

Abstract
Fascioscapulohumeral muscular dystrophy (FSHD) is caused by a unique genetic mechanism that relies on contraction and hypomethylation of the D4Z4 macrosatellite array on the chromosome 4q telomere allowing ectopic expression of the DUX4 gene in skeletal muscle. Genetic analysis is difficult due to the large size and repetitive nature of the array, a nearly identical array on the 10q telomere, and the presence of divergent D4Z4 arrays scattered throughout the genome. Here, we combine nanopore long-read sequencing with Cas9-targeted enrichment of 4q and 10q D4Z4 arrays for comprehensive genetic analysis including determination of the length of the 4q and 10q D4Z4 arrays with base-pair resolution. In the same assay, we differentiate 4q from 10q telomeric sequences, determine A/B haplotype, identify paralogous D4Z4 sequences elsewhere in the genome, and estimate methylation for all CpGs in the array. Asymmetric, length-dependent methylation gradients were observed in the 4q and 10q D4Z4 arrays that reach a hypermethylation point at approximately 10 D4Z4 repeat units, consistent with the known threshold of pathogenic D4Z4 contractions. High resolution analysis of individual D4Z4 repeat methylation revealed areas of low methylation near the CTCF/insulator region and areas of high methylation immediately preceding the DUX4 transcriptional start site. Within the DUX4 exons, we observed a waxing/waning methylation pattern with a 180-nucleotide periodicity, consistent with phased nucleosomes. Targeted nanopore sequencing complements recently developed molecular combing and optical mapping approaches to genetic analysis for FSHD by adding precision of the length measurement, base-pair resolution sequencing, and quantitative methylation analysis.
INTRODUCTION
Fascioscapulohumeral muscular dystrophy (FSHD) is one of the most common forms of muscular dystrophy with a prevalence of 1:15,000-1:20,000 1–3. As suggested in the name, FSHD results in weakness of the muscles of the face, shoulder stabilizers, and upper arm but also affects proximal and distal muscles of the legs. Unlike other muscular dystrophies, weakness can be asymmetric, and onset and severity are variable even between members of the same family with the same mutation 1, 4. Weakness is progressive, leading to significant morbidity with ~20% requiring a wheelchair over 50 years of age, and 10% developing restrictive respiratory disease 5, 6. Complicating diagnosis, FSHD has a mechanism combining both genetic and epigenetic factors in a region that is notoriously difficult to analyze due to its length and repetitive elements.
FSHD maps to the subtelomeric end of chromosome 4q, in a region that contains tandem copies of D4Z4, a 3.3kb CpG-rich macrosatellite with an intronless retrogene encoding the DUX4 homeobox domain protein. DUX4 is an important regulator of early genome activation, but expression is heavily repressed by epigenetic mechanisms after the first few cell divisions 7. In FSHD patients, chromatin relaxation accompanied by hypomethylation of the D4Z4 array leads to ectopic expression of DUX4 which is toxic to skeletal muscle 8. Hypomethylation of the array is asymmetric, involving the proximal more than distal D4Z4 repeats, and is inversely correlated with the number of repeats 9. DUX4 expression in skeletal muscle arises from the distal D4Z4 repeat and requires a short flanking sequence, pLAM, which contains a 3’-terminal exon and polymorphic signals for pre-mRNA cleavage and polyadenylation 9, 10. pLAM and related polymorphic sequences are found in FSHD permissive 4qA haplotypes. Non-FSHD permissive haplotypes include the nearly identical sequences on chromosome 10q which lack the polyadenylation signal, 4qA haplotypes that have pLAM but lack the polyadenylation signal, and 4qB haplotypes which lack pLAM altogether. These non-permissive haplotypes are not known to result in FSHD despite hypomethylation of the contracted array 8.
Most individuals have 11-100 D4Z4 repeats in the array, but contraction of the array to 10 or less copies on a permissive 4qA haplotype results in FSHD1 (MIM158900) 8. FSHD1 is the most common form of FSHD, accounting for 95% of cases 11. Contraction length of the D4Z4 array is correlated to severity of disease but does not explain all the variability. Individuals with 1-3 D4Z4 repeats usually have severe disease, individuals with 4-7 repeats typically have intermediate disease manifestations, and individuals with 8-10 repeats are usually mildly affected 6, 12, 13. FSHD2 (MIM 158901) is phenotypically identical to FSHD1, but has a digenic mechanism involving both a permissive 4qA haplotype and mutation in one of several chromatin modifier genes (SMCHD1, DNMT3B, and LRIF1) 14–17. The combined permissive 4qA haplotype and additional mutation in a chromatin modifier gene, results in hypomethylation of the D4Z4 array and expression of the DUX4 gene, even without a full contraction of the array. However, the number of repeats in the D4Z4 array is generally smaller in FSHD2 patients than controls suggesting that repeat length is an important factor in pathogenesis in addition to hypomethylation due to a mutation in the second gene 9, 11.
Analysis of the FSHD related D4Z4 array on 4q is complicated by the large size of the array, a nearly identical array on telomeric chromosome 10q, and the presence of divergent D4Z4 arrays scattered throughout the genome 18–20. The gold-standard diagnostic test for FSHD1 is Southern blot after restriction digest which can differentiate the chromosome 4q and 10q D4Z4 arrays and estimate their length 21. More recently, molecular combing and optical mapping techniques have emerged to estimate the size of the array 22–24. These methods represent a significant advance, but like Southern blotting they are technically demanding and are only available at a few select laboratories. Neither method provides detailed information about the methylation state or background haplotype (4qA/4qB) without additional testing. Epigenetic analysis using targeted bisulfite sequencing can differentiate between FSHD and healthy controls using a direct measurement of methylation in critical regions of the D4Z4 array and has been proposed as a novel diagnostic tool that does not require determination of the length of the array 25, 26. Genetic testing is further complicated by relatively common translocations between the 4q and 10q arrays, duplication arrays, and somatic mosaicism 11, 27.
