

Abstract
Acute lung injury (ALI) is an acute inflammatory process arises from a wide range of lung insults. A major cause of ALI is dysfunction of the pulmonary vascular endothelial barrier but the mechanisms involved are incompletely understood. The therapeutic potential of histone deacetylase (HDAC) inhibitors for the treatment of cardiovascular and inflammatory diseases is increasingly apparent, but the mechanisms by which HDACs regulate pulmonary vascular barrier function remain to be resolved. We found that specific Class IIa HDACs inhibitor, TMP269, significantly attenuated the lipopolysaccharide (LPS)-induced human lung microvascular endothelial cells (HLMVEC) barrier compromise in vitro and improved vascular barrier integrity and lung function in murine model of ALI in vivo. TMP269 decreased LPS-induced myosin light chain phosphorylation suggesting the role for Class IIa HDACs in LPS-induced cytoskeleton reorganization. TMP269 did not affect microtubule structure and tubulin acetylation in contrast to the HDAC6-specific inhibitor, Tubastatin A suggesting that Class IIa HDACs and HDAC6 (Class IIb) regulate endothelial cytoskeleton and permeability via different mechanisms. Furthermore, LPS increased the expression of ArgBP2 which has recently been attributed to HDAC-mediated activation of Rho. Depletion of ArgBP2 abolished the ability of LPS to disrupt barrier function in HLMVEC and both TMP269 and Tubastatin A decreased the level of ArgBP2 expression after LPS stimulation suggesting that both Class IIa and IIb HDACs regulate endothelial permeability via ArgBP2-dependent mechanism. Collectively, our data strongly suggest that Class IIa HDACs are involved in LPS-induced ALI in vitro and in vivo via specific mechanism which involved contractile responses, but not microtubule reorganization.
Keywords: acute lung injury, barrier protection, Class IIA HDACs inhibition, LPS, pulmonary endothelium, TMP269
1 |. INTRODUCTION
Acute lung injury (ALI) is an acute systemic inflammatory process that can originate from a wide range of lung insults including corrosive chemicals, toxins or inflammatory mediators (Skerrett, Niederman, & Fein, 1989). ALI has a high mortality rate with more than 200,000 cases each year (Johnson & Matthay, 2010; Rubenfeld et al., 2005) but no effective therapeutic options. One of the key causes of ALI is a dysfunction of the pulmonary vascular endothelial barrier, but the mechanisms involved are incompletely understood (Johnson & Matthay, 2010). Endothelial cells (ECs) provide a semi-selective barrier between the blood and the interstitial space that regulates the passage of both inflammatory cells and fluid. While ECs barrier integrity is critically important for the pathogenesis of ALI, the mechanisms regulating ECs barrier under edemagenic and barrier-protective conditions remain incompletely understood and require further investigation.
Lipopolysaccharide (LPS), a proinflammatory mediator derived from the cell walls of G− bacteria, compromises EC barrier function in vitro and in vivo (Catravas et al., 2010; Chatterjee et al., 2008). Murine lung injury induced by LPS is characterized by neutrophil infiltration into the lung, increased production of inflammatory mediators, formation of a protein-rich alveolar fluid and alveolar flooding, structural damage to the alveolar–capillary membrane, and lung dysfunction (Chatterjee et al., 2008; de Souza Xavier Costa et al., 2017). LPS-induced disruption of the ECs barrier is critically dependent upon pro-contractile changes in the ECs cytoskeleton, including the activation of Rho signaling resulting in the inhibition of myosin light chain (MLC) phosphatase (MLCP) followed by an increase in MLC phosphorylation, initiation of contraction, and opening of the paracellular avenues of permeability (Kasa, Csortos, & Verin, 2015; Kovacs-Kasa et al., 2016; Kovacs et al., 2018).
Arg kinase binding protein 2 (ArgBP2) is an adapter protein that plays an important role in cell adhesion and the regulation of cytoskeletal structure (Cestra, Toomre, Chang, & De Camilli, 2005). Recent studies suggested that Rho signaling can be regulated, at least in part, via ArgBp2 expression in ECs (Martin et al., 2013). The role of ArgBP2 in LPS-induced-disruption of the EC barrier is currently unknown.
Epigenetic changes elicited by the histone deacetylases (HDACs) have emerged as a major driver of enduring changes in cell behavior and vascular function (Galan et al., 2016; Hull, Montgomery, & Leyva, 2016; New et al., 2016; Qu et al., 2016; Regna et al., 2015). The therapeutic potential of HDAC inhibitors for the treatment of cardiovascular and inflammatory diseases is increasingly apparent, but the mechanisms by which specific HDACs regulate pulmonary vascular barrier function remain to be resolved (Joshi et al., 2015; Saito et al., 2011). The best known function of HDACs is their ability to modify core histone function by deacetylating ε-amino acids in histones in the nucleus leading to transcriptional repression (de Ruijter, van Gennip, Caron, Kemp, & van Kuilenburg, 2003). Recent discoveries have revealed that HDACs are able to deacetylate several nonhistone proteins including some outside the nucleus and contribute to the regulation of a series of cellular functions such as gene transcription and signal transduction (Glozak & Seto, 2007; Halili, Andrews, Sweet, & Fairlie, 2009; Ito, Barnes, & Adcock, 2000).
Classical HDACs are comprised 11 different isoforms with a highly conserved deacetylase domain. HDACs are subdivided into four classes (I, IIa, IIb, and IV) based on structural similarity, enzymatic activity, subcellular localization, and expression patterns (Haberland, Montgomery, & Olson, 2009; Kovacs et al., 2018). While Class I HDACs 1/2/3/8 are primarily localized in the nucleus and Class IIb HDACs 6/10/11 are localized in the cytoplasm, Class IIa HDACs 4/5/7/9 are nuclear-cytoplasmic proteins with the ability to shuttle between the nucleus and the cytoplasm in response to various signals (Kao et al., 2001; McKinsey, Zhang, & Olson, 2001). In contrast to other HDACs, Class IIa enzymes have minimal deacetylase activity on acetylated histones and primarily act as corepressors of transcriptional events in the nucleus (Di Giorgio & Brancolini, 2016; Jones et al., 2008; Lahm et al., 2007; Parra, 2015).
Recent studies support the involvement of HDACs in the regulation of cytoskeleton and ECs permeability via the deacetylation of cytoskeletal targets (Fontaine et al., 2015; Kuhlmann et al., 2016; Song & Brady, 2015). HDAC6-mediated deacetylation of α-tubulin, which leads to microtubule (MT) destabilization, is involved in ALI induced by cigarette smoke (Borgas et al., 2016) and tumor necrosis factor-α (Yu, Ma, Shetty, Ma, & Fu, 2016). It has also been reported that the Class IIa enzyme, HDAC7, is a critical regulator of vascular stability in embryogenesis (Chang et al., 2006) and regulates cytoskeleton function (Joo & Yamada, 2014; Martin et al., 2008). Furthermore, a newly published study reported on the involvement of HDAC7 in Escherichia coli-induced lung vascular barrier compromise in a murine model of ALI, but the mechanisms by which HDAC7-regulates ECs barrier function were not described (Kasotakis et al., 2019).
In the present study, we evaluated the role of Class IIa HDACs in LPS-induced cytoskeletal rearrangement and barrier dysfunction in ECs in vitro and in a murine model of ALI using a specific Class IIa HDAC inhibitor, TMP269 (Lobera et al., 2013). We demonstrated that TMP269 significantly attenuated the loss of ECs barrier function in primary human lung microvascular ECs and preserved lung function, and barrier integrity in a murine LPS-induced ALI model implicating the involvement of Class IIa HDACs in LPS-induced ALI. A systematic comparison of the effects of TMP269 with those of a specific HDAC6 inhibitor, Tubastatin A (Edwards & Pender, 2011; Robey et al., 2011) indicated that while both HDAC6 and Class IIa HDACs are involved in LPS-induced loss of ECs barrier function via activation of Rho signaling and increased ArgBP2 expression, Class IIa HDACs do not mediate LPS-induced changes in MT destabilization. These results suggest the existence of specific roles for structurally different HDAC isoforms in the regulation of ECs cytoskeleton and barrier function.
2 |. MATERIALS AND METHODS
2.1 |. Mice
All animal studies were approved and carried out according to the rules and regulations by the Augusta University Animal Care and Use Committee and National Institutes of Health. Black C57BL/6 mice were from Jackson Laboratory.
2.2 |. Cell culture
Freshly isolated human lung microvascular endothelial cells (HLMVEC) were used in all in vitro experiments, isolated in our laboratory (Catravas et al., 2010). Lung specimens were provided by Cardiothoracic Surgery that would otherwise be discarded. These existing samples are unidentified and no personal information was obtained that could be traced back to the patients. Waiver of informed consent was approved by the Augusta University Institutional Review Board.
2.3 |. Immunofluorescence
HLMVEC monolayers were fixed in 3.7% paraformaldehyde (PFA), then were permeabilized with 0.25% Triton X-100 in Tris-buffered saline with Tween for 5 min at room temperature, and then blocked with 2% bovine serum albumin in Tris-buffered saline with Tween O/N. Between each step, cells were rinsed three times with 1 × phosphate-buffered saline (PBS). Cells were incubated with the acetylated tubulin mAb (1:300) and then were incubated with the secondary antibody Alexa488 (Molecular Probes) 1:300 diluted in blocking solution for 1 h at room temperature. Coverslips were rinsed and mounted in ProLong® Gold Antifade Reagent and observed with ×63 immersion oil objective lenses using a microscope (Axiolab; Carl Zeiss).
2.4 |. Silencing RNA transfection
About 50 nM of ArgBP2 small interfering RNA (siRNA; GE Dharmacon) was used to transfect human lung microvascular ECs with SiPort transfection reagent (Thermo Fisher Scientific) following the manufacturer’s instructions.
