

Abstract
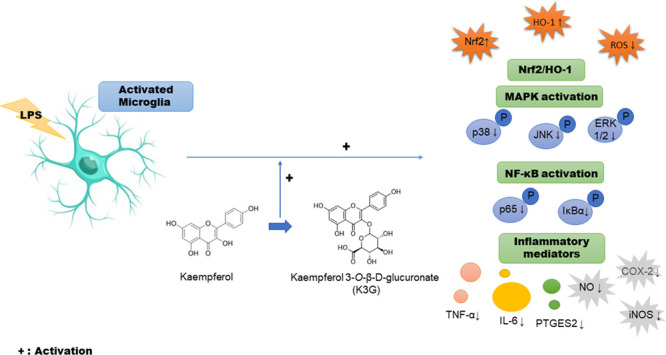
Aglycone- and glycoside-derived forms of flavonoids exist broadly in plants and foods such as fruits, vegetables, and peanuts. However, most studies focus on the bioavailability of flavonoid aglycone rather than its glycosylated form. Kaempferol-3-O-β-d-glucuronate (K3G) is a natural flavonoid glycoside obtained from various plants that have several biological activities, including antioxidant and anti-inflammatory effects. However, the molecular mechanism related to the antioxidant and antineuroinflammatory activity of K3G has not yet been demonstrated. The present study was designed to demonstrate the antioxidant and antineuroinflammatory effect of K3G against lipopolysaccharide (LPS)-stimulated BV2 microglial cells and to evaluate the underlying mechanism. Cell viability was determined by MTT assay. The inhibition rate of reactive oxygen species (ROS) and the production of pro-inflammatory mediators and cytokines were measured by DCF-DA assay, Griess assay, enzyme-linked immunosorbent assay (ELISA), and western blotting. K3G inhibited the LPS-induced release of nitric oxide, interleukin (IL)-6, and tumor necrosis factor-α (TNF-α) as well as the expression of prostaglandin E synthase 2. Additionally, K3G reduced the expression of inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), and nuclear factor-kappa B (NF-κB) related proteins. Mechanistic studies found that K3G downregulated phosphorylated mitogen-activated protein kinases (MAPKs) and upregulated the Nrf2/HO-1 signaling cascade. In this study, we demonstrated the effects of K3G on antineuroinflammation by inactivating phosphorylation of MPAKs and on antioxidants by upregulating the Nrf2/HO-1 signaling pathway through decreasing ROS in LPS-stimulated BV2 cells.
Introduction
Microglia is the first line of immunological defense in the central nervous system (CNS) against stimuli such as infectious pathogens and debris.1,2 However, the overactivation of microglia is also closely associated with the release of neurotoxic pro-inflammatory cytokine or protein to initiate neurodegenerative disease.3−5 BV2 murine microglial cells are widely used as an experimental model because they possess characteristics similar to primary microglia.6 During neuroinflammatory reactions, microglial cells produce pro-inflammatory mediators such as nitric oxide (NO), interleukin (IL)-6, tumor necrosis factor-α (TNF-α), inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), IL-1β, and prostaglandin E2 (PGE2) and activate nuclear factor-kappa B (NF-κB) and mitogen-activated protein kinases (MAPKs) pathways.7,8 Because excessive induction of neurotoxic pro-inflammatory cytokine or mediator can result in brain diseases, the modulation of microglia activation is an important therapeutic method to reduce neuronal injury and neuroinflammation.
Flavonoids found in various fruits, peanuts, and tea have been shown to be effective in neurodegeneration and cognitive decline as well as brain immune modulation.9 Kaempferol-3-O-β-d-glucuronate (K3G) is a flavonoid glycoside, which has various pharmacological effects. Originally, kaempferol suppressed the levels of inflammatory mediators10 and increased antioxidants,11 as well as attenuating neuroinflammation by blocking oxidative stress and inflammatory response through modulating reactive oxygen species (ROS)-dependent MAPKs and NF-κB pathways in microglia.12 Also, K3G exhibits anti-inflammatory effects through the inhibition of pro-inflammatory mediators, such as IL-6, IL-1β, NO, PGE2, and LTB4, as well as phosphorylation of NF-κB in RAW 264.7 cells.13 Moreover, kaempferol and its glycosides, including kaempferol-3-O-β-d-galactopyranoside, kaempferol-7-O-β-d-glucopyranoside, kaempferol-3-O-β-d-glucopyranoside, and kaempferol-3-O-α-l-rhamnopyranosyl-(1 → 6)-β-d-glucopyranoside, proved to be potent aldose reductase inhibitors.14 Active glucuronide such as K3G sodium has anti-HIV-1 activity.15
Although it has been reported previously that K3G has pharmacological effects that include antioxidant and anti-inflammatory properties, the specific antineuroinflammatory mechanism of K3G in lipopolysaccharide (LPS)-stimulated BV2 cells has not been investigated. Moreover, most studies focus on the effects of flavonoid aglycone rather than the glycosylated form. This study aimed to concentrate on the antineuroinflammatory effect of K3G via the inhibition of NO, iNOS, PTGES2, COX-2, and pro-inflammatory cytokines (IL-6 and TNF-α) and inactivating the phosphorylation of MAPKs, the NF-κB pathway, and antioxidants by upregulating the Nrf2/HO-1 signaling pathway through decreasing ROS in LPS-stimulated BV2 cells.
Results
Effects of K3G on BV2 Cell Viability
K3G was obtained from the stamens of Nelumbo nucifera as described in our published report.14 The chemical structure of K3G is shown in Figure 1. Before demonstrating the antineuroinflammatory activity of K3G, we investigated cell viability using an MTT assay in BV2 microglial cells following treatment with K3G at indicated concentrations from 1.56 to 50 μΜ for 24 h incubation. The results showed that BV2 microglial cells treated with K3G did not induce cytotoxic effects in the presence or absence of LPS when compared with the control (Figure 2). Therefore, up to 50 μΜ of K3G seemed to be nontoxic to BV2 microglial cells, and these concentrations were used for respective assays.
Figure 1.

Chemical structure of kaempferol-3-O-β-d-glucuronate (K3G).
Figure 2.

Effects of K3G on viability in BV2 microglial cells. Data are presented as the mean ± SD. One-way analysis of variance (ANOVA) followed by Turkey’s multiple comparison test was used for comparing differences between multiple groups. n.s., no significance.
Effects of K3G on Intracellular ROS Production
Activated microglial cells have been reported to increase oxidative stress, including the production of ROS, and reduce the antioxidant enzymes that prevent cell or tissue damage. Therefore, we measured whether K3G could prevent ROS production in LPS-stimulated BV2 cells using a DCFH-DA assay. Cells were pretreated with indicated concentrations (25 and 50 μM) of K3G for 2 h and stimulated with LPS (1 μg/mL) for 24 h. Figure 3 indicates that LPS-stimulated BV2 cells exhibited a high ROS level. However, K3G effectively attenuated ROS levels when compared with the LPS-treated group. As a positive control drug, Trolox (50 μM) significantly inhibited ROS release.
Figure 3.

Effects of K3G on the production of ROS in LPS-stimulated BV2 cells. Data are the mean ± SD. Significance: ###P < 0.001 vs non LPS treated cell values; ***P < 0.001 vs LPS-treated cell values.
Effects of K3G on Expression of iNOS and COX-2
NO is induced from l-arginine by iNOS in the brain, and this activation is related to the progression of neurodegenerative disease. COX-2 is an indicator of neuroinflammation. Moreover, iNOS and COX-2 are well-characterized indicators of NF-κB-related inflammation.16 Therefore, the blocking of iNOS and COX-2 can verify the antineuroinflammatory potency. Cells were pretreated with K3G (25 and 50 μM) for 2 h and stimulated with LPS (1 μg/mL) for 24 h. The expression of iNOS, COX-2, and β-actin was detected by western blotting. According to Figure 4A and B, LPS significantly increased the levels of iNOS and COX-2 in cells compared to the control group. However, K3G downregulated the expression of iNOS and COX-2 in a dose-dependent manner. Interestingly, 50 μM of K3G exhibited promising downregulatory effects against iNOS and COX-2.
Figure 4.

Effects of K3G on the expression of iNOS (A) and COX-2 (B) in LPS-stimulated BV2 cells. Data are the mean ± SD. Significance: #P < 0.05 and ###P < 0.001 vs non LPS treated cell values; *P < 0.05 and ***P < 0.001 vs LPS-treated cell values.
Effects of K3G on NO and PTGES2 Levels
LPS-stimulated NO production is an important part of the neuroinflammatory pathway. As an endotoxin material, LPS is widely used in the stimulation of microglia and then activates a neuroinflammatory response. Overproduction of NO by microglia is related to the initiation of neuronal cell death.17 Therefore, the reduction of NO is regarded as an effective method in the treatment of disease. In neuroinflammation, activated microglia produce prostaglandin E2, which causes neuronal cell death via the activation of the EP2 receptor.18 Prostaglandin E synthase 2 (PTGES2) catalyzes the conversion of prostaglandin H2 to prostaglandin E2.
