

Abstract
Hepatic stellate cells (HSC) are non-parenchymal cells within the liver that are the main drivers of fibrosis in a variety of diseases of the liver. Upon liver injury, HSC can transdifferentiate into myofibroblasts that migrate to sites of injury and produce the extracellular matrix (ECM) that make up fibrotic lesions. Prior work has suggested that inhibitors of the mitochondrial pyruvate carrier (MPC) can suppress markers of HSC activation and reduce fibrosis in a mouse model of nonalcoholic steatohepatitis (NASH). Treatment of isolated HSC with the MPC inhibitor 7ACC2 resulted in decreased expression of several markers of HSC activation. To determine the effects of MPC deletion on HSC activation we generated mice with HSC-specific deletion of the MPC using the lecithin retinol acyltransferase promoter-driven Cre (Lrat-Mpc2−/− mice). HSC isolated from Lrat-Mpc2−/− mice had diminished activation in vitro compared to HSC from wild-type mice. Furthermore, when wild-type and Lrat-Mpc2−/− littermate mice were placed on a NASH-inducing diet (high in fat, fructose, and cholesterol), circulating transaminase levels were significantly lower in Lrat-Mpc2−/− mice, along with a reduction of hepatic gene expression for several markers of HSC activation. Bulk RNAseq analyses revealed a decrease in pathways associated with inflammatory signaling, immune cell activation and migration, and ECM remodeling in Lrat-Mpc2−/− mice compared to wild-type mice on the NASH diet. Finally, in a second model of NASH, both wild-type and Lrat-Mpc2−/− littermate mice were placed on a western-type diet (42% kcal fat, 0.2% cholesterol) and housed at thermoneutrality for a period of 20 weeks. Again Lrat-Mpc−/− mice had reduced circulating transaminase levels and decreased expression of several HSC activation markers. These data suggest that MPC inhibition modulates HSC metabolism to prevent activation and illuminate another mechanism by which MPC inhibitors could prove therapeutically beneficial for people with NASH.
BACKGROUND and AIMS:
Nonalcoholic steatohepatitis (NASH) is a progressive liver disease that dramatically increases the risk of developing cirrhosis, liver failure, and hepatocellular carcinoma [1–3]. Despite being a leading cause of liver-related morbidity and mortality, there are no licensed drug therapies for NASH. NASH is characterized by hepatocyte injury, inflammation, and fibrosis, but the molecular mechanisms driving progression to more severe forms of the disease are unclear. The liver is composed of a variety of cell types including parenchymal (hepatocytes) and non-parenchymal cells (endothelial, immune, and stellate cells). While multiple cell lineages contribute to NASH progression, hepatic stellate cells (HSC) are the primary drivers of fibrosis in a variety of liver diseases [4].
At the basal (quiescent) state, HSC display an adipogenic-like transcriptional program (PPARγ, SREBP1c) and are one of the body’s major storage sites for vitamin A in the form of retinyl esters [5]. Upon hepatic injury, HSC can proliferate and differentiate into myofibroblasts that can migrate to sites of injury. HSC are activated in response to a variety of stimuli including factors released upon cell death/injury, changes in the extracellular matrix, immune cells, and numerous growth factors (TGFβ, PDGF, CTGF, etc.) [6]. Although important for tissue repair, chronic activation of HSC leads to excess extracellular matrix deposition in the liver, resulting in hepatic scarring (fibrosis). Importantly, activated HSC undergo dramatic changes in metabolism to meet the high demand for energy that is necessary to facilitate increased rates of proliferation and ECM production [7]. Glucose utilization is enhanced and there are marked increases in rates of both mitochondrial oxidation and anaerobic glycolysis upon HSC activation [8]. Additional evidence suggests that activated HSC have increased mitochondrial abundance in activated versus quiescent HSC in vitro [8]. Furthermore, interventions to suppress these metabolic changes have been shown to result in reduced HSC activation [8, 9]. Based on the importance of energy metabolism in HSC function, it seems plausible that these pathways may be targeted therapeutically to limit the progression of NASH to fibrosis.