2.5 |. Western blot analysis
HLMVEC lysates were collected after treatments and lysed by modified radioimmunoprecipitation assay buffer (Sigma-Aldrich) and clear supernatants were combined with Laemmli buffer. Proteins were separated by polyacrylamide gel electrophoresis and blotted to polyvinylidene difluoride membrane. Diphospho-MLC (T18/S19), acetyl-α-tubulin, phospho-MYPT (T853), and horseradish peroxidase-conjugated actin antibodies (Cell Signaling Technology, Inc.) and ArgBP2 antibody (Santa Cruz Biotechnology) were used to stain the membranes.
2.6 |. Electric cell impedance sensing system
HLMVECs were plated on gold microelectrodes in special electric cell impedance sensing system (ECIS) arrays (Applied Biophysics, Troy, NY). Transendothelial electrical resistance (TER) was measured after the baseline was set. Either Class IIa HDACs inhibitor (TMP269) or HDAC6 inhibitor (Tubastatin A) was used as a pretreatment for 2 h, then cells were challenged with LPS using an ECIS, described in (Kovacs-Kasa et al., 2016, 2018). HLMVEC were either transfected with control siRNA or with ArgBP2 siRNA. TER was measured after 72 h posttransfection and monitored for 20 h.
2.7 |. Lung histology
Lungs from vehicle (0.9% saline), TMP269, LPS, or TMP269/LPS treated animals were collected after injecting them with 4% PFA solution under 15cmH2O pressure. Lungs were paraffin embedded and sections were mounted on slides. After deparafinization steps, slides were incubated with Target Retrieval Solution to unmask the antibody epitopes. Lung sections were stained for hematoxylin and eosin (H&E) and myeloperoxidase (MPO).
2.8 |. Measurement of MPO activity
MPO staining kit was purchased from Dako. The MPO activity was measured according to the manufacturer’s instructions and as it was previously described (Kovacs-Kasa et al., 2016).
2.9 |. Lung injury model in mice
Mice were anesthetized with ketamine (100 mg/kg ip) and xylazine-HCl (10 mg/kg ip). A neck midline incision was performed, and the trachea and jugular vein were exposed 15 min before instillation, mice received either TMP269 (2.5 mg/kg; Selleckchem) or vehicle (0.9% saline) intravenously through the right jugular vein. Then, mice were intubated and injected with LPS (1 mg/kg it) or 0.9% sterile saline. Body weight of the mice was measured at the beginning of and at the termination of the experiment. Mice were killed 16 h after endotoxin challenge with ketamine (500 mg/kg ip) and xylazine-HCl (50 mg/kg ip). Bronchoalveolar lavage fluid (BALF) was collected, then lungs were perfused with normal saline solution and then either harvested and fixed in 4% PFA for histopathological examination or excised, snap-frozen in liquid nitrogen, and stored at −80°C until used.
2.10 |. Isolation of BALF
BALF was conducted by injecting and withdrawing 1 ml normal saline via endotracheal catheter as we described previously (Kovacs-Kasa et al., 2016). BALF was centrifuged at 2500g for 10 min, and the clear supernatant was further analyzed. Protein amount was determined by Bicinchoninic Acid Protein Assay Kit (Thermo Fisher Scientific). Water was added to the cell pellet to lyse red blood cells, then 20 times PBS was added to normalize saline concentration and then the neutrophil numbers were determined in each sample.
2.11 |. Lung function measurement
Mice were pretreated either with vehicle, TMP269, LPS, or TMP269/LPS. About 16 h later mice were anesthetized with ketamine (100 mg/kg ip) and xylazine-HCl (10 mg/kg ip). The trachea was exposed and the O2 saturation and heart rate were measured with Mouseox Plus Pulse-Oximeter (STARR Life Sciences Corporation) as it was previously described (Aggarwal et al., 2014). Then mice were intubated and connected to a ventilator system (FlexiVent, Scireq). Lung function measurements were performed as it was described previously (Gross et al., 2018).
2.12 |. Statistical analysis
Results are presented as mean ± SEM for 3–5 experiments. One-way analysis of variance or Student’s t test was used to determine significance. p < .05 was considered statistically significant.
3 |. RESULTS
3.1 |. Pharmacological inhibition of Class IIa HDACs attenuates LPS-induced lung injury in a murine model
To evaluate whether Class IIa HDACs are involved in LPS-induced vascular barrier disruption in vivo, we employed the novel, Class IIa selective HDACs inhibitor, TMP269 (Lobera et al., 2013). TMP269 was recently used in a murine model of spinal cord injury to inhibit Class IIa HDACs (Qi & Wang, 2017). To assess the role of Class IIa HDACs in an LPS-induced in vivo model of ALI, C57BL/6 mice were injected intravenous with TMP269 (2.5 mg/kg) and then LPS (1 mg/kg) was instilled intratracheally to induce ALI. Sixteen hour later mice were killed, and lung sections evaluated for inflammatory changes using H&E staining (Figure 1a), or for neutrophil infiltration, via MPO staining (Figure 1b). Figure 1 shows that exposure to LPS for 16 h induced a robust proinflammatory response that was characterized by prominent neutrophil infiltration and edematous disruption of alveolar structure. These changes were attenuated in mice pretreated with TMP269 suggesting the involvement of Class IIa HDACs in LPS-induced ALI in murine model. Lung injury scores were also calculated from the lung sections as it was described before (Aggarwal et al., 2014; Figure 1c).
FIGURE 1.
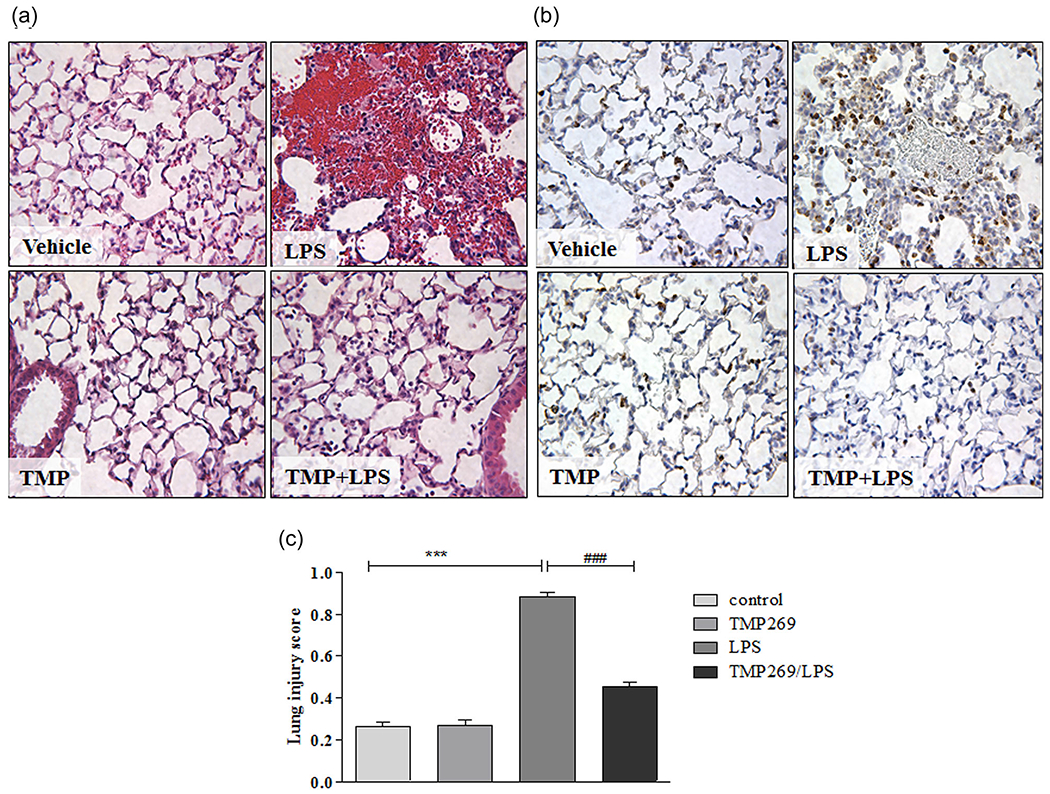
Effect of Class IIa HDACs inhibition on LPS-induced lung inflammation in vivo. Class IIa HDACs inhibitor, TMP269 (2.5 mg/kg final plasma concentration) was instilled intravenously through the right jugular vein (RJV) 15 min before LPS (1 mg/kg) was injected (intratracheally). Sixteen hour later lungs were collected. Control, LPS, TMP269, and TMP269/LPS samples were paraffin embedded and sections were further analyzed with (a) hematoxylin and eosin staining or (b) MPO staining. (c) Lung injury scores, calculated from five independent parameters (neutrophils in the alveolar space, neutrophils in the interstitial space, the presence of hyaline, membranes, the presence of proteinaceous debris filling the airspaces, and alveolar septal thickening; Matute-Bello et al., 2011) on the lung sections. Values are mean ± SEM, number of lung sections = 5/mice. HDACs, histone deacetylases; LPS, lipopolysaccharide; MPO, myeloperoxidase. ***p < .0001, **p < .001; *p < .05 vehicle versus LPS ###p < .0001, ##p < .001, #p < .05 LPS versus TMP269/LPS
3.2 |. Inhibition of Class IIa HDACs decreases lung permeability and inflammation in LPS-induced ALI
To further investigate the role of Class IIa HDACs in LPS-induced ALI, we measured LPS-induced pulmonary vascular leakage and inflammation in mice treated with and without TMP269. To assess changes in LPS-induced inflammatory processes, BALF was collected and protein levels measured. As shown in Figure 2a, LPS significantly increased protein extravasation into the alveolar airspace, which was markedly reduced by TMP269. Accordingly, the number of inflammatory cells in BALF was significantly decreased in mice pretreated with the Class IIa HDACs inhibitor as compared with vehicle (Figure 2b). The vascular capillary leak was measured using Evans Blue Dye-conjugated albumin. LPS increased the leakage of the pulmonary vessels in vehicle-treated mice, whereas mice administered TMP269 were significantly protected (Figure 2c). TMP269 also attenuated the loss of body weight following LPS treatment (Figure 2d). Overall, these data indicate that inhibition of class IIa HDACs reduces the inflammatory and edemagenic effects of LPS in a murine model of ALI.
