

Summary
Aberrant neurodevelopment is a core deficit of autism spectrum disorder (ASD). Here we ask whether a non-genetic factor, prenatal exposure to the environmental pollutant methylmercury (MeHg), is a contributing factor in ASD onset. We showed that adult mice prenatally exposed to non-apoptotic MeHg exhibited key ASD characteristics, including impaired communication, reduced sociability, and increased restrictive repetitive behaviors, whereas in the embryonic cortex, prenatal MeHg exposure caused premature neuronal differentiation. Further single-cell RNA sequencing (scRNA-seq) analysis disclosed that prenatal exposure to MeHg resulted in cortical radial glial precursors (RGPs) favoring asymmetric differentiation to directly generate cortical neurons, omitting the intermediate progenitor stage. In addition, MeHg exposure in cultured RGPs increased CREB phosphorylation and enhanced the interaction between CREB and CREB binding protein (CBP). Intriguingly, metformin, an FDA-approved drug, can reverse MeHg-induced premature neuronal differentiation via CREB/CBP repulsion. These findings provide insights into ASD etiology, its underlying mechanism, and a potential therapeutic strategy.
Subject areas: Drugs, Molecular neuroscience, Neurotoxicology, Transcriptomics
Graphical abstract
Highlights
-
•
Prenatal MeHg exposure causes autism spectrum disorder-like behaviors in adulthood
-
•
MeHg exposure increases neuronal differentiation of fetal radial glial precursor
-
•
MeHg exposure enhances CREB phosphorylation/activation in fetal radial glial cells
-
•
Metformin reverses MeHg-induced differentiation via CREB/CBP repulsion
Drugs; Molecular neuroscience; Neurotoxicology; Transcriptomics
Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental disorder that is characterized by impairments in language, communication, and sociability, combined with restricted, repetitive behaviors.1 Aberrant embryonic cerebral cortex development has been shown to contribute to ASD pathophysiology2 which is caused by both genetic and non-genetic components. Although a majority of past research focuses on the genetic contribution to ASD pathogenesis,3,4,5,6 the role of environmental insults in ASD development remains unclear.
According to the World Health Organization (WHO), mercury (Hg) is considered one of the top ten chemicals of major public health concern (https://www.who.int/news-room/fact-sheets/detail/mercury-and-health). Among the different forms of Hg, methylmercury (MeHg) is the most common form to which humans and animals are exposed.7 MeHg is a bioaccumulated environmental toxicant that can pass through the blood-brain barrier and the placenta by mimicking methionine to affect both the developing and mature CNS.8 Prenatal exposure to MeHg is associated with cognitive deficits and motor dysfunction in children, which has been observed in both human populations9,10 and rodent studies.11 However, it remains controversial whether prenatal MeHg exposure can lead to ASD onset,12,13 possibly because of the limitations of epidemiological studies as most of the reports are either cross-sectional or case-control studies,14 uncertainty associated with estimating exposure using different biomarkers,15 and multifactorial outcomes of MeHg exposure with varying dosages.16
Embryonic cerebral cortex development is vulnerable to environmental insults (such as drugs, alcohol, and environmental toxicants), potentially leading to neurodevelopmental disorders, including ASD.17,18 Although most work investigating the relationship between MeHg exposure and perturbed neurological function has been focused on cell apoptosis, oxidative stress, and mitochondrial dysfunction,19,20,21 the effects of prenatal low-dose MeHg exposure on cortical development and its underlying mechanism remain elusive. Several studies have shown that passaging neural progenitor cells (NPCs) are highly sensitive to MeHg.22,23,24 Our recent work, using a culture model that mimics in vivo cortical development in the dish,25,26 reveals that MeHg treatments at sub-micromolar (nanomolar) levels induced premature neuronal differentiation of cultured embryonic cortical precursors without causing cell death.16 These findings suggest that MeHg, when given at non-apoptotic doses in vivo, might perturb cortical neurogenesis in the fetal period which could have life-long impacts on neuro-performance. Of interest, overproduction of neurons has been found in mouse models of ASD,27,28 ASD organoid models,28,29 and human ASD patients.30,31
cAMP-response element-binding protein (CREB) is a key transcription factor that plays an important role in neurogenesis, synaptogenesis, and cell survival.32,33,34,35 Of interest, human neuroblastoma cells treated with MeHg at nanomolar concentrations showed a significant increase in CREB phosphorylation at Ser133 (pS133-CREB), an activated form of CREB, which protected the cells against apoptosis.36 We dosed cultured embryonic cortical precursors with similar nanomolar concentrations of MeHg and found that it promoted neuronal differentiation of the cortical precursors without causing cell death.16 These findings suggest that MeHg might activate CREB in cortical precursors to promote neuronal differentiation. Our earlier work showed that metformin, an FDA-approved drug, was able to promote the differentiation of cortical precursors through the phosphorylation of CREB-binding protein (CBP), a co-activator of CREB, at serine 436 (p-S436 CBP).37 Other work has also shown that p-S133 CREB and p-S436 CBP are repulsive of each other and cannot co-exist in the brain and the liver.38,39 Therefore, we propose that metformin treatment could reverse the premature neuronal differentiation caused by low-dose MeHg by initiating protein-protein repulsion between CREB and CBP.
Here, we tested this hypothesis using embryonic cortical precursor cell culture and single-cell RNA sequencing (scRNA-seq) analysis of embryonic cortex tissue. We showed that non-apoptotic low-dose MeHg exposure enhanced CREB activity to promote differentiation of cultured embryonic cortical precursors. scRNA-seq analysis revealed that prenatal exposure to low-dose MeHg resulted in cortical radial glial precursors (RGPs) favoring asymmetric differentiation to directly overproduce cortical neurons, thereby omitting the intermediate progenitor stage. In addition, we found that metformin treatment both in culture and in vivo could restore the premature neurogenesis caused by low-dose MeHg exposure through repulsion between CREB and CBP. Importantly, prenatal exposure to low-dose MeHg can induce ASD-like behaviors including impaired communication, social interaction deficits, repetitive behavior, and cognitive inflexibility. Our study raises awareness of the life-long outcomes caused by low-dose MeHg on fetal neurodevelopment and provides insight into its underlying cellular and molecular mechanisms. More importantly, we have identified a possible therapeutic target for low-dose prenatal MeHg exposure.
Results
Prenatal treatment with 0.2ppm MeHg causes ASD-like behavior in young adult mice
To determine the long-term behavioral consequences of prenatal exposure to low-dose MeHg, we administered 0 and 0.2ppm MeHg (an amount at least 10 times lower than previous in vivo studies40 via drinking water from E0 to birth). At postnatal day 7 (P7) and P9 we probed for impaired ultrasonic vocalizations (USV) and at 2 months old, a battery of behavioral tests was performed on the offspring to probe for additional ASD-related behaviors (Figure 1A).
Figure 1.

Prenatal treatment with 0.2ppm MeHg causes ASD-like behaviour in young adult mice
(A) Schematic of prenatal exposure to MeHg in pregnant mice, created with BioRender.com. 0ppm (control) and 0.2ppm MeHg was administered through drinking water to pregnant mice starting at E0 until birth. (B–D) USV analysis was conducted on P7 and P9 pups.
(B) Representative spectrograms of P9 control and MeHg-treated pups.
(C and D) The total number of calls, n = 11 animals/group, Mann–Whitney U test, ∗p < 0.05, and (D) average call length per call was analyzed, n = 10 animals/group, Student’s t test, ∗p < 0.05.
(E and F) The Marble Burying test assessed the number of marbles buried after 30 min interaction time. (E) Representative images for both control and 0.2ppm MeHg. (F) Quantitative analysis of number of marbles buried in the cage. n = 9–15 animals/group, Student’s t test, ∗∗p < 0.01.
(G) The adult social interaction test assessed the amount of time that testing mice spent in the interaction zone with either an empty wire-mesh cage or the cage with a novel mouse of the same strain, sex, age.
(H) The time spent in interaction zone for female mice, n = 12 animals/group, Student’s t test, ∗∗p < 0.01, ns (non-significant).
(I and J) Acquisition of the initial platform location in the MWM task was performed across 4 days of training with latency to reach the platform as a measurement of learning. n = 15–16 animals/group.
(K and L) After acquisition, the platform was removed at day 11, and mice were given a 60 s probe trial. (K) Representative heat maps from each group shown in L. (L) The percentage of time spent in the target quadrant between control and 0.2ppm MeHg was analyzed as a measurement of memory. n = 15–16 animals/group, Student’s t test, ns (non-significant).
(M and N) Following the probe, the platform was moved to the opposite quadrant. Acquisition of the new platform location in the MWM task was performed across 2 days of training with latency to reach the platform as a measurement of learning. n = 15 animals/group.
(O and P) After acquisition, the platform was removed at day 14, and mice were given a 60-s probe trial. (O) Representative heat maps from each group shown in P. (P) The percentage of time spent in the new target quadrant and old target quadrant was analyzed as a measurement of reversal memory. n = 15 animals/group, Student’s t test, ∗p < 0.05, ns (non-significant). Error bars indicate the standard error of the mean (SEM). See also Figure S1.
The triad of impairment makes up ASD diagnosis, and it consists of restrictive repetitive behaviors and deficits in social interaction and communication.1 To probe for impaired communication that is typically observed in individuals with ASD, we assessed USVs at P7 and P9. We found that P9 pups following prenatal MeHg exposure exhibited a reduced number of USVs (Figures 1B and 1C) and decreased length per call (Figures 1B and 1D). This indicates that prenatal treatment with low-dose MeHg impairs communication at early postnatal. To probe for repetitive behaviors, we performed the Marble Burying test. Here, 20 marbles were arranged-on top of loosely packed woodchips, and the mice were given 30 min to interact with the environment. A marble was scored as buried if two-thirds of its surface area was covered by bedding41 (Figure 1E). We observed significantly more marbles buried by the 0.2ppm MeHg-treated mice (Figure 1F), with no changes in locomotion (Figures S1A and S1B) or anxiety (Figures S1C–S1E) measured using the open-field test. In addition, we performed the Nestlet shredding test, another task used to indicate repetitive behavior. Here, mice prenatally treated with either 0.2ppm MeHg or 0ppm MeHg (control) were given 1.5h to interact with one cotton nestlet in an empty cage. The MeHg-treated group showed a trend to increase the percentage of nestlet shredded at the end of 1.5h (Figure S1F). In combination, these tests reveal that prenatal MeHg exposure increases repetitive behaviors, a key indicator of ASD. To assess sociability, we performed the adult social interaction test (Figures 1G and 1H). The control females spent significantly more time in the interaction zone with the novel mouse relative to an empty cage. In contrast, the MeHg-treated females did not show a significant change in interest in the novel mouse (Figure 1H). MeHg-treated females also spent significantly less time interacting with the novel mouse when compared to control females (Figure S1G). This reveals that prenatal MeHg exposure reduces the sociability of mice, a key behavioral feature of ASD. Of interest, this test is highly sex-dependent, both control and MeHg-treated male mice did not show an increased interaction time with a novel mouse (Figure S1H). Morris water maze (MWM) is a behavioral task typically used to assess learning and spatial memory. It is based on the principle that mice are highly motivated to escape from the water. Following 4 days of acquisition training to a submerged platform in a pool of water (Figures 1I and 1J), the memory probe was performed on day 11 after removing the platform from the pool. Both the control and MeHg-treated mice showed no difference in time spent in the target quadrant (Figures 1K–1L), and a similar overall specificity of memory (Figures 1K andS1I–S1J). We then used reversal learning in MWM to examine cognitive flexibility. In reversal learning, the platform was moved from the initial target location (old target) to the opposite quadrant (new target). Acquisition to the novel location was performed over 2 days of training with latency to reach the platform as a measurement of learning (Figures 1M and 1N). The reversal probe was performed to measure cognitive flexibility on day 14. Here, the control mice showed a significant preference toward the new target location over the old target location, whereas the MeHg-treated mice failed to show significant quadrant preference in the new target zone (Figures 1O, 1P, and S1K–S1L). Although there was no significant difference in % of time spent in a new target quadrant if we directly compared the control group with the MeHg group, the precision of the new memory, particularly in the new target quadrant, was impaired in the MeHg-treated group, which reflects spatial cognitive inflexibility (Figures S1K and S1L). Overall, these results indicate that prenatal 0.2ppm MeHg exposure impairs communication, increases restrictive and repetitive behaviors, and decreases sociability for the grown-up off-springs. This is the first time low-dose MeHg has been correlated with ASD-like behaviors in mice.
To examine Hg accumulation in mice, we analyzed the feces, urine, and blood of all the mice undergoing behavioral testing at P22, P42, and at sacrifice (Figures S1M−S1P). We found that the highest concentration of Hg was detected in the feces at P22 (Figure S1N), which is consistent with the known route of MeHg excretion.42 Following behavioral testing, the mice were sacrificed, and major Hg target organs were assessed. Hg was detected in the liver and kidneys of MeHg-treated mice. In the liver, MeHg was roughly 6 times more concentrated than in control mice, and in the kidneys, MeHg was roughly 3 times more concentrated than in control mice (Figure S1Q). To determine the distribution of Hg in the brain following prenatal exposure, various brain regions were isolated for Hg analysis. We found that very little Hg was present in the adult brain of the prenatally exposed mice (Figure S1R), suggesting that the behavioral deficits observed in early adulthood are residual effects of aberrant embryonic neurodevelopment that is elicited by prenatal MeHg exposure.