In this study, we evaluated whether K3G could attenuate the production of NO and PTGES2 in LPS-stimulated BV2 microglial cells. Since NO and PGE2 are products of iNOS and COX-2 enzymes, respectively, we analyzed the effect of K3G treatment on the products of NO and PTGES2 in LPS-stimulated BV2 cells (Figure 5). Cells were pretreated with different concentrations of K3G for 2 h and then stimulated with LPS (1 μg/mL) for 18 h. The culture media was used to measure the amount of nitrite to determine NO production. As shown in Figure 5A, the LPS-treated cells significantly induced NO production compared to the control cells. However, significant concentration-dependent suppression by K3G (6.25, 12.5, 25, and 50 μM) in NO production was observed in BV2 cells (Figure 5A). As a positive control drug, AMT significantly inhibited LPS-stimulated NO release from BV2 cells. In addition, K3G concentration dependently diminished the production of PTGES2 in cells treated with LPS (Figure 5B).
Figure 5.

Effects of K3G on the levels of NO (A) and PTGES2 (B) in LPS-stimulated BV2 cells. Data are the mean ± SD. Significance: ##P < 0.01 and ###P < 0.001 vs non LPS treated cell values; **P < 0.01 and ***P < 0.001 vs LPS-treated cell values. AMT is used as a positive control.
Effects of K3G on Pro-Inflammatory Cytokines
Pro-inflammatory cytokines play a crucial role in the pathogenesis of various inflammatory diseases.19 Therefore, the reduction of pro-inflammatory cytokine is regarded as an effective method to treat neuroinflammatory disease. K3G was examined for its antineuroinflammatory activity on the level of pro-inflammatory cytokines, such as IL-6 and TNF-α, in LPS-stimulated BV2 cells using an enzyme-linked immunosorbent assay (ELISA) kit. Cells were pretreated with K3G (25 and 50 μM) for 2 h and then stimulated with LPS (1 μg/mL) for 18 h. The level of cytokines in the culture media was measured. As indicated in Figure 6A and B, LPS stimulation led to a significant increase in cytokine production. Pretreatment with K3G mildly inhibited the LPS-induced cytokine production in a dose-dependent manner. As a positive control drug, AMT inhibited pro-inflammatory cytokine release.
Figure 6.

Effects of K3G on the levels of IL-6 (A) and TNF-α (B) in LPS-stimulated BV2 cells. Data are the mean ± SD. Significance: ##P < 0.01 and ###P < 0.001 vs non LPS treated cell values; *P < 0.05, **P < 0.01, and ***P < 0.001 vs LPS-treated cell values. AMT is used as a positive control.
Effects of K3G on MAPKs Pathway
MAPKs are a family of serine/threonine protein kinases that play an important role in the inflammatory process, as well as in neuroinflammation,20 to produce inflammatory mediators, including iNOS, COX-2, and NF-κB.21 Moreover, the phosphorylation of MAPKs is related to the regulation of the NF-κB pathway. Inhibition of the MAPK pathway is involved in the control of neurodegenerative diseases. The MAPK pathway is known to play an important role in the transcriptional regulation of LPS-induced iNOS and COX-2 expression. Cells were pretreated with the indicated concentrations of K3G for 2 h and stimulated with LPS (1 μg/mL) for 18 h. The expression of MAPKs was detected by western blotting. As shown in Figure 7A–C, K3G (25 and 50 μM) strongly inhibited LPS-induced activation of p-JNK, p-ERK, and p-p38 MAPK in the BV2 cells. The inhibition rate of the p-p38 MAPK by K3G is higher than that of p-ERK and p-JNK.
Figure 7.

Effects of K3G on the MAPKs expressions in LPS-stimulated BV2 cells. Total ERK MAPK and phosphorylated ERK MAPK (A), total JNK MAPK and phosphorylated JNK MAPK (B), and total p38 MAPK and phosphorylated p38 MAPK (C). Data are presented as the mean ± SD. Significance: ##P < 0.01 and ###P < 0.001 vs non LPS treated cell values; *P < 0.05 and **P < 0.01 vs LPS-treated cell values.
Effects of K3G on the NF-κB Pathway
The translocation of NF-κB from the cytosol to the nucleus is regarded as an essential condition for the activation of inflammatory gene transcription. The activated NF-κB pathway is based on the degradation of IκBα, which activates nuclear translocation. The expression of inflammatory mediators in microglia is activated by the NF-κB pathway. Therefore, the inhibition of NF-κB transcriptional activity is a potential strategy in the treatment of neuroinflammatory disease.22 We investigated whether K3G would contribute to the attenuation of the NF-κB pathway in LPS-stimulated BV2 microglial cells. Cells were pretreated with the indicated concentrations of K3G for 4 h and stimulated with LPS (1 μg/mL) for 0.5 h. The expression of NF-κB related proteins was detected by western blotting. As shown in Figure 8A,B, the phosphorylation of IκBα is increased, while IκBα is decreased in LPS treatment group.
Figure 8.

Effects of K3G on the NF-κB related proteins in LPS-stimulated BV2 cells. IκBα (A), phosphorylated IκBα (B), cytosol NF-κB p65 (C), and nucleus NF-κB p65 (D). Data are the mean ± SD. Significance: #P < 0.05 vs non treated cell values; *P < 0.05 and **P < 0.01 vs LPS-treated cell values.
However, phosphorylation of IκBα is decreased in a dose-dependent manner after K3G treatment.
As shown in Figure 8C,D, the level of NF-κB p65 in the cytosol was decreased, whereas in the nucleus, it was increased after LPS treatment. However, the changes induced by LPS were inhibited by K3G pretreatment, and the nuclear translocation of NF-κB p65 decreased in a dose-dependent manner in LPS-stimulated BV2 microglial cells. This result suggests that K3G could inhibit NF-κB activation in BV2 microglial cells by blocking IκBα degradation and the nuclear translocation of NF-κB.
Effects of K3G on Regulation of the Nrf2/HO-1 Pathway
It is widely accepted that the induction of HO-1 expression can execute an antineuroinflammatory response by protecting neurons against neurotoxins. HO-1 is an important antioxidant against oxidative stress such as ROS. HO-1 was reported to be modulated by Nrf2, which is an antioxidant transcription factor. Furthermore, Nrf2 has antineuroinflammatory activity by the direct inhibition of the NF-κB pathway.23,24 In this study, we examined the effects of K3G on HO-1 and Nrf2 expression in LPS-stimulated BV2 microglia. Cells were pretreated with the indicated concentrations (25 and 50 μM) of K3G for 2 h and stimulated with LPS (1 μg/mL) for 18 h. The expression of HO-1 and Nrf2 was detected by western blotting (Figure 9). As shown in Figure 9A,B, pretreatment with up to 25 and 50 μM of K3G increased HO-1 expression. As expected, the expression of Nrf2 in microglia was dose-dependently increased when treated with K3G. It was indicated that the protective effect of K3G against ROS was related to an increase in Nrf2/HO-1 protein expression.
Figure 9.

Effects of K3G on the expression of HO-1 (A) and Nrf2 (B) in LPS-stimulated BV2 cells. Data are the mean ± SD. Significance: #P < 0.05 vs non treated cell values; *P < 0.05 and **P < 0.01 vs LPS-treated cell values.
Discussion
Inflammation is a host’s defense system against harmful stimuli, which leads to the induction of cytokine and chemokine to promote an inflammatory response.25 Although inflammation has a beneficial effect in the defense of the host, prolonged inflammation can induce the pathogenesis of many diseases, including chronic disorders, multiple sclerosis, arthritis, and neurodegenerative disease.26 In recent years, chronic inflammation has been related to several types of progressive diseases such as cancer, aging, neurological problems, and diabetes.27 Among the diseases, neurodegenerative disease is characterized by inflammation in the brain during the aging process.28 In particular, aging contributes to neurodegenerative disease by the induction of oxidative stress, loss of endogenous antioxidant defense, and production of free radicals. Degradation of the antioxidant system with increased ROS and production of reactive nitrogen species activates transcription factors such as NF-κB and other genes involved in neurodegeneration.29
LPS, which is the major outer surface membrane component of Gram-negative bacteria, could initiate major cellular responses such as the pathogenesis of inflammatory responses.30 LPS has been used to investigate an inflammatory model since it stimulates the production of cytokines, including IL-8, IL-6, TNF-α, and IL-1β.31,32 Microglial cells can be overactivated with LPS, resulting in a neuroinflammation response.33 Also, LPS induces inflammation by binding to the toll-like 4 receptor on the microglia, which can provoke transduction pathways, including JNK and NF-κB.
The CNS consists of neurons and glial cells. Glial cells are divided into oligodendrocytes, astrocytes, and microglial cells.34 Microglia constitute up to 20% of the cell population in regions of the brain.35 Homeostasis of microglia in the brain is important because moderate levels of microglia activation are related to immune defense. However, activated microglial cells have an important role in inflammation within the CNS because they can cause neuroinflammation and neurodegeneration. LPS-induced microglial cells are considered to be useful cellular and molecular models of neuroinflammation. Specifically, BV2 cells are a well-characterized model system for microglia. BV2 cells are a valid substitute for primary microglia in many experimental systems36 and are activated with LPS to establish a cellular model of neuroinflammation.
LPS induces an increase in the levels of pro-inflammatory cytokine in BV2 cells. COX-2 and iNOS are increased after LPS stimulation in microglia.37 LPS rapidly phosphorylates ERK, JNK, and p38 MAPK,7 leading to NF-κB activation in glial cells.20 This activation of glial cells leads to an increase in the production of pro-inflammatory mediators such as NO and PGE2,38 resulting in neuroinflammation.