Prior work has suggested that inhibitors of the mitochondrial pyruvate carrier (MPC) can suppress markers of HSC activation and reduce fibrosis in a mouse model of NASH [10]. The mitochondrial pyruvate carrier (MPC) plays a pivotal role in mitochondrial metabolism by transporting pyruvate, a product of glycolysis, into the mitochondrial matrix to enter the TCA cycle. The MPC is a heterodimeric complex of the proteins MPC1 and MPC2 [11, 12]. Both proteins are required for MPC stability, as the deletion of one protein destabilizes the complex [11–13]. Previous work has shown that MPC inhibition in multiple tissues provides metabolic benefits [13, 14] and reduces NASH progression in mice [10], but little is known about whether MPC inhibitors regarding their effects on HSC directly. To investigate this, we examined how disrupting the MPC affects stellate cell activation in vitro, and generated mice with stellate cell-specific Mpc2 deletion, to understand the effects on NASH progression in vivo.
RESULTS:
Inhibition of the MPC reduces hepatic stellate cell activation in vitro.
In order to test the role of MPC inhibition on HSC activation, we first isolated HSC from the liver of wild-type mice and treated them with the MPC inhibitor (MPCi) 7ACC2. Culturing isolated stellate cells for a period of seven days resulted in a substantial increase in several markers of HSC activation including multiple collagen isoforms, and non-collagenous genes that participate in ECM remodeling including Acta2, Spp1, and Timp1, compared to cells harvested at day one (Figure 1). However, treatment with 7ACC2, at doses previously shown to be specific towards the MPC [15], led to a significant reduction in in several activation markers including Col1a1, Col1a2, Spp1, and Timp1 (Figure 1A).
Figure 1: The MPC inhibitor 7ACC2 reduces hepatic stellate cell activation in vitro.
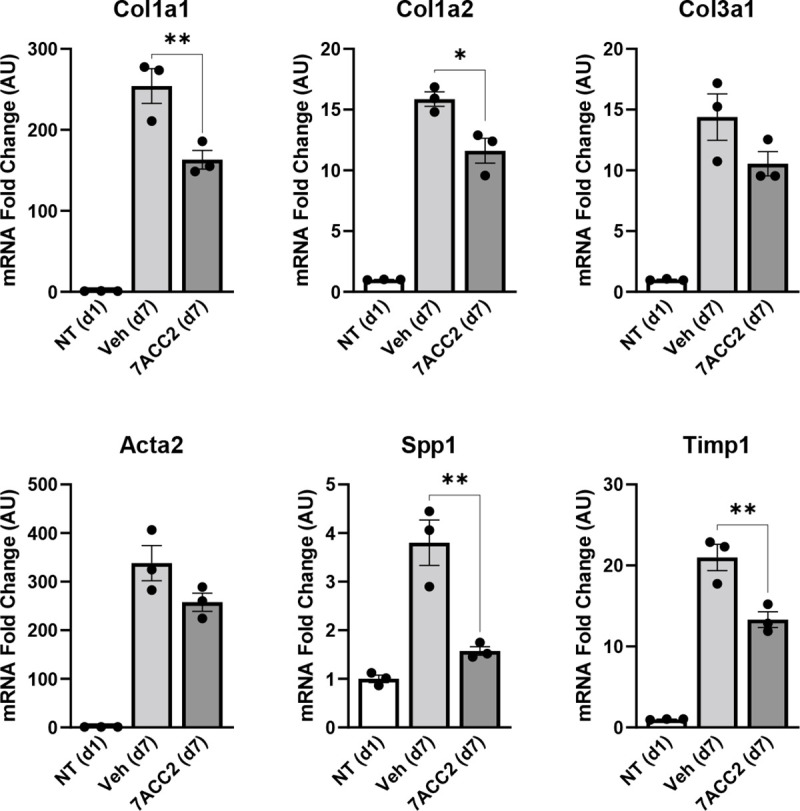
Hepatic stellate cells (HSC) were isolated from wild-type Mpc2fl/fl mice and cultured for up to 7 days (d7) with or without the addition of vehicle (DMSO) or 7ACC2 (1 μM). A portion were harvested after 1 day of culture (non-treated, NT d1) for quiescent HSC. Gene expression was measured by qRT-PCR and data are expressed as mean ± SEM, relative to d1 HSC. *p<0.05, **p<0.01.
To further investigate the role of the MPC in HSC, we generated mice with stellate cell-specific deletion of Mpc2 by crossing Mpc2fl/fl mice with mice expressing the Cre recombinase under the lecithin-retinol acyltransferase (LratCre) promoter [4] to generate Lrat-Mpc2−/− mice. First, we isolated HSC from both Lrat-Mpc2−/− and wild-type littermate mice and cultured. As before, seven days in culture led to a considerable increase in several HSC activation markers in both groups, but the expression of these markers were substantially reduced in the Lrat-Mpc2−/− HSC compared to WT cultures (Figure 2). Importantly, we saw a significant reduction in Mpc2 gene expression in Lrat-Mpc2−/− mice relative to wild-type littermates at day seven (Figure 2), but there was no difference between groups at day one, possibly suggesting that Lrat-Cre promoter was not yet active or the presence of other cell types in the HSC isolation. Collectively, our results demonstrate that genetic or pharmacological inhibition of the MPC reduces HSC activation in vitro.