Prenatal exposure to MeHg increases neuronal differentiation of cortical precursors at the expense of their proliferation in vivo
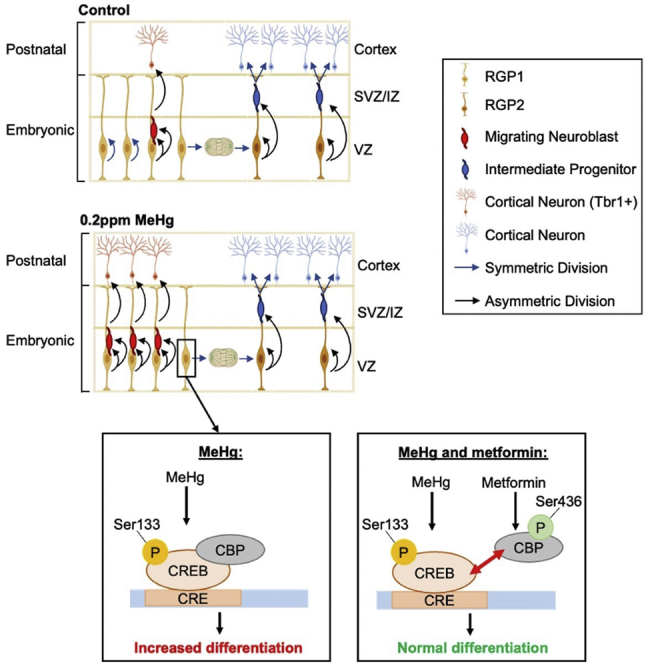
Our early culture work showed that a low-dose of MeHg promoted premature neuronal differentiation of E12 cortical precursors.16 Here, we further asked whether exposure to low-dose MeHg in pregnant mice can cause similar impacts on embryonic cortex development in vivo. To test this, we administered 0 and 0.2ppm MeHg via drinking water to pregnant mice from E0 until E15 and sacrificed the dams to collect E15 brain samples from embryos for immunohistochemical analysis (Figure 2A). We found that 0.2ppm MeHg significantly reduced the number and proportion of proliferating Ki67+/Pax6+ cortical precursors, also known as radial glial precursors (RGPs), in the ventricular zone (VZ) without alteration of the total number of Pax6+ cortical precursors (Figures 2B–2D and S2A). Although the number of Tbr2+ intermediate progenitors was not significantly changed by 0.2ppm MeHg exposure (Figures 2E and 2F), the number of DCX+ neuroblasts was robustly increased specifically in the VZ (Figures 2G, 2H, S2B, and S2C). Although we did not observe an increase in newborn neuron density within the cortical plate (CP), there was a significant increase in the thickness of the CP (Figures 2G, 2I, S2D, and S2E). We further assessed deep layer Tbr1+ mature cortical neurons in the CP and did not observe apparent changes in the proportion of Tbr1+ neurons over total live cells after 0.2ppm MeHg exposure (Figures 2J and 2K). We also reported that 0.2ppm MeHg did not increase apoptotic cell death in the E15 cortex in vivo (Figures S2F–S2G). Thus, prenatal exposure of 0.2ppm MeHg promotes premature differentiation of cortical precursors in the VZ to generate newborn neurons at the expense of their proliferation in vivo, without altering cell death.
Figure 2.

Prenatal exposure to MeHg increases neuronal differentiation of cortical precursors at the expense of their proliferation in vivo
(A) Schematic of prenatal exposure to MeHg in pregnant mice, created with BioRender.com. 0ppm (control) and 0.2ppm MeHg were administered through drinking water to pregnant mice starting at E0 until E15. At E15 brains were collected for immunohistochemistry.
(B, E) Images of E15 cerebral cortex sections from embryos receiving 0 and 0.2ppm MeHg treatment, immunostained for Pax6 (B, green), Ki67 (B, red) or Tbr2 (E, red) and counterstained for Hoechst (blue). Scale bar: 25 μm.
(C, D, and F) Quantitative analysis of the number and proportion of Ki67+/Pax6+ proliferating cortical precursors (C and D) and Tbr2+ intermediate progenitors (F) within VZ and SVZ/IZ, determined from sections similar to those shown in (B, E). n = 3 embryos/group, Student’s t test ∗p < 0.05.
(G) Images of E15 cerebral cortex sections from embryos receiving 0 and 0.2ppm MeHg treatment, immunostained for DCX (G, green) and counterstained for Hoechst (blue). Scale bar: 25 μm.
(H) Quantitative analysis of the number of DCX+ immature neurons within VZ, determined from sections similar to those shown in (G). n = 4 embryos/group, Student’s t test, ∗p < 0.05.
(I) Quantitative analysis of thickness for the CP was determined from coronal sections similar to those shown in (G). n = 3 embryos/group, Student’s t test, ∗∗p < 0.01.
(J) Images of E15 cerebral cortex sections from embryos receiving 0 and 0.2ppm MeHg treatment, immunostained for Tbr1 (red) and counterstained for Hoechst (blue). Scale bar: 25 μm.
(K) Quantitative analysis of the number of Tbr1+ deep layer mature neurons within CP, determined from sections similar to those shown in (J). n = 4 embryos/group. Error bars indicate the standard error of the mean (SEM). See also Figure S2.
Perinatal exposure to MeHg increases the genesis of deep layer cortical neurons in vivo
To ask whether prolonged exposure of 0.2ppm MeHg in pregnant mice through the perinatal stage from E0 to P7 has impacts on subventricular zone (SVZ) neural stem cell development and postnatal cortex development, we administered 0 and 0.2ppm MeHg via drinking water to pregnant mice starting from E0 to P7, at which point we sacrificed pups for immunohistochemical analysis and Hg measurements (Figures 3A and S3A). We measured Hg concentrations in the brains and found that P7 brains from the perinatal treatment of MeHg had a 2-fold higher Hg accumulation than E15 brains from the prenatal treatment of MeHg (Figure S3B). Immunohistochemical analysis showed that both proliferating Ki67+/Pax6+ cortical precursor and Tbr2+ intermediate progenitor populations within the SVZ in the P7 brain were not altered by 0.2ppm MeHg treatment (Figures 3B, 3C, and 3E–3G). However, the number of DCX+ neuroblasts within the SVZ was significantly increased after the 0.2ppm MeHg treatment (Figures 3D and 3H). We further examined deep and superficial layer cortical neurons and found that 0.2ppm MeHg treatment increased the total number of Tbr1+ deep layer cortical neurons throughout the cortex, with a robust increase, particularly in layer V (Figures 3I–3K). In contrast, the number of Satb2+ superficial layer cortical neurons was not altered throughout the cortex (Figures 3L–3N) by 0.2ppm MeHg treatment. In addition, 0.2ppm MeHg treatment did not alter the thickness of the cortex in the P7 brains (Figures S3C and S3D). In summary, perinatal exposure to 0.2ppm MeHg significantly increases postnatal neurogenesis without depleting the postnatal neural stem/progenitor cell pool and specifically enhances the genesis of deep layer cortical neurons.
Figure 3.

Perinatal exposure to MeHg increases the genesis of deep layer cortical neurons in vivo
(A) Schematic of perinatal exposure to MeHg in pregnant mice, created with BioRender.com. 0ppm (control) and 0.2ppm MeHg were administered through drinking water to pregnant mice starting at E0 until P7. At P7 brains were collected for immunohistochemistry.
(B–D) Images of P7 SVZ sections from pups receiving 0 and 0.2ppm MeHg treatment since E0, immunostained for Pax6 (B, green), Ki67 (B, red), Tbr2 (C, red) and DCX (D, green) and counterstained for Hoechst (blue). Scale bar: 50 μm.
(E–H) Quantitative analysis of the number and proportion of of Ki67+/Pax6+proliferating cortical precursors (E-F), the number of Tbr2+ intermediate progenitors (G) and DCX+ immature neurons (H) within SVZ of P7 brains, determined from sections similar to those shown in (B, C, D). n = 4 embryos/group, Student’s t test, ∗∗p < 0.01.
(I and L) Images of P7 cerebral cortex sections from pups receiving 0 and 0.2ppm MeHg treatment since E0, immunostained for Tbr1 (I, red) and Satb2 (L, red) and counterstained for Hoechst (blue). Scale bar: 50 μm.
(J, K, M, and N) Quantitative analysis of the number of Tbr1+ deep layer neurons (J-K) and Satb2+ superficial layer mature neurons (M−N) at all cortical neuron layers (layer I-layer VI), determined from sections similar to those shown in (I, L). n = 4 embryos/group, Student’s t test, ∗∗p < 0.01. Error bars indicate the standard error of the mean (SEM). See also Figure S3.
Prenatal exposure to MeHg results in cell trajectory alterations in E13.5 cortices
To understand the underlying cellular mechanisms that contribute to MeHg-induced neuronal differentiation during embryonic cortex development, we performed multiplexed scRNA-seq43 using cortex collected from E13.5 embryos that have been exposed to 0ppm or 0.2ppm MeHg as described above to profile dissected and dissociated E13.5 murine cortical cells (Figure 4A). In total, we collected 6 embryonic cortices from each group receiving 0ppm or 0.2ppm MeHg treatment. The dissociated cortical cells were randomized to be barcoded in four groups before being combined for the MULTI-seq (Figures 4A and S4A–S4F). Following demultiplexing, we transcriptionally profiled a total of 4,947 mouse cortical cells from the control group (0ppm MeHg) and 5460 cells from the MeHg group (0.2ppm) (Figure 4A). After initial processing (see experimental procedures), cells were clustered based on their expression profile, and annotation of cell types was based on established cell markers available from the published scRNA-seq dataset for embryonic cortex at E13.5-E14 age44,45 (Figures 4B–4D and S4G, Tables 1 and 2). A total of 10 clusters were identified: RGP1, RGP2, intermediate progenitors, immature neurons, mature neurons, interneurons, endothelial cells, layer I cortical neurons, pericytes, and a cluster of transitional cells (Figures 4B–4D). The transitional cell population did not highly express any known marker genes used to annotate specific cell types, but universally expressed feature genes shown in RGP1, RGP2, and immature neurons at minimal levels (Figure S4G) and was named to reflect this. Of interest, we observed that MeHg treatment increased the proportion of transitional cells (Figure 4E). Sub-clustering showed that the transitional cells were comprised of 4 subpopulations of cells: RGP1, immature/mature neurons, layer I cortical neurons, and pericytes (Figures 4F–4H). The composition of these cells suggests that the transitional cell population represents the asymmetrical division of RGP1 cells to directly generate cortical neurons. MeHg treatment increased the total population of transitional cells but did not alter the cell distribution within the 4 subpopulations of cell clusters (Figure 4I). To further examine asymmetric division within the transitional cell population we classified cells into three populations using canonical markers, IP+, RG+, or N+ (Tables 1 and 2). We focused on the percentage of cells in the transitional population that co-express two major cell-type markers. We showed that within the transitional population, roughly 30% of cells co-express marker genes for both RG+ and N+ (RG+/N+). This is the largest population of co-expressing cells, which confirms that the majority of differentiation that takes place in the transitional population is asymmetric (Figures S5A–S5D). We then removed the transitional cell population from the total cell population and found that only 1.5% of cells are RG+/N+, which confirms that asymmetric division is specific to the transitional population (Figures S5E–S5H).
Figure 4.

scRNA-seq characterization of the E13.5 cortex prenatally exposed to MeHg
(A) Schematic of prenatal exposure of MeHg to pregnant mice, created with BioRender.com. 0ppm (control) and 0.2ppm MeHg were administered through drinking water to pregnant mice starting at E0 until E13.5 at which point the dorsal cortical cells were collected for multiplexing. Two technical replicates from both 0 and 0.2ppm MeHg treated groups were tagged and pooled for multiplexing RNA-seq. Following demultiplexing, downstream analysis and trajectory inference were performed.
(B) Visualization cells from 0 to 0.2ppm MeHg groups after PCA and UMAP, colored by Seurat clustering and annotated by cell type.
(C) UMAP visualization of 0.2ppm MeHg (red) and 0ppm (blue) treatment groups.
(D) Visualization of the total cell population after PCA and UMAP, colored by expression of key marker genes (Mki67, Pax6, Neurod1, Bcl11b, Reln, GAD2, Clnd5, Rgs5).
(E) Proportion of cells in each cluster for 0 and 0.2ppm MeHg.
(F) Visualization of subclusters of the transitional cell population after PCA and UMAP, colored by Seurat clustering and annotated by cell type.
(G) UMAP visualization of 0.2ppm MeHg (red) and 0ppm (blue) treatment groups in transitional cell population subclusters.
(H) Heatmap of top ten differentially up-regulated genes over four clusters within the transitional cell population.
(I) Proportion of cells in each subcluster within the transitional cell population for both 0 and 0.2ppm MeHg. See also Figure S4 and Tables 1 and 2.
Table 1.
Annotations of each cell cluster used for UMAP visualization of the total cell population, along with the abundance of each cluster, and specific marker genes for each cluster
| Cluster # | Cell Type | % Abundance Control | % Abundance MeHg | Marker Genes |
|---|---|---|---|---|
| 1 | RGP1 | 16.33 | 15.64 | Top2a, Ube2c, Cenpf, Fabp7, Mki67 |
| 2 | RGP2 | 18.71 | 19.15 | Sox2, Pax6, Hes1, Hes5, Pcna, Mcm3/6, Fabp7 |
| 3 | Intermediate Progenitors | 11.84 | 11.19 | Eomes, Sstr2, Gadd45g, Mfap4 |
| 5 | Immature Neurons | 26.78 | 23.86 | Neurod1, Sox11, Neurod6, Dcx |
| 6 | Mature Neurons | 12.93 | 12.34 | Bcl11b, Sox5, Stmn2 |
| 7 | Layer 1 Neurons | 0.38 | 0.58 | Reln, Trp73, Lhx1, and Lhx5 |
| 8 | Interneurons | 2.62 | 3.55 | GAD1, GAD2, Sst, Dlx1, Dlx2 |
| 9 | Endothelial Cells | 0.8 | 0.69 | Igfbp7, Cldn5, Col4a1, Col4a2 Pecam1, Vwa1 |
| 10 | Pericytes | 0.1 | 0.32 | Igfbp7, Rgs5, PdgfrB |
Table 2.
