

Abstract
The sequencing of the human genome has allowed the study of the genetic architecture of common diseases; the number of genomic variants that contribute to risk of disease and their joint frequency and effect size distribution. Common diseases are polygenic, with many loci contributing to phenotype, with the cumulative burden of risk alleles determining individual risk in conjunction with environmental factors. The majority of risk loci occur in non-coding regions of the genome regulating cell- and context-specific gene expression. Although the effect sizes of most risk alleles are small, their cumulative effects in individuals, quantified as a polygenic (risk) score, can identify people at increased risk of disease, thereby facilitating prevention or early intervention.
Competing genetic models of disease
Paradoxically, many common diseases occur in individuals with no family history of the disease, yet relatives of those affected have an increased risk compared to an average person from the population (1). These empirical observations can be reconciled with several genetic models which differ in the expected number of contributing causal variants. The ability to directly measure from DNA the degree of genetic variation among individuals has allowed for tests that could support or refute proposed genetic models. Initial experimental designs mimicked approaches that had been successful in identifying causal variants of single gene Mendelian disorders, that assume simple recessive or dominant inheritance, driven by the hypothesis that the genetics of common disease would be explained by relatively few genes harboring major mutations that individually or in combination cause disease. However, by-and-large, these experimental designs were not successful in leading to robust and replicable associations for polygenic traits, despite their success in mapping mutations causing Mendelian disease.
Sequencing of the human genome (2, 3) has led to the characterization of genomic variation between individuals and populations and expanded our genomic technologies, computational methods and experimental designs. One important innovation has been the development of the SNP-array (single nucleotide polymorphism array or SNP-chip) technology, whereby hundreds of thousands of DNA variants (mostly SNPs) can be scored robustly in a single assay (4). The low cost of this technology drove widespread uptake of the experimental design of genome-wide association studies (GWAS), which test for SNP allele frequency differences between affected (cases) and unaffected (controls) individuals and have revolutionized our understanding of the genetic basis of common polygenic disease (Box).
Box: Genome-wide Association studies (GWAS).
GWAS is an experimental design to detect population-wide associations between genetic markers and trait loci for applications in plant and animal breeding and for human disease (22). In human GWAS, the association between each of the hundreds of thousands to millions of SNPs identified within a sequenced pool of individuals and a complex trait is tested for statistical significance from a sample of individuals in the population. For common disease, the sample consists of cases and controls, whereas for a quantitative trait it can comprise a random set of individuals spanning the distribution of the trait.
GWAS relies on population-level linkage disequilibrium (LD, the non-random association between alleles at different genomic loci), between alleles at observed genetic markers and unobserved disease loci on the same chromosome. LD is created by geographic and evolutionary forces, in humans particularly those due to finite ancestral population size and natural selection (56). The amount of LD in most human populations is such that scoring a few hundred thousand well-chosen loci is sufficient to ‘tag’ the entire genome for unobserved common variants, even when the total number of common variants is of the order of tens of million. By design, GWAS leads to statistically significant associations between loci and traits. However, in practice neither the variant or variants that cause the statistical association signal nor the gene (or other functional unit) that is their target is identified, because the causal variant(s) may not be genotyped or imputed, because by chance a non-causal variant in LD (correlated) with a causal variant may have a stronger association signal, and because the actual causal variants may be far from the target gene, e.g. 1Mb for a SNP associated with body-mass index (48). This is a known but important limitation of the design.
GWAS has illuminated the polygenic nature of common diseases. Hundreds to thousands of genomic loci with a robust association with any one disease or trait have been identified (5). As GWAS sample sizes increase, the number of loci detected with statistical significance also increases in a linear manner (6). However, as sample sizes grow, the effect sizes of the new significantly associated loci tend to be become very small. In combination with the known limitations of GWAS (see Box), this complicates efforts to use raw results from GWAS to gain insights into disease pathogenesis and pathophysiology.
In addition to GWAS, genome-wide surveys of rare variants have been conducted either by population studies of the protein coding regions of the genome, the exomes, in case-control studies, or by enrichment of de novo variants in cases from family studies (7). Empirical results are consistent with those from GWAS, in that rare deleterious mutations in many genes can contribute to disease risk and that on average the exome contributes only a small proportion of risk in the population. Since every individual harbors de novo mutations, disease-relevance of the mutations requires the affected gene to have been identified in several families together with annotations of functional relevance, with loss of function variants of most relevance (8). The GWAS design is not restricted to disease and has become a powerful tool to study phenotypes such as gene expression, DNA methylation and histone modification, at the level of bulk tissue and individual cells.
Two decades ago, the debate about the genetic architecture of common disease was divided between proponents of a Mendelian model, in which a few rare variants of large effect size were segregating in the population, and advocates of a many-gene polygenic model, whereby the cumulative burden of risk variants in a person determines their risk (9). Halfway models postulated that effect sizes would be intermediate and that such variants would likely be found among protein altering polymorphisms (9). Many proponents of sequencing the human genome anticipated a rapid identification of disease genes with a subsequent fast track in developing therapeutics. These predictions were mostly wrong. Instead, GWAS studies provided empirical support for a highly polygenic architecture of common disease and evidence for a role of polygenic variation in rare diseases (Table 1).
Table 1.