Figure 2: Stellate cell-specific deletion of Mpc2 blunts HSC activation in vitro.

Mice with stellate cell-specific deletion of Mpc2 were generated by crossing Mpc2fl/fl mice with mice expressing the Cre-recombinase driven by a lecithin retinol acyltransferase promotor (Lrat-Mpc2−/−). HSC were isolated from littermate wild-type (WT) and Lrat-Mpc2−/− (KO) mice and cultured for up to 7 days (d7), while a portion of cells were harvested after 1 day of culture (d1) for quiescent HSC. Gene expression was measured by qRT-PCR and data are expressed as mean ± SEM, relative to WT d1 HSC. **p<0.01, ****p<0.0001.
Lrat-Mpc2−/− mice are protected from NASH-inducing diet.
Next, we sought to examine how MPC inhibition in stellate cells affects NASH progression in vivo. Lrat-Mpc2−/− and wild-type littermate mice were placed on either a low-fat diet (LFD) or a NASH-inducing diet (high in fat, fructose, and cholesterol; HFC) for a period of 12 weeks. To exacerbate NASH progression, we treated mice with a one-time dose of carbon tetrachloride after four weeks on diet. After 12 weeks of diet, Lrat-Mpc2−/− mice had an average body weight that was ~17% less than wild-type mice on the HFC diet, but both genotypes gained a similar amount of weight in either diet group (Figure 3A). Lrat-Mpc2−/− mice on the HFC diet had a significant reduction in liver weight, fat mass, and lean mass, compared to wild-type; however, when normalized to total body weight, there was no difference in between groups (Figure 3B–D). Next, we assessed plasma transaminases, ALT and AST, as markers of liver injury and found that on the HFC diet Lrat-Mpc2−/− mice had a significant reduction in both ALT and AST levels by ~58% and ~48%, respectively, compared wild-type mice (Figure 3E). There were no changes in plasma lipid concentrations (Supplementary Figure 1A). Histological examination of H&E-stained liver sections revealed a slight trend towards reduced macrosteatosis, inflammation, and NAFLD activity score, but these did not reach statistical significance (Figure 3F, Supplementary Figure 1B). Lastly, we measured hepatic gene expression and found that Lrat-Mpc2−/− mice had significantly decreased expression of several markers of HSC activation, including Acta2, multiple collagen isoforms, Timp1, and Spp1 (Figure 3G). Taken together, our results suggest that Mpc2 deletion in hepatic stellate cells are protective from NASH development in mice.
Figure 3: Lrat-Mpc2−/− mice are protected from NASH-inducing diet.

At about 8 weeks of age, littermate wild-type (WT) and Lrat-Mpc2−/− (KO) mice were placed on either a low-fat diet (LFD) or a NASH-inducing diet (high in fat, fructose, and cholesterol; HFC) for a period of 12 weeks. To exacerbate NASH progression, we treated mice with a one-time dose of carbon tetrachloride after four weeks on diet. (A) Terminal body weight, expressed as total mass and percent body weight gain from initiation of diet. (B) Liver weight measured at sacrifice, expressed as total mass and percent body weight. Body composition determined by EchoMRI for both (C) fat mass and (D) lean mass, represented as total mass and percent body weight. (E) Plasma levels of circulating transaminases ALT and AST collected at sacrifice. (F) Representative liver sections with H&E staining. (G) Hepatic gene expression measured by qRT-PCR and expressed relative to WT LFD control group. All data expressed as mean ± SEM (n=7–11/group). *p<0.05, **p<0.01.