Emerging long-range sequencing technologies allow for sequencing across complex repetitive regions and address existing gaps in genetic analysis for FSHD. Recent publication of the first complete genome relied heavily on these new long-range sequencing tools to successfully sequence highly repetitive centromeric and telomeric sequences including both 4q, 10q, and other D4Z4 arrays 28. Nanopore sequencing is a single-molecule long-range sequencing tool that can routinely produce read lengths of 30 kb or more 29. Nanopore sequencing has been successfully used to sequence the 4q D4Z4 array from a BAC clone, but an attempt to sequence the array from genomic DNA as part of a whole genome analysis resulted in only a small number of full-length reads from the D4Z4 array limiting utility of this technique 30. Nanopore Cas9-Targeted Sequencing (nCATS) is a recently developed tool to enrich target sequences for nanopore sequencing allowing marked increase in read depth for targeted sequences 31, and has been proposed as a novel tool to sequence the D4Z4 array in FSHD 32. We used nCATS as a comprehensive tool for genetic analysis of FSHD to determine the length of the 4q and 10q D4Z4 arrays, differentiate the 4q from 10q arrays, determine A/B haplotype, identify divergent D4Z4 elsewhere in the genome, and estimate methylation across all CpGs in the array.
RESULTS
Targeted nanopore sequencing of D4Z4 repeat arrays
We used multiple guide RNAs (gRNAs) to target centromeric (p13E-11) and telomeric (pLAM) sequences flanking D4Z4 arrays on chromosomes 4qA and 10q (Fig. 1A, 1B). Human peripheral blood DNA was cut with a Cas9 p13E-11/pLAM gRNA mixture, followed by adapter ligation and nanopore sequencing on MinION flow cells. Targeted enrichment was observed at both 4q and 10q, as well as consistent enrichment peaks on other chromosomes with divergent D4Z4 arrays (Fig. 1C). The average 4q/10q on-target percentage was 0.7% from eight MinION libraries using the p13E-11/pLAM gRNA mixture (Table 1). FSHD1 participant P2 had a known 4qA 6U D4Z4 allele confirmed with multiple 17.5 kb, p13E-11 to pLAM, full length, cut-to-cut reads covering the full array (39 6U reads, Table 1, Fig. 1D). The enrichment was reproducible with 50, 68, and 78 reads confirming 4qA 6U D4Z4 alleles from participants P1, P3, and P4. Two additional FSHD1 participants had their pathogenic 4qA alleles confirmed, including an 8.0 kb 3U allele (P6) and compound heterozygous 9U alleles from participant P5, who was heterozygous for a 27.6 kb 4qAS 9U allele and a 29.0 kb 4qAL 9U allele. In FSHD2 participants P7 and P8, we identified 60.1 kb (19U) and 50.2 kb (16U) p13E-11 to pLAM reads spanning their shortest 4qA allele, respectively. In each of the eight samples, we identified reads that spanned 4qA or 10q D4Z4 arrays (Table 1). Full-length reads spanning shorter arrays were recovered at greater efficiency, consistent with a size bias operating at the nanopores. The ability to specifically map 4q and 10q D4Z4 reads was also evident by the ratio of 4q-specific (XapI) and 10q-specific (BlnI) restriction sites (Fig. 1D) in reads aligned to D4Z4 repeats.
Figure 1. Cas9-targeted nanopore sequencing of D4Z4 repeats.
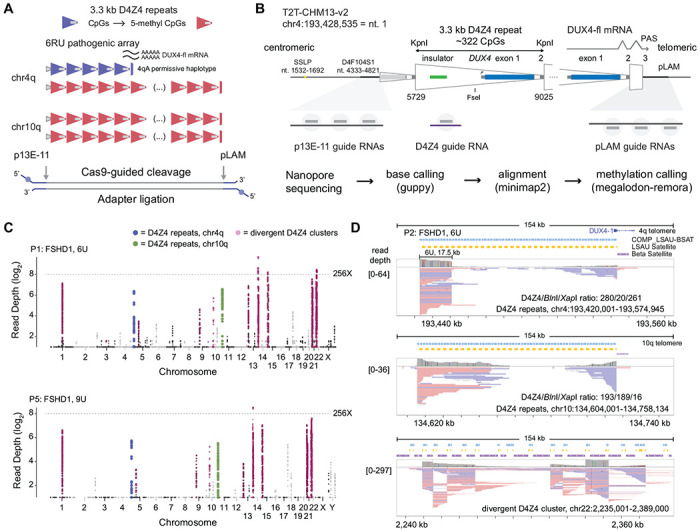
(A) Schematic of D4Z4 repeat arrays found in sub-telomeric regions of human chr4q and 10q. A 6U D4Z4 array is shown on the permissive 4qA haplotype, and blue/red shading of individual repeats indicate CpG methylation. (B) Locations of Cas9-guide RNA cleavage sites at the centromeric (p13E-11) end, telomeric (pLAM) end, and within each repeat (D4Z4). Steps used in the sequencing pipeline for identifying targeted reads are shown. (C) Read depth Manhattan plots from two FSHD1 participants (P1, P5) mapped to the T2T CHM13 v2.0 reference genome (log2 scale, minimum depth = 2 reads, summarized in 2 kb, non-overlapping windows). Colors indicate reads mapped to the chr4q (blue) and 10q (green) D4Z4 arrays, as well as divergent DUX4 clusters (purple). (D) IGV browser alignments of targeted reads from FSHD1 participant P5 with a 4qA 6U contraction. The D4Z4/BlnI/XapI ratio shows the number of 3.3 kb D4Z4 repeats contained with the aligned reads and the number of 10q-specific (BlnI) and 4q-specific (XapI) restriction sites found within those repeats.