Polygenic contributions in a range of genetic architecture contexts
| Genetic architecture context | Disease examples | Detail of polygenic role |
|---|---|---|
| Dominant Mendelian disease: a single variant (rare in the population) leads to high frequency of disease in some families | Huntington: a progressive neurodegeneration disorder defined by elevated number of copies of a CAG trinucleotide repeat located in the first exon of the HTT gene. | Age of onset is associated with the number of copies of the CAG repeat. The Genetic Modifiers of Huntington’s Disease Consortium have used the GWAS paradigm studying age of onset (42), identifying to date 21 associated loci, providing evidence of natural compensatory mechanisms with a polygenic architecture which are considered the most likely avenue to identify drug target opportunities for this classic Mendelian disease. |
| Lynch syndrome: a colorectal cancer defined by pathogenic variant in one of four genes: MLH1, MSH2, MSH6, PMS2. | In the UK Biobank the probability of colorectal cancer by age 75 was 2% for those without Lynch syndrome pathogenic variants and 35% for those with variants (0.15% in UKB). For those with Lynch syndrome variants the probability of colorectal cancer by age 75 ranged from 11% to 80% for the lowest to highest percentile of PRS constructed from colorectal cancer GWAS (57). | |
| Familial hypercholesterolemia (FH): a high cholesterol disease leading to high risk of coronary artery disease (CAD) defined by pathogenic variant in one of three genes: LDLR, APOB, PCSK9. | In the UKB the probability of CAD by age 75 was 13% for those without FH pathogenic variants and 41% for those with variants (0.67% of CAD cases). For those with FH variants the probability of CAD by age 75 ranged from 18% to 78% for the lowest to highest percentile of PRS constructed from CAD GWAS (57). | |
| Presumed monogenic mutations | Developmental delay disorder: children with neurodevelopmental disorders with clinical features sufficiently severe to implicate a de novo or inherited monogenic mutation. | As well as reporting severe/moderate/mild intellectual disability and high rates of ASD (17%), patients in the Deciphering Developmental Delay (DDD) study (n = 6,987) were on average 0.72 SD shorter, weighed 0.15 SD less and had a head circumference that was 1.20 SD smaller than the age and sex-adjusted population (58). In addition to these mean differences associated with developmental delay, variation in birth weight, height and ASD within the DDD cohort was highly associated with their corresponding PRS. Finally, common variants, which are not correlated with monogenic mutations, explained about 8% of risk of DDD. Therefore, polygenic variation can affect both the risk and clinical features in a disorder that is usually considered to be monogenic. |
| Large effect variants in common disease | Breast, ovarian and prostate cancers: pathogenic/likely pathogenic variants in BRCA1 and BRCA2 are found in a small proportion of those with these cancers. | In the UKB the probability of breast cancer (BC) by age 75 was 10% in those without pathogenic or likely BC pathogenic variants (found in 3.1% of the 17,344 controls), compared to 35% in those with such variants (found in 9.1% of the 1,1920 women with breast cancer). For those with pathogenic variants the probability of BC by age 75 ranged from 13% to 76% for the lowest to highest percentile of PRS constructed from BC GWAS (57). Qualitatively similar results have been reported for ovarian and prostate cancers (59, 60). |
| Glaucoma: Pathogenic variants in the myocilin MYOC gene account for 2–4% of primary open-angle glaucoma, the most common subtype of glaucoma; p.Gln368Ter is the most common variant | In the UKB, of those with the pathogenic Gln368Ter MYOC variant (n=965), those in the lowest tertile of PRS for glaucoma had 2% risk of glaucoma by age 60, but the risk was six-fold higher in those in the top tertile of PRS (55). | |
| Disease definition impacts genetic architecture | ASD/developmental delay | In psychiatric and behavioural disorders, diagnoses are allocated on the observable clinical symptoms which overlap only partially with genetic architecture. For example, ASD is 3–4 times more common in boys, indicating a female protective effect. As a result, girls with ASD have higher mean ASD PRS than boys. Girls with ASD are more likely to have large effect or de novo variants than boys, as boys with these variants are more likely to be diagnosed with developmental delay. |
| Common disorders of onset in early adulthood, such as schizophrenia and type 2 diabetes | While large effect variants are observed in late onset (post-reproductive years) disorders such as Alzheimer Disease (APOE), ALS (SOD1) and age-related macular degeneration (CFH), such variants are notably absent from common adult onset behavioural/psychiatric (61) or metabolic disorders (62). Large effect variants may lead to earlier clinical presentations resulting in childhood-onset syndromic diagnoses. |
ASD: autism spectrum disorders; PRS: polygenic risk score; SD: standard deviation; UKB: UK biobank – a volunteer cohort of 500,000 people recruited aged 50–70 years.
A spectrum of genetic architectures
Genetic architecture encompasses the total number of functional variants and the frequency and effect size of each of them that affect a trait or disease. Causal variants of common diseases have frequencies across a wide allelic spectrum, from rare to common. Many genetic effects act in an additive manner, with respect to an underlying disease liability, with disease resulting from the accumulated burden of risk factors (Fig. 1), resulting in non-additive (e.g., multiplicative) determination of whether disease occurs or not. Empirical results from GWAS have shown that although common diseases are highly polygenic, their genetic architecture differ. In particular, some diseases (e.g., coronary artery disease, type 2 diabetes) have risk variants of large effects that segregate at appreciable frequency in some populations (10) combined with a polygenic tail, whereas for other diseases (e.g., schizophrenia) no such variants have been detected (Table 1). The disease burden (liability) model is consistent with observed risk for diseases where there are variants of large effect. For example, the consequence of large effect variants can be amplified or attenuated by polygenic risk background and other factors contributing to risk (Table 1). In general, common adult-onset disorders are not due to rare large effect variants, as such mutations often lead to more severe and early clinical symptoms, diagnosed in childhood. A wide range of polygenic genetic architectures underlie diseases (11). Thus, the genetic architecture of disease, whether rare or common, is consistent with the hypothesis that all diseases are polygenic, but some have mutations of large effect (Table 1).
Fig. 1. Visualizing the polygenicity of common disease.