To gain a better understanding of the changes taking place in the hepatic transcriptome of Lrat-Mpc2−/− and wild-type mice, we performed bulk RNA sequencing (RNAseq) analysis on livers from both genotypes on either the LFD or HFC diet. Analysis of differentially expressed genes (DEG) revealed Lrat-Mpc2−/− mice had a decrease in the expression of 123 genes while 91 genes were increased compared to wild-type mice on the LFD (Figure 4A). Comparison within the HFC groups revealed that Lrat-Mpc2−/− livers exhibited reduced expression of 343 genes, while the expression of 133 genes was elevated, compared to wild-type mice (Figure 4B). Gene set enrichment analysis a decrease in immune responses and apoptosis in Lrat-Mpc2−/− relative to wild-type mice in both diet groups (Figure 4C–D). Examination of perturbed KEGG signaling and metabolism pathways revealed a decrease in retinol metabolism and cell adhesion, while oxidative phosphorylation and protein processing were increased in Lrat-Mpc2−/− mice on the LFD (Figure 4E). On the HFC diet, Lrat-Mpc2−/− mice again displayed a decrease in expression of immune signaling pathways while pathways associated with steroid hormone biosynthesis and amino acid metabolism were increased (Figure 4F). Collectively, these results indicate that stellate cell-specific deletion of Mpc2 results in a shift in global hepatic transcriptome that is associated with a decreased immune response and possibly altered metabolism.
Figure 4: RNAseq reveals reduced immune responses in Lrat-Mpc2−/− mice.

RNA sequencing was performed on liver tissue from both wild-type (WT) and Lrat-Mpc2−/− (KO) mice on placed on either a LFD or HFC diet (n=5/group). Volcano plot of differentially expressed genes with p-value <0.05 comparing (A) KO versus WT mice on LFD and (B) KO versus WT mice on HFC diet. Differentially expressed genes with Log fold change (LogFC) less than −0.5, or greater than 0.5, were highlighted in either blue or red, respectively. Analysis of perturbations in Hallmark gene set collections when comparing (C) KO versus WT mice on LFD and (D) KO versus WT mice on HFC diet. Changes in KEGG signaling and metabolism pathways when comparing (C) KO versus WT mice on LFD and (D) KO versus WT mice on HFC diet.
Lrat-Mpc2−/− mice are protected from NASH exacerbated by thermoneutral housing.
To further confirm our findings and add scientific rigor, we used a second model to induce NASH by placing both Lrat-Mpc2−/− and wild-type littermate mice in thermoneutral housing while feeding a western-type diet (42% kcal fat, 0.2% cholesterol) for a period of 20 weeks. Previous reports have demonstrated that housing at thermoneutrality, while feeding mice a western diet, exacerbates NASH development and is associated with activation of inflammatory pathways that are like those found in human disease development [16].
Like our previous experiment, Lrat-Mpc2−/− mice had a modest but significant 15% decrease in body weight compared to wild-types, yet both groups gained a similar amount of weight over time (Figure 5A). We again found that both ALT and AST levels were 57% and 43% lower, respectively, in Lrat-Mpc2−/− mice compared to wild-type littermates (Figure 5B). Total liver and fat mass were lower in Lrat-Mpc2−/− mice, but there was no difference in total lean mass compared to wild-type (Figure 5C–D, Supplementary Figure 2). As with our previous experiment, when liver and fat mass were normalized to body weight, there was no difference in liver or fat mass between genotypes (Figure 5C–D). Lastly, Lrat-Mpc2−/− mice had reduced hepatic expression of several markers of HSC activation including multiple collagen isoforms, Timp1, Spp1, and Lgals3 (Figure 5E). Collectively, our data demonstrate that stellate cell-specific disruption of the MPC blunts HSC activation and reduces NASH progression.
Figure 5: Lrat-Mpc2−/− mice are protected from NASH exacerbated by thermoneutral housing.

At about 8 weeks of age, littermate wild-type (WT) and Lrat-Mpc2−/− (KO) mice were placed in thermoneutral housing (30°C) for 20 weeks. (A) Terminal body weight, expressed as total mass and percent body weight gain from initiation of diet. (B) Plasma levels of circulating transaminases ALT and AST collected at sacrifice. (C) Liver weight measured at sacrifice, expressed as total mass and percent body weight. (D) Analysis of fat mass determined by EchoMRI, represented as total mass and percent body weight. (G) Hepatic gene expression measured by qRT-PCR and expressed relative to WT group. All data expressed as mean ± SEM (n=5–9/group). *p<0.05, **p<0.01.
