

Abstract
Alzheimer’s disease (AD), a persistent neurological dysfunction, has an increasing prevalence with the aging of the world and seriously threatens the health of the elderly. Although there is currently no effective treatment for AD, researchers have not given up, and are committed to exploring the pathogenesis of AD and possible therapeutic drugs. Natural products have attracted considerable attention owing to their unique advantages. One molecule can interact with multiple AD-related targets, thus having the potential to be developed in a multi-target drug. In addition, they are amenable to structural modifications to increase interaction and decrease toxicity. Therefore, natural products and their derivatives that ameliorate pathological changes in AD should be intensively and extensively studied. This review mainly presents research on natural products and their derivatives for the treatment of AD.
Keywords: Natural product, Alzheimer’s disease, cholinease inhibitor
Introduction
Alzheimer’s disease is an insidious and progressive neuro- degenerative disease1. Clinical symptoms include memory impairment, aphasia, apraxia, agnosia, impairment of visuospatial skills, executive dysfunction, personality and behavioural changes, and other manifestations of dementia. Numerous complications lead to a high number of deaths. By 2025, AD patients aged 65 and over will reach 7.1 million in developed countries, an increase of nearly 29% from the 5.5 million patients of the same age in 2018. Unless there is a medical breakthrough, the number of AD patients may nearly triple from 5.5 million to 13.8 million by 20502. The severity of the disease does not allow us to ignore it. An increasing number of scientists are conducting comprehensive research on the pathological mechanism of AD, but the cause has not been clearly determined.
Based on the pathological conditions of patients, various hypotheses have been proposed for the study of the aetiology of AD, including microtubule-associated protein aggregation, Aβ cascade, neuro-inflammation, and gene mutation. Some researchers believe that the central cholinergic hypothesis is the most relevant to the pathogenesis of AD. The nucleus basalis of Meynert in the basal forebrain of patients with AD is severely damaged3, and the content of choline is significantly increased. Severe depletion of presynaptic acetylcholine leads to severe memory loss, therefore, cholinergic agonists and cholinesterase inhibitors are consider for ameliorating cognitive deficits4; Others that tubulin and Aβ cascades work together leading to a series of pathological changes. Excessive Aβ in the brain of patients cannot be degraded in time, and aggregates and precipitates in the brain to form insoluble amyloids. These insoluble amyloids induce neurotoxicity5. It also enhances the hyperphosphorylation of tau protein, causing its abnormal aggregation intracellular neurofibrillary tangles (NFTs), resulting in the loss of normal nerve cells function and even death6. With further research, it has been found that homeostasis of metal ion metabolism in the body can also cause neuronal dysfunction and death7. Some studies have shown that excessive iron mediates the formation of Aβ plaques in the brain. And causes hyperphosphorylation of tau protein in the brain. In addition, oxidative stress is a pathogenic factor in AD. The imbalance of oxidants and antioxidants in the body leads to the increase of reactive oxygen species (ROS) and reactive nitrogen species (RNS), including H2O2 and NO8, which at high levels can promote Aβ aggregation and phosphorylation of tau protein9, and can induce mutations in neuronal DNA and RNA10, thereby aggravating the nerve damage. The multiple pathogenetic mechanisms are interconnected, making it impossible to accurately identify the aetiology of AD. These pathogenic mechanisms derive from pathological changes of patients with AD, and have been suggested by various studies.
The current clinical treatment drugs for AD are mainly cholinesterase inhibitors, including: donepezil, rivastigmine, galantamine; and the N-methyl-D-aspartic acid (NMDA) receptor antagonists: memantine and amantadine. These drugs relieve the symptoms of AD but lack efficacy. Researchers are now turning their attention to the development of therapeutics using natural products. Studies have shown that the structural complexity and chemical properties of natural products give them value in drug development. With the progress in scientific research, more natural product structures and properties have been confirmed, and natural products have become an important source of new lead compounds in drug research and development11. For example, A series of novel Lappaconitine derivatives enhanced its anti-inflammatory activity, and dihydroartemisinin derivatives and ursolic acid improved its anti-T.gondii activity12–14. Furthermore, natural products and their derivatives, and foodstuffs account for more than one-third of all new molecular entities approved by the FDA15,16. Natural products have also shown efficacy in neurodegenerative diseases. Phenylpropanoids, flavonoids, terpenoids, alkaloids, and their derivatives have been shown to improve the pathological changes of AD by acting on multiple targets, such as inhibiting acetylcholinesterase, reducing tau and amyloid aggregation, reducing neuroinflammation, reducing Aβ1-42-induced neurotoxicity, and inhibiting β-site amyloid precursor protein cleaving enzyme 1 (BACE-1). In this review, we summarise the recent five years literature on the effects of natural products on AD.
Phenylpropanoids and their derivatives
Ferulic acid and its derivatives
Ferulic acid (FA) is a phenylpropionic acid compound that has attracted the attention of many scientists for the treatment of AD. FA was found to ameliorate AD-like pathology and inhibit cognitive decline by preventing capillary hypofunction in APP/PS1 mice17. Treatment with the combination of two nutraceuticals, epigallocatechin gallate (EGCG) and FA, reduced brain parenchymal and cerebrovascular beta-amyloid deposition, neuroinflammation, oxidative stress, and synaptic toxicity in APP/PS1 mice18. In vivo experiments have shown that FA inhibits cholinesterase activity. Inhibition of cholinesterase can enhance cholinergic neurotransmission, thereby relieving the symptoms of AD (Figure 1).
Figure 1.

The chemical structure of ferulic acid and its derivatives 1–8.
Yash et al. synthesised several FA derivatives, and showed that compound 1 inhibited acetylcholinesterase (AChE) (IC50 = 5.74 μM) more strongly than the lead compound FA (IC50 = 15.19 μM) in vitro and in vivo. It showed good in vivo activity in an AD mouse model and improved spatial memory in mice with cognitive impairment in the Y-maze model. In addition, Compound 1 can effectively improve the levels of AChE and BChE in mouse models. and the introduction of an indole ring and quinoline ring improved selectivity compared to a benzene ring. They suggested that compound 1 has the potential to be developed for the treatment of AD and other neurodegenerative diseases19 (Figure 1, Table 1).
Table 1.
Effect of ferulic acid derivatives on the various pathological pathways involve in AD.
| Compound No. | Classification | The major targets | Activity (µM) | References |
|---|---|---|---|---|
| 1 | Ferulic acid derivatives | AChE inhibitory activity | IC50 (hAChE) = 5.74 | 19 |
| 2 | MAO-B inhibitory activity Inhibits self-induced aggregation of Aβ1-42 |
IC50 (hMAO-B) = 0.32 | 20 | |
| 3 | IC50 (hMAO-B) = 0.56 | |||
| 4 | IC50 (hMAO-B) = 0.54 | |||
| 5 | IC50 (hMAO-B) = 0.73 | |||
| 6 | IC50 (hMAO-B) = 0.86 | |||
| 7 | Inhibits self-induced Aβ Aggregation and the efficacy of AChE | IC50 (hAChE) = 0.0192 IC50 (hBChE)= 0.66 |
21 | |
| 8 | HDAC6 inhibitory activity DPPH radical scavenging ability Cu2+chelating ability Preventing Aβ25 − 35 induced spatial work and long-term memory dysfunction |
IC50 (HDAC6)=0.0307 | 22 | |
| 9 | Coumarin derivatives | MAO-B inhibitory activity | IC50 (hMAO-B) = 0.00507 | 23 |
| 10 | IC50 (hMAO-B) =0.0042 | |||
| 11 | IC50 (hMAO-B) =0.00394 | |||
| 12 | Inhibit AChE and BChE | IC50 (hBChE) = 30.3 | 24 | |
| 13 | IC50 (hBChE) = 29.2 | |||
| 14 | IC50 (hBChE) = 37.2 | |||
| 15 | IC50 (hBChE) = 50.1 | |||
| 16 | Inhibit AChE and MAO-B Reversing cognitive dysfunction in AD mice induced by scopolamine Inhibit AChE and BChE |
IC50 (hAChE) = 0.0068 | 25 | |
| 17 | IC50 (hAChE) = 0.114 | |||
| 18 | IC50 (hBChE) = 30.2 | 26 | ||
| 19 | IC50 (hBChE) = 0.363 | |||
| 20 | Eugenol derivatives | MAO-A inhibitory activity | IC50 (hMAO-A) = 5.989 | 27 |
| 21 | IC50 (hMAO-A) = 7.348 | |||
| 22 | MAO-B inhibitory activity | IC50 (hMAO-B) = 7.494 | ||
| 23 | IC50 (hMAO-B) = 9.183 |
Gaofeng et al. synthesised novel O-alkyl ferulamide derivatives and evaluated their activity against monoamine oxidase B (MAO-B). Compounds 2, 3, 4, 5, and 6 exhibited significant selective MAO-B inhibition (IC50 = 0.32, 0.56, 0.54, 0.73, and 0.86 μM, respectively), significantly inhibited Aβ1-42 aggregation, and effectively protected PC12 cells Aβ1-42-induced damage, all of which have a good blood-brain barrier permeability. This is consistent with the properties required for the development of new drugs20 (Figure 1, Table 1).
Jin-Shuai et al. designed and synthesised 15 FA derivatives. Through in vitro experiments, compound 7 was found to effectively inhibit AChE and BChE (IC50 = 19.7 nM and 0.66 μM). respectively; when a benzylamino group with a five-carbon spacer was attached to FA, the inhibition rate of AChE was improved. Furthermore, the introduction of an electron-absorbing methoxy group to the benzene ring of FA enhanced its inhibitory activity. hAChE and hBuChE were inhibited to a greater extent when the carbamate substituent on the benzylamino group was an N-methyl group. At 20 μM, the inhibition rate of Aβ aggregation was 49.2 and PC12 cells were well protected from hydrogen peroxide-induced oxidative cell damage. Molecular docking data indicated that compound 7 is a multi-target inhibitor that interacts with both the catalytic anion site and peripheral anion site of AChE, thus exerting a dual inhibitory effect21 (Figure 1, Table 1).
The structure of FA and melatonin was assembled into a histone deacetylase 6 (HDAC6) inhibitor to obtain a hybrid compound 8 that can inhibit multiple targets. which can have multiple targets. It had obvious inhibitory activity against HDAC6 (IC50 = 30.7 nM), DPPH free radical scavenging ability similar to that of FA, and Cu2+ chelating ability similar to that of melatonin. Aβ25-35-induced memory impairment was reversed at lower doses. In addition, simultaneous immunomodulatory, HDAC6-selective inhibitory, and antioxidant properties were observed. Thus, it is a potential drug candidate for the treatment of neurodegenerative diseases22 (Figure 1, Table 1).
Coumarin and its derivatives
The numerous pharmacological activities of coumarins depend on their core structure (e.g. simple coumarins, fused polycyclic coumarins, and dicoumarins). At present, there have been more in-depth studies on the role of coumarin in AD. Coumarins exhibit highly diverse activities against various pathways in AD. They act as Aβ42 aggregation inhibitors, dopamine and serotonin mediators, gamma-aminobutyric acid (GABA) receptor agonists, NMDA receptor antagonists, and monoamine oxidase (MAO) inhibitors (Figure 2).
Figure 2.