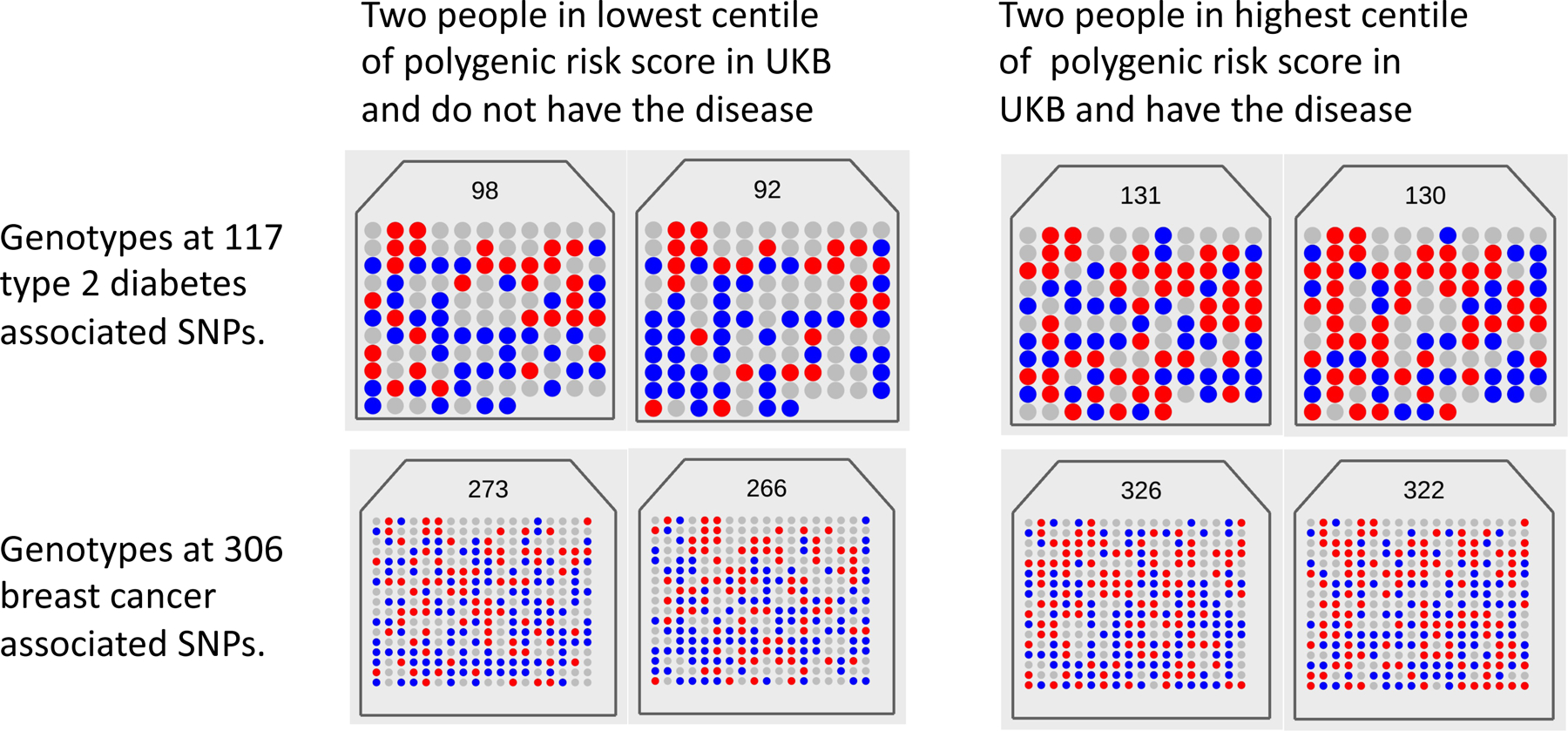
Each panel shows the number of risk alleles (0 = gray, 1 = blue, 2 = red) for an individual from the UK Biobank (UKB) at each of 117 type 2 diabetes (T2D) and 306 breast cancer (BC) disease-associated loci. The total counts of risk alleles for the individual is listed at the top of each panel. For each disease, two individuals were selected with disease and in the top 1% of the count of the number of risk alleles, and two controls without disease from the bottom 1%. The panels show that (i) every individual has a unique risk profile, (ii) controls carry risk alleles at many loci and (iii) cases have on average only a small increase in the risk variant burden. Selecting the top and bottom 1% of this population on the number of risk variants gives mean numbers of risk variants of 131 and 98 for T2D and 325 and 269 for BC, respectively, with disease prevalence of 7% versus 2% (T2D) and 19% versus 2% (BC). Hence, although the mean number of risk variants between cases and controls is generally small, individuals who have a high burden of risk variants have greatly increased risk of disease. Polygenic scores are not diagnostic; most individuals in the top 1% of the PRS distribution do not have disease and, conversely, some in the bottom 1% do.
Polygenicity and natural selection
Recent studies (11–13) have used theory, simulations and empirical analyses to highlight negative selection as a prominent evolutionary force shaping the degree of polygenicity of diseases as well as its variation across genomic loci. Negative selection primarily acts by removing alleles with a strong deleterious effect on fitness from the population. Therefore, negative selection directly controls the number of mutations segregating in the population, thus imposing an upper bound on disease polygenicity depending on a variant’s relation to fitness. More generally, negative selection also prevents mildly deleterious alleles on fitness from reaching high frequency in the population (14). Thus, a signature of negative selection induces an inverse relationship between the magnitude of SNP effects and their minor allele frequency (13, 15). Statistical methods using GWAS summary statistics have exploited this property to show evidence of negative selection for a wide range of traits and disease, including late onset disease (12, 13, 16). However, as GWAS only partially inform on causal variants, those methods have limited ability to quantify a direct relationship between the strength of selection and the degree of polygenicity. Indeed, the estimated number of SNPs with a non-zero effects on a trait (12, 17, 18), another commonly used statistic to quantify polygenicity, is correlated with but is not an unbiased estimator of the number of causal variants. Importantly, the bias of that estimator depends on the disease heritability but also on the strength of selection (12). Other factors influencing bias are properties of the study design, such as sample size and criteria for selecting SNP density and frequency (17). Despite all those limitations, estimates of the number of non-zero effect SNPs suggests that common diseases are, on average, less polygenic than other traits including anthropometric and brain-related phenotypes. Schizophrenia is a well-recognized outlier, with one of the largest degree of polygenicity reported across traits and diseases (11, 18). One feature of the observed polygenicity of complex traits, including common disease, is that it tends to be distributed widely across the entire genome. It implies that there are many sites in the genome that, when perturbed through mutation, can contribute to a particular trait or disease.