Annotations of each cell cluster used for UMAP visualization of the transitional cell population, along with the abundance of each cluster, and specific marker genes for each cluster
| Cluster # | Cell Type | % Abundance Control | % Abundance MeHg | Marker Genes |
|---|---|---|---|---|
| 1 | RGP1 | 52.99 | 53.18 | Dbi, Fabp7, Vim, Cenpf, Ube2c |
| 2 | Immature/Mature Neurons | 44.01 | 43.76 | Tubb3, Sox11, Neurod6, Stmn2 |
| 3 | Layer 1 Neurons | 1.70 | 0.86 | Reln |
| 4 | Pericytes/Endothelial Cells | 1.28 | 2.17 | Igfbp7, Rgs5, Col4a1, Col4a2 |
We then conducted trajectory inference using RNA velocity from scVelo46 to study the full transcriptional dynamics of splicing kinetics. To do this, scVelo utilizes the relative abundance of nascent (unspliced) and mature (spliced) mRNA in each cell as an indicator of the future state of the cell. This method allowed us to describe the direction and speed with which cells transition between clusters. Of interest, RNA velocity revealed an altered cell lineage trajectory caused by MeHg exposure (Figures 5A–5C). In control (0ppm MeHg) cortical cells, the RGP1 population shows a 90% positive velocity directed toward RGP2, following symmetrical division, and then differentiating into intermediate progenitors, immature, and mature neurons (Figures 5A and 5B). In contrast, RGP1 cells in MeHg-exposed cortices showed approximately 50% positive velocity directed toward RGP2 that further differentiate into intermediate progenitors, and 50% negative velocity directed toward the transitional cells, which directly give rise to the population of immature neurons (Figures 5A–5C). These results provide evidence to support that prenatal MeHg exposure enhances the asymmetric division of RGP1 to generate neurons directly through the transitional cell stage.
Figure 5.

Prenatal exposure to MeHg results in cell trajectory alterations in E13.5 cortices
(A) Velocity vectors for total cell populations in both 0ppm (control) and 0.2ppm MeHg, visualized and calculated from RNA velocity using the stochastic model, projected onto UMAP visualizations.
(B) Enhanced image of velocity vectors in the RGP1 and RGP2 population for both 0 and 0.2ppm MeHg.
(C) Quantitative analysis of velocity vectors in RGP1 directed toward the RGP2 population between 45 and 180°. Quantitative analysis of velocity vectors in RGP1 directed toward the transitional cell population between 270 and 360°. Parameters used to differentiate vector direction.
(D) Pseudotime analysis for 0 and 0.2ppm MeHg.
(E and F) Phase portraits of marker genes (Pax6, Tubb3, and Tbr1) in 0ppm (E) and 0.2ppm MeHg (F), calculated from RNA velocity.
(G) Velocity vectors for transitional cell populations in both 0 and 0.2ppm MeHg treatment groups were visualized and calculated from RNA velocity using the stochastic model, projected onto UMAP visualizations.
(H) Pseudotime analysis for the 0 and 0.2ppm MeHg treatments within the transitional cell population.
(I and J) Phase portraits of marker genes (Pax6, Tubb3, and Tbr1) in the transitional cell population for 0ppm (I) and 0.2ppm MeHg (J) treatments, calculated from RNA velocity. See also Figure S5.
We further utilized RNA velocity to order cells along a trajectory that corresponds to their underlying biological process. Pseudotime analysis of the total cell population revealed that clusters including RGP1, RGP2, and the transitional cells in MeHg-exposed cortical cells were in a much later developmental stage than those in control cortical cells (Figure 5D), suggesting that prenatal MeHg exposure may accelerate the differentiation process. Next, we used phase portraits generated through scVelo to visualize RNA velocity for specific key cortical development marker genes, Pax6, a marker for cortical precursors; βIII-tubulin (Tubb3), a marker for newborn neurons; and Tbr1, a marker for deep layer cortical neurons. A gene with positive RNA velocity is predicted to be up-regulated. In opposition, a negative velocity indicates that the gene is predicted to be down-regulated.46 In the total cell population, we observed that Pax6 is downregulated in MeHg-exposed RGP1, RGP2, and intermediate progenitors compared to the control. Tubb3 showed a strong trend toward upregulation in the MeHg-exposed RGP2 population. A more obvious downregulated state for Tubb3 was observed in the MeHg-exposed transitional cells, immature neurons, and mature neurons relative to the control. Of interest, corresponding to downregulated Tubb3 gene, we found that the Tbr1 gene showed a more up-regulated state in MeHg-exposed transitional cells, immature neurons, and mature neurons relative to the control (Figures 5E and 5F). These specific alterations in the key marker genes support our pseudotime analysis results, indicating that MeHg accelerates the neuronal differentiation process.
We then performed RNA velocity and pseudotime analysis on the sub-populated transitional cells as described above. We showed that MeHg also advanced the differentiation process of transitional cells from RGPs toward immature neurons (Figures 5G and 5H). Similar to the total cell population, we confirmed this by visualizing RNA velocity for specific key cortical development marker genes, Pax6, Tubb3, and Tbr1 in the transitional cells. Pax6 trended toward downregulation in both RGP1 and immature neuron subpopulations in MeHg-exposed transitional cells relative to that in the control group. In contrast, the Tubb3 gene trended toward upregulation in MeHg-exposed transitional cells. Of interest, the Tbr1 gene was not expressed in the control, whereas the Tbr1 gene was present in the MeHg-exposed transitional cells (Figures 5I and 5J). This result provides further evidence that prenatal MeHg exposure results in premature neuronal differentiation of RGP1 through an increased transitional cell population.
MeHg exposure stimulates CREB activity specifically in RGPs
To test whether CREB activity plays a role in MeHg-induced neuronal differentiation, we investigated the regulation of downstream CREB-targeted genes, including Jund, Fos, Dusp1, and Egr1.47,48 Both Jund and Fos were significantly up-regulated in the RGP1 from the MeHg-exposed cortical cells (Figures 6A and 6B), but not in the other cell clusters (Figures S6A and S6B). To further test whether MeHg can increase CREB activity in cortical precursors, we performed a culture experiment using cortical precursors isolated from E11-12 embryos and cultured these cortical precursors in the absence and presence of 250 nM MeHg for 4 hours (Figure 6C). Western blotting analysis showed that p-S133 CREB was significantly up-regulated in the cortical precursors exposed to 250 nM MeHg, a concentration used in our previous work16 (Figures 6D–6F). Of interest, we also observed that MeHg significantly reduced the phosphorylation of atypical protein kinase C (aPKC) at threonine 410/403 (pT410/403-aPKC), an activated form of aPKC (Figures 6G–6I). Also, MeHg affected CREB and aPKC phosphorylation/activation without altering total protein expression. These results together with those reported in earlier works37,38,39 support the model (Figure S6C) that MeHg and metformin exposure alone increases p-S133 CREB and p-S436 CBP, respectively, to enhance neuronal differentiation of cortical precursors. On the other hand, co-treatment of metformin and MeHg can reverse the premature differentiation caused by MeHg, by initiating repulsion between p-S133 CREB and p-S436 CBP. To test this model, we used a PLA to assess intranuclear interactions between CBP and p-S133 CREB at the single-cell level. Our results showed that the interactions between CBP and p-S133 CREB were significantly enhanced by 250 nM MeHg treatment alone, whereas co-treatment with metformin eliminated the increased interactions (Figures 6J and 6K). Thus, metformin, an FDA-approved drug, has the potential to reverse MeHg-induced neuronal differentiation through repulsive interaction between CREB and CBP in cultured cortical precursors.
Figure 6.

Prenatal MeHg exposure stimulates CREB activity specifically in RGPs
(A) Dot plot of downstream CREB target genes (Jund, Fos, Dusp1, Egr1) and their expression from scRNA-seq data in RGP1 and RGP2 cell populations for control (0ppm) and 0.2ppm MeHg groups.
(B) Violin plot from scRNA-seq showing Fos (upper panels) and Jund (lower panels) expressions in RGP1 and RGP2 cell populations in control (0ppm) and 0.2ppm MeHg treatment groups, n > 800 cells for each group, Mann–Whitney U test, ∗∗∗∗p < 0.0001, ns (non-significant).
(C) Schematic of cortical percussors isolated from E11-12 CD1 mice, created with BioRender.com. Cells were exposed to 0 nM (control) or 250 nM MeHg with no metformin or metformin (500 μM) for 4 h, at which point they were lysed for western blot or PLA analysis.
(D–I) Western blot analysis of E12 cortical precursors cultured for 4 hours in the absence or presence of 250 nM MeHg. (D, G) Blots images were probed for pS133-CREB (D), pT410/403-aPKC (G), total CREB (D), total aPKC (G) and GAPDH (D, G) as a loading control. (E-F) Quantitative analysis of pS133-CREB (E) and total CREB (F) over GAPDH. n = 5 independent experiments, Student’s t test, ∗p < 0.05, ns (non-significant) (H-I) Quantitative analysis of pT410/403-aPKC(H) and aPKC (I) over GAPDH, n = 4 independent experiments, Student’s t test, ∗p < 0.05, ns (non-significant).
(J) Images of PLA+ interactions (lower panels, red) between pS133-CREB and CBP, counterstained with Hoechst (blue) in cultured cortical precursors of four treatment groups i) control (0ppm MeHg and 0 μM metformin), ii) 250 nM MeHg, iii) 500 μM metformin, and iv) co-treatment of 500 μM metformin +250 nM MeHg, scale bar: 5 μm. Images of negative control PLA (upper panels) in the absence of anti-pS133-CREB and anti-CBP (upper left) or absence of PLA probes (upper right).
(K) Quantitative analysis of pS133- CREB and CBP interaction signals (red) within nuclei (blue). n = 100 cells/group, n = 4 independent experiments, two-way ANOVA (group × metformin interaction F(1, 396) = 11.34, p = 0.0008, group F(1, 396) = 1.463, p = 0.2272, metformin F(1, 396) = 25.01, p < 0.0001, n = 400) with post hoc test, ∗p < 0.05, ∗∗p < 0.01. Error bars indicate the standard error of the mean (SEM). See also Figure S6.
Metformin eliminates premature neuronal differentiation caused by MeHg exposure
To test whether metformin can eliminate MeHg-induced neuronal differentiation, we first conducted E11-E12 cortical precursor culture for 3 days in the absence and presence of 250 nM MeHg,16 and with or without 500 μM metformin37 (Figure 7A). Treatment with 250 nM MeHg alone increased cortical precursor differentiation, manifested by an increased percentage of newborn neurons (βIII-tubulin+) and a reduced proportion of cortical precursors (Pax6+ and Sox2+) in culture (Figures 7B–7F). In addition, MeHg treatment reduced the proliferation of cortical precursors (Ki67+) (Figure 7G). Similarly, treatment with 500 μM metformin alone increased the percentage of newborn neurons (βIII-tubulin+) and reduced the percentage of cortical precursors (Figures 7B–7F), marked by Pax6 and Sox2, as previously reported.37 Of interest, the co-treatment with both 250 nM MeHg and 500 μM metformin reversed the increased percentage of newborn neurons, as well as the reduced cortical precursor population and proliferation caused by MeHg alone (Figures 7B–7G).
Figure 7.

Metformin eliminates premature neuronal differentiation caused by MeHg exposure
(A) Schematic of cortical percussor isolated from E11-12 CD1 mice, created with BioRender.com. Cells were exposed to four conditions: i) control (0ppm MeHg+0 μM metformin), ii) 250 nM MeHg, iii) 500 μM metformin, iv) co-treatment of 250 nM MeHg +500 μM metformin for 3 days, followed by immunocytochemical analysis.
(B and E) Images of cortical percussors after 3 days in culture treated with four conditions mentioned in (A), immunostained for Pax6 (B, red), βIII-tubulin (B, green), Ki67 (E, green), Sox2 (E, red) and counterstained for Hoechst (blue). Scale bar: 30 μm.
(C) Quantitative analysis of βIII-tubulin+ cells, n = 6 independent experiments, two-way ANOVA(group × metformin interaction F(1, 20) = 15.28, p = 0.0009, group F(1, 20) = 0.3370, p = 0.5680, metformin F(1, 20) = 0.09295, p = 0.7636, n = 24).
(D) Quantitative analysis of Pax6+ cells, n = 6 independent experiments, two-way ANOVA(group × metformin interaction F(1, 20) = 13.99, p = 0.0013, group F(1, 20) = 6.652, p = 0.0179, metformin F(1, 20) = 0.04803, p = 0.8287, n = 24).
(F) Quantitative analysis of Sox2+ cells, n = 3 independent experiments, two-way ANOVA(group × metformin interaction F(1, 8) = 10.05, p = 0.0132, group F(1, 8) = 7.171, p = 0.0280, metformin F(1, 8) = 0.0002320, p = 0.9882, n = 12).
(G) Quantitative analysis of Ki67+ cells. n = 3 independent experiments, two-way ANOVA(group × metformin interaction F(1, 8) = 12.89, p = 0.0071, group F(1, 8) = 3.335, p = 0.1053, metformin F(1, 8) = 0.2942, p = 0 0.6023, n = 12) with post hoc test, ∗∗p < 0.01, ∗p < 0.05, ns (non-significant).