The chemical structure of coumarin and its derivatives 9–19.
Fonseca et al. modified the structure of coumarin and chromone, and evaluated their MAO-A and MAO-B inhibitory activities. The results showed that there was no significant difference between the two natural product derivatives in their activities. They had inhibitory effect on MAO-B and no significant activity on MAO-A. The MAO-B inhibitory activity of coumarin derivative 9 (IC50=5.07 nM), chromone derivative 10 (IC50=4.2 nM) and compound 11 (IC50=3.94 nM) is strong. The introduction of aniline has a good effect. The introduction of chlorine substituents at the same time can enhance the MAO-B inhibitory activity. The results of pharmacokinetics showed that compounds 9 and 10 had good binding power and selectivity through non-competitive inhibition23 (Figure 2, Table 1).
Orhan et al. evaluated the activity of 17 natural coumarin derivatives on AChE and BChE. Compounds 12, 13, 14 and 15 (IC50 = 30.3, 29.2, 37.2, and 50.1 μM, respectively) for butyrylcholine Esterase was more inhibited than positive control galantamine (IC50 = 60.2 μM), structural groups and oxyanion cavities in these coumarins and peripheral anion site residues of AChE/BChE interaction, potent inhibition of BChE and AChE24 (Figure 2, Table 1).
He et al. studied the structure-activity relationship between coumarin and dithiocarbamate and synthesised several novel coumarin derivatives with dual targets. Compound 16 was the most effective inhibitor of AChE (IC50 = 68 nM). Compound 17 is a dual binding site inhibitor of AChE and a competitive inhibitor of MAO-B (0.114 µM for hAChE; 0.101 µM for hMAO-B) with a good blood-brain barrier permeability. The o-methyl substitution of piperidine on the nitrogen atom in 16 plays a key role in inhibiting the activity of AChE and MAO-B. Adding another methyl group on the piperidine ring gives compound 17, which acquires significant MAO-B inhibitory activity. It was shown to reverse scopolamine-induced cognitive impairment in AD mice without having acute toxicity in mice at higher doses25. Thus, compound 17 can be used as a potential multi-target drug for the treatment of AD (Figure 2, Table 1).
Lee et al. synthesised warfarin-diethyl phosphate and coumarin-organophosphate compounds and evaluated their selectivity in inhibiting BChE in vitro, and concluded that the introduction of organophosphate increased ChE inhibitory activity. The selectivity of coumarin-organophosphate compounds is generally stronger than that of warfarin-diethyl phosphate for BChE inhibition; in particular,compound 19 showed a stronger inhibitory effect on BChE than 18. A 100-fold enhanced inhibitory activity (IC50 = 0.363 μM) was observed26. It was shown that the organophosphate attached to the coumarin structure rather than the benzyl structure is more favourable for faster phosphorylation of the leaving group. The introduction of organophosphates plays a key role in enhancing the activity and increasing the binding force of the ligands (Figure 2, Table 1).
Eugenol and its derivatives
Eugenol is used to prevent and treat AD. Inhibits AChE and BChE activities in a dose-dependent manner, s showing higher antioxidant activity28. Studies have shown that eugenol can significantly reduce the total area of hippocampal amyloid plaques and enhance memory in male Wistar rats at both 0.01 and 0.02 mg/kg doses29. It also shows inhibitory activity against the MAO-B enzyme and free-radical scavenging activity30. The eugenol-calamus combination attenuated lipid peroxide (LPO) and AChE levels in the hippocampus and significantly improved cognitive function in streptozotocin (STZ)-induced rats31. Numerous studies have shown that eugenol has the potential to be developed as a multi-target directed ligand (MTDL) for the treatment of AD (Figure 3).
Figure 3.

The chemical structure of eugenol and its derivatives 20–23.
Based on the inhibitory effect of eugenol monomers on MAO, a series of eugenol derivatives was designed and synthesised, and their activities were evaluated. Compounds 20 and 21 were found to be the most effective hMAO-A inhibitors, with IC50 values of 5.989 µM and 7.348 µM, respectively, which were stronger than the positive control drug Chlorgyline (IC50 = 18.74 µM); selectivity index values were 0.19 and 0.14, respectively. The semicarbazide group in compound 20 was found to be important for MAO inhibition. Furthermore, the effectiveness of compound 20, and compound 21 demonstrated the importance of the ester bond in targeting the active site of h-MAOA. Compounds 22 and 23 were more effective in inhibiting hMAO-B, with IC50 values of 7.494 µM and 9.183 µM, respectively, which were stronger than the control drug Pargyline (IC50 = 20.04 µM); selectivity indices were 5.14 and 5.72 µM, respectively; The introduction of 4-hydroxy-3- methoxybenzoate in the structure of compound 22 clearly enhanced the inhibitory activity against hMAO-B, and the smaller molecular structure of compound 23 makes it more active; therefore, it can be assumed that the introduction of the methoxy group slightly reduced the activity. The inhibition rates of the above four derivatives against the two monoamine oxidases were stronger than that of eugenol27. Reducing the activity of monoamine oxidase can significantly reduce oxidative stress (Figure 3, Table 1).
Flavonoids and their derivatives
Quercetin, kampferol and their derivatives
Natural flavonoids show a large number of biological activities. At present, there is evidence that these compounds can be used as MAO inhibitors, such as Homoisoflavonoids can be used as natural scaffolds for strong and selective MAO-B inhibition32,33, as well as natural flavonols Quercetin and Kampferol are flavonoid alcohols that widely exist in the plant kingdom. At present, Quercetin is an expectorant drug used clinically, and also has functions such as lowering blood pressure. The similarity of their structures determines the similarity of their pharmacological effects. At present, researchers have found that Quercetin and Kampferol can be used as effective MAO-A inhibitors. Among the six compounds isolated from H.hircine, Quercetin is the only one that selectively inhibits MAO-A, with an IC50 value of 0.010 µM34. Gidaro’s research shows that Kampferol also has a strong inhibitory effect on MAO-A35. As effective MAO inhibitor, they can effectively protect nerve cells (Figure 4).
Figure 4.

The chemical structure of quercetin /kampferol and their derivatives 24–26.
A series of quercetin derivatives were designed and synthesised, and their inhibitory and antioxidant activities on hMAO were screened. The results of hMAO inhibition in vitro showed that compound 24 was an effective hMAO-A inhibitor, while compounds 25 and 26 were effective hMAO-B inhibitors. DPPH radical scavenging activity data showed that compounds 25 and 26 showed enhanced antioxidant capacity, IC50 values were 5.931 µM and 6.421 µM, respectively. In addition, compounds 25 and 26 also showed significant H2O2 scavenging capacity, with IC50 values of 5.80 µM and 6.20 µM, respectively. In the docking study, compounds 25 and 26 have good correlation with experimental MAO. This explains the specific inhibition of MAO activity and antioxidant activity. These compounds can be used as effective neuroprotective agents and antioxidants to treat AD, so further experimental research is particularly important for clinical application36 (Figure 4, Table 2).
Table 2.
Effect of flavonoids derivatives on the various pathological pathways involve in AD.
| Compound NO. | Classification | The major targets | Activity (µM) | References |
|---|---|---|---|---|
| 24 | Quercetin derivatives | MAO-A inhibitory activity MAO-B inhibitory activity DPPH radical scavenging activity The hydrogen peroxide scavenging activity |
IC50 (hMAO-A) = 13.1 IC50 (hMAO-B) = 46.11 |
36 |
| 25 | IC50 (hMAO-A) = 32.16 IC50 (hMAO-B) = 18.87 |
|||
| 26 | IC50 (hMAO-A) = 36.92 IC50 (hMAO-B) = 12.70 |
|||
| 27 | Isoliquiritigenin derivatives | 5-LO inhibitory activity Inhibits self induced aggregation of Aβ1-42 |
IC50 (Aβ1-42) = 3.2 | 37 |
| 28 | IC50 (Aβ1-42) = 2.2 | |||
| 29 | IC50 (Aβ1-42) = 5.9 | |||
| 30 | IC50 (Aβ1-42) = 14.6 | |||
| 31 | Liquiritigenin derivatives | Selectively inhibit AChE and BChE The radical scavenging effects |
IC50 (hAChE) = 0.03 IC50 (hBChE) = 5.46 |
38 |
| 32 | Chalcone derivatives | Inhibit hMAO-A and hMAO-B | IC50 (hMAO-B) = 0.0044 | 39 |
| 33 | IC50 (hMAO-A) = 4.9 IC50 (hMAO-B) = 0.0051 |
|||
| 34 | Inhibit AChE and BChE Metal chelating agent Inhibit Aβ1-42 aggregation Improve memory impairment induced by scopolamine |
IC50 (hAChE) = 1.3 IC50 (hBChE) = 1.2 |
40 | |
| 35 | AChE inhibitory activity BACE-1 inhibitory activity |
IC50 (hAChE) = 0.08 IC50 (BACE-1) = 2.71 |
41 |
Liquiritigenin and isoliquiritigenin and their derivatives
Both liquiritigenin and isoliquiritigenin are dihydro- flavonoid monomer compounds extracted from the natural plant liquorice. As these two compounds have similar structures, their pharmacological activities are also similar. In recent years, researchers have found that liquiritigenin and isoliquiritigenin are promising agents for the treatment of AD. Studies have shown that liquiritigenin can be a potent inhibitor of tau amyloid fibril formation by preventing structural transitions in its structure and exposure of hydrophobic groups. Therefore, reducing tau aggregation-mediated neurotoxicity42. Aβ levels can also be reduced by modulating the M1/M2 phenotype transition in microglia, thereby reducing memory decline during AD43. Isoliquiritigenin attenuated Aβ25-35 induced neuronal damage in rat cortical neurons by interfering with [Ca2+]i and ROS production44. Isoliquiritigenin reduces neuronal damage by inhibiting 5-lipoxygenase (5-LO). The activity of 5-LO is regulated by 5-lipoxygenase-activating protein (FLAP), and targeting the 5-LO/FLAP pathway is considered an effective strategy for the treatment of AD. 5-LO is involved in AD pathological changes, and its activity is significantly enhanced. Studies have shown that activation of 5-LO promotes Aβ amyloid deposition and tau hyperphosphorylation45 (Figure 5, Table 2).
Figure 5.