Other methods (19–21) have also leveraged the availability of large sets of GWAS associated SNPs to quantify small changes in risk allele frequencies in response to new selection pressures. These methods have shown evidence of directional selection on health-related traits such as blood pressure and body mass index (20). Furthermore, the refined characterization of genes that are constrained by natural selection has provided new ways to annotate GWAS findings, thereby improving their biological interpretation.
In summary, recent contributions shed new light on the causal link between natural selection and polygenicity, and improved GWAS-based approaches, to refine detection and quantification of the effect of natural selection on the genetic architecture of complex diseases. We anticipate that quantification of polygenicity from whole-genome sequence (WGS) data will reveal more detailed resolution and more reliable predictions regarding the evolution of complex disease (7).
Polygenic (risk) scores continue to mature
An exciting development in human genetics has been the use of GWAS data to create polygenic predictors for complex traits and quantify their association with disease and other outcomes in independent data. A polygenic (risk) score (PRS) is a sum of the number of risk variants at multiple genomic loci, weighted by their effect size which have been estimated from independent data. Harnessing genome-wide linkage disequilibrium between marker genotypes and trait loci and perform a genomic prediction is was first suggested in the context of plant and animal breeding and has been used widely in agriculture (22). Polygenic prediction is entirely based upon association, just like GWAS. The increase in sample sizes in GWAS has facilitated polygenic predictors that have effect sizes of risk as large as known monogenic mutations that are clinically actionable (10) (Fig. 2). Since genetic factors are not the only contributors to risk of common disease, polygenic predictors are not expected to be diagnostic. Nonetheless, they have utility similar to other predictors used in medicine. When more of the trait variance is accounted for in polygenic predictors, the differences in lifetime prevalence between individuals at high and low predicted risk should increase and improve the accuracy of polygenic prediction, by better modelling of the distribution of effect sizes (23–25). Resources with polygenic score information have been created to ensure reproducibility in research and translation (26, 27).
Fig. 2. Risk as a function of polygenic predictor percentiles.
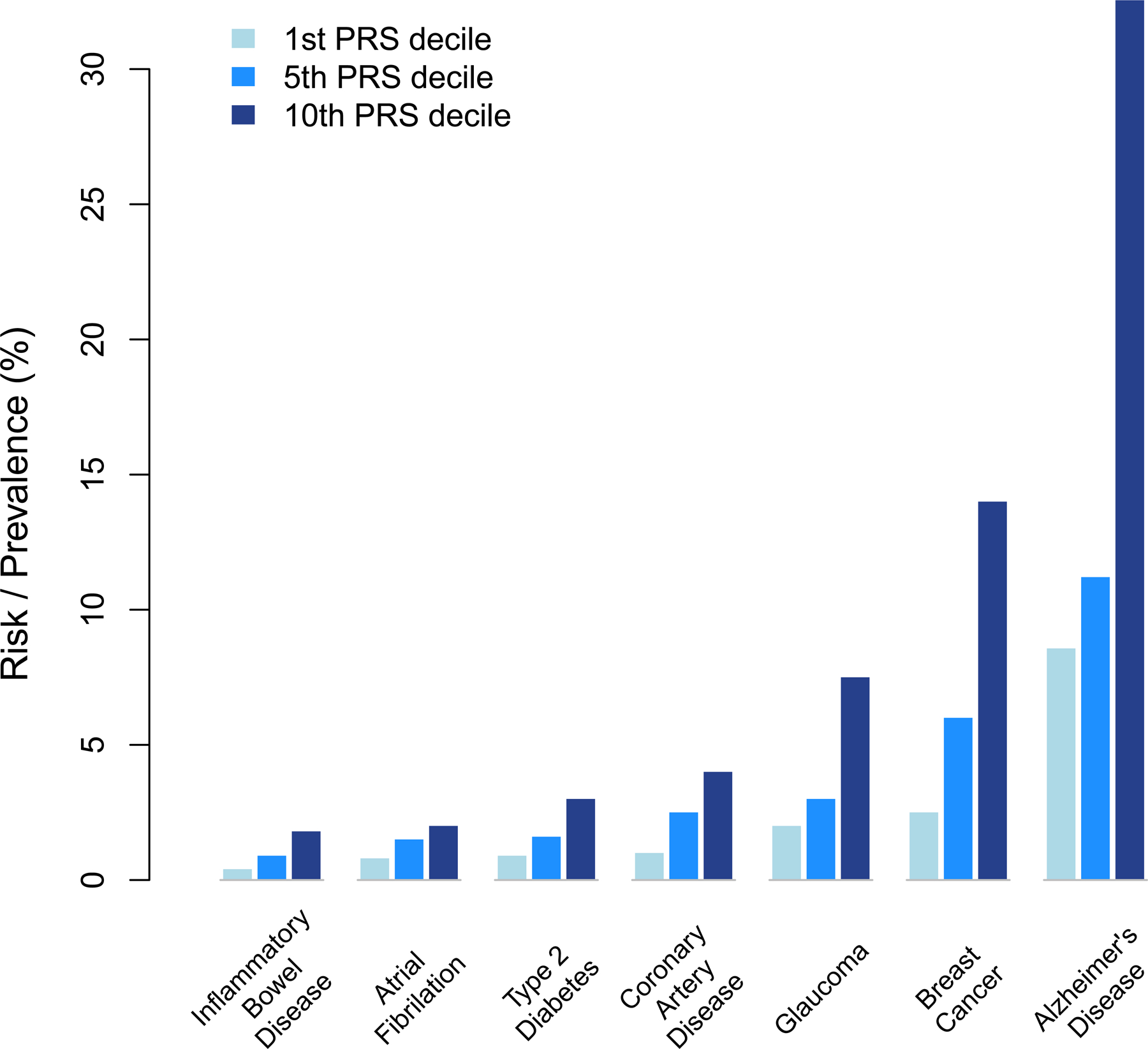
Risk or prevalence of inflammatory bowel disease, atrial fibrillation, type 2 diabetes, coronary artery disease, glaucoma, breast cancer and Alzheimer’s Disease are shown as function of the 1th, 5th and 10th decile of their polygenic risk score (PRS) predictor. Glaucoma and breast cancer prevalence were measured at age 70 and 60, respectively. Prevalence of inflammatory bowel disease, atrial fibrillation, type 2 diabetes, and coronary artery disease were measured in the UK Biobank at age ranging from 40 to 70; and prevalence of Alzheimer’s disease from 65 years. Improvements in the accuracy of polygenic scores will increase the difference in lifetime risks across the percentiles. Data from (10, 53–55). Rare BCRA1/BRAC2 mutations affecting breast cancer risk are not captured in the common variant polygenic score. For Alzheimer’s Disease, the common APOE e4 variant explains approximately half of the risk in the 10th decile group (53).
