

Summary
Recombination is crucial for crop breeding because it can break linkage drag and generate novel allele combinations. However, the high‐resolution recombination landscape and its driving forces in soybean are largely unknown. Here, we constructed eight recombinant inbred line (RIL) populations and genotyped individual lines using the high‐density 600K SoySNP array, which yielded a high‐resolution recombination map with 5636 recombination sites at a resolution of 1.37 kb. The recombination rate was negatively correlated with transposable element density and GC content but positively correlated with gene density. Interestingly, we found that meiotic recombination was enriched at the promoters of active genes. Further investigations revealed that chromatin accessibility and active epigenetic modifications promoted recombination. Our findings provide important insights into the control of homologous recombination and thus will increase our ability to accelerate soybean breeding by manipulating meiotic recombination rate.
Keywords: soybean, recombination landscape, chromatin accessibility, epigenetic modification, high‐density 600K SoySNP array
Introduction
Meiotic recombination is a fundamental biological process for organisms, which is initiated by DNA double‐strand breaks (DSBs) catalysed by the topoisomerase‐like protein SPO11 (Keeney et al., 1997) and can result in two divergent recombination outcomes: crossovers (COs) and non‐crossovers (NCOs) (Zelkowski et al., 2019). COs are involved in the reciprocal exchange of genetic material between homologous chromosomes; in contrast, NCOs involve no exchange of genetic material or gene conversion. In Arabidopsis, most COs are generated through the class I pathway, also known as the ZMM pathway (Yelina et al., 2015). Some key proteins involved in the ZMM pathway have been identified and functionally characterized, such as MSH4, MSH5, MER3, HEI10, ZIP4, SHOC1, PTD and ZYP1 (Osman et al., 2011; Wang and Copenhaver, 2018; Yelina et al., 2015).
The occurrence of meiotic recombination could be influenced by many genomic features and epigenetic factors. For instance, several specific DNA sequence motifs, such as poly‐A, CTT‐repeat and CNN‐repeat motifs, are overrepresented at recombination sites in plants and are suspected to act as cis‐elements to confer recombination pattern (Lawrence et al., 2017). In many mammalian species, recombination hotspots are mainly determined by PRDM9, which targets specific DNA sequence motifs and deposits H3K4me3 and H3K36me3 in surrounding regions (Paigen and Petkov, 2018). Although PRDM9 homologues have not been found in plants, an increasing number of studies have revealed that epigenetic marks and chromatin accessibility play a crucial role in recombination formation (Choi et al., 2013; Marand et al., 2019; Yelina et al., 2015).
Recombination is critical for crop breeding because it can disrupt linkage drag between advantageous and deleterious alleles and generate novel combinations of valuable and improved alleles (Lambing et al., 2017; Wijnker and de Jong, 2008). Previously, a number of studies focus on the pattern of meiotic recombination, and its regulatory factors were investigated in important crops, including rice (Ma et al., 2016; Wu et al., 2003), maize (Bauer et al., 2013; Pan et al., 2016; Rodgers‐Melnick et al., 2015) and wheat (Gardiner et al., 2019; Jordan et al., 2018), which helped breeders accelerate the creation of new varieties. As a major economic crop, soybean (Glycine max [L]. Merr.) supplies over 59% and 70% of plant oil and vegetable protein in the world respectively (Fang et al., 2021). However, comprehensive and systematic investigations of the pattern of recombination at a genome‐wide and high‐resolution level in soybean are lacking, and the recombination landscape and its driving forces are largely unknown.
To this end, we customized an ultra‐high‐density 600K SoySNP array for soybean, genotyped eight soybean recombinant inbred line (RIL) populations and constructed a high‐resolution recombination map. Our systematic investigations revealed that recombination events were significantly enriched in genic regions and that both genomic features and epigenetic forces play important roles in determining the recombination landscapes of soybean.
Results
Development of the high‐density 600K SoySNP array
To genotype individual lines of RIL populations for the construction of a high‐resolution recombination map in soybean, we first developed a high‐density single‐nucleotide polymorphism (SNP) array that at most covers the representative SNPs. To this end, previously obtained resequencing data of 854 representative wild soybeans, soybean landraces and cultivated soybean accessions collected from different regions and countries (Fang et al., 2017; Zhou et al., 2015) were used. Following a series of stringent selection procedures (Figure 1a), a total of 600 009 SNPs were chosen for incorporation into the array (Figure 1b). The interval distance between each pair of markers was ~1.6 kb, and an interval distance greater than 5 kb accounted for only 2.4% of the pairs (Figure 1c).
Figure 1.
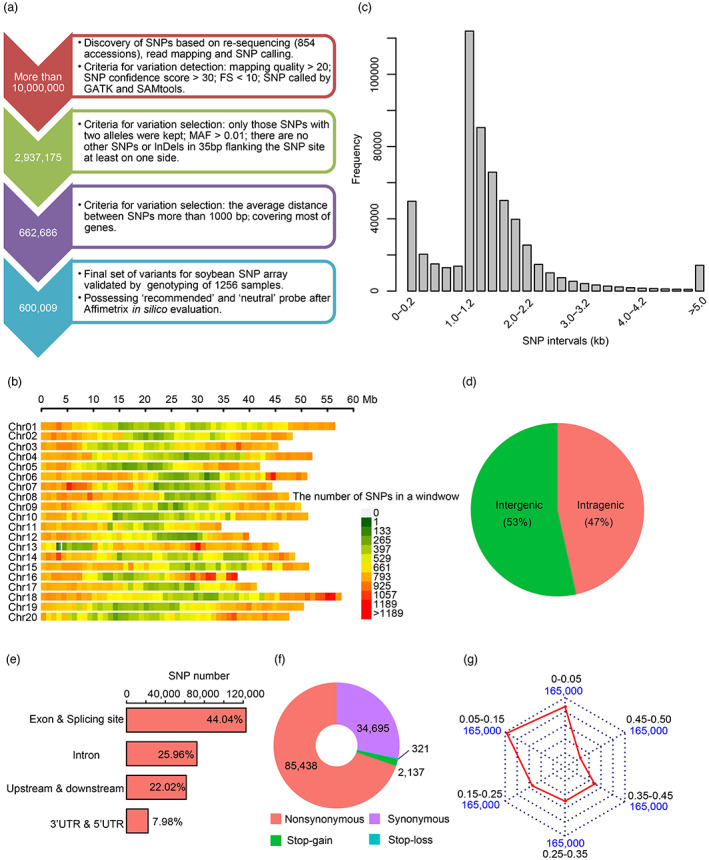
Construction of the high‐density 600K SoySNP array and its characteristics. (a) The design process of the 600K SoySNP array in soybean. MAF, minor allele frequency. (b) The distribution of SNPs along the 20 chromosomes. The number of SNPs in the non‐overlapping window with the size of 1 Mb was counted along the chromosome. (c) The distribution of SNP intervals. (d) The proportions of SNPs in intergenic and intragenic regions. (e) The classification of SNPs in intragenic regions. According to the genome annotation, SNPs in intragenic regions were classed in exonic regions (locating in a coding exon), splicing sites (within 2 bp of a splicing junction), 5′ untranslated regions (UTRs) and 3′ UTRs, intronic regions (overlapping with an intron), upstream (within a 1 kb region upstream from the transcription start site) and downstream regions (locating in a 1 kb region downstream from the transcription termination site). (f) Impacts of high‐quality SNPs in coding regions. (g) The distribution of SNPs with different minor allele frequency (MAF) in the natural soybean populations. Red lines represent the number of SNPs in various intervals for MAF.
Compared to intergenic SNPs, SNPs located in genic regions are more promising as functional makers. In the 600K SoySNP array, approximately 47% of SNPs were located in intragenic regions (Figure 1d), especially the exon regions and splicing sites (Figure 1e), resulting in most of the protein‐encoding genes in the genome being covered (50 659/55 589, 91.1%). Of the SNPs in the coding regions, 85 438 were in non‐synonymous sites, 34 695 were in synonymous sites, and the remaining 2458 SNPs had a strong predicted impact on gene function, such as a stop‐loss or stop‐gain impact (Figure 1f). Additionally, the majority of SNPs (76.0%) had a minor allele frequency (MAF) of more than 0.05 in the natural resequencing populations (Figure 1g), indicating that enough polymorphic markers could be detected between the two soybean accessions.
To evaluate the repeatability of the 600K SoySNP array, six soybean accessions were randomly selected and genotyped two times using this array. The total call rates of the six lines were up to 99.4%, and the error rate varied from 0.1% to 1.4% (Figure S1a). To examine the accuracy of this array, high‐depth resequencing with an average depth of ~20.5× was carried out for parental soybean accessions (Table S1). Genotypes called from resequencing and the SNP array were compared. The pairwise concordance rates of the two methods ranged from 94.7% (PI 518664) to 97.2% (Sui Nong No.14) (Figure S1b). The results suggested that the 600K SoySNP array had high stability and accuracy for genotyping of soybean populations.
Constructing and genotyping soybean RIL populations
To capture a comprehensive recombination landscape in soybean, we developed and genotyped eight RIL populations. The eight RIL populations comprised up to 1243 RILs from crosses between 13 soybean accessions, including one wild soybean accession, four soybean landrace accessions and eight soybean cultivars (Figure 2a; Tables S1 and S2). To assess whether the genotyping results met the requirements for recombination event identification, we evaluated the SNPs based on their clustering performance (Figure S2). The investigation revealed that 61% of the total SNPs in the array were grouped into the ‘PolyHighResolution’ (PHR) cluster (Figure S2), which showed better performance than previous study (Wang et al., 2016) and the robustness of the SNP genotyping results.