The chemical structure of liquiritigenin/isoliquiritigenin and its derivatives 27–31.
Yi-Ping et al. designed and synthesised a series of isoliquiritigenin derivatives, most of which were more potent than the positive control drugs (resveratrol and nordihydroguaiaretic acid) on Aβ1-42 aggregation and 5-LO. More potent and similar to the lead compound isoliquiritigenin. In addition, it is interesting that compounds 27–30 have a strong inhibitory effect on the above two targets, and the structure-activity relationship shows that substitution at the 4-position of the A ring can improve the inhibitory activity of the compound, especially the six-membered ring amino side chain substituent. Among them, compound 27 (Figure 5) has the strongest inhibitory effect on the two targets, and the IC50 value of Aβ1-42 aggregation inhibition was 3.2 µM, which was stronger than that of the positive control drugs resveratrol (IC50 = 15.9 µM) and isoliquiritigenin (IC50=19.7 µM); the inhibition IC50 of 5-LO was 6.1 µM, which was stronger than that of the positive control drugs Noidihydroguaisretic acid and isoliquiritigenin, with IC50 of 12.4 µM and 18.6 µM, respectively. Molecular docking results showed that van der Waals forces and hydrogen bonding play important roles in the stability of 27/Aβ1-42 complexes and the intermolecular hydrogens bonding interaction between compound 27 and 5-LO. Therefore, compound 27 can be further studied as a potential drug for the treatment of AD37 (Figure 5, Table 2).
Selectivity of AChE and BuChE Based on Liquiritigenin, 7-prenyloxy-2,3-dihydroflavanone derivatives and 5-hydroxy-7- prenyloxy-2,3-dihydroflavanone derivatives, among 32 synthesised derivatives, compound 31 .It was confirmed to inhibit AChE (IC50 = 0.09 µM) and BChE (IC50 value of 5.46 µM) significantly stronger than donepezil (IC50 = 0.48 and 10.60 µM), through dual-site binding ability38. The structure-activity relationship of the two derivatives was discussed. The structure-activity relationships of the two derivatives are discussed. The introduction of the 5-position hydroxyl group may enhance the hydrophilicity of the molecule, and the introduction of the methoxy group improved inhibitory activity. Dihydroflavanones can bind to peripheral anion sites, causing some molecules to tilt and bind more tightly with the catalytic active site of acetylcholinesterase, effectively inhibiting its activity (Figure 5, Table 2).
Chalcone and its derivatives
Chalcone, also known as diphenylpropenone, is a flavonoid composed of phenolic compounds, one of the largest groups of bioactive natural products. Its unique chemical structural features have inspired the synthesis of numerous chalcone derivatives. To explore their ability to reduce intracellular amyloid and associated oxidative damage, more than ten natural products, including chalcone, were assayed, and trans-chalcone and baicalein were found to be the most potent compounds46. All compounds were flavonoids. Six natural chalcones were assayed, and most compounds exhibited inhibitory effects against both MAO-B and AChE47. Chalcone has a lower molecular weight, structural modification on its skeleton can increase the interaction with the target. Chalcone is more suitable for multi-target drug molecular design. This can greatly improve biological activity (Figure 6).
Figure 6.

The chemical structure of chalcone and its derivatives 32–35.
Based on the inhibitory activity of monoamine oxidase, Chimenti et al. designed and synthesised a series of chalcone derivatives, all of which showed inhibitory activity. Compound 32 (IC50=4.4 nmol) showed the strongest inhibitory effect on MAO-B; Compound 33 showed inhibitory activity on MAO-A and MAO-B, IC50 was respectively 4.9 μM and 0.0051 μM. The results of molecular docking confirmed the experimental results in vitro. Compound 32 interacts effectively with tyrosine amino acid residues, and compound 33 shows interaction through its chlorine phenyl ring and two hydrogen bonds with other related residues. This also explains the source of the inhibitory effect of the two compounds. It is also confirmed that chalcone can be used as an important source of hMAOs inhibitors skeleton39 (Figure 6, Table 2).
Ping et al. designed and synthesised a series of novel chalcone-O-alkylamine derivatives with anti- AD activity based on the concept of multi-target therapeutic drugs. The experimental results showed that the compounds have multi-target therapeutic effects: (1) Inhibit both AChE and BChE; (2)Inhibit the expression of MAO-B; (3)Inhibit self-induced Aβ1-42 aggregation; (4) Be used as selective metal chelators. Among them, the comprehensive experimental results found that compound 34 had the highest potential. The IC50 values for the inhibition of AChE and BuChE were 1.3 μM and 1.2 μM, respectively, and inhibitory activity on the expression of MAO-B was the strongest (IC50 = 0.57 μM). It exhibits significant inhibition and depolymerisation of Aβ1-42 aggregation and can also inhibit and break Cu2+-induced Aβ1-42 aggregation. Structure-activity relationship analysis showed that the length of the methylene chain has a key effect on the inhibition of AChE and BuChE, and elongation of the carbon chain significantly increases the inhibitory effect40 (Figure 6, Table 2).
Renata et al. synthesised a series of 2′-aminochalcone derivatives for the inhibition of AChE and BACE-1 enzymes and Aβ. All amino-modified chalcones were found to have a better inhibitory effect on BACE-1 enzyme, which can reduce the production and deposition of amyloid. Furthermore; compound 35 had a strong inhibitory effect on multiple targets; the IC50 value for ACHE was 0.08 μM, and the IC50 value for BACE-1 was 2.71 μM. The molecular docking results verified the in vitro experiments, and the compound can be used as a dual-target drug candidate for AD41 (Figure 6, Table 2).
Terpenoids and their derivatives
Triptolide and its derivatives
Triptolide is an epoxy diterpene lactone and one of the main active components of Tripterygium wilfordii. Triptolide can inhibit the activation and proliferation of microglia and astrocytes in an APP/PS1 double-transgenic AD mouse model reduce neurotoxicity48. Furthermore, it was found to inhibit the expression of β-site amyloid precursor protein cleaving enzyme 1 and reduce Aβ production and deposition in the brain thus reversing the pathological changes in AD49. In vivo experiments have shown that triptolide increases the expression of insulin-degrading enzyme, a major Aβ-degrading enzyme in the brain50, which can reduce AD pathological symptoms by increasing Aβ degradation. A number of studies have confirmed that the natural product triptolide is an effective and multifunctional lead compound that can act on multiple targets relevant to AD and has the potential to be developed for the treatment of AD (Figure 7).
Figure 7.

The chemical structure of triptolide and its derivatives 36–44.
Some researchers have synthesised 9 triptolide derivatives based on structure-activity relationship analysis. Cell culture experiments were carried out, and it was concluded that compound 43 had the greatest potential for further study. Compounds 36–38 are modified by ring-opening C12 and C13-epoxy structure of triptolide; compound 39 is modified by ring-opening C-7 and C8-β-epoxy structure; compounds 40–44 are modified by ring-opening of 14 -β-OH for modification. In vitro experiments showed that triptolide could protect cells from Aβ1-42 damage, and the ring-opening of its epoxy group enhanced its cell damage. indicating that the neuroprotective pharmacophore of triptolide is epoxy group rather than 14-β-OH. Compound 43 dose-dependently enhanced the survival rate of Aβ1-42-treated cells. It also had the strongest anti-inflammatory activity, similar to triptolide, and completely inhibited LPS-induced TNF-α expression at a concentration of 10 nM, which warrants further study51 (Figure 7, Table 3).
Table 3.
Effect of terpenoids derivatives on the various pathological pathways involve in AD.
| Compound No. | Classification | The major targets | Activity (µM) | References |
|---|---|---|---|---|
| 36–44 | Triptolide derivatives | Protect nerve cells from Aβ1-42 damage Inhibits LPS induce TNF |
_ | 51 |
| 45/46 | Andrographolide derivatives | Inhibits LPS induced NO production, iNOS expression and proinflammatory cytokine TNF- α and IL-6 Protect neurons from microglia mediated neurotoxicity Promotes NGF induced axon growth in PC12 cells |
_ | 52 |
| 47-49 | β-Secretase inhibitors AChE inhibitory activity |
_ | 53 | |
| 50 | Glycyrrhetinic acid derivatives | AChE inhibitory activity | IC50 (hAChE) = 3.43 | 54 |
| 51 | IC50 (hAChE) = 5.39 | |||
| 52 | IC50 (hAChE) = 6.27 | |||
| 53 | IC50 (hAChE) = 8.68 | |||
| 54 | BChE inhibitory activity | IC50 (hBChE) = 5.43 | 55 | |
| 55 | IC50 (hBChE) = 9.81 | |||
| 56 | Safranal derivatives | MAO-A inhibitory activity MAO-B inhibitory activity |
IC50 (hMAO-A) = 9.93 IC50 (hMAO-B) = 0.0091 |
56 |
Andrographolide and its derivatives
Andrographolide (ANDRO) is a diterpene lactone compound extracted from Andrographis paniculata, which is one of the main components of the traditional Chinese medicine Andrographis paniculata, and is known as a natural antibiotic drug. Therefore, it is widely used in clinical practice57. However, many studies have shown that ANDRO can be a drug for AD through multiple pathways. For example, ANDRO activated autophagy-related genes and proteins (Beclin-1 and LC3), while it also enhanced the expression of Nrf2 and p62 at the mRNA and protein levels, and decreased p-tau and p-tau in Aβ1-42 stimulated cells. Thus improving cell damage caused by Aβ1-42 injury58; ANDRO has also been shown to stimulate neurogenesis in the hippocampus and improve memory performance by inducing the proliferation of neural precursor cells; ANDRO induces glucosamine-nitrosourea compound behavioural disorders and changes in hippocampal biochemistry, neuroinflammatory mediators, and neurotransmitters have protective effects59. In addition, ANDRO reduced the levels of Aβ1-42 and p-tau in the rat hippocampus, thereby exerting neuroprotective effects and ameliorating cognitive impairment and AD-like symptoms60. ANDRO can improve the pathological symptoms of AD in various ways, and more in-depth and comprehensive research may bring ANDRO to the clinic (Figure 8).
Figure 8.