FIGURE 2.
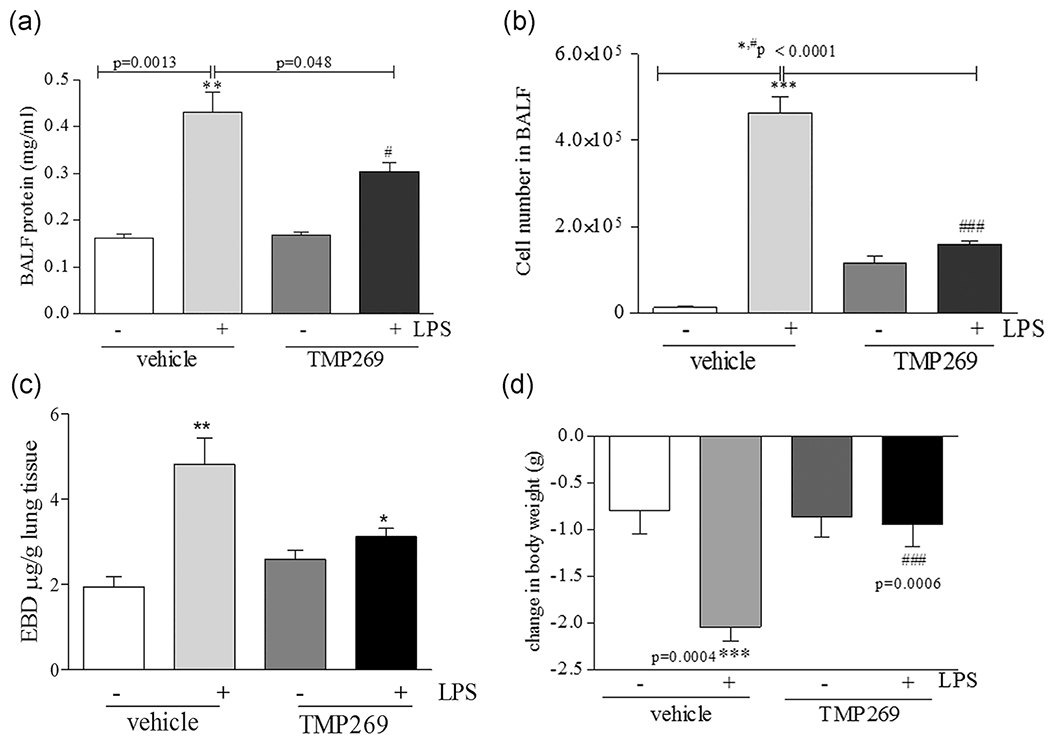
TMP269 pretreatment significantly attenuates LPS-mediated lung inflammatory processes in the murine model of ALI. (a) TMP269 (2.5 mg/kg), or saline (vehicle) was instilled intravenously 15 min before LPS (intratracheally) challenge. The protein content (mg/ml) was determined from the lavage fluid in the supernatants using bicinchoninic acid protein assay kit. Values are mean ± SEM, n = 4–6. **p < .005 vehicle versus LPS, #p < .05 LPS versus TMP269/LPS. (b) BALF was collected from all groups and it was centrifuged. Total numbers of cells were counted using a hemocytometer. Values are mean ± SEM, n = 4–6. ***p < .0001, **p < .001 vehicle versus LPS; *p < .05 LPS versus TMP269/LPS. (c) EBD-conjugated albumin was administered via tail vein 2 h prior the termination of the experiment. Then lungs were collected and EBD was measured. (d) TMP269 (2.5 mg/kg), or saline (vehicle) was instilled intravenously 15 min before LPS (intratracheally) challenge. Body weight was measured before and at the termination of the experiment. Mice lost weight after LPS exposure; values are mean ± SEM, n = 4–6. ***p = .0004 vehicle versus LPS; ###p = .0006 LPS versus TMP269/LPS. ALI, acute lung injury; EBD, Evans Blue Dye; LPS, lipopolysaccharide
3.3 |. TMP269 improves lung function, oxygen saturation, and heart rate in LPS-induced ALI
TMP269 (2.5 mg/kg) or saline (vehicle) were administered intravenously via the R.J.V. and 15 min later, mice were given 1 mg/kg LPS intratracheally. After 16 h of LPS exposure, mice were anesthetized and transcutaneous oxygen saturation (Figure 3a) and heart rate (Figure 3b) monitored via placement of a small animal pulse oximeter noninvasively on the neck. Mice were subsequently intubated and connected to a Flexivent system (Scireq) to measure lung function. LPS promoted shift of the pressure–volume (PV) curve to lower lung volumes in vehicle, but not in TMP269-treated mice (Figure 3c). Data from the PV curve indicate that mice treated with vehicle and then exposed to LPS experienced a lower tidal volume as compared with mice treated with TMP269 on the same amount of pressure.
FIGURE 3.
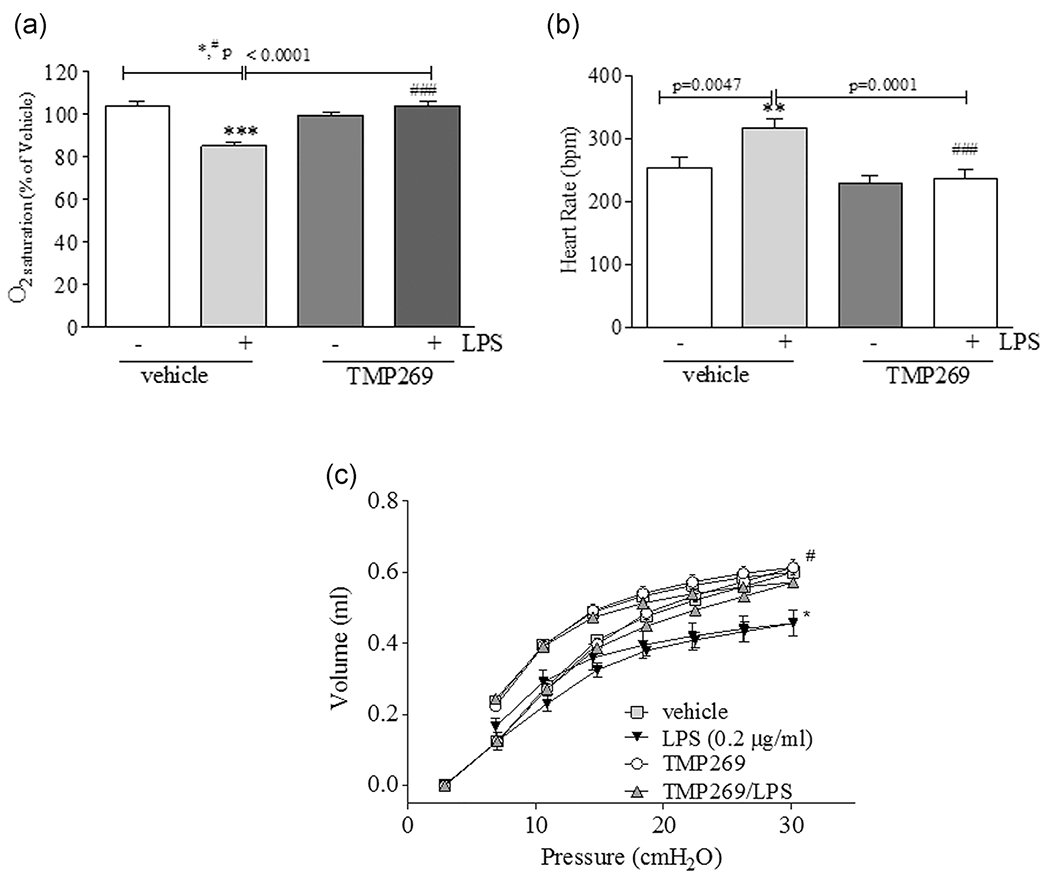
TMP269 pretreatment significantly improves lung function and heart rate in LPS challenged mice. (a) and (b) Mice were intravenously injected either with TMP269 (2.5 mg/kg) or saline (vehicle) followed by intratracheal injection of 1 mg/kg LPS. After 16 h, transcutaneous oxygen saturation (a) and heart rate (b) were monitored via a small animal pulse oximeter by placing the noninvasive sensor on the neck. (c) Lung function was measured after TMP and LPS treatment using Felxivent (Scireq). Values are mean ± SEM, n = 4–6. ***p < .0001, **p < .005; *p < .05 vehicle versus LPS; ###p < .0001, #p < .05 LPS versus TMP269/LPS. LPS, lipopolysaccharide
3.4 |. Inhibition of Class IIa HDACs significantly attenuated LPS-induced endothelial permeability in HLMVEC
The anti-inflammatory role of nonspecific HDAC inhibitors is well established (Adcock, 2007; Halili et al., 2010). It has also been convincingly demonstrated that Class I HDAC3 and Class IIb HDAC6 play critical roles in LPS-induced ALI (Joshi et al., 2015). It is also well established that HDAC6 is involved in EC barrier compromise in vitro and in vivo via a mechanism that primarily involves tubulin deacetylation followed by destabilization of the MT network (Borgas et al., 2016; Hubbert et al., 2002; Karki et al., 2019). In contrast, the role of Class IIa HDACs in LPS-induced cytoskeletal rearrangement and endothelial barrier regulation has not yet been investigated. To clarify the role of Class IIa HDACs in regulating EC permeability in vitro, HLMVEC were treated with TMP269 for 2 h before LPS challenge and TER (an inverse index of permeability) was monitored for 12 h. As shown in Figure 4a, TMP269 completely abolished the decrease in TER induced by LPS, supporting a critical role of Class IIa HDACs in mediating the ability of LPS to disrupt the lung micro-vascular endothelial cell barrier. Interestingly, HDAC6 inhibition with Tubastatin A had a similar effect on LPS-mediated decrease in TER, confirming the important role of HDAC6 activity in endotoxin caused EC barrier dysfunction. As the mechanisms of HDAC6-mediated EC barrier regulation are relatively well defined and involved cytoskeletal rearrangement (Borgas et al., 2016; Hubbert et al., 2002; Karki et al., 2019), we next compared the roles of Class IIb and Class IIa HDACs in regulating the EC cytoskeleton in vitro.
