

Significance
This study describes a model for the severe phenotype of patients with Neurofibromatosis Type 1 (NF1) caused by specific missense mutations. We show, that these mutations act as dominant negative mutants, through dimerization with wild-type neurofibromin, and use our newly solved cryo-EM structure of the neurofibromin dimer to explain why these mutations disrupt protein structure and to predict and validate other patient variants. Until now, there have been very few genotype–phenotype relationships for the NF1 disease, despite the identification of more than two thousand pathogenic variants. Mutations at codons 844 to 848 exhibit a severe phenotype, but the mechanism for their action was not known. This work has important implications for clinical management and understanding neurofibromin loss-of-function in disease.
Keywords: NFI, cryo-EM, neurofibromatosis type I
Abstract
The majority of pathogenic mutations in the neurofibromatosis type I (NF1) gene reduce total neurofibromin protein expression through premature truncation or microdeletion, but it is less well understood how loss-of-function missense variants drive NF1 disease. We have found that patient variants in codons 844 to 848, which correlate with a severe phenotype, cause protein instability and exert an additional dominant-negative action whereby wild-type neurofibromin also becomes destabilized through protein dimerization. We have used our neurofibromin cryogenic electron microscopy structure to predict and validate other patient variants that act through a similar mechanism. This provides a foundation for understanding genotype–phenotype correlations and has important implications for patient counseling, disease management, and therapeutics.
Neurofibromin is a GTPase activating protein (GAP) that accelerates the intrinsic GTPase activity of RAS proteins, converting them from an active GTP (guanosine-5'-triphosphate)-bound state to an inactive GDP (guanosine diphosphate)-bound one (1). It is targeted to the membrane through interaction with SPRED (Sprouty-related, EVH1 domain-containing) proteins (2). In the absence of this negative regulation, levels of RAS-GTP and activity of downstream pathways are elevated, leading to profound effects on cell proliferation, differentiation state, and cell survival. Dysregulation of RAS signaling through NF1 loss is commonly found in a range of malignancies including those of skin, lung, and myeloid origin (3).
NF1 is part of the RASopathies group of developmental syndromes which are driven by hyperactivation of the RAS-ERK (extracellular signal-related kinase) pathway (4) NF1 patients exhibit a range of symptoms which usually begin during infancy and develop over time into adulthood (5) Common features include café au lait modules (CALMs), cutaneous neurofibromas, and Lisch nodules. Some patients develop more severe manifestations including intellectual disability, skeletal abnormalities, cardiac defects, plexiform neurofibromas, optic pathway gliomas, and other malignancies. There are over 2,800 individual mutations identified in NF1 patients but relatively few reports have correlated genotype with phenotype. More notably, patients with p.Met992del exhibit a mild phenotype, (6, 7) those with p.Arg1276, p.Lys1423, and p.Arg1809 substitutions present Noonan syndrome-like features including cardiovascular anomalies (8, 9), while patients carrying NF1 microdeletions exhibit a more severe phenotype including increased prevalence of neurofibromas and skeletal abnormalities (10). Substitution mutations clustering at codons 844 to 848 are linked to a particularly severe phenotype and cause an increased incidence of major external and spinal plexiform neurofibromas, skeletal abnormalities, and Noonan syndrome features amongst other symptoms (11).
Results
In our efforts to better understand how neurofibromin is regulated, we discovered that neurofibromin exists as a high-affinity dimer in cells (12); however, the biological implications of this complex were more difficult to explain. Hypothesizing that neurofibromin dimers may have a role in disease, we tested several pathogenic point mutations in NF1 but found that they were all capable of binding to wild-type protein. We noticed that missense mutations in residues 844 to 848 (Fig. 1A) reduce protein expression when compared to wild type (Fig. 1B) in NF1 knockout 293T cells. This is consistent with previous reports of lower mutant protein expression observed in a G848R mouse model of NF1 (13, 14) and in cells transiently expressing mouse Nf1 cDNA (complementary DNA) with L847P and G848R (15). Despite lower levels of expression, these mutants are still able to form heterodimers with wild-type protein (Fig. 1C) and maintain their interaction with SPRED1 (Fig. 1D). Levels of RAS-GTP in NF1−/− cells were reduced upon expression of these mutants indicating that they retain GAP activity (Fig. 1E). We wondered whether the mechanism behind low protein expression was due to targeting for degradation. We observed that 844 to 848 mutants are highly ubiquitylated compared to wild-type neurofibromin and coimmunoprecipitate with increased amounts of HSP70 (Fig. 1F), an indicator that the cell is responding to protein misfolding (16). To verify that the effects on expression levels were not an artifact of transient transfection, we generated a doxycycline (Dox)-inducible expression system in the NF1 knockout 293T cells. We previously thought that it was not possible to efficiently transduce full-length NF1 lentivirus due to its size. However, we successfully introduced a fluorescent epitope-tagged (HA-mScarlet) WT and L847P NF1 (SI Appendix, Fig. S1) which was expressed in a Dox dose-dependent manner (Fig. 1G). We observed that the L847P mutant expressed considerably less than the WT under the same conditions, confirming that the previous response was not transient-transfection-specific. To rule out the possibility that NF1 mutant effects on stability were 293T-specific and to examine the phenomenon in an NF1 disease-relevant biological system, we transduced a previously characterized immortalized peripheral neurofibroma-derived Schwann cell line (ipNF95.11b C) (17) in which we had knocked out NF1. We saw the same phenotype as in the 293T, and given the corroborating data from the aforementioned mouse model, we concluded that 844 to 848 mutants are less stable in a variety of biological scenarios. We also found that these mutations completely abolish soluble recombinant protein expression from baculovirus-infected insect cells (Fig. 1H). We concluded that these hotspot mutations primarily function through protein instability rather than impaired GAP activity or disruption of well-established protein–protein interactions (i.e., SPRED1).