Figure 2.
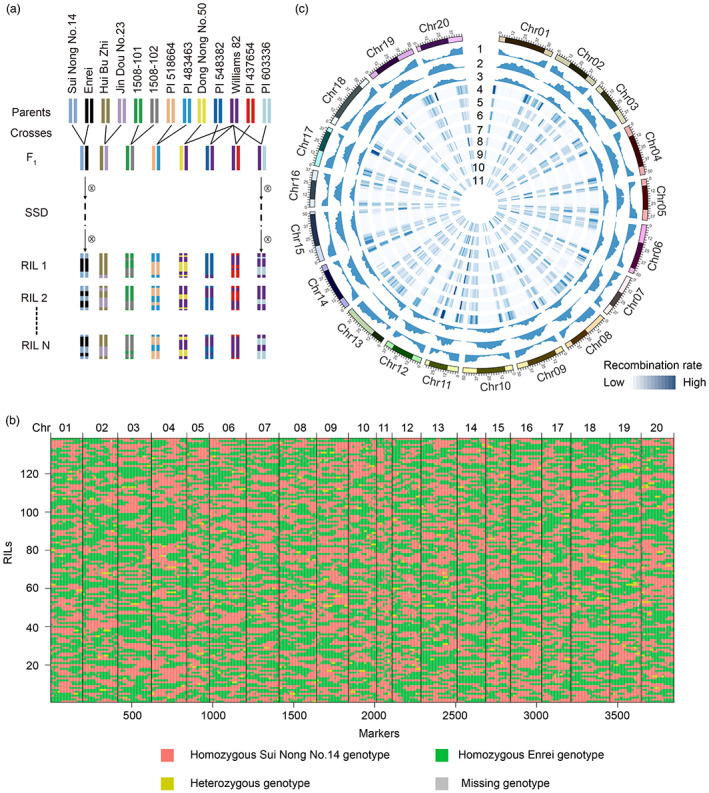
Overview of meiotic recombination in soybean RIL populations. (a) Construction of eight soybean RIL populations. Different colours represent various parental genotypes. SSD, single‐seed descent. (b) Recombination map of Sui Nong No.14 × Enrei. The colour red represents the homozygous Sui Nong No.14 genotype, green indicates the homozygous Enrei genotype, yellow represents the heterozygous genotype, and grey indicates a missing genotype. (c) Recombination landscape in the soybean genome. Tracks from outer to inner circles represent the distribution of gene density (1), TE density (2), GC content (3) and recombination rate for eight soybean RIL populations (4‐11), including Sui Nong No.14 × Enrei (4), Hui Bu Zhi × Jin Dou No.23 (5), 1508‐101 × 1508‐102 (6), PI 518664 × PI 483463 (7), Dong Nong No.50 × Williams 82 (8), PI 548382 × Williams 82 (9), PI 437654 × Williams 82 (10) and PI 603336 × Williams 82 (11). The black blocks indicate pericentromeric regions.
Given the small proportion of missing data and genotype calling errors in the SNP array, we corrected the genotype errors or inferred missing genotypes by using relatively large flanking genotype blocks to further improve the accuracy of recombination breakpoint prediction. Based on these recombination breakpoints, we constructed a recombination map for each of the eight RIL populations (Figures 2b and S3; Table S3). To reduce redundant genotype information, a bin map was developed for each population, with the numbers of bins ranging from 2625 to 5700 (Table S4). The majority of bins (77.1%) were shorter than 200 kb (Figure S4a) and 59.4% of bins contained no more than 10 genes (Figure S4b), suggesting a high resolution of the bin maps.
Using bins as markers, we further estimated the genetic distance of each bin map. Similar to results in previous studies (Lee et al., 2020; Qi et al., 2014), the genetic map length per RIL population varied from 2212.6 to 2840.5 cM (Figure S5; Table S4). The average distance between adjacent bins was 0.7 cM (Table S4). The recombination rate was defined as the rate of total genetic distance estimated versus the physical length of the Williams 82 reference genome. The average recombination rate ranged from 2.3 cM/Mb in the PI 437654 × Williams 82 population to 3.0 cM/Mb in the 1508‐101 × 1508‐102 population, with an average of 2.6 cM/Mb (Table S4).
Construction of the recombination landscape in soybean
Based on the inferred genotypes, a total of 64 912.5 recombination events were detected in the eight RIL populations, with an average number of 8114.1 events per population (Table S4). Subsequently, we calculated the total number of recombination events (TRE) for RIL in each population. We determined that the average TRE remained relatively stable among the eight RIL populations, ranging from 48.4 in the PI 437654 × Williams 82 population to 58.6 in the 1508‐101 × 1508‐102 population (Figure S6; Table S4). In addition, overall, the TRE showed a normal distribution in each population (Figure S6).
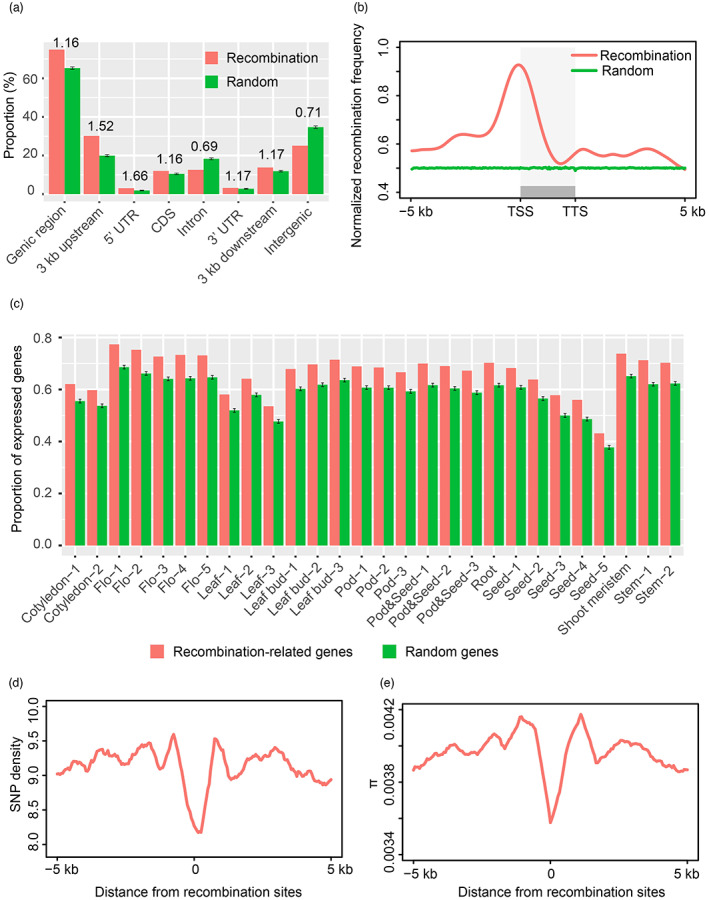
We found that the recombination was significantly suppressed in pericentromeric regions compared with chromosome arms (Figure 2c). Further investigation on the centromere and telomere regions demonstrated that the recombination rate showed a decline pattern from telomere to centromere across the chromosome (Figure S7a), which is consistent with previous studies (Rodgers‐Melnick et al., 2015; Zelkowski et al., 2019), indicating a conservation of recombination rate in different species. Additionally, the recombination rate was negatively correlated with transposable element (TE) density and GC content but positively correlated with gene density (Figures 2c and S7b–d). These patterns were consistent with findings from other plants (Choi et al., 2013; Jordan et al., 2018; Lambing et al., 2017; Pan et al., 2016; Rowan et al., 2019), indicating that recombination was related to well‐known genomic features in plants (Lambing et al., 2017).
The number of recombination events has been reported to be associated with chromosome size in some plants, such as Arabidopsis thaliana (Rowan et al., 2019), rice (Ma et al., 2016), maize (Kianian et al., 2018) and potato (Marand et al., 2017). However, we did not observe an obvious linear relationship between the number of recombination events and chromosome size (Figure S7e). In soybean, approximately half the length of the chromosome is occupied by pericentromeric regions (Schmutz et al., 2010). To eliminate the suppressing effects of the large pericentromeric regions on recombination, we calculated the correlation between recombination events and chromosome arm length and found an increasing but not significant positive correlation (Figure S7f).
Non‐reciprocal recombination events, also called non‐crossovers (NCOs), also popularly occur (Gardiner et al., 2019; Wijnker et al., 2013; Yang et al., 2012). We tried to identify the NCOs according to previous methods (Gardiner et al., 2019; Li et al., 2019) using our SoySNP array genotyping data for each line. Overall, an average of 25 events for each RIL across the eight soybean populations was detected (Table S5). The event number is significantly lower than that of CO, which may be partly resulted from the limitation of our genotyping method. Consistent with that of CO events, NCOs were unevenly distributed along the genome but more prevalent in pericentromere regions (Figure S8a), indicating NCO played an important role in generation of genetic diversity and breaking linkages in these regions. Nevertheless, consistent to that of CO events, NCOs also showed negative correlation to GC content and TE density, but positive correlation to gene density (Figure S8b–d).