DISCUSSION:
Upon activation by liver injury, HSC undergo complex changes in intermediary metabolism that allow them to proliferate, transdifferentiate into myofibroblasts, and migrate to sites of injury. These changes include increased glycolysis and reliance on glutamine, and ultimately a shift towards producing greater amounts of ECM products that can result in hepatic scarring (fibrosis). Prior work has demonstrated that inhibition of glycolysis or glutaminolysis can attenuate HSC activation in response to activating stimuli [8, 9]. In the current work, we focused our studies on the MPC, which connects anaerobic glycolysis with mitochondrial metabolism by transporting pyruvate, a major end-product of glycolysis, into the mitochondrial matrix where it can participate in the TCA cycle. Use of MPCi has been previously shown to reduce markers of HSC activation and fibrosis in mouse models of NASH, but the direct effect on HSC has been little studied. Both MPC pharmacological inhibition and genetic deletion of Mpc2, resulted in reduced activation of HSC in vitro. Importantly, we found that LratCre-Mpc2−/− mice were also protected from progression of NAFLD to NASH in two separate mouse models. Overall, these studies point towards a novel therapeutic target to reduce HSC activation to decrease NASH progression.
Activated HSC undergo a dramatic shift in metabolism whereby there is an increase in glucose uptake and glycolysis, a phenomenon that is often found in cancer cells [7]. Previous work has demonstrated that inhibition of glycolysis reduces the activation of HSC [8]. Since the MPC sits at a critical hub connecting glycolysis and the TCA cycle, we hypothesized that inhibition of this important node would reduce the ability of HSC to undergo the metabolic changes necessary to sustain activation. Indeed, our studies found that direct treatment of isolated HSC with either MPCi or genetic deletion of Mpc2, resulted in a reduction in several HSC activation markers. Importantly, our in vitro findings were recapitulated in multiple mouse models of NASH where mice with stellate cell-specific deletion of Mpc2 were protected NASH progression as evidenced by reduced levels of ALT and AST, as well as decreased expression of numerous markers of HSC activation. Analysis of RNAseq data revealed a striking decrease in gene sets associated with both innate and adaptive immune responses, a key finding since immune cell infiltration is one of the early signs of disease development [17]. Although these findings provide evidence that MPC inhibition limits the activation of HSC, the exact mechanisms driving this are not clear.
While enhanced glucose utilization has been shown to be a defining feature of HSC activation, enhanced glutamine metabolism also plays a significant role. Glutamine has long been known to stimulate collagen production in fibroblasts [18, 19] and hepatic stellate cells in vitro [9, 20]. Evidence has suggested that enhanced glutaminolysis, the process of converting glutamine into TCA cycle intermediates, is a key feature of fibrosis in people with NASH [21]. Glutamine regulates collagen production by enhancing the transcription of the genes encoding collagens and other components of the ECM and by serving as a substrate for collagen synthesis. Du et al demonstrated that, compared to HSC supplied with both glucose and glutamine, glucose deprivation alone had little to no effect while glutamine deprivation substantially impaired HSC activation and function in isolated HSC [9]. Inhibition of enzymes involved in glutaminolysis (glutaminase, GLS; glutamate dehydrogenase, GDH; or the transaminases GOT2 and GPT2) limited HSC proliferation, activation, and migration in vitro [9]. A key metabolite produced by glutaminolysis is alpha-ketoglutarate (AKG), and the addition of dimethyl AKG, a cell permeable form of AKG, was able to rescue HSC activation in the presence of the various glutaminolysis inhibitors [9].
Although glutamine can serve as an important metabolite to replenish TCA intermediates for the downstream production of ATP to support the energetic demands of fibroblast activation, there are also biosynthetic fates of glutamine that may be important in HSC activation. Recent work in lung fibroblasts has illustrated that rather than simply generating TCA intermediates, glutamine supplementation contributes to collagen production as a substrate for synthesis of glycine and proline, two of the most abundant amino acids in collagen [22]. Along these lines, cancer associated fibroblasts are reliant on glutamine for the synthesis of proline, mediated by the enzyme PYCR1, to promote excess collagen production [23]. Reduction of PYCR1 levels reduced collagen production and authors found that PYCR1 was upregulated epigenetically via acetylation facilitated by acetyl-CoA produced by the pyruvate dehydrogenase complex [23]. Since MPC inhibition would limit intramitochondrial pyruvate available for generation of acetyl-CoA, inhibition of the MPC may limit HSC activation by altering epigenetic regulation. However, additional work is required to support this hypothesis. Collectively, we postulate that inhibition of the MPC limits HSC activation by reducing the ability of cells to use pyruvate as a metabolic substrate, which impacts signaling and biosynthetic processes. Alternatively, or in addition, limiting mitochondrial pyruvate metabolism may alter the metabolic fate of glutamine to impact the process of activation.
