

Summary
Background
Venetoclax combined with intensive chemotherapy has been shown to be safe with promising activity in fit patients with newly diagnosed acute myeloid leukaemia. The aim of this study was to compare the activity of venetoclax plus intensive chemotherapy with intensive chemotherapy alone.
Methods
This was a post-hoc propensity score matched analysis of prospective clinical trials (NCT03214562, NCT02115295, and NCT01289457) in patients at The University of Texas MD Anderson Cancer Center, Texas, USA between March 29, 2010, and June 15, 2021. Eligible patients were aged 18 years and older, and had newly diagnosed acute myeloid leukaemia or high-risk myelodysplastic syndrome, and were treated within trials incorporating purine analogues with an anthracycline and cytarabine either with venetoclax plus intensive chemotherapy or with intensive chemotherapy alone. Patients in the venetoclax plus intensive chemotherapy cohort were matched with patients in the intensive chemotherapy cohort. Morphological response and measurable residual disease (MRD) was assessed using bone marrow aspiration and biopsy and eight-colour multiparameter flow cytometry. The primary objectives were rate of MRD negative composite complete response and cumulative incidence of transition to allogeneic haematopoietic stem-cell transplantation (HSCT). All patients who had response within two treatment cycles (induction and re-induction) were included in the analyses. Secondary objectives included assessment of event-free and overall survival.
Findings
The propensity matched cohort included 279 patients (median age 49 years [IQR 39–57]; 131 [47%] were men and 148 [53%] were women); 85 in the venetoclax plus intensive chemotherapy cohort and 194 in the intensive chemotherapy cohort. After a median follow up of 30 months (95% CI 26–36), 64 (86%) of 74 patients in the venetoclax plus intensive chemotherapy cohort had an MRD-negative composite complete response rate compared with 86 [61%] of 140 patients in the intensive chemotherapy cohort (odd ratio 3.2 [95% CI 1.5–6.7]; p=0.0028). The overall cumulative incidence of allogeneic HSCT in responding patients was higher with venetoclax plus intensive chemotherapy than intensive chemotherapy (79% [95% CI 67–88] vs 57% [49–65]; hazard ratio [HR] 1.52 [95% CI 1.11–2.08]; p=0.012). Venetoclax plus intensive chemotherapy improved event-free survival (median not reached [NR; 95% CI NR–NR] vs 14.3 months [10.7–33.5]; HR 0.57 [95% CI 0.34–0.95]; p=0.030), but overall survival did not significantly differ between the two cohorts (median NR [95% CI 24–NR] vs 32 months [19–NR]; HR 0.63 [95% CI 0.35–1.1], p=0.13).
Interpretations
Venetoclax combined with intensive induction chemotherapy induced deep MRD-negative remissions, allowing transition to allogeneic HSCT in first remission, and improvement in event-free survival. These results highlight the incremental benefit of venetoclax added to intensive induction chemotherapy across European LeukemiaNet risk groups, and serve as a benchmark to inform enrolment on future confirmatory prospective clinical trials.
Introduction
Frontline acute myeloid leukaemia treatment historically relied on anthracyclines and cytarabine based regimens with resultant complete remission rates of approximately 55%.1 Anthracycline selection and dose augmentation increased complete response rates to 65–78%,2, 3, 4 although the overall survival benefit was seen mainly in younger patients (younger than 50 years) with favourable or intermediate-risk cytogenetics.2 Unfortunately, relapse remains common, often occurring within 24 months following diagnosis.1, 2, 3, 4
Strategies to mitigate relapse, including incorporation of multi-drug chemotherapy regimens,5, 6 high-dose cytarabine consolidation,7 or increasing consolidation cycles,8 further improved complete response rates to approximately 75–85%.4, 5, 6 Nonetheless, long-term event-free survival remained at around 45%.4, 5, 6 Allogeneic haematopoietic stem-cell transplantation (HSCT) in remission remains standard for patients with relapse risk exceeding 35–40%,9, 10 but improved treatment strategies are needed.
Cytogenetic and molecular risk stratification identifies patients at highest risk of relapse,11, 12 enabling individualisation of therapy.13, 14, 15 Along with prognostic information provided by molecular and cytogenetic analysis, measurable residual disease (MRD) assessed using multiparameter flow cytometry or molecular methods is a key predictor of relapse and survival in acute myeloid leukaemia,16, 17, 18 serving as a marker of depth of response and enabling dynamic risk assessment throughout treatment.9 Multi-drug intensive chemotherapy regimens result in MRD-negative remission in approximately 50–60% of patients.6, 17
In unfit (ie, ineligible for intensive chemotherapy due to coexisting conditions) patients aged 75 years or older with acute myeloid leukaemia in the phase 3 VIALE-A study,19 the BCL2 inhibitor, venetoclax combined with azacitidine improved MRD-negative complete response rates compared with placebo plus azacitidine (23.4% [95% CI 18.6–28.8] vs 7.6% [3.8–13.2]). Whether venetoclax combined with intensive chemotherapy can improve outcomes in younger, fit patients remains an active inquiry. Single-arm phase 2 investigations of venetoclax plus intensive chemotherapy reported composite complete response rates of 72–94%, with MRD-negative composite complete response rates of 82–96%.20, 21, 22 Corresponding 12-month event-free survival for patients treated with chemotherapy with cladribine, idarubicin, and high-dose cytarabine (CLIA) plus venetoclax was 68% (95% CI 54–85),20 and with chemotherapy with fludarabine, cytarabine, idarubicin, and granulocyte-colony stimulating factor (FLAG-IDA) plus venetoclax was 85% (72–100).21
Although clinical and molecular outcomes in patients with acute myeloid leukaemia treated with venetoclax combined with lower intensity therapies (ie, azacitidine or low-dose cytarabine) have been reported,19, 22 no comparisons of venetoclax plus intensive chemotherapy and intensive chemotherapy alone exist, and identification of molecular subgroups predictive of response or resistance to venetoclax plus intensive chemotherapy remain undefined. We aimed to determine the outcomes of a cohort of patients treated with venetoclax plus intensive chemotherapy compared with a well-matched historical cohort of patients receiving intensive chemotherapy and characterise clinical and molecular subgroups of patients who might benefit from treatment with venetoclax plus intensive chemotherapy.
Methods
Study design and participants
In this post-hoc propensity score matched cohort study, we analysed data of patients aged 18 years or older with newly diagnosed, de novo, secondary acute myeloid leukaemia, therapy-related acute myeloid leukaemia, or high-risk myelodysplastic syndrome (defined as patients with ≥10% bone marrow blasts) treated as part of clinical trials with frontline purine analogue-based therapy in combination with an anthracycline and cytarabine at The University of Texas MD Anderson Cancer Center, Texas, USA between March 29, 2010, and June 15, 2021.
Of these patients, those who were treated with FLAG-IDA plus venetoclax (aged >18 to <65 years; NCT03214562) between Jan 11, 2019, and June 15, 2021, or treated with CLIA plus venetoclax (aged 18–65 years; NCT02115295) between Feb 25, 2019, and March 23, 2021, comprised the venetoclax plus intensive chemotherapy cohort. Patients treated with CLIA (aged 18–65 years; NCT02115295) between May 30, 2014, and June 27, 2019; treated with fludarabine, idarubicin, and cytarabine (FIA) between Aug 23, 2011, and Sept 26, 2016 (NCT01289457); and treated with clofarabine, idarubicin, and cytarabine (CIA; aged ≥18 years; NCT01289457) between March 29, 2010, and Aug 25, 2011, comprised the intensive chemotherapy cohort. Preliminary activity and toxicity data for these trials have previously been published.6, 20, 21, 23, 24, 25 Enrolled patients who received at least one cycle in each study were included in the present analysis.