In most species, recombination is concentrated in specific genomic regions, also known as recombination hotspots (Baudat et al., 2013; Choi and Henderson, 2015). In this study, we detected a total of 132 hotspots distributed along 20 chromosomes via 1000 permutation tests (Figure 3a; Table S6). Among these 132 hotspots, only two were shared among all populations, and furthermore, 46.2% of the hotspots belonged to only one population, demonstrating that these hotspots were population‐ or genotype‐specific (Figure 3b). We further detected QTLs for recombination hotspot usage (the proportions of recombination events occurring in hotspots relative to the whole genome) and found only one QTL detected in two populations (Figure 3c; Table S7). In addition, we identified a total of 13 QTLs controlling TRE from individual RIL populations. Nevertheless, no overlapping QTL was found among different populations (Table S8). These results indicated that recombination activities might be subjected to specific genotype background in soybean RIL populations and crosses between proper soybean accessions could be used to elevate the recombination rate in specific genomic regions which would facilitate the breeding process by breaking linkage drag.
Figure 3.
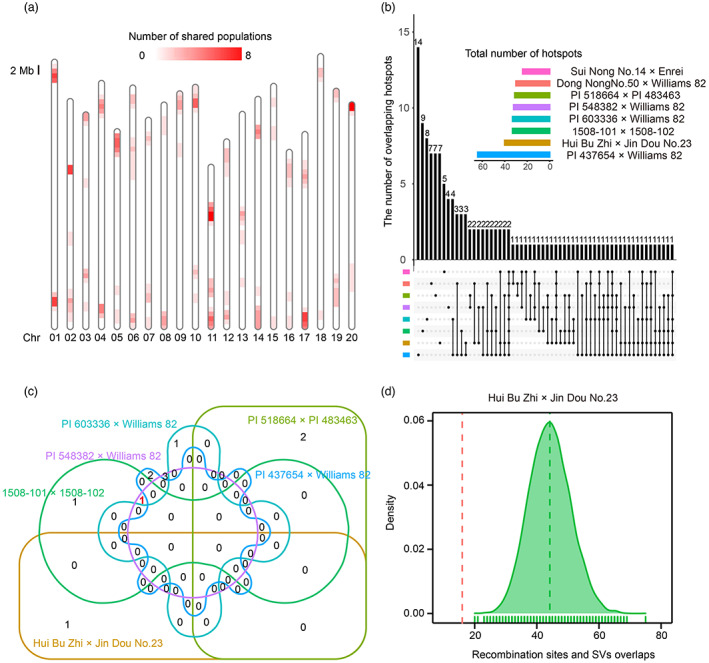
Genome‐wide and local recombination rate variation in the soybean genome. (a) The distribution of recombination hotspots identified from eight soybean RIL populations in the soybean genome. The gradient colours from white to red indicate an increasing number of RIL populations shared by a given hotspot. (b) Upset plot demonstrating the intersection of recombination hotspots between different RIL populations. Horizontal bars represent total number of recombination hotspots detected in each RIL population; numbers above vertical bars represent the count of hotspots shared by a given intersection of various RIL populations. (c) QTLs for recombination hotspot usage shared by eight RIL populations. Red number indicates the number of QTLs detected in two RIL populations (PI 548382 × Williams 82 and 1508‐101 × 1508‐102). (d) Overlaps between recombination events and SVs in the Hui Bu Zhi × Jin Dou No.23 population. The red dashed line indicates average number of overlaps between observed recombination events and SVs. The green dashed line represents the average number of overlaps between SVs and random recombination events that matched the distributions of observed recombination events.
It has been reported that structural variants (SVs) could influence the distribution of recombination events (Choi et al., 2013; Rowan et al., 2019). Our previous study detected extensive SVs among soybean natural population (Liu et al., 2020; Liu and Tian, 2020). To determine the effect of SVs on recombination events, we investigated the SVs between the parents of the eight RIL populations and identified an average of 5867 SVs for each parent pair (Figure S9). Interestingly, we found that the regions with SVs in each parent pair had fewer internal recombination events than expected (Figures 3d and S10), indicating that recombination events tended not to occur in the regions of SVs.
Meiotic recombination preferentially occurs in the promoter regions of transcriptionally active genes
Benefiting from the high density of SNP markers in this assay and the large population size, we obtained a comprehensive high‐resolution recombination map that contained 5636 recombination sites (defined as the midpoint of the recombination interval) with a recombination interval length ≤2 kb (average: 1.37 kb). The high‐resolution events greatly mirrored the distribution of all events (Figures 2c and S11). The number of high‐resolution recombination events identified in the study is much larger than that in Arabidopsis thaliana (Shilo et al., 2015), maize (Kianian et al., 2018) and potato (Marand et al., 2017), thus laying a solid foundation for further investigations of the fine‐scale genomic features associated with recombination events in soybean.
Interestingly, we found that 75.5% of high‐resolution recombination events overlapped with genic regions (including 6 kb flanking regions of a gene and the gene body) (Figure 4a). To test whether this overlap was by chance, we performed a simulation of the random sites that matched the distribution of high‐resolution recombination events across the whole genome via a Monte Carlo (MC) procedure (10 000×; the random simulation sites are referred to as MC random sites) approach and then calculated the overlap rate between genic/intergenic regions and recombination events and MC random sites. In contrast to that in intergenic regions, the occurrence frequency of recombination events was higher in genic regions than expected by chance, suggesting that recombination events in soybean preferentially occur in genic regions (Figure 4a). To investigate the relationship between recombination and genes in more detail, genic regions was subdivided into the six categories: 3 kb upstream of the gene start, 5′ untranslated region (UTR), coding sequence (CDS), intron, 3′ UTR and 3 kb downstream of the gene end. The simulation results showed the highest frequency of events in 5′ UTRs (1.66 times that in the random dataset; empirical, P < 1.0 e−4), followed by 3 kb upstream of the gene start, 3 kb downstream of the gene end, 3′ UTRs, CDSs and introns (Figure 4a). Furthermore, an obvious peak was observed near transcription start sites (TSSs) (Figure 4b). Therefore, these results indicated recombination tended to occur in gene promoter in soybean.
Figure 4.
Relationship of fine‐scale genomic features with high‐resolution recombination events in soybean. (a) Association of high‐resolution recombination events with different genomic features against random sets (permutation tests). Error bars indicate the standard deviation of 10 000 permutation tests. Genic region includes 3 kb upstream of a gene start, 5′ UTR, CDS, intron, 3′ UTR and 3 kb downstream of a gene end. The number above vertical bars represents the ratio of high‐resolution recombination events to random sites. The midpoint of a high‐resolution recombination interval was used to calculate the number of overlap with genomic features. (b) The distribution of recombination frequency relative to all protein‐coding genes in soybean. TSS, transcription start site. TTS, transcription termination site. (c) Comparisons of the proportion of expressed genes between recombination‐related gene set and randomly selected gene set. Genes with FPKM values >0.5 were considered as expressed genes. Error bars indicate the standard deviation of 10 000 permutations for random genes. (d) The distribution of SNP density across high‐resolution recombination sites and in flanking 5 kb regions. (e) Nucleotide diversity distribution across high‐resolution recombination sites and in flanking 5 kb regions.
We defined these genes or their flanking 3 kb regions that overlapped with recombination sites as recombination‐related genes in this study. To determine the potential functions of these genes, we first performed Gene Ontology (GO) analysis, and we did not find significant terms that were overrepresented among these genes. Subsequently, we investigated the transcriptional activities of these recombination‐related genes using our previously reported transcriptome dataset of 28 samples from different tissues (Shen et al., 2014) and surprisingly found that the proportion of expressed genes was higher than expected by chance (Figure 4c), indicating that recombination was more likely to target transcriptionally active genes in soybean.
Genic regions usually are highly evolutionarily conserved (Monroe et al., 2022), and we inferred that genomic regions spanning recombination sites were under purifying selection. We then investigated the distribution of SNPs across the recombination sites using previous resequencing data of 302 representative soybean accessions (Zhou et al., 2015). We determined that the SNP density at recombination sites was much lower than that in flanking regions (Figure 4d). Furthermore, genomic regions near recombination sites possessed lower genetic diversity than their vicinity (Figure 4e). These results indicated that recombination sites were relatively conserved and under purifying selection in soybean.
Poly‐A and AT‐rich motifs are associated with recombination frequency at gene promoters
Recombination sites may be associated with specific DNA motifs in different species (Choi et al., 2013; Kianian et al., 2018; Lawrence et al., 2017; Shilo et al., 2015; Wijnker et al., 2013; Zelkowski et al., 2019). To investigate whether specific DNA motifs were enriched at recombination sites in soybean, de novo motif discovery was performed for high‐resolution recombination events using MEME software (Bailey et al., 2009). The analysis showed that two distinct motifs were overrepresented among recombination sites: poly‐A and AT‐rich motifs (Figure 5a). Investigation of the global distribution of these two motifs showed that both exhibited preferential distribution in euchromatic regions at the chromosome level, similar to that of recombination events (Figures 5b and S11). In addition, interestingly, we found that all these motifs were significantly overrepresented at gene promoters (Figure 5c,d), consistent with the enrichment of recombination frequency at gene promoters. These results indicated that to some extent poly‐A and AT‐rich motifs might promote the formation of recombination at gene promoters.
Figure 5.