The chemical structure of andrographolide and its derivatives 45–49.
Xu et al. designed and synthesised 12 Andrographis paniculata derivatives, all of which were non-toxic. Screening for inhibitors of LPS-induced NO production in BV-2 cells showed that compounds 45 and 46 had stronger inhibitory rates than the lead compound ANDRO, suggesting that the introduction of C-3 and C-19 acetyl groups has a major effect. compounds 45 and 46 on LPS-induced proinflammatory factors TNF-α and IL-6 significantly attenuated the levels of LPS-induced pro-inflammatory factors TNF-α and IL-6 in a dose-dependent manner. These results suggest that compounds 45 and 46 can reduce the production of pro-inflammatory factors and protect neuronal cells from microglia-mediated neurotoxicity52 (Figure 8, Table 3).
Dey et al. synthesised 24 andrographolide derivatives, which can be divided into two series according to their conformation at the C-14 position. BACE-1 cleaves β-amyloid precursor protein (βAPP) to produce Aβ, while α-secretase prevents Aβ production and triggers the release of neuroprotective sAPPα61. ANDRO and compound 47 were found to activate α-secretase, and compounds 48 and 49 to effectively inhibit BACE-1, thus all inhibiting the production of Aβ53. Therefore, other cascade reactions caused by Aβ are reduced and damage to nerve cells is prevented (Figure 8, Table 3).
Glycyrrhetinic acid and its derivatives
Glycyrrhetinic acid also has potential for the treatment of AD. Both 18α- and 18β-glycyrrhetinic acids exert different degrees of inhibition of BACE1. 18β-Glycyrrhetinic acid showed a strong inhibitory effect with IC50 of 8.93 µM62. Furthermore, it decreased the expression levels of BACE-1 and reduced Aβ production Studies have shown that the coumarins glycerol and liquiritigenin, isolated from liquorice, have cholinesterase inhibitory effects63. Some triterpenoids, such as oleanolic acid and ursolic acid, have been shown to inhibit acetylcholinesterase64, Based on this, many researchers have modified the structure of glycyrrhetinic acid to produce acetylcholine inhibitors with higher inhibitory activities (Figure 9).
Figure 9.

The chemical structure of glycyrrhetinic acid and its derivatives 50–55.
Fatma et al. synthesised a series of glycyrrhizic acid derivatives, and evaluated the AChE inhibitory effect of 13 derivatives and glycyrrhizin using the Ellman method. They found that 50, 51, 52 and 53 had higher inhibitory effects than glycyrrhizin (IC50 = 3.43, 5.39, 6.27 and 8.68 μM, respectively). From these data, The inhibition rate of C-30 forming amide bond is stronger than that of forming ester bond. No cytotoxicity was observed in the normal cells (WI-38). Pharmacological experiments indicated that the compounds had insignificant antioxidant capacity; thus, their effect is not through the antioxidant pathway54. The complete mechanism of action is not yet clear, and further in-depth research is required (Figure 9, Table 3).
Stefan et al. synthesised a series of aminoglycyrrhetinic acid derivatives and discussed the structure-activity relationship at C-3 and C-30 positions. The first group of derivatives is to synthesise 27 amino acids-glycyrrhetinic acid by esterification of N-Boc-amino acids with C-3 position. The second group is glycyrrhetinic acid and the corresponding brominated and alkyl diamines to form ester and amide derivatives; the third group is the reaction of glycyrrhetinic acid C-30 and nitrogen-containing heterocycles with larger groups, and two derivatives were synthesised. In vitro experiments showed that the inhibitory effect of the first group of derivatives on AChE was stronger than that of BChE. Compounds 54 and 55 had strong selective inhibition on BChE, with IC50 of 5.43 and 9.81 μM, respectively. There was an interaction between the two compounds and the active site of BChE55. The results of molecular docking confirmed the results of cell experiments (Figure 9, Table 3).
Safranal and its derivatives
Saffron is a perennial flower of crocus in Iridaceae, also known as saffron, which is most commonly used in spices. Saffron and its main components can improve mental diseases and neurodegenerative diseases, among which Safranal is the main component of saffron, a monoterpenoid aldehyde. At present, Safranal can reverse the neurotoxicity of PC12 cells induced by OGD by regulating oxidation and apoptosis65; Reduced Aβ and H2O2 induced PC12 cytotoxicity and oxidative damage66; Interacted with cholinergic, glutamic and dopaminergic systems67 (Figure 10).
Figure 10.
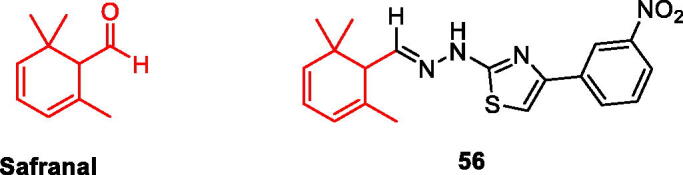
The chemical structure of safranal and its derivatives 56.
Carradori et al. evaluated the hMAO activity of a series of saffron aldehyde derivatives, and found that safranal had no inhibitory effect on hMAO, but its derivatives showed good inhibitory effect, especially compound 56 on hMAO-A (IC50 = 9.93 μM) And hMAO-B (IC50 = 0.091 μM) All showed inhibition, especially for MAO-B, its inhibition effect could be compared with lazabemide (IC50 = 0.01 μM). It’s on a par. Compound 56 can be used as a new skeleton of hMAO inhibitors. It can be seen that the structure of Safranal may be transformed into a highly effective hMAO-B inhibitor and a moderately effective hMAO-A inhibitor56 (Figure 10, Table 3).
Saponin and its derivatives
Diosgenin and its derivatives
Diosgenin is an important natural steroidal sapogenin derived from a wide range of sources, including fenugreek. It is an important raw material for the synthesis of steroid hormone drugs such as cortisone. Diosgenin protected a rat model of AD from Aβ1-42-induced neurotoxicity68. Diosgenin was found to reduce heat shock homologs in Aβ-induced injury in 5XFAD mice69, HSC70 belongs to the family of heat shock proteins and can promote neuronal axonal degeneration and pathology in the brain of AD patients when expressed at high levels70, Τhus, effectively inhibiting HSC70 as a new target for the treatment of AD (Figure 11).
Figure 11.

The chemical structure of diosgenin and its derivatives 57–59.
Based on the fact that diosgenin can interact with multiple targets, many scientists have modified its structure, hoping to obtain anti-AD drug candidates with stronger activity and fewer side effects. Gui-Xiang et al. added carbamates to the diosgenin structure and examined the structure-activity relationship. Studies have shown that carbamate fragments can significantly improve AD pathology. Therefore, this topic has become a research hotspot. Numerous studies have reported on the treatment of AD71. Antioxidative, anti-inflammatory, and anti-Aβ1-42 activities of the obtained diosgenin carbamate derivatives were evaluated. Compound 57 protected nerve cells from H2O2-induced damage. A ten-fold stronger protective effect than the lead compound diosgenin was observed; it significantly reduced the production of pro-inflammatory factors such as TNF-α, IL-6, IL-1β, and NO, and reduced Aβ1-42-induced nerve cell damage in a dose-dependent manner72 (Figure 11, Table 4).
Table 4.
Effect of saponin derivatives on the various pathological pathways involve in AD.
| Compound NO. | Classification | The major targets | Activity | References |
|---|---|---|---|---|
| 57 | Diosgenin derivatives | Protect the nerve cells of ageing mice with subcutaneous injection of D-galactose Reduced IL-1β, IL-6 and TNF-a levels |
_ | 72 |
| 58 | Reduced ROS production induced by H2O2 Reduces 6-OHDA, H2O2, Aβ1-42 induced cell damage |
Neuroprotective effects against H2O2: 52.9% Neuroprotective effects against 6-OHDA: 38.4% Neuroprotection activity against Aβ1-42 damage: 54.4% |
73 | |
| 59 | Reduces neurotoxicity induced by oxygen glucose deprivation and NO production induced by LPS | NO inhibition %(10 μM): 40.4% | 74 | |
| 60 | Sarsasapogenin derivatives | Improved learning and memory impairment of mice injected with Aβ1-42 Protect PC12 cells from Aβ1-42induced damage |
The viability of H2O2-treated PC12 cells (10 µM): 36.6% | 75 |
| 61 | The viability of H2O2-treated PC12 cells (10 µM): 43.9% | |||
| 62 | Inhibit Aβ1-42 aggregation Reduce H2O2 induced SH-SY5Y cell neurotoxicity |
Inhibition of Aβ1-42 aggregation: 84.74% | 76 | |
| 63 | Inhibition of Aβ1-42 aggregation: 75.06% |
Given that indole skeleton has been widely considered to have neuroprotective effects, Li-Cheng et al. synthesised 19 diosgenin-indole derivatives. Some compounds were modified by the introduction of carbamate fragments, while others used ester bonds as linkers, and the data showed that carbamate fragments attributed more neuroprotective properties than the ester bonds. All compounds were tested for neuroprotective effects against H2O2 (hydrogen peroxide), 6-OHDA (6-hydroxydopamine), and Aβ1-42. It is known that compound 58 can protect against nerve damage caused by the three agents, and compound 58 can reverse the cell damage of H2O2 (52.9%), 6-OHDA (38.4%), and Aβ1-42 (54.4%). was the most potent compound. Molecular docking also showed that 58 had good affinity for Aβ1-42 and could interact with it. In the water maze experiment, Compound 58 significantly improved cognition and memory in Aβ1-42-injured mice. Therefore, Compound 58 can be considered a potential multi-target drug for AD73 (Figure 11, Table 4).
The introduction of triazole fragments significantly enhanced the neuroprotective activity of diosgenin. Diosgenin-phenyl/benzyltriazole derivatives were designed and synthesised, and their structure-activity relationship was examined. The activity of benzyl triazole was stronger than that of phenyl triazole. Compound 59 showed the strongest inhibitory effect on NO production at 10 μM, reducing neuroinflammation. Neuroprotective activity reached 73.9 μM, which was stronger than diosgenin (3.5 μM). Compound 59 has the potential for further research in the fight against AD74 (Figure 11, Table 4).
Sarsasapogenin and its derivatives
Sarsasapogenin is a steroidal sapogenin isolated from the Chinese herbal medicine Anemarrhena. It is similar in structure to diosgenin and has a similar structure-activity relationship. Current studies have shown that sarsasapogenin significantly inhibits key enzymes involved in AD pathogenesis, namely AChE, BuChE, BACE1, and MAO-B77,78, in a concentration-dependent manner. It also inhibits neuroinflammation and Aβ amyloid production79,80. It can exert neuroprotective effects in many ways, thereby improving pathological symptoms. Derivative design has also received considerable attention. Based on the activity of lead compounds, the synthesis of potential AD drugs with stronger activity and minimal side effects has also been extensively studied (Figure 12).
Figure 12.