Table 1.
Read depth and targeting efficiency from nanopore Cas9 targeted sequencing in FSHD1 and FSHD2 participants and healthy controls
| Participant ID | Total Mapped Reads | All On-target Reads | 4q On-target Reads | 10q On-target Reads | D4Z4 On-target Reads (3.3kb) | Divergent D4Z4 Reads | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
||||||||||
| Total | % p13-pLAM | % D4Z4 | p13 | pLAM | Full Array | p13 | pLAM | Full Array | 4q | 10q | Reads | % Total | ||
| Cas9 gRNA:p13E-11+pLAM a | ||||||||||||||
|
| ||||||||||||||
| P1 | 35,019 | 392 | 1.1% | - | 91 | 78 | 68 (6U) | 109 | 114 | 38 (11U) 34 (13U) |
- | - | 16,368 | 46.7% |
| P2 | 30,954 | 207 | 0.7% | - | 64 | 70 | 39 (6U) | 35 | 38 | 5 (13U) | - | - | 17,631 | 57.0% |
| P3 | 116,981 | 613 | 0.5% | - | 137 | 183 | 78 (6U) | 143 | 150 | 36 (9U) | - | - | 42,885 | 36.7% |
| P4 | 77,047 | 319 | 0.4% | - | 83 | 107 | 50 (6U) | 59 | 70 | 7 (13U) 1(19U) |
- | - | 23,604 | 30.6% |
| P5 | 18,256 | 179 | 1.0% | - | 62 | 36 | 15 (9U-S) 10 (9U-L) |
56 | 25 | 0 | - | - | 8,381 | 45.9% |
| P6 | 27,317 | 139 | 0.5% | - | 48 | 54 | 36 (3U) | 14 | 23 | 1 (13U) | - | - | 9,044 | 33.1% |
| P7 | 53,538 | 385 | 0.7% | - | 30 | 34 | 4 (19U) | 171 | 150 | 63 (8U) 34 (12U) |
- | - | 13,583 | 25.4% |
| P8 | 52,066 | 254 | 0.5% | - | 33 | 42 | 11 (16U) 1 (26U) |
87 | 92 | 39 (10U) 1 (25U) |
- | - | 20,093 | 38.6% |
|
| ||||||||||||||
| Cas9 gRNA:p13E-11 a | ||||||||||||||
|
| ||||||||||||||
| P2 | 30,734 | 77 | 0.3% | - | 27 | 0 | 0 | 50 | 0 | 0 | - | - | 42 | 0.1% |
|
| ||||||||||||||
| Cas9 gRNA:D4Z4 b | ||||||||||||||
|
| ||||||||||||||
| C1 | 9,042 | 2,741 | - | 30.3% | - | - | - | - | - | - | 1,077 | 1,664 | 512 | 5.7% |
| C2 | 10,345 | 2,555 | - | 24.7% | - | - | - | - | - | - | 1,424 | 1,131 | 321 | 3.1% |
| P9 | 6,431 | 410 | - | 6.4% | - | - | - | - | - | - | 217 | 193 | 138 | 2.1% |
| P10 | 1,418 | 476 | - | 33.6% | - | - | - | - | - | - | 258 | 218 | 66 | 4.7% |
| P1 | 6,977 | 826 | - | 11.8% | - | - | - | - | - | - | 372 | 454 | 246 | 3.5% |
| P1 | 3,348 | 339 | - | 10.1% | - | - | - | - | - | - | 162 | 177 | 117 | 3.5% |
|
| ||||||||||||||
| Cas9 gRNA:p13E-11+pLAM+D4Z4 | ||||||||||||||
|
| ||||||||||||||
| P9a | 116,799 | 3,582 | 0.5% | 2.6% | 115 | 210 | 40 (6U) | 107 | 109 | 0 | 1,524 | 1,517 | 37,467 | 32.1% |
| P10a | 39,605 | 1,979 | 0.7% | 4.3% | 63 | 91 | 42 (6U) | 55 | 50 | 8 (13U) | 888 | 832 | 16,052 | 40.5% |
| C3c | 9,952 | 1,219 | 0.2% | 12.0% | 3 | 10 | 0 | 5 | 4 | 0 | 806 | 391 | 4,152 | 41.7% |
|
| ||||||||||||||
| Ultra -Long kit V14 d | ||||||||||||||
|
| ||||||||||||||
| C4 | 6,140,482 | 253 | 0.004% | 0.037% | 74 | 51 | 14 (16U) 4 (41U) |
62 | 66 | 10 (20U) 5 (21U) |
- | - | - | - |
Sequencing on MinION R9.4.1 flowcell;
Sequencing on Flongle R9.4.1 flowcell;
Sequencing on MinION R10.4.1 flowcell;
Sequencing on PromethION 10.4.1 flowcell.
Additional enriched peaks outside of 4q and 10q telomeres (Fig. 1C) mapped to regions annotated in the Telomere-to-Telomere CHM13v2.0 (T2T) reference genome as LSAU-beta-satellite (BSAT) composite repeats that included homologous D4Z4 repeats 33. The location of these reads suggested targeting by the pLAM guide RNAs adjacent to divergent D4Z4 sequence (Fig. 1D). The 20 largest genomic regions with LSAU-BSAT annotations (average span = 200 kb, range 20 kb – 621 kb, total span = 4.0 Mb; Table S1) accounted for, on average, 39.2% of the total reads, ranging from 25.4 to 57.0% (Table 1, Divergent D4Z4 Reads). We confirmed that these reads were due to Cas9 cleavage at pLAM-related targets by removing the pLAM guide RNAs from a second library prepared from participant P2. This library retained the on-target p13E-11 reads but eliminated the LSAU-BSAT region reads (Table 1).