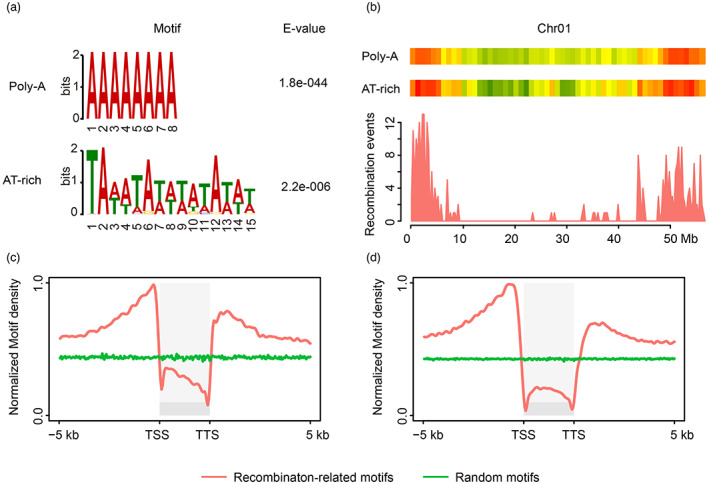
DNA motifs identified in the vicinity of high‐resolution recombination sites. (a) Enrichment of poly‐A and AT‐rich motifs was found in high‐resolution recombination intervals using MEME. (b) The distribution of the two motifs and high‐resolution recombination events on chromosome 1. (c, d) The distribution of Poly‐A (c) and AT‐rich (d) motifs over transcription units and their flanking regions. TSS, transcription start site. TTS, transcription termination site. Red lines indicate the distribution of recombination‐related motifs over gene bodies and their flanking regions. Green lines represent the distribution of random motifs via simulation.
Chromatin accessibility promotes recombination formation in soybean
It is well known that chromatin accessibility is significantly associated with gene expression in plants (Lu et al., 2017, 2019). The phenomenon in which recombination events tended to occur in genic regions with active gene transcriptional states indicated that recombination may be associated with chromatin signatures. To test this hypothesis, we carried out a state‐of‐the‐art assay for transposase‐accessible chromatin using sequencing (ATAC‐seq) to infer DNA accessibility near recombination sites (Table S9) and investigated the relationship between recombination and DNA accessibility at the genome‐wide level. The result showed a strong correlation between the recombination events and DNA accessibility (Figure 6a,b).
Figure 6.
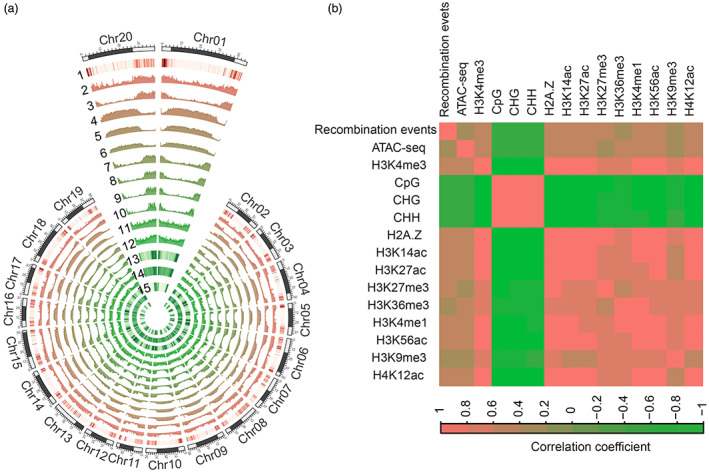
Relationship between recombination events and epigenetic modifications at the chromosome level. (a) Circos plot of high‐resolution recombination events and epigenetic modifications. Tracks from outer to inner circles represent the distribution of high‐resolution recombination events (1), ATAC‐seq (2), H3K4me3 (3), CpG (4), CHG (5), CHH (6), H2A.Z (7), H3K14ac (8), H3K27ac (9), H3K27me3 (10), H3K36me3 (11), H3K4me1 (12), H3K56ac (13), H3K9me3 (14) and H4K12ac (15). Reads counts and DNA methylation levels were counted based on the non‐overlapping 200 kb genomic regions. The black blocks indicate pericentromeric regions. (b) Correlation coefficient heatmap for recombination events with chromatin accessibility and epigenetic modifications.
Accessible chromatin regions (ACRs) are regions that show significantly reduced nucleosome occupancy compared with that in the whole genome. In this study, we identified 35 865 ACRs and, interestingly, found significant overlaps (empirical, P < 1.0 e−4) between high‐resolution recombination sites and ACRs relative to randomly sampled sites (Figure 7a). Furthermore, the shortest distance between recombination sites and ACRs was much shorter than random expectation (empirical, P < 1.0 e−4; Figure 7b,c), indicating that recombination events occurred much closer to nucleosome‐depleted regions.
Figure 7.
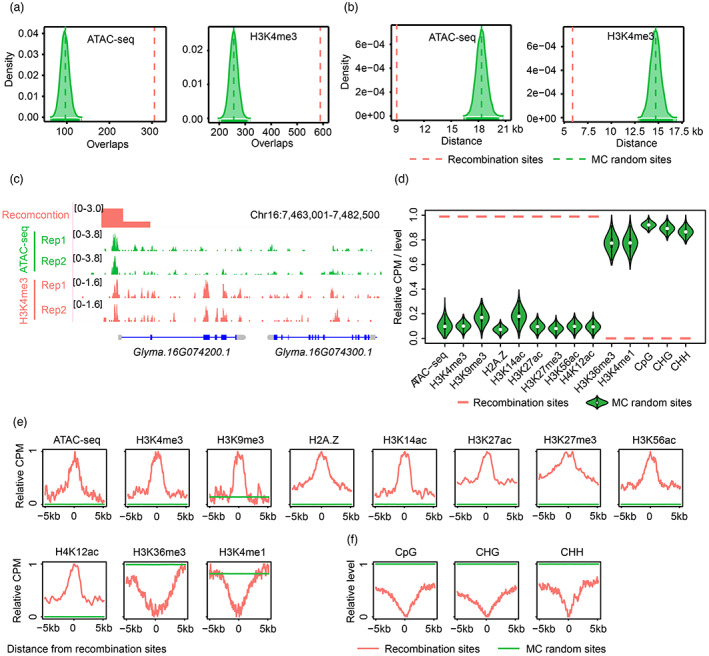
Fine‐scale epigenomic features are associated with high‐resolution recombination events in soybean. (a) Overlap analyses for accessible chromatin regions (ACRs) and H3K4me3 peaks with recombination sites. Red dashed lines indicate the average number of overlaps between observed high‐resolution recombination events and ACRs or H3K4me3 peaks. Green dashed lines indicate the average number of overlaps between randomly sampled sites and ACRs or H3K4me3 peaks. (b) The shortest distance from recombination sites to ACRs and H3K4me3 peaks. Red dashed lines indicate the average value of the shortest distance from recombination sites to ACRs or H3K4me3 peaks. Green dashed lines indicate the average value of the shortest distance from randomly sampled sites to ACRs or H3K4me3 peaks. (c) An ACR and an H3K4me3 peak co‐localized with recombination events in regions near the transcription start site (TSS) of Glyma.16G074200.1. (d) Epigenetic status across high‐resolution recombination sites and randomly selected sites. Total read counts or methylation level for 2 kb genomic regions centred on recombination sites were compared to random sites. CPM, counts per million. (e) The distribution of normalized read counts for ATAC‐seq, different histone modifications and histone variant H2A.Z across recombination sites and in flanking regions compared to random sites. (f) Average profiles of normalized DNA methylation levels across recombination sites and in their flanking regions compared to random sites.
To determine whether the accessible chromatin state was significantly associated with recombination, we calculated normalized counts per million (CPM) for mapped reads in 10 kb flanking regions of recombination sites and MC random sites. The result indicated that recombination sites have higher CPM than random sites (empirical, P < 1.0 e−4; Figure 7d). In addition, ATAC‐seq signals were significantly elevated at recombination sites compared with their flanking regions (Wilcoxon signed‐rank test, P < 2.2 e−16; Figure 7e), indicating that decreasing nucleosome occupancy was strongly associated with the occurrence of recombination. Therefore, chromatin accessibility appears to have significant effects on meiotic recombination in soybean.
Epigenetic context of meiotic recombination
As the number of ACRs was far greater than that of recombination events in each RIL (Figure S6), chromatin accessibility may be necessary but not sufficient for recombination formation. We speculated that other epigenetic factors are likely involved in recombination pathways in soybean. H3K4me3 is known to be an active epigenetic mark and shows a close relationship with recombination activities in both animals (Baudat et al., 2013) and plants (Yelina et al., 2015). To precisely determine the relationship of H3K4me3 modifications with recombination in soybean, Cleavage Under Targets and Tagmentation (CUT&Tag) technology was applied to investigate the enrichment of H3K4me3 (Table S9). A positive correlation between recombination and H3K4me3 level was observed at the chromosome level (Figure 6). Further investigation identified a total of 69 040 H3K4me3 peaks that significantly overlapped with recombination sites, which exhibited a shorter distance to recombination sites than MC random sites (empirical, P < 1.0 e−4; Figure 7a–c). We further compared H3K4me3 levels between high‐resolution recombination sites and random sites. A higher H3K4me3 level was found at recombination sites (empirical, P < 1.0 e−4; Figure 7d). Next, we profiled the distribution of H3K4me3 centred on recombination sites and found significant enrichment of H3K4me3 at recombination sites compared to flanking regions (Wilcoxon signed‐rank test, P < 2.2 e−16; Figure 7e). Collectively, these results demonstrated that the H3K4me3 level was strongly positively correlated with recombination rate at the fine scale.
In contrast to H3K4me3, DNA methylation acts as a classical repressing epigenetic mark when located in gene promoters and has been reported to play a crucial role in controlling meiotic recombination. To uncover the DNA methylation level at recombination sites, whole‐genome bisulphite sequencing (WGBS) was performed with an average depth of >64× (Table S9). At the genome‐wide level, DNA methylation was negatively correlated with recombination frequency (Figure 6). As expected, we found that the DNA methylation level of all three contexts (CpG, CHG and CHH, H = A, T or C) in the recombination sites was lower than that expected by chance (empirical, P < 1.0 e−4; Figure 7d) and was lower than that of flanking regions (Wilcoxon signed‐rank test, P < 2.2 e−16; Figure 7f), indicating that DNA methylation may inhibit homologous recombination in soybean. Observations for these three sequence contexts in this study were in agreement with those in maize (Kianian et al., 2018) but different from those in rice except CpG context (Marand et al., 2019), suggesting dissimilarities among soybean, maize and rice recombination maps.