Fig. 1.

844 to 848 neurofibromin mutants are less stable than wild-type protein. (A) Domain structure of neurofibromin showing the location of mutation hotspot. (B) Lysates from 293T NF1−/− cells expressing FLAG-tagged neurofibromin. Expression was analyzed via western blot. (C) FLAG-WT and mutant neurofibromin immunoprecipitates (IPs) from 293T NF1−/− were tested for dimerization with cotransfected HA-WT neurofibromin. The amount of mutant NF1 plasmid was increased to even levels of expression with wild type. (D) HA-tagged WT and mutant neurofibromin coimmunoprecipitated with FLAG-SPRED1. NF1 ΔM1215 is used as a control for the absence of SPRED1 binding. (E) Levels of RAS-GTP by GST-RAF1-RBD (RAS Binding Domain) pulldown from lysates indicate the level of neurofibromin GAP activity in 293T NF1−/−. (F) Ubiquitin (Ubi) and HSP70 were detected on FLAG immunoprecipitants of mutant neurofibromin. (G) Lysates from HA-mScarlet-NF1-transduced 293T and ipNF95.11b C Schwann cells after 48 h Dox treatment at the indicated concentrations. (H) Recombinant mutant neurofibromin proteins are highly insoluble in baculovirus-infected insect cells. Images of Coomassie-stained SDS-PAGE. T = Total, S = Soluble.
We noticed that, when coexpressed with 844 to 848 mutants, wild-type neurofibromin expression levels were also reduced. To examine whether the mutant was causing lower expression of wild type, we transfected increasing amounts of either FLAG-tagged wild-type, G848E or L847P with constant amounts of HA-tagged wild-type neurofibromin. Increasing amounts of FLAG-wild type has no effect on HA-wild type (HA-WT) expression, whereas increasing amounts of transfected G848E or L847P caused wild-type neurofibromin levels to decrease in a dose-dependent manner (Fig. 2 A and B). To examine protein turnover rates, we treated cells with cycloheximide to inhibit protein synthesis and found that mutant proteins are turned over at a higher rate compared to wild type (Fig. 2C). Neurofibromin degradation by the proteasome has been reported to be mediated by CUL3 (18), Hypoxia-Associated Factor (19), and SAG-CUL1-FBXW7 (20) under certain circumstances. We treated NF1-transfected 293T cells with proteasome inhibitors to validate that the mechanism of degradation was via the proteasome, but we were not able to consistently observe any effect of the drugs at the incubation times and concentrations used whilst preserving cell viability, likely due to the half-life of the protein being over 48 h in normal cell culture conditions (20). The failure to produce soluble recombinant neurofibromin point mutants in our insect cell system (Fig. 1G), though, is consistent with a proteasomal degradation mechanism, as baculovirus infection of insect cells leads to proteotoxicity and accumulation of ubiquitylated proteins in aggresomes, leading to a dramatic loss of protein solubility (21).
Fig. 2.