The high percentage of near-target reads at divergent D4Z4 arrays implied pLAM gRNAs were effectively cleaving multiple targets per genome equivalent. This suggested that a single gRNA designed within the D4Z4 repeat may yield a high percentage of monomer-sized, 3.3 kb D4Z4 reads arising from the 4q and 10q telomeres. We generated six additional libraries using a single D4Z4-gRNA (Fig. 1B) and observed a high yield of on-target, 4q and 10q, 3.3 kb monomer, D4Z4 reads using the smaller Flongle flow cell (average = 19.5% on target D4Z4 reads, Table 1). The ratio of monomer (3.3 kb) to dimer (6.6 kb) D4Z4 reads averaged 19:1, indicating almost complete cleavage of the D4Z4 arrays by the D4Z4 gRNA. Three additional samples had p13E-11/pLAM and D4Z4 gRNA libraries prepared separately and combined for sequencing onto single MinION flow cells. These mixed libraries (p13E-11/pLAM and D4Z4) showed comparable yields for their component gRNA targets (Table 1). Finally, we compared the yield of targeted reads from the nCATs libraries sequenced on single MinION flow cells to a public dataset derived from a peripheral blood DNA library using ultra-long protocols and sequenced on three PromethION flow cells. This library had a total read yield 120X greater than our average MinION nCATs library, and a total Gigabase yield that was 450X greater (Table 1, sample C4). Despite this large difference in the overall yield of the PromethION dataset, comparable reads counts localized to the 4q and 10q D4Z4 arrays (Table 1) suggesting that the lower cost nCATs methodology is currently on par with whole genome nanopore sequencing for specifically studying the D4Z4 regions.
D4Z4 arrays have asymmetric CpG methylation gradients
To evaluate the extent of hypomethylation that occurs as a result of contraction of the D4Z4 array, we extracted the 5-methyl CpG (5mC) base modifications from the nanopore reads. The methylation status of individual p13E-11 to pLAM cut-to-cut reads were summarized at the D4Z4 repeat level, from the ~320 CpG dinucleotides per 3.3 kb repeat. The results were quantified as the percentage of 5mC per repeat and the distributions for 4qA and 10q arrays from FSHD1 participants P1 and P3 are shown as box plots in Fig. 2A. Repeat-level methylation increases in a step-wise fashion from the centromeric (p13E-11) to telomeric (pLAM) repeat for 6U, 9U, 11U, and 13U arrays. Smoothed methylation frequency plots and single-molecule methylation plots (Fig. 2B) using the same data displayed a characteristic striped methylation pattern within each repeat indicating a recurring pattern of higher and lower methylation, and a gradual, step-wise methylation increase across the arrays.
Figure 2. CpG methylation gradients traverse chr4q and 10q D4Z4 arrays.
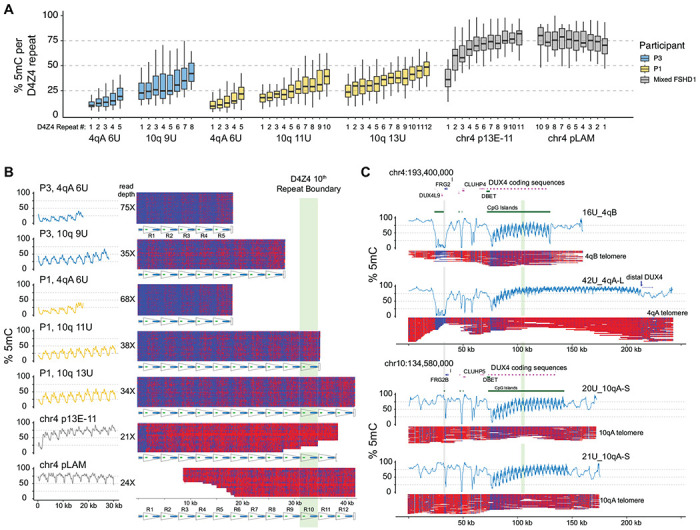
(A) Methylation levels of p13E-11 to pLAM targeted reads summarized as % 5mC sites per D4Z4 repeat (3.3 kb KpnI-to-KpnI region with ~320 CpG sites per repeat). Repeat numbering begins at the centromeric end (p13E-11) and is reversed for the chr4 pLAM (telomeric) reads. (B) Single-read plots of targeted reads from (A) using modbamtools with unmethylated CpGs represented in blue and 5-methyl CpGs in red. The schematic representation of D4Z4 repeats shows DUX4 exons 1 and 2 and the CTCF insulator region, and the green stripe indicates the 10th centromeric D4Z4 repeat. (C) Alignments and methylation plots of untargeted reads (R10.4.1_e8.2 nanopores) from a control subject (C4) mapped to the 4qB 16U haplotype, the 4qA 43U haplotype, the 10q 20U haplotype, and the 10q 21U haplotype. The grey stripe delineates the start of the syntenic telomeric region shared between 4q and 10q, and the green stripe marks the 10th repeat.