To obtain the most comprehensive views of the relationships between more histone modifications and the recombination rate, we conducted a CUT&Tag experiment for H3K9me3 (Table S9) and collected additional publicly available next‐generation sequencing (NGS) data of histone variant and different histone modifications (Lu et al., 2019; Wang et al., 2021), including H2A.Z, H3K14ac, H3K27ac, H3K27me3, H3K56ac, H4K12ac, H3K36me3 and H3K4me1. At the chromosome scale, we found positive correlations between all these modifications and recombination frequency (Figure 6). Of these modifications, H3K36me3 and H3K4me1 may confer a negative effect on the formation of meiotic recombination at the fine scale (Figure 7d,e), which was different from the observations at the chromosome scale, indicating that a high‐resolution recombination map may provide a more precise picture of the relationship between epigenetic context and recombination rate.
To further reveal the correlation between recombination in genic regions and epigenetic state, we profiled the chromatin accessibility, DNA methylation, histone variant and histone modification level across genic regions. As expected, elevated chromatin accessibility, the histone variant H2A.Z and histone modification level, including that for H3K4me3, H3K9me3, H3K14ac, H3K27ac, H3K27me3, H3K56ac and H4K12ac, were found in recombination‐related genes compared to randomly selected genes (Wilcoxon signed‐rank test, P < 3.0 e−9; Figure S12a). However, DNA methylation, H3K36me3 and H3K4me1 showed lower levels (Wilcoxon signed‐rank test, P < 1.8 e−19; Figure S12), indicating that these modifications may prevent recombination occurrence in genic regions.
The recombination events and epigenetic marks tended to occur in genic regions, which suggest that the correlations between recombination and epigenetic modifications may be artefacts. To eliminate this possibility, we extracted only the high‐resolution recombination sites (1382 sites) that located in intergenic regions (genomic regions >3 kb from protein‐coding genes) and investigated the chromatin state of intergenic recombination sites. We found, except H3K4me1, that the distributions of the epigenetic marks in intergenic recombination sites were similar to that from all high‐resolution recombination sites (Figure S13), indicating that the active chromatin state and epigenetic modifications are natural characteristics of recombination sites in soybean. In addition, given that the majority of recombination sites located in genic regions, we thus asked whether these intergenic recombination events also happened in loci that expressed non‐coding RNA (ncRNA) transcripts. We collected soybean long non‐coding RNA (lncRNA) transcripts from two previous studies (Golicz et al., 2018; Lin et al., 2020) and found that a total of 70 lncRNA transcripts overlapped with intergenic recombination sites (Table S10), indicating a potential relationship between meiotic recombination and lncRNA transcription.
Discussion
In general, meiotic recombination is non‐randomly distributed along the chromosome and tends to cluster in certain DNA regions, forming recombination hotspots. In multiple mammalian species, the location of recombination hotspots is determined by PRMD9 action (Paigen and Petkov, 2018). Although plants have no clear PRMD9 homologues, recombination hotspots are also frequently detected in their genomes (Choi et al., 2013; Marand et al., 2019; Pan et al., 2016). Similarly, we identified 132 hotspots in the soybean genome (Figure 3a), suggesting that the mechanism underlying the formation of these recombination hotspots is independent of PRMD9 and not clear. In addition, these hotspots showed obvious local recombination variations among various soybean populations (Figure 3b). Our results further revealed that extensive SVs between soybean accessions could impact local recombination patterns via suppression of adjacent recombination events (Figures 3d and S10). In addition to SVs, the diversity of nucleolus organizer regions (NORs) across maize lines results in a locally reduced recombination rate (Bauer et al., 2013). Thus, the extent to which other genomic structural elements, such as NORs, shape the recombination landscape needs to be comprehensively investigated in soybean.
Recombination events tend to occur in open chromatin regions in soybean (Figure 7a–c) as well as Arabidopsis (Choi et al., 2013; Wijnker et al., 2013), maize (Kianian et al., 2018), potato (Marand et al., 2017) and rice (Marand et al., 2019). In this study, we further revealed the comprehensive and detailed relationships of the recombination rate with DNA methylation and chromatin structure at both the chromosome scale and fine scale (Figures 6, 7, S12 and S13), showing that the combinatorial interaction of these factors shaped the recombination landscape in genic regions, especially at promoters, in soybean. Recently, chromosome organization was reported to be linked to meiotic recombination (Patel et al., 2019; Zuo et al., 2021), suggesting the complexity of the regulatory mechanism for recombination in eukaryotes. In Arabidopsis, the recombination rate was manipulated in large‐scale and non‐specific regions by genetic disruption of the DNA methylation and histone modification pathway (Underwood et al., 2018). Here, our presentation of the comprehensive epigenetic profile at recombination sites in soybean will enhance our knowledge of the relationship between epigenetic modifications and recombination events, which will facilitate the utilization and modification of recombination in soybean breeding.
In the crop breeding process, the phenomenon in which desirable traits are tightly linked to one or more undesirable traits is frequently observed, which might be caused by linkage between adjacent alleles. For instance, our previous study revealed widespread genetic interactions among various alleles in soybean (Fang et al., 2017). In the process of crop breeding and improvement, breeders desire to break linkage drags between two or more specific genetic loci. Increasing knowledge of how genomic and epigenetic contexts influence recombination frequency would enhance our ability to control recombination activities and thus help overcome this difficult problem.
In this study, two motifs, namely, poly‐A and AT‐rich motifs, were overrepresented at recombination sites and gene promoters (Figure 5). These motifs facilitated the occurrence of recombination at gene promoters possibly by serving as binding sites of transcription factors involved in the recombination pathway. In addition, it has been reported that poly‐A and AT‐rich sequences facilitate the formation of local accessible regions by decreasing nucleosome occupancy (Field et al., 2008; Segal and Widom, 2009). In Arabidopsis, A‐rich and CTT repeat motifs were repeatedly identified in two independent studies (Choi et al., 2013; Shilo et al., 2015) and respectively overrepresented at promoters and gene bodies (Shilo et al., 2015), indicating the potential consistency and inconsistency of the recombination mechanism between these two species.
Methods
Plant materials and growing conditions
A total of eight soybean RIL populations were derived from crosses between eight soybean cultivars (Sui Nong No.14, Enrei, Jin Dou No.23, 1508‐101, 1508‐102, PI 518664, Dong Nong No.50 and Williams 82), four soybean landraces (Hui Bu Zhi, PI 548382, PI 437654 and PI 603336) and one wild soybean (PI 483463) followed by nine generations of self‐fertilization (Figure 2a; Tables S1 and S2). These RILs and their parental lines were planted in the experimental fields in Beijing (39° N, 116° E).
Development of the 600K SoySNP array and genotyping of the populations
We selected high‐quality SNPs from a previous study (Fang et al., 2017; Zhou et al., 2015) according to several criteria described in Figure 1a, and a total of 600 009 SNPs were used to develop the 600K SoySNP array by CapitalBio Technology. The variant effects of these high‐quality SNPs were determined on the basis of gene annotations of the Williams 82 reference genome (Wm82.a2.v1) (Schmutz et al., 2010) via ANNOVAR with default parameters (Wang et al., 2010).
Young leaves were sampled to extract high‐quality genomic DNA through the modified cetyl‐trimethyl ammonium bromide (CTAB) method. DNA samples from the parents and their derived populations were diluted to 20 ng/μL and genotyped with the 600K SoySNP array. The signal intensity data of each probe in the array were reported in CEL files, which were analysed with Axiom Analysis Suite (v.1.1.0.616) for quality control and genotype calling. Samples with a Dish value >0.85 and SNP call rates >95% were retained for subsequent analysis.
Genome resequencing and identification of nucleotide variations between parental accessions
Resequencing data from 13 parents were mapped to the soybean reference genome (Wm82.a2.v1) (Schmutz et al., 2010) by BWA‐MEM (parameter: ‐M) (Li and Durbin, 2009). We further filtered low quality (mapping quality score < 20) using SAMtools v.1.3.1 (Li et al., 2009) and duplicated read through Sambamba (Tarasov et al., 2015). SNPs were called using the HaplotypeCaller module in the GATK software (McKenna et al., 2010) according to a previously developed pipeline (Liu et al., 2020).
Based on the high‐quality alignment results, we further identified candidate SVs, including deletions, insertions and inversions, between the two parents for each RIL population by delly v.0.8.7 (Rausch et al., 2012), lumpy v.0.3.1 (Layer et al., 2014) and manta v.1.6.0 (Chen et al., 2016) with at least three supported reads and lengths >50 bp. For each parent pair, SV calls from these three software programs were merged using SURVIVOR v.1.0.7 (parameters: minimum distance of 1 kb and same SV type) (Jeffares et al., 2017). Finally, SVs identified by two or three tools were retained.
Generation of recombination maps and linkage maps
In order to generate recombination maps, SNPs that belonged to homozygous genotype but were different between parental lines were selected as candidate markers. Among these markers, SNPs with the ratio of missing genotypes >0.2 and the ratio of heterozygous genotypes >0.2 in each population were further filtered. In addition, we also performed chi‐square tests for each SNP with the expected segregation ratios of 1:1 in the RIL population and discarded SNPs that significantly deviated from this expected ratio (P < 0.01). In final, an average of 139 490 high confidence polymorphic SNPs between the two parents were kept for identification of recombination events (Table S4).