(H) Schematic of prenatal exposure of MeHg to pregnant mice, created with BioRender.com. Pregnant mice were exposed to four conditions: i) control (0ppm MeHg + 0 mg metformin, ii) 0.2ppm MeHg, iii) 4 mg/ml metformin, iv) 0.2ppm MeHg + 4 mg/mL metformin. Treatment was administered through drinking water to pregnant mice starting at E0 until E15. At E15 brains were collected for immunohistochemistry.
(I, L, N) Images of E15 cerebral cortex sections from embryos receiving four condition treatments mentioned in (H), immunostained for Pax6 (I, green), Ki67 (I, red) or Sox2 (L, green), Mcm2 (L, red), or DCX (N, green) and counterstained for Hoechst (blue). Scale bar: 20 μm.
(J and K) Quantitative analysis of the number and proportion of Pax6+/Ki67+ proliferating cortical precursors in the VZ. J: two-way ANOVA(group × metformin interaction F(1, 12) = 4.060, p = 0.0669, group F(1, 12) = 5.253, p = 0.0408, metformin F(1, 12) = 6.156, p = 0.0289, n = 16). K: two-way ANOVA(group × metformin interaction F(1, 12) = 6.461, p = 0.0259, group F(1, 12) = 2.961, p = 0.1109, metformin F(1, 12) = 20.79, p = 0.0007, n = 16) with post hoc test, ∗p < 0.05, ns (non-significant).
(M) Quantitative analysis of the number of Sox2+/Mcm2+proliferating radial glial cells within VZ and SVZ/IZ, two-way ANOVA(group × metformin interaction F(1, 14) = 3.167, p = 0.0969, group F(1, 14) = 4.683, p = 0.0482, metformin F(1, 14) = 1.379, p = 0.2599, n = 18) with post hoc test, ∗p < 0.05, ns (non-significant).
(O) Quantitative analysis of the number of DCX+ cells in the VZ, two-way ANOVA(group × metformin interaction F(1, 14) = 4.817, p = 0.0455, group F(1, 14) = 4.858, p = 0.0448, metformin F(1, 14) = 0.1290, p = 0.7248, n = 18) with post hoc test, ∗p < 0.05, ns (non-significant). These quantifications (J-K, M, O) were determined from cortical sections similar to those shown in (I, L, N). n = 4–5 embryos per group. Error bars indicate the standard error of the mean (SEM). See also Figure S7.
Second, we performed in vivo experiments to test whether metformin (4 mg/ml) co-treatment with 0.2ppm MeHg via drinking water from E0 to E15 can eliminate MeHg-induced premature neuronal differentiation (Figure 7H). We measured Hg levels in fetal brains to confirm that E15 brains from the MeHg treatment alone group and a group co-treated with MeHg, and metformin have a similar level of Hg accumulation within the brains (Figures S7A and S7B). These results support that metformin treatment via drinking water does not affect Hg accumulation in the fetal brains but directly counteracts MeHg-perturbed cortical development. We examined the number of Pax6+/Ki67+ and Sox2+/Mcm2+ proliferating cortical precursors. We found that co-treatment of MeHg with metformin restored MeHg-reduced proliferating cortical precursors, marked by Pax6+/Ki67+ and Sox2+/Mcm2+ populations (Figures 7I–7M, S7C, and S7D). Furthermore, co-treatment with metformin and MeHg erased MeHg-increased DCX+ immature neurons within the VZ (Figures 7N–7O). Thus, metformin treatment was able to eliminate the premature neuronal differentiation caused by MeHg exposure. We then measured the cortical thickness at the VZ, SVZ/IZ, and CP in all four groups. Of interest, we observed that MeHg alone group showed a significant increase in the thickness of the CP relative to the control group. At the same time, co-treatment with metformin and MeHg exhibited a similar thickness of CP as the control group (Figures S7E and S7F).
Discussion
Currently, ASD prevalence is increasing because of a variety of factors including enhanced awareness and improved screening. However, the etiology of ASD remains largely unknown because there is extreme variability in the appearance, severity of symptoms, and the underlying cause of onset.49 The core features of ASD symptoms are defined as defects in communication, sociability, and restrictive, repetitive behaviors and interests.1 Increasing evidence shows that ASD is frequently associated with cerebral overgrowth,50 and enhanced neuronal production.27,28,29,30,51 Recent work from ASD patient-derived organoid models reveals aberrant overproduction of deep-layer neurons and a thicker immature cortex.29 Thus, our findings that prenatal MeHg-promoted deep-layer cortical neurogenesis could potentially contribute to the onset of ASD-like behaviors. Our findings also highlight the impact of low-dose prenatal MeHg exposure on aberrant cerebral cortex development and susceptibility to ASD onset.
This study is the first to show that prenatal low-dose MeHg exposure triggers ASD-like behaviors in a rodent model. Individuals with ASD experience deficits in verbal language. To test this in a rodent model we performed USV analysis at early postnatal. Mouse pups emit calls during their first two weeks of life when separated from their mother and littermates.52 We found that mice prenatally exposed to low-dose MeHg vocalized significantly less at P9 than control mice. This is consistent with other genetic mouse models of ASD53,54,55 and other studies which show a relationship between prenatal MeHg and changes in USVs.56 Children with ASD typically perform repetitive behaviors such as tapping, hand flapping, and rocking.1 To test this in mice, we performed the Marble Burying test and found that mice prenatally treated with low-dose MeHg buried significantly more marbles than control mice. Another key component of ASD is poor sociability, which we also observed in MeHg-treated mice using the adult social interaction test. The third common characteristic in individuals with ASD is rigidity in routine and interests. To test this, we used the MWM test. This behavioral task is typically used to assess learning and spatial memory; however, we utilized the reversal learning/memory component of MWM to specifically examine cognitive rigidity. Here, cognitive rigidity was measured by the inability to alter target zone preference which has been shown in other ASD mouse models.57 We found that mice prenatally treated with low-dose MeHg showed poor reversal memory similar to genetic ASD mouse models. Overall, we have demonstrated the three key components of ASD within MeHg-treated mice. Of interest, recent studies have shown that MeHg exposure to high-dose MeHg in adulthood exasperates the effects of a genetic ASD model (BTBR T + Itpr3tf/J (BTBR) mouse model).58,59 Our study provides further evidence for the potential link between ASD and MeHg.
Embryonic RGPs are known to perform asymmetric division to produce intermediate progenitor/cortical neurons or symmetric division to expand the cortical precursor cell pool.60 We used our scRNA-seq dataset from E13.5 cortices to identify a cluster of transitional cell populations in which RGPs undergo asymmetrical division to directly produce cortical neurons. This provides evidence at the single-cell level that embryonic RGPs can take two trajectories during cortical development, either symmetric division to expand the cortical precursor pool that ultimately produces the sequential generation of intermediate progenitors, immature neurons, and mature neurons, or asymmetric division to directly generate cortical neurons through the transitional cell population. This phenomenon was also exhibited in human neocortical development in mid-gestation disclosed by a scRNA-seq data.61 Importantly, we found that fetal MeHg exposure can alter RGP trajectory, prone to a transitional cell state. This accelerates the differentiation process by increasing asymmetrical division of cortical precursors to directly generate cortical neurons. This process was further confirmed by our immunohistochemical analysis showing that prenatal MeHg exposure significantly increased the number of DCX+ neuroblasts, specifically in the VZ, at the expense of proliferating Pax6+ cortical precursors.
This study demonstrated an underlying cellular mechanism that mediates MeHg-induced premature neuronal differentiation and showed that CREB activation was an essential molecular mechanism to modulate MeHg-induced neuronal differentiation of embryonic RGPs. CREB is a key transcription factor that plays a multifaceted role in survival, proliferation, and differentiation in the different neurogenic zones depending on the stage of brain development.62,63,64 Phosphorylation at Ser133 constitutes the rate-limiting step in the activation of CREB.65 Intriguingly, it has been shown that p-S133 CREB is increased in the embryonic cortex during neurogenesis and is constitutively expressed in immature neurons in the adult neurogenic zones.33,34,64,66 Here, our scRNA-seq results showed that direct CREB-targeted genes, Jund and Fos, were specifically up-regulated in the RGP1 cell population but not in other neural cell populations following prenatal MeHg exposure. These results suggest that MeHg-induced CREB activation in RGP1 may initiate the process that alters their trajectory toward the transitional cell population to generate cortical neurons. This process was further confirmed using our E12 cortical precursor cell culture model, showing that MeHg significantly increased p-S133 CREB level and promoted premature neuronal differentiation at the expense of cortical precursor proliferation. We conducted a series of experiments to understand the role of p-S133-driven CREB activity in modulating MeHg-induced premature differentiation, and to identify a potential therapeutic strategy to reverse the developmental defects. We used the knowledge obtained from early work on metformin37,38,39 to propose the model that metformin treatment can eliminate the premature differentiation caused by MeHg, by initiating repulsion between p-S133 CREB and p-S436 CBP. We have successfully validated this model using results from both cell culture and in vivo studies. Therefore, the potential use of metformin as a therapeutic treatment for MeHg-induced neurotoxicity may be explored.
MeHg is a potent neurotoxicant that poses health risks to the global population.67 There is consistent epidemiological evidence showing that low levels of prenatal MeHg exposure may cause neuro-cognitive effects during early childhood (3–7 years),68 youth,69 and/or adulthood.70 It has been estimated that the accumulated health effects associated with Hg exposure from 2010 to 2050 are projected to be $19 trillion (95% confidence interval: 4.7–54) (2020 USD).71 Our findings on MeHg-perturbed cortical development open a new avenue to rethink how low-dose fetal exposure to MeHg through moderate consumption of MeHg-contaminated fish can impair fetal brain development, potentially leading to neural developmental disorders. The results emphasize the importance of managing the risk of MeHg exposure by controlling Hg emissions through international treaties like the Minamata Convention (http://www.mercuryconvention.org/), or by issuing regional fish consumption advice to sensitive populations, e.g. Government of Canada, 2019 (https://www.canada.ca/en/health-canada/services/food-nutrition/food-safety/chemical-contaminants/environmental-contaminants/mercury/mercury-fish.html) and U.S. FDA (https://www.fda.gov/food/consumers/advice-about-eating-fish).
Overall, we put forward four major conclusions. First, prenatal low-dose MeHg exposure triggers ASD-like behaviors including impaired communication, reduced sociability, and increasingly restrictive and repetitive behaviors in young adult mice. Second, we show that prenatal MeHg exposure in vivo enhances neuronal differentiation of cortical precursors and increases cortical thickness. Third, we discover the underlying cellular and molecular mechanisms that contribute to MeHg-induced premature neuronal differentiation. Finally, we demonstrate that metformin treatment can eliminate the MeHg-induced premature neuronal differentiation of cortical precursors both in culture and in vivo and that metformin restores the MeHg-increased thickness of the cortex. These findings reveal the long-term consequences of low-dose prenatal MeHg exposure, the underlying cellular and molecular mechanisms contributing to MeHg-induced premature neuronal differentiation, and a potential therapeutic strategy to reverse the developmental defects.
Limitations of the study
Results from rodent studies are inherently limited in extrapolating to the human condition. It remains unclear how low-dose MeHg affects human embryonic brain development. Future human brain organoid models will be implemented to study the impact of low-dose MeHg on human cerebral cortex development. Electrophysiological recordings will be employed as well in human brain organoid models to understand aberrant neural development caused by low-dose MeHg exposure.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse anti-βIII-tubulin | BioLegend | Cat#801201; RRID:AB_2313773 |
| rabbit anti-Sox2 | Millipore | Cat#AB5603; RRID:AB_2286686 |
| mouse anti-Ki67 | Abcam | Cat#ab15580; RRID:AB_443209 |
| rabbit anti-Pax6 | Biolegend | Cat#901301; RRID:AB_2565003 |
| rabbit anti-Tbr2 | Abcam | Cat#ab23345; RRID:AB_778267 |
| mouse anti-Ki67 | BD Pharmingen | Cat#550609; RRID:AB_393778 |
| rabbit anti-Mcm2 | Abcam | Cat#ab4461; RRID:AB_304470 |
| mouse anti-Sox2 | Abcam | Cat#ab79351; RRID:AB_10710406 |
| goat anti-DCX | Santa Cruz biotechnology | Cat#sc-8066; RRID:AB_2088494 |
| rabbit anti-Tbr1 | Abcam | Cat#ab31940; RRID:AB_2200219 |
| mouse anti-Satb2 | Abcam | Cat#ab51502, RRID:AB_882455 |
| donkey anti-rabbit Alexa Fluor 555 | Thermo Fisher Scientific | Cat#A-31572; RRID:AB_162543 |
| goat anti-mouse Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-32723; RRID:AB_2633275 |
| goat anti-mouse Alexa Fluor 555 | Thermo Fisher Scientific | Cat#A-21422; RRID:AB_2535844 |
| donkey anti-rabbit Alexa Fluor 647 | Thermo Fisher Scientific | Cat#A-31573; RRID:AB_2536183 |
| goat anti-mouse Alexa Fluor 647 | Thermo Fisher Scientific | Cat#A-32728; RRID:AB_2633277 |
| donkey anti-goat Alexa Fluor 647 | Thermo Fisher Scientific | Cat#A-21447; RRID:AB_141844 |
| Hoechst 33342 | Cell Signaling | Cat#4082; RRID:AB_10626776 |
| rabbit anti-p-CREB (S133) | Cell Signaling | Cat#9198S; RRID:AB_2561044 |
| mouse anti-CBP | Santa Cruz Biotechnology | Cat#sc-7300; RRID:AB_626817 |
| rabbit anti-paPKCζ/ι (T410/403) | Cell Signaling | Cat#9378; RRID:AB_2168217 |
| mouse anti-CREB | Cell Signaling | Cat#9104S; RRID:AB_490881 |
| mouse anti-aPKCζ/ι | BD Pharmingen | Cat#610175; RRID:AB_397574 |
| HRP-conjugated goat anti-mouse | Cell Signaling | Cat#7076; RRID:AB_330924 |
| HRP-conjugated goat anti-rabbit | Cell Signaling | Cat# 7074; RRID:AB_2099233 |
| Chemicals, peptides, and recombinant proteins | ||
| 1000ppm MeHg(II)Cl | Alfa Aesar | Cat#33553 |
| 1,1-Dimethylbiguanide hydrochloride | Sigma-Aldrich | Cat#D150959 |
| Metformin hydrochloride | Sigma-Aldrich | Cat#PHR1084 |
| Critical commercial assays | ||
| Proximity ligation assay | Sigma-Aldrich | Cat#DUO92101 |
| In Situ Cell Death Detection Kit | Sigma-Aldrich | Cat#11684817910 |
| Deposited data | ||
| scRNA-seq datasets | This paper | GEO: GSE178929 |
| Published code | This paper | Zenodo: https://doi.org/10.5281/zenodo.7468729 |
| Experimental models: Organisms/strains | ||
| CD-1® IGS Mouse | Charles River | Crl: CD1(ICR) |
| C57BL/6J mouse (JAX™) | Jacksons Lab | JAX: 000664 |
| Software and algorithms | ||
| Image J | NIH | https://imagej.nih.gov/ij/download.html |
| Zeiss Axiovision | Carl Zeiss Microscopy | https://carl-zeiss-vision-axiovision-viewer.software.informer.com/4.8/ |
| UltraVox XT | Noldus | https://www.noldus.com/ultravox-xt |
| Ethovision XT | Noldus | https://www.noldus.com/ethovision-xt |
| R studio | Posit | https://posit.co/ |
| Pycharm | Jetbrains | https://www.jetbrains.com/pycharm/ |
| Graphpad | Prism | https://www.graphpad.com/scientific-software/prism/ |
Resource availability
Lead contact
Correspondence should be addressed to the lead contact, Jing Wang, jiwang@ohri.ca.