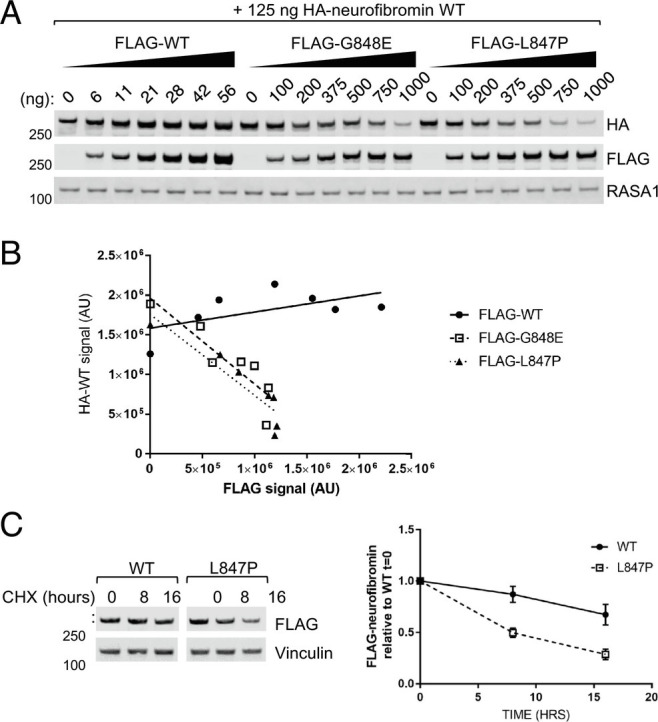
844 to 848 mutants cause wild-type neurofibromin to be destabilized. (A) Lysates from 293T NF1−/− where HA-neurofibromin WT was cotransfected with increasing levels of either WT or mutant (FLAG-tagged). (B) Quantification of A with linear regression trendline. (C) Lysates collected from 293T NF1−/−transfected of WT or mutant neurofibromin and treated with 100 μg/mL cycloheximide for the indicated times. Band intensity was quantified (error bars represent SD from 3 independent experiments).
We sought to elucidate the structural basis for the severe phenotypic effects of mutations affecting residues 844 to 848. Using cryogenic electron microscopy (cryo-EM), we determined the 3.6 Å resolution structure of the 2818 amino acid isoform I (Uniprot P21359-2) which lacks 21 exon 23A-encoded residues within the GAP-related Domain (GRD). Our structure shows that 844Leu-Cys-Ala-Leu-Gly848 forms the C-terminal half of α-helix 34 (Fig. 3A and SI Appendix, Fig. S3A and Table S1), which is spatially juxtaposed to the functionally important GRD. The structural overlay confirms that the segment adopts the same fold in our structure as in the previously determined neurofibromin structures (22–24). Except for Gly848, the side chains of 844 to 848 coat the hydrophobic interhelical surface of the α33 to α36 HEAT repeat (Fig. 3 B and C), thus stabilizing the repeat itself and presumably the distal half of the N-HEAT domain of neurofibromin. Of note, the hydrophobic core residues are highly conserved across species (SI Appendix, Fig. S3B) implying the preservation of structural integrity in neurofibromin orthologues.
Fig. 3.

Cryo-EM structure of the intact short isoform of human neurofibromin. (A) Top view of the final model of NF1 dimer (cartoon) and the corresponding 3D cryo-EM density (transparent grey surface) showing NF1 protomers in dark and light gray, GRD in purple, and Sec-PH (pleckstrin homology) domain in blue. Boxed area indicates the location of the GRD (purple) and Sec-PH (blue) domains in one of the NF1 protomers. PDB: 8E20, EMDB: EMD-27826. (B) A close-up and rotated view of the boxed area from A shows that helices α33 to α36 (red, yellow, green, and light blue) are located directly below the GRD domain (purple). The view is rotated ~90° clockwise around the horizontal axis when compared to A. (C) Side view of the α33 to α36 helical bundle (ribbon cartoon colored red, yellow, green, and light blue). Severe phenotype mutations map to the region encompassing residues 844 to 847 (side chains shown in ball-and-stick representation). (D) Top view of the same bundle. For clarity, helix α33 is not shown. The view is rotated ~90° clockwise around the horizontal axis relative to C. (E) Residues affected by pathogenic mutations are colored using Kyte-Doolittle amino acid hydrophobicity scale where hydrophobic and polar residues are red and blue, respectively.
The above-described mutations mapping to α34 are substitutions wherein shorter hydrophobic residues are replaced with either rigid Pro ring, bulky hydrophobic Phe and Tyr, or bulky polar His, Arg, and Glu. The introduction of any of the bulkier side chains would induce a conformational change of the backbone atoms, which would break the regular pitch of the α-helix and destabilize the rigid α33 to α36 HEAT interface. Likewise, placing Pro in this region would likely compromise both the fold and structural rigidity of α34. Thus, each of these mutations, causatively linked to severe phenotypes, is proposed to disrupt the tight packing of a-helical repeats, exposing otherwise buried interhelical hydrophobic segments to solvent. The uncovered hydrophobic residues would be recognized by the protein quality control system, ultimately leading to degradation of the entire neurofibromin dimer and lowering of both mutant and wild-type protein levels in the cell. These structural predictions align well with our biochemical analyses and rationalize why wild-type neurofibromin expresses poorly in the presence of a destabilizing pathogenic variant.