FIGURE 4.
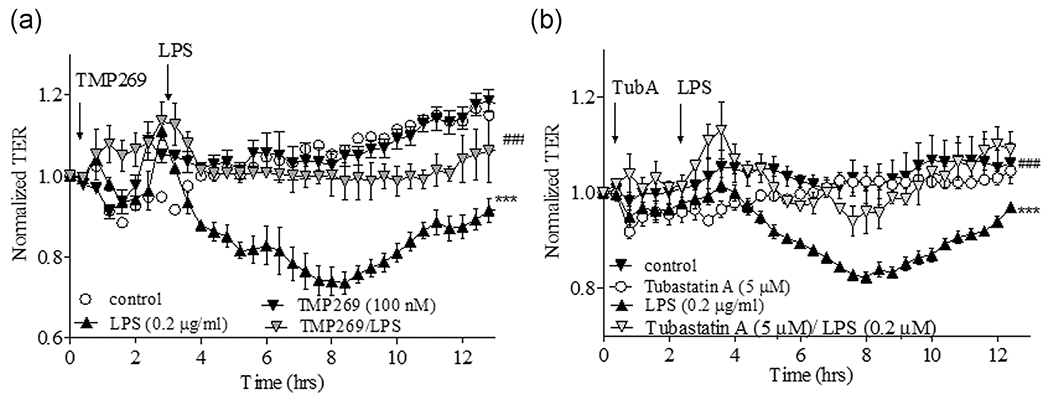
The effect of Class IIa HDACs and HDAC6 inhibition on LPS-induced TER increase of HLMVEC monolayer. HLMVEC were plated in ECIS arrays (8W10E). Baseline was set, then cells were challenged with either vehicle (0.1% dimethyl sulfoxide) or (a) TMP269 (100 nM) or (b) Tubastatin A (5 μM) for 2 h, then LPS (0.2 μg/ml) was added and TER was monitored for 12 h. Results are expressed as mean ± SEM (n = 4 experiments). ECIS, electric cell impedance sensing system; HDACs, histone deacetylases; HLMVEC, human lung microvascular endothelial cell; LPS, lipopolysaccharide. ***,###p < .0001
3.5 |. In contrast to Class IIb HDAC6, Class IIa HDACs activate Rho signaling in a MT-independent manner
LPS activates the Rho pathway and Rho kinase resulting in inactivation of MLCP via phosphorylation of its targeting subunit 1 (MYPT1) on T853. Loss of MLCP activity leads to increased phospho-MLC levels, cell contraction, and ECs barrier dysfunction (Hartshorne, 1998; Matsumura & Hartshorne, 2008). We have recently reported that MLCP plays a critical role in ECs barrier protection (Kim et al., 2012; Kovacs-Kasa et al., 2016). Here we next compared the involvement of Class IIb HDAC6 and Class IIa HDACs in mediating LPS-induced Rho-kinase mediated ECs barrier disruption via changes in MYPT1 and MLC phosphorylation. As shown in Figure 5, LPS significantly increased MYPT1 phosphorylation in ECs strongly suggesting Rho activation. This effect was attenuated by both Class IIa HDACs and Class IIb HDAC6 inhibitors. LPS also induced a significant increase in MLC phosphorylation which was attenuated by both TMP269 and Tubastatin A. Since the changes in MYPT1 and MLC phosphorylation are well-known manifestations of changes in the Rho activity, these data strongly suggested that both HDAC6 and Class IIa HDACs are involved in LPS-induced ECs cytoskeletal rearrangement via activation of the Rho pathway. Surprisingly, while Tubastatin A robustly increased the level of acetylated tubulin reflecting MT stabilization, TMP269 has no effect on it (Figures 5a,b and S1). These data strongly suggested that in contrast to HDAC6, Class IIa HDACs regulate Rho signaling in a MT-independent fashion. To confirm these findings, we next compared MT structure in TMP269- and Tubastatin A-pretreated HLMVEC with and without LPS using immunofluorescence staining with an antibody specific for acetylated (stable) MTs. As shown in Figure 6, LPS predictably decreased the amount of acetylated MTs reflecting a generalized disorganization of MT structure. Pretreatment with TMP269 modestly modified MT structure in control cells and had no effect on the MT destabilization induced by LPS. In contrast, pretreatment with tubastatin A increased acetylated tubulin staining and restored and even enhanced the MT network following exposure to LPS. These data support the ability of Class IIa HDACs to promote Rho-mediated cytoskeletal reorganization in a MT-independent manner and suggests that Class IIa HDACs and HDAC6 regulate the function of the endothelial cytoskeleton and paracellular permeability via different mechanisms.
FIGURE 5.
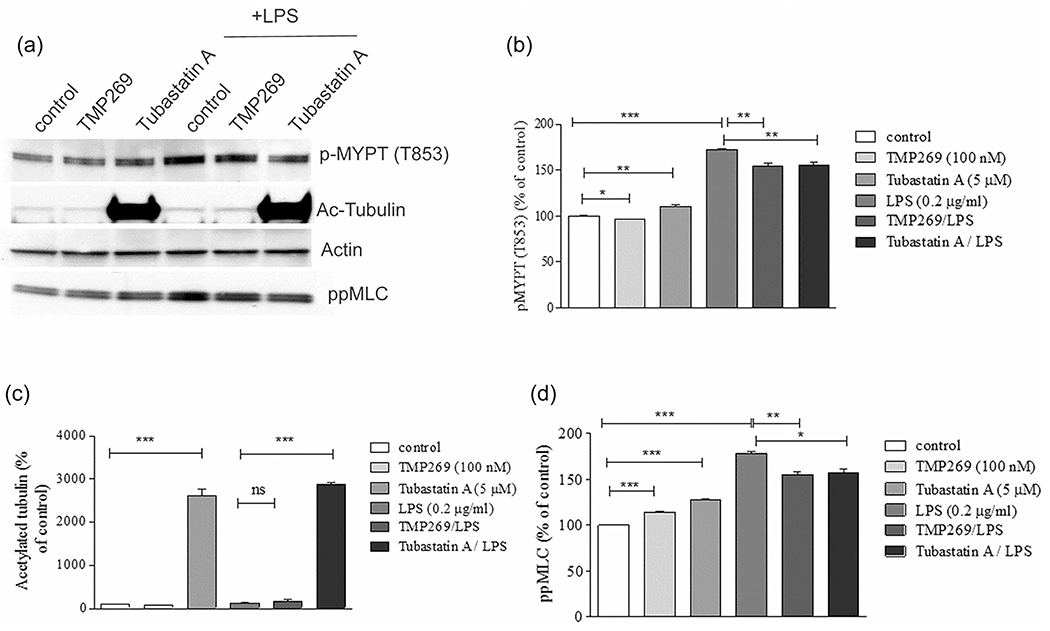
Distinct effects of HDAC6 and Class IIa HDACs inhibition on LPS-induced cytoskeletal signaling. (a) HLMVEC were pretreated either with Class IIa HDAC inhibitor (TMP269, 100 nM), HDAC6 inhibitor (Tubastatin A, 5 μM), or vehicle (control, 0.1% dimethyl sulfoxide) for 2 h then challenged with LPS (0.2 mg/ml) for 4 h. (a) Western blot image represents the changes in p-MYPT1 and pp-MLC levels, in conjunction with changes in tubulin acetylation. (b–d) Quantitative analysis of p-MYPT1, pp-MLC, and acetylated tubulin with densitometry. Data are shown as mean ± SEM (n = 4 experiments). HDACs, histone deacetylases; HLMVEC, human lung microvascular endothelial cell; LPS, lipopolysaccharide. *p < .05, **p < .01, ***p < .0001 versus control and versus LPS treatment
FIGURE 6.