All patients provided written informed consent for treatment on prospective clinical trial protocols and provided informed consent for retrospective research involving their clinical treatment history; full inclusion and exclusion criteria, dosing, and treatment schedules for included regimens are in the appendix (pp 1–7). Investigations were done with approval from the institutional review committee and in accordance with the declaration of Helsinki.
Procedures
All included patients enrolled on clinical protocols were followed prospectively until death. We did cytogenetic and molecular analyses within respective trial protocols.20 Over the study period, we expanded the next-generation sequencing platform from a 28-gene panel to an 81-gene panel. We assessed MRD with eight-colour multiparameter flow cytometry using leukaemia associated immunophenotype or deviation from normal assessment20, 26 with a minimum sensitivity of 10−3 (range 10−3 to 10−4 0.1–0.01%).
We assessed morphological response (defined using revised international working group criteria27) and MRD by bone marrow aspiration and biopsy at the end of induction (approximately cycle 1 day 28) or at the end of re-induction if morphological response was not attained following induction as defined per specific study protocols. As some patients had their best response later during consolidation (eg, a complete response with incomplete haematological recovery after cycle 1 might have evolved to a complete response after cycle 2) and the exact number of cycles before best response was not collected within our dataset, we provided median time (days) to best response (including MRD-negativity since MRD was assessed at the same time as morphological response) as well as the percentage of patients having best response within 30 or 60 days of cycle 1 day 1. Non-responding patients were removed from protocol if no documented response occurred within two treatment cycles (induction and re-induction).
Outcomes
The dual primary outcomes assessed between study cohorts were MRD-negative composite complete response rate and the cumulative incidence of transition to allogeneic HSCT; secondary outcomes were response outcomes, event-free survival, and overall survival. Overall response rate included a composite of complete response, complete response with partial haematological recovery, complete response with incomplete haematological recovery, morphological leukaemia free state, and partial response. Composite complete response rate included complete response, complete response with partial haematological recovery, and complete response with incomplete haematologic recovery. Survival endpoints included overall survival (defined as the time from treatment initiation to death or last follow up if alive at last follow up) and event-free survival (defined as time from study enrolment until no response, date of relapse, or death from any cause, whichever came first); patients not responding were counted as having an event-free survival event on cycle 1 day 1. We also recorded the rate of patients with a response who transitioned to allogeneic HSCT. Landmarking was done for survival analyses pertaining to HSCT.
Statistical analysis
Propensity score matching for selected variables (age, European LeukemiaNet [ELN] risk, acute myeloid leukaemia type, and baseline bone marrow blast percentage) used logistic regression with the nearest-neighbour method (control patients are matched with treated patients that are closest based upon a distance measure). Calliper settings were set to less than 0.2 to restrict distance between matched units. We used clustering to account for differences between patients with similar propensity scores. A standardised mean difference (SMD) threshold of less than 0.1 was determined as an indicator of reduced bias (based upon commonly accepted SMD values in propensity-score matched analysis28) between treatment cohorts.
We used the Fisher’s exact test or χ2 test to compare categorical variables and the Kruskal-Wallis or Wilcoxon rank sum test to compare continuous variables as appropriate. Adjustment for multiple comparisons was done using the Benjamini-Horchberg procedure when necessary. We used weighted logistic regression modelling to calculate the odds ratio (OR) for the primary outcome of MRD-negative composite complete response rate. We used cumulative incidence functions to determine the incidence of allogeneic HSCT with death as competing risk. Time-to-event outcomes within the propensity score matching population were analysed using the stratified log-rank method and Cox proportional hazard modelling. Time-to-event outcomes for exploratory subgroups within the venetoclax plus intensive chemotherapy cohort were analysed using unstratified log-rank and Cox proportional hazard modelling. Clinically relevant exploratory subgroup analyses were restricted to subgroups with 12 or more patients and included outcomes based on ELN risk group, type of acute myeloid leukaemia (de novo vs secondary or therapy related acute myeloid leukaemia), patient age at treatment initiation (<60 years vs ≥60 years), and cytogenetic and molecular markers of sensitivity within the propensity score matched population and venetoclax plus intensive chemotherapy cohort. Statistical analyses were done using R version 4.17.
Role of the funding source
The funder of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report.
Results
312 patients were eligible for inclusion (appendix p 8). After propensity score matching, 33 (11%) patients were excluded because the matching criteria were not fulfilled, leaving 279 patients for the final analysis. Median age was 49 years (IQR 39–57), and 131 (47%) were men and 148 (53%) were women. 194 (70%) of 279 patients were treated with intensive chemotherapy (67 [35%] of 194 with FIA, 98 [51%] with CLIA, and 29 [15%] with CIA) and 85 (30%) of 279 patients were treated with venetoclax plus intensive chemotherapy (40 [47%] of 85 with FLAG-IDA plus venetoclax and 45 [53%] with CLIA plus venetoclax; appendix p 9). No significant clinical or molecular differences were observed between cohorts, although the venetoclax plus intensive chemotherapy cohort contained more patients with secondary or therapy-related acute myeloid leukaemia than the intensive chemotherapy cohort (table 1; appendix pp 8, 12). Patients aged 60 years or older represented 13 (15%) of 85 patients treated with venetoclax plus intensive chemotherapy and 24 (12%) of 194 patients treated with intensive chemotherapy. FLT3-ITD mutations were enriched in patients treated with intensive chemotherapy compared with venetoclax plus intensive chemotherapy, who accordingly received more FLT3-directed therapy with the addition of an FLT3 inhibitor.
Table 1:
Patient Demographics
| Characteristic* | Overall, N = 279 | IC, N = 194 | VEN, N = 85 | p-value |
|---|---|---|---|---|
| Age | 49 (39-57) | 51 (41-57) | 45 (37-56) | 0.063 |
| Sex, male | 131 / 279 (47%) | 87 / 194 (45%) | 44 / 85 (52%) | 0.28 |
| BM Blast ** , % | 55 (32 - 75) | 56 (33 - 77) | 55 (27 - 72) | 0.11 |
| AML Type | 0.0088 | |||
| De novo | 243 / 279 (87%) | 176 / 194 (91%) | 67 / 85 (79%) | |
| High-risk MDS | 3 / 279 (1%) | - | 3 / 85 (4%) | |
| sAML | 16 / 279 (6%) | 8 / 194 (4%) | 8 / 85 (9%) | |
| tAML | 17 / 279 (6%) | 10 / 194 (5%) | 7 / 85 (8%) | |
| ELN Risk Group | 0.23 | |||
| Favorable | 53 / 279 (19%) | 32 / 194 (16%) | 21 / 85 (25%) | |
| Intermediate | 96 / 279 (34%) | 67 / 194 (35%) | 29 / 85 (34%) | |
| Adverse | 130 / 279 (47%) | 95 / 194 (49%) | 35 / 85 (41%) | |
| Cytogenetics | 0.57 | |||
| Adverse/Complex | 80 / 279 (29%) | 58 / 194 (30%) | 22 / 85 (26%) | |
| Diploid | 133 / 279 (48%) | 93 / 194 (48%) | 40 / 85 (47%) | |
| Favorable | 1 / 279 (1%) | - | 1 / 85 (1%) | |
| Insufficient/Unknown | 11 / 279 (4%) | 7 / 194 (4%) | 4 / 85 (5%) | |
| Other intermediate | 53 / 279 (19%) | 35 / 194 (18%) | 18 / 85(21%) | |
| Molecular mutations | ||||
| NPM1 Mutated | 71 / 276 (26%) | 51 / 191 (27%) | 20 / 85 (24%) | 0.57 |
| IDH1 Mutated | 19 / 250 (8%) | 12 / 165 (7%) | 7 / 85 (8%) | 0.78 |
| IDH2 Mutated | 33 / 250 (13%) | 23 / 165 (14%) | 10 / 85 (12%) | 0.63 |
| FLT3 mutated† | 88/ 279 (32) | 66 / 194 (34%) | 22 / 85 (26%) | |
| FLT3-D835 | 24 / 277 (9%) | 12 / 192 (6%) | 12 / 85 (14%) | 0.032 |
| FLT3- ITD | 70 / 278 (25%) | 58 / 193 (30%) | 12 / 85 (14%) | 0.0048 |
| FLT3 inhibitor | 60 / 88 (68%) | 51 /66 (77%) | 9 / 22 (41%) | 0.0015 |
| RUNX1 Mutated | 20 / 221 (9%) | 9 / 136 (7%) | 11 / 85 (13%) | 0.11 |
| ASXL1 Mutated | 28 / 248 (11%) | 20 / 163 (12%) | 8 / 85 (9%) | 0.49 |
| TP53 Mutated | 20 / 241 (8%) | 15 / 156 (10%) | 5 / 85 (6%) | 0.32 |
All variables reported as N(%) or median (IQR);
Includes patients with extramedullary AML;
Concurrent FLT3-ITD and TKD mutations were present in 6 patients.