Polygenicity spurs new experimental designs
Past studies in human disease genetics relied on family data, whereas the current focus is on populations (e.g., GWAS). However, the distinction is artificial since populations consist of individuals that are related to each in some way, either with recent or more distant common ancestors. Combining the power of population genomics studies with the naturally occurring controls that pedigree studies offer, can address the degree of heritable vs. environmental impacts on phenotype (nature-nurture) with new designs, when previously environment and genetic effects were difficult to disentangle. For example, GWAS data on families were used to separate a direct (causal) genetic effect from an indirect genetic effect that reflects the parental “nurturing” environment (28). We anticipate that such designs are important for understanding common diseases and their risk factors, especially those with a behavioral component. The availability of tens of thousands of GWAS-associated SNPs offers opportunities to answer scientific questions about natural selection, human behavior and causality. For example, as the predictive accuracy of polygenic scores increase, we can now quantify the flux of trait-associated alleles between geographical areas and thereby elucidate some of the correlates of population migration (29, 30). More widely, there is a growing trend to embed genomics into social sciences research (31), requiring careful consideration of the implications and applications of such studies (32).
Another exciting new experimental design facilitated by GWAS discoveries is Mendelian Randomization, which attempt to mimic a randomized control trial by using SNPs that are associated with an modifiable risk factor (e.g., body-mass index) to quantify its causal effect on disease risk (e.g., type 2 diabetes) (33). Testing for putative causality between exposure and disease is important in epidemiology and public health because it leads to evidence-based policy interventions and health advice. Mendelian randomization is a cost-effective way to prioritize hypotheses that could then be addressed by formal but expensive clinical trials, or to provide causal evidence when such trials would be unethical. More generally, the widespread availability of GWAS summary statistics for diseases and other complex traits, including molecular omics phenotypes, has led to new powerful analysis paradigms by mimicking experiments where all traits would be measured on all individuals, yet without the necessity of individual-level data (34).
Association studies to date have mostly relied on SNP-chip technology combined with statistical imputation using a sequenced reference panel. While successful for populations of European ancestry, they have been less so for other less sampled populations (35); likely because of differences in allele frequencies and linkage disequilibrium (LD) patterns between human populations (35). Large biobank-style studies across multiple ancestries are needed to understand human gene-disease associations within and between populations and improve polygenic predictors across ancestries.
Whole genome sequencing offers more information for GWAS analysis to discover disease loci because all genomic variants in a sample are observed, in contrast to GWAS-by-chip where only a small fraction of all possible variants are genotyped, and additional variants are imperfectly imputed using a statistical algorithm. Therefore, GWAS-by-WGS can, in principle, improve the ability to detect associations between rare sequence variants and disease. However, the relationship between effect size and allele frequency (12) implies that sample sizes to detect rare variants from WGS data would need to be very much larger than the detection of common variants through GWAS-by-chip, because detection power is a function of both the effect size and allele frequency and also on the total number of variants tested, which is much larger in GWAS-by-WGS. Burden tests, whereby the number and composition of multiple rare variants in a gene are jointly assessed for disease association, may overcome some of these statistical limitations. Sufficiently powered studies performing GWAS-by-WGS will improve polygenic risk scores, because the combination of rare and common variants will explain more of the genetic variation contributing to the trait.
From genetic to biological models
Researchers have, understandably, tried to simplify the complexity of polygenicity and proposed biological models that are compatible with a large number of genomic sites that are associated with common disease. One such model proposes that the diagnostic label of a given disease can reflect multiple distinct underlying biological pathways, each polygenic (36). In this way, having high polygenic risk for any one of the underlying biological pathways is sufficient to lead to disease. This model implies that gene-gene interactions on the scale of liability for the labelled disease are important, which appears inconsistent with subsequent empirical results which point to additivity on this scale. Additivity on the liability scale is an important property as it implies that effect sizes are transferable across individuals within a population, so that disease can be studied and predicted irrespective of the specific combination of risk alleles in a person (Fig. 1).
More recently, the omnigenic model was proposed to reconcile polygenicity with biological function (37). The model proposes a mechanistic explanation for why so many variants, spread across the genome can be responsible for the genetic variation between people observed for complex traits and common disease. In brief, genetic variants with proximal, cis-effects on peripheral genes perturb the regulation of a smaller class of core genes via distal, trans-regulatory networks. Thus, studying peripheral genes help unravel the biology of a trait, because they explain the trans-regulatory context within which core genes sit, but the disease-specific core genes are the most important for disease-specific research. One component of the hypothesis, the identification of core genes has been tested for three molecular traits measured in blood, urate, IGF-1 and testosterone concluding that association signals were interpretable in terms of the physiology of the traits and concentrated near core genes and core pathways (38). However, even for these molecular traits it was not possible to test the trans-regulatory network component of the hypothesis which requires larger sample sizes than currently available, suggesting that it may be impossible to test the omnigenic model for complex traits such as common disease.