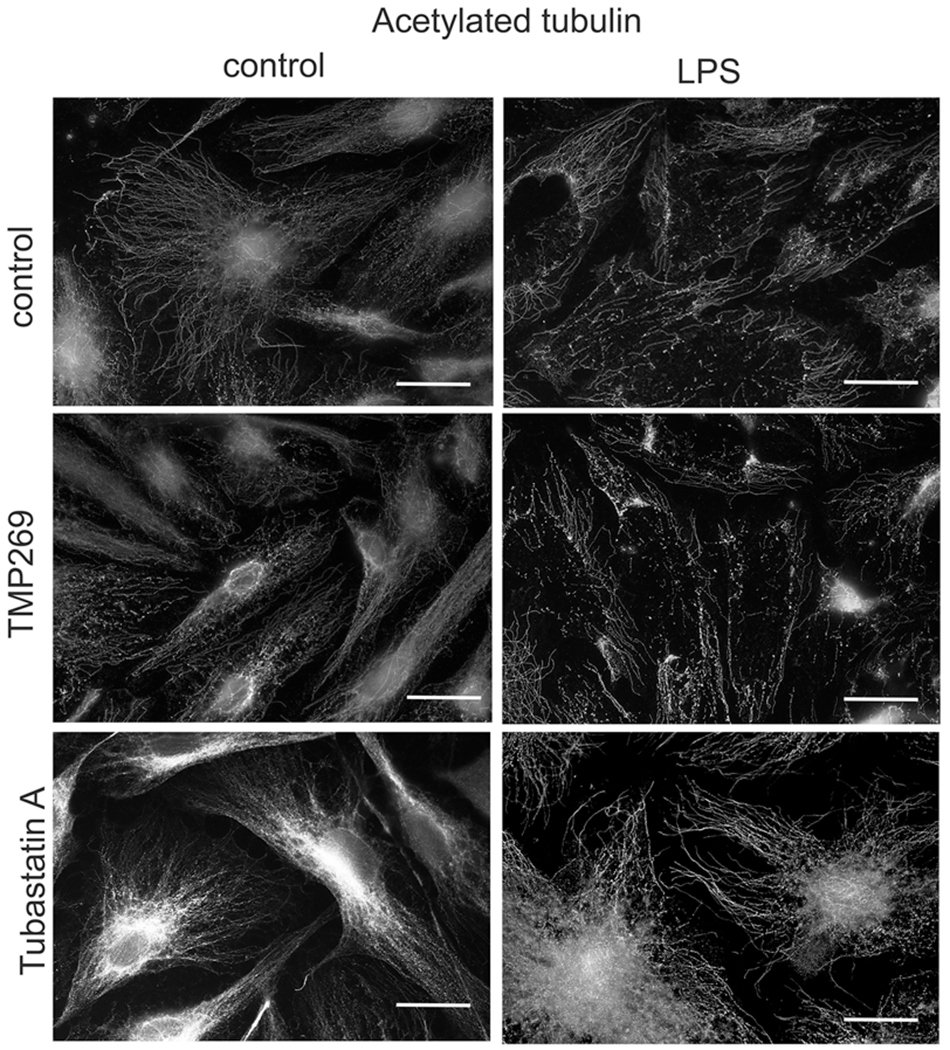
Inhibition of HDAC6, but not Class IIa HDACs increased tubulin acetylation and restored microtubule structure after LPS treatment. HLMVEC were plated in 12 well plates on glass coverslips until grown to confluence. Cells were pretreated either with Class IIa HDAC inhibitor (TMP269, 100 nM), HDAC6 inhibitor (Tubastatin A, 5 μM) or vehicle (control, 0.1% dimethyl sulfoxide) for 2 h then challenged with LPS (0.2 mg/ml) for 4 h and immunofluorescent staining was performed to visualize the acetylated tubulin. n = 3. Scale bar = 100 μm. HDACs, histone deacetylases; HLMVEC, human lung microvascular endothelial cell; LPS, lipopolysaccharide
3.6 |. ArgBP2 is involved in LPS-induced HDAC-mediated EC barrier dysfunction
Previous studies have shown that Rho signaling can be activated by HDAC7 (Class IIa HDAC) via increased expression of ArgBP2 (Martin et al., 2013). To analyze the role of ArgBP2 in mediating LPS-induced ALI, we first analyzed ArgBP2 expression after LPS challenge. We confirmed that ArgBP2 expression was significantly increased after LPS treatment, with a peak of expression at 2–4 h in HLMVEC (Figure 7a,b). Next, we examined the role of ArgBP2 in regulating EC barrier function by performing TER measurements. HLMVEC were transfected with specific siRNA against ArgBP2 or a nontargeting (control). Seventy-two hour posttransfection, TER was measured in the presence or absence of LPS. As shown in Figure 7c, silencing of ArgBP2 abrogated the LPS-induced decrease in TER suggesting that ArgBP2 plays a critical role in mediating the effects of LPS on endothelial barrier function. Inhibition of either Class IIa HDACs or the Class IIb HDAC6 had no effect on the baseline level of ArgBP2, but significantly attenuated the LPS-induced increase in ArgBP2 expression. These data support roles for Class IIa HDACs and HDAC6 in the activation of Rho signaling and increased EC permeability via transcriptional regulation of ArgBP2 expression.
FIGURE 7.
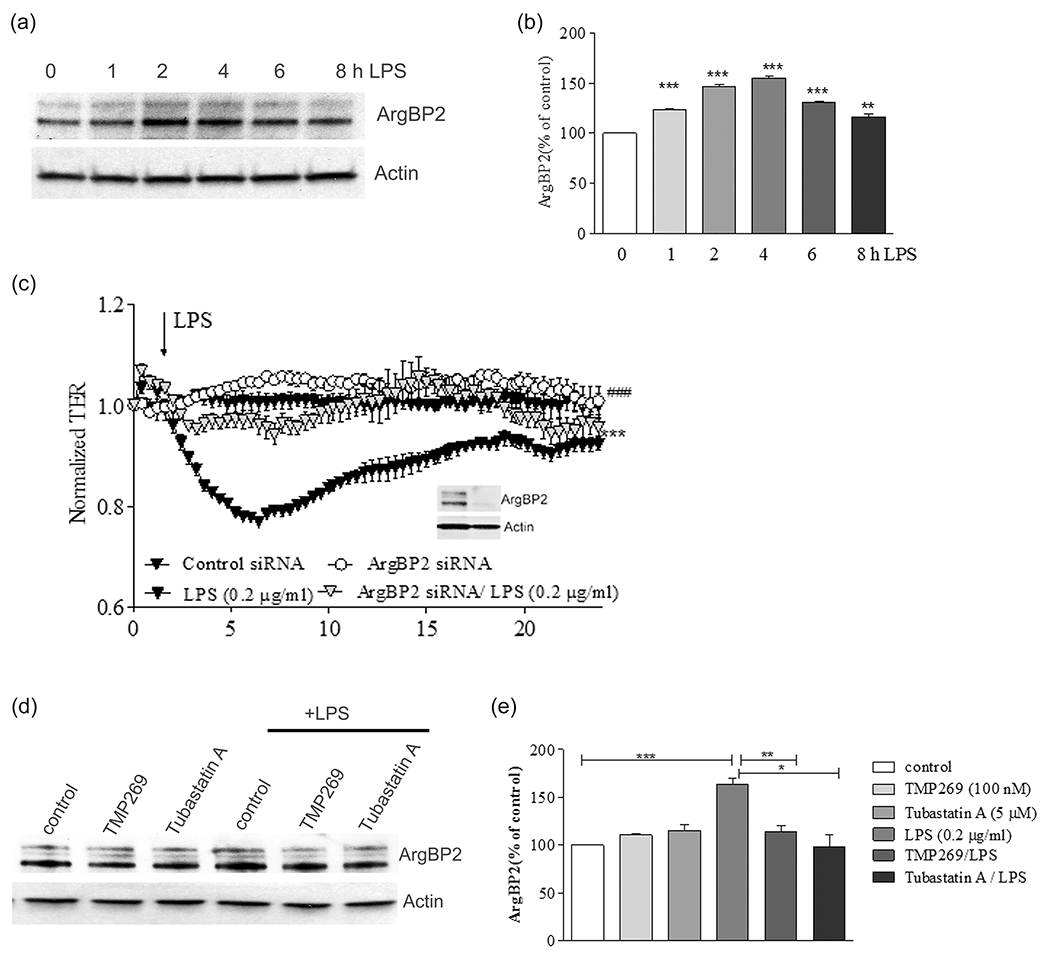
Effect of TMP269 and Tubastatin A on ArgBP2 expression in LPS treated endothelial cells. (a) HLMVEC were treated with LPS for 1, 2, 4, 6, and 8 h, then cells were collected to quantify ArgBP2 expression by western blot. Representative immunoblots show the change in the expression level of ArgBP2. Actin was used as a loading control. (b) Bar graphs show the changes in the expression of ArgBP2 quantified with scanning densitometry. (c) TER measurements were performed on HLMVEC treated with specific silencing RNA for ArgBP2 and control siRNA. Resistance of the cells was measured in the presence or absence of LPS. Inset shows representative immunoblots demonstrating silencing efficiency. (d) HLMVEC were pretreated either with Class IIa HDAC inhibitor (TMP269, 100 nM), HDAC6 inhibitor (Tubastatin A, 5 μM), or vehicle (control, 0.1% dimethyl sulfoxide) for 2 h then challenged with LPS (0.2 mg/ml) for 4 h. (e) Quantification of ArgBP2 expression. Results are expressed as mean ± SEM (n = 4 experiments). HDAC, histone deacetylase; HLMVEC, human lung microvascular endothelial cell; LPS, lipopolysaccharide; ns, not significant; siRNA, small interfering RNA. **p < .01, ***p < .0001
4 |. DISCUSSION
The specific roles of HDAC isoforms in ECs barrier regulation remain incompletely defined and at times, contradictory. While recent studies have shown that inhibition/downregulation of HDACs 3 and 6 attenuate ECs hyperpermeability in murine ALI models (Borgas et al., 2016; Joshi et al., 2015), other studies have suggested that inhibition of HDACs 1–3 promotes stress fiber formation and the loss of barrier function (Rolando et al., 2009, 2015). These discrepancies suggest that the roles of individual HDACs in various models of EC permeability differs significantly and highlights a critical need to assess the importance of HDAC isoforms in lung microvascular EC (HLMVEC) and their roles in pulmonary ECs barrier regulation.
To evaluate whether Class IIa HDACs are involved in LPS-induced ECs barrier compromise in vitro and in vivo we employed specific Class IIa inhibitor, TMP269 (Lobera et al., 2013). While the selectivity of TMP269 toward Class IIa HDACs was not tested in ECs, it has been confirmed using cell-based assays in cancer cells (Kikuchi et al., 2015). The IC50 for cell toxicity is in the range of 20–40 μM, which greatly exceeds our working concentration (100 nM). Our in vitro studies showed that at concentrations of 100 nM, TMP269 preferentially inhibited HDAC9 and HDAC7 and to a lesser extent, HDACs 4 and 5 with no effect on other HDAC isoforms (IC50 ≥ 2 μM; Lobera et al., 2013). TMP269 was developed to target the catalytic region of Class IIa HDACs (Lobera et al., 2013; which includes HDAC 4, 5, 7, and 9) and its inhibitory profile is consistent with selective actions on Class IIa HDACs activity.
TMP269 significantly attenuated LPS-induced ALI in the murine model and almost completely abolished LPS-induced decrease in TER in HLMVEC demonstrating the involvement of Class IIa HDACs in mediating LPS-induced disruption of the EC barrier in vitro and in vivo. Interestingly, previous studies have indicated that Class IIa HDACs have low deacetylase activity (Jones et al., 2008) and our study is the first report to show the involvement of Class IIa HDACs enzymatic activity in the regulation of lung vascular barrier. It has been reported that other HDACs such as HDAC3 (Class I HDAC) can specifically interact with HDAC7 (Class IIa HDAC) in vitro and in vivo (Fischle et al., 2001). Furthermore, HDAC3 serves as a one of the cofactors for Class II HDACs 7 and 4, which enhances their nuclear activity (Fischle et al., 2001). It was recently shown that HDAC3 is involved in LPS-induced HLMVEC barrier compromise and lung injury in a murine ALI model (Joshi et al., 2015). Whether HDAC3 interacts with Class II HDACs to enhance their enzymatic activity and ability to disrupt the ECs barrier remains to be determined.