Abnormally activated microglia can produce enzymes, including COX-2 and iNOS, which are responsible for neuroinflammation. Because COX-2 and iNOS are correlated with the pathophysiological condition of neuroinflammation,39 inhibition of these proteins is a crucial therapeutic method in the prevention of neurodegenerative disease. Neuroinflammation is characterized by an imbalance of the antioxidant system in the brain. Eventually, homeostatic imbalance leads to disease pathogenesis in brain immune cells which produce pro-inflammatory cytokines40 such as IL-1β, IL-6, TNF-α, and interferon (IFN)-γ. These mediators provoke neuronal dysfunction, loss of neurons, and cell death. If progress is more severe and ongoing, neurodegenerative disease occurs.41 Therefore, inhibition of overactivated microglia is an important strategy for the treatment of neuronal disease.42
Flavonoids have antioxidant43,7 and anti-inflammatory effects9,44 by controlling mediators involved in inflammation. They possess neuroprotective effects related to their ability to modulate neurodegenerative diseases. Various pure flavonoids, including quercetin, genistein, epigallocatechin-3-gallate, or total extracts, can decrease pro-inflammatory cytokines, prevent neuronal damage, and upregulate antioxidant ability. For example, flavonol glucuronide significantly inhibits ROS levels and elastase production, which may result in anti-inflammatory activity.45 Also, a previous study demonstrated that flavonoids isolated from N. nucifera exhibit both excellent metal chelating and radical scavenging properties in DPPH, ROS, and ONOO– model systems.46 The variety of flavonoids is dependent on their structure as well as the arrangement of the functional groups, including hydroxyl, methoxyl, and glycosidic units. The differences in the structures of flavonoids affect antioxidant activity. For example, a hydroxyl group at the C-3 positions is a structural requirement for the antioxidant ability of the flavonoids.
Among the flavonoids, kaempferol possesses good activity in all three antioxidant model systems. The major biological activities of kaempferol-3-O-β-d-glucuronide reported so far include antioxidant,47,48 rat lens aldose inhibition,14 anti-inflammatory,13 and antiHIV15 activities. K3G has been reported to exhibit anti-inflammatory effects in LPS-stimulated RAW264.7 cells and mice models.13 In addition, kaempferol glycosides have an antineuroprotective effect against neuroinflammation by inhibiting NF-κB and STAT3.47 Other flavonoids such as quercetin and catechin possess neuroprotective potential by ameliorating redox stress and modulating antiapoptotic pathways.48 Spinasterol was shown to produce HO-1 in mouse hippocampal HT22 and BV2 microglial cells.49 Quercetin-3-rutinoside exhibits the attenuation of neuroinflammation and oxidative stress in the spinal cord.50 Flavonoid-rich extracts, including quercetin-3-rhamnoside and isorhamnetin-3-O-β-rutinoside, have the potential for treating stroke because of their anti-inflammatory and antioxidant properties.51 However, most studies concentrate on the effects of flavonoid aglycone rather than its glycosylated form.
In this study, we demonstrated the effects of K3G on antineuroinflammation by inactivating the phosphorylation of MPAKs and upregulating the Nrf2/HO-1 signaling pathway through decreasing ROS in LPS-stimulated BV2 cells.
ROS production is important in the development of several inflammatory diseases. Excessive ROS-derived oxidative stress may induce the progression of the inflammatory, neuroinflammatory, and neurodegenerative response52,53 even though ROS have some biological benefits. Oxidative stress is a main factor in various neurodegenerative diseases. Alzheimer’s disease (AD), Huntington’s disease, and amyotrophic lateral sclerosis show increased levels of ROS in the brain.54,55 ROS can act as a stimulator of pro-inflammatory gene expression by activating MAPKs.12 Inhibition of ROS production regulates the activation of microglia by decreasing the phosphorylation of MAPKs. Similarly, LPS-induced ROS generation can affect the LPS-stimulated activation of MAPKs.30 It is well known that ROS have an important influence on the activation of Nrf2. ROS production is related to the activation of the Nrf2/HO-1 signaling pathway56 because Nrf2/HO-1 signaling acts as an antioxidant to reduce the level of ROS.57 Therefore, the blocking of ROS generation may have potential as a therapeutic target to reduce neuronal degeneration.
In the present study, we demonstrated that pretreatment with K3G significantly scavenged LPS-stimulated ROS production in BV2 cells. The level of ROS was decreased in a dose-dependent manner (Figure 3). This result indicated that K3G induced an antioxidative effect by scavenging ROS.
NO is the product of inducible nitric oxide synthase, which has an important role in neuroinflammation. A high level of NO, which is produced from l-arginine by iNOS, can lead to an inflammatory reaction, neurodegenerative disorder,58 and activates microglial cells. During the inflammatory processes in the CNS, pro-inflammatory mediators such as NO can recruit microglial cells at the inflammatory site.59 It is well known that activated microglia induce NO, which contributes to neurodegenerative diseases such as AD. In addition, PGE2 is derived from arachidonic acid by COX-2. COX-2 inhibition could be the reason for the downregulation of PGE2 in microglial cells. Our study showed that iNOS and COX-2 are downregulated in cells after up to 50 μΜ of K3G treatment (Figure 4) without showing any signs of toxicity (Figure 2). The iNOS and COX-2 inhibition could be the reason for the downregulation of NO and PTGES2 production in the activated microglial cells (Figure 5A,B). From these results, we revealed that the inhibitory effects of K3G on NO and PTGES2 production were mediated by the inhibition of iNOS and COX-2 protein expression, respectively.
ROS are recognized as mediators in neuronal damage because of the production of pro-inflammatory cytokines. The increase in the level of pro-inflammatory cytokines follows a similar pattern to the production of ROS.60 Moreover, pro-inflammatory cytokines such as IL-6, IL-1β, and TNF-α play an important role in inducing neurodegeneration in the CNS.61,62 IL-6 and TNF-α are pro-inflammatory cytokines that are produced at an early stage, and their raised levels can be found in a variety of inflammatory diseases.63 Overexpression of pro-inflammatory cytokines can be regarded as an indicator of neurological disease in the brain.64,65 Using a natural product such as K3G is a promising therapy in neuroinflammation because it could block pro-inflammatory cytokines. Our results suggest that in LPS-stimulated BV2 microglial cells, K3G has a mildly decreasing effect in the suppression of pro-inflammatory cytokines, including IL-6 and TNF-α (Figure 6A,B).
MAPKs play an important role in the regulation of cell growth, differentiation, and the control of cellular responses as well as the modulation of inflammatory processes. As protein kinases, MAPKs, which include ERK, JNK, and p38 MAPK, have received attention due to their role in the phosphorylation of transcription factors, activation of inflammatory response,43,66 and neuroinflammation.20
Microglia are resident immune cells in the brain and important regulators in a variety of neurodegenerative diseases.67 Activation of microglia produces pro-inflammatory mediators, including cytokines, chemokines, and NO.68 Above all, ERK signaling is an important regulator of pro-inflammatory cytokine production in microglia69 and a critical regulator of IFNγ-mediated pro-inflammatory activation of microglia.70 In addition, ERK signaling is related to the promotion of neural cell death and can be involved in neurodegeneration because the phosphorylation of ERK is related to neuronal disease-associated microglial gene expression and neurotoxic molecules. The inhibition of ERK signaling decreases brain injuries, including cerebral ischemia trauma, since chronic activation of ERK can contribute to the pathogenesis of neural disease. Phosphorylated ERK was found in aggregates in the substantia nigra in the brain of patients with Parkinson’s disease.71 Also, the relationship between β-amyloid (Aβ) and ERK activation is involved in AD. Increased phosphorylation of ERK was found in brain extracts from patients with AD.72 Therefore, the blocking of ERK signaling in microglia could be a critical target in the treatment of neurodegenerative disease.