For each RIL, continuous SNPs originating either from the one of two parents or both parents were merged into a single genotype block. Missing or error genotypes were further imputed on the basis of flanking genotype blocks. According to genotype information, recombination events for each line were calculated for two scenarios according to a previous study (Pan et al., 2016): (i) the two genotypes of adjacent blocks were homozygous but different, and one recombination event was considered to occur; and (ii) either genotype was heterozygous, and a 0.5 recombination event was assumed to happen.
According to the recombination breakpoint assumed at the transition between adjacent and different genotype blocks, we constructed a recombination map and bin map for each RIL population. Considering bins as genetic markers, the linkage maps were created using the est.map function in R/qtl package (Broman et al., 2003) with the Kosambi mapping method.
Identification of centromeric regions and telomeres
We identified centromere and telomere regions for each chromosome by mapping the two centromere‐specific satellite repeats (CentGm‐1 and CentGm‐2) (Gill et al., 2009; Tek et al., 2010) and the telomeric repeat (AAACCCT and AGGGTTT) (Valliyodan et al., 2019) by BLASTN respectively.
QTL mapping for recombination hotspot usage and TRE
To identify genetic factors influencing recombination variation in soybean, the recombination hotspot usage and TRE for each RIL were considered as traits and QTL analyses were performed with the scanning step of 1.0 cM and IM‐ADD method implemented in QTL IciMapping v.4.2.53 (Li et al., 2007). The threshold for LOD was set at 2.5. The confidence interval (CI) of each QTL was determined by a 1.0 LOD‐drop support region.
Nucleotide diversity analysis
SNP density across recombination sites was calculated with a window size of 500 bp and a step length of 50 bp using SNP datasets from our previous study (Zhou et al., 2015). In addition, nucleotide diversity analysis with a sliding window of 1 kb and a step size of 50 bp was performed with PopGenome (Pfeifer et al., 2014).
Motif de novo analysis
To identify motifs in recombination sites, high‐resolution recombination intervals with the lengths <= 2 kb were kept. All these intervals were extracted from the Williams 82 reference genome (Wm82.a2.v1) and then provided to MEME software (Bailey et al., 2009) with the random recombination events as a background. Motifs with an E‐value cut‐off <1 e−5 and at least 50 sites detected were retained. In order to determine the distribution of these motifs along chromosomes, we mapped the positional weight matrix for each motif onto soybean genome by the MOODS tool (Korhonen et al., 2009) with P value <0.001.
Estimation of recombination rates and identification of recombination hotspots
The recombination rate (cM/Mb) was defined as the genetic distance (cM) per 1 Mb of physical distance. When calculating the local recombination rate, a window size of 2 Mb with a walking distance of 1 Mb was adopted. Under the null hypothesis that the occurrence of recombination events across the whole soybean genome was random, 1000 permutation tests were conducted to determine the threshold for recombination hotspots at a significance level of 0.05.
MethylC‐seq library construction and DNA methylation analysis
The MethylC‐seq libraries were constructed according to a previous report (Urich et al., 2015) with two biological replicates and sequenced on the Illumina NovaSeq 6000 platform. The raw sequencing reads were filtered and trimmed by Trimmomatic (Bolger et al., 2014). Through Bismark (Krueger and Andrews, 2011), trimmed reads were aligned to the reference genome (Wm82.a2.v1). After removing duplicate reads using Sambamba (Tarasov et al., 2015), the methylation level was calculated based on the methylation information for each cytosine site.
ATAC‐seq experiments, CUT&Tag assays and data processing
ATAC‐seq experiments were performed as described previously (Lu et al., 2017) with two biological replicates. For each replicate, approximately 100 mg of flower buds with a length of 2–4 mm from the soybean accession Williams 82 were harvested and immediately chopped with a razor blade and placed in 0.2 mL pre‐chilled lysis buffer (25 mM Tris–HCl pH 7.6, 0.44 M sucrose, 10 mM MgCl2, 0.1% Triton‐X, 10 mM beta‐mercaptoethanol). We further filtered the chopped slurry using a 40‐μm cell strainer and washed the nuclei with lysis buffer. The sorted nuclei (50 000 nuclei per reaction) were incubated with purified Tn5 at 37 °C for 30 min. The integration products were purified using a Qiagen MinElute PCR Purification Kit and then amplified using Q5® High‐Fidelity DNA Polymerase (NEB, M0491L).
The CUT&Tag assay was performed as described previously (Tao et al., 2020). For each replicate, approximately 100 mg of flower buds with a length of 2–4 mm from the soybean accession Williams 82 were prepared, and the nuclei were immediately extracted, similar to the step in the ATAC‐seq experiments. The sorted nuclei (10 000 nuclei per reaction) were bound to the primary antibody, and the reaction was conducted for each antibody (anti‐H3K4me3, Abcam, ab8580 and anti‐H3K9me3, Abcam, ab8898) at 4 °C overnight. Then, nuclei were centrifuged, incubated with secondary antibody, guinea pig anti‐rabbit IgG (heavy and light chain) (Antibodies‐Online ABIN101961), at 4 °C for approximately 1–2 h, and washed twice with wash buffer. Subsequently, we resuspended nuclei in 100 μL of CT‐300 buffer and performed transposase incubation using 0.75 μL of pA‐Tn5/pG‐Tn5 for 2–3 h at 4 °C. We conducted tagmentation at 37 °C for 1 h and added 10 μL of 0.5 M EDTA, 3 μL of 10% SDS and 2.5 μL of 20 mg/mL protease K to stop the tagmentation reaction. DNA was purified by standard phenol‐chloroform extraction followed by ethanol precipitation and then amplified using Q5® High‐Fidelity DNA Polymerase (NEB, M0491L).
Libraries were constructed using the standard Illumina protocols. In the process of library construction, amplified libraries were purified with AMPure beads. Sequencing of these libraries was performed on the Illumina NovaSeq 6000 platform. The raw sequencing reads were filtered and trimmed using Trimmomatic (Bolger et al., 2014) and were subjected to Bowtie2 analysis (Langmead and Salzberg, 2012) with default parameters for alignment to the reference genome. We discarded reads mapped to the chloroplast and mitochondrial genomes from downstream analysis. Peaks were detected by MACS3 algorithms (Feng et al., 2012) with parameters (ATAC‐seq: ‐g 1.1e+9 ‐q 0.05 ‐‐nomodel ‐‐SPMR ‐‐shift ‐100 ‐‐extsize 200 ‐‐keep‐dup all; H3K4me3: ‐g 1.1e+9 ‐q 0.05 ‐‐nomodel ‐‐SPMR ‐‐extsize 147 ‐‐keep‐dup all) using uniquely mapped reads with high mapping quality (mapping quality score > 20). Overlapped peaks between replicates were kept for subsequent analyses. The signal densities for ATAC‐seq, DNA methylation and histone modifications across high‐resolution recombination sites were calculated using deepTools v.3.5.1 (Ramirez et al., 2016).
Conflict of interest
The authors declare that they have no competing interests.
Author contributions
Z.T. designed and supervised this project. X.M., S.L., Y.L., G.Z., Y.P. and M.Z. performed the data analyses. X.M., S.L., L.F., Z.Z. and X.Y. performed the experiments. Y.M., H.N., F.K. and J.M. constructed RIL populations. Z.T. and X.M. wrote the manuscript.
Supporting information
Figure S1 Evaluation of the repeatability and accuracy of SNPs in the 600K SoySNP array.
Figure S2 Classification of SNPs in the 600K SoySNP array.
Figure S3 Recombination maps for soybean RIL populations.
Figure S4 Features of bins in the recombination bin map.
Figure S5 Linkage maps for eight soybean RIL populations using bins as genetic markers.
Figure S6 Distribution of recombination events identified in eight soybean RIL populations.
Figure S7 Genomic features of recombination events in soybean.
Figure S8 Non‐crossover (NCO) events and their genomic features in soybean RIL populations.
Figure S9 SVs detected between the parental lines of each RIL population.
Figure S10 Overlaps between SVs and recombination events for each population.
Figure S11 Poly‐A and AT‐rich motifs associated with the distribution of high‐resolution recombination events along Chr02 to Chr20.
Figure S12 Epigenetic modifications across recombination‐related genes and random genes obtained from permutations and in their flanking regions.
Figure S13 The distribution of read counts for chromatin states, histone modifications, histone variant H2A.Z and DNA methylation levels along intergenic recombination sites and their flanking regions compared to random sites.
Table S1 Summary of 13 representative soybean accessions for deep resequencing
Table S2 Eight soybean RIL populations constructed in our study
Table S4 Summary of genetic maps for eight soybean RIL populations
Table S5 Summary for non‐crossover (NCO) events detected in soybean RIL populations
Table S6 Recombination hotspots identified in eight soybean RIL populations
Table S7 QTL mapping for recombination hotspot usage
Table S8 QTL mapping for the total number of recombination events for each RIL in soybean RIL populations
Table S9 Sequencing information in this study
Table S10 Summary of long non‐coding RNAs (lncRNAs) overlapping intergenic recombination sites.
Table S3 The high‐resolution genotypic map for eight soybean RIL populations.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant nos. 31788103 and 32201832), the Strategic Priority Research Program of the Chinese Academy of Sciences (grant no. XDA24030501), Support Plan for Innovation and Development of Key Industries in South Xinjiang (2022DB015) and Xplorer Prize Award.
Contributor Information
Shulin Liu, Email: slliu@genetics.ac.cn.
