doughty J. R. Bohacek R. S. Knap A. K. J. Med. Chem. 2001;44:4524–4534. doi: 10.1021/jm010206q. [DOI] [PubMed] [Google Scholar]
- Löser R. Schilling K. Dimmig E. Gütschow M. J. Med. Chem. 2005;48:7688–7707. doi: 10.1021/jm050686b. [DOI] [PubMed] [Google Scholar]
- Cianni L. Sartori G. Rosini F. de Vita D. Pires G. Lopes B. R. Leitão A. Burtoloso A. C. B. Montanari C. A. Bioorg. Chem. 2018;79:285–292. doi: 10.1016/j.bioorg.2018.04.006. [DOI] [PubMed] [Google Scholar]
- Asaad N. Bethel P. A. Coulson M. D. Dawson J. E. Ford S. J. Gerhardt S. Grist M. Hamlin G. A. James M. J. Jones E. v. Karoutchi G. I. Kenny P. W. Morley A. D. Oldham K. Rankine N. Ryan D. Wells S. L. Wood L. Augustin M. Krapp S. Simader H. Steinbacher S. Bioorg. Med. Chem. Lett. 2009;19:4280–4283. doi: 10.1016/j.bmcl.2009.05.071. [DOI] [PubMed] [Google Scholar]
- World Health Organization, Treating Malaria, https://www.who.int/activities/treating-malaria, (accessed May 2022)
- Tse E. G. Korsik M. Todd M. H. Malar. J. 2019;18:93. doi: 10.1186/s12936-019-2724-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marella A. Akhter M. Shaquiquzzaman M. Tanwar O. Verma G. Alam M. M. Med. Chem. Res. 2015;24:1018–1037. doi: 10.1007/s00044-014-1188-5. [DOI] [Google Scholar]
- Nizi E. Sferrazza A. Fabbrini D. Nardi V. Andreini M. Graziani R. Gennari N. Bresciani A. Paonessa G. Harper S. Bioorg. Med. Chem. Lett. 2018;28:1540–1544. doi: 10.1016/j.bmcl.2018.03.069. [DOI] [PubMed] [Google Scholar]
- Oliveira R. Guedes R. C. Meireles P. Albuquerque I. S. Gonçalves L. M. Pires E. Bronze M. R. Gut J. Rosenthal P. J. Prudêncio M. Moreira R. O'Neill P. M. Lopes F. J. Med. Chem. 2014;57:4916–4923. doi: 10.1021/jm5004528. [DOI] [PubMed] [Google Scholar]
- Bermudez J. Davies C. Simonazzi A. Pablo Real J. Palma S. Acta Trop. 2016;156:1–16. doi: 10.1016/j.actatropica.2015.12.017. [DOI] [PubMed] [Google Scholar]
- Silva D. G. Ribeiro J. F. R. Vita D. Cianni L. Franco C. H. Freitas-Junior L. H. Moraes C. B. Rocha J. R. Burtoloso A. C. B. Kenny P. W. Leitão A. Montanari C. A. Bioorg. Med. Chem. Lett. 2017;27:5031–5035. doi: 10.1016/j.bmcl.2017.10.002. [DOI] [PubMed] [Google Scholar]
- Gomes J. C. Cianni L. Ribeiro J. Rocho F. R. Silva S. C. M. Batista P. H. J. Moraes C. B. Franco C. H. Freitas-Junior L. H. G. Kenny P. W. Leitão A. Burtoloso A. C. B. Vita D. Montanari C. A. Bioorg. Med. Chem. 2019;27:115083. doi: 10.1016/j.bmc.2019.115083. [DOI] [PubMed] [Google Scholar]
- Lameiro R. F. Shamim A. Rosini F. Cendron R. Batista P. H. J. Montanari C. A. Future Med. Chem. 2020;13:25–43. doi: 10.4155/fmc-2020-0057. [DOI] [PubMed] [Google Scholar]
- Alves L. Santos D. A. Cendron R. Rocho F. R. Matos T. K. B. Leitão A. Montanari C. A. Bioorg. Med. Chem. 2021;41:116211. doi: 10.1016/j.bmc.2021.116211. [DOI] [PubMed] [Google Scholar]
- Ribeiro J. F. R. Cianni L. Li C. Warwick T. G. Vita D. Rosini F. Rocho F. R. Martins F. C. P. Kenny P. W. Lameira J. Leitão A. Emsley J. Montanari C. A. Bioorg. Med. Chem. 2020;28:115743. doi: 10.1016/j.bmc.2020.115743. [DOI] [PubMed] [Google Scholar]
- Büscher P. Cecchi G. Jamonneau V. Priotto G. Lancet. 2017;390:2397–2409. doi: 10.1016/S0140-6736(17)31510-6. [DOI] [PubMed] [Google Scholar]
- Ettari R. Previti S. Tamborini L. Cullia G. Grasso S. Zappalà M. Mini-Rev. Med. Chem. 2016;16:1374–1391. doi: 10.2174/1389557515666160509125243. [DOI] [PubMed] [Google Scholar]
- Ettari R. Tamborini L. Angelo I. C. Micale N. Pinto A. de Micheli C. Conti P. J. Med. Chem. 2013;56:5637–5658. doi: 10.1021/jm301424d. [DOI] [PubMed] [Google Scholar]
- di Chio C. Previti S. Amendola G. Ravichandran R. Wagner A. Cosconati S. Hellmich U. A. Schirmeister T. Zappalà M. Ettari R. Eur. J. Med. Chem. 2022;236:114328. doi: 10.1016/j.ejmech.2022.114328. [DOI] [PubMed] [Google Scholar]
- Giroud M. Kuhn B. Saint-Auret S. Kuratli C. Martin R. E. Schuler F. Diederich F. Kaiser M. Brun R. Schirmeister T. Haap W. J. Med. Chem. 2018;61:3370–3388. doi: 10.1021/acs.jmedchem.7b01870. [DOI] [PubMed] [Google Scholar]