To examine the methylation levels of longer D4Z4 arrays in individuals with FSHD1, we aggregated the p13E-11 or pLAM reads from non-contracted 4qA alleles. The results showed that the centromeric end (p13E-11) had higher levels of methylation and a steeper increase than the shorter arrays, while the telomeric end (pLAM) had a uniformly high level of methylation with a slight gradual decrease distally (Fig. 2A, 2B). To ensure that these patterns were not specific to individuals with FSHD1, we also analyzed whole-genome data from a publicly available healthy control (C4) sequenced on high-density PromethION flow cells, where both alleles on chromosome 4 and 10 could be resolved. The whole genome nanopore data allowed us to assemble reads across the entire syntenic chromosome 4 and 10 region, and asymmetric methylation gradients were observed on all four haplotypes: the 4qB 16U, 4qA 42U, 10q 20U, and 10q 21U (Fig. 2C). The data confirmed the presence of an asymmetric methylation pattern in this control individual, indicating that these asymmetric methylation gradients are not specific to FSHD1.
Methylation gradient modeling and methylation spreading
Despite the consistent gradient from low methylation in the proximal end of the array and higher methylation in the distal end, we observed a high level of variability in overall methylation between reads, and similar variation within reads from one repeat to the next (Fig 3A). To better quantify the methylation gradient, we used a linear mixed-effects model (LMM) based on read-level D4Z4 repeat unit methylation as the repeated measure. The LMM estimated the %5mC level from the first D4Z4 repeat (intercept) and the change per repeat (slope). For participant P1, the LMM estimated an intercept of 9.7% 5mC on the first D4Z4 unit with 3.7% (slope) based on 68 full 4qA 6U reads (Fig. 3A). The LMM also allowed for the calculation of separate intercept and slope for individual reads, which varied in overall %5mC from 7.6 to 38.1%. These individual regression lines are plotted with the observed repeat-level %5mC values together with reads ordered by overall %5mC (Fig. 3A). The 4q and 10q arrays from FSHD1 participants with full-length reads (4q: 3U, 6U, 9U and 10q: 9U, 11U, 13U) had intercepts from 9.6 to 26.1% and slopes from 1.9 to 3.9% (Table 2, Fig. 3B). FSHD2 participants (4q:16U, 19U and 10q 8U, 10U, 12U) showed intercepts ranging from 9.1 to 14.3%, similar to FSHD1 participants, but showed lower slopes, ranging from 0.7 to 1.6% (Table 2, Fig. 3B).
Figure 3. Methylation gradient modeling.
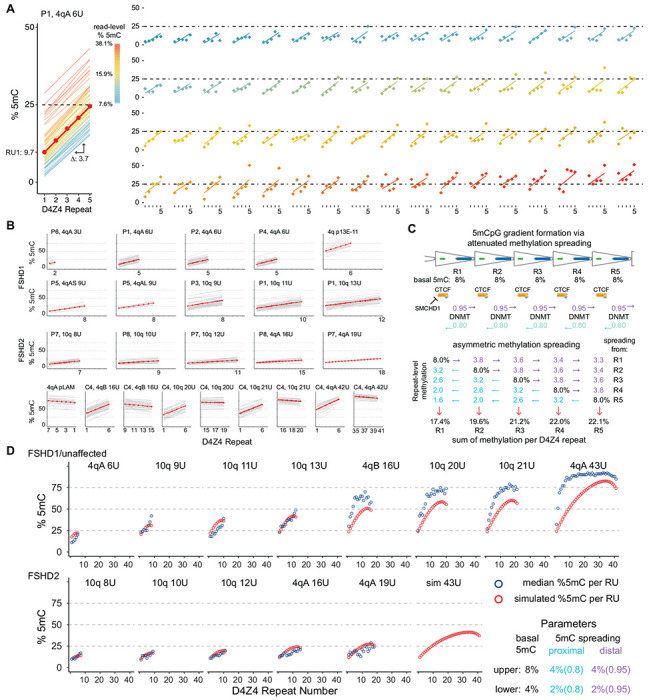
(A) Intercept and slope estimates from 68 p13E-11 to pLAM 6U reads from participant P1. The red line is the model estimate and the thin lines are the individual read estimates with a color scale for read-level % 5mC. The right panel shows the D4Z4 methylation frequencies for each read (points) with their individual regression line. The 68 plots are ordered by read-level % 5mC. (B) Intercept and slope estimates as in (A) from FSHD1, FSHD2 participants, and control subject C4 (see Table 2). (C) Model for gradient formation via basal methylation followed by asymmetric spreading. (D) Predicted D4Z4 methylation levels from simulated basal methylation and spreading (red points) compared to observed values (blue points, mean methylation per repeat, data from (B)). In the lower panel, the basal methylation and spreading parameters were reduced by a factor of 2.
Table 2:
D4Z4 methylation gradient analysis using a linear mixed-effects model to estimate intercept and slope.