Overall, 247 (89%) of 279 patients responded to treatment. 26 patients (three in the venetoclax plus intensive chemotherapy group and 23 in the intensive chemotherapy group) discontinued study protocols due to having had no response; one patient in the intensive chemotherapy group was not evaluable. The remaining five patients were early deaths. Overall response rate was higher in the venetoclax plus intensive chemotherapy cohort than the intensive chemotherapy group (table 2; responses in the overall population before matching are shown in the appendix [p 10]). Median time to best response (including MRD-negativity) with venetoclax plus intensive chemotherapy was 29 days (IQR 27–40) versus 32 days (27–40) with intensive chemotherapy. Most patients attained their best response within 60 days of induction, and more patients treated with venetoclax plus intensive chemotherapy than with intensive chemotherapy attained their best response within 30 days of induction (table 2). Similar composite complete response rates were observed in both groups (77 [91%] of 85 patients in the venetoclax plus intensive chemotherapy group vs 166 [86%] of 194 patients in the intensive chemotherapy group), with no significant difference with respect to ELN risk group, although patients with adverse-risk disease had numerically higher composite complete response rates with venetoclax plus intensive chemotherapy (table 2). Overall, early (30-day and 60-day) mortality was low across cohorts (table 2).
Table 2:
Patient outcomes
| Characteristic* | Overall, N = 279 | IC, N = 194 | VEN, N = 85 | p-value |
|---|---|---|---|---|
| Overall Response Rate | 247 / 279 (89%) | 166 / 193 (86%) | 81 / 85 (95%) | 0.019 |
| Composite CR | 243 / 279 (87%) | 166 / 194 (86%) | 77 / 85 (91%) | 0.2 |
| CR | 222 / 279 (80%) | 153 / 194 (79%) | 69 / 85 (81%) | |
| CRh | 4 / 279 (1%) | - | 4 / 85 (5%) | |
| CRi | 17 / 279 (6%) | 13 / 194 (6.7%) | 4 / 85 (5%) | |
| MRD-negative CRc | 0.0028 | |||
| Negative | 150 / 214 (70%) | 86 / 140 (61%) | 64 / 74 (86%) | |
| Positive | 64 / 214 (30%) | 54 / 140 (39%) | 10 / 74 (14% | |
| MLFS | 4 / 279 (1%) | 0 / 194 (0%) | 4 / 85 (5%) | |
| No Response | 26 / 279 (9%) | 23 / 194 (12%) | 3 / 85 (4%) | |
| Early Mortality | 5 / 279 (2%) | 4 / 194 (2%) | 1 / 85 (1%) | |
| 30-day mortality | 4 / 279 (1%) | 3/194 (2%) | 1/85 (1%) | |
| 60-day mortality | 5 / 279 (2%) | 4/194 (2%) | - | |
| Not Evaluable | 1 / 279 (0.4%) | 1 / 194 (0.5%) | - | |
| Time to best response | 31 (27-40) | 32 (27-40) | 29 (27-40) | 0.12 |
| Response by Day 30 | 117 / 247 (47%) | 69 / 166 (42%) | 48 / 81 (58%) | 0.0089 |
| Response by Day 60 | 219 / 247 (89%) | 146 / 166 (88%) | 73 / 81 (90%) | 0.61 |
| ELN Favorable Risk | ||||
| Composite CR | 51 / 53 (96%) | 32 / 32 (100%) | 19 / 21 (90%) | 0.15 |
| MRD-Negative CRc | 40 / 47 (85%) | 23 / 28 (82%) | 17 / 19 (89%) | 0.43 |
| ELN Intermediate Risk | ||||
| Composite CR | 84 / 96 (88%) | 58 / 67 (87%) | 26 / 29 (90%) | 1.0 |
| MRD-Negative CRc | 52 / 71 (73%) | 32 / 47 (68%) | 20 / 24 (83%) | 0.14 |
| ELN Adverse Risk | ||||
| Composite CR | 108 / 130 (83%) | 76 / 95 (80%) | 32 / 35 (91%) | 0.12 |
| MRD-Negative CRc | 58 / 96 (60%) | 31 / 65 (48%) | 27 / 31 (87%) | 0.0059 |
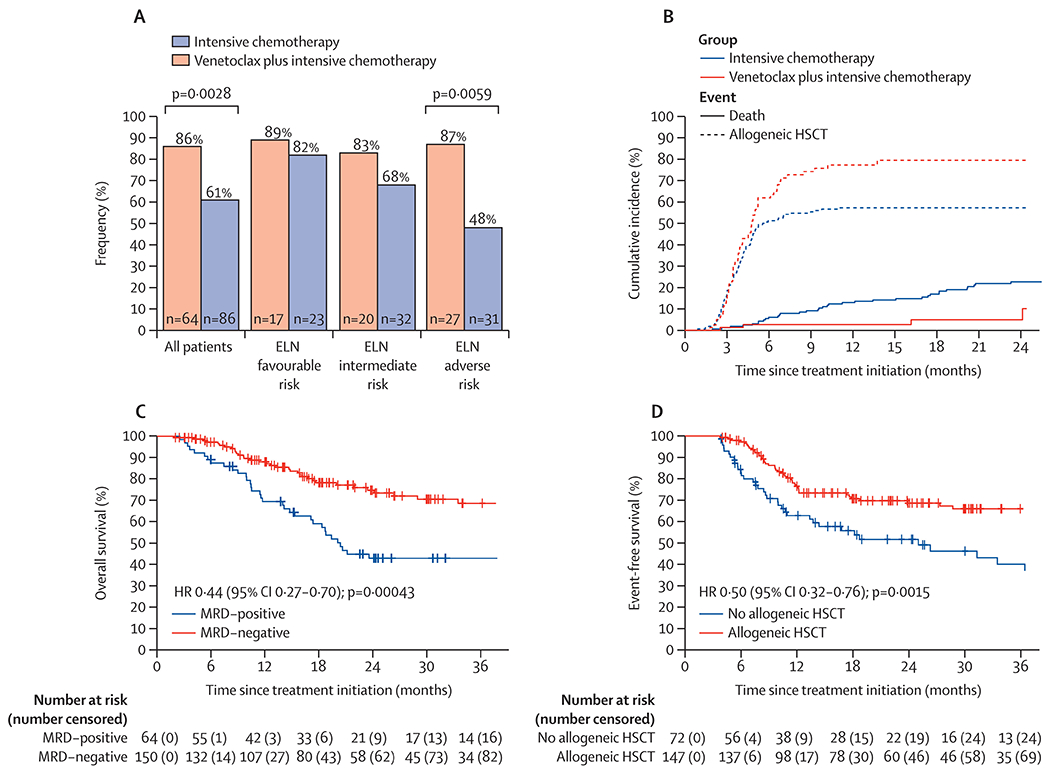
In MRD-evaluable patients (214 (88%) of 243), the primary outcome of attaining MRD-negative composite complete response occurred more often in patients receiving venetoclax plus intensive chemotherapy than in patients receiving intensive chemotherapy (64 [86%] of 74] vs 86 [61%] of 140; OR 3.2 [95% CI 1.5–6.7]; p=0.0028; figure 1A). MRD-negative composite complete response rates were similar in patients with ELN favourable risk irrespective of treatment, as they were in patients with ELN intermediate risk (table 2). Treatment with venetoclax plus intensive chemotherapy improved MRD-negative composite complete response rates in patients with ELN adverse risk acute myeloid leukaemia (OR 5.0 [95% CI 1.6–15.8]; p=0.0059). Responses by molecular subgroups between cohorts are shown in the appendix (p 11).