To test our structure-driven hypothesis, we predicted substitutions in the 844 to 848 segment that would be less disruptive to the structure of α34 and the α33 to α36 HEAT repeat. Indeed, mutations L847I and G848A do not diminish protein expression levels in 293T cells like their pathogenic counterparts (Fig. 4A) and do rescue protein solubility in the baculovirus expression system (Fig. 4B). These “suppressor” mutants, which maintain hydrophobicity, are not only less ubiquitylated than the pathogenic variants, but their level of coimmunoprecipitation with HSP70 is similarly decreased (SI Appendix, Fig. S3B). We further speculated that N-terminal pathogenic variants mapping to conserved regions flanking the 844 to 848 segment (SI Appendix, Fig. S3B) may also be detrimental to the neurofibromin structure. We used our cryo-EM model to identify variants W837F, F894S, and L898R [present in the ClinVar database (www.ncbi.nlm.nih.gov/clinvar)] that may cause disruption to the helices and at the same time predicted ‘less-disruptive’ versions as we had done for L847 and G848 (Fig. 4C). All 3 pathogenic substitutions express less well than wild type and can be ‘suppressed’ by less disruptive mutations W837F, F894L, and L898I in 293T (Fig. 4A) and insect cells (Fig. 4D). We observe the same, reversible pattern of ubiquitylation and HSP70-binding (SI Appendix, Fig. S4A) as we had for codons 844 to 848. Perhaps more importantly, W837F, F894S, and L898R retain GAP activity (SI Appendix, Fig. S4B) and can still form a complex with SPRED1 (Extended Data Fig. 4C), arguing that the GRD functionality is not impaired. Given the observation that mutations mapping to 844 to 848 act in a dominant negative manner by destabilizing wild-type protein, we repeated a dose–response curve of cotransfected wild-type and mutant plasmids. In this regard, F894S resembles a severe pathogenic variant (Fig. 4E and SI Appendix, Fig. S4D), supporting our observations that severe mutations, which disrupt neurofibromin protein structure, reduce cellular levels of neurofibromin by targeting the wild-type protein for degradation through heterodimerization.
Fig. 4.

Loss of stability in α-helix 34 of the N-terminal HEAT domain can be predicted and rescued with hydrophobic residue substitution. (A) Lysates from 293T NF1−/− cells expressing FLAG-tagged neurofibromin wild type or with pathogenic and alternative ‘suppressor’ residues. (B) Solubility and purification of His6-tagged WT, L847P or L847I and G848E or G848A neurofibromin mutants from baculovirus lysates. T = Total, S = Soluble. (C) Cryo-EM structure reveals the location of additional residues mutated in human patients. These residues (red ball-and-stick) map to the α33-α36 HEAT repeat (gray cartoon) and are juxtaposed to the functionally pivotal HEAT:GRD:Sec-PH interface. GRD is colored purple and Sec-PH is blue. (D) Solubility and purification of W837R, W837F, F894S, F894L, L898R, and L898I mutants as in B. (E) Lysates from 293T NF1−/− where HA-neurofibromin WT was cotransfected with increasing levels of either wild type or mutant (FLAG-tagged).
Discussion
Our discovery that neurofibromin exists as a high affinity dimer (12) and our and other cryo-EM structures (22–24) changed our understanding of how it may be regulated and led us to examine whether it was indicated in the molecular mechanisms of NF1 disease. Our results have provided a basis for rationalizing why missense mutations in residues 844 to 848 of neurofibromin cause severe phenotypes in patients, including an increased incidence of major plexiform and spinal neurofibromas (11). These mutations also reduce the level of protein expression through targeted protein degradation and exert a dominant-negative effect on wild-type protein through the heterodimerization. This is expected to cause a reduction in total neurofibromin levels in haploinsufficient cells, below a threshold observed for other genotypes and subsequently, a larger increase in RAS signaling (Fig. 5A). As an example, plexiform neurofibromas initiate when Schwann cells lose NF1 heterozygosity through the loss of the wild-type allele by either acquired deletion or mutation. Studies have demonstrated the importance of the heterozygous neurofibroma cells (e.g., mast cells, fibroblasts, macrophages) in creating and sustaining a supportive inflammatory microenvironment for Schwann cell survival and proliferation (25). In patients with 844 to 848 mutations, haploinsufficient cells may have increased RAS signaling and would therefore contribute to disease progression (26) (Fig. 5B). This may include enhanced responsiveness to signaling by other cells, secretion of SCF (stem cell factor) by NF1−/− Schwann cells that recruits and supports NF1−/+ mast cells, and increased cytokine and growth factor secretion that would stimulate other cells within the tumour (5). It is likely that there are other pathogenic mutations in NF1 that affect protein stability and we have been able to use structural information to identify some of those that behave in the same way as 844 to 848 residues. There is little to no phenotypic information for variants at W837, F894, and L898. Independent studies describe symptoms of CALMs (27), CALMs and bone abnormalities (28), neurofibromas and CALMs (29), and cutaneous neurofibromas (30) in different patients. It may be possible to use this technique to further characterize genotype–phenotype correlations which are not only important for understanding the mechanisms of disease but also to be able to offer patient counselling or more informed therapeutic decisions.