Recent studies have demonstrated that the severity of E. coli-induced ALI may be linked to changes in HDAC7 expression (Kasotakis et al., 2019). While the experiments with HDAC7 depletion are convincing, the data on pharmacological inhibition of expression in response to the broad spectrum HDAC inhibitor, Trichostatin (TSA) are not so clear given the ability of TSA to target the catalytic sites of multiple HDACs (Finnin et al., 1999). Therefore, how TSA specifically decreases HDAC7 expression is not clear (Verin, 2020). However, our data in concert with published studies mentioned above (Kasotakis et al., 2019) clearly demonstrated the involvement of Class IIa HDACs in lung injury in sterile (LPS) and nonsterile (E. coli)-induced murine ALI models (Kasotakis et al., 2019).
The underlying mechanisms by which Class IIa HDACs compromise the function of the ECs barrier are not yet described. Published data suggest that they may activate the Rho pathway (Martin et al., 2013) which leads to increases in MYPT1 and MLC phosphorylation (Martin et al., 2013). Our data showing that TMP269 decreases LPS-induced MLC and MYPT1 phosphorylation strongly suggests the involvement of Class IIa HDACs in mediating LPS-induced activation of Rho-dependent contractile mechanisms.
The Rho pathway can be activated through HDAC6 (Class IIb HDAC)-mediated tubulin deacetylation, which promotes MT destabilization and loss of ECs barrier function (Hubbert et al., 2002; Karki et al., 2019; Lee et al., 2008; Miyake et al., 2016). Consistent with these published data, specific inhibition of HDAC6 by Tubastatin A (Butler et al., 2010) almost completely abolished LPS-induced ECs permeability, and attenuated LPS-induced increase in MYPT1 and MLC phosphorylation, reflective of Rho activation and increased tubulin acetylation, consistent with MT stabilization. Accordingly, inhibition of HDAC6 restored and even enhanced the MT network following LPS administration. In contrast to these data, while TMP269 produced similar effects on LPS-induced MLC and MYPT1 phosphorylation, and EC permeability, it did not affect tubulin acetylation or MT structure in the presence or absence of LPS. Collectively, these results strongly suggest that Class IIa HDACs activate the Rho pathway and increase EC permeability in a MT-independent manner. Interestingly, TMP269 and Tubastatin A alone have very modest effect (if any) on MLC and MYPT1 phosphorylation under basal conditions, but they substantially attenuate the effects of LPS, suggesting that LPS activates both HDAC6 and Class II HDACs. In contrast, Tubastatin A robustly increased the expression of acetyl tubulin independent of LPS treatment, suggesting that HDAC6 is constitutively active against tubulin.
The absence of effect of TMP269 on MT structure is somewhat surprising since it was shown that HDAC5 (Class IIA HDAC) can deacetylate tubulin in neurons (Cho & Cavalli, 2012). The apparent discrepancy may be explained by the following reasons: first, in contrast to other classes of HDACs, Class II HDACs are tissue selective and unlike HDAC7, HDAC5 is not abundantly expressed in the endothelium (Haberland et al., 2009); second, HDAC5 is not involved in LPS-induced lung injury; third, at the given concentration of TMP269 (2.5 mg/kg) we did not achieve complete HDAC5 inhibition. Whether or not HDAC 5 is involved in LPS-induced ALI will require further studies.
It was recently proposed that nuclear export of HDAC7 may release transcriptional repression of the adapter protein, ArgBP2, leading to activation of Rho signaling in EC (Martin et al., 2013). Our data indicate that LPS increases ArgBP2 expression in a time-dependent manner and that inhibition of Class IIa HDACs by TMP269 decreases its expression supporting the involvement of Class IIa HDACs in the activation of Rho signaling and ECs permeability via transcriptional regulation of ArgBP2 expression. Importantly, depletion of ArgBP2 abrogated LPS-induced decrease in TER, supporting a functional role in mediating LPS-induced EC barrier disruption. Interestingly, HDAC6 inhibition also decreased ArgBP2 expression. This was somewhat surprising because HDAC6 is predominantly located in the cytosol. However, recent studies have demonstrated that HDAC6 is able to deacetylate the transcription factor EB leading to its nuclear import and activation of transcriptional activity in kidney cells (Brijmohan et al., 2018). The precise mechanisms of HDAC6-mediated regulation of ArgBP2 expression will require additional studies.
Class IIa HDACs may also participate in LPS-induced ECs barrier compromise via direct deacetylation (destabilization) of cytoskeletal targets. HDAC7 binds to cytoskeletal proteins such as MLCP, (β-catenin, and filamin B in functional complexes in various cell types including EC (Margariti et al., 2010; Parra, Mahmoudi, & Verdin, 2007; Su et al., 2013), supporting the concept that Class IIa HDACs may have direct effect on the cytoskeleton. Whether Class IIa HDACs are involved in ECs barrier compromise through direct deacetylation/destabilization of cytoskeletal targets remains to be determined.
While our study is focused on the role of Class IIa HDAC activity in ECs barrier regulation, we cannot exclude the possibility that Class IIa HDACs affect lung epithelial barrier in a similar model. Upregu-lation of pan-HDAC activity was demonstrated in nasal epithelial cells of patients with allergic rhinitis. Furthermore, pan-HDAC inhibition restored epithelial integrity in cells and in mice by promoting expression of tight junction proteins (Steelant et al., 2019). In addition, HDAC1 and HDAC9 (Class IIa HDAC) were upregulated in bronchial epithelial cells from asthmatic patients and the inhibition of HDAC activity restores compromised barrier by increasing tight junction expression (Wawrzyniak et al., 2017). Surprisingly, some data of literature showed that LPS decreases HDAC activity in alveolar epithelial cells (Chen, Luo, Li, & Ran, 2009). Whether Class IIa HDACs participate in alveolar–epithelial barrier dysfunction induced by LPS remains to be determined.
Collectively, our studies herein demonstrate that Class IIa HDACs are directly involved in LPS-induced ECs barrier disruption in vitro and in the pulmonary vascular injury seen in the LPS-induced murine ALI model. In vitro studies on human lung microvascular ECs demonstrate that in contrast to Class IIb HDACs such as HDAC6, Class IIa HDACs-mediated LPS-induced ECs hyperpermeability involves ArgBP2-mediated activation of Rho signaling, but not MT destabilization (Figure 8). These data highlight the functional differences between two classes of HDACs in the regulation of ECs permeability.
FIGURE 8.
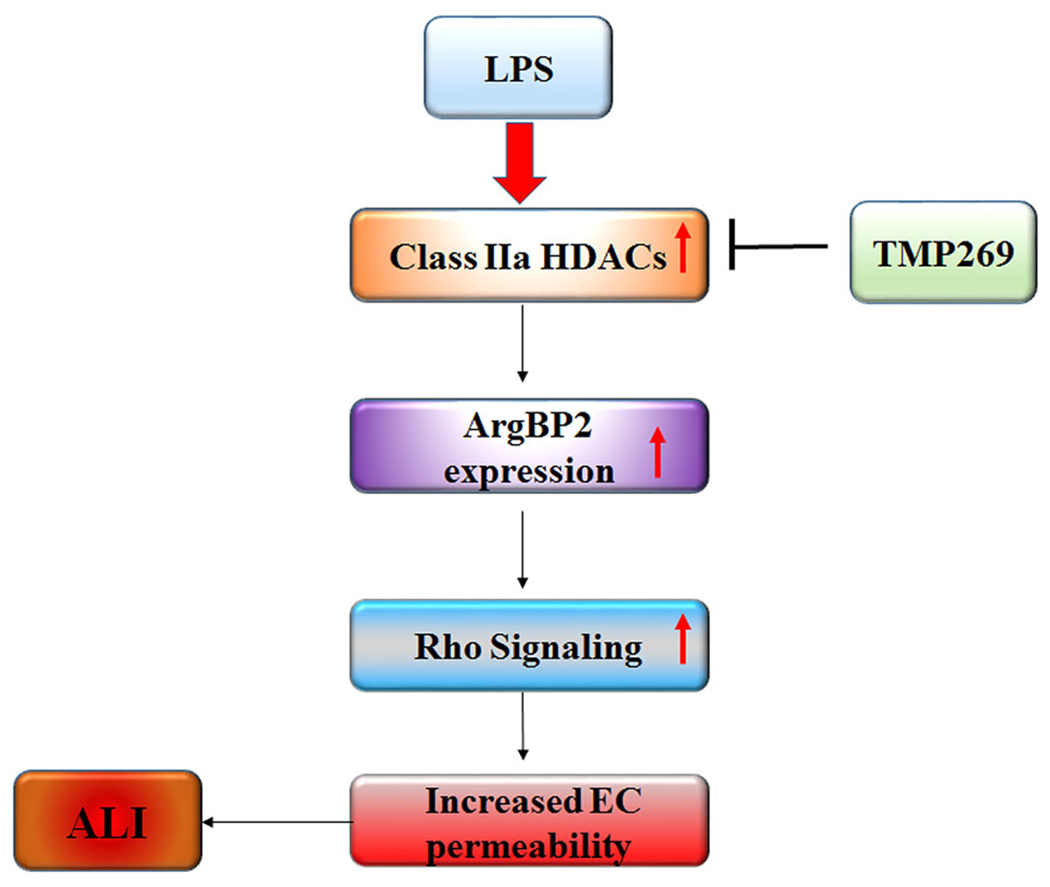
Schematic illustration of proposed mechanisms of Class IIa HDACs involvement in LPS-induced ALI. The Class IIa HDAC inhibitor attenuates the effect of LPS-induced ECs hyperpermeability which involves ArgBP2-mediated activation of Rho signaling. ALI, acute lung injury; EC, endothelial cell; HDACs, histone deacetylases; LPS, lipopolysaccharide
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the National Institute of Health Program Project grant HL101902 (A.V. and D.F.), and NIH/NHLBI grant R01 HL134934 (Y.S.), and AHA Postdoctoral Fellowship 18POST33990193 (A.K.), and Career Development Award 18CDA34110225 (L.K.). The authors thank Dr D. Xu and Mrs M. Palombo for technical support.