The chemical structure of sarsasapogenin and its derivatives 60–63.
The introduction of N-substituted piperazine carboxylic acid groups significantly enhanced anti-inflammatory and antioxidant activities. Sarsasapogenin can be considered a potential building block for AD drug design81. Gui-Xian et al. evaluated sarsasapogenin-N-substituted piperazine carboxylic acid derivatives and found that compounds 60 and 61 significantly increased the viability of H2O2-treated PC12 cells by 36.6 and 43.9%, respectively; compounds 60 and 61 of 10 μM showed high NO inhibition rates of 15.2 and 24.41%, respectively75 (Figure 12, Table 4).
Wenbao designed and synthesised a novel sarsasapogenin-triazole hybrid and examined its structure-activity relationship. The introduction of 1,4-substituted triazolyl groups significantly enhanced their activity. Compounds 62 and 63 had a good inhibitory effect on Aβ aggregation, with inhibition rates of 84.74 and 75.06%, respectively, which were higher than those of the positive control drug curcumin (55.87%), and reduced Aβ-induced neuronal damage. In addition, the learning and memory abilities of model mice improved76. Therefore, the sarsasapogenin skeleton can serve as a basic structure for the development of anti-AD drugs (Figure 12, Table 4).
Alkaloids and their derivatives
Piperine and its derivatives
Piperine is a piperidine alkaloid with a relatively simple structure and one of the main physiologically active components of piperine and longan. Piperine is currently used clinically as a spectral anticonvulsant. In addition, Masoomeh et al. showed that long-term treatment with piperine reduced oxidative damage of rats intracerebroventricularly (ICV) injected with STZ82. Furthermore, it reduced the synaptic toxicity of STZ in the hippocampus, thereby exerting a neuroprotective effect. Suresh et al. showed that intraperitoneal injection of piperine in diabetic rats not only improved memory in diabetic rats, but also reduce AD-related BACE1, PSEN1, APAF1, CASPASE3, and CATALASE gene expression83. Hsieh et al. also found that piperine protects hippocampal neurons by upregulating protein kinase B (Akt) and glycogen synthase kinase 3β (GSK-3β) signalling pathways in the hippocampus and reducing kainate (KA)- induced excitotoxicity84. Studies have also shown that piperine can inhibit the pathological changes in AD in multiple ways and can be used as a potential anti-AD drug (Figure 13).
Figure 13.

The chemical structure of piperine and its derivatives 64–65.
Xiping et al. synthesised Compound 64. Through in vitro experiments, it was shown that Compound 64 binds strongly to Keap-1 and activate the Keap1-Nrf2-ARE signalling pathway in vitro85. This signalling pathway is important targets of AD86,87. The activation of Nrf2/ARE and inhibition of Keap1 can play a strong role in the treatment of AD. The activation of Nrf2 increases the expression levels of superoxide dismutase, catalase, and glutathione peroxidase, thereby reducing oxidative stress88. Inhibition of Keap1 increases Nrf2 expression thus reducing oxidative stress, which plays a key role in AD pathology. Ibotenic acid (IBO), an NMDA receptor agonist, induces excitatory cholinergic dysfunction89. Compound 64 effectively prevented IBO-induced cognitive impairment, elevated acetylcholine levels, increased neurotransmission, and reversed cholinergic neuronal damage85 (Figure 13, Table 5).
Table 5.
Effect of alkaloid derivatives on the various pathological pathways involve in AD.
| Compound NO. | Classification | The major targets | Activity | References |
|---|---|---|---|---|
| 64 | Piperine derivatives | Inhibiting apoptotic cell death induced by IBO Inhibited the interaction between Keap1 and Nrf2 |
_ | 85 |
| 65 | Inhibits the increase of Bax/Bcl2 ratio protects hippocampal neurons Inhibits the activation of NLRP3 inflammatory bodies in hippocampus Reduces Aβ1-42 Induced neurotoxicity in SH-SY5Y cells |
_ | 90 | |
| 66 | Harmine derivatives | Inhibits GSK-3β and DYRK1A, Inhibits oka induced tau hyperphosphorylation in SH-SY5Y cells, |
IC50 (GSK-3β) = 71 nM IC50 (DYRK1A) = 103 nM |
91 |
| 67 | AChE inhibitory activity BChE inhibitory activity Inhibits GSK-3β |
IC50 (hAChE) = 0.27 µM IC50 (hBChE) = 20.82 µM IC50 (GSK-3β) = 6.78 µM |
92 | |
| 68 | Oxymatrine derivatives | Protect PC12 cells from Aβ induced damage Inhibition of LPS induced NO |
_ | 93 |
| 69 | ||||
| 70 |
The research group also synthesised a piperine derivative compound 65. In vivo and in vitro experiments showed that compound 65 (1) Inhibited the formation of the Keap1-Nrf2 complex and reduced oxidative stress; (2) Reduced neurotoxicity induced by Aβ1–42 in SH-SY5Y cells and improved Aβ-induced cognitive impairment; and (3) Reduced expression levels of NLRP3, IL-1β and TNF-α inflammatory factors and resolved neuroinflammation. Therefore, compound 65 can be used to treat AD through various pathways, such as reducing oxidative stress, neurotoxicity, and neuroinflammation90 (Figure 13, Table 5).
Harmine and its derivatives
Harmine alkaloids (HARs) are β-carboline alkaloids extracted from HAR officinalis seeds, and their molecular skeleton contains a pyridoindole structure. HARs have a variety of pharmacological activities, including antitumor, anti-inflammatory, antiviral, and anti-AD effects. Research has shown that HARs can act on multiple targets to improve the symptoms of AD. In vivo experiments, HARs were found to selectively inhibit AChE and MAO-A94. Dandan et al. showed that HAR has good permeability across the blood-brain barrier. In vitro experiments have shown that it can inhibit AChE activity. Molecular docking data also showed that it is tightly bound to the catalytic site of AChE95. A study by Yunpeng et al. showed that the combination of HAR and memantine (MEM) increases efficacy in the treatment of AD. The results from the pharmacokinetic experiments showed that the elimination rate of HARs, MEM, and HOL (the main metabolite of HARs) was slowed down after combination treatment, improving bioavailability and achieving therapeutic concentrations96 (Figure 14).
Figure 14.

The chemical structure of harmine and its derivatives 66–67.
Wenwu et al. obtained two series of compounds with significantly enhanced neuroprotective effects by the structural modification of HAR. Among these, compounds 66 and 67 exhibited the best activity. Pharmacological experiments showed that the compound 66 exhibited strong inhibitory effects on GSK-3β and dual-specificity tyrosine phosphorylation-regulated kinase 1 A (DYRK1A), with IC50 values of 71 and 103 nM, respectively91. GSK-3β is involved in tau hyperphosphorylation97, and DYRK1A is an upstream kinase of GSK-3β signalling. First, DYRK1A is phosphorylated and GSK-3β phosphorylates tau protein. Therefore, inhibiting the expression of GSK-3β and DYRK1A can reduce tau hyperphosphorylation, effectively preventing the formation of NFTs98, and reducing nerve damage. Experiments have shown that compound 66 is a dual-target inhibitor, which greatly reduces the hyperphosphorylation of tau protein and prevents nerve cell damage. It can be used as a dual-target drug candidate for the effective treatment of AD (Figure 14, Table 5).
The second series was developed by introducing the N-benzylpiperidine group. All compounds were found to have significant anti-AChE activity and good inhibitory activity against BChE. Among them, compound 67 had the best effect, with IC50 of 0.27 and 20.82 μM for AChE and BChE, respectively. It also inhibited GSK-3β with an IC50 of 6.78 μM. From these data, it can be concluded that the linkage of two carbon atoms between the 7-position β-carboline and N-benzylpiperidine is more suitable for increasing the inhibitory activity towards AChE. Molecular docking data showed that compound 67 was tightly bound to both the catalytic active site and peripheral anionic site of AChE, and also formed a stable interaction with GSK-3β. It has been demonstrated that 67 can exert anti-AD effects by inhibiting AChE, BChE, and GSK-3β92 (Figure 14, Table 5).
Oxymatrine and its derivatives
Oxymatrine (OMT) is a quinazoline alkaloid extracted from the root of Sophora flavescens, which proves that OMT can reduce Aβ1–42-induced neuronal damage and inhibit the activation of microglia by inhibiting NF-κB and MAPK signalling pathways and anti-neuroinflammatory effects99. In addition, OMT can improve behavioural performance and reduce the density of Aβ plaques and astrocyte clusters in model mice100 (Figure 15).
Figure 15.