JNKs are involved in regulatory physiological processes, including brain development, memory repair, and function.73 They are activated by stress such as pro-inflammatory mediators.74 JNK is an important indicator of inflammation and stress responses in the pathogenesis of Parkinson’s disease, AD, and stroke75,76 because increased JNK phosphorylation may be related to the mechanism of neuroinflammation. Neurodegeneration could be induced by JNK phosphorylation followed by activator protein 1 (AP-1) and inflammatory mediators, including NO, iNOS, COX-2, and PGE2, and pro-inflammatory cytokines, such as IL-6, IL-1β, and TNF-α, leading to subsequent neurotoxicity.33,77−79 The JNK pathway, which is activated by cellular stimuli, is related to an increased level of Aβ plaques and neurofibrillary tangles as well as neuron cell death in AD patients.80 JNKs include JNK1, JNK2 (which is expressed in many tissues), and JNK3, which is abundant in the brain. Among the JNKs, JNK3 and pro-inflammatory cytokines, such as IL-6, IL-1β, and TNF-α, are elevated in chronic social defeat stressed mice.81 Therefore, the inhibition of JNK3 may alleviate symptoms of neuroinflammation and neurodegeneration. Among JNK inhibitors, the development of a JNK3 inhibitor may have the potential in preventing neuroinflammation because JNK3 is expressed in the brain.82 SP600125, which is a JNK inhibitor, blocks the production of IL-6, IL-8, and IL-1β.83
P38 MAPK is activated by LPS stimulation and may play an important role in the increase of iNOS and TNF-α gene expression.84 As a member of the MAPK family, p38 MAPK participates in inflammation and other physiological responses.85 For example, p38 MAPK plays an important role in neuronal diseases, including Parkinson’s disease and AD, because it can induce neuroinflammation and degeneration. In addition, p38 MAPK has a central role in modulating chronic pain, microglial activation, rheumatoid arthritis, and cardiovascular disease.85−88 Moreover, p38 MAPK could regulate the induction of endothelial vascular cell adhesion molecule-1 (VCAM-1), which is involved in cell proliferation and differentiation of the immune reaction.89 The p38 MAPK is rapidly phosphorylated at a tyrosine residue in response to LPS and other stimulants, including inflammatory cytokines, ultraviolet irradiation, toxicants, and ROS. The phosphorylation of p38 MAPK leads to an increase in pro-inflammatory mediators such as COX-2 and PGE2.90 However, the blocking of p38 MAPK induces suppression of pro-inflammatory cytokines, such as TNF-α, IL-6 and IL-1β, iNOS, COX-2, and PGE2 in RAW 264.7 cells treated with LPS.91 Because p38 MAPK is involved in glial activation and neuroinflammation, inhibition of p38 MAPK leads to the alleviation of neurotoxicity.92 Anti-inflammatory drugs, such as GSK-681323 to treat rheumatoid arthritis, SCIO-469 to treat multiple myeloma, and RWJ67657 to treat inflammatory diseases, are designed to inhibit p38 MAPK activity.93 Also, natural plant extracts or plant-derived compounds that can block p38 MAPK activity and exhibit strong anti-inflammatory properties are potential therapeutic drug candidates. It has been reported that p38 MAPK inhibitors have been used as a potential therapy for neurodegenerative disease.94
In this study, we found that K3G could attenuate neuroinflammation by inhibiting the MAPK and NF-κB signaling pathways (Figures 7 and 8). These findings demonstrate that K3G has a fundamental role in the antineuroinflammation effect of MAPK because it can scavenge ROS, which can activate MAPKs to trigger neuroinflammation.
Recent studies have demonstrated that LPS-induced inflammation is related to complicated cellular signaling pathways, including NF-κB and MAPKs.22,25 The activation of NF-κB is necessary for the release of iNOS, COX-2, pro-inflammatory cytokines in the inflammatory response,95 and related MAPKs.8,96 Specifically, the phosphorylation of the IκB protein is an important step in the activation of NF-κB. It is well known that the activity of NF-κB is inhibited in the cytoplasm while combined with an IκB protein.97 The phosphorylation or degradation of the IκB protein by IκB kinase results in the activation of NF-κB and the transcription of inflammatory mediators, including IL-1β, TNF-α, and IL-6.98 Because the expression of inflammatory mediators in microglia is increased through the phosphorylation of NF-κB,99 it is an important target for the treatment of neuroinflammatory disease. In our study, we demonstrated that K3G decreased the phosphorylation of IκBα (Figure 8A,B). Furthermore, western blot analysis confirmed that K3G suppressed the translocation of NF-κB p65. NF-κB p65, which is translocated from the cytosol to the nucleus after LPS treatment, was also blocked (Figure 8C,D). Consistent with these findings, the MPAK and NF-κB signaling pathway was shown to be related to the K3G mediated inhibitory effect on neuroinflammation in microglial BV2 cells.
The Nrf2/HO-1 signaling pathway is believed to have neuroprotective potential by activating antioxidant enzymes and decreasing oxidative stress. It has a cytoprotective reaction in the brain because it can activate the expression of neuroprotective genes that code for antioxidant and anti-inflammation proteins.100 However, the inactivation of HO-1 and dysfunction of Nrf2 increased the production of ROS and neurological problems.101,102 In addition, the downregulation of Nrf2/HO-1 decreases antioxidant enzymes and results in the degeneration of 5-hydroxytryptamine (5-HT),103 dopamine (DA), and acetylcholinesterase (AchE) in neurons. Finally, it can provoke neurological problems such as cognitive dysfunction and neuropsychological disturbances. Nrf2 promotes the expression of HO-1 and can protect against oxidative stress by blocking apoptosis and inflammation in the brain.101 When HO-1 is deficient, the risk of neuroinflammation rises and the level of ROS is increased. Activation of HO-1 and Nrf2 decreased LPS-induced pro-inflammatory mediator NO and pro-inflammatory related genes and lowered JNK, whereas anti-inflammatory cytokines such as IL-10 and IL-4 are increased in BV2 microglial cells.33 HO-1 expression is closely mediated by Nrf2 in inhibiting the inflammatory response. To demonstrate the antioxidative potential of K3G, the expression of HO-1 and Nrf2 has been demonstrated and results showed strong antioxidative effects of K3G at the indicated concentrations. Both HO-1 and Nrf2 were upregulated in response to K3G in LPS-stimulated BV2 cells (Figure 9A,B). Our results suggest that K3G induced the activation of Nrf2/HO-1 via a ROS-dependent MAPK signaling pathway.
Conclusions
The present study is the first to report the inhibitory effects of K3G on neuroinflammation by suppressing inflammatory mediators, including iNOS and COX-2, and pro-inflammatory cytokines, such as IL-6, PGE2, and TNF-α, in LPS-stimulated BV2 cells. These beneficial effects could be linked to the MAPKs and NF-κB pathway to inhibit neuroinflammation. We also found that K3G suppresses ROS generation through the inhibition of MAPK phosphorylation in BV2 cells. K3G effectively suppressed ROS production by increasing the Nrf2/HO-1 signaling pathway. Although K3G exhibited potent antineuroinflammatory effects in vitro, its function in vivo has yet to be clarified. Verification of the antineuroinflammatory effects of K3G and demonstration of relative mechanism in vivo models will be useful for the application of K3G as a therapeutic agent for neuroinflammatory diseases.
Materials and Methods
Chemicals and Reagents
Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Welgene (Gyeongsan, Korea). Fetal bovine serum (FBS) and antibiotics were obtained from Gibco BRL (Rockville, MD, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), 2-amino-5,6-dihydro-6-methyl-4H-1,3-thiazine hydrochloride (AMT), 2′,7′-dichlorofluorescein diacetate (DCFH-DA), and (±)-6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) were acquired from Sigma-Aldrich (St. Louis, MO, USA). Primary antibodies (iNOS, COX-2, phospho-NF-κB p65, NF-κB p65, phospho-IκBα, IκBα, phospho-ERK, ERK, phospho-JNK, JNK, phospho-p38 MAPK, p38 MAPK, HO-1, Nrf2, PTGES2, Lamin B1, and β-actin) and secondary antibodies were obtained from Cell Signaling Technology Inc. (Beverly, MA, USA) and Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Polyvinylchloride fluoride (PVDF) membrane was obtained from Millipore (Billerica, MA, USA), and Super-Signal West Pico Chemiluminescent Substrate was purchased from Pierce Biotechnology (Rockford, IL, USA). Total protein of the cell was extracted using PRO-PREP solution (iNtRON Biotechnology, Seongnam, Korea). NE-PER nuclear and cytoplasmic extraction reagents were purchased from Thermo Scientific (Carlsbad, CA, USA). ELISA for cytokine detection was purchased from BD Biosciences (San Jose, CA, USA). All other chemicals and solvents were purchased from Sigma-Aldrich unless otherwise stated.
Cell Culture and LPS Stimulation
The BV2 immortalized murine microglial cells were obtained from Prof. Yung Hyun Choi (Department of Biochemistry, College of Oriental Medicine, Dong-Eui University, Busan, Korea). The BV2 microglial cells were cultured in DMEM supplemented with 10% FBS, penicillin (100 units/mL), and streptomycin (100 μg/mL) at 37 °C in a humidified incubator containing 5% CO2. K3G dissolved in DMSO as a 50 mM stock solution, and dilutions were made in DMEM. To stimulate cells, the medium was replaced with fresh DMEM and 1 μg/mL LPS was added in the presence or absence of K3G for the indicated time periods.
Cell Viability
Cell viability was assessed using the MTT assay. Briefly, BV2 cells were seeded into 96-well plates at a density of 5 × 103 cells/well and incubated at 37 °C for 24 h. The cells were then treated with K3G at various concentrations (1.56, 3.12, 6.25, 12.5, 25, and 50 μM) in the presence or absence of LPS. After incubation for an additional 24 h at 37 °C, 100 μL of MTT solution (0.5 mg/mL in PBS) was added to each well and the incubation continued for another 3 h. Then, the supernatant was removed and crystal formazan was dissolved in 100% DMSO. The resulting color was measured at 540 nm using a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Measurement of Intracellular ROS
The intracellular ROS scavenging activity was measured using a ROS-sensitive fluorescence indicator DCFH-DA.104 In the presence of ROS, DCFH is oxidized to fluorescent 2′,7′-dichlorofluorescein (DCF), which is detected using a fluorescent spectrophotometer. BV2 cells were incubated with the indicated concentrations (25 and 50 μM) of K3G for 2 h and stimulated with LPS (1 μg/mL) for another 24 h. The DCF fluorescence intensity was measured at an excitation wavelength of 485 nm and an emission wavelength of 528 nm using a VICTOR X multilabel reader (Perkin-Elmer, Wellesley, MA, USA). The ROS inhibitor Trolox was used as a positive control.