| Subject ID | Chromosome | Length D4Z4 | % Methylated CpG RU1 |
Change in % methylated CpG per RUa |
||
|---|---|---|---|---|---|---|
| Intercept | 90% CI | Slope | 90% CI | |||
| FSHD1 | ||||||
|
| ||||||
| P6 | chr4qA-S | 3U | 14.2 | 10.6 - 17.7 | 0 | −2.1 - 2.1 |
| P1 | chr4qA-S | 6U | 10.3 | 7.3 - 13.2 | 2.8 | 2.3 - 3.4 |
| P2 | chr4qA-S | 6U | 15.3 | 12.0 - 18.5 | 2.1 | 1.5 - 2.8 |
| P3 | chr4qA-S | 6U | 11.5 | 8.6 - 14.5 | 2.3 | 1.8 - 2.8 |
| P4 | chr4qA-S | 6U | 10.4 | 7.2 - 13.7 | 2.9 | 2.3 - 3.5 |
| P5 | chr4qA-S | 9U | 10.1 | 5.6 - 14.5 | 2 | 1.3 - 2.7 |
| P5 | chr4qA-L | 9U | 18 | 12.8 - 23.2 | 2.3 | 1.4 - 3.1 |
| P3 | chr10q | 9U | 24.9 | 21.5 - 28.3 | 2.3 | 1.7 - 2.8 |
| P1 | chr10q | 11U | 17.4 | 15.1 - 19.7 | 2.2 | 1.9 - 2.6 |
| P1 | chr10q | 13U | 25.4 | 22.0 - 28.8 | 1.9 | 1.3 - 2.4 |
| P1, P2, P3, P4, P5, P6 | chr4qA-S | >10U, centromeric | 46.6 | 42.5 - 50.7 | 6.1 | 5.3 - 6.8 |
| P1, P2, P3, P4, P5, P6 | chr4qA-S | >10U, telomeric | 76.2 | 72.1 - 80.3 | −1 | −1.6 - −0.3 |
|
| ||||||
| FSHD2 | ||||||
|
| ||||||
| P7 | chr10q | 8U | 9.5 | 6.5 - 12.5 | 1.2 | 0.7 - 1.7 |
| P8 | chr10q | 10U | 11.9 | 8.6 - 15.2 | 0.7 | 0.2 - 1.3 |
| P7 | chr10q | 12U | 11.8 | 8.4 - 15.3 | 0.8 | 0.2 - 1.3 |
| P8 | chr4qA-S | 16U | 13.3 | 8.4 - 18.2 | 0.5 | −0.3 - 1.2 |
| P7 | chr4qA-S | 19U | 14.4 | 7.0 - 21.8 | 0.6 | −0.5 - 1.7 |
|
| ||||||
| Unaffected Control | ||||||
|
| ||||||
| C4 | chr4qB | 16U, prox 6U | 33.7 | 30.4 - 37.0 | 6.2 | 5.6 - 6.8 |
| C4 | chr4qB | 16U, distal 6U | 64.1 | 60.7 - 67.4 | −1.3 | −1.9 - −0.7 |
| C4 | chr10q | 20U, prox 6U | 29.4 | 25.9 - 33.0 | 5.7 | 5.1 - 6.4 |
| C4 | chr10q | 20U, distal 6U | 71.4 | 67.9 - 74.9 | −0.5 | −1.2 - 0.1 |
| C4 | chr10q | 21U, prox 6U | 34.5 | 31.0 - 37.9 | 5.7 | 5.1 - 6.4 |
| C4 | chr10q | 21U, distal 6U | 77.1 | 73.5 - 80.6 | −0.9 | −1.6 - −0.2 |
| C4 | chr4qA-L | 42U, prox 6U | 42.5 | 38.4 - 46.6 | 7.1 | 6.3 - 7.9 |
| C4 | chr4qA-L | 42U, distal 6U | 89.8 | 86.3 - 93.4 | −1 | −1.6 - −0.4 |
RU-repeat unit
Variation in the overall per read methylation was evident, as well as a trend for increased methylation of the first D4Z4 repeat with longer arrays with notable differences between 3U and 6U versus the 9U, 11U, and 13U arrays (Fig. 3B, Table 2). Centromeric (p13E-11) reads from longer arrays had the highest intercepts, including the >10U, 4q reads from FSHD1 participants (52%) and the 16U to 42U arrays from the control subject, which ranged from 32.7 to 48.5%. Telomeric reads (pLAM) from the longer arrays consistently showed negative slopes, ranging from −1.1 to −0.2, suggesting a tapering from the high %5mC levels at internal repeats in very large arrays.
These patterns suggested a simple model combining basal D4Z4 methylation and spreading that could lead to the formation of asymmetric gradients. Fig. 3C shows a model based on ad hoc parameters for basal methylation rates (8% per D4Z4 repeat) and asymmetric spreading (a directional rate of 95% distal spreading versus 80% proximal spreading) that results in gradient formation. We used these parameters to simulate D4Z4 methylation levels for different length arrays. The modeled findings were consistent with the observed median D4Z4 repeat levels from FSHD1 and control participants, including the tapering of methylation in the longer arrays (Fig. 3D). When the basal parameter was reduced by a factor of 2, from 8% to 4%, the simulated gradient more closely paralleled the observed methylation levels in FSHD2 participants.
Fine-scale D4Z4 methylation patterns suggest nucleosome positioning
In addition to methylation gradients across the D4Z4 array, we observed consistent methylation patterns within individual repeats, including areas of low and high methylation. The single-read and smoothed methylation frequency plots of the 4qA 6U haplotype reads from participant P1 (Fig. 4A) demonstrate this characteristic pattern of repeating across the array from R1 to R5. Aligning methylation frequencies of individual CpGs within the 6U haplotype from participant P1 with the annotated features of the 3.3 kb KpnI-KpnI repeat showed recurring low and high methylation levels across repeats R1-R5 (Fig. 4B). The smoothed methylation frequency aligned across repeats R1-R5 showed a consistently rising pattern of methylation with repeat number. The CpGs in the CTCF/insulator region were among the least methylated residues. This region contains the DR1 amplicon previously identified as relatively hypomethylated 26, 34. CpGs near the DUX4 transcription start site, especially the CpG at nucleotide (nt) 1704, had high methylation levels ranging from 28-90%. A phasic methylation pattern with ~180 nt periodicity started near this region and propagated through DUX4 exons 1 and 2 (Fig. 4B).
Figure 4. Fine-scale D4Z4 repeat methylation patterns.