Materials availability
This study did not generate new unique reagents. The materials underlying this article will be shared upon reasonable request to the lead contact, Jing Wang.
Experimental model and subject details
Animals
All animal use was approved by the Animal Care Committee of the University of Ottawa in accordance with the Canadian Council of Animal Care policies. CD-1 mice were purchased from Charles River and C57BL/6J mouse were bred in house. CD-1 mice were specifically used to study embryonic cortical development because they produce large litters. However, CD-1 mice go blind with age, therefore, C57BL/6J mice were used for all subsequent behavioural testing. All mice were maintained on a 12h light/12 hour dark cycle with ad libitum access to food and water. Experimental animals were kept in both group and single housed cages dependent on litter size. The weight of early postanal mice ranged from 3-6g. The weights of adult C57BL/6J mice ranged from 15-30g. Weights of embryonic mice were not reported. Both male and female mice were used for all tests. Sex differences of C57BL/6J mice used during behavioural testing were assessed and notable sex differences were reported. The sex of the experimental animals used during embryo and early postnatal were not reported.
Primary cell culture
Primary cortical precursors were obtained from E11-12 cortices dissected from CD-1 mice (Charles River Laboratories) as previously described.26 Briefly, embryos were transferred to ice-cold Hanks’ balanced salt solution (HBSS) (cat#14175103, Fisher Scientific), and the cerebral cortices were isolated from the brain after the meninges were removed. The cortical tissue was mechanically triturated with a plastic pipette and seeded on coverslips in a 24-well plate or directly into a 6-well plate (Thermo Fisher Scientific), both pre-coated with 15% poly-L-ornithine (PLO) (cat#72302, Sigma-Aldrich) and 5% laminin (cat#CB40232, Thermo Fisher Scientific). For immunocytochemical experiments, cells were plated in a 24-well plate at a density of 200,000 cells/ml. For western blotting experiments, 1,000,000 cells were seeded in each well of a 6-well plate. The cortical precursors were cultured in a neurobasal medium (cat#21103049, Thermo Fisher Scientific) containing 500uM GlutaMAX supplement (cat#35050061, Thermo Fisher Scientific), 2% B27 supplement (cat#17504044, Thermo Fisher Scientific), 1% penicillin-streptomycin (cat#15140122, Thermo Fisher Scientific), and 40 ng/mL FGF2 (cat#10018B, PeproTech). The sex of cells is not reported as cell isolation occurred in embryo.
Method details
Experimental treatments
To study the long-term consequences of prenatal MeHg exposure we dosed pregnant C57BL/6 mice with 0ppm, or 0.2ppm MeHg via drinking water starting from the first day of their pregnancy (confirmed with vaginal copulation plug as embryonic day 0 (E0)) until birth. At postnatal day 7 (P7) and P9 we performed Ultrasonic Vocalization testing (USV). Starting at P60, we performed all subsequent behavioural testing on the offspring. The behavioural tests were performed in order from the least stressful assessment to the most to reduce stress-related interference.
To study the effects of prenatal low-dose MeHg exposure on embryonic cortical development, we dosed pregnant CD-1 mice with 0ppm, or 0.2ppm MeHg via drinking water starting from the first day of their pregnancy E0 until E15. On E15, pregnant mice were sacrificed, and embryos were collected. The brains of the embryos were harvested for further analysis. To study the effects of perinatal exposure of MeHg on postnatal cortical development, we dosed pregnant CD-1 mice with 0ppm or 0.2ppm MeHg via drinking water from E0 until P7. The P7 pups were sacrificed on the last day of treatment, and their brains were collected. To study whether metformin could reverse the effects of MeHg on embryonic cortical development, we dosed pregnant CD-1 mice with 0ppm MeHg (control), 0.2ppm MeHg, 4 mg/mL metformin, or 0.2ppm MeHg+4 mg/mL metformin via drinking water from E0 to E15. Control received drinking water only. At the end of the treatment, brains were collected from the E15 embryos for immunohistochemistry. For all immunohistochemical analysis, the 3–4 embryos in each condition came from two biological different pregnant mice. 0.2ppm MeHg drinking water was prepared by diluting a stock solution of 1000ppm MeHg(II)Cl (cat#33553, Alfa Aesar) in distilled water. For the preparation of metformin solution, metformin powder (cat#D150959, Sigma-Aldrich) was dissolved in distilled water to make a final concentration of 4 mg/mL.
Primary cultured cells were exposed to 0 nM, 250 nM MeHg, 500uM metformin, or 250 nM MeHg +500uM metformin for 4 or 72 hours. 250 nM MeHg was achieved by a 1:250 dilution of 62.5 μM MeHg that was freshly prepared from a stock solution of 4 mM MeHg(II)Cl (cat#33553, Alfa Aesar) through serial dilution with the culture medium. 500 μM metformin was made by 1:400 dilution of a stock solution of 0.2M metformin (cat#PHR1084, Sigma-Aldrich) in the culture medium.
Behavioral testing
Ultrasonic vocalization (USV) test
The test was done at the University of Ottawa Behavior Core. Prior to the expected date of birth, female mice were checked once a day, day of birth was recorded as P0. At P7 and P9 pups were isolated from their mother and littermates and placed inside a soundproof container for 4 min. The microphone was set to 5 cm distance from pups, illumination set at 100 lux, gain set at 45. After 4 min the pups were sexed, weighed, and returned to their home cage. USVs were analyzed using UltraVox XT. Calls were recorded and quantified based on call characterization shown below (adapted from Vogel et al., 2019).72 Max peak frequency and call duration were then measured for each recorded call.
Call characterization
We used the following criteria to characterize and quantify each call:
-
1.
Short: Call shorter than 5 ms.
-
2.
Chevron: Call resembled an ‘inverted-U’, which was identified by a continuous increase in pitch ≥12.5 kHz followed by a decrease that was ≥6.25 kHz
-
3.
Complex: Included one component containing two or more directional changes in frequency, each ≥6.25 kHz
-
4.
Flat: Call ≤3 kHz in change from start to end
-
5.
Upward: single harmonic that increases in frequency >3 kHz from beginning to end
-
6.
Downward: single harmonic that decreases in frequency >3 kHz from beginning to end
-
7.
Two-Component: Call has two parts: a main call (flat or downward) with an additional shorter component at a different frequency towards the end
-
8.
Frequency-Step: Three or more instantaneous frequency harmonics appearing as a vertically discontinuous “step” on a spectrogram, but with no interruption in time
-
9.
Composite: Two independent components, emitted simultaneously at different frequencies
Open-field test
The test was done at the University of Ottawa Behavior Core. Experimental mice were placed at the center of an open-field box (35 cm × 35 cm x 27 cm) with illumination set at 100 lux for 5 min. Ethovision tracking software was used to record the behaviour of mice during testing and calculate velocity and distance travelled.
Marble Bury test
The test was done at the University of Ottawa Behavior Core. The mice were placed in a disposable cage (31 cm × 19.5 cm x 11 cm) with woodchip bedding (Sani Chips) to a depth of 6.5 cm with illumination set at 100 lux for a 5 min habituation period. After 5 min the mice are removed and returned to their home cage. 20 marbles were then evenly spaced on top of the woodchip bedding. After 5 min the mice are returned to the disposable cage where they were left undisturbed for 30 min to interact with the 20 marbles. The number of marbles buried in 30 min was recorded by hand. A marble was scored as buried if two-thirds of its surface area is covered by bedding.
Nestlet Shredding test
Nestlet shredding test was done at the University of Ottawa Behavior Core. Here, mice were given 1.5 hours to interact with one cotton nestlet in an empty cage with illumination set to 100 lux. Each nestlet was weighed before the experiment, and the nestlet that was shredded was weighed after the 1.5h interaction time. The percentage of nestlet shredded was recorded for each animal.
Adult Social Interaction test
The test was done at the University of Ottawa Behavior Core. To assess social interaction, the mice were placed in an open-field box with illumination limited to red lights only to initially explore an empty wire-mesh enclosure for a period of 5 min. Interaction with the enclosure was measured using Ethovision software as the duration of time spent in close proximity to the enclosure. Testing mice were removed, and an age, sex, and strain-matched novel mouse was placed inside the wire-mesh enclosure 5 min later. Testing mice were then placed back in the open-field box for 5 min and interaction time with the novel mouse was measured. Ethovision tracking software was used to record the behaviour of mice during testing.
Morris Water Maze (MWM)
The test was done at the University of Ottawa Behavior Core. The mice were trained on the hidden platform version of the water maze using a circular pool (122 cm diameter, 83.5 cm depth, 22°C) filled with 74.2 cm of water and made opaque with nontoxic white paint. The escape platform (10 cm diameter) was submerged 0.5 cm below the water surface. All testing was conducted under 120 lux lighting and an extra maze visual cue with “X” printed in black ink (2.9 cm thickness) on a white paper (13.5 × 15 cm) was located on one wall. Acquisition was measured as latency to reach the platform and four possible start locations were pseudo-randomly assigned to each trial. Each animal was given four 60 s trials to find the platform with a 20 min inter-trial interval across 4 days. A probe trial was then completed on day 11 after the 4-day training, leaving the to mice swim in the pool for 60 s when the platform was removed. Immediately after this, the platform was moved to the opposite location and animals were trained in the same manner for 2 additional days. A probe trial was then completed on day 14 after the 2-day training, leaving the mice to swim in the pool for 60 s when the platform was removed. Ethovision tracking software was used to record the behaviour of mice during testing.
scRNA-seq
Multiplexing samples for scRNA-seq
Pregnant CD-1 mice were treated with 0ppm, or 0.2ppm MeHg via drinking water starting from E0 to E13.5. Since RNA proceeds protein, E13.5 embryos were used in order to validate E15 immunohistochemistry results. Six embryos from 2 pregnant mice for each treatment (0ppm or 0.2ppm MeHg) were dissected, and cerebral cortices were collected after the removal of meninges from the brains of embryos. The cortical tissues were incubated in papain solution at 37°C for 20 min. Afterward, papain solution was removed, and the tissues were dissociated by a BSA-coated Pasteur pipette in the neurobasal medium (cat#21103049, Thermo Fisher Scientific) containing 500uM GlutaMAX Supplement (cat#35050061, Thermo Fisher Scientific), 2% B27 supplement (cat#17504044, Thermo Fisher Scientific), 1% penicillin-streptomycin (cat#15140122, Thermo Fisher Scientific), and 40 ng/mL FGF2 (cat#10018B, PeproTech). The tissue debris was then allowed to settle for 1 min. The supernatant was collected and was centrifuged at 300g for 2 min. After centrifugation, the supernatant was removed, and the cell pellet was resuspended in the medium by gently pipetting 20 times. The cell suspension was finally passed through a 40 μM sterile cell strainer (Fisherbrand).
Multiplexing was performed according to the MULTI-seq protocol.43 Dissociated cortical cells from 0ppm (control) and 0.2ppm MeHg treatment were labeled with different DNA barcodes using lipid-modified oligonucleotides (LMOs) to stabilize the barcodes within the plasma membrane. Briefly, cells from each group (0ppm/0.2ppm MeHg) were duplicated and divided into two samples (250,000 cells/sample). The cells were then resuspended and pelleted in a 1.5 mL Eppendorf tube at 500 × g for 5 min. Then the four samples were labeled with four different barcodes. For the barcode labelling, the resulting pellet was resuspended in 80 μL of 250 nM anchor mix and 2uL of a 10uM unique barcode (kindly provided by Prof. Zev Gartner from the University of California, San Francisco). Samples were then incubated on ice for 10 min. Afterward, 10 μL of 2 μM the co-anchor LMO was added to each sample and incubated for 5 min. Then 200 μL of PBS with 1% BSA was added to each sample, and the samples were pelleted at 500 × g for 5 min. Each sample was resuspended and pelleted in 200uL PBS with 1% BSA at 500 × g for another 5 min. Viability and cell counts were assessed using a Countess II Cell Counter and Viability Analyzer (Life Technologies). Only samples with a viability ≥80% were further processed. Subsequently, the four samples were pooled together into a new Eppendorf tube and were pelleted at 500 × g for 5 min. After centrifugation, the cell pellet was resuspended by PBS containing 1% BSA. The final concentration of cell suspension was 500–1000 cells/μL.