Figure 1:
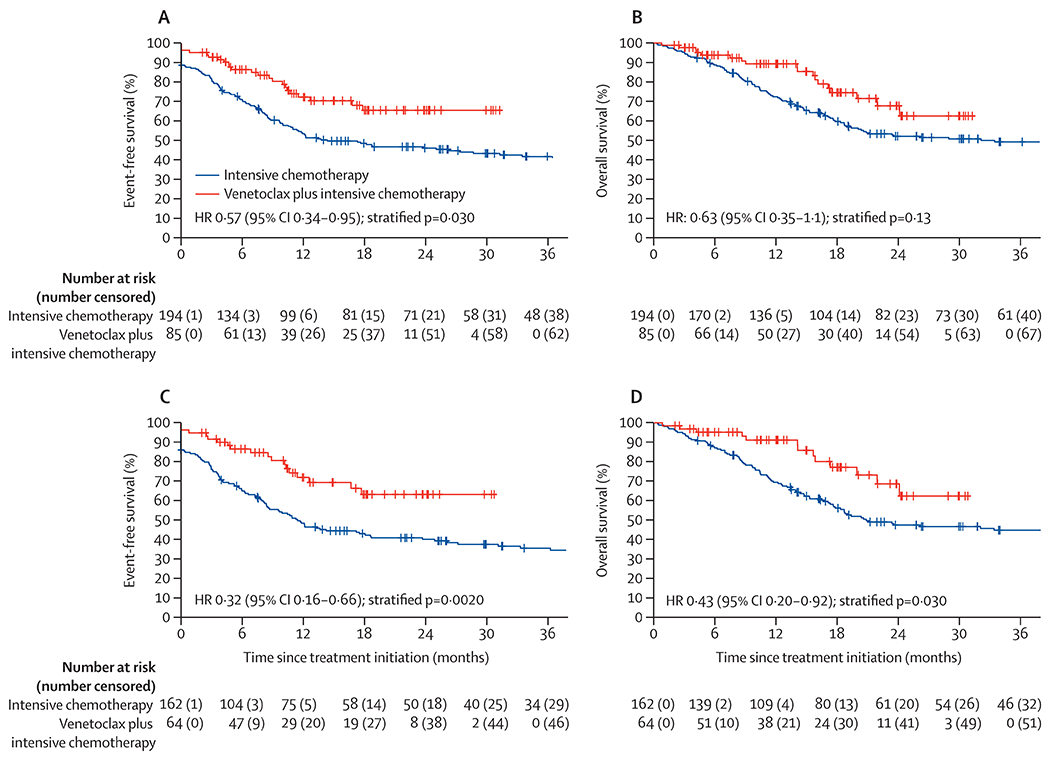
MRD-negative CRc rates with VEN+IC compared to IC in all propensity score matched patients and across ELN risk groups(A). Cumulative incidence of HSCT with VEN+IC compared to IC with death as a competing risk (B). Overall survival in MRDevaluable patients attaining a composite CR (C). Event-free survival within the landmarked population of patients proceeding to HSCT in first remission (D).
154 (55%) of 279 patients transitioned to allogeneic HSCT following induction (150 [97%] of 154 in first complete remission), including 58 (72%) of 81 patients treated with venetoclax plus intensive chemotherapy after a median of 3.9 months (IQR 3.1–5.1) and 96 (58%) of 166 patients treated with intensive chemotherapy after a median of 3.8 months (2.9–4.8) months. Of the 154 patients proceeding to allogeneic HSCT, ELN risk was favourable for 34 (22%) patients, intermediate for 50 (32%) patients, and adverse for 70 (45%) patients, with no significant differences between venetoclax plus intensive chemotherapy and intensive chemotherapy cohorts (12 [21%] of 58 vs 22 [23%] of 96 for favourable risk, 20 [34%] of 58 vs 30 [31%] of 96 for intermediate risk, and 26 [45%] of 58 vs 44 [46%] of 96 for adverse risk). With death as a competing risk, the overall cumulative incidence of allogeneic HSCT in patients who responded was higher with venetoclax plus intensive chemotherapy than intensive chemotherapy (79% [95% CI 67–88] vs 57% [49–65]; HR 1.52 [95% CI 1.11–2.08]; p=0.012), and the overall incidence of death was lower with venetoclax plus intensive chemotherapy than intensive chemotherapy (5% [95% CI 1–13] vs 26% [18–35]; HR 0.24 [0.08–0.66]; p=0.018; figure 1B). No significant difference in the cumulative incidence of allogeneic HSCT with venetoclax plus intensive chemotherapy versus intensive chemotherapy at 12 months was observed in patients in the favourable ELN risk group (60% vs 69%; p=0.33) or in the intermediate risk group (71% vs 43%; p=0.053), although an increased incidence of allogeneic HSCT was observed in patients in the adverse ELN risk group treated with venetoclax plus intensive chemotherapy compared with intensive chemotherapy (82% vs 46%; p<0.0001).
After a median follow up of 30 months (95% CI 26–36), 138 events (23 in the venetoclax plus intensive chemotherapy group and 115 in the intensive chemotherapy group) and 116 deaths (18 in the venetoclax plus intensive chemotherapy group and 98 in the intensive chemotherapy group) had occurred. Median event-free survival for the study population was 26 months (95% CI 14–56) and median overall survival was 59 months (95% CI 23–not reached [NR]). Median event-free and overall survival were NR in the venetoclax plus intensive chemotherapy cohort (24-month event-free survival 65% [95% CI 54–79]; 24-month overall survival 67% [95% CI 55–83]). No significant difference between treatment groups was seen with respect to 24-month event-free survival (CLIA plus venetoclax 70% [95% CI 57–86]; FLAG-IDA plus venetoclax 61% [45–83]) or 24-month overall survival (CLIA plus venetoclax 62% [95% CI 44–86]; FLAG-IDA plus venetoclax 74% [57–95]). Within the intensive chemotherapy cohort, median event-free survival was 14.3 months (95% CI 11–34) and median overall survival was 32 months (95% CI 19–NR). Similar to the venetoclax plus intensive chemotherapy cohort, no significant difference was observed between treatment groups with respect to median event-free survival (13 months [95% CI 6–78] in the CIA group, 14 months [10–NR] in the CLIA group, and 18 months [9–NR] in the FIA group), or median overall survival (21 months [95% CI 17–NR] in the CIA group, 34 months [17–NR] in the CLIA group, and 47 months [19–NR] in the FIA group; appendix p 13).