Fig. 5.

Reduced levels of neurofibromin in patients with 844 to 848/destabilizing variants may cause severe phenotype through enhanced RAS pathway signalling in haploinsufficient tissues. (A) Schematic to demonstrate the predicted effect of different NF1 allele combinations in NF1 disease and unaffected cells. Loss of expression by premature truncation or microdeletion of 1 allele of NF1 reduces total protein expression by half. In the presence of 844 to 848 codon variants or other destabilizing substitutions, the total level of neurofibromin is predicted to be lower due to the dominant negative effect of the mutant on the wild type, through dimerization and targeting for degradation. (B) Hypothetical consequences of a destabilizing-variant microenvironment. Using plexiform neurofibroma as an example, loss of heterozygosity in a Schwann cell or precursor initiates recruitment of other cell types. Haploinsufficient cells of the microenvironment, such as fibroblasts, macrophages and mast cells, are predicted to be hyperresponsive to signals from the cells of origin (Schwann cells), such as cytokines and growth factors, and may create an enhanced tumor-supportive environment/inflammatory response.
Operable plexiform neurofibromas are given surgical intervention, and although the MEK inhibitor Selumetinib is approved for the treatment of inoperable tumours (31), MEK inhibition is not without side effects or the potential for acquired resistance. There are also no approved therapies for many of the other symptoms of NF1 such as behavioral or bone and cardiac defects. Here, we provide a mechanism of neurofibromin protein homeostasis in disease. Once we understand the mechanisms of protein turnover, we may be able to design therapeutic approaches that restore neurofibromin function through the stabilization of the protein. This could be either by preventing posttranslational modifications and degradation or through modulation of neurofibromin dimerization. We have also developed a methodology for the transduction of inducible tagged full-length NF1 into cell lines, including NF1−/− 293T and immortalized Schwann cell lines. Our prior attempts to stably reexpress NF1 were hindered by apparent TRE3G promoter silencing and loss of inducibility of expression. We have overcome this by redesigning the expression vectors to select for cells that remain inducible, perhaps due to their integration in regions of chromatin resistant to epigenetic silencing. This system has numerous applications for the study of NF1 disease and is particularly relevant in the field of gene therapy.
Materials and Methods
Cloning and Mutagenesis.
Gateway LR recombination according to the manufacturer’s protocol to insert NF1, SPRED1, and RAS entry clones into transient mammalian expression vectors pCAN-FLAG-DEST and pCAN-HA-DEST. Point mutations in NF1 isoform-2 were generated by PCR mutagenesis of Addgene plasmid 70423.
Mammalian Cell Culture, Immunoprecipitation, and Western Blot.
Generation of NF1-null 293T cells has been previously described (12). ipNF95.11b C was purchased from ATCC, and CRISPR technology was used to generate a homozygous NF1 knockout clone (methodology to be published elsewhere). All cells were maintained in DMEM with 10% fetal bovine serum and penicillin–streptomycin (10 U/mL) and were tested regularly for mycoplasma contamination with MycoAlert (Lonza). Plasmids were transfected using JetOPTIMUS reagent (Polyplus) according to the manufacturer’s protocol. Cells were lysed 48 h later in a lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA (or 5 mM MgCl2 where RAS activity was concerned), 1% Triton-X-100, 1 mM DTT, EDTA-free protease inhibitors (Roche), and phosphatase inhibitor cocktails (Sigma)). Supernatants from cleared lysates were incubated with Anti-FLAG M2 affinity gel (Sigma) to immunoprecipitated FLAG-tagged protein complexes, or recombinant GST-RAF1 RBD beads to pull down GTP-bound RAS, for 2 h at 4 °C followed by 3 washes with lysis buffer (without protease and phosphatase inhibitors). Beads and lysates were mixed with NuPAGE LDS (ThermoFisher) sample buffer and heated at 70 °C for 10 min prior to loading on either 3 to 8% Tris-Acetate or 4 to 12% Bis-Tris NuPAGE gels. Standard Western Blotting procedures were followed and visualized using 680- and 800-conjugated secondary antibodies (Pierce, Jackson Laboratories) on an Odyssey Li-COR scanner or HRP-conjugated antibodies and development on film using Crescendo substrate (Millipore). Primary antibodies used were FLAG-M2 and KRAS (Sigma), HA, HSP70, and Ubiquitin (Cell Signaling), pan-RAS (Abcam), GST and Vinculin (Bethyl Laboratories), and RASA1 (Santa Cruz Biotechnology). Quantification of Western Blot bands was performed using Image-Studio (Li-COR) and visualized in Prism (GraphPad).