Funding information
National Institute of Health Program Project, Grant/Award Number: NIH: HL101902; NIH/NHLBI, Grant/Award Number: R01 HL134934; American Heart Association, Grant/Award Numbers: 18CDA34110225, 18POST33990193
Footnotes
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
DATA AVAILABILITY STATEMENT
The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- Adcock IM (2007). HDAC inhibitors as anti-inflammatory agents. British Journal of Pharmacology, 150(7), 829–831. 10.1038/sj.bjp.0707166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal S, Gross CM, Kumar S, Dimitropolou C, Sharma S, Gorshkov BA, … Black SM. (2014). DDAH II over-expression attenuates lipopolysaccharide mediated lung leak in acute lung injury. American Journal of Respiratory Cell and Molecular Biology, 50(3), 614–625. 10.1165/rcmb.2013-0193OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgas D, Chambers E, Newton J, Ko J, Rivera S, Rounds S, & Lu Q (2016). Cigarette smoke disrupted lung endothelial barrier integrity and increased susceptibility to acute lung injury via histone deacetylase 6. American Journal of Respiratory Cell and Molecular Biology, 54(5), 683–696. 10.1165/rcmb.2015-0149OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brijmohan AS, Batchu SN, Majumder S, Alghamdi TA, Thieme K, McGaugh S, … Advani A. (2018). HDAC6 inhibition promotes transcription factor EB activation and is protective in experimental kidney disease. Frontiers in Pharmacology, 9, 34. 10.3389/fphar.2018.00034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, & Kozikowski AP (2010). Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. Journal of the American Chemical Society, 132(31), 10842–10846. 10.1021/ja102758v [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catravas JD, Snead C, Dimitropoulou C, Chang AS, Lucas R, Verin AD, & Black SM (2010). Harvesting, identification and barrier function of human lung microvascular endothelial cells. Vascular Pharmacology, 52(5-6), 175–181. 10.1016/j.vph.2009.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cestra G, Toomre D, Chang S, & De Camilli P (2005). The Abl/Arg substrate ArgBP2/nArgBP2 coordinates the function of multiple regulatory mechanisms converging on the actin cytoskeleton. Proceedings of the National Academy of Sciences of the United States of America, 102(5), 1731–1736. 10.1073/pnas.0409376102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Young BD, Li S, Qi X, Richardson JA, & Olson EN (2006). Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell, 126(2), 321–334. 10.1016/j.cell.2006.05.040 [DOI] [PubMed] [Google Scholar]
- Chatterjee A, Snead C, Yetik-Anacak G, Antonova G, Zeng J, & Catravas JD (2008). Heat shock protein 90 inhibitors attenuate LPS-induced endothelial hyperpermeability. American Journal of Physiology: Lung Cellular and Molecular Physiology, 294(4), L755–L763. 10.1152/ajplung.00350.2007 [DOI] [PubMed] [Google Scholar]
- Chen J, Luo J, Li B, & Ran P (2009). E1A has no effect on LPS-induced IL-6 secretion in rat alveolar epithelial cells. Respiration, 78(1), 84–92. 10.1159/000209743 [DOI] [PubMed] [Google Scholar]
- Cho Y, & Cavalli V (2012). HDAC5 is a novel injury-regulated tubulin deacetylase controlling axon regeneration. EMBO Journal, 31(14), 3063–3078. 10.1038/emboj.2012.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AJ, & Pender SL (2011). Histone deacetylase inhibitors and their potential role in inflammatory bowel diseases. Biochemical Society Transactions, 39(4), 1092–1095. 10.1042/BST0391092 [DOI] [PubMed] [Google Scholar]
- Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, … Pavletich NP. (1999). Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature, 401(6749), 188–193. 10.1038/43710 [DOI] [PubMed] [Google Scholar]
- Fischle W, Dequiedt F, Fillion M, Hendzel MJ, Voelter W, & Verdin E (2001). Human HDAC7 histone deacetylase activity is associated with HDAC3 in vivo. Journal of Biological Chemistry, 276(38), 35826–35835. 10.1074/jbc.M104935200 [DOI] [PubMed] [Google Scholar]
- Fontaine SN, Sabbagh JJ, Baker J, Martinez-Licha CR, Darling A, & Dickey CA (2015). Cellular factors modulating the mechanism of Tau protein aggregation. Cellular and Molecular Life Science, 72(10), 1863–1879. 10.1007/s00018-015-1839-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan M, Varona S, Orriols M, Rodriguez JA, Aguilo S, Dilme J, … Rodriguez C. (2016). Induction of histone deacetylases (HDACs) in human abdominal aortic aneurysm: Therapeutic potential of HDAC inhibitors. Disease Models & Mechanisms, 9(5), 541–552. 10.1242/dmm.024513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio E, & Brancolini C (2016). Regulation of class IIa HDAC activities: It is not only matter of subcellular localization. Epigenomics, 8(2), 251–269. 10.2217/epi.15.106 [DOI] [PubMed] [Google Scholar]
- Glozak MA, & Seto E (2007). Histone deacetylases and cancer. Oncogene, 26(37), 5420–5432. 10.1038/sj.onc.1210610 [DOI] [PubMed] [Google Scholar]
- Gross CM, Kellner M, Wang T, Lu Q, Sun X, Zemskov EA, … Black SM. (2018). LPS-induced Acute Lung Injury Involves NF-kappaB-mediated downregulation of SOX18. American Journal of Respiratory Cell and Molecular Biology, 58(5), 614–624. 10.1165/rcmb.2016-0390OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, & Olson EN (2009). The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nature Reviews Genetics, 10(1), 32–42. 10.1038/nrg2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halili MA, Andrews MR, Labzin LI, Schroder K, Matthias G, Cao C, … Sweet MJ. (2010). Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. Journal of Leukocyte Biology, 87(6), 1103–1114. 10.1189/jlb.0509363 [DOI] [PubMed] [Google Scholar]
- Halili MA, Andrews MR, Sweet MJ, & Fairlie DP (2009). Histone deacetylase inhibitors in inflammatory disease. Current Topics in Medicinal Chemistry, 9(3), 309–319. [DOI] [PubMed] [Google Scholar]
- Hartshorne DJ (1998). Myosin phosphatase: Subunits and interactions. Acta Physiologica Scandinavica, 164(4), 483–493. 10.1046/j.1365-201X.1998.00447.x [DOI] [PubMed] [Google Scholar]
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, … Yao TP. (2002). HDAC6 is a microtubule-associated deacetylase. Nature, 417(6887), 455–458. 10.1038/417455a [DOI] [PubMed] [Google Scholar]
- Hull EE, Montgomery MR, & Leyva KJ (2016). HDAC inhibitors as epigenetic regulators of the immune system: Impacts on cancer therapy and inflammatory diseases. BioMed Research International, 2016, 8797206–8797215. 10.1155/2016/8797206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Barnes PJ, & Adcock IM (2000). Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Molecular and Cellular Biology, 20(18), 6891–6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ER, & Matthay MA (2010). Acute lung injury: Epidemiology, pathogenesis, and treatment. Journal of Aerosol Medicine and Pulmonary Drug Delivery, 23(4), 243–252. 10.1089/jamp.2009.0775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, Altamura S, De Francesco R, Gallinari P, Lahm A, Neddermann P, … Steinkuhler C. (2008). Probing the elusive catalytic activity of vertebrate class IIa histone deacetylases. Bioorganic & Medicinal Chemistry Letters, 18(6), 1814–1819. 10.1016/j.bmcl.2008.02.025 [DOI] [PubMed] [Google Scholar]
- Joo EE, & Yamada KM (2014). MYPT1 regulates contractility and microtubule acetylation to modulate integrin adhesions and matrix assembly. Nature Communications, 5, 3510. 10.1038/ncomms4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi AD, Barabutis N, Birmpas C, Dimitropoulou C, Thangjam G, Cherian-Shaw M, … Catravas JD (2015). Histone deacetylase inhibitors prevent pulmonary endothelial hyperpermeability and acute lung injury by regulating heat shock protein 90 function. American Journal of Physiology: Lung Cellular and Molecular Physiology, 309(12), L1410–L1419. 10.1152/ajplung.00180.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao HY, Verdel A, Tsai CC, Simon C, Juguilon H, & Khochbin S (2001). Mechanism for nucleocytoplasmic shuttling of histone deacetylase 7. Journal of Biological Chemistry, 276(50) ,47496–47507. 10.1074/jbc.M107631200 [DOI] [PubMed] [Google Scholar]
- Karki P, Ke Y, Tian Y, Ohmura T, Sitikov A, Sarich N, … Birukova AA. (2019). Staphylococcus aureus-induced endothelial permeability and inflammation are mediated by microtubule destabilization. Journal of Biological Chemistry, 294(10), 3369–3384. 10.1074/jbc.RA118.004030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasa A, Csortos C, & Verin AD (2015). Cytoskeletal mechanisms regulating vascular endothelial barrier function in response to acute lung injury. Tissue Barriers, 3(1-2), e974448. 10.4161/21688370.2014.974448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasotakis G, Kintsurashvili E, Galvan MD, Graham C, Purves JT, Agarwal S, … Remick DG. (2019). Histone deacetylase 7 inhibition in a murine model of gram-negative pneumonia-induced acute lung injury. Shock, 53(3), 344–351. 10.1097/SHK.0000000000001372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi S, Suzuki R, Ohguchi H, Yoshida Y, Lu D, Cottini F, … Anderson KC. (2015). Class IIa HDAC inhibition enhances ER stress-mediated cell death in multiple myeloma. Leukemia, 29(9), 1918–1927. 10.1038/leu.2015.83 [DOI] [PubMed] [Google Scholar]