Western Blot Analysis
To detect the level of protein, a western blot technique was used. BV2 cells were cultured in 6-well plates and pretreated with K3G in the presence or absence of LPS. Afterward, the cells were washed twice with cold PBS and lysed using PRO-PREP solution. The concentration of protein in the supernatants was determined using a BCA protein assay kit (Thermo Fisher Scientific, Rockford, IL, USA). After quantification, the protein of the cell was denatured by boiling at 94 °C for 5 min and it was separated using 8–15% gels. After gel electrophoresis, the protein was transferred to PVDF membranes. The PVDF membrane was soaked in 5% skim milk or 3% bovine serum albumin solution to block nonspecific responses at room temperature for 1 h. Next, the membrane was incubated with each specific primary antibody at 4 °C overnight incubation. After washing with TBST buffer, the membrane was incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 1 h. After another washing step, the protein was visualized with a Super-Signal West Pico Chemiluminescent Substrate according to the manufacturer’s instructions. Densitometric analysis of the bands was performed using GeneGnome 5 image analysis (Synoptics, Cambridge, U.K.) software.
NO Production
The NO concentration in the medium was measured using Griess reagent as a marker of NO production. Briefly, BV2 cells (10 × 104 cells/well) in a 24-well plate with 500 μL culture medium were pretreated with various concentrations (6.25, 12.5, 25, and 50 μM) of K3G for 2 h and incubated for 18 h with LPS (1 μg/mL). After incubation, the supernatant was collected and stored at −80 °C until use. The absorbance values of the mixture were determined using a microplate spectrophotometer (Molecular Devices Sunnyvale, CA, USA) at 540 nm. The NO inhibitor AMT was used as a positive control.
Measurement of Cytokine
Following the manufacturer’s instructions, the levels of cytokines, TNF-α and IL-6, were measured using ELISA kits (BD Biosciences, San Jose, CA, USA). Absorbance was determined at 450 nm using a microplate spectrophotometer (Molecular Devices Sunnyvale, CA, USA).
Extraction of Nuclear and Cytosol Protein
Nuclear and cytosolic proteins were separated using a NE-PER nuclear and cytoplasmic extraction kit. Cytoplasmic extraction reagents I and II and nuclear extraction reagent were prepared immediately before separating the proteins in the nucleus and cytosol. Briefly, BV2 microglial cells were harvested with trypsin–EDTA and then centrifuged at 500 × g for 5 min. The cells were washed with PBS. After removal of the supernatant, leaving the cell pellet as dry as possible, ice-cold cytoplasmic extraction reagent I was added to the cell pellet to lyse the cells. After incubation on ice for 10 min, ice-cold cytoplasmic extraction reagent II was added to the tube. The cells were again centrifuged at 16,000 × g for 5 min at 4 °C. Cytosolic protein extract was obtained by collecting the supernatant and storing it at −80 °C until use. The remaining cell pellets were resuspended in a nuclear extraction reagent for nuclear protein extract. After incubation on ice for 40 min, the cells were centrifuged at 16,000 × g at 4 °C for 10 min. The nuclear protein extract was obtained by collecting the supernatant.
Statistics Analysis
Data were expressed as the means ± standard deviations (SD) of at least three independent experiments unless otherwise indicated. Data were compared using one-way ANOVA. Data analysis was carried out using GraphPad Prism version 5 program. P values <0.05, 0.01, and 0.001 were considered significant. All analyses were performed using GraphPad Prism version 5.
Acknowledgments
The authors are thankful to Professor Yung Hyun Choi (Department of Biochemistry, College of Oriental Medicine, Dongeui University, Busan, Korea) for providing necessary materials. This research was supported by a National Research Foundation of Korea (NRF) grant funded by the Ministry of Science and ICT (No. 2020R1C1C1008331 [HNIBR202100303]).
Author Contributions
J.S.C. contributed to the conception and design of the experiment. H.J.L., R.P., and S.H.S. performed all experiments and verified the analytical data. J.S.C. and H.A.J. supervised the experiments in discussion with H.J.L., and H.J.L. wrote the manuscript. All authors discussed the final results and approved the final manuscript.
The authors declare no competing financial interest.
References
- Amor S.; Puentes F.; Baker D.; van der Valk P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar-Colucci S.; Landreth G. E. Microglia and inflammation in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 156–167. 10.2174/187152710791012071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D.; Baker T. J.; Shih L. C.; Lachman L. B. Interleukin 1 of the central nervous system is produced by ameboid microglia. J. Exp. Med. 1986, 164, 594–604. 10.1084/jem.164.2.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung W. K.; Ahn Y. W.; Lee S. H.; Choi Y. H.; Kim S. K.; Yea S. S.; Choi I. H.; Park S. G.; Seo S. K.; Lee S. W.; Choi I. W. Ecklonia cava ethanolic extracts inhibit lipopolysaccharide-induced cyclooxygenase-2 and inducible nitric oxide synthase expression in BV2 microglia via the MAP kinase and NF-κB pathways. Food Chem. Toxicol. 2009, 47, 410–417. 10.1016/j.fct.2008.11.041. [DOI] [PubMed] [Google Scholar]
- Stephenson J.; Nutma E.; van der Valk P.; Amor S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. 10.1111/imm.12922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocchini V.; Mazzolla R.; Barluzzi R.; Blasi E.; Sick P.; Kettenmann H. An immortalized cell line expresses properties of activated microglial cells. J. Neurosci. Res. 1992, 31, 616–621. 10.1002/jnr.490310405. [DOI] [PubMed] [Google Scholar]
- Guo C.; Yang L.; Wan C. X.; Xia Y. Z.; Zhang C.; Chen M. H.; Wang Z. D.; Li Z. R.; Li X. M.; Geng Y. D.; Kong L. Y. Anti-neuroinflammatory effect of sophoraflavanone G from sophora alopecuroides in LPS-activated BV2 microglia by MAPK, JAK/STAT and Nrf2/HO-1 signaling pathways. Phytomedicine 2016, 23, 1629–1637. 10.1016/j.phymed.2016.10.007. [DOI] [PubMed] [Google Scholar]
- Yuan L.; Wu Y. C.; Ren X.; Liu Q.; Wang J.; Liu X. Isoorientin attenuates lipopolysaccharide-induced pro-inflammatory responses through down-regulation of ROS-related MAPK/NF-κB signaling pathway in BV-2 microglia. Mol. Cell. Biochem. 2014, 386, 153–165. 10.1007/s11010-013-1854-9. [DOI] [PubMed] [Google Scholar]
- Spencer J. P.; Vafeiadou K.; Williams R. J.; Vauzour D. Neuroinflammation: modulation by flavonoids and mechanisms of action. Mol. Aspects Med. 2012, 33, 83–97. 10.1016/j.mam.2011.10.016. [DOI] [PubMed] [Google Scholar]
- Park M. J.; Lee E. K.; Heo H. S.; Kim M. S.; Sung B.; Kim M. K.; Lee J.; Kim N. D.; Anton S.; Choi J. S.; Yu B. P.; Chung H. Y. The anti-inflammatory effect of kaempferol in aged kidney tissues: the involvement of nuclear factor-kappa B via nuclear factor inducing kinase/Ikappa B kinase and mitogen-activated protein kinase pathways. J. Med. Food 2009, 12, 351–358. 10.1089/jmf.2008.0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Sun W.; Sun X.; Wang Y.; Zhou M. Kaempferol ameliorates cisplatin induced nephrotoxicity by modulating oxidative stress, inflammation and apoptosis via ERK and NF-κB pathways. AMB Express 2020, 10, 58. 10.1186/s13568-020-00993-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Yao X.; Sun B.; Jiang W.; Liao C.; Dai X.; Chen Y.; Chen J.; Ding R. Pretreatment with kaempferol attenuates microglia-mediate neuroinflammation by inhibiting MAPKs-NF-κB signaling pathway and pyroptosis after secondary spinal cord injury. Free Radical Biol. Med. 2021, 168, 142–154. 10.1016/j.freeradbiomed.2021.03.037. [DOI] [PubMed] [Google Scholar]
- Khajuria V.; Gupta S.; Sharma N.; Tiwari H.; Bhardwaj S.; Dutt P.; Satti N.; Nargotra A.; Bhagat A.; Ahmed Z. Kaempferol-3-o-β-d-glucuronate exhibit potential anti-inflammatory effect in LPS stimulated RAW 264.7 cells and mice model. Int. Immunopharmacol. 2018, 57, 62–71. 10.1016/j.intimp.2018.01.041. [DOI] [PubMed] [Google Scholar]