Generation of Inducible Expression Constructs and Transduction.
To generate constructs for the Tet-inducible expression of NF1 WT and mutants, the lentiviral attR1-attR2 Gateway destination vector pLIX403 (Addgene 41395) was modified to attR4-attR2 by Gibson assembly. Gateway multisite reactions into pLIX403 attR4-attR2 were then performed with entry clones for the TRE3G promoter (attL4-attL5), HA(hemagglutinin)-m(monomeric)Scarlet (attR5-attR1), and NF1 WT or mutants (attL1-attL2) using LR Clonase II (Thermo Fisher Scientific). Lentiviral particles were then generated by equimolar cotransfection of these NF1 expression vectors with psPAX2 (Addgene 12260) and pMD2.G (Addgene 12259) packaging plasmids in HEK293T cells. Forty-eight hours later, the supernatant viral titer was measured using Lenti-X GoStix Plus (Takara Bio) following the manufacturer’s instructions. Normalized viral titers for NF1 WT and mutants were then mixed with 5 µg/mL hexadimethrine bromide (Polybrene, MedChemExpress), 0.45 µm filtered, and applied to target NF1-null HEK293T and ipNF95.11b C cells with centrifugation at 1,000 × g for 45 min. Forty-eight hours after infection, cells were selected and then subsequently maintained in a medium containing 0.4 µg/mL puromycin. Expression of HA-mScarlet-NF1 WT and mutants was induced with 1 µg/mL Dox for 48 h, and cells exhibiting robust expression were sorted by flow cytometry using a Sony SH800 Cell Sorter (Sony). Following cell sorting, Dox was withdrawn for at least 10 d before the commencement of experiments.
Baculovirus.
Constructs for recombinant production of full-length neurofibromin in insect cells were previously described (12). Point mutants were generated using the Agilent QuickChange site-directed mutagenesis kit per the manufacturer’s instructions, and constructs were completely sequence verified to ensure that no additional mutations were introduced. Baculovirus production and insect cell expression in Tni-FNL cells were done as previously described (12).
Purification of Full-Length Human NF1 Isoform 2.
Full-length NF1 (short isoform), preceded by a hexa-histidine tag and a Tev protease recognition site, was produced in Tni-FNL cells and purified as described (12). Briefly, 250 mL of frozen cells were thawed, resuspended, and homogenized using a Dounce homogenizer. Following centrifugation at 235,000 × g, the supernatant was supplemented with 40 mM imidazole and loaded onto a 5 mL HisTrap HP Ni-Sepharose column (Cytiva). After rinsing the column with 50 mM Tris-HCl pH 8.0, 300 mM NaCl, 4 mM TCEP, 40 mM imidazole, and Sigma protease inhibitors in DMSO at a dilution of 1 in 500, the protein was eluted with the same buffer supplemented with 500 mM imidazole. Fractions containing the neurofibromin dimer were further purified on a preparative HiLoad 16/600 Superose 6 pg column (Cytiva) in 50 mM Tris-HCl pH 8.0, 300 mM NaCl, and 5 mM TCEP, supplemented with Sigma protease inhibitors in DMSO at a dilution of 1:500. The neurofibromin dimer fractions were combined, concentrated, and repurified on a Superose 6 10/300 GL column (Cytiva) in 20 mM Tris-HCl pH 8.0, 300 mM NaCl, 5 mM TCEP. The neurofibromin-dimer peak fraction was cleared by centrifugation at 21,000 × g for 10 min at +10 °C. The pure sample was diluted to ~0.3 mg/mL and used for grid preparation.
Cryo-EM Sample Preparation and Data Acquisition.
UltraAuFoil grids (300 mesh, Electron Microscopy Sciences) were plasma cleaned using a SOLARUS plasma cleaner (Gatan Inc.). Then, 3 mL of purified neurofibromin was subsequently deposited onto the grids, which were then plunge frozen in liquid ethane using a Leica EM GP plunge freezer (Leica Microsystems Inc.). Grid screening and data collection were carried out on a CryoARM200 microscope (JEOL Inc.) equipped with a K3 camera (Gatan) operated in super-resolution mode and correlated double sampling mode. A total of 42,049 movies were collected over five separate data collection sessions. Each image was fractioned into 60 frames with an exposure time of 6.232 s and a dose rate of 9.6 electrons/Å2. All data collection was performed with SerialEM.
Cryo-EM Data Processing.