A disease-specific palette model was proposed for type 2 diabetes (39), as a simple visualization of the polygenic model. This model suggests that although there are multiple biological processes involved in diabetes (such as insulin secretion, insulin resistance, dyslipidemia), with multiple underlying environmental risk factors, individuals with diabetes are likely to have a risk burden across many biological and environmental categories. Many common diseases are chronic, relapsing and remitting. A network dyshomeostasis model has been proposed to describe the cumulative contribution of genetic variants and the environmental factors impacting the gut microbiome that together lead to risk of inflammatory bowel disease (40). Statistically, the omnigenic, palette and dyshomeostasis models are consistent with the additive liability threshold model of common disease, in which probability of disease is multiplicative across all risk factors, both genetic and environmental.
From variant discovery to disease biology
Many GWAS results confirm biology that is already known or suspected, for example the role of sodium / potassium ATPase in type 2 diabetes (41), which drives the expectation that GWAS results can add to our understanding of biology. In general, the biological inference is more straightforward for individual molecular traits such as blood metabolites than for common diseases. We currently have few maps linking mechanism to medicine. For example, while the causal role of Huntington’s Disease (HD) was identified as an expanded genomic repeat in 1993, this knowledge has still not led to new treatments. However, GWAS studies are now being used to identify polygenic variants that provide protection for onset of HD on the basis of the age of onset (42). GWAS of Crohn’s Disease (CD) (43, 44) helped define the role of autophagy in this disease and catalyzed research to define the roles of key genes in autophagy and the role of autophagy in the pathophysiology of CD. Inflammatory bowel diseases (IBD) GWAS led the field in biological understanding and translation (40), perhaps because the diseases are well-defined and less heterogeneous with affected tissue easier to biopsy than in other common diseases. Such research has identified genetic variants converging on a multitude of cellular and molecular pathways that regulate homeostasis of the mucosal immune system interacting with the gut microbiome (40).
In general, however, it has been difficult to learn new biology solely from GWAS results, because of the large number of associated variants, their small effect sizes, and the inherent challenges in moving from the associated SNPs to causal variants to causal genes (or other functional units) and towards the driving disease biology. Also, the effects from associated SNPs to trait may transit through intermediate phenotypes, such as transcript levels (8) (Fig. 3). Effect sizes of SNPs estimated in GWAS have been integrated with molecular phenotypes such as gene expression, chromatin accessibility and other functional annotations (45) in enrichment analyses, as a way to learn more about the most proximal molecular mechanisms affected by GWAS signals (Fig. 3). Greater understanding of the nature of DNA secondary structures (46) have provided insights into how a SNP thought to be causal for an established association in or near one gene may exert effects on a gene located at a considerable distance within the genome (47, 48). Furthermore, the impacts and interactions of a panoply of RNA-based functional units is still lacking. Thus, the transition from causal SNP to causal gene or protein remains challenging.
Fig. 3: Causal path from a genotype to disease via multiple molecular mediators.
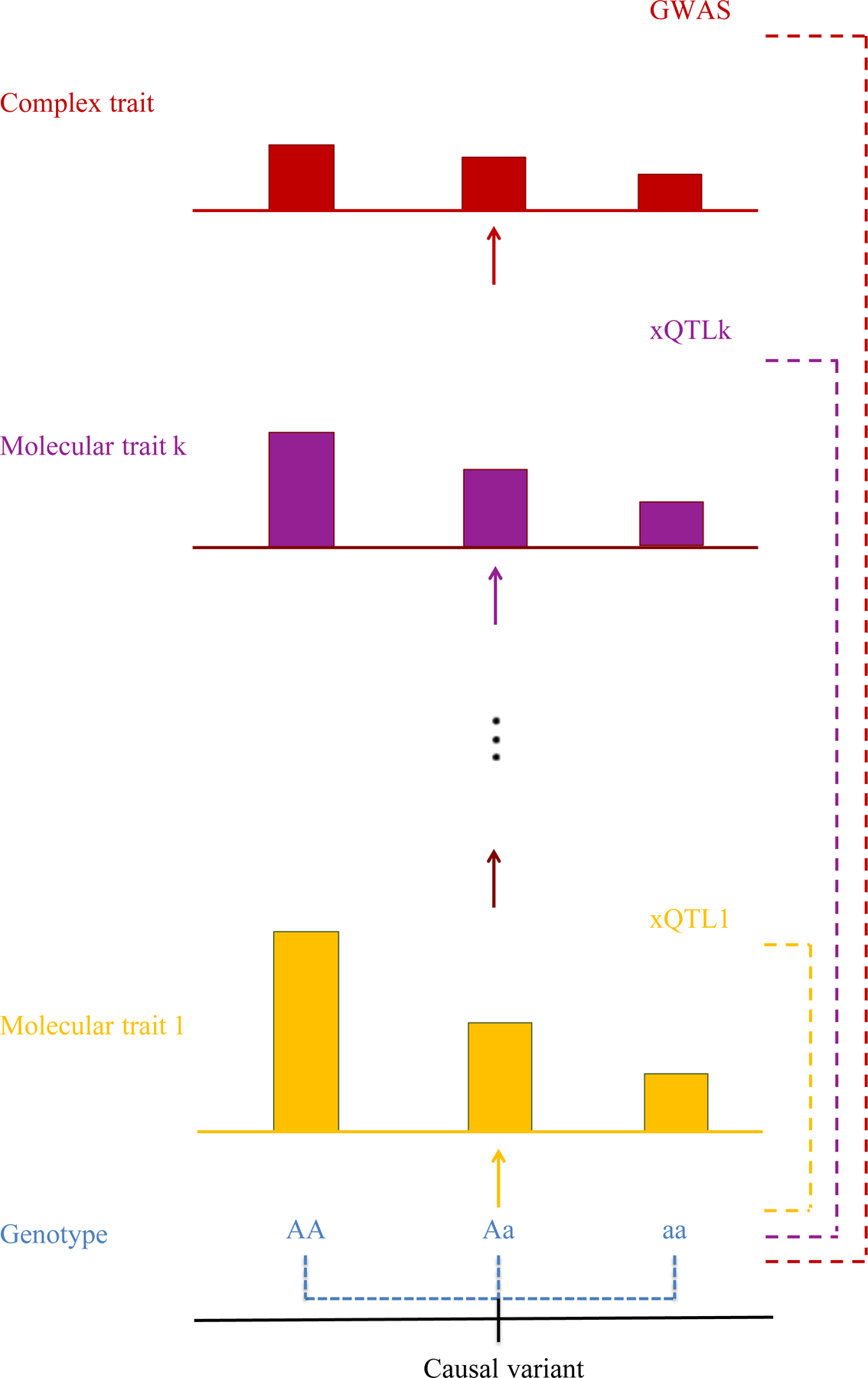
The red dotted line shows that a genotype is associated with disease (hypertension), the primary observation from GWAS. The yellow and blue dotted lines show that the same genotype is associated with multiple molecular mediators (1 to k). The genotype is a quantitative trait locus (QTL) for moderators x, where x can encompass a wide range of mechanisms, included gene expression (eQTL), splicing (sQTL), epigenetic marks such as methylation (mQTL) and protein levels (pQTL), both in cis and trans. The schematic shows how a causal variant can contribute to disease risk through many different routes. There could be a single molecular mediator (k = 1, e.g., gene expression) or multiple ones, and if there are multiple molecular traits involved the order may be different for different causal variants. Finally, as observed with empirical data, effect sizes tend to become smaller (from top to bottom, as shown) because at each step additional factors come into play that dilute the contribution from the genetic variant. Researchers use statistical co-localization and mediation analyses to find the most likely causal paths using GWAS data on disease and moderators. Data on additional molecular mediators are being collected to fill in missing links in the genotype to disease causal pathway.