ScRNA-seq library preparation and sequencing
The scRNA-seq library preparation and sequencing were performed at StemCore laboratories from the OHRI. The multiplexed suspension was processed for 3′ scRNA-seq (v3) with the 10× Genomics Chromium System, targeting to capture 20,000 cells. As described in McGinnis et al.,43 barcode cDNA libraries were isolated from the gene expression library during initial size exclusion and processed independently. Gene expression libraries were prepared according to the manufacturer’s protocol. Libraries were sequenced using the high-output 75 cycle kit on the NextSeq500 (Illumina), achieving an average coverage of 20,558 reads per cell. The resulting 20,535 cells had an average of 2248 unique genes detected and 5120 UMI counts per cell.
Processing of raw sequencing reads
Raw sequencing reads were processed using cellranger v4.0.0 and the mm10 build of the mouse genome. Except for explicitly setting–expect-cells = 20,000, default parameters were used for all samples. MULTI-seq barcode libraries were simply trimmed to 28bp using Timmomatic v0.36 prior to demultiplexing.73
Demultiplexing expression data
Barcode reads were matched to the reference sequences, and the incorporated cell barcode was used to assign the barcode count to an individual cell. The distribution of counts for each barcode across all cells was assessed, and a threshold was defined to deem a cell positive for the barcode by identifying the minima between the bimodal (signal in negative/positive populations) distribution. Cells positive for more than one barcode are classified as multiplets. Only cells positive for a single barcode are retained for downstream analysis.
ScRNA-seq analysis
Quality control was first performed on all four samples concurrently, including multiplets and negatives. The expression matrix was loaded into R as a Seurat (v4.0.1) object. First, cells with less than 200 genes detected were removed. Second, contaminated cells (red blood cells) were removed by correlating library size and gene detection in each cell and removing outliers.30 Finally, cells with more than 10% mitochondrial reads were removed. The top 2000 variable genes were detected in Seurat using the “vst” selection method. Expression values were scaled, and the following technical factors were regressed out: percentage of mitochondrial reads and number of RNA molecules detected. Principal component analysis (PCA) was performed on the highly variable genes, and UMAP embeddings were calculated from the first 30 principal components with 0.2 resolution.
Any additional cell populations that highly expressed red blood cell markers Hbb-y and Hba-x were subset out. Clusters were identified by the expression of known cell-type markers. The markers Fabp7 and Cenpf, Fabp7 and Hes5, Eomes, Dcx, Sox5, Reln, Gad1, Igfbp7, and Rgs5 were used to identify the basic cell-types, radial glial precursors 1 (RGP1), radial glial precursors 2 (RGP2), intermediate progenitors, immature neurons, mature neurons, layer 1 cortical neurons, interneurons, endothelial cells, and pericytes, respectively. We also assessed additional genes discriminating these clusters by performing a simple Wilcoxon rank-sum test with the FindAllMarkers() function in Seurat. We next isolated the transitional cell subset and reprocessed the subset using the same normalization and integration approach. After data visualization for both the total cell population and the transition cell population, multiplets and negatives were subset out, and 0ppm and 0.2ppm were separated for further downstream analysis.
Transitional state analysis
For UMAP visualizations cells were considered positive for markers of a cell-type if the mean expression of a group of cell type marker genes was >0.5 log normalized expression. Canonical markers were used to differentiate cell types. Radial glial-positive (RG+) markers used were Sox2, Fabp7, Pax6, Hes1, and Hes5. Intermediate progenitors-positive (IP+) markers used were Eomes, Sstr2, Gadd45g, and Mfap4. Neuronal-positive cells (N+) markers used were Tubb3, Neurod1, Sox11, Neurod6, Dcx, Bcl11b, and Sox5.
RNA velocity
PCA and UMAP embeddings that were produced during the initial analysis step were embedded using Seurat. Velocity estimates were calculated using the scVelo (v0.2.2.) python package with default parameters. Velocity vectors and velocity graphs were computed and then projected on UMAP embeddings. Velocity vector analysis was conducted using Image J.
Tissue preparation and immunohistochemistry
The collected brain samples were fixed in 4% paraformaldehyde (PFA) in 0.1M phosphate-buffered saline (PBS) at room temperature for 24 hours. They were then immersed in 30% sucrose solution in 0.1M PBS and stored at 4°C until use. Brain samples were embedded in optimal cutting temperature compound (OCT) (cat#95057-838, VWR) and frozen for cryosectioning. Serial 16μm brain sections were obtained using a cryostat and sequentially mounted on glass slides. Glass slides were allowed to air dry before storage at −80°C for later use.
For the detection of Mcm2, Sox2, Tbr2, doublecortin (DCX), and Tbr1, brain sections stored in the −80°C freezer were first washed with 1× PBS 3 times (5 min each). Section slides were then incubated in 0.01 M citric acid (pH 6.0) at 90°C for 10 min for antigen retrieval. Afterwards, brain sections were blocked/permeabilized for at least 1 hour at room temperature in 10% normal goat serum (NGS) (cat#16050122, Thermo Fisher Scientific) or 3% BSA (cat#9998S, Cell Signalling Technology) in 1× PBS with 0.3% Triton X-100 (PBST). Subsequently, the brain sections were incubated with each primary antibody overnight, followed by secondary antibody treatment at room temperature for 1 h. The brain sections were washed with 1× PBS three times (5 min each) before incubation of secondary antibodies.
For the detection of Ki67 and Pax6, brain sections were blocked for 1 hour at room temperature using the mouse IgG blocking reagent from the mouse on mouse (M.O.M.) immunodetection kit basic (cat#BMK-2202, Vector Laboratories). Brain sections were then washed with 1× PBS 2 times (2 min each) and further incubated with M.O.M. diluent prepared according to the manufacturer’s protocol. After 5-min incubation with the M.O.M. diluent, slides were incubated with primary antibodies in the M.O.M. diluent overnight at room temperature. Subsequently, slides were washed with 1× PBS 2 times (2 min each) and incubated with the M.O.M. biotinylated anti-mouse IgG reagent for 10 min. After 2 washes with 1× PBS (2 min each), brain sections were incubated with secondary antibodies, Streptavidin Alexa Fluor 555 conjugate- (cat#S32355, Invitrogen) and donkey anti-rabbit Alexa Fluor 647 (cat#A31573, Invitrogen). Brain sections were mounted using PermaFluor™ solution (cat#TA-030-FM, Thermo Fisher Scientific).
TUNEL assay
In Situ Cell Death Detection Kit, POD (Sigma-Aldrich, cat#11684817910) was used for detecting TUNEL+ cells. Brain sections were first washed with PBS three times (5 min each) and then incubated with blocking solution (3% H2O2 in methanol) for 10 min. After rinsing the slides with PBS, brain sections were further incubated with permeabilization solution (0.1% Triton X–100 and 0.1% sodium citrate) for 2 min. The slides were then washed with PBS. Afterwards, TUNEL reaction mixture was prepared by mixing the label solution and enzyme solution according to the manufacturer’s instructions. Brain sections were incubated in the TUNEL reaction mixture for 60 min at room temperature, and nuclear counterstaining was performed with Hoechst 33342 (cat#4082, Cell Signalling Technology, 1:1000). The slides were finally mounted with coverslips using PermaFluor™ solution (cat#TA-030-FM, Thermo Fisher Scientific).
Immunocytochemistry (primary cell culture)
Cultured cells were fixed in 4% paraformaldehyde for 10 min after 72 hours in culture, they were then blocked with 10% normal goat serum (NGS) (cat#16050122, Thermo Fisher Scientific), diluted in 1x PBS with 0.3% Triton X-100. The cells were incubated within primary antibodies diluted in 10% NGS in PBS with 0.3% Triton X-100 and then incubated in a humid chamber at 4 °C overnight. Following this, secondary antibodies were diluted in PBST and incubated for 1 hour at room temperature. After rinsing with PBS, the coverslips were mounted in a Lab Vision PermaFluor Aqueous Mounting Medium (cat#TA-030-FM, Thermo Fisher Scientific). The culture was washed three times for 5 min/time with 1× PBS between each step.
Proximity ligation assay (PLA)
Cells were fixed in 4% paraformaldehyde for 10 min after 4h in culture and were incubated in PBST for 1h at room temperature for permeabilization. Cells were blocked in Duolink blocking solution for 1hat 37°C. Primary antibodies were diluted in Duolink antibody diluent and incubated in a humid chamber at 4 °C overnight. Slides were then washed with Duolink 1× wash buffer A two times for 5 min. PLUS and MINUS PLA probes were diluted 1:5 in the Duolink antibody diluent and added to the coverslips which were incubated for 1hat 37°C. The coverslips were then washed with Duolink 1× wash buffer A two times for 5 min. The coverslips were incubated in 5× Duolink Ligation buffer diluted 1:5 in water with ligase added at a 1:40 dilution for 30minat 37°C. Coverslips were then washed with Duolink 1× wash buffer A two times for 5 min. Coverslips were incubated in 5× Duolink amplification buffer diluted 1:5 in water with polymerase added at a 1:80 dilution for 100min at 37°C. Coverslips were then washed with Duolink 1× wash buffer B two times for 10 min and Duolink 0.01× wash buffer B for 1minat room temperature. Coverslips were mounted with Duolink In Situ Mounting Medium containing DAPI staining and stored in −20°C (cat#DUO92101-1KT, Sigma-Aldrich). Digital image acquisition was performed using a Zeiss Axioplan 2 fluorescent microscope with Zeiss Axiovision software containing z axis capability (Carl Zeiss Microscopy, Thornwood, NY, USA). Ten to fifteen image sections at a maximum of 1μm apart were captured in the z axis for each section and then processed as an optical stack for quantification. 25 cells per sample over at least four independent experiments were analyzed. PLA+ dots (each dot represents one interaction) were quantified. Quantification was conducted in Image J.
Image acquisition and quantification
For cultured cells, digital image acquisition was performed using a Zeiss Axioplan M.2 fluorescent microscope with Zeiss Axiovision software (Carl Zeiss Microscopy, Thornwood, NY, USA). Six random images per experiment were taken for all conditions, and at least 700 cells per condition were counted for each experiment. At least four independent experiments from four pregnant mice were conducted for all conditions. Quantification was performed using Image J.
For brain sections, digital images of brain sections were taken by a Zeiss Imager M.1 fluorescent microscopy with Zeiss Axiovision software containing z-axis capability. Ten to fifteen image sections at a maximum of 1μm apart were captured in the Z-axis for each brain section and were processed as an optical stack for quantification. Quantification was performed throughout the cortex, including the ventricular zone (VZ), subventricular zone (SVZ)/intermediate zone (IZ), and cortical plate (CP). At least three anatomically matched brain sections were quantified for each embryo or pup among different treatment groups.
Western blot analysis and densitometry
After culturing for 4 hours in the neurobasal medium, the cortical precursors were lysed in cold lysis buffer containing 1mM sodium orthovanadate (Sigma-Aldrich S6508-10G), 20 mM sodium fluoride (cat#AC201295000, Fisher Scientific), 1mM PMSF (cat#P7626-5G, Sigma-Aldrich), 10 μg/mL aprotinin (Fisher Scientific, cat#PI78432) and 20 μg/mL leupeptin (cat#PI78436, Fisher Scientific). Protein concentration was determined using the bicinchoninic acid assay (cat#23225, Fisher Scientific) and BSA as a standard. Equal amounts of protein (10–20μg) were boiled in sample buffer, separated by 12% SDS-PAGE gels, and transferred to 0.2μm PVDF membrane 1 hour at 100V. Membranes were blocked in 3% BSA in TBS-T for 1 hour at room temperature and then incubated overnight at 4°C with primary antibodies. After washing with TBS-T, membranes were incubated with secondary antibodies in blocking solution for 1hat room temperature. Detection was performed using the ECL chemiluminescence reagent (cat#PI32209, Fisher Scientific) and autoradiography films (cat#E3018, Denville Scientific). Membranes were subsequently stripped washed with TBS-T and re-probed as described above. Densitometry was performed using Image J.
Hg analysis
Hg in the feces, urine, and blood of mice undergoing behavioural testing was analyzed at P22 and P42. Feces, urine, and blood were collected and immediately stored at −20°C for Hg analysis. Following behavioural testing at 3 months old, the mice were sacrificed, and the kidneys, liver, and brain were collected and immediately frozen with liquid nitrogen and stored at −80°C for Hg analysis. The brains were dissected, and the cerebellum, occipital lobe, cerebral cortex, hypothalamus, hippocampus, pons/medulla, and olfactory bulb were isolated and immediately frozen with liquid nitrogen and stored at −80°C for Hg analysis. The right hemispheres of E15 and P7 brains (after removal of the cerebellum) were collected and immediately stored at −80°C for future Hg analysis.
For the measurement of Hg levels a direct thermal decomposition mercury analyzer (MA-3000, Nippon Instrument, Bryan, TX, USA) was used. Quality assurance/quality control methods included blanks samples and standard reference material DOLT5. The MA-3000 used for Hg analysis has a detection limit of 0.01 to 0.02ng.