- Kim KM, Csortos C, Czikora I, Fulton D, Umapathy NS, Olah G, & Verin AD (2012). Molecular characterization of myosin phosphatase in endothelium. Journal of Cellular Physiology, 227(4), 1701–1708. 10.1002/jcp.22894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs-Kasa A, Gorshkov BA, Kim KM, Kumar S, Black SM, Fulton DJ, … Verin AD (2016). The protective role of MLCP-mediated ERM dephosphorylation in endotoxin-induced lung injury in vitro and in vivo. Scientific Reports, 6, 39018. 10.1038/srep39018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs-Kasa A, Kim KM, Cherian-Shaw M, Black SM, Fulton DJ, & Verin AD (2018). Extracellular adenosine-induced Rac1 activation in pulmonary endothelium: Molecular mechanisms and barrier-protective role. Journal of Cellular Physiology, 233(8), 5736–5746. 10.1002/jcp.26281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann N, Wroblowski S, Knyphausen P, Boor S, Brenig J, Zienert AY, … Lammers M. (2016). Structural and mechanistic insights into the regulation of the fundamental Rho regulator RhoGDIalpha by lysine acetylation. Journal of Biological Chemistry, 291(11), 5484–5499. 10.1074/jbc.M115.707091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, … Gallinari P. (2007). Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proceedings of the National Academy of Sciences of the United States of America, 104(44), 17335–17340. 10.1073/pnas.0706487104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs L, Kovacs-Kasa A, Verin AD, Fulton D, Lucas R, & Su Y (2018). Histone deacetylases in vascular permeability and remodeling associated with acute lung injury. Vessel Plus, 2, 15. 10.20517/2574-1209.2018.06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Lim KH, Guo X, Kawaguchi Y, Gao Y, Barrientos T, … Yao TP. (2008). The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Research, 68(18), 7561–7569. 10.1158/0008-5472.CAN-08-0188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobera M, Madauss KP, Pohlhaus DT, Wright QG, Trocha M, Schmidt DR, … Nolan MA. (2013). Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nature Chemical Biology, 9(5), 319–325. 10.1038/nchembio.1223 [DOI] [PubMed] [Google Scholar]
- Margariti A, Zampetaki A, Xiao Q, Zhou B, Karamariti E, Martin D, … Zeng L. (2010). Histone deacetylase 7 controls endothelial cell growth through modulation of beta-catenin. Circulation Research, 106(7), 1202–1211. 10.1161/CIRCRESAHA.109.213165 [DOI] [PubMed] [Google Scholar]
- Martin M, Geudens I, Bruyr J, Potente M, Bleuart A, Lebrun M, … Dequiedt F. (2013). PP2A regulatory subunit Balpha controls endothelial contractility and vessel lumen integrity via regulation of HDAC7. EMBO Journal, 32(18), 2491–2503. 10.1038/emboj.2013.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Potente M, Janssens V, Vertommen D, Twizere JC, Rider MH, … Dequiedt F. (2008). Protein phosphatase 2A controls the activity of histone deacetylase 7 during T cell apoptosis and angiogenesis. Proceedings of the National Academy of Sciences of the United States of America, 105(12), 4727–4732. 10.1073/pnas.0708455105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura F, & Hartshorne DJ (2008). Myosin phosphatase target subunit: Many roles in cell function. Biochemical and Biophysical Research Communications, 369(1), 149–156. 10.1016/j.bbrc.2007.12.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, … Kuebler WM, Acute Lung Injury in Animals Study, G. (2011). An official American Thoracic Society workshop report: Features and measurements of experimental acute lung injury in animals. American Journal of Respiratory Cell and Molecular Biology, 44(5), 725–738. 10.1165/rcmb.2009-0210ST [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, & Olson EN (2001). Control of muscle development by dueling HATs and HDACs. Current Opinion in Genetics & Development, 11(5), 497–504. [DOI] [PubMed] [Google Scholar]
- Miyake Y, Keusch JJ, Wang L, Saito M, Hess D, Wang X, … Matthias P. (2016). Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nature Chemical Biology, 12(9), 748–754. 10.1038/nchembio.2140 [DOI] [PubMed] [Google Scholar]
- New M, Sheikh S, Bekheet M, Olzscha H, Thezenas ML, Care MA, … Thangue NB. (2016). TLR adaptor protein MYD88 mediates sensitivity to HDAC inhibitors via a cytokine-dependent mechanism. Cancer Research, 76(23), 6975–6987. 10.1158/0008-5472.CAN-16-0504 [DOI] [PubMed] [Google Scholar]
- Parra M (2015). Class IIa HDACs: New insights into their functions in physiology and pathology. FEBS Journal, 282(9), 1736–1744. 10.1111/febs.13061 [DOI] [PubMed] [Google Scholar]
- Parra M, Mahmoudi T, & Verdin E (2007). Myosin phosphatase dephosphorylates HDAC7, controls its nucleocytoplasmic shuttling, and inhibits apoptosis in thymocytes. Genes and Development, 21(6), 638–643. 10.1101/gad.1513107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, &Wang P. (2017). Class IIa HDACs inhibitor TMP269 promotes M1 polarization of macrophages after spinal cord injury. Journal of Cellular Biochemistry, 119(4), 3081–3090. 10.1002/jcb.26446 [DOI] [PubMed] [Google Scholar]
- Qu X, Yu H, Jia B, Yu X, Cui Q, Liu Z, & Chu Y (2016). Association of downregulated HDAC 2 with the impaired mitochondrial function and cytokine secretion in the monocytes/macrophages from gestational diabetes mellitus patients. Cell Biology International, 40(6), 642–651. 10.1002/cbin.10598 [DOI] [PubMed] [Google Scholar]
- Regna NL, Vieson MD, Gojmerac AM, Luo XM, Caudell DL, & Reilly CM (2015). HDAC expression and activity is upregulated in diseased lupus-prone mice. International Immunopharmacology, 29(2), 494–503. 10.1016/jjntimp.2015.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey RW, Chakraborty AR, Basseville A, Luchenko V, Bahr J, Zhan Z, & Bates SE (2011). Histone deacetylase inhibitors: emerging mechanisms of resistance. Molecular pharmaceutics, 8(6), 2021–2031. 10.1021/mp200329f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolando M, Munro P, Stefani C, Auberger P, Flatau G, & Lemichez E (2009). Injection of Staphylococcus aureus EDIN by the Bacillus anthracis protective antigen machinery induces vascular permeability. Infection and Immunity, 77(9), 3596–3601. 10.1128/IAI.00186-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolando M, Stefani C, Doye A, Acosta MI, Visvikis O, Yevick HG, … Lemichez E. (2015). Contractile actin cables induced by Bacillus anthracis lethal toxin depend on the histone acetylation machinery. Cytoskeleton, 72(10), 542–556. 10.1002/cm.21256 [DOI] [PubMed] [Google Scholar]
- Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, … Hudson LD. (2005). Incidence and outcomes of acute lung injury. New England Journal of Medicine, 353(16), 1685–1693. 10.1056/NEJMoa050333 [DOI] [PubMed] [Google Scholar]
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, & van Kuilenburg AB (2003). Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochemical Journal, 370(Pt 3), 737–749. 10.1042/BJ20021321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S, Lasky JA, Guo W, Nguyen H, Mai A, Danchuk S, … Shan B. (2011). Pharmacological inhibition of HDAC6 attenuates endothelial barrier dysfunction induced by thrombin. Biochemical and Biophysical Research Communications, 408(4), 630–634. 10.1016/j.bbrc.2011.04.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerrett SJ, Niederman MS, & Fein AM (1989). Respiratory infections and acute lung injury in systemic illness. Clinics in Chest Medicine, 10(4), 469–502. [PubMed] [Google Scholar]
- Song Y, & Brady ST (2015). Post-translational modifications of tubulin: Pathways to functional diversity of microtubules. Trends in Cell Biology, 25(3), 125–136. 10.1016/j.tcb.2014.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza Xavier Costa N, Ribeiro Junior G, Dos Santos Alemany AA, Belotti L, Zati DH, Frota Cavalcante M, dddd dddd, & Ferraz da Silva LF. (2017). Early and late pulmonary effects of nebulized LPS in mice: An acute lung injury model. PLoS One, 12(9):e0185474de. 10.1371/journal.pone.0185474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steelant B, Wawrzyniak P, Martens K, Jonckheere AC, Pugin B, Schrijvers R, … Hellings PW. (2019). Blocking histone deacetylase activity as a novel target for epithelial barrier defects in patients with allergic rhinitis. Journal of Allergy and Clinical Immunology, 144(5), 1242–1253e1247. 10.1016/j.jaci.2019.04.027 [DOI] [PubMed] [Google Scholar]
- Su YT, Gao C, Liu Y, Guo S, Wang A, Wang B, & Kao HY (2013). Monoubiquitination of filamin B regulates vascular endothelial growth factor-mediated trafficking of histone deacetylase 7. Molecular and Cellular Biology, 33(8), 1546–1560. 10.1128/MCB.01146-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verin AD (2020). Letter to the Editor: Regarding the Manuscript of Kasotakis et al “Histone Deacetylase 7 Inhibition in a Murine Model of Gram-negative Pneumonia-induced Acute Lung Injury.” Shock 53: 344-351, 2020. Shock, 53(3), 375–377. 10.1097/SHK.0000000000001430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawrzyniak P, Wawrzyniak M, Wanke K, Sokolowska M, Bendelja K, Rückert B, … Akdis CA. (2017). Regulation of bronchial epithelial barrier integrity by type 2 cytokines and histone deacetylases in asthmatic patients. Journal of Allergy and Clinical Immunology, 139(1), 93–103. 10.1016/j.jaci.2016.03.050 [DOI] [PubMed] [Google Scholar]
- Yu J, Ma Z, Shetty S, Ma M, & Fu J (2016). Selective HDAC6 inhibition prevents TNF-alpha-induced lung endothelial cell barrier disruption and endotoxin-induced pulmonary edema. American Journal of Physiology: Lung Cellular and Molecular Physiology, 311(1), L39–L47. 10.1152/ajplung.00051.2016 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.