Data processing was done using the RELION (32) software package. Collected movie frames were motion-corrected and dose-weighed using MotionCorr (33) followed by contrast transfer function estimation using CTFFIND4 (34) Initial particles were picked using Laplacian-of-Guassian-based auto-picker. Obtained representative 2D classes were used for another round of template-based auto-picking that yielded 692,875 particles. Following several rounds of 2D classifications, a cleaned particle stack was used to generate an initial model without imposing symmetry. Subsequent processing steps involved Refine3D and CTF Refine and particle polishing, resulting in a cryo-EM map at 3.6Å overall resolution with C2 symmetry. Particle subtraction with a mask around one Neurifibromin lobe, followed by multibody refinement using four masks (SI Appendix, Fig. S3A), yielded the map at the overall resolution of 3.3Å with improved density for the most of the Neurofibromin core domains.
Model Building and Refinement.
Published models of GRD and Sec-PH (PDB: 1nf1 and 2d4q, respectively) were rigid-body fitted in the density using Chimera. The core of the NF1 dimer was completely manually modeled in Coot (35) and then compared with the AlphaFold2 (36) predictions of separate domains. Structure refinement was done in Phenix (37). Iterative manual inspection and rebuilding of the 3D model in Coot relied on real-space difference maps calculated in Servalcat (38).
PDB and EMDB Accession Codes.
Neurofibromin full dimer pdb: 8E20, EMDB: EMD-27826
Body1 pdb:8EDO, EMDB: EMD-28038
Body2 pdb: 8EDL, EMDB: EMD-28035
Illustrations.
Figs. 1A and 5 and SI Appendix, Fig. S1 were created with BioRender.com. Fig. 2 B and C and SI Appendix, Fig. S4D were made in GraphPad Prism.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Hannah Ambrose and Ashley Mitchell for their technical expertise in insect cell production work and Jennifer Mehalko for assistance in the production of neurofibromin proteins. We thank Antonio Cuevas-Navarro and Richard Van for their technical assistance. This project was funded in part with federal funds from the National Cancer Institute, the NIH, contract no. 75N91019D00024, The Department of Defence CDMRP grant number W81XWH2010129, and the Neurofibromatosis Therapeutic Acceleration Program (NTAP).
Author contributions
L.C.Y., D.E., J.O., and F.M. designed research; L.C.Y., R.G.d.S, S.-W.H., Z.H., A.M., M.D., J.D., V.W., R.G., A.C., M.R.A., M.J.S., and J.O. performed research; L.C.Y., R.G.d.S, S.-W.H., Z.H., A.M., M.D., J.D., D.E., J.O., and F.M. analyzed data; F.M. and D.V.N. supervised study; and L.C.Y., D.E., J.O., and F.M. wrote the paper.
Competing interest
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Jana Ognjenovic, Email: jana.ognjenovic@nih.gov.
Frank McCormick, Email: frank.mccormick@ucsf.edu.
Data, Materials, and Software Availability
Coordinates and maps have been deposited in PDB www.rcsb.org and EMDB www.ebi.ac.uk/emdb/ (see accession codes above), respectively, and will be released upon publication. All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Martin G. A., et al. , The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 63, 843–849 (1990). [DOI] [PubMed] [Google Scholar]
- 2.Stowe I. B., et al. , A shared molecular mechanism underlies the human rasopathies Legius syndrome and Neurofibromatosis-1. Gene. Dev. 26, 1421–1426 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Philpott C., Tovell H., Frayling I. M., Cooper D. N., Upadhyaya M., The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genomics 11, 13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rauen K. A., The RASopathies. Genom. Hum. Genet. 14, 355–369 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gutmann D. H., et al. , Neurofibromatosis type 1. Nat. Rev. Dis. Primers 3, nrdp20174 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Upadhyaya M., et al. , An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): Evidence of a clinically significant NF1 genotype-phenotype correlation. Am. J. Hum. Genet. 80, 140–151 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koczkowska M., et al. , Expanding the clinical phenotype of individuals with a 3-bp in-frame deletion of the NF1 gene (c.2970_2972del): An update of genotype–phenotype correlation. Genet. Med. 21, 867–876 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koczkowska M., et al. , Clinical spectrum of individuals with pathogenic NF1 missense variants affecting p.Met1149, p.Arg1276, and p.Lys1423: Genotype–phenotype study in neurofibromatosis type 1. Hum. Mutat. 41, 299–315 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rojnueangnit K., et al. , High incidence of noonan syndrome features including short stature and pulmonic stenosis in patients carrying NF1 missense mutations affecting p.Arg1809: Genotype-phenotype correlation. Hum. Mutat. 36, 1052–1063 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kehrer-Sawatzki H., Mautner V.-F., Cooper D. N., Emerging genotype–phenotype relationships in patients with large NF1 deletions. Hum. Genet. 136, 349–376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koczkowska M., et al. , Genotype-phenotype correlation in NF1: Evidence for a more severe phenotype associated with missense mutations affecting NF1 codons 844–848. Am. J. Hum. Genet. 