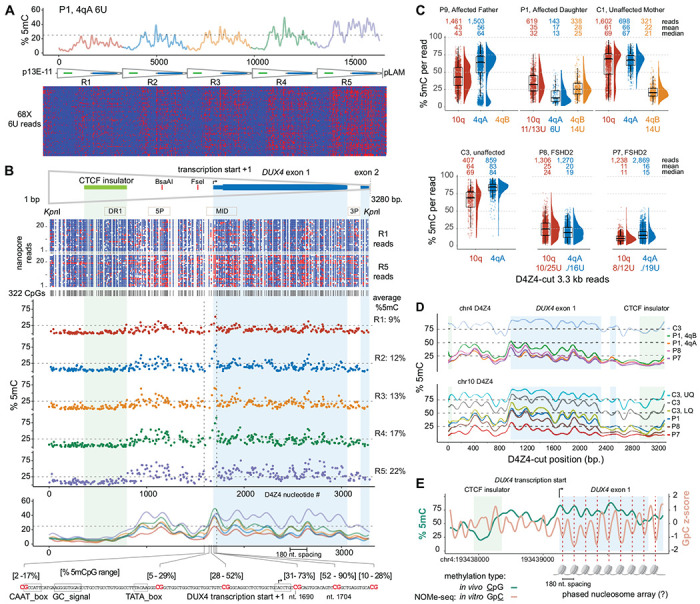
(A) Methylation plot of the 68 4qA 6U reads from participant P1 using the “smooth_ksmooth” function (smoothness parameter = 10) and single-read plots using modbamtools. (B) Annotated D4Z4 features including DUX4 exons 1 and 2 (mRNA from AF117653.3), the CTCF insulator (Blp1-Blp1 fragment from Ottaviani et al.,2010), BsaAI and FseI methylation-sensitive restriction sites, and PCR amplicons commonly used for bisulfite sequencing. A methylartist plot shows 322 CpG residues represented as blue (unmethylated) or red (methylated) dots for the R1 and R5 repeats from a subset of reads in (A). The middle panel shows the methylation level averaged from 68 chr4 6U reads in (A) for each of the 322 CpGs, aligned from R1 through R5. The lower panel shows the methylation frequency plot for each repeat, smoothed as in (A), with the location and methylation frequency range of six CpG residues in the region containing the DUX4 proximal promoter and transcription start from Dixit et al. (AF117653.3). (C) Methylation levels of individual 3.3 kb D4Z4-cut reads with % 5mC per read on the y-axis, with the number of mapped reads and mean/median methylation shown for each chromosome. Upper plot = FSHD1 trio (affected father, affected daughter, unaffected mother) segregating a pathogenic 6U repeat; lower plot = unaffected versus FSHD2 participants. (D) Methylation frequency plots of D4Z4-cut reads from (C), smoothed as in (A). (E) % 5mC and Nucleosome Occupancy and Methylation sequencing (NOMe-seq) GpC z-scores plotted for the second chr4 D4Z4 repeat (centromeric) using in vivo/in vitro methylation data from the HG002/NA24385 lymphoblastoid cell line.
D4Z4 3.3 kb reads from an enrichment using a Cas9-gRNA targeting individual D4Z4 repeats (Fig. 1B, Table 1) were used to examine the fine-scale methylation structure in additional FSHD1, FSHS2, and control participants. Although these reads lacked positional information for the individual D4Z4 repeat, sequence alignments enabled discrimination between 4qA, 4qB, and 10q repeats. The paternally inherited 4qA reads from the 6U haplotype carried by participant P1 were distinguishable from the 4qB reads inherited from her mother (Fig. 4C). The lower methylation range of the 4qA-specific reads and the intermediate methylation range of the 10q reads from the 11U/13U haplotypes were similar to the repeat-level summaries from full haplotype reads (Fig. 4B). The 6U haplotype methylation status in her father was evident from the bi-phasic profiles of his 4qA-specific reads, emphasizing the distinction in repeat-level methylation between small and large arrays. The relative absence of higher methylation levels from the two FSHD2 participants was apparent, although the mean and median methylation levels of the FSHD2 4qA reads were comparable to the 4qA reads from the 6U haplotype from FSHD1 participant P1. The smoothed methylation frequency plots of the 4qA D4Z4 reads demonstrated similarity of the FSHD1 and FSHD2 methylation profiles (Fig. 4D). This result suggests that reductions overall methylation levels specific for FSHD2 does not alter the fine-scale D4Z4 methylation pattern. From both 4q and 10q profiles, the maintenance of the fine-scale pattern with increasing methylation is evident, and the phasic methylation pattern beginning near the start of DUX4 exon 1 is variably seen with repeats from both chromosomes and with increasing overall methylation levels.
The 180-nt periodicity of CpG methylation suggested the possibility that this phasic pattern corresponds to nucleosome occupancy. To test this hypothesis, we examined the D4Z4 region for nucleosome protection utilizing a nanopore NOMe-seq data set (Nucleosome Occupancy and Methylome Sequencing) generated from HG002 lymphoblastoid cells by the T2T consortium 35. The CpG methylation frequency in the extended 4qA region of the HG002 cell line was comparable to the 4qA 42U allele from peripheral blood of control sample C4 and an asymmetric gradient was seen at the centromeric end of the HG002 array (Fig. S1). This methylation gradient was also observed in the profile from HG002 PacBio HiFi long-read sequencing, confirming that these methylation gradients can be detected with an orthogonal long-read sequencing chemistry. The smoothed frequency plot from the 4q centromeric D4Z4 repeat R2 displayed an intermediate level of methylation and a smoothed frequency plot displayed phasing similar to the profiles observed in peripheral blood (green line, Fig. 4E). The NOMe-seq GpC z-score, which measures the relative protection of GpC dinucleotides from in vitro methylation of isolated chromatin with a GpC methyltransferase 36, also showed a ~180 nt GpC phasic pattern across the exon 1 and 2 region (orange line, Fig. 4E). The GpC z-score valleys were coincident with CpG methylation peaks indicating that CpG methylation is higher in more protected regions. The endogenous phasic CpG methylation across exon 1 was offset by ~90 nt from the GpC z-score, consistent with the methylation status of CpGs in this region influencing the positioning of nucleosomes.