- Lim S. S.; Jung Y. J.; Hyun S. K.; Lee Y. S.; Choi J. S. Rat lens aldose reductase inhibitory constituents of Nelumbo nucifera stamens. Phytother. Res. 2006, 20, 825–830. 10.1002/ptr.1847. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Huang N.; Lu J. C.; Li X.; Wang Y. H.; Yang L. M.; Zheng Y. T.; Xiao A. K. Water-soluble phenolic compounds and their anti-HIV-1 activities from the leaves of Cyclocarya paliurus. J. Food Drug Anal. 2010, 18, 2. 10.38212/2224-6614.2230. [DOI] [Google Scholar]
- Sharif O.; Bolshakov V. N.; Raines S.; Newham P.; Perkins N. D. Transcriptional profiling of the LPS induced NF-kappaB response in macrophages. BMC Immunol. 2007, 8, 1. 10.1186/1471-2172-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste J. E.; Tarragon E.; Campuzano C. M.; Ros-Bernal F. Implications of glial nitric oxide in neurodegenerative diseases. Front. Cell Neurosci. 2015, 9, 322. 10.3389/fncel.2015.00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagishi H.; Kosuge Y.; Yoneoka Y.; Ozone M.; Endo M.; Osada N.; Ishige K.; Kusama-Eguchi K.; Ito Y. Prostaglandin E2-induced cell death is mediated by activation of EP2 receptors in motor neuron-like NSC-34 cells. J. Pharmacol. Sci. 2013, 121, 347–350. 10.1254/jphs.12274SC. [DOI] [PubMed] [Google Scholar]
- Fujiwara N.; Kobayashi K. Macrophages in inflammation. Curr. Drug. Targets Inflamm. Allergy 2005, 4, 281–286. 10.2174/1568010054022024. [DOI] [PubMed] [Google Scholar]
- Velagapudi R.; Aderogba M.; Olajide O. A. Tiliroside, a dietary glycosidic flavonoid, inhibits TRAF-6/NF-κB/p38-mediated neuroinflammation in activated BV2 microglia. Biochim. Biophys. Acta 2014, 1840, 3311–3319. 10.1016/j.bbagen.2014.08.008. [DOI] [PubMed] [Google Scholar]
- Suh S. J.; Chung T. W.; Son M. J.; Kim S. H.; Moon T. C.; Son K. H.; Kim H. P.; Chang H. W.; Kim C. H. The naturally occurring biflavonoid, ochnaflavone, inhibits LPS-induced iNOS expression, which is mediated by ERK1/2 via NF-κB regulation, in RAW264.7 cells. Arch. Biochem. Biophys. 2006, 447, 136–146. 10.1016/j.abb.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Li J.; Kim K. W.; Oh H. C.; Kim Y. C. Anti-inflammatory effects of Sanhuang-Siwu-Tang in lipopolysaccharide-stimulated RAW264.7 macrophages and BV2 microglial cells. Biol. Pharm. Bull. 2021, 44, 535–543. 10.1248/bpb.b20-00871. [DOI] [PubMed] [Google Scholar]
- Innamorato N. G.; Rojo A. I.; García-Yagüe Á. J.; Yamamoto M.; de Ceballos M.; Guadrado A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J. Immunol. 2008, 181, 680–689. 10.4049/jimmunol.181.1.680. [DOI] [PubMed] [Google Scholar]
- Wang M.; Wang K.; Gao X.; Zhao K.; Chen H.; Xu M. Anti-inflammatory effects of isoalantolactone on LPS-stimulated BV2 microglia cells through activating GSK-3β-Nrf2 signaling pathway. Int. Immunopharmacol. 2018, 65, 323–327. 10.1016/j.intimp.2018.10.008. [DOI] [PubMed] [Google Scholar]
- Roy A.; Park H. J.; Abdul Q. A.; Jung H. A.; Choi J. S. Pulegone exhibits anti-inflammatory activities through the regulation of NF-κB and Nrf-2 signaling pathways in LPS-stimulated RAW264.7 cells. Nat. Prod. Sci. 2018, 24, 28–35. 10.20307/nps.2018.24.1.28. [DOI] [Google Scholar]
- Choudhari A. S.; Raina P.; Deshpande M. M.; Wali A. G.; Zanwar A.; Bodhankar S. L.; Kaul-Ghaneka R. Evaluating the anti-inflammatory potential of Tectaria cicutaria L. rhizome extract in vitro as well as in vivo. J. Ethnopharmacol. 2013, 150, 215–222. 10.1016/j.jep.2013.08.025. [DOI] [PubMed] [Google Scholar]
- Libby P. Inflammatory mechanisms: the molecular basis of inflammation and disease. Nutr. Rev. 2007, 65, S140–S146. 10.1111/j.1753-4887.2007.tb00352.x. [DOI] [PubMed] [Google Scholar]
- Akiyama H.; Barger S.; Barnum S.; Bradt B.; Bauer J.; Cole G. M.; Cooper N. R.; Eikelenboom P.; Emmerling M.; Fiebich B. L.; Finch C. E.; Frautschy S.; Griffin W. S.; Hampel H.; Hull M.; Landreth G.; Lue L.; Mrak R.; Mackenzie I. R.; McGeer P. L.; O’Banion M. K.; Pachter J.; Pasinetti G.; Plata-Salaman C.; Rogers J.; Rydel R.; Shen Y.; Streit W.; Strohmeyer R.; Tooyoma I.; Van Muiswindel F. L.; Veerhuis R.; Walker D.; Webster S.; Wegrzyniak B.; Wenk G.; Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. 10.1016/S0197-4580(00)00124-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catani M. V.; Gasperi V.; Bisogno T.; Maccarrone M. Essential dietary bioactive lipids in neuroinflammatory diseases. Antioxid. Redox Signaling 2018, 29, 37–60. 10.1089/ars.2016.6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.; Min J. S.; Kim B.; Chae U. B.; Yun J. W.; Choi M. S.; Kong I. K.; Chang K. T.; Lee D. S. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-κB pathways. Neurosci. Lett. 2015, 584, 191–196. 10.1016/j.neulet.2014.10.016. [DOI] [PubMed] [Google Scholar]
- Meng F.; Lowell C. A. Lipopolysaccharide (LPS)-induced macrophage activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. J. Exp. Med. 1997, 185, 1661–1670. 10.1084/jem.185.9.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S.; Chen G.; Qi M.; El-Assaad F.; Wang Y.; Dong S.; Chen L.; Yu D.; Weaver J. C.; Beretov J.; Krilis S. A.; Giannakopoulos B. Gram negative bacterial inflammation ameliorated by the plasma protein beta 2-Glycoprotein I. Sci. Rep. 2016, 6, 33656. 10.1038/srep33656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subedi L.; Lee J. H.; Yumnam S.; Ji E.; Kim S. Y. Anti-inflammatory effect of sulforaphane on LPS-activated microglia potentially through JNK/AP-1/NF-κB inhibition and Nrf2/HO-1 activation. Cell 2019, 8, 194. 10.3390/cells8020194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen N. J.; Lyons D. A. Glia as architects of central nervous system formation and function. Science 2018, 362, 181–185. 10.1126/science.aat0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrenis K. Microglia in cell culture and in transplantation therapy for central nervous system disease. Methods 1998, 16, 320–344. 10.1006/meth.1998.0688. [DOI] [PubMed] [Google Scholar]
- Henn A.; Lund S.; Hedtjärn M.; Schrattenholz A.; Pörzgen P.; Leist M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX 2009, 26, 83–94. 10.14573/altex.2009.2.83. [DOI] [PubMed] [Google Scholar]
- Dai J. N.; Zong Y.; Zhong L. M.; Li Y. M.; Zhang W.; Bian L. G.; Ai Q. L.; Liu Y. D.; Sun J.; Lu D. Gastrodin inhibits expression of inducible NO synthase, cyclooxygenase-2 and proinflammatory cytokines in cultured LPS-stimulated microglia via MAPK pathways. PLoS One 2011, 6, e21891 10.1371/journal.pone.0021891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lull M. E.; Block M. L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. 10.1016/j.nurt.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vane J. R.; Mitchell J. A.; Appleton I.; Tomlinson A.; Bishop-Bailey D.; Croxtall J.; Willoughby D. A. Inducible isoforms of cyclooxygenase and nitric-oxide synthase in inflammation. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 2046–2050. 10.1073/pnas.91.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkley K. S.; Popichak K. A.; Afzali M. F.; Legare M. E.; Tjalkens R. B. Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. J. Neuroinflammation 2017, 14, 99. 10.1186/s12974-017-0871-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dansokho C.; Heneka M. T. Neuroinflammatory responses in Alzheimer’s disease. J. Neural. Transm. 2018, 125, 771–779. 10.1007/s00702-017-1831-7. [DOI] [PubMed] [Google Scholar]
- Choi Y. H.; Park H. Y. Anti-inflammatory effects of spermidine in lipopolysaccharide-stimulated BV2 microglial cells. J. Biomed. Sci. 2012, 19, 31. 10.1186/1423-0127-19-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D. S.; Kim K. S.; Ko W.; Li B.; Jeong G. S.; Jang J. H.; Oh H.; Kim Y. C. The cytoprotective effect of sulfuretin against tert-butyl hydroperoxide-induced hepatotoxicity through Nrf2/ARE and JNK/ERK MAPK-mediated heme oxygenase-1 expression. Int. J. Mol. Sci. 2014, 15, 8863–8877. 10.3390/ijms15058863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnuolo C.; Moccia S.; Russo G. L. Anti-inflammatory effects of flavonoids in neurodegenerative disorders. Eur. J. Med. Chem. 2018, 153, 105–115. 10.1016/j.ejmech.2017.09.001. [DOI] [PubMed] [Google Scholar]