In summary, the current work provides evidence that inhibition of the MPC ameliorates HSC activation. Using Cre-LoxP mediated recombination, we generated novel stellate cell-specific Mpc2 knockout mice, which were protected from NASH development in two separate dietary models. Analysis of hepatic RNAseq data demonstrated that HSC-specific Mpc2 deletion resulted in a decreased in pathways associated with immune cell activation and extracellular matrix remodeling. It is possible that MPC inhibition could mediate these changes through epigenetic regulation and/or altering cellular energetics and use of other important metabolic substrates. However, further studies are required to determine the precise mechanisms at play. Overall, these data highlight an alternative mechanism by which MPC inhibitors could be a novel therapeutic option for people afflicted with NASH.
MATERIALS AND METHODS:
Animal Studies:
All animal experiments were approved by the Institutional Animal Care and Use Committee of Washington University in St. Louis (animal protocol number 20–0004). To generate HSC-specific deletion of Mpc2, we crossed Mpc2fl/fl mice with lecithin retinol acyltransferase LratCre mice [4]. Littermates that did not express Cre were used as controls in all studies. For high fat diet studies, at 8 weeks of age, male mice were switched from standard chow to either a low-fat diet (Research diets D09100304) or a NASH-inducing diet high in fat, fructose, and cholesterol (Research diets D09100310) and maintained on diet for 12 weeks. After 4 weeks of diet, mice were given one-time intraperitoneal injection of carbon tetrachloride (CCL4) dissolved in corn oil (0.5 μL CCL4 per gram body weight). After 12 weeks of diet feeding, animals were fasted for 5 h, then sacrificed for tissue and blood collection.
For thermoneutral studies, mice were switched from a standard chow diet to a western-type diet (42% kcal fat, 0.2% cholesterol; TD88137). At the same time, mice were placed in thermoneutral housing (30°C). Mice were maintained on dietary feeding and thermoneutral housing for 20 weeks then sacrificed for tissue and blood collection following a 5 h fast.
Body composition:
Total body composition of fat and lean mass was determined by EchoMRI-100H (EchoMRI LLC).
Plasma analyses:
Plasma levels of alanine transaminase (ALT) and aspartate transaminase (AST) were determined using kinetic absorbance assays (Teco Diagnostics) as described previously [10]. Analysis of plasma triglycerides (Thermo Fisher Scientific), total cholesterol (Thermo Fisher Scientific), and non-esterified fatty acids (FUJIFILM Wako) were determined using colorimetric based assay as previously reported [24, 25].
Histological Analyses:
Liver tissue was fixed in 10% neutral buffered formalin for 24 hours then paraffin embedded, then sections were cut and stained with hematoxylin and eosin (H&E). H&E sections were assessed for steatosis score, lobular inflammation, and NAFLD total score, by a board-certified pathologist (M.H.) who was blinded from treatment groups, as previously reported [10, 25].
Hepatic Stellate Cell Isolation:
HSC were isolated from mice as previously described [10]. Briefly, following collagenase perfusion, HSC were purified using a density gradient centrifugation Optiprep (Sigma), then cells were seeded on to standard tissue culture dishes in DMEM media (Gibco) containing 10 % fetal bovine serum, and 1x Pen/Strep. After 24 hours, a subset of cells was harvested for use as a quiescent HSC control group. In the remaining cells, media was switched to media containing either vehicle (DMSO) or the MPCi 7ACC2 (1 μM). Media was replenished every other day.
RNA isolation qRT-PCR:
RNA was isolated as previously described [26]. For in vitro studies, RNA was harvested from tissue culture dishes using TRIzol Reagent (Ambion) and Purelink™ RNA Mini Kit (Invitrogen™). For liver tissue, ~30 mg of frozen liver tissue was homogenized in TRIzol Reagent (Ambion) using the TissueLyser II (Qiagen) followed by isolation using the Purelink™ RNA Mini Kit (Invitrogen™). RNA was reverse transcribed into complimentary DNA (cDNA) using High-Capacity reverse transcriptase (Life Technologies). Quantitative real-time polymerase chain reaction was performed using Power SYBR Green (Thermo Fisher Scientific) and an optical 384-Well Reaction Plate (Applied Biosystems) using ViiA 7 Real-Time PCR System (Applied Biosystems). Relative gene expression was determined by the ΔΔCt method 36b4 as a reference gene.