Antibodies
The primary antibodies used for immunocytochemistry were mouse anti-βIII-tubulin (cat#801201, BioLegend, 1:1000), rabbit anti-Pax6 (cat#901301, BioLegend, 1:1000), rabbit anti-Sox2 (cat#AB5603MI, Millipore, 1:500), and mouse anti-Ki67 (1:500, Abcam, cat#ab15580). The secondary antibodies used were donkey anti-rabbit Alexa Fluor 555 (cat#A31572, Thermo Fisher Scientific, 1:500) and goat anti-mouse Alexa Fluor 488 (cat#A32723, Thermo Fisher Scientific, 1:500). Nuclear counterstaining was performed with Hoechst 33342 (cat#4082, Cell Signalling Technology, 1:1000).
The primary antibodies used for immunohistochemistry were rabbit anti-Pax6 (cat#901301, Biolegend, 1:2000), rabbit anti-Tbr2 (cat#ab233456, Abcam, 1:500), mouse anti-Ki67 (cat#550609, BD Pharmingen, 1:200), rabbit anti-Mcm2 (cat#ab4461, Abcam, 1:500) mouse anti-Sox2 (cat#ab79351, Abcam, 1:400), goat anti-DCX (cat#sc-8066, Santa Cruz biotechnology, 1:500) and rabbit anti-Tbr1 (cat#ab31940, Abcam, 1:500), and mouse anti-Satb2 (cat#ab51502, Abcam, 1:500). The secondary antibodies used were donkey anti-rabbit Alexa Fluor 555 (cat#A31572, Thermo Fisher Scientific, 1:1000), goat anti-mouse Alexa Fluor 555 (cat#A21422, Thermo Fisher Scientific, 1:1000), donkey anti-rabbit Alexa Fluor 647 (cat#A31573, Thermo Fisher Scientific, 1:1000), goat anti-mouse Alexa Fluor 647 (cat#A32728, Thermo Fisher Scientific, 1:1000) and donkey anti-goat Alexa Fluor 647 (cat#A21447, Thermo Fisher Scientific, 1:1000). Hoechst 33342 (cat#4082, Cell Signalling Technology, 1:1000) was used for the nuclear staining.
For PLA, the primary antibodies were rabbit anti-p-CREB (S133) (cat#9198S, Cell Signaling 1:500) and mouse anti-CBP (cat#sc-7300, Santa Cruz Biotechnology, 1:200).
For western blots, the primary antibodies were rabbit anti-paPKCζ/ι (T410/403) (cat#9378, Cell Signaling, 1:500), mouse anti-aPKCζ/ι (cat#610175, BD Pharmingen, 1:500), rabbit anti-p-CREB (S133) (cat#9198S, Cell Signaling, 1:500), mouse anti-CREB (cat#9104S, Cell Signaling, 1:500). Secondary antibodies for western blots were HRP-conjugated goat anti-mouse, anti-rabbit (cat#7074, cat#7076, Cell Signalling, 1:3000).
Quantification and statistical analysis
Statistical analyses were performed with a two-tailed Student’s t test, one-way, or two-way ANOVA with Tukey’s multiple comparisons as post hoc analysis. Normalization was assessed by the Shapiro-Wilk (p< 0.05) test and conducted on all control data. On non-normally distributed data, a non-parametric statistical test was performed. Error bars indicate the standard error of the mean (SEM). p-values presented as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
A priori, the study sample size for all immunohistochemical analyses was calculated by a statistical power analysis primarily to compare the difference between the two study groups. In the absence of available pilot data, a pragmatic decision was taken with the following assumptions: a difference of 30% between the control and MeHg treatment group, a 10% standard deviation from the mean, and a significance level of 5%, and a minimum statistical power of 80%. The calculated effective sample size of 3 or greater per condition was targeted. For all behavioural testing, we utilized the Mead’s resource equation to determine sample size.
Acknowledgments
This work was supported by NSERC Discovery Grant (06605/RGPIN/2019) and Canada Research Chair Grant (950-225645) to H.M. Chan. A. Loan was supported by an NSERC-CREATE grant (CREATE-449153) and the Queen Elizabeth II Graduate Scholarship in Science and Technology. The authors would like to thank Dr. Emmanuel Yumvihoze, Alena Kalinina, OHRI StemCore Facility, University of Ottawa Behavior Core, and the Animal Care Veterinary Services (ACVS) at the University of Ottawa for technical support, advice, and discussions. The authors would also like to thank Cecilia Liu for providing advice on establishing an early pipeline for scRNA-seq analysis.
Author contributions
A.L. performed behavioral testing, embryonic cortical precursor culture, immunocytochemistry, immunohistochemistry, and PLA assay. C.K. performed USV analysis. J.L. performed experiments related to in vivo animal work including MeHg drinking water preparation, tissue preparation, sectioning, and immunohistochemistry. A.L., J.L., and D.C. performed multiplexing single-cell RNA-seq experiments. D.C. and A.L. performed bioinformatic analysis for single-cell RNA-seq dataset. B.V. provided the resource for multiplexing single-cell RNA-seq. J.W. and H.M.C. contributed to the conceptualization, funding, and project management. J.W., H.M.C., J.L., and A.L. contributed to experimental design, data interpretation, and writing the paper.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: January 31, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106093.
Contributor Information
Jing Wang, Email: jiwang@ohri.ca.
Hing Man Chan, Email: laurie.chan@uottawa.ca.
Supplemental information
Data and code availability
-
•
scRNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
All code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Lord C., Bishop S.L. Recent advances in autism research as reflected in DSM-5 criteria for autism spectrum disorder. Annu. Rev. Clin. Psychol. 2015;11:53–70. doi: 10.1146/annurev-clinpsy-032814-112745. [DOI] [PubMed] [Google Scholar]
- 2.Packer A. Neocortical neurogenesis and the etiology of autism spectrum disorder. Neurosci. Biobehav. Rev. 2016;64:185–195. doi: 10.1016/J.NEUBIOREV.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Uddin M., Unda B.K., Kwan V., Holzapfel N.T., White S.H., Chalil L., Woodbury-Smith M., Ho K.S., Harward E., Murtaza N., et al. OTUD7A regulates neurodevelopmental phenotypes in the 15q13.3 microdeletion syndrome. Am. J. Hum. Genet. 2018;102:278–295. doi: 10.1016/J.AJHG.2018.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.C Yuen R.K., Merico D., Bookman M., L Howe J., Thiruvahindrapuram B., Patel R.v., Whitney J., Deflaux N., Bingham J., Wang Z., et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 2017;20:602–611. doi: 10.1038/NN.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Portmann T., Yang M., Mao R., Panagiotakos G., Ellegood J., Dolen G., Bader P.L., Grueter B.A., Goold C., Fisher E., et al. Behavioral abnormalities and circuit defects in the basal ganglia of a mouse model of 16p11.2 deletion syndrome. Cell Rep. 2014;7:1077–1092. doi: 10.1016/J.CELREP.2014.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nordenbæk C., Jørgensen M., Kyvik K.O., Bilenberg N. A Danish population-based twin study on autism spectrum disorders. Eur. Child Adolesc. Psychiatr. 2013;23:35–43. doi: 10.1007/S00787-013-0419-5. [DOI] [PubMed] [Google Scholar]
- 7.Clarkson T.W., Magos L., Myers G.J. The toxicology of mercury — current exposures and clinical manifestations. N. Engl. J. Med. 2003;349:1731–1737. doi: 10.1056/nejmra022471. [DOI] [PubMed] [Google Scholar]
- 8.Kajiwara Y., Yasutake A., Adachi T., Hirayama K. Methylmercury transport across the placenta via neutral amino acid carrier. Arch. Toxicol. 1996;70:310–314. doi: 10.1007/S002040050279. [DOI] [PubMed] [Google Scholar]
- 9.Grandjean P., Weihe P., White R.F., Debes F., Araki S., Yokoyama K., Murata K., Sørensen N., Dahl R., Jørgensen P.J. Cognitive deficit in 7-year-old children with prenatal exposure to methylmercury. Neurotoxicol. Teratol. 1997;19:417–428. doi: 10.1016/S0892-0362(97)00097-4. [DOI] [PubMed] [Google Scholar]
- 10.Prpić I., Milardović A., Vlašić-Cicvarić I., Špiric Z., Radić Nišević J., Vukelić P., Snoj Tratnik J., Mazej D., Horvat M. Prenatal exposure to low-level methylmercury alters the child’s fine motor skills at the age of 18 months. Environ. Res. 2017;152:369–374. doi: 10.1016/j.envres.2016.10.011. [DOI] [PubMed] [Google Scholar]
- 11.Bisen-Hersh E.B., Farina M., Barbosa F., Rocha J.B.T., Aschner M. Behavioral effects of developmental methylmercury drinking water exposure in rodents. J. Trace Elem. Med. Biol. 2014;28:117–124. doi: 10.1016/j.jtemb.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jafari T., Rostampour N., Fallah A.A., Hesami A. The association between mercury levels and autism spectrum disorders: a systematic review and meta-analysis. J. Trace Elem. Med. Biol. 2017;44:289–297. doi: 10.1016/J.JTEMB.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 13.Saghazadeh A., Rezaei N. Systematic review and meta-analysis links autism and toxic metals and highlights the impact of country development status: higher blood and erythrocyte levels for mercury and lead, and higher hair antimony, cadmium, lead, and mercury. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2017;79:340–368. doi: 10.1016/J.PNPBP.2017.07.011. [DOI] [PubMed] [Google Scholar]
- 14.Jafari Mohammadabadi H., Rahmatian A., Sayehmiri F., Rafiei M. The relationship between the level of copper, lead, mercury and autism disorders: a meta-analysis. Pediatr. Health Med. Therapeut. 2020;11:369–378. doi: 10.2147/PHMT.S210042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berglund M., Lind B., Björnberg K.A., Palm B., Einarsson Ö., Vahter M. Inter-individual variations of human mercury exposure biomarkers: a cross-sectional assessment. Environ. Health. 2005;4:20. doi: 10.1186/1476-069X-4-20/FIGURES/4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan X., Wang J., Chan H.M. Correction: yuan et al. Sub-micromolar methylmercury exposure promotes premature differentiation of murine embryonic neural precursor at the expense of their proliferation. Toxics. 2021;9:322. doi: 10.3390/TOXICS9120322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishii S., Hashimoto-Torii K. Impact of prenatal environmental stress on cortical development. Front. Cell. Neurosci. 2015;9:207. doi: 10.3389/fncel.2015.00207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson B.L., Levitt P., Stanwood G.D. Prenatal exposure to drugs: effects on brain development and implications for policy and education. Nat. Rev. Neurosci. 2009;10:303–312. doi: 10.1038/NRN2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sokolowski K., Falluel-Morel A., Zhou X., DiCicco-Bloom E. Methylmercury (MeHg) elicits mitochondrial-dependent apoptosis in developing hippocampus and acts at low exposures. Neurotoxicology. 2011;32:535–544. doi: 10.1016/j.neuro.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Falluel-Morel A., Sokolowski K., Sisti H.M., Zhou X., Shors T.J., DiCicco-Bloom E. Developmental mercury exposure elicits acute hippocampal cell death, reductions in neurogenesis, and severe learning deficits during puberty. J. Neurochem. 2007;103:1968–1981. doi: 10.1111/j.1471-4159.2007.04882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sokolowski K., Obiorah M., Robinson K., Mccandlish E., Buckley B., Dicicco-Bloom E. Neural stem cell apoptosis after low-methylmercury exposures in postnatal hippocampus produce persistent cell loss and adolescent memory deficits. Dev. Neurobiol. 2013;73:936–949. doi: 10.1002/dneu.22119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edoff K., Raciti M., Moors M., Sundström E., Ceccatelli S. Gestational age and sex influence the susceptibility of human neural progenitor cells to low levels of MeHg. Neurotox. Res. 2017;32:683–693. doi: 10.1007/s12640-017-9786-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tamm C., Duckworth J., Hermanson O., Ceccatelli S. High susceptibility of neural stem cells to methylmercury toxicity: effects on cell survival and neuronal differentiation. J. Neurochem. 2006;97:69–78. doi: 10.1111/j.1471-4159.2006.03718.x. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe J., Nakamachi T., Ohtaki H., Naganuma A., Shioda S., Nakajo S. Low dose of methylmercury (MeHg) exposure induces caspase mediated-apoptosis in cultured neural progenitor cells. J. Toxicol. Sci. 2013;38:931–935. doi: 10.2131/jts.38.931. [DOI] [PubMed] [Google Scholar]
- 25.Qian X., Shen Q., Goderie S.K., He W., Capela A., Davis A.A., Temple S. Timing of CNS cell generation: a programmed sequence of neuron and glial cell production from isolated murine cortical stem cells. Neuron. 2000;28:69–80. doi: 10.1016/S0896-6273(00)00086-6. [DOI] [PubMed] [Google Scholar]
- 26.Wang J., Weaver I.C.G., Gauthier-Fisher A., Wang H., He L., Yeomans J., Wondisford F., Kaplan D.R., Miller F.D. CBP histone acetyltransferase activity regulates embryonic neural differentiation in the normal and rubinstein-taybi syndrome brain. Dev. Cell. 2010;18:114–125. doi: 10.1016/j.devcel.2009.10.023. [DOI] [PubMed] [Google Scholar]