Similar challenges are faced in understanding how genes alter human biology to drive disease susceptibility. Given the number of genes whose functions are unknown or poorly annotated, the nature of some of these challenges are clear. Knowledge of when and where each gene is most uniquely needed for maintenance of healthy homeostasis is required (49, 50). Studies in Mendelian diseases with large-effect mutations have revealed complexities in cell- and tissue-specific effects of genes on human biology affecting disease risk; greater challenges in elucidating similar mechanisms for genes in which the genetic variation contributing to disease risk are substantially smaller await.
From our current vantage point it is clear that genetic studies of common human diseases need more tools to directly investigate the biological links between genetic variants, environment and disease. When individually small effects coalesce to have organism-wide consequences for health and disease, they do so by working through biological systems that we are learning to characterize at all levels of scale from nuclear, to cellular, to tissue, and to organism.
Given the preponderance of non-coding variation associated with polygenic liability to human disease, research is needed to elaborate and measure, over a variety of contexts and at scale, the many ways that RNA affects protein abundance. These approaches are already starting to emerge. For example, through an integrated analysis of genome, transcription and DNA methylome of kidneys, 479 GWAS associations between SNPs and blood pressure could be connected to 1,038 genes expressed in kidneys, identifying druggable targets for hypertension (51).
There are many drugs currently used with success for common disorders, such as lithium for bipolar disorder, for which the biological mechanism is not understood. Causal inference through Mendelian Randomization, particularly with regard to using genetic information to identify potential drug targets (52), could lead to drug repurposing and partially sidestep the need to link GWAS SNPs to mechanism.
As genetics and genomics partner with medical investigations in the diagnosis and treatment of patients, we anticipate many medical centers becoming a source of data to probe how genetic variation relates to the broad phenotypic spectrum of humanity. Challenges lay ahead, but tackling these complex problems provides opportunities to improve all human health. Despite the all-too-human tendency to oversimplify the complexity of biology, recent progress reminds us that we can improve not just our global understanding of the biology of common human diseases, but also our practical ability to contribute meaningfully to their treatment even as we discover and acknowledge greater complexity in the underlying genetic models.
Funding:
The Australian Research Council (FL180100072 and DE200100425), the Australian National Health and Medical Research Council (1113400 and 1173790) and the National Institutes of Health (U01HG0090086, U54MD010722, R01MH113362).
We thank Tian Lin and Yang Wu for help with the figures, and three reviewers for helpful comments and suggestions. Figures 1 and 2 were created using data from the UK Biobank, under Project number 12505.
Footnotes
Competing interests: none. Due to space limitations, we were not able to cite many other relevant papers in the literature.