During the more contemporary time periods (ie, 2019–22), more patients were enrolled on venetoclax-based trials than on intensive chemotherapy protocols; however, in a post-hoc analysis no significant overall survival difference was observed with respect to enrolment year, even within the intensive chemotherapy group that enrolled patients over a longer period than protocols incorporating venetoclax (appendix p 14).
Improved median event-free survival was observed in the venetoclax plus intensive chemotherapy group compared with the intensive chemotherapy group (NR [95% CI NR–NR] vs 14.3 months [11–34]; HR 0.57 [95% CI 0.34–0.95]; p=0.030). Estimated 24-month event-free survival with venetoclax plus intensive chemotherapy was 66% (95% CI 54–79) and with intensive chemotherapy was 46% (39–54). Median overall survival did not significantly differ between the two cohorts (median NR [95% CI 24–NR] vs 32 months [19–NR]; HR 0.63 [95% CI 0.35–1.1]; p=0.13), with an estimated 24-month overall survival of 68% [95% CI 55–83] versus 52% (45–60; figure 2). Median event-free survival for venetoclax plus intensive chemotherapy versus intensive chemotherapy across ELN risk groups was NR versus NR (24-month event-free survival 72% [95% CI 54–97] vs 75% [61–92]; HR 1.2 [95% CI 0.4–3.7]; p=0.72) for favourable, NR (CI 18–NR) versus 24 months (13–NR; HR 0.61 [95% CI 0.15–2.5]; p=0.50) for intermediate, and NR (95% CI 17–NR) versus 8 months (6–12; HR 0.2 [95% CI 0.07–0.58]; p=0.0027) for adverse-risk acute myeloid leukaemia. Median overall survival was NR versus NR for favourable (24-month overall survival 66% [95% CI 45–97] vs 74% [60–91%]; HR 1.4 [95% CI 0.45–4.2]; p=0.58), NR versus NR for intermediate (24-month overall survival 74% [95% CI 56–99] vs 58% [47–72]; HR 0.39 [95% CI 0.04–3.8]; p=0.42), and NR [95% CI 20–NR] versus 17 months (95% CI 13–34; HR 0.31 [95% CI 0.1–0.90]; p=0.029) for adverse-risk acute myeloid leukaemia.
Figure 2:
Event-free survival within propensity score matched patients treated with VEN+IC compared to IC(A). Overall survival in patients treated with VEN+IC compared to IC(B). Event-free survival in patients with ELN intermediate or adverse risk AML treated with VEN+IC compared to IC(C). Overall survival in patients with intermediate or adverse risk AML treated with VEN+IC compared to IC (D).
Excluding patients in the ELN favourable risk group who appeared to derive minimal benefit from the addition of venetoclax, venetoclax plus intensive chemotherapy showed significant benefit compared with intensive chemotherapy in patients with intermediate or adverse ELN risk acute myeloid leukaemia, improving median event-free survival (NR [95% CI NR–NR] vs 12 months [9–19]; HR 0.32 [95% CI 0.16–0.66]; p=0.0020) and overall survival (NR [95% CI 24–NR] vs 21 months [18–72]; HR 0.43 [95% CI 0.20–0.92]; p=0.030; figure 2). After multivariate analysis with adjustment for variables used in propensity score matching (ELN risk, age, baseline bone marrow blast percentage, and acute myeloid leukaemia type), treatment with venetoclax plus intensive chemotherapy continued to show improvement in overall survival (HR 0.40 [95% CI 0.18–0.90] p=0.026).
Among MRD-evaluable patients in remission (n=214), those who had an MRD-negative composite complete response (150 [70%]) by multiparameter flow cytometry, compared with patients who were MRD-positive, had improved event-free survival (NR [95% CI 51–NR] vs 12 months [9–25]; HR 0.36 [95% CI 0.24–0.56]; p<0.0001) and overall survival (NR [95% CI 59–NR] vs 20 months [17–NR]; HR 0.44 [95% CI 0.27–0.70]; p=0.00043; figure 1C). Similarly, in a landmark analysis at the median time to allogeneic HSCT (3.8 months), event-free survival was improved in responding patients proceeding with allogeneic HSCT in first composite complete response compared with patients who did not transition (78 months [95% CI 51–NR] vs 25 months [14–NR]; HR 0.50 [95% CI 0.32–0.76]; p=0.0015; figure 1D).
A differential benefit for allogeneic HSCT was observed across ELN risk groups. No significant benefit was observed in patients in the ELN favourable risk group (HR 0.87 [95% CI 0.3–2.6]; p=0.80) or intermediate risk group (HR 0.58 [0.28–1.2]; p=0.15); however, patients with ELN adverse-risk acute myeloid leukaemia showed a significant reduction in risk of relapse or death with allogeneic HSCT (HR 0.31 [95% CI 0.17–0.58]; p=0.0002), even in the subset of patients who were MRD-negative (HR 0.28 [0.11–0.72]; p=0.0075). Median overall survival with allogeneic HSCT was 102 months (95% CI 59–NR) and without allogeneic HSCT it was NR (23–NR; HR 0.78 [95% CI 0.48–1.27]; p=0.31); however, a substantial proportion of patients who relapsed received allogeneic HSCT (11 [44%] of 25 patients) following successful salvage therapy, decreasing the accuracy of formal assessment on survival. No significant survival difference was observed for venetoclax plus intensive chemotherapy versus intensive chemotherapy treatment in patients aged 60 years and older (median overall survival NR [95% CI 17–NR] vs 13 months [9–NR]; HR 0.93 [95% CI 0.1–5.8]; p=0.94), or in patients with diploid or other intermediate risk cytogenetics (24-month overall survival was 70% [95% CI 55–88] vs 59% [50–68]; HR 0.93 [95% CI 0.40–2.0]; p=0.85; figure 3).
Figure 3:
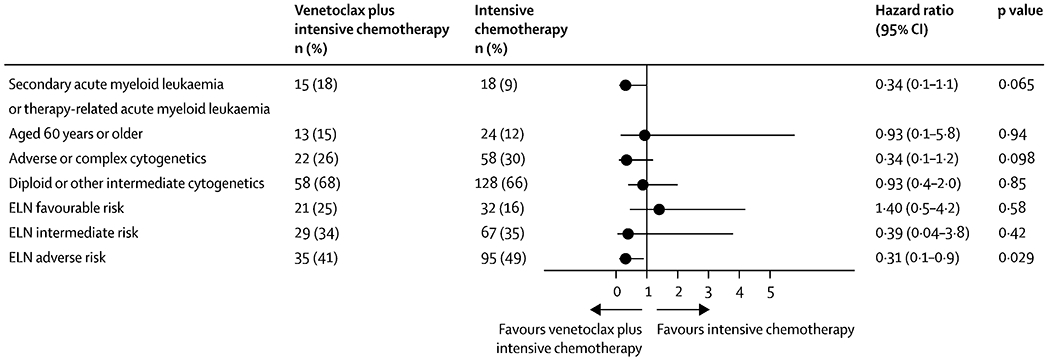
Univariate cox proportional hazard ratios for pre-specified exploratory subgroup analyses of patients treated with VEN+IC compared to IC. Patients with sAML/tAML, adverse or complex cytogenetic, or ELN adverse-risk disease.
Overall survival did not significantly differ in patients with secondary or therapy related acute myeloid leukaemia treated with venetoclax plus intensive chemotherapy versus intensive chemotherapy (median overall survival NR [95% CI 24–NR] vs 13 months [11–NR]; HR 0.34 [95% CI 0.11–1.07]; p=0.065) or in patients with adverse or complex cytogenetics (median overall survival NR [95% CI 20–NR] vs 16 months [11–47]; HR 0.34 [95% CI 0.09–1.2]; p=0.098). No survival difference was observed based on FLT3 mutation status (ITD and TKD) or FLT3 inhibitor use between cohorts (appendix p 17).