102, 69–87 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sherekar M., et al. , Biochemical and structural analyses reveal that the tumor suppressor neurofibromin (NF1) forms a high-affinity dimer. J. Biol. Chem. 295, 1105–1119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toonen J. A., et al. , NF1 germline mutation differentially dictates optic glioma formation and growth in neurofibromatosis-1. Hum. Mol. Genet. 25, 1703–1713 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li K., et al. , Mice with missense and nonsense NF1 mutations display divergent phenotypes compared with human neurofibromatosis type I. Dis. Model Mech. 9, 759–767 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Long A., et al. , Analysis of patient-specific NF1 variants leads to functional insights for Ras signaling that can impact personalized medicine. Hum. Mutat. 43, 30–41 (2022). [DOI] [PubMed] [Google Scholar]
- 16.Rosenzweig R., Nillegoda N. B., Mayer M. P., Bukau B., The Hsp70 chaperone network. Nat. Rev. Mol. Cell Bio. 20, 665–680 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Li H., Chang L.-J., Neubauer D. R., Muir D. F., Wallace M. R., Immortalization of human normal and NF1 neurofibroma Schwann cells. Lab Invest. 96, 1105–1115 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Hollstein P. E., Cichowski K., Identifying the ubiquitin ligase complex that regulates the NF1 tumor suppressor and ras. Cancer Discov. 3, 880–893 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Green Y. S., et al. , Hypoxia Associated Factor (HAF) mediates neurofibromin ubiquitination and degradation leading to Ras-ERK pathway activation in hypoxia. Mol. Cancer Res. 17, molcanres.1080.2018 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan M., et al. , SAG/RBX2/ROC2 E3 ubiquitin ligase is essential for vascular and neural development by targeting NF1 for degradation. Dev. Cell 21, 1062–1076 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lyupina Y. V., et al. , Proteotoxic stress induced by Autographa californica nucleopolyhedrovirus infection of Spodoptera frugiperda Sf9 cells. Virology 436, 49–58 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Lupton C. J., et al. , The cryo-EM structure of the human neurofibromin dimer reveals the molecular basis for neurofibromatosis type 1. Nat. Struct. Mol. Biol. 28, 982–988 (2021). [DOI] [PubMed] [Google Scholar]
- 23.Naschberger A., Baradaran R., Rupp B., Carroni M., The structure of neurofibromin isoform 2 reveals different functional states. Nature 599, 315–319 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaker-Margot M., et al. , Structural basis of activation of the tumor suppressor protein neurofibromin. Mol. Cell 82, 1288–1296.e5 (2022). [DOI] [PubMed] [Google Scholar]
- 25.Liao C.-P., et al. , Contributions of inflammation and tumor microenvironment to neurofibroma tumorigenesis. J. Clin. Invest. 128, 2848–2861 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frayling I. M., et al. , Breast cancer risk in neurofibromatosis type 1 is a function of the type of NF1 gene mutation: A new genotype-phenotype correlation. J. Med. Genet. 56, 209–219 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Boyd P. K., et al. , Phenotypic variability among café-au-lait macules in neurofibromatosis type 1. J. Am. Acad. Dermatol. 63, 440–447 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balla B., et al. , FAst and robust next-generation sequencing technique using ion torrent personal genome machine for the screening of neurofibromatosis type 1 (NF1) gene. J. Mol. Neurosci. 53, 204–210 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Montani D., et al. , Pulmonary hypertension in patients with neurofibromatosis type I. Medicine 90, 201–211 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Cannon A., et al. , Cutaneous neurofibromas in Neurofibromatosis type I: A quantitative natural history study. Orphanet. J. Rare Dis. 13, 31 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gross A. M., Dombi E., Widemann B. C., Current status of MEK inhibitors in the treatment of plexiform neurofibromas. Child’s Nerv. Syst. 36, 2443–2452 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Zivanov J., et al. , New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X., et al. , Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–590 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rohou A., Grigorieff N., CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta. Crystallogr. Sect. D. Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Jumper J., et al. , Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liebschner D., et al. , Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta. Crystallogr. Sect. D. 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamashita K., Palmer C. M., Burnley T., Murshudov G. N., Cryo-EM single-particle structure refinement and map calculation using Servalcat. Acta. Crystallogr. Sect. D. Struct. Biol. 77, 1282–1291 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
Coordinates and maps have been deposited in PDB www.rcsb.org and EMDB www.ebi.ac.uk/emdb/ (see accession codes above), respectively, and will be released upon publication. All study data are included in the article and/or SI Appendix.