The chemical structure of oxymatrine and its derivatives 68–70.
Pei-Liang et al. measured the neuroprotective effect of 13 OMT derivatives on Aβ1–42-treated PC12 cells and found that compounds 68, 69, and 70 (Figure 15) reduced more efficiently IL-1β levels and reversed cellular damage. The introduction of the benzyl group enhanced the inhibitory activity. Compound 68 exhibited the best neuroprotective effect among all derivatives, increasing cell viability by 98.37% at 1 μM and 103.13% at 5 μM. Compound 68 (5 μM) significantly reduced IL-1β levels in PC12 cells thus reducing damage caused by neuroinflammation93 (Figure 15, Table 5).
Conclusion and outlook
According to the findings in the 2018 World Alzheimer’s Disease report, there is one case of AD worldwide every three seconds. The sharp increase in incidence has aroused global concern. A variety of natural products for the prevention and treatment of AD have been studied in vivo and in vitro, and many natural products can improve the pathological state of AD through multi-targets, so natural products are more suitable for multi-target drug molecular design, which is currently recognised as the most effective method. Natural products also have some defects, such as poor water solubility, high lipid dissolution, strong cytotoxicity and so on. Some shortcomings can be improved by structural modification of structure-activity relationship. For example, the introduction of amino acids, short peptides and electron-absorbing groups into natural products such as betulinic acid, 3-oxyoleanolic acid, glycyrrhizic acid and homoaconitine can greatly improve their solubility and biological activity and significantly reduce toxic reactions. However, the clinical application of natural products is limited because the active groups in the structure of some natural products cannot be confirmed. Therefore, the development of clinical drugs based on the pharmacological properties of natural products is a primary goal. This review summarises the recent five years studies on natural products and their derivatives that have been shown to ameliorate AD pathology. We hope to provide potential lead drug candidates for developing drugs against AD.
Funding Statement
This work was financially supported by the Higher Education Discipline Innovation Project (111 Project, D18012), National Natural Science Foundation of China [No. 81960626, 82060628, 82204310, 82260002, 81460001], Natural Science Research Foundation of Jilin Province for Sciences and Technology [No. 20220101353JC], Education Department Project of Jilin [No. JJKH20220563KJ, JJKH20220546KJ]. Doctoral Research Start-up Fundation of Yanbian University [No. ydbq202215, ydbq202217].
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Liu X-Y, Wang X-J, Shi L, Liu Y-H, Wang L, Li K, Bu Q, Cen X-B, Yu X-Q.. Rational design of quinoxalinone-based red-emitting probes for high-affinity and long-term visualizing amyloid-β in vivo. Anal Chem. 2022;94(21):7665–7673. [DOI] [PubMed] [Google Scholar]
- 2.Zhu Z, Lu S, Wang S-H, Gorriz JM, Zhang Y-D.. DSNN: a dense net-based SNN for explainable brain disease classification. Front Syst Neurosci. 2022;16:838822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ostrovskaya RU, Belnik AP, Storozheva ZI.. Noopept efficiency in experimental Alzheimer disease (cognitive deficiency caused by β-amyloid25-35 injection into Meynert basal nuclei of rats). Bull Exp Biol Med. 2008;146(1):77–80. [DOI] [PubMed] [Google Scholar]
- 4.Sultzer DL, Lim AC, Gordon HL, Yarns BC, Melrose RJ.. Cholinergic receptor binding in unimpaired older adults, mild cognitive impairment, and Alzheimer’s disease dementia. Alzheimers Res Ther. 2022;14(1):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skovronsky DM, Doms RW, Lee VMY.. Detection of a novel intraneuronal pool of insoluble amyloid β protein that accumulates with time in culture. J Cell Biol. 1998;141(4):1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou X-W, Li X, Bjorkdahl C, Sjogren MJ, Alafuzoff I, Soininen H, Grundke-Iqbal I, Iqbal K, Winblad B, Pei J-J, et al. Assessments of the accumulation severities of amyloid β-protein and hyperphosphorylated tau in the medial temporal cortex of control and Alzheimer’s brains. Neurobiol Dis. 2006;22(3):657–668. [DOI] [PubMed] [Google Scholar]
- 7.Wang C, Liu P, Wang XL, Chen C, Fu XQ, Zhang B. Metal ions and Alzheimer’s disease. Sichuan Yixue. 2008;29:467–469. [Google Scholar]
- 8.Panda P, Verma HK, Lakkakula S, Merchant N, Kadir F, Rahman S, Jeffree MS, Lakkakula BVKS, Rao PV.. Biomarkers of oxidative stress tethered to cardiovascular diseases. Oxid Med Cell Longev. 2022;2022:9154295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rivas-Arancibia S, Rodriguez-Martinez E, Mendez-Garcia A, Moctezuma-Salgado M, Jimenez-Espindola P, Lopez-Gonzales U. Oxidative stress, inflammation, and formation of beta- amyloid 1-42 in brain. Free Radicals and Diseases 2016;113–130. [Google Scholar]
- 10.Hoye AT, Davoren JE, Wipf P, Fink MP, Kagan VE.. Targeting mitochondria. Acc Chem Res. 2008;41(1):87–97. [DOI] [PubMed] [Google Scholar]
- 11.Newman DJ, Cragg GM.. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J Nat Prod. 2020;83(3):770–803. [DOI] [PubMed] [Google Scholar]
- 12.Deng H, Huang X, Jin C, Jin C-M, Quan Z-S.. Synthesis, in vitro and in vivo biological evaluation of dihydroartemisinin derivatives with potential anti-Toxoplasma gondii agents. Bioorg Chem. 2020;94:103467. [DOI] [PubMed] [Google Scholar]
- 13.Pang L, Liu C-Y, Gong G-H, Quan Z-S.. Synthesis, in vitro and in vivo biological evaluation of novel lappaconitine derivatives as potential anti-inflammatory agents. Acta Pharm Sin B. 2020;10(4):628–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang YL, Jin LL, Cheng X, Yan WF, Deng H, Shen QK, Quan ZS, Jin CM, Zhang CH. Synthesis and evaluation of in vitro and in vivo anti-Toxoplasma gondii activity of tetraoxane-substituted ursolic acid derivatives. Nat Prod Res. 2022. [DOI] [PubMed] [Google Scholar]
- 15.de la Torre BG, Albericio F.. The pharmaceutical industry in 2020. An analysis of FDA drug approvals from the perspective of molecules. Molecules. 2021; 26:627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patridge E, Gareiss P, Kinch MS, Hoyer D.. An analysis of FDA-approved drugs: natural products and their derivatives. Drug Discov Today. 2016;21(2):204–207. [DOI] [PubMed] [Google Scholar]
- 17.Wang N-Y, Li J-N, Liu W-L, Huang Q, Li W-X, Tan Y-H, Liu F, Song Z-H, Wang M-Y, Xie N, et al. Ferulic acid ameliorates Alzheimer’s disease-like pathology and repairs cognitive decline by preventing capillary hypofunction in aPP/PS1 mice. Neurotherapeutics. 2021;18(2):1064–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mori T, Koyama N, Tan J, Segawa T, Maeda M, Town T.. Combined treatment with the phenolics (-)-epigallocatechin-3-gallate and ferulic acid improves cognition and reduces Alzheimer-like pathology in mice. J Biol Chem. 2019;294(8):2714–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh YP, Tej GNVC, Pandey A, Priya K, Pandey P, Shankar G, Nayak PK, Rai G, Chittiboyina AG, Doerksen RJ, et al. Design, synthesis and biological evaluation of novel naturally-inspired multifunctional molecules for the management of Alzheimer’s disease. Eur J Med Chem. 2020;198:112257. [DOI] [PubMed] [Google Scholar]
- 20.Zhu G, Bai P, Wang K, Mi J, Yang J, Hu J, Ban Y, Xu R, Chen R, Wang C, et al. Design, synthesis, and evaluation of novel O-alkyl ferulamide derivatives as multifunctional ligands for treating Alzheimer’s disease. J Enzyme Inhib Med Chem. 2022;37(1):1375–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lan J-S, Zeng R-F, Jiang X-Y, Hou J-W, Liu Y, Hu Z-H, Li H-X, Li Y, Xie S-S, Ding Y, et al. Design, synthesis and evaluation of novel ferulic acid derivatives as multi-target-directed ligands for the treatment of Alzheimer’s disease. Bioorg Chem. 2020;94:103413. [DOI] [PubMed] [Google Scholar]
- 22.He F, Chou CJ, Scheiner M, Poeta E, Yuan Chen N, Gunesch S, Hoffmann M, Sotriffer C, Monti B, Maurice T, et al. Melatonin- and ferulic acid-based HDAC6 selective inhibitors exhibit pronounced immunomodulatory effects in vitro and neuroprotective effects in a pharmacological Alzheimer’s disease mouse model. J Med Chem. 2021;64(7):3794–3812. [DOI] [PubMed] [Google Scholar]
- 23.Fonseca A, Reis J, Silva T, Matos MJ, Bagetta D, Ortuso F, Alcaro S, Uriarte E, Borges F.. Coumarin versus chromone monoamine oxidase B inhibitors: quo vadis? J Med Chem. 2017;60(16):7206–7212. [DOI] [PubMed] [Google Scholar]
- 24.Orhan IE, Tosun F, Senol Deniz FS, et al. Butyrylcholinesterase-inhibiting natural coumarin molecules as potential leads. Phytochem Lett. 2021;44:48–54. [Google Scholar]
- 25.He Q, Liu J, Lan J-S, Ding J, Sun Y, Fang Y, Jiang N, Yang Z, Sun L, Jin Y, et al. Coumarin-dithiocarbamate hybrids as novel multitarget AChE and MAO-B inhibitors against Alzheimer’s disease: design, synthesis and biological evaluation. Bioorg Chem. 2018; 81:512–528. [DOI] [PubMed] [Google Scholar]
- 26.Macklin LJ, Schwans JP.. Synthesis, biochemical evaluation, and molecular modeling of organophosphate-coumarin hybrids as potent and selective butyrylcholinesterase inhibitors. Bioorg Med Chem Lett. 2020;30(13):127213. [DOI] [PubMed] [Google Scholar]
- 27.Dhiman P, Malik N, Khatkar A.. Lead optimization for promising monoamine oxidase inhibitor from eugenol for the treatment of neurological disorder: synthesis and in silico based study. BMC Chem. 2019;13(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adefegha SA, Okeke BM, Oboh G.. Antioxidant properties of eugenol, butylated hydroxylanisole, and butylated hydroxyl toluene with key biomolecules relevant to Alzheimer’s diseases-in vitro. J Food Biochem. 2021;45(3):e13276. [DOI] [PubMed] [Google Scholar]