The current cohort represents the largest prospective cohort of patients treated with venetoclax plus intensive chemotherapy, thus molecular predictors of response within the full cohort (n=91) were assessed (appendix p 15). Signaling pathway aberrations were most prevalent (49 [54%] of 91), represented by mutations in N/KRAS (27 [30%]), FLT3-ITD (13 [14%]), FLT3-D835 (12 [13%]), and PTPN11 (8 [9%]). Methylation pathway aberrations were second most prevalent (45 [49%] of 91), represented by mutations in DNMT3A (24 [26%]), IDH2 (12 [13%]), TET2 (9 [10%]), and IDH1 (7 [8%]).
The effect on survival was assessed across pathways (ie, methylation [n=45], signaling [n=49], transcription factors [n=26], cohesin [n=6], chromatin [n=20], splicing [n=14], and tumour suppressors [n=12]. Given the small number of patients with cohesin mutations and patient overlap with chromatin mutations, this group was combined for increased power (n=22). No significant difference in response or survival was observed based upon mutations in NPM1, methylation, signaling, transcription factor, or cohesin-chromatin genes. Specific adverse-risk molecular mutations, including ASXL1, RUNX1, splicing mutations (SRSF2, U2AF1, SF3B1, or ZRSR2), signaling mutations (K/NRAS, FLT3, KIT, or CBL), or cytogenetic abnormalities (KMT2A-rearranged), had no negative effect on event-free or overall survival with venetoclax plus intensive chemotherapy treatment (appendix p 16). Compared with patients with wild-type mutations, patients with tumour suppressor mutations had inferior event-free survival (12 months [95% CI 5–NR] vs NR [NR–NR]; HR 3.4 [95% CI 1.3–8.8]; p=0.010), but overall survival did not significantly differ (20 months [95% CI 17–NR] vs NR [NR–NR]; HR 2.6 [95% CI 0.92–7.3]; p=0.070). Inferior event-free survival was driven solely by five patients with TP53 mutations, who, compared with patients with TP53 wild-type mutations, showed inferior event-free survival (5 months [95% CI 4–NR] vs NR [NR–NR]; HR 7.4 [95% CI 2.7–20]; p<0.0001) and inferior overall survival (17 months [95% CI 9–NR] vs NR [NR–NR]; HR 5.9 [95% CI 2.1–16.6]; p=0.0008).
Discussion
In this post-hoc propensity score matched analysis of younger (median age 49 years [IQR 39–47]) patients with newly diagnosed acute myeloid leukaemia, venetoclax plus intensive chemotherapy increased MRD-negative remission rates and enabled a significant portion of patients to transition safely to consolidative HSCT. Compared to intensive chemotherapy alone, venetoclax plus intensive chemotherapy improved event-free survival in the entire study population and overall survival in patients with ELN intermediate or adverse risk acute myeloid leukaemia. The effect of venetoclax plus intensive chemotherapy on event-free survival and overall survival outcomes was most prominent in adverse risk patients.
Previous analyses of low-intensity venetoclax-based regimens compared with intensive chemotherapy have varied results, with similar or nominally inferior outcomes observed compared to intensive chemotherapy.29 These analyses are particularly relevant because no comparison yet exists evaluating young, fit patients treated with venetoclax plus intensive chemotherapy or intensive chemotherapy alone. However, such analyses are limited by imbalances in patient age, comorbidities, and other covariates confounding outcomes within respective treatment populations.29 After propensity score matching, low-intensity venetoclax-based therapies showed improved survival compared with intensive chemotherapy.30, 31
MRD is a strong indicator of disease status and relapse risk, resulting in adoption of MRD assessment in remission to guide further acute myeloid leukaemia directed therapy.10, 16, 17 Venetoclax plus intensive chemotherapy treatment increased rates of MRD-negative composite complete response compared with chemotherapy alone (86% vs 61%), with improved event-free and overall survival in MRD-negative patients. These data highlight the importance of MRD as a marker of successful induction therapy and the advantage of incorporating venetoclax with intensive chemotherapy to elicit clinically meaningful response and survival endpoints in specific subgroups of patients with acute myeloid leukaemia.32
Incremental increases in MRD-negative response rates were observed between patients treated with venetoclax plus intensive chemotherapy compared with intensive chemotherapy across ELN risk groups, suggesting that addition of venetoclax had a differential benefit. Consistent with this differential treatment effect, venetoclax plus intensive chemotherapy enabled transition to HSCT in more patients with ELN intermediate risk or adverse risk acute myeloid leukaemia than intensive chemotherapy did. Allogeneic HSCT in first remission reduced the risk of relapse or death in the entire study population, particularly in patients with ELN adverse risk acute myeloid leukaemia.
Improved event-free survival was observed with venetoclax plus intensive chemotherapy compared with intensive chemotherapy. In the overall population, survival did not statistically differ between venetoclax plus intensive chemotherapy and intensive chemotherapy. Patients in the ELN favourable risk group had similar response and survival rates with venetoclax plus intensive chemotherapy compared with intensive chemotherapy. When analysing patients with only intermediate or adverse risk acute myeloid leukaemia, improved overall survival was observed with venetoclax plus intensive chemotherapy compared with intensive chemotherapy, suggesting a survival benefit in these patients.
Our study had limitations, including the retrospective nature and time over which patients were enrolled. We used propensity score matching to minimise the risk of bias, and no survival effect was observed based upon enrolment period. Additionally, information pertaining to post-protocol therapies were not readily available. Although none of the patients in the venetoclax plus intensive chemotherapy cohort received venetoclax-based maintenance following allogeneic HSCT on protocol, it is possible a minority received off-label venetoclax-based maintenance outside of a clinical trial. Conversely, patients in the intensive chemotherapy cohort might have received venetoclax-based regimens as salvage therapy.
Within the venetoclax plus intensive chemotherapy cohort, tumour suppressor mutations correlated with decreased event-free and overall survival, driven by the relatively few patients with mutated TP53. Although the small sample size within the venetoclax plus intensive chemotherapy cohort (n=5) prevents definitive conclusions, outgrowth of TP53 mutated leukaemic clones under the selective pressure of venetoclax is well characterised,33 resulting in clinical relapse with decreased intensity venetoclax-based therapies.22
Aside from patients with TP53 mutations, encouraging outcomes were observed with venetoclax plus intensive chemotherapy across other adverse risk molecular subgroups including patients with signalling mutations (ie, RAS, FLT3-ITD or FLT3-TKD, and PTPN11) or splicing mutations (SRSF2, U2AF1, SF3B1, and ZRSR2)—the latter associated with outcomes akin to those observed in secondary acute myeloid leukaemia.23 In support of this notion, patients with secondary acute myeloid leukaemia had improved overall survival with venetoclax plus intensive chemotherapy compared with intensive chemotherapy. Although notable, these results must be interpreted with caution because of the small number of included patients. Longer follow up and larger prospective, randomised investigations of venetoclax plus intensive chemotherapy versus intensive chemotherapy are necessary to support these results and derive definitive conclusions within molecular and clinical subgroups.
Supplementary Material
Research in context.