Zhixi Tian, Email: zxtian@genetics.ac.cn.
Data availability
The sequencing data generated in this study have been uploaded to the Genome Sequence Archive (GSA) database in the BIG Data Center (https://ngdc.cncb.ac.cn/) under accession number PRJCA008111. The resequencing data for 13 parental soybean accessions were deposited into Sequence Read Archive (SRA) database in NCBI under accession number PRJNA394629 or the GSA database in BIG Data Center under accession number PRJCA000205. The previously reported NGS data for H2A.Z, H3K36me3, H3K4me1 and H3K56ac were from the Gene Expression Omnibus (GEO) database through accession number GSE128434 and for H3K14ac, H3K27ac, H3K27me3 and H4K12ac were from SRA database via accession number PRJNA657728.
References
- Bailey, T.L. , Boden, M. , Buske, F.A. , Frith, M. , Grant, C.E. , Clementi, L. , Ren, J. et al. (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat, F. , Imai, Y. and de Massy, B. (2013) Meiotic recombination in mammals: localization and regulation. Nat. Rev. Genet. 14, 794–806. [DOI] [PubMed] [Google Scholar]
- Bauer, E. , Falque, M. , Walter, H. , Bauland, C. , Camisan, C. , Campo, L. , Meyer, N. et al. (2013) Intraspecific variation of recombination rate in maize. Genome Biol. 14, R103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman, K.W. , Wu, H. , Sen, S. and Churchill, G.A. (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics, 19, 889–890. [DOI] [PubMed] [Google Scholar]
- Chen, X. , Schulz‐Trieglaff, O. , Shaw, R. , Barnes, B. , Schlesinger, F. , Kallberg, M. , Cox, A.J. et al. (2016) Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics, 32, 1220–1222. [DOI] [PubMed] [Google Scholar]
- Choi, K. and Henderson, I.R. (2015) Meiotic recombination hotspots ‐ a comparative view. Plant J. 83, 52–61. [DOI] [PubMed] [Google Scholar]
- Choi, K.H. , Zhao, X.H. , Kelly, K.A. , Venn, O. , Higgins, J.D. , Yelina, N.E. , Hardcastle, T.J. et al. (2013) Arabidopsis meiotic crossover hot spots overlap with H2A. Z nucleosomes at gene promoters. Nat. Genet. 45, 1327–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, C. , Ma, Y.M. , Wu, S.W. , Liu, Z. , Wang, Z. , Yang, R. , Hu, G.H. et al. (2017) Genome‐wide association studies dissect the genetic networks underlying agronomical traits in soybean. Genome Biol. 18, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, X. , Sun, X. , Yang, X. , Li, Q. , Lin, C. , Xu, J. , Gong, W. et al. (2021) MS1 is essential for male fertility by regulating the microsporocyte cell plate expansion in soybean. Sci. China Life Sci. 64, 1533–1545. [DOI] [PubMed] [Google Scholar]
- Feng, J. , Liu, T. , Qin, B. , Zhang, Y. and Liu, X.S. (2012) Identifying ChIP‐seq enrichment using MACS. Nat. Protoc. 7, 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field, Y. , Kaplan, N. , Fondufe‐Mittendorf, Y. , Moore, I.K. , Sharon, E. , Lubling, Y. , Widom, J. et al. (2008) Distinct modes of regulation by chromatin encoded through nucleosome positioning signals. PLoS Comp. Biol. 4, e1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner, L.J. , Wingen, L.U. , Bailey, P. , Joynson, R. , Brabbs, T. , Wright, J. , Higgins, J.D. et al. (2019) Analysis of the recombination landscape of hexaploid bread wheat reveals genes controlling recombination and gene conversion frequency. Genome Biol. 20, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill, N. , Findley, S. , Walling, J.G. , Hans, C. , Ma, J. , Doyle, J. , Stacey, G. et al. (2009) Molecular and chromosomal evidence for allopolyploidy in soybean. Plant Physiol. 151, 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golicz, A.A. , Singh, M.B. and Bhalla, P.L. (2018) The long intergenic noncoding RNA (lincRNA) landscape of the soybean genome. Plant Physiol. 176, 2133–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffares, D.C. , Jolly, C. , Hoti, M. , Speed, D. , Shaw, L. , Rallis, C. , Balloux, F. et al. (2017) Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 8, 14061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan, K.W. , Wang, S. , He, F. , Chao, S. , Lun, Y. , Paux, E. , Sourdille, P. et al. (2018) The genetic architecture of genome‐wide recombination rate variation in allopolyploid wheat revealed by nested association mapping. Plant J. 95, 1039–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney, S. , Giroux, C.N. and Kleckner, N. (1997) Meiosis‐specific DNA double‐strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88, 375–384. [DOI] [PubMed] [Google Scholar]
- Kianian, P.M.A. , Wang, M. , Simons, K. , Ghavami, F. , He, Y. , Dukowic‐Schulze, S. , Sundararajan, A. et al. (2018) High‐resolution crossover mapping reveals similarities and differences of male and female recombination in maize. Nat. Commun. 9, 2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korhonen, J. , Martinmäki, P. , Pizzi, C. , Rastas, P. and Ukkonen, E. (2009) MOODS: fast search for position weight matrix matches in DNA sequences. Bioinformatics, 25, 3181–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger, F. and Andrews, S.R. (2011) Bismark: a flexible aligner and methylation caller for Bisulfite‐Seq applications. Bioinformatics, 27, 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambing, C. , Franklin, F.C. and Wang, C.R. (2017) Understanding and manipulating meiotic recombination in plants. Plant Physiol. 173, 1530–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. and Salzberg, S.L. (2012) Fast gapped‐read alignment with Bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, E.J. , Griffin, C.H. and Henderson, I.R. (2017) Modification of meiotic recombination by natural variation in plants. J. Exp. Bot. 68, 5471–5483. [DOI] [PubMed] [Google Scholar]
- Layer, R.M. , Chiang, C. , Quinlan, A.R. and Hall, I.M. (2014) LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 15, R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, K. , Kim, M.S. , Lee, J.S. , Bae, D.N. , Jeong, N. , Yang, K. , Lee, J.D. et al. (2020) Chromosomal features revealed by comparison of genetic maps of Glycine max and Glycine soja . Genomics, 112, 1481–1489. [DOI] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Ye, G. and Wang, J. (2007) A modified algorithm for the improvement of composite interval mapping. Genetics, 175, 361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, R. , Bitoun, E. , Altemose, N. , Davies, R.W. , Davies, B. and Myers, S.R. (2019) A high‐resolution map of non‐crossover events reveals impacts of genetic diversity on mammalian meiotic recombination. Nat. Commun. 10, 3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, X. , Lin, W. , Ku, Y.S. , Wong, F.L. , Li, M.W. , Lam, H.M. , Ngai, S.M. et al. (2020) Analysis of soybean long non‐coding RNAs reveals a subset of small peptide‐coding transcripts. Plant Physiol. 182, 1359–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Du, H. , Li, P. , Shen, Y. , Peng, H. , Liu, S. , Zhou, G.A. et al. (2020) Pan‐Genome of wild and cultivated soybeans. Cell 182, 162–176. [DOI] [PubMed] [Google Scholar]
- Liu, Y. and Tian, Z. (2020) From one linear genome to a graph‐based pan‐genome: a new era for genomics. Sci. China Life Sci. 63, 1938–1941. [DOI] [PubMed] [Google Scholar]
- Lu, Z. , Hofmeister, B.T. , Vollmers, C. , DuBois, R.M. and Schmitz, R.J. (2017) Combining ATAC‐seq with nuclei sorting for discovery of cis‐regulatory regions in plant genomes. Nucleic Acids Res. 45, e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Z. , Marand, A.P. , Ricci, W.A. , Ethridge, C.L. , Zhang, X. and Schmitz, R.J. (2019) The prevalence, evolution and chromatin signatures of plant regulatory elements. Nat. Plants, 5, 1250–1259. [DOI] [PubMed] [Google Scholar]
- Ma, X. , Fu, Y.C. , Zhao, X.H. , Jiang, L.Y. , Zhu, Z.F. , Gu, P. , Xu, W.Y. et al. (2016) Genomic structure analysis of a set of Oryza nivara introgression lines and identification of yield‐associated QTLs using whole‐genome resequencing. Sci. Rep. 6, 27425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marand, A.P. , Jansky, S.H. , Zhao, H.N. , Leisner, C.P. , Zhu, X.B. , Zeng, Z.X. , Crisovan, E. et al. (2017) Meiotic crossovers are associated with open chromatin and enriched with Stowaway transposons in potato. Genome Biol. 18, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marand, A.P. , Zhao, H. , Zhang, W. , Zeng, Z. , Fang, C. and Jiang, J. (2019) Historical meiotic crossover hotspots fueled patterns of evolutionary divergence in rice. Plant Cell 31, 645–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe, J.G. , Srikant, T. , Carbonell‐Bejerano, P. , Becker, C. , Lensink, M. , Exposito‐Alonso, M. , Klein, M. et al. (2022) Mutation bias reflects natural selection in Arabidopsis thaliana . Nature 602, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman, K. , Higgins, J.D. , Sanchez‐Moran, E. , Armstrong, S.J. and Franklin, F.C. (2011) Pathways to meiotic recombination in Arabidopsis thaliana . New Phytol. 190, 523–544. [DOI] [PubMed] [Google Scholar]
- Paigen, K. and Petkov, P.M. (2018) PRDM9 and its role in genetic recombination. Trends Genet. 34, 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, Q. , Li, L. , Yang, X. , Tong, H. , Xu, S. , Li, Z. , Li, W. et al. (2016) Genome‐wide recombination dynamics are associated with phenotypic variation in maize. New Phytol. 210, 1083–1094. [DOI] [PubMed] [Google Scholar]