- Cianni L. Feldmann C. W. Gilberg E. Gütschow M. Juliano L. Leitão A. Bajorath J. Montanari C. A. J. Med. Chem. 2019;62:10497–10525. doi: 10.1021/acs.jmedchem.9b00683. [DOI] [PubMed] [Google Scholar]
- Patel S. Homaei A. El-Seedi H. R. Akhtar N. Biomed. Pharmacother. 2018;105:526–532. doi: 10.1016/j.biopha.2018.05.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk B. Nat. Rev. Drug Discovery. 2006;5:785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- Mohamed M. M. Sloane B. F. Nat. Rev. Cancer. 2006;6:764–775. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- Siklos M. BenAissa M. Thatcher G. R. J. Acta Pharm. Sin. B. 2015;5:506–519. doi: 10.1016/j.apsb.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein H.-G. Keilhoff G. Neural Regener. Res. 2018;13:2100–2101. doi: 10.4103/1673-5374.241457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi H. Neural Regener. Res. 2020;15:25–29. doi: 10.4103/1673-5374.264444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake M. T. Clarke B. L. Oursler M. J. Khosla S. Endocr. Rev. 2017;38:325–350. doi: 10.1210/er.2015-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlewood-Evans A. Kokubo T. Ishibashi O. Inaoka T. Wlodarski B. Gallagher J. A. Bilbe G. Bone. 1997;20:81–86. doi: 10.1016/S8756-3282(96)00351-1. [DOI] [PubMed] [Google Scholar]
- Liu T. Luo S. Libby P. Shi G.-P. Pharmacol. Ther. 2020;213:107587. doi: 10.1016/j.pharmthera.2020.107587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou X. Liu Y. Lei X. Li P. Mi D. Ren L. Guo L. Guo R. Chen T. Hu J. Xiang Z. Mu Z. Chen X. Chen J. Hu K. Jin Q. Wang J. Qian Z. Nat. Commun. 2020;11:1620. doi: 10.1038/s41467-020-15562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz J. Li T. Bartz U. Gütschow M. ACS Med. Chem. Lett. 2016;7:211–216. doi: 10.1021/acsmedchemlett.5b00474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardegger L. A. Kuhn B. Spinnler B. Anselm L. Ecabert R. Stihle M. Gsell B. Thoma R. Diez J. Benz J. Plancher J.-M. Hartmann G. Banner D. W. Haap W. Diederich F. Angew. Chem., Int. Ed. 2011;50:314–318. doi: 10.1002/anie.201006781. [DOI] [PubMed] [Google Scholar]
- Lameira J. Bonatto V. Cianni L. Rocho F. R. Leitão A. Montanari C. A. Phys. Chem. Chem. Phys. 2019;21:24723–24730. doi: 10.1039/C9CP04820K. [DOI] [PubMed] [Google Scholar]
- Li Y.-Y. Fang J. Ao G.-Z. Expert Opin. Ther. Pat. 2017;27:643–656. doi: 10.1080/13543776.2017.1272572. [DOI] [PubMed] [Google Scholar]
- Rocho F. R. Bonatto V. Lameiro R. F. Lameira J. Leitão A. Montanari C. A. Expert Opin. Ther. Pat. 2022;32:561–573. doi: 10.1080/13543776.2022.2040480. [DOI] [PubMed] [Google Scholar]
- Lu J. Wang M. Wang Z. Fu Z. Lu A. Zhang G. J. Enzyme Inhib. Med. Chem. 2018;33:890–904. doi: 10.1080/14756366.2018.1465417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benýšek J. Buša M. Rubešová P. Fanfrlík J. Lepšík M. Brynda J. Matoušková Z. Bartz U. Horn M. Gütschow M. Mareš M. J. Enzyme Inhib. Med. Chem. 2022;37:515–526. doi: 10.1080/14756366.2021.2024527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankovic Z. Cai J. Kerr J. Fradera X. Robinson J. Mistry A. Hamilton E. McGarry G. Andrews F. Caulfield W. Cumming I. Dempster M. Waller J. Scullion P. Martin I. Mitchell A. Long C. Baugh M. Westwood P. Kinghorn E. Bruin J. Hamilton W. Uitdehaag J. van Zeeland M. Potin D. Saniere L. Fouquet A. Chevallier F. Deronzier H. Dorleans C. Nicolai E. Bioorg. Med. Chem. Lett. 2010;20:1524–1527. doi: 10.1016/j.bmcl.2010.01.100. [DOI] [PubMed] [Google Scholar]
- Hanlim Pharmaceutical Co., LTD., US Pat., 20210047301, 2021
- Smith P. A. Koehler M. F. T. Girgis H. S. Yan D. Chen Y. Chen Y. Crawford J. J. Durk M. R. Higuchi R. I. Kang J. Murray J. Paraselli P. Park S. Phung W. Quinn J. G. Roberts T. C. Rougé L. Schwarz J. B. Skippington E. Wai J. Xu M. Yu Z. Zhang H. Tan M.-W. Heise C. E. Nature. 2018;561:189–194. doi: 10.1038/s41586-018-0483-6. [DOI] [PubMed] [Google Scholar]