Evidence before this study
A literature review of the PubMed database between Jan 1, 2021, and Dec 31, 2021, was done for prospective reports of venetoclax combined with intensive induction chemotherapy in younger, fit patients with newly diagnosed acute myeloid leukaemia using key terms including “venetoclax”, “induction chemotherapy”, and “AML”. No language restrictions were included. Articles were limited to prospective clinical trials reporting outcomes of venetoclax in combination with intensive chemotherapy. A formal literature search was not done given the scarce available literature. Three single-arm phase 2 studies incorporating venetoclax with intensive chemotherapy were identified that reported encouraging efficacy without serious safety signals. However, there was no comparative effectiveness study of venetoclax in combination with intensive chemotherapy measured against intensive chemotherapy alone.
Added value of this study
The present study reports outcomes in a large cohort of younger (median age 49 years) adult patients with acute myeloid leukaemia treated with venetoclax combined with intensive chemotherapy compared with a well-matched contemporary cohort treated with intensive chemotherapy alone. The report provides the first evidence that combining venetoclax with intensive chemotherapy improves rates of measurable residual disease-negative composite complete response, transition to allogeneic haematopoietic stem-cell transplantation, and event-free survival compared with intensive chemotherapy.
Implications of all the available evidence
Venetoclax combined with intensive chemotherapy resulted in favorable response rates and survival in single-arm, prospective clinical investigations, suggesting the combination could improve outcomes compared to intensive chemotherapy alone. These data support the activity of venetoclax added to intensive chemotherapy, providing an early benchmark that should be substantiated by prospective phase 3 trials.
Acknowledgments
CDD is supported by Leukemia & Lymphoma Society Career Scholar Award and V Foundation Lloyd Family Clinical Scholar Award. CAL and PKR are supported by a T32 training grant from the National Institutes of Health (T32CA009666). PKR is supported by a T32 training grant from the National Institutes of Health (T32CA009666).
Declaration of interests
GB reports consultancy fees from Protagonist Therapeutics, research funding from Ryvu, membership on an entity’s Board of Directors or advisory committees from Takeda, research funding from Astex, membership on an entity’s Board of Directors or advisory committees from ArgenX, current employment by University of Texas MD Anderson Cancer Center, consultancy and membership on an entity’s Board of Directors or advisory committees from Novartis, and consultancy from GSK. MY reports research funding from Daiichi-Sankyo and Pfizer. NP reports consultancy from DAVA Oncology, LFB Biotechnologies, MustangBio, Stemline Therapeutics, Affymetrix, Roche Diagnostics, Blueprint Medicines, Clearview Healthcare Partners, Novartis Pharmaceuticals, Celgene Corporation, Incyte, Protagonist Therapeutics, Abbvie Pharmaceuticals, Aptitude Health, Bristol-Myers Squibb, ImmunoGen, Pacylex Pharmaceuticals, and CareDx; membership on the Board of Directors or advisory committees of ASCO Leukemia Advisory Panel, Dan’s House of Hope, Stemline Therapeutics, Abbvie Pharmaceuticals, ASH Communications Committee, and HemOnc Times/Oncology Times; and grants or contracts from Sager Strong Foundation, MustangBio, Daiichi Sankyo, Springer Science + Business Media, Stemline Therapeutics, Samus, Novartis Pharmaceuticals, Abbvie Pharmaceuticals, Plexxicon, and Cellectis S.A. ADR; research funding from Cellectis S.A. ADR, Daiichi Sankyo, Affymetrix, Samus, Novartis Pharmaceuticals, Abbvie Pharmaceuticals, Plexxicon, and Stemline Therapeutics; and research support from Novartis Pharmaceuticals. UP reports research funding from Bayer, Abbvie, Novartis, and Incyte. ES reports honoraria from Magenta, Novartis, and Bayer HealthCare Pharmaceuticals; consultancy from Navan, Axio, Adaptimmune, Magenta, and Novartis; and patents and royalties from Affimed and Takeda. CDD reports membership on an entity’s Board of Directors or advisory committees from GlaxoSmithKline and Notable Labs; current stock options in a privately-held company in Notable Labs, honoraria from ImmuneOnc, Bristol Myers Squibb, Agios/Servier, Takeda, Novartis, Foghorn, Celgene a Bristol Myers Squibb company, and Forma; research funding from ImmuneOnc, and Bristol Myers Squibb, AbbVie, Agios/Servier, Foghorn, Celgene a Bristol Myers Squibb company, and Forma; and consultancy for AbbVie and Agios/Servier. TMK reports consultancy from Abbvie, Sanofi-Aventis, Liberum, Jazz, Genentech, Daiichi Sankyo, Novartis, Pfizer, and Aglos; grant or research support from Abbvie, Genentech, Bristol Myers Squibb, and Amgen; speaker’s bureau fees from Cure, and research support from Genfleet, Cellonkos, Astellas, AstraZeneca, Ascentage, and Pulmotech. ND reports consultancy for Novartis, Trovagene, Abbvie, Genentech, Amgen, Pfizer, Bristol Myers Squibb, Astellas, Daiichi Sankyo, Sevier, ImmunoGen, Gilead Sciences, Trillium, Dava Oncology (Arog), Celgene, Syndax, Shattuck Labs, Agios, Kite Pharmaceuticals, SOBI, STAR Therapeutics, and Jazz Pharmaceuticals; member of the Data Monitoring Committee for Jazz Pharmaceuticals; research funding from Trovagene, Genentech, FATE Therapeutics, Hanmi, Amgen, Pfizer, Bristol Myers Squibb, Novimmune, Astellas, Daiichi Sankyo, Sevier, ImmunoGen, Glycomimetics, Gilead Sciences, Trillium, Karyopharm, Newave, and Abbvie. NJS reports consultancy for Takeda Oncology, Jazz Pharmaceuticals, NGMBio, AstraZeneca, and Amgen; research funding from Takeda Oncology and Astellas; honoraria from Amgen, and Novartis. MK reports grant support from Agios, Rafael Pharmaceuticals, Cellectis, Ascentage, AstraZeneca, Sanofi, Ablynx, Genentech, Forty Seven, AbbVie, F Hoffmann-La Roche, and Calithera; research funding from Agios, Rafael Pharmaceuticals, KisoJi, Eli Lilly, Ascentage, AstraZeneca, Sanofi, Ablynx, Genentech, Forty Seven, AbbVie, Stemline Therapeutics, and Calithera; intellectual property rights from Eli Lilly, Novartis, and Reata Pharmaceuticals; current stock options in Reata Pharmaceuticals; pending research funding from Novartis; consultancy for Genentech, F Hoffmann-La Roche, and AbbVie; honoraria from Genentech, F Hoffmann-La Roche, and AbbVie. FR reports honoraria from Astex, Taiho, Bristol Myers Squibb, Xencor, Agios, AstraZeneca, Novartis, AbbVie, Celgene, Jazz, Syros Pharmaceuticals, and Amgen; research funding from Astex, Taiho, Bristol Myers Squibb, Xencor, Agios, Prelude, AbbVie, Celgene, Jazz, Syros Pharmaceuticals, and Amgen; membership on the Board of Directors or advisory committees of Bristol Myers Squibb and Celgene; consultancy for Syros Pharmaceuticals. KT reports consultancy for Novartis, Celgene/BMS, GSK, and Symbio Pharmaceuticals; and membership on Symbio Pharmaceuticals’ Board of Directors or advisory committee. SL reports current equity in publicly-traded Abbvie; honoraria from Curio Sciences and Peerview; and consultancy for Gerson Lehrman Group, Guidepoint, and Qualworld. KS reports consultancy for Novartis; research funding from Novartis; membership on the Board of Directors or advisory committees of Pfizer and Daiichi-Sankyo. HK reports honoraria from AbbVie, Ipsen Pharmaceuticals, AstraZeneca, KAHR Medical, Aptitude Health, Pfizer, Astellas Health, Novartis, NOVA Research, Precision Biosciences, Taiho Pharmaceutical Canada, and Amgen; research funding from AbbVie, Immunogen, BMS, Jazz Pharmaceuticals, Ascentage, Daiichi-Sankyo, Pfizer, Novartis, and Amgen. All other authors declare no competing interests.