- 29.Taheri P, Yaghmaei P, Tehrani HS, et al. Effects of eugenol on Alzheimer’s disease-like manifestations in insulin- and Aβ-induced rat models. Neurophysiology. 2019;51:114–119. [Google Scholar]
- 30.Chowdhury S, Kumar S.. Inhibition of BACE1, MAO-B, cholinesterase enzymes, and anti-amyloidogenic potential of selected natural phytoconstituents: multi-target-directed ligand approach. J Food Biochem. 2021;45(1):e13571. [DOI] [PubMed] [Google Scholar]
- 31.James I, Manisha C, Choephel T, et al. Ethanolic extract of acorus calamus linn and eugenol combination improved the cognitive functions of streptozotocin administrated rats through attenuation of lipid peroxidation and acetylcholine esterase level. J Global Trends Pharm Sci. 2019;10:5870–5881. [Google Scholar]
- 32.Carradori S, Gidaro MC, Petzer A, Costa G, Guglielmi P, Chimenti P, Alcaro S, Petzer JP.. Inhibition of human monoamine oxidase: biological and molecular modeling studies on selected natural flavonoids. J Agric Food Chem. 2016;64(47):9004–9011. [DOI] [PubMed] [Google Scholar]
- 33.Desideri N, Bolasco A, Fioravanti R, Monaco LP, Orallo F, Yáñez M, Ortuso F, Alcaro S.. Homoisoflavonoids: natural scaffolds with potent and selective monoamine oxidase-B inhibition properties. J Med Chem. 2011;54(7):2155–2164. [DOI] [PubMed] [Google Scholar]
- 34.Chimenti F, Cottiglia F, Bonsignore L, Casu L, Casu M, Floris C, Secci D, Bolasco A, Chimenti P, Granese A, et al. Quercetin as the active principle of Hypericum hircinum exerts a selective inhibitory activity against MAO-A: extraction, biological analysis, and computational study. J Nat Prod. 2006;69(6):945–949. [DOI] [PubMed] [Google Scholar]
- 35.Gidaro MC, Astorino C, Petzer A, Carradori S, Alcaro F, Costa G, Artese A, Rafele G, Russo FM, Petzer JP, et al. Kaempferol as selective human MAO-A inhibitor: analytical detection in calabrian red wines, biological and molecular modeling studies. J Agric Food Chem. 2016;64(6):1394–1400. [DOI] [PubMed] [Google Scholar]
- 36.Dhiman P, Malik N, Khatkar A.. In silico design, synthesis of hybrid combinations: quercetin based MAO inhibitors with antioxidant potential. Curr Top Med Chem. 2019;19(2):156–170. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y-P, Zhang Z-Y, Li Y-P, Li D, Huang S-L, Gu L-Q, Xu J, Huang Z-S.. Syntheses and evaluation of novel isoliquiritigenin derivatives as potential dual inhibitors for amyloid-beta aggregation and 5-lipoxygenase. Eur J Med Chem. 2013; 66:22–31. [DOI] [PubMed] [Google Scholar]
- 38.Guan L, Peng D, Zhang L, Jia J, Jiang H.. Design, synthesis, and cholinesterase inhibition assay of liquiritigenin derivatives as anti-Alzheimer’s activity. Bioorg Med Chem Lett. 2021;52:128306. [DOI] [PubMed] [Google Scholar]
- 39.Chimenti F, Fioravanti R, Bolasco A, Chimenti P, Secci D, Rossi F, Yáñez M, Orallo F, Ortuso F, Alcaro S. Chalcones: a valid scaffold for monoamine oxidases inhibitors. J Med Chem. 2009;52:2818–2824. [DOI] [PubMed] [Google Scholar]
- 40.Bai P, Wang K, Zhang P, Shi J, Cheng X, Zhang Q, Zheng C, Cheng Y, Yang J, Lu X, et al. Development of chalcone-O-alkylamine derivatives as multifunctional agents against Alzheimer’s disease. Eur J Med Chem. 2019;183:111737. [DOI] [PubMed] [Google Scholar]
- 41.Sakata RP, Antoniolli G, Lancellotti M, Kawano DF, Guimarães Barbosa E, Almeida WP.. Synthesis and biological evaluation of 2'-aminochalcone: a multi-target approach to find drug candidates to treat Alzheimer’s disease. Bioorg Chem. 2020;103:104201. [DOI] [PubMed] [Google Scholar]
- 42.Yuan X, Wang Z, Zhang L, Sui R, Khan S.. Exploring the inhibitory effects of liquiritigenin against tau fibrillation and related neurotoxicity as a model of preventive care in Alzheimer’s disease. Int J Biol Macromol. 2021;183:1184–1190. [DOI] [PubMed] [Google Scholar]
- 43.Du Y, Luo M, Du Y, Xu M, Yao Q, Wang K, He G.. Liquiritigenin decreases Aβ levels and ameliorates cognitive decline by regulating microglia M1/M2 transformation in AD mice. Neurotox Res. 2021;39(2):349–358. [DOI] [PubMed] [Google Scholar]
- 44.Lee HK, Yang E-J, Kim JY, Song K-s, Seong YH.. Inhibitory effects of glycyrrhizae radix and its active component, isoliquiritigenin, on Aβ(25-35)-induced neurotoxicity in cultured rat cortical neurons. Arch Pharm Res. 2012;35(5):897–904. [DOI] [PubMed] [Google Scholar]
- 45.Shekhar S, Yadav SK, Rai N, Kumar R, Yadav Y, Tripathi M, Dey AB, Dey S.. 5-LOX in Alzheimer’s disease: potential serum marker and in vitro evidences for rescue of neurotoxicity by its inhibitor YWCS. Mol Neurobiol. 2018;55(4):2754–2762. [DOI] [PubMed] [Google Scholar]
- 46.Dhakal S, Ramsland PA, Adhikari B, et al. Trans-chalcone plus baicalein synergistically reduce intracellular amyloid beta (Abeta42) and protect from Abeta42 induced oxidative damage in yeast models of Alzheimer’s disease. Int J Mol Sci. 2021;22:9456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oh JM, Kang M-G, Hong A, Park J-E, Kim SH, Lee JP, Baek SC, Park D, Nam S-J, Cho M-L, et al. Potent and selective inhibition of human monoamine oxidase-B by 4-dimethylaminochalcone and selected chalcone derivatives. Int J Biol Macromol. 2019;137:426–432. [DOI] [PubMed] [Google Scholar]
- 48.Li J-M, Zhang Y, Tang L, Chen Y-H, Gao Q, Bao M-H, Xiang J, Lei D-L.. Effects of triptolide on hippocampal microglial cells and astrocytes in the APP/PS1 double transgenic mouse model of Alzheimer’s disease. Neural Regen Res. 2016;11(9):1492–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Q, Xiao B, Cui S, Song H, Qian Y, Dong L, An H, Cui Y, Zhang W, He Y, et al. Triptolide treatment reduces Alzheimer’s disease (AD)-like pathology through inhibition of BACE1 in a transgenic mouse model of AD. Dis Model Mech. 2014;7(12):1385–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng S, LeBlanc KJ, Li L.. Triptolide preserves cognitive function and reduces neuropathology in a mouse model of Alzheimer’s disease. PLOS One. 2014;9(9):e108845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ning C, Mo L, Chen X, Tu W, Wu J, Hou S, Xu J.. Triptolide derivatives as potential multifunctional anti-Alzheimer agents: synthesis and structure-activity relationship studies. Bioorg Med Chem Lett. 2018;28(4):689–693. [DOI] [PubMed] [Google Scholar]
- 52.Xu Y, Wei H, Wang J, Wang W, Gao J.. Synthesis of andrographolide analogues and their neuroprotection and neurite outgrowth-promoting activities. Bioorg Med Chem. 2019;27(11):2209–2219. [DOI] [PubMed] [Google Scholar]
- 53.Dey A, Chen R, Li F, et al. Synthesis and characterization of andrographolide derivatives as regulators of beta APP processing in human cells. Molecules. 2021;26:7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abdel Bar FM, Elimam DM, Mira AS, El-Senduny FF, Badria FA.. Derivatization, molecular docking and in vitro acetylcholinesterase inhibitory activity of glycyrrhizin as a selective anti-Alzheimer agent. Nat Prod Res. 2019;33(18):2591–2599. [DOI] [PubMed] [Google Scholar]
- 55.Schwarz S, Lucas SD, Sommerwerk S, Csuk R.. Amino derivatives of glycyrrhetinic acid as potential inhibitors of cholinesterases. Bioorg Med Chem. 2014;22(13):3370–3378. [DOI] [PubMed] [Google Scholar]
- 56.De Monte C, Carradori S, Chimenti P, Secci D, Mannina L, Alcaro F, Petzer A, N'Da CI, Gidaro MC, Costa G, et al. New insights into the biological properties of Crocus sativus L.: chemical modifications, human monoamine oxidases inhibition and molecular modeling studies. Eur J Med Chem. 2014;82:164–171. [DOI] [PubMed] [Google Scholar]
- 57.Burgos RA, Alarcon P, Quiroga J, et al. Andrographolide, an anti-inflammatory multitarget drug: all roads lead to cellular metabolism. Molecules. 2021;26:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gu LL, Yu QQ, Li Q, et al. Andrographolide protects PC12 cells against β-amyloid-induced autophagy-associated cell death through activation of the Nrf2-mediated p62 signaling pathway. Int J Mol Sci. 2018;19:2844/1–2844/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arredondo SB, Reyes DT, Herrera-Soto A, Mardones MD, Inestrosa NC, Varela-Nallar L.. Andrographolide promotes hippocampal neurogenesis and spatial memory in the APPswe/PS1ΔE9 mouse model of Alzheimer’s disease. Sci Rep. 2021;11(1):22904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Patel R, Kaur K, Singh S.. Protective effect of andrographolide against STZ induced Alzheimer’s disease in experimental rats: possible neuromodulation and Aβ(1-42) analysis. Inflammopharmacology. 2021;29(4):1157–1168. [DOI] [PubMed] [Google Scholar]
- 61.Miranda A, Montiel E, Ulrich H, Paz C.. Selective secretase targeting for Alzheimer’s disease therapy. J Alzheimers Dis. 2021;81(1):1–17. [DOI] [PubMed] [Google Scholar]
- 62.Wagle A, Seong SH, Zhao BT, Woo MH, Jung HA, Choi JS.. Comparative study of selective in vitro and in silico BACE1 inhibitory potential of glycyrrhizin together with its metabolites, 18 alpha- and 18 beta-glycyrrhetinic acid, isolated from Hizikia fusiformis. Arch Pharm Res. 2018;41(4):409–418. [DOI] [PubMed] [Google Scholar]
- 63.Jeong GS, Kang MG, Lee JY, et al. Inhibition of butyrylcholinesterase and human monoamine oxidase-B by the coumarin glycyrol and liquiritigenin isolated from Glycyrrhiza uralensis. Molecules. 2020;25:3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wei JC, Huang HH, Zhong NF, Gao YN, Liu XL, Long GQ, Hu GS, Wang AH, Jia JM.. Euphorfistrines A-G, cytotoxic and AChE inhibiting triterpenoids from the roots of Euphorbia fischeriana. Bioorg Chem. 2021;116:105395. [DOI] [PubMed] [Google Scholar]