REFERENCES AND NOTES
- 1.Yang J, Visscher PM, Wray NR, Sporadic cases are the norm for complex disease. Eur J Hum Genet 18, 1039–1043 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Venter JC et al. , The sequence of the human genome. Science 291, 1304–1351 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Lander ES et al. , Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Kim S, Misra A, SNP genotyping: technologies and biomedical applications. Annu Rev Biomed Eng 9, 289–320 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Visscher PM et al. , 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet 101, 5–22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Canela-Xandri O, Rawlik K, Tenesa A, An atlas of genetic associations in UK Biobank. Nat Genet 50, 1593–1599 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karczewski KJ et al. , The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lappalainen T, MacArthur D, From variant to function in human disease genetics. Science, (2021). [DOI] [PubMed] [Google Scholar]
- 9.Risch NJ, Searching for genetic determinants in the new millennium. Nature 405, 847–856 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Khera AV et al. , Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 50, 1219–1224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Connor LJ et al. , Extreme Polygenicity of Complex Traits Is Explained by Negative Selection. The American Journal of Human Genetics 105, 456–476 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeng J et al. , Widespread signatures of natural selection across human complex traits and functional genomic categories. Nat Commun 12, 1164 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schoech AP et al. , Quantification of frequency-dependent genetic architectures in 25 UK Biobank traits reveals action of negative selection. Nat Commun 10, 790 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eyre-Walker A, Genetic architecture of complex traits and its implications for fitness and genome-wide association studies. Proc Natl Acad Sci U S A 107, 1752–1756 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gazal S et al. , Functional architecture of low-frequency variants highlights strength of negative selection across coding and non-coding annotations. Nat Genet 50, 1600–1607 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Speed D et al. , Reevaluation of SNP heritability in complex human traits. Nat Genet 49, 986–992 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Qi G, Park JH, Chatterjee N, Estimation of complex effect-size distributions using summary-level statistics from genome-wide association studies across 32 complex traits. Nat Genet 50, 1318–1326 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Holland D et al. , Beyond SNP heritability: Polygenicity and discoverability of phenotypes estimated with a univariate Gaussian mixture model. PLoS Genet 16, e1008612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Field Y et al. , Detection of human adaptation during the past 2000 years. Science 354, 760–764 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Speidel L, Forest M, Shi S, Myers SR, A method for genome-wide genealogy estimation for thousands of samples. Nat Genet 51, 1321–1329 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stern AJ, Speidel L, Zaitlen NA, Nielsen R, Disentangling selection on genetically correlated polygenic traits via whole-genome genealogies. Am J Hum Genet 108, 219–239 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wray NR, Kemper KE, Hayes BJ, Goddard ME, Visscher PM, Complex Trait Prediction from Genome Data: Contrasting EBV in Livestock to PRS in Humans: Genomic Prediction. Genetics 211, 1131–1141 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kichaev G et al. , Leveraging Polygenic Functional Enrichment to Improve GWAS Power. Am J Hum Genet 104, 65–75 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lloyd-Jones LR et al. , Improved polygenic prediction by Bayesian multiple regression on summary statistics. Nat Commun 10, 5086 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ge T, Chen CY, Ni Y, Feng YA, Smoller JW, Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat Commun 10, 1776 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lambert SA et al. , The Polygenic Score Catalog as an open database for reproducibility and systematic evaluation. Nat Genet 53, 420–425 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Becker J et al. , Resource profile and user guide of the Polygenic Index Repository. Nat Hum Behav, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kong A et al. , The nature of nurture: Effects of parental genotypes. Science 359, 424–428 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Abdellaoui A et al. , Genetic correlates of social stratification in Great Britain. Nat Hum Behav 3, 1332–1342 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Haworth S et al. , Apparent latent structure within the UK Biobank sample has implications for epidemiological analysis. Nat Commun 10, 333 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harden KP, Koellinger PD, Using genetics for social science. Nat Hum Behav 4, 567–576 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turley P et al. , Problems with Using Polygenic Scores to Select Embryos. N Engl J Med 385, 78–86 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davies NM, Holmes MV, Davey Smith G, Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ (Clinical research ed.) 362, k601 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pasaniuc B, Price AL, Dissecting the genetics of complex traits using summary association statistics. Nat Rev Genet 18, 117–127 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin AR et al. , Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet 51, 584–591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zuk O, Hechter E, Sunyaev SR, Lander ES, The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci U S A 109, 1193–1198 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boyle EA, Li YI, Pritchard JK, An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 169, 1177–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sinnott-Armstrong N, Naqvi S, Rivas M, Pritchard JK, GWAS of three molecular traits highlights core genes and pathways alongside a highly polygenic background. Elife 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCarthy MI, Painting a new picture of personalised medicine for diabetes. Diabetologia 60, 793–799 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Graham DB, Xavier RJ, Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature 578, 527–539 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sweeney G, Klip A, Regulation of the Na+/K+-ATPase by insulin: why and how? Mol Cell Biochem 182, 121–133 (1998). [PubMed] [Google Scholar]
- 42.Genetic Modifiers of Huntington’s Disease Consortium, CAG Repeat Not Polyglutamine Length Determines Timing of Huntington’s Disease Onset. Cell 178, 887–900 e814 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hampe J et al. , A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet 39, 207–211 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Rioux JD et al. , Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 39, 596–604 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finucane HK et al. , Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ho JW et al. , Comparative analysis of metazoan chromatin organization. Nature 512, 449–452 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smemo S et al. , Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507, 371–375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Claussnitzer M et al. , FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N Engl J Med 373, 895–907 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gonzalez-Serrano LE, Chihade JW, Sissler M, When a common biological role does not imply common disease outcomes: Disparate pathology linked to human mitochondrial aminoacyl-tRNA synthetases. J Biol Chem 294, 5309–5320 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sissler M, Gonzalez-Serrano LE, Westhof E, Recent Advances in Mitochondrial Aminoacyl-tRNA Synthetases and Disease. Trends Mol Med 23, 693–708 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Eales JM et al. , Uncovering genetic mechanisms of hypertension through multi-omic analysis of the kidney. Nat Genet 53, 630–637 (2021). [DOI] [PubMed] [Google Scholar]
- 52.Hyman MC et al. , Genetically Predicted Blood Pressure and Risk of Atrial Fibrillation. Hypertension 77, 376–382 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Q et al. , Risk prediction of late-onset Alzheimer’s disease implies an oligogenic architecture. Nat Commun 11, 4799 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mavaddat N et al. , Polygenic Risk Scores for Prediction of Breast Cancer and Breast Cancer Subtypes. Am J Hum Genet 104, 21–34 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Craig JE et al. , Multitrait analysis of glaucoma identifies new risk loci and enables polygenic prediction of disease susceptibility and progression. Nat Genet 52, 160–166 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Slatkin M, Linkage disequilibrium--understanding the evolutionary past and mapping the medical future. Nat Rev Genet 9, 477–485 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fahed AC et al. , Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat Commun 11, 3635 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Niemi MEK et al. , Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature 562, 268–271 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuchenbaecker KB et al. , Evaluation of Polygenic Risk Scores for Breast and Ovarian Cancer Risk Prediction in BRCA1 and BRCA2 Mutation Carriers. J Natl Cancer Inst 109, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lecarpentier J et al. , Prediction of Breast and Prostate Cancer Risks in Male BRCA1 and BRCA2 Mutation Carriers Using Polygenic Risk Scores. J Clin Oncol 35, 2240–2250 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gandal MJ, Leppa V, Won H, Parikshak NN, Geschwind DH, The road to precision psychiatry: translating genetics into disease mechanisms. Nat Neurosci 19, 1397–1407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fuchsberger C et al. , The genetic architecture of type 2 diabetes. Nature 536, 41–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]