RNA sequencing:
RNA was isolated from liver tissue as described above and bulk RNA sequencing was performed at the Genomic Technologies and Access Center at the McDonald Genomic Institute of Washington University School of Medicine in St. Louis. An Agilent Bioanalyzer was used to assess RNA integrity and ribosomal RNA depletion was performed with RiboErase (HMR). Samples were prepared according to library kit manufacturer’s protocol, then indexed, pooled, and sequenced on a NovaSeq S4 2×150, targeting ~30 million reads per sample. Detailed methods of RNA sequencing analysis are previously reported [26].
Statistical Analyses:
GraphPad Prism Software, version 9.5.0 for windows, was used to generate figures. All data and are presented as the mean ±SEM. Statistical significance was calculated using an unpaired Student’s t-test, two-way analysis of variance (ANOVA), or one-way ANOVA with Tukey’s multiple comparisons test, with a statistically significant difference defined as p<0.05.
Supplementary Material
ACKNOWLEDGMENTS:
This work was funded by NIH grant R01 DK104735 (to B.N.F.). The Core services of the Diabetes Research Center (P30 DK020579), Digestive Diseases Research Cores Center (P30 DK052574), and the Nutrition Obesity Research Center (P30 DK56341) at the Washington University School of Medicine also supported this work. D.F was supported by a training grant (T32 DK007120). We would like to thank Robert F. Schwabe, MD for kindly providing Lrat-Cre transgenic mice.
Footnotes
DISCLOSURES
Brian Finck is a member of the Scientific Advisory Board and owns stock in Cirius Therapeutics, which is developing an MPC inhibitor for clinical use in treating NASH.
References
- [1].Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64(1):73–84. [DOI] [PubMed] [Google Scholar]
- [2].Younossi Z. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2018;15. [DOI] [PubMed] [Google Scholar]
- [3].Taylor RS, Taylor RJ, Bayliss S, Hagstrom H, Nasr P, Schattenberg JM, Ishigami M, Toyoda H, Wai-Sun Wong V, Peleg N, Shlomai A, Sebastiani G, Seko Y, Bhala N, Younossi ZM, Anstee QM, McPherson S, Newsome PN. Association Between Fibrosis Stage and Outcomes of Patients With Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Gastroenterology 2020. [DOI] [PubMed] [Google Scholar]
- [4].Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, Pradere JP, Schwabe RF. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nature communications 2013;4:2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hendriks HF, Verhoofstad WA, Brouwer A, de Leeuw AM, Knook DL. Perisinusoidal fat-storing cells are the main vitamin A storage sites in rat liver. Experimental cell research 1985;160(1):138–49. [DOI] [PubMed] [Google Scholar]
- [6].Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Advanced drug delivery reviews 2017;121:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Trivedi P, Wang S, Friedman SL. The Power of Plasticity-Metabolic Regulation of Hepatic Stellate Cells. Cell metabolism 2021;33(2):242–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chen Y, Choi SS, Michelotti GA, Chan IS, Swiderska-Syn M, Karaca GF, Xie G, Moylan CA, Garibaldi F, Premont R, Suliman HB, Piantadosi CA, Diehl AM. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology 2012;143(5):1319–29.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Du K, Hyun J, Premont RT, Choi SS, Michelotti GA, Swiderska-Syn M, Dalton GD, Thelen E, Rizi BS, Jung Y, Diehl AM. Hedgehog-YAP Signaling Pathway Regulates Glutaminolysis to Control Activation of Hepatic Stellate Cells. Gastroenterology 2018;154(5):1465–79.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].McCommis KS, Hodges WT, Brunt EM, Nalbantoglu I, McDonald WG, Holley C, Fujiwara H, Schaffer JE, Colca JR, Finck BN. Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology 2017;65(5):1543–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ERS, Martinou JC. Identification and functional expression of the mitochondrial pyruvate carrier. Science 2012;336(6090):93–6. [DOI] [PubMed] [Google Scholar]