- Patel, L. , Kang, R. , Rosenberg, S.C. , Qiu, Y. , Raviram, R. , Chee, S. , Hu, R. et al. (2019) Dynamic reorganization of the genome shapes the recombination landscape in meiotic prophase. Nat. Struct. Mol. Biol. 26, 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer, B. , Wittelsburger, U. , Ramos‐Onsins, S.E. and Lercher, M.J. (2014) PopGenome: an efficient Swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 31, 1929–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, X. , Li, M.W. , Xie, M. , Liu, X. , Ni, M. , Shao, G. , Song, C. et al. (2014) Identification of a novel salt tolerance gene in wild soybean by whole‐genome sequencing. Nat. Commun. 5, 4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez, F. , Ryan, D.P. , Gruning, B. , Bhardwaj, V. , Kilpert, F. , Richter, A.S. , Heyne, S. et al. (2016) deepTools2: a next generation web server for deep‐sequencing data analysis. Nucleic Acids Res. 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch, T. , Zichner, T. , Schlattl, A. , Stutz, A.M. , Benes, V. and Korbel, J.O. (2012) DELLY: structural variant discovery by integrated paired‐end and split‐read analysis. Bioinformatics, 28, i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers‐Melnick, E. , Bradbury, P.J. , Elshire, R.J. , Glaubitz, J.C. , Acharya, C.B. , Mitchell, S.E. , Li, C. et al. (2015) Recombination in diverse maize is stable, predictable, and associated with genetic load. Proc. Natl. Acad. Sci. U. S. A. 112, 3823–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan, B.A. , Heavens, D. , Feuerborn, T.R. , Tock, A.J. , Henderson, I.R. and Weigel, D. (2019) An ultra high‐density Arabidopsis thaliana crossover map that refines the influences of structural variation and epigenetic features. Genetics, 213, 771–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutz, J. , Cannon, S.B. , Schlueter, J. , Ma, J. , Mitros, T. , Nelson, W. , Hyten, D.L. et al. (2010) Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183. [DOI] [PubMed] [Google Scholar]
- Segal, E. and Widom, J. (2009) Poly(dA:dT) tracts: major determinants of nucleosome organization. Curr. Opin. Struct. Biol. 19, 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, Y. , Zhou, Z. , Wang, Z. , Li, W. , Fang, C. , Wu, M. , Ma, Y. et al. (2014) Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell 26, 996–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilo, S. , Melamed‐Bessudo, C. , Dorone, Y. , Barkai, N. and Levy, A.A. (2015) DNA crossover motifs associated with epigenetic modifications delineate open chromatin regions in Arabidopsis . Plant Cell 27, 2427–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao, X. , Feng, S. , Zhao, T. and Guan, X. (2020) Efficient chromatin profiling of H3K4me3 modification in cotton using CUT&Tag. Plant Methods, 16, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov, A. , Vilella, A.J. , Cuppen, E. , Nijman, I.J. and Prins, P. (2015) Sambamba: fast processing of NGS alignment formats. Bioinformatics, 31, 2032–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tek, A.L. , Kashihara, K. , Murata, M. and Nagaki, K. (2010) Functional centromeres in soybean include two distinct tandem repeats and a retrotransposon. Chromosome Res. 18, 337–347. [DOI] [PubMed] [Google Scholar]
- Underwood, C.J. , Choi, K. , Lambing, C. , Zhao, X. , Serra, H. , Borges, F. , Simorowski, J. et al. (2018) Epigenetic activation of meiotic recombination near Arabidopsis thaliana centromeres via loss of H3K9me2 and non‐CG DNA methylation. Genome Res. 28, 519–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urich, M.A. , Nery, J.R. , Lister, R. , Schmitz, R.J. and Ecker, J.R. (2015) MethylC‐seq library preparation for base‐resolution whole‐genome bisulfite sequencing. Nat. Protoc. 10, 475–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valliyodan, B. , Cannon, S.B. , Bayer, P.E. , Shu, S. , Brown, A.V. , Ren, L. , Jenkins, J. et al. (2019) Construction and comparison of three reference‐quality genome assemblies for soybean. Plant J. 100, 1066–1082. [DOI] [PubMed] [Google Scholar]
- Wang, J. , Chu, S. , Zhang, H. , Zhu, Y. , Cheng, H. and Yu, D. (2016) Development and application of a novel genome‐wide SNP array reveals domestication history in soybean. Sci. Rep. 6, 20728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. , Li, M. and Hakonarson, H. (2010) ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Jia, G. , Jiang, X. , Cao, S. , Chen, Z.J. and Song, Q. (2021) Altered chromatin architecture and gene expression during polyploidization and domestication of soybean. Plant Cell 33, 1430–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. and Copenhaver, G.P. (2018) Meiotic recombination: mixing it up in plants. Annu. Rev. Plant Biol. 69, 577–609. [DOI] [PubMed] [Google Scholar]
- Wijnker, E. and de Jong, H. (2008) Managing meiotic recombination in plant breeding. Trends Plant Sci. 13, 640–646. [DOI] [PubMed] [Google Scholar]
- Wijnker, E. , Velikkakam James, G. , Ding, J. , Becker, F. , Klasen, J.R. , Rawat, V. , Rowan, B.A. et al. (2013) The genomic landscape of meiotic crossovers and gene conversions in Arabidopsis thaliana . Elife 2, e01426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J. , Mizuno, H. , Hayashi‐Tsugane, M. , Ito, Y. , Chiden, Y. , Fujisawa, M. , Katagiri, S. et al. (2003) Physical maps and recombination frequency of six rice chromosomes. Plant J. 36, 720–730. [DOI] [PubMed] [Google Scholar]
- Yang, S. , Yuan, Y. , Wang, L. , Li, J. , Wang, W. , Liu, H. , Chen, J.Q. et al. (2012) Great majority of recombination events in Arabidopsis are gene conversion events. Proc. Natl. Acad. Sci. U. S. A. 109, 20992–20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelina, N. , Diaz, P. , Lambing, C. and Henderson, I.R. (2015) Epigenetic control of meiotic recombination in plants. Sci. China Life Sci. 58, 223–231. [DOI] [PubMed] [Google Scholar]
- Zelkowski, M. , Olson, M.A. , Wang, M. and Pawlowski, W. (2019) Diversity and determinants of meiotic recombination landscapes. Trends Genet. 35, 359–370. [DOI] [PubMed] [Google Scholar]
- Zhou, Z. , Jiang, Y. , Wang, Z. , Gou, Z. , Lyu, J. , Li, W. , Yu, Y. et al. (2015) Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 33, 408–414. [DOI] [PubMed] [Google Scholar]
- Zuo, W. , Chen, G. , Gao, Z. , Li, S. , Chen, Y. , Huang, C. , Chen, J. et al. (2021) Stage‐resolved Hi‐C analyses reveal meiotic chromosome organizational features influencing homolog alignment. Nat. Commun. 12, 5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Evaluation of the repeatability and accuracy of SNPs in the 600K SoySNP array.
Figure S2 Classification of SNPs in the 600K SoySNP array.
Figure S3 Recombination maps for soybean RIL populations.
Figure S4 Features of bins in the recombination bin map.
Figure S5 Linkage maps for eight soybean RIL populations using bins as genetic markers.
Figure S6 Distribution of recombination events identified in eight soybean RIL populations.
Figure S7 Genomic features of recombination events in soybean.
Figure S8 Non‐crossover (NCO) events and their genomic features in soybean RIL populations.
Figure S9 SVs detected between the parental lines of each RIL population.
Figure S10 Overlaps between SVs and recombination events for each population.
Figure S11 Poly‐A and AT‐rich motifs associated with the distribution of high‐resolution recombination events along Chr02 to Chr20.
Figure S12 Epigenetic modifications across recombination‐related genes and random genes obtained from permutations and in their flanking regions.
Figure S13 The distribution of read counts for chromatin states, histone modifications, histone variant H2A.Z and DNA methylation levels along intergenic recombination sites and their flanking regions compared to random sites.
Table S1 Summary of 13 representative soybean accessions for deep resequencing
Table S2 Eight soybean RIL populations constructed in our study
Table S4 Summary of genetic maps for eight soybean RIL populations
Table S5 Summary for non‐crossover (NCO) events detected in soybean RIL populations
Table S6 Recombination hotspots identified in eight soybean RIL populations
Table S7 QTL mapping for recombination hotspot usage
Table S8 QTL mapping for the total number of recombination events for each RIL in soybean RIL populations
Table S9 Sequencing information in this study
Table S10 Summary of long non‐coding RNAs (lncRNAs) overlapping intergenic recombination sites.
Table S3 The high‐resolution genotypic map for eight soybean RIL populations.
Data Availability Statement
The sequencing data generated in this study have been uploaded to the Genome Sequence Archive (GSA) database in the BIG Data Center (https://ngdc.cncb.ac.cn/) under accession number PRJCA008111. The resequencing data for 13 parental soybean accessions were deposited into Sequence Read Archive (SRA) database in NCBI under accession number PRJNA394629 or the GSA database in BIG Data Center under accession number PRJCA000205. The previously reported NGS data for H2A.Z, H3K36me3, H3K4me1 and H3K56ac were from the Gene Expression Omnibus (GEO) database through accession number GSE128434 and for H3K14ac, H3K27ac, H3K27me3 and H4K12ac were from SRA database via accession number PRJNA657728.