- 27.Fang W.Q., Chen W.W., Jiang L., Liu K., Yung W.H., Fu A.K.Y., Ip N.Y. Overproduction of upper-layer neurons in the neocortex leads to autism-like features in mice. Cell Rep. 2014;9:1635–1643. doi: 10.1016/J.CELREP.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 28.Zhang W., Ma L., Yang M., Shao Q., Xu J., Lu Z., Zhao Z., Chen R., Chai Y., Chen J.F. Cerebral organoid and mouse models reveal a RAB39b-PI3K-mTOR pathway-dependent dysregulation of cortical development leading to macrocephaly/autism phenotypes. Genes Dev. 2020;34:580–597. doi: 10.1101/GAD.332494.119/-/DC1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schafer S.T., Paquola A.C.M., Stern S., Gosselin D., Ku M., Pena M., Kuret T.J.M., Liyanage M., Mansour A.A., Jaeger B.N., et al. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat. Neurosci. 2019;22:243–255. doi: 10.1038/s41593-018-0295-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Casanova M.F., van Kooten I.A.J., Switala A.E., van Engeland H., Heinsen H., Steinbusch H.W.M., Hof P.R., Trippe J., Stone J., Schmitz C. Minicolumnar abnormalities in autism. Acta Neuropathol. 2006;112:287–303. doi: 10.1007/S00401-006-0085-5. [DOI] [PubMed] [Google Scholar]
- 31.Courchesne E., Mouton P.R., Calhoun M.E., Semendeferi K., Ahrens-Barbeau C., Hallet M.J., Barnes C.C., Pierce K. Neuron number and size in prefrontal cortex of children with autism. JAMA. 2011;306:2001–2010. doi: 10.1001/JAMA.2011.1638. [DOI] [PubMed] [Google Scholar]
- 32.Barco A., Alarcon J.M., Kandel E.R. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell. 2002;108:689–703. doi: 10.1016/S0092-8674(02)00657-8. [DOI] [PubMed] [Google Scholar]
- 33.Merz K., Herold S., Lie D.C. CREB in adult neurogenesis - master and partner in the development of adult-born neurons? Eur. J. Neurosci. 2011;33:1078–1086. doi: 10.1111/j.1460-9568.2011.07606.x. [DOI] [PubMed] [Google Scholar]
- 34.Nakagawa S., Kim J.E., Lee R., Chen J., Fujioka T., Malberg J., Tsuji S., Duman R.S. Localization of phosphorylated cAMP response element-binding protein in immature neurons of adult hippocampus. J. Neurosci. 2002;22:9868–9876. doi: 10.1523/jneurosci.22-22-09868.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walton M.R., Dragunow I. Is CREB a key to neuronal survival? Trends Neurosci. 2000;23:48–53. doi: 10.1016/S0166-2236(99)01500-3. [DOI] [PubMed] [Google Scholar]
- 36.Unoki T., Abiko Y., Toyama T., Uehara T., Tsuboi K., Nishida M., Kaji T., Kumagai Y. Methylmercury, an environmental electrophile capable of activation and disruption of the Akt/CREB/Bcl-2 signal transduction pathway in SH-SY5Y cells. Sci. Rep. 2016;6:1–10. doi: 10.1038/srep28944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J., Gallagher D., Devito L.M., Cancino G.I., Tsui D., He L., Keller G.M., Frankland P.W., Kaplan D.R., Miller F.D. Metformin activates an atypical PKC-CBP pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem Cell. 2012;11:23–35. doi: 10.1016/j.stem.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 38.Syal C., Seegobin M., Sarma S.N., Gouveia A., Hsu K., Niibori Y., He L., Wondisford F.E., Frankland P.W., Wang J. Ectopic expression of aPKC-mediated phosphorylation in p300 modulates hippocampal neurogenesis, CREB binding and fear memory differently with age. Sci. Rep. 2018;8:13489. doi: 10.1038/s41598-018-31657-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He L., Chang E., Peng J., An H., McMillin S.M., Radovick S., Stratakis C.A., Wondisford F.E. Activation of the cAMP-PKA pathway antagonizes metformin suppression of hepatic glucose production. J. Biol. Chem. 2016;291:10562–10570. doi: 10.1074/JBC.M116.719666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antunes dos Santos A., Appel Hort M., Culbreth M., López-Granero C., Farina M., Rocha J.B.T., Aschner M. Methylmercury and brain development: a review of recent literature. J. Trace Elem. Med. Biol. 2016;38:99–107. doi: 10.1016/j.jtemb.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang Y.C., Cole T.B., Costa L.G. Behavioral phenotyping for autism spectrum disorders in mice. Curr. Protoc. Toxicol. 2017;72:11.22.1–11.22.21. doi: 10.1002/CPTX.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.National Research Council (US) 2000. Toxicological effects of methylmercury. [DOI] [Google Scholar]
- 43.McGinnis C.S., Patterson D.M., Winkler J., Conrad D.N., Hein M.Y., Srivastava V., Hu J.L., Murrow L.M., Weissman J.S., Werb Z., et al. MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nat. Methods. 2019;16:619–626. doi: 10.1038/s41592-019-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuzwa S.A., Borrett M.J., Innes B.T., Voronova A., Ketela T., Kaplan D.R., Bader G.D., Miller F.D. Developmental emergence of adult neural stem cells as revealed by single-cell transcriptional profiling. Cell Rep. 2017;21:3970–3986. doi: 10.1016/J.CELREP.2017.12.017. [DOI] [PubMed] [Google Scholar]
- 45.Loo L., Simon J.M., Xing L., McCoy E.S., Niehaus J.K., Guo J., Anton E.S., Zylka M.J. Single-cell transcriptomic analysis of mouse neocortical development. Nat. Commun. 2019;10:134–211. doi: 10.1038/s41467-018-08079-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bergen V., Lange M., Peidli S., Wolf F.A., Theis F.J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 2020;38:1408–1414. doi: 10.1038/s41587-020-0591-3. [DOI] [PubMed] [Google Scholar]
- 47.Conkright M.D., Guzmán E., Flechner L., Su A.I., Hogenesch J.B., Montminy M. Genome-wide analysis of CREB target genes reveals a core promoter requirement for cAMP responsiveness. Mol. Cell. 2003;11:1101–1108. doi: 10.1016/S1097-2765(03)00134-5. [DOI] [PubMed] [Google Scholar]
- 48.Impey S., McCorkle S.R., Cha-Molstad H., Dwyer J.M., Yochum G.S., Boss J.M., McWeeney S., Dunn J.J., Mandel G., Goodman R.H. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell. 2004;119:1041–1054. doi: 10.1016/j.cell.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 49.Cheroni C., Caporale N., Testa G. Autism spectrum disorder at the crossroad between genes and environment: contributions, convergences, and interactions in ASD developmental pathophysiology. Mol. Autism. 2020;11:69. doi: 10.1186/S13229-020-00370-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schafer S.T., Gage F.H. The when and where: molecular and cellular convergence in autism. Biol. Psychiatr. 2021;89:419–420. doi: 10.1016/j.biopsych.2020.12.016. [DOI] [PubMed] [Google Scholar]
- 51.Courchesne E., Pramparo T., Gazestani V.H., Lombardo M.v., Pierce K., Lewis N.E. The ASD Living Biology: from cell proliferation to clinical phenotype. Mol. Psychiatr. 2019;24:88–107. doi: 10.1038/S41380-018-0056-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caruso A., Ricceri L., Scattoni M.L. Ultrasonic vocalizations as a fundamental tool for early and adult behavioral phenotyping of Autism Spectrum Disorder rodent models. Neurosci. Biobehav. Rev. 2020;116:31–43. doi: 10.1016/J.NEUBIOREV.2020.06.011. [DOI] [PubMed] [Google Scholar]
- 53.Sungur A.Ö., Schwarting R.K.W., Wöhr M. Early communication deficits in the Shank1 knockout mouse model for autism spectrum disorder: developmental aspects and effects of social context. Autism Res. 2016;9:696–709. doi: 10.1002/AUR.1564. [DOI] [PubMed] [Google Scholar]
- 54.Fröhlich H., Rafiullah R., Schmitt N., Abele S., Rappold G.A. Foxp1 expression is essential for sex-specific murine neonatal ultrasonic vocalization. Hum. Mol. Genet. 2017;26:1511–1521. doi: 10.1093/HMG/DDX055. [DOI] [PubMed] [Google Scholar]
- 55.Fujita E., Tanabe Y., Imhof B.A., Momoi M.Y., Momoi T. Cadm1-Expressing synapses on purkinje cell dendrites are involved in mouse ultrasonic vocalization activity. PLoS One. 2012;7:e30151. doi: 10.1371/JOURNAL.PONE.0030151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Biamonte F., Latini L., Giorgi F.S., Zingariello M., Marino R., de Luca R., D’Ilio S., Majorani C., Petrucci F., Violante N., et al. Associations among exposure to methylmercury, reduced Reelin expression, and gender in the cerebellum of developing mice. Neurotoxicology. 2014;45:67–80. doi: 10.1016/J.NEURO.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 57.Moy S.S., Nadler J.J., Young N.B., Perez A., Holloway L.P., Barbaro R.P., Barbaro J.R., Wilson L.M., Threadgill D.W., Lauder J.M., et al. Mouse behavioral tasks relevant to autism: phenotypes of ten inbred strains. Behav. Brain Res. 2007;176:4–20. doi: 10.1016/J.BBR.2006.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Attia S.M., Ahmad S.F., Nadeem A., Attia M.S.M., Ansari M.A., Al-Hamamah M.A., Hussein M.H., Alameen A.A., Alasmari A.F., Bakheet S.A. Multiple exposure to methylmercury aggravates DNA damage in the BTBR T + Itpr3 tf/J autistic mouse model: the role of DNA repair efficiency. Toxicology. 2022;477:153277. doi: 10.1016/J.TOX.2022.153277. [DOI] [PubMed] [Google Scholar]
- 59.Al-Mazroua H.A., Nadeem A., Ansari M.A., Attia S.M., Albekairi T.H., Bakheet S.A., Alobaidi A.F., Alhosaini K., Alqarni S.A., Ibrahim K.E., et al. Methylmercury chloride exposure exacerbates existing neurobehavioral and immune dysfunctions in the BTBR T+ Itpr3tf/J mouse model of autism. Immunol. Lett. 2022;244:19–27. doi: 10.1016/J.IMLET.2022.03.001. [DOI] [PubMed] [Google Scholar]
- 60.Matsuzaki F., Shitamukai A. Cell division modes and cleavage planes of neural progenitors during mammalian cortical development. Cold Spring Harbor Perspect. Biol. 2015;7:a015719. doi: 10.1101/cshperspect.a015719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Polioudakis D., de la Torre-Ubieta L., Langerman J., Elkins A.G., Shi X., Stein J.L., Vuong C.K., Nichterwitz S., Gevorgian M., Opland C.K., et al. A single-cell transcriptomic atlas of human neocortical development during mid-gestation. Neuron. 2019;103:785–801.e8. doi: 10.1016/j.neuron.2019.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Giachino C., de Marchis S., Giampietro C., Parlato R., Perroteau I., Schütz G., Fasolo A., Peretto P. cAMP response element-binding protein regulates differentiation and survival of newborn neurons in the olfactory bulb. J. Neurosci. 2005;25:10105–10118. doi: 10.1523/JNEUROSCI.3512-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jagasia R., Steib K., Englberger E., Herold S., Faus-Kessler T., Saxe M., Gage F.H., Song H., Lie D.C. GABA-cAMP response element-binding protein signaling regulates maturation and survival of newly generated neurons in the adult hippocampus. J. Neurosci. 2009;29:7966–7977. doi: 10.1523/JNEUROSCI.1054-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Landeira B.S., Santana T.T.D.S., Araújo J.A.d.M., Tabet E.I., Tannous B.A., Schroeder T., Costa M.R. Activity-independent effects of CREB on neuronal survival and differentiation during mouse cerebral cortex development. Cerebr. Cortex. 2018;28:538–548. doi: 10.1093/cercor/bhw387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gonzalez G.A., Montminy M.R. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 66.Gouveia A., Hsu K., Niibori Y., Seegobin M., Cancino G.I., He L., Wondisford F.E., Bennett S., Lagace D., Frankland P.W., Wang J. The aPKC-CBP pathway regulates adult hippocampal neurogenesis in an age-dependent manner. Stem Cell Rep. 2016;7:719–734. doi: 10.1016/j.stemcr.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Driscoll C.T., Mason R.P., Chan H.M., Jacob D.J., Pirrone N. Mercury as a global pollutant: sources, pathways, and effects. Environ. Sci. Technol. 2013;47:4967–4983. doi: 10.1021/ES305071V. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karagas M.R., Choi A.L., Oken E., Horvat M., Schoeny R., Kamai E., Cowell W., Grandjean P., Korrick S. Evidence on the human health effects of low-level methylmercury exposure. Environ. Health Perspect. 2012;120:799–806. doi: 10.1289/ehp.1104494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choi A.L., Mogensen U.B., Bjerve K.S., Debes F., Weihe P., Grandjean P., Budtz-Jørgensen E. Negative confounding by essential fatty acids in methylmercury neurotoxicity associations. Neurotoxicol. Teratol. 2014;42:85–92. doi: 10.1016/j.ntt.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Debes F., Weihe P., Grandjean P. Cognitive deficits at age 22 years associated with prenatal exposure to methylmercury. Cortex. 2016;74:358–369. doi: 10.1016/j.cortex.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang Y., Song Z., Huang S., Zhang P., Peng Y., Wu P., Gu J., Dutkiewicz S., Zhang H., Wu S., et al. Global health effects of future atmospheric mercury emissions. Nat. Commun. 2021;12:3035. doi: 10.1038/s41467-021-23391-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vogel A.P., Tsanas A., Scattoni M.L. Quantifying ultrasonic mouse vocalizations using acoustic analysis in a supervised statistical machine learning framework. Sci. Rep. 2019;9:8100. doi: 10.1038/s41598-019-44221-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/BIOINFORMATICS/BTU170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
scRNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
All code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.