Footnotes
Data sharing
Qualified researchers can request access to individual patient-level data reported in this Article after its publication. No identifying data will be provided. All requests for data must include a description of the research proposal and be submitted to the corresponding author.
References
- 1.Rai KR, Holland JF, Glidewell OJ, et al. Treatment of acute myelocytic leukemia: a study by cancer and leukemia group B. Blood 1981; 58(6): 1203–12. [PubMed] [Google Scholar]
- 2.Fernandez HF, Sun Z, Yao X, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med 2009; 361(13): 1249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohtake S, Miyawaki S, Fujita H, et al. Randomized study of induction therapy comparing standard-dose idarubicin with high-dose daunorubicin in adult patients with previously untreated acute myeloid leukemia: the JALSG AML201 Study. Blood 2011; 117(8): 2358–65. [DOI] [PubMed] [Google Scholar]
- 4.Burnett AK, Russell NH, Hills RK, et al. A randomized comparison of daunorubicin 90 mg/m2 vs 60 mg/m2 in AML induction: results from the UK NCRI AML17 trial in 1206 patients. Blood 2015; 125(25): 3878–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holowiecki J, Grosicki S, Giebel S, et al. Cladribine, but not fludarabine, added to daunorubicin and cytarabine during induction prolongs survival of patients with acute myeloid leukemia: a multicenter, randomized phase III study. J Clin Oncol 2012; 30(20): 2441–8. [DOI] [PubMed] [Google Scholar]
- 6.Jabbour E, Short NJ, Ravandi F, et al. A randomized phase 2 study of idarubicin and cytarabine with clofarabine or fludarabine in patients with newly diagnosed acute myeloid leukemia. Cancer 2017; 123(22): 4430–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mayer RJ, Davis RB, Schiffer CA, et al. Intensive Postremission Chemotherapy in Adults with Acute Myeloid Leukemia. New England Journal of Medicine 1994; 331(14): 896–903. [DOI] [PubMed] [Google Scholar]
- 8.Burnett AK, Russell NH, Hills RK, et al. Defining the Optimal Total Number of Chemotherapy Courses in Younger Patients With Acute Myeloid Leukemia: A Comparison of Three Versus Four Courses. J Clin Oncol 2021; 39(8): 890–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hourigan CS, Dillon LW, Gui G, et al. Impact of Conditioning Intensity of Allogeneic Transplantation for Acute Myeloid Leukemia With Genomic Evidence of Residual Disease. J Clin Oncol 2020; 38(12): 1273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017; 129(4): 424–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med 2016; 374(23): 2209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mrózek K, Marcucci G, Nicolet D, et al. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J Clin Oncol 2012; 30(36): 4515–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med 2017; 377(5): 454–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DiNardo CD, Stein EM, de Botton S, et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N Engl J Med 2018; 378(25): 2386–98. [DOI] [PubMed] [Google Scholar]
- 15.Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017; 130(6): 722–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Short NJ, Zhou S, Fu C, et al. Association of Measurable Residual Disease With Survival Outcomes in Patients With Acute Myeloid Leukemia: A Systematic Review and Meta-analysis. JAMA Oncol 2020; 6(12): 1890–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jongen-Lavrencic M, Grob T, Hanekamp D, et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. New England Journal of Medicine 2018; 378(13): 1189–99. [DOI] [PubMed] [Google Scholar]
- 18.Ivey A, Hills RK, Simpson MA, et al. Assessment of Minimal Residual Disease in Standard-Risk AML. N Engl J Med 2016; 374(5): 422–33. [DOI] [PubMed] [Google Scholar]
- 19.DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. New England Journal of Medicine 2020; 383(7): 617–29. [DOI] [PubMed] [Google Scholar]
- 20.DiNardo CD, Lachowiez CA, Takahashi K, et al. Venetoclax Combined With FLAG-IDA Induction and Consolidation in Newly Diagnosed and Relapsed or Refractory Acute Myeloid Leukemia. J Clin Oncol 2021; 39(25): 2768–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kadia TM, Reville PK, Borthakur G, et al. Venetoclax plus intensive chemotherapy with cladribine, idarubicin, and cytarabine in patients with newly diagnosed acute myeloid leukaemia or high-risk myelodysplastic syndrome: a cohort from a single-centre, single-arm, phase 2 trial. Lancet Haematol 2021; 8(8): e552–e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DiNardo CD, Tiong IS, Quaglieri A, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood 2020; 135(11): 791–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu J, Jorgensen JL, Wang SA. How Do We Use Multicolor Flow Cytometry to Detect Minimal Residual Disease in Acute Myeloid Leukemia? Clin Lab Med 2017; 37(4): 787–802. [DOI] [PubMed] [Google Scholar]
- 24.Cheson BD, Bennett JM, Kopecky KJ, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol 2003; 21(24): 4642–9. [DOI] [PubMed] [Google Scholar]
- 25.Zeidan AM, Pollyea DA, Borate U, et al. Venetoclax Plus Azacitidine (VEN-AZA) Vs. Intensive Chemotherapy (IC) As Induction for Patients with Acute Myeloid Leukemia (AML): Retrospective Analysis of an Electronic Medical Records (EMR) Database in the United States. Blood 2021; 138(Supplement 1): 277-. [Google Scholar]
- 26.Cherry EM, Abbott D, Amaya M, et al. Venetoclax and azacitidine compared with induction chemotherapy for newly diagnosed patients with acute myeloid leukemia. Blood Adv 2021; 5(24): 5565–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maiti A, Qiao W, Sasaki K, et al. Venetoclax with decitabine vs intensive chemotherapy in acute myeloid leukemia: A propensity score matched analysis stratified by risk of treatment-related mortality. Am J Hematol 2021; 96(3): 282–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Norsworthy KJ, Gao X, Ko CW, et al. Response Rate, Event-Free Survival, and Overall Survival in Newly Diagnosed Acute Myeloid Leukemia: US Food and Drug Administration Trial-Level and Patient-Level Analyses. J Clin Oncol 2021: Jco2101548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thijssen R, Diepstraten ST, Moujalled D, et al. Intact TP-53 function is essential for sustaining durable responses to BH3-mimetic drugs in leukemias. Blood 2021; 137(20): 2721–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chua CC, Roberts AW, Reynolds J, et al. Chemotherapy and Venetoclax in Elderly Acute Myeloid Leukemia Trial (CAVEAT): A Phase Ib Dose-Escalation Study of Venetoclax Combined With Modified Intensive Chemotherapy. J Clin Oncol 2020; 38(30): 3506–17. [DOI] [PubMed] [Google Scholar]
- 31.Abuasab T, Kantarjian H, Garcia-Manero G, Montalban-Bravo G, Alvarado Y, Yilmaz M, Pemmaraju N, Chien KS, Mohamed SF, Daver N, Kornblau SM. Phase II Study of Cladribine, Idarubicin, Cytarabine (CLIA) Plus Gilteritinib in Patients with FLT3 Mutated Acute Myeloid Leukemia (AML). Blood. 2021. Nov 23;138:2330. [Google Scholar]
- 32.Kadia T, Cortes JE, Jabbour EJ, Daver N, Pemmaraju N, Ravandi F, Jain N, Verstovsek S, DiNardo C, Alvarado Y, Ferrajoli A. Phase II study of cladribine, idarubicin, and cytarabine (araC) in patients with acute myeloid leukemia (AML). Blood. 2015. Dec 3;126(23):2541.26500341 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.