- Granica S.; Czerwińska M. E.; Żyżyńska-Granica B.; Kiss A. K. Antioxidant and anti-inflammatory flavonol glucuronides from Polygonum aviculare L. Fitoterapia 2013, 91, 180–188. 10.1016/j.fitote.2013.08.026. [DOI] [PubMed] [Google Scholar]
- Jung H. A.; Kim J. E.; Chung H. Y.; Choi J. S. Antioxidant principles of Nelumbo nucifera stamens. Arch. Pharmacal Res. 2003, 26, 279–285. 10.1007/BF02976956. [DOI] [PubMed] [Google Scholar]
- Ye L.; Chen C.; Wang L. F.; Kuang X.; Liu K.; Zhang H.; Du J. R. Neuroprotective effect of kaempferol glycosides against brain injury and neuroinflammation by inhibiting the activation of NF-κB and STAT3 in transient focal stroke. PLoS One 2013, 8, e55839 10.1371/journal.pone.0055839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josiah S. S.; Famusiwa C. D.; Crown O. O.; Lawal A. O.; Olaleye M. T.; Akindahunsi A. A.; Akinmoladun A. C. Neuroprotective effects of catechin and quercetin in experimental Parkinsonism through modulation of dopamine metabolism and expression of IL-1β, TNF-α, NF-κB, IκKB and p53 genes in male Wistar rats. Neurotoxicology 2022, 90, 158–171. 10.1016/j.neuro.2022.03.004. [DOI] [PubMed] [Google Scholar]
- Jeong G. S.; Li B.; Lee D. S.; Kim K. H.; Lee I. K.; Lee K. R.; Kim Y. C. Cytoprotective and anti-inflammatory effects of spinasterol via the induction of heme oxygenase-1 in murine hippocampal and microglial cell lines. Int. Immunopharmacol. 2010, 10, 1587–1594. 10.1016/j.intimp.2010.09.013. [DOI] [PubMed] [Google Scholar]
- Wu J.; Maoqiang L.; Fan H.; Zhenyu B.; Qifang H.; Xuepeng W.; Liulong Z. Rutin attenuates neuroinflammation in spinal cord injury rats. J. Surg. Res. 2016, 203, 331–337. 10.1016/j.jss.2016.02.041. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Qi Y.; Xu Y.; Han X.; Peng J.; Liu K.; Sun C. K. Protective effect of flavonoid-rich extract from rosa laevigata michx on cerebral ischemia-reperfusion injury through suppression of apoptosis and inflammation. Neurochem. Int. 2013, 63, 522–532. 10.1016/j.neuint.2013.08.008. [DOI] [PubMed] [Google Scholar]
- Koppula S.; Kumar H.; Kim I. S.; Choi D. K. Reactive oxygen species and inhibitors of inflammatory enzymes, NADPH oxidase, and iNOS in experimental models of Parkinson’s disease. Mediat. Inflamm. 2012, 2012, 823902 10.1155/2012/823902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElroy P. B.; Liang L. P.; Day B. J.; Patel M. Scavenging reactive oxygen species inhibits status epilepticus-induced neuroinflammation. Exp. Neurol. 2017, 298, 13–22. 10.1016/j.expneurol.2017.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerit J.; Edeas M.; Bricaire F. Neurodegenerative diseases and oxidative stress. Biomed. Pharmacother. 2004, 58, 39–46. 10.1016/j.biopha.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Barnham K. J.; Masters C. L.; Bush A. I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug. Discov. 2004, 3, 205–214. 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- Ren J.; Su D.; Li L.; Cai H.; Zhang M.; Zhai J.; Li M.; Wu X.; Hu K. Anti-inflammatory effects of aureusidin in LPS-stimulated RAW264.7 macrophages via suppressing NF-κB and activating ROS- and MAPKs-dependent Nrf2/HO-1 signaling pathways. Toxicol. Appl. Pharmacol. 2020, 387, 114846 10.1016/j.taap.2019.114846. [DOI] [PubMed] [Google Scholar]
- Ramsey C. P.; Glass C. A.; Montgomery M. B.; Lindl K. A.; Ritson G. P.; Chia L. A.; Hamilton R. L.; Chu C. T.; Jordan-Sciutto K. L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. 10.1097/nen.0b013e31802d6da9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce J. A. Eicosanoids in asthma, allergic inflammation, and host defense. Curr. Mol. Med. 2008, 8, 335–349. 10.2174/156652408785160989. [DOI] [PubMed] [Google Scholar]
- Chen A.; Kumar S. M.; Sahley C. L.; Muller K. J. Nitric oxide influences injury-induced microglial migration and accumulation in the leech CNS. J. Neurosci. 2000, 20, 1036–1043. 10.1523/JNEUROSCI.20-03-01036.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulua A. C.; Simon A.; Maddipati R.; Pelletier M.; Park H.; Kim K. Y.; Sack M. N.; Kastner D. L.; Siegel R. M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. 10.1084/jem.20102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman-Martinez L.; Maccioni R. B.; Andrade V.; Navarrete L. P.; Pastor M. G.; Ramos-Escobar N. Neuroinflammation as a common feature of neurodegenerative disorder. Front. Pharmacol. 2019, 10, 1008. 10.3389/fphar.2019.01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L.; He D.; Bai Y. Microglia-mediated inflammation and neurodegenerative disease. Mol. Neurobiol. 2016, 53, 6709–6715. 10.1007/s12035-015-9593-4. [DOI] [PubMed] [Google Scholar]
- Mustafa G.; Mahrosh H. S.; Arif R. In silico characterization of growth differentiation factors as inhibitors of TNF-Alpha and IL-6 immune-mediated inflammatory disease rheumatoid arthritis. Biomed. Res. Int. 2021, 2021, 5538535 10.1155/2021/15538535. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Subedi L.; Kwon O. W.; Pak C.; Lee G.; Lee K.; Kim H.; Kim S. Y. N,N-disubstituted azines attenuate LPS-mediated neuroinflammation in microglia and neuronal apoptosis via inhibiting MAPK signaling pathways. BMC Neurosci. 2017, 18, 82. 10.1186/s12868-017-0399-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellner A.; Barhum Y.; Angel A.; Perets N.; Steiner I.; Offen D.; Lev N. Toll-like receptor-4 inhibitor TAK-242 attenuates motor dysfunction and spinal cord pathology in an amyotrophic lateral sclerosis mouse model. Int. J. Mol. Sci. 2017, 18, 1666. 10.3390/ijms18081666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy-from molecular mechanisms to therapeutic benefits. Biochim. Biophys. Acta 2005, 1754, 253–262. 10.1016/j.bbapap.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Xu X.; Zhang A.; Zhu Y.; He W.; Di W.; Fang Y.; Shi X. MFG-E8 reverses microglial-induced neurotoxic astrocyte (A1) via NF-κB and PI3K-Akt pathways. J. Cell. Physiol. 2018, 234, 904–914. 10.1002/jcp.26918. [DOI] [PubMed] [Google Scholar]
- Ray S. N.; Dilnashin H.; Birla H.; Singh S. S.; Zahra W.; Rathore A. S.; Singh B. K.; Singh S. P. The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox. Res. 2019, 35, 775–795. 10.1007/s12640-019-0003-y. [DOI] [PubMed] [Google Scholar]
- Gao T.; Jernigan J.; Raza S. A.; Dammer E. B.; Xiao H.; Seyfried N. T.; Levey A. I.; Rangaraju S. Transcriptional regulation of homeostatic and disease-associated-microglial genes by IRF1, LXRβ, and CEBPα. Glia 2019, 67, 1958–1975. 10.1002/glia.23678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M. J.; Ramesha S.; Weinstock L. D.; Gao T.; Ping L.; Xiao H.; Dammer E. B.; Duong D. D.; Levey A. I.; Lah J. J.; Seyfried N. T.; Wood L. B.; Rangaraju S. Extracellular signal-regulated kinase regulates microglial immune responses in Alzheimer’s disease. J. Neurosci. Res. 2021, 99, 1704–1721. 10.1002/jnr.24829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J. H.; Kulich S. M.; Oury T. D.; Chu C. T. Cytoplasmic aggregates of phosphorylated extracellular signal-regulated protein kinases in Lewy body diseases. Am. J. Pathol. 2002, 161, 2087–2098. 10.1016/S0002-9440(10)64487-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo C.; Dolcini V.; Salis S.; Venezia V.; Zambrano N.; Russo T.; Schettini G. Signal transduction through tyrosine-phosphorylated C-terminal fragments of amyloid precursor protein via an enhanced interaction with Shc/Grb2 adaptor proteins in reactive astrocytes of Alzheimer’s disease brain. J. Biol. Chem. 2002, 277, 35282–35288. 10.1074/jbc.M110785200. [DOI] [PubMed] [Google Scholar]