- [12].Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, Cox JE, Cardon CM, Van Vranken JG, Dephoure N, Redin C, Boudina S, Gygi SP, Brivet M, Thummel CS, Rutter J. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 2012;337(6090):96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].McCommis KS, Chen Z, Fu X, McDonald WG, Colca JR, Kletzien RF, Burgess SC, Finck BN. Loss of Mitochondrial Pyruvate Carrier 2 in the Liver Leads to Defects in Gluconeogenesis and Compensation via Pyruvate-Alanine Cycling. Cell metabolism 2015;22(4):682–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vacanti NM, Divakaruni AS, Green CR, Parker SJ, Henry RR, Ciaraldi TP, Murphy AN, Metallo CM. Regulation of substrate utilization by the mitochondrial pyruvate carrier. Molecular cell 2014;56(3):425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hodges WT, Jarasvaraparn C, Ferguson D, Griffett K, Gill LE, Chen Y, Ilagan MXG, Hegazy L, Elgendy B, Cho K, Patti GJ, McCommis KS, Finck BN. Mitochondrial pyruvate carrier inhibitors improve metabolic parameters in diet-induced obese mice. The Journal of biological chemistry 2022;298(2):101554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Giles DA, Moreno-Fernandez ME, Stankiewicz TE, Graspeuntner S, Cappelletti M, Wu D, Mukherjee R, Chan CC, Lawson MJ, Klarquist J, Sünderhauf A, Softic S, Kahn CR, Stemmer K, Iwakura Y, Aronow BJ, Karns R, Steinbrecher KA, Karp CL, Sheridan R, Shanmukhappa SK, Reynaud D, Haslam DB, Sina C, Rupp J, Hogan SP, Divanovic S. Thermoneutral housing exacerbates nonalcoholic fatty liver disease in mice and allows for sex-independent disease modeling. Nature medicine 2017;23(7):829–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gadd VL. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014;59. [DOI] [PubMed] [Google Scholar]
- [18].Bellon G, Monboisse JC, Randoux A, Borel JP. Effects of preformed proline and proline amino acid precursors (including glutamine) on collagen synthesis in human fibroblast cultures. Biochimica et biophysica acta 1987;930(1):39–47. [DOI] [PubMed] [Google Scholar]
- [19].Bellon G, Chaqour B, Wegrowski Y, Monboisse JC, Borel JP. Glutamine increases collagen gene transcription in cultured human fibroblasts. Biochimica et biophysica acta 1995;1268(3):311–23. [DOI] [PubMed] [Google Scholar]
- [20].Li J, Ghazwani M, Liu K, Huang Y, Chang N, Fan J, He F, Li L, Bu S, Xie W, Ma X, Li S. Regulation of hepatic stellate cell proliferation and activation by glutamine metabolism. PLoS One 2017;12(8):e0182679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Du K, Chitneni SK, Suzuki A, Wang Y, Henao R, Hyun J, Premont RT, Naggie S, Moylan CA, Bashir MR, Abdelmalek MF, Diehl AM. Increased Glutaminolysis Marks Active Scarring in Nonalcoholic Steatohepatitis Progression. Cellular and molecular gastroenterology and hepatology 2020;10(1):1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hamanaka RB, O’Leary EM, Witt LJ, Tian Y, Gökalp GA, Meliton AY, Dulin NO, Mutlu GM. Glutamine Metabolism Is Required for Collagen Protein Synthesis in Lung Fibroblasts. American journal of respiratory cell and molecular biology 2019;61(5):597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kay EJ, Paterson K, Riera-Domingo C, Sumpton D, Däbritz JHM, Tardito S, Boldrini C, Hernandez-Fernaud JR, Athineos D, Dhayade S, Stepanova E, Gjerga E, Neilson LJ, Lilla S, Hedley A, Koulouras G, McGregor G, Jamieson C, Johnson RM, Park M, Kirschner K, Miller C, Kamphorst JJ, Loayza-Puch F, Saez-Rodriguez J, Mazzone M, Blyth K, Zagnoni M, Zanivan S. Cancer-associated fibroblasts require proline synthesis by PYCR1 for the deposition of pro-tumorigenic extracellular matrix. Nature metabolism 2022;4(6):693–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics (Oxford, England) 2014;30(7):923–30. [DOI] [PubMed] [Google Scholar]
- [26].Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nature methods 2017;14(4):417–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics (Oxford, England) 2012;28(16):2184–5. [DOI] [PubMed] [Google Scholar]
- [28].Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford, England) 2010;26(1):139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu R, Holik AZ, Su S, Jansz N, Chen K, Leong HS, Blewitt ME, Asselin-Labat ML, Smyth GK, Ritchie ME. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic acids research 2015;43(15):e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Luo W, Friedman MS, Shedden K, Hankenson KD, Woolf PJ. GAGE: generally applicable gene set enrichment for pathway analysis. BMC bioinformatics 2009;10:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhao S, Guo Y, Sheng Q, Shyr Y. Advanced heat map and clustering analysis using heatmap3. BioMed research international 2014;2014:986048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Luo W, Brouwer C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics (Oxford, England) 2013;29(14):1830–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC bioinformatics 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics : a journal of integrative biology 2012;16(5):284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.