- 65.Forouzanfar F, Asadpour E, Hosseinzadeh H, Boroushaki MT, Adab A, Dastpeiman SH, Sadeghnia HR.. Safranal protects against ischemia-induced PC12 cell injury through inhibiting oxidative stress and apoptosis. Naunyn Schmiedebergs Arch Pharmacol. 2021;394(4):707–716. [DOI] [PubMed] [Google Scholar]
- 66.Rafieipour F, Hadipour E, Emami SA, Asili J, Tayarani-Najaran Z.. Safranal protects against beta-amyloid peptide-induced cell toxicity in PC12 cells via MAPK and PI3 K pathways. Metab Brain Dis. 2019;34(1):165–172. [DOI] [PubMed] [Google Scholar]
- 67.Nanda S, Madan K.. The role of Safranal and saffron stigma extracts in oxidative stress, diseases and photoaging: a systematic review. Heliyon. 2021;7(2):e06117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Som S, Antony J, Dhanabal S, Ponnusankar S.. Neuroprotective role of Diosgenin, a NGF stimulator, against Aβ (1-42) induced neurotoxicity in animal model of Alzheimers disease. Metab Brain Dis. 2022;37(2):359–372. [DOI] [PubMed] [Google Scholar]
- 69.Yang XM, Tohda C.. Diosgenin restores Aβ-induced axonal degeneration by reducing the expression of heat shock cognate 70 (HSC70). Sci Rep. 2018;8:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang XM, Tohda C.. Heat shock cognate 70 inhibitor, VER-155008, reduces memory deficits and axonal degeneration in a mouse model of Alzheimer’s disease. Front Pharmacol. 2018;9:48/1–48/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Matosevic A, Bosak A.. Carbamate group as structural motif in drugs: a review of carbamate derivatives used as therapeutic agents. Arh Hig Rada Toksikol. 2020;71(4):285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang G-X, Huang Y, Zheng L-L, Zhang L, Su L, Wu Y-H, Li J, Zhou L-C, Huang J, Tang Y, et al. Design, synthesis and evaluation of diosgenin carbamate derivatives as multitarget anti-Alzheimer’s disease agents. Eur J Med Chem. 2020;187:111913. [DOI] [PubMed] [Google Scholar]
- 73.Zhou L-C, Liang Y-F, Huang Y, Yang G-X, Zheng L-L, Sun J-M, Li Y, Zhu F-L, Qian H-W, Wang R, et al. Design, synthesis, and biological evaluation of diosgenin-indole derivatives as dual-functional agents for the treatment of Alzheimer’s disease. Eur J Med Chem. 2021;219:113426. [DOI] [PubMed] [Google Scholar]
- 74.Huang Y, Huang W, Yang G, Wang R, Ma L.. Design and synthesis of novel diosgenin-triazole hybrids targeting inflammation as potential neuroprotective agents. Bioorg Med Chem Lett. 2021;43:128092. [DOI] [PubMed] [Google Scholar]
- 75.Yang G-X, Ge S-L, Wu Y, Huang J, Li S-L, Wang R, Ma L.. Design, synthesis and biological evaluation of 3-piperazinecarboxylate sarsasapogenin derivatives as potential multifunctional anti-Alzheimer agents. Eur J Med Chem. 2018; 156:206–215. [DOI] [PubMed] [Google Scholar]
- 76.Wang W, Wang W, Yao G, Ren Q, Wang D, Wang Z, Liu P, Gao P, Zhang Y, Wang S, et al. Novel sarsasapogenin-triazolyl hybrids as potential anti-Alzheimer’s agents: design, synthesis and biological evaluation. Eur J Med Chem. 2018;151:351–362. [DOI] [PubMed] [Google Scholar]
- 77.Feng B, Zhao X-Y, Song Y-Z, Liang W-N, Liu J-L.. Sarsasapogenin reverses depressive-like behaviors and nicotinic acetylcholine receptors induced by olfactory bulbectomy. Neurosci Lett. 2017;639:173–178. [DOI] [PubMed] [Google Scholar]
- 78.Kashyap P, Muthusamy K, Niranjan M, Trikha S, Kumar S.. Sarsasapogenin: a steroidal saponin from Asparagus racemosus as multi target directed ligand in Alzheimer’s disease. Steroids. 2020;153:108529. [DOI] [PubMed] [Google Scholar]
- 79.Wang L, Jin Y, Sui HJ, et al. Protection of sarsasapogenin against amyloid beta-protein induced neurotoxicity in primary cultured hippocampal neurons of neonatal rats. Zhongguo Yaolixue Yu Dulixue Zazhi. 2013;27:629–634. [Google Scholar]
- 80.Wang Z-D, Yao G-D, Wang W, Wang W-B, Wang S-J, Song S-J.. Synthesis and evaluation of 26-amino acid methyl ester substituted sarsasapogenin derivatives as neuroprotective agents for Alzheimer’s disease. Steroids. 2017;125:93–106. [DOI] [PubMed] [Google Scholar]
- 81.Colabufo NA, Leopoldo M, Perrone R, et al. Preparation of cyclohexyl-substituted piperazine compounds for treating Alzheimer’s disease. EP2703387A1, 2014.
- 82.Nazifi M, Oryan S, Esfahani DE, Ashrafpoor M.. The functional effects of piperine and piperine plus donepezil on hippocampal synaptic plasticity impairment in rat model of Alzheimer’s disease. Life Sci. 2021;265:118802. [DOI] [PubMed] [Google Scholar]
- 83.Kumar S, Chowdhury S, Razdan A, Kumari D, Purty RS, Ram H, Kumar P, Nayak P, Shukla SD.. Downregulation of candidate gene expression and neuroprotection by piperine in streptozotocin-induced hyperglycemia and memory impairment in rats. Front Pharmacol. 2020;11:595471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hsieh TY, Chang Y, Wang SJ.. Piperine provides neuroprotection against kainic acid-induced neurotoxicity via maintaining NGF signalling pathway. Molecules. 2022;27:2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang X, Ji J, Liu C, Zhou M, Li H, Ye S, Hu Q.. HJ22, a Novel derivative of piperine, Attenuates ibotenic acid-induced cognitive impairment, oxidativestress, apoptosis and inflammation via inhibiting the protein-protein interaction of Keap1-Nrf2. Int Immunopharmacol. 2020;83:106383. [DOI] [PubMed] [Google Scholar]
- 86.Fakhri S, Pesce M, Patruno A, Moradi SZ, Iranpanah A, Farzaei MH, Sobarzo-Sánchez E.. Attenuation of Nrf2/Keap1/ARE in Alzheimer’s disease by plant secondary metabolites: a mechanistic review. Molecules. 2020;25:4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.He L, Sun Y.. The potential role of Keap1-Nrf2 pathway in the pathogenesis of Alzheimer’s disease, type 2 diabetes, and type 2 diabetes-related Alzheimer’s disease. Metab Brain Dis. 2021;36:1469–1479. [DOI] [PubMed] [Google Scholar]
- 88.Qi G, Mi Y, Wang Y, Li R, Huang S, Li X, Liu X.. Neuroprotective action of tea polyphenols on oxidative stress-induced apoptosis through the activation of the TrkB/CREB/BDNF pathway and Keap1/Nrf2 signaling pathway in SH-SY5Y cells and mice brain. Food Funct. 2017;8(12):4421–4432. [DOI] [PubMed] [Google Scholar]
- 89.Nag S, Tang F.. Cholinergic lesions of the rat brain by ibotenic acid and 192 IgG-saporin: effects on somatostatin, substance P and neuropeptide Y levels in the cerebral cortex and the hippocampus. Neurosci Lett. 1998;252(2):83–86. [DOI] [PubMed] [Google Scholar]
- 90.Yang X, Zhi J, Leng H, Chen Y, Gao H, Ma J, Ji J, Hu Q.. The piperine derivative HJ105 inhibits Abeta1-42-induced neuroinflammation and oxidative damage via the Keap1-Nrf2-TXNIP axis. Phytomedicine. 2021;87:153571. [DOI] [PubMed] [Google Scholar]
- 91.Liu W, Liu X, Tian L, Gao Y, Liu W, Chen H, Jiang X, Xu Z, Ding H, Zhao Q, et al. Design, synthesis and biological evaluation of harmine derivatives as potent GSK-3beta/DYRK1A dual inhibitors for the treatment of Alzheimer’s disease. Eur J Med Chem. 2021;222:113554. [DOI] [PubMed] [Google Scholar]
- 92.Liu W, Liu X, Liu W, Gao Y, Wu L, Huang Y, Chen H, Li D, Zhou L, Wang N, et al. Discovery of novel beta-carboline derivatives as selective AChE inhibitors with GSK-3beta inhibitory property for the treatment of Alzheimer’s disease. Eur J Med Chem. 2022;229:114095. [DOI] [PubMed] [Google Scholar]
- 93.Dong P-L, Li Z, Teng C-L, Yin X, Cao X-K, Han H.. Synthesis and evolution of neuroprotective effects of oxymatrine derivatives as anti-Alzheimer’s disease agents. Chem Biol Drug Des. 2021;98(1):175–181. [DOI] [PubMed] [Google Scholar]
- 94.Jiang B, Meng L, Zou N, Wang H, Li S, Huang L, Cheng X, Wang Z, Chen W, Wang C, et al. Mechanism-based pharmacokinetics-pharmacodynamics studies of harmine and harmaline on neurotransmitters regulatory effects in healthy rats: challenge on monoamine oxidase and acetylcholinesterase inhibition. Phytomedicine. 2019;62:152967. [DOI] [PubMed] [Google Scholar]
- 95.He D, Wu H, Wei Y, Liu W, Huang F, Shi H, Zhang B, Wu X, Wang C.. Effects of harmine, an acetylcholinesterase inhibitor, on spatial learning and memory of APP/PS1 transgenic mice and scopolamine-induced memory impairment mice. Eur J Pharmacol. 2015;768:96–107. [DOI] [PubMed] [Google Scholar]
- 96.Zhang Y, Li S, Wang Y, et al. Potential pharmacokinetic drug(-)drug interaction between harmine, a cholinesterase inhibitor, and memantine, a non-competitive N-methyl-d-aspartate receptor antagonist. Molecules. 2019;24(7):1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jiang X-Y, Chen T-K, Zhou J-T, He S-Y, Yang H-Y, Chen Y, Qu W, Feng F, Sun H-P.. Dual GSK-3beta/AChE inhibitors as a new strategy for multitargeting anti-Alzheimer’s disease drug discovery. ACS Med Chem Lett. 2018;9(3):171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pradeepkiran JA, Munikumar M, Reddy AP, Reddy PH.. Protective effects of a small molecule inhibitor ligand against hyperphosphorylated tau-induced mitochondrial and synaptic toxicities in Alzheimer disease. Hum Mol Genet. 2021;31(2):244–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dong P, Ji X, Han W, Han H.. Oxymatrine attenuates amyloid beta 42 (A beta(1-42))-induced neurotoxicity in primary neuronal cells and memory impairment in rats. Can J Physiol Pharmacol. 2019;97(2):99–106. [DOI] [PubMed] [Google Scholar]
- 100.Chen Y, Qi Z, Qiao B, Lv Z, Hao Y, Li H.. Oxymatrine can attenuate pathological deficits of Alzheimer’s disease mice through regulation of neuroinflammation. J Neuroimmunol. 2019;334:576978. [DOI] [PubMed] [Google Scholar]