- Nguyen P. L.; Bui B. P.; Duong M. T. H.; Lee K.; Ahn H. C.; Cho J. Suppression of LPS-induced inflammation and cell migration by azelastine through inhibition of JNK/NF-κB pathway in BV2 microglial cells. Int. J. Mol. Sci. 2021, 22, 9061. 10.3390/ijms22169061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip Y. T.; Davis R. J. Signal transduction by the c-Jun N-terminal Kinase (JNK)-from inflammation to development. Curr. Opin. Cell Biol. 1998, 10, 205–219. 10.1016/S0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- Javadov S.; Jang S.; Agostini B. Crosstalk between mitogen-activated protein kinases and mitochondria in cardiac diseases: therapeutic perspectives. Pharmacol. Ther. 2014, 144, 202–225. 10.1016/j.pharmthera.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waetzig V.; Herdegen T. Context-specific inhibition of JNKs: overcoming the dilemma of pretection and damage. Trends Pharmacol. Sci. 2005, 26, 455–461. 10.1016/j.tips.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Ventura J. J.; Cogswell P.; Flavell R. A.; Baldwin A. S. Jr.; Davis R. J. JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev. 2004, 18, 2905–2915. 10.1101/gad.1223004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neniskyte U.; Vilalta A.; Brown G. C. Tumor necrosis factor alpha-induced neuronal loss is mediated by microglial phagocytosis. FEBS Lett. 2014, 588, 2952–2956. 10.1016/j.febslet.2014.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T.; Morris P. L. Interleukin-1beta signals through a c-Jun N-terminal kinase dependent inducible nitric oxide synthase and nitric oxide production pathway in Sertoli epithelial cells. Endocrinology 2006, 147, 5424–5430. 10.1210/en.2006-0643. [DOI] [PubMed] [Google Scholar]
- Yarza R.; Vela S.; Solas M.; Ramirez M. J. c-Jun N-terminal Kinase (JNK) signaling as a therapeutic target for Alzheimer’s disease. Front. Pharmacol. 2016, 6, 321. 10.3389/fphar.2015.00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z.; Yuan C.; Yang J.; Peng W.; Wang Y.; Gao W. Behavioral defects induced by chronic social defeat stress are protected by Momordica charantia polysaccharides via attenuation of JNK3/PI3K/AKT neuroinflammatory pathway. Ann. Transl. Med. 2019, 7, 6. 10.21037/atm.2018.12.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anfinogenova N. D.; Quinn M. T.; Schepetkin I. A.; Atochin D. N. Alarmins and c-Jun N-Terminal Kinase (JNK) signaling in neuroinflammation. Cell 2020, 9, 2350. 10.3390/cells9112350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez K.; Kennedy A.; Mclntosh M. K. JNK inhibition by SP600125 attenuates trans-10, cis-12 conjugated linoleic acid-mediated regulation of inflammatory and lipogenic gene expression. Lipids 2011, 46, 885–892. 10.1007/s11745-011-3587-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J.; Zhong W.; Zhang M.; Zhang R.; Hu W. P38 mitogen-activated protein kinase and Parkinson’s disease. Transl. Neurosci. 2018, 9, 147–153. 10.1515/tnsci-2018-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. K.; Kim N. J. Recent advances in the inhibition of p38 MAPK as a potential strategy for the treatment of Alzheimer’s disease. Molecules 2017, 22, 1287. 10.3390/molecules22081287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marber M. S.; Rose B.; Wang Y. The p38 mitogen-activated protein kinase pathway-a potential target for intervention in infarction, hypertrophy, and heart failure. J. Mol. Cell. Cardiol. 2011, 51, 485–490. 10.1016/j.yjmcc.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. B.; Cheng T. T.; Chindalore V.; Damjanov N.; Burgos-Vargas R.; Delora P.; Zimany K.; Travers H.; Caulfield J. P. Evaluation of the efficacy and safety of pamapimod, a p38 MAP kinase inhibitor, in a double-blind, methotrexate-controlled study of patients with active rheumatoid arthritis. Arthritis Rheum. 2009, 60, 335–344. 10.1002/art.24266. [DOI] [PubMed] [Google Scholar]
- Lin X.; Wang M.; Zhang J.; Xu R. p38 MAPK: a potential target of chronic pain. Curr. Med. Chem. 2014, 21, 4405–4418. 10.2174/0929867321666140915143040. [DOI] [PubMed] [Google Scholar]
- Pietersma A.; Tilly B. C.; Gaestel M.; Jong N. D.; Lee J. C.; Koster J. F.; Sluiter W. P38 mitogen activated protein kinase regulates endothelial VCAM-1 expression at the post-transcriptional level. Biochem. Biophys. Res. Commun. 1997, 230, 44–48. 10.1006/bbrc.1996.5886. [DOI] [PubMed] [Google Scholar]
- Teismann P.; Ferger B. Inhibition of the cyclooxygenase isoenzymes COX-1 and COX-2 provide neuroprotection in the MPTP-mouse model of Parkinson’s disease. Synapse 2001, 39, 167–174. . [DOI] [PubMed] [Google Scholar]
- Kim E. H.; Shim B.; Kang S.; Jeong G.; Lee J. S.; Yu Y. B.; Chun M. Anti-inflammatory effects of Scutellaria baicalensis extract via suppression of immune modulators and MAP kinase signaling molecules. J. Ethnopharmacol. 2009, 126, 320–331. 10.1016/j.jep.2009.08.027. [DOI] [PubMed] [Google Scholar]
- Mounz L.; Ralay Ranaivo H.; Roy S. M.; Hu S.; Wenhui H.; Craft J. M.; McNamara L. K.; Chico L. W.; Van Eldik L. J.; Watterson D. M. A novel p38 alpha MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer’s disease mouse model. J. Neuroinflammation 2007, 4, 21. 10.1186/1742-2094-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amirouche A.; Tadesse H.; Lunde J. A.; Bélanger G.; Côté J.; Jasmin B. J. Activation of p38 signaling increases utrophin A expression in skeletal muscle via the RNA-binding protein KSRP and inhibition of AU-rich element-mediated mRNA decay: implications for novel DMD therapeutics. Hum. Mol. Genet. 2013, 22, 3093–3111. 10.1093/hmg/ddt165. [DOI] [PubMed] [Google Scholar]
- Mounz L.; Ammit A. J. Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology 2010, 58, 561–568. 10.1016/j.neuropharm.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Islam M. N.; Choi R. J.; Jin S. E.; Kim Y. S.; Ahn B. R.; Zhao D.; Jung H. A.; Choi J. S. Mechanism of anti-inflammatory activity of umbelliferone 6-carboxylic acid isolated from Angelica decursiva. J. Ethnopharmacol. 2012, 144, 175–181. 10.1016/j.jep.2012.08.048. [DOI] [PubMed] [Google Scholar]
- Kim A. R.; Lee M. S.; Shin T. S.; Hua H.; Jang B. C.; Choi J. S.; Byun D. S.; Utsuki T.; Ingram D.; Kim H. R. Phlorofucofuroeckol A inhibits the LPS-stimulated iNOS and COX-2 expressions in macrophages via inhibition of NF-κB, Akt, and p38 MAPK. Toxicol. In Vitro 2011, 25, 1789–1795. 10.1016/j.tiv.2011.09.012. [DOI] [PubMed] [Google Scholar]
- Wang J.; Guo C.; Wei Z.; He X.; Kou J.; Zhou E.; Yang Z.; Fu Y. Morin suppresses inflammatory cytokine expression by downregulation of nuclear factor-κB and mitogen-activated protein kinase (MAPK) signaling pathways in lipopolysaccharide-stimulated primary bovine mammary epithelial cells. J. Dairy Sci. 2016, 99, 3016–3022. 10.3168/jds.2015-10330. [DOI] [PubMed] [Google Scholar]
- Im E. J.; Kim S. J.; Hong S. B.; Park J. K.; Rhee M. H. Anti-inflammatory activity of bee venom in BV2 microglial cells: mediation of MyD88-dependent NF-κB signaling pathway. Evid. Based Complement. Alternat. Med. 2016, 2016, 3704764 10.1155/2016/3704764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onasanwo S. A.; Velagapudi R.; El-Bakoush A.; Olajide O. A. Inhibition of neuroinflammation in BV2 microglia by the biflavonoid kolaviron is dependent on the Nrf2/ARE antioxidant protective mechanism. Mol. Cell. Biochem. 2016, 414, 23–36. 10.1007/s11010-016-2655-8. [DOI] [PubMed] [Google Scholar]
- Upadhayay S.; Mehan S. Targeting Nrf2/HO-1 anti-oxidant signaling pathway in the progression of multiple sclerosis and influences on neurological dysfunctions. Brain Disord. 2021, 3, 100019 10.1016/j.dscb.2021.100019. [DOI] [Google Scholar]
- Loboda A.; Damulewicz M.; Pyza E.; Jozkowicz A.; Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. 10.1007/s00018-016-2223-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv C.; Maharjan S.; Wang Q.; Sun Y.; Han X.; Wang S.; Mao Z.; Xin Y.; Zhang B. α-Lipoic acid promotes neurological recovery after ischemic stroke by activating the Nrf2/HO-1 pathway to attenuate oxidative damage. Cell. Physiol. Biochem. 2017, 43, 1273–1287. 10.1159/000481840. [DOI] [PubMed] [Google Scholar]
- Bansal Y.; Singh R.; Parhar I.; Kuhad A.; Soga T. Quinolinic acid and nuclear factor erythroid 2-related factor 2 in depression: role in neuroprogression. Front. Pharmacol. 2019, 10, 452. 10.3389/fphar.2019.00452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel C. P.; Bondy S. C. Sensitive and rapid quantitation of oxygen reactive species formation in rat synaptosomes. Neurochem. Int. 1990, 17, 435–440. 10.1016/0197-0186(90)90025-O. [DOI] [PubMed] [Google Scholar]