Divergent D4Z4 repeats including a paralogous chr22 cluster are highly methylated
The abundance of pLAM-related reads, as shown in Table 1, facilitated a genome-wide analysis of the sequence and methylation status of D4Z4-related sequences. We examined the copy number of a 4 kb query sequence encompassing the distal 3.3 kb D4Z4 repeat linked to the flanking 0.7 kb of 4qAS-specific sequence that includes the terminal ~0.3 kb D4Z4 truncation, DUX4 exon 3, and polymorphic PAS region that permits DUX4 expression from the distal D4Z4 repeat 8. The T2T reference genome contained 13 occurrences of this full 4 kb sequence, including the two copies on 4q and 10q. A ~3.1 kb sequence that deleted ~0.9 kb beginning at the 5’ KpnI site including the CTCF/insulator region, was the most common D4Z4-related sequence with 319 copies (Fig. 5A). Both the full and 5’ truncated copies were primarily located in the LSAU-BSAT rich regions of acrocentric chromosomes (chr13, 14, 15, 21, and 22) and in the pericentric region of chr1 (Table S2) and accounted for approximately 98% of the full-length, divergent DUX4 ORFs in the T2T reference genome (Table S3). A high methylation state of a full D4Z4-pLAM sequence from chr22 is shown in Fig. 5B, where a percent-identity plot indicates the sequence conservation with the D4Z4-4qAS-pLAM region.
Figure 5. Methylation profiles of divergent D4Z4 repeats.
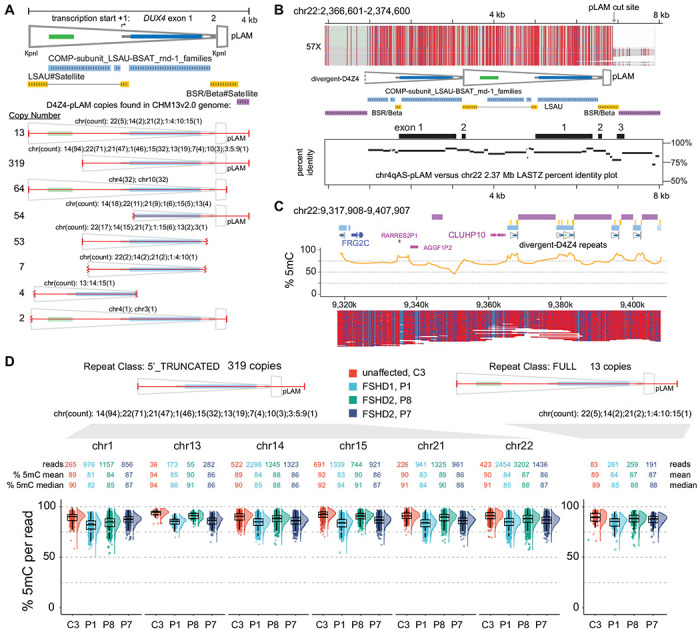
(A) Annotated structure and copy number of the distal 4 kb D4Z4 repeat beginning at the 5’ KpnI site through the 4qAS pLAM sequence/β-satellite junction (see Table S2 for list of genomic locations). (B) Methylation profile of a full-length, divergent D4Z4 repeat found on chr 22. The LASTZ percent identity (PIP) plot shows the distribution of sequence identity along the length of this chr22 D4Z4 repeat compared to the distal chr 4qAS D4Z4 repeat. (C) Methylation plots of untargeted reads mapping to chr22:9,317,908-9,717,807 region from the C4 control (R10.4.1_e8.2 nanopores). (D) Distributions of CpG methylation levels of divergent D4Z4 repeats where each point represents the % 5mC of an individual read within the region that aligns completely with a divergent repeat. The C3 sample was sequenced using R10.4.1_e8.2 nanopores.
The T2T reference genome resolved numerous regions that contain segmental duplications of FSHD region gene 1 (FRG1) and FSHD region gene 2 (FRG2) 28. We examined a T2T chr22 region with a divergent D4Z4 cluster that retains paralogous sequence through the FRG2 gene and the inverted single D4Z4 repeat found on 4q but absent from 10q. The methylation plots reveal a highly methylated state for both full and 5’ truncated D4Z4 repeats in this region (Fig. 5C). A dot plot alignment of the 4q region and the entire ~380 kb chr22 D4Z4 cluster demonstrates high sequence conservation that extends beyond the FRG2 paralog (Fig. S2). The divergent D4Z4 repeats in this cluster are interspersed with BSAT repeats and can be found in both forward and reverse orientations relative to 4q D4Z4 repeats. Moreover, the methylation frequency and single-read plots of the entire ~380 kb chr22 D4Z4 cluster support a uniformly high level of methylation without evidence for methylation gradients (Fig. S2).
In contrast to arrays on 4q and 10q, methylation levels for these divergent D4Z4 arrays were uniformly high. We conducted an analysis of methylation levels derived from reads aligned to the full and 5’ truncated class of divergent repeats across multiple chromosomal regions for FSHD1 participant P1, FSHD2 participants P7 and P8, and control participant C3. A total of 24,645 read alignments (803 full and 23,851 5’ truncated) were utilized and the median methylation levels for all four participants ranged from 81% to 94% (Fig. 5D). Notably, there were no read alignments with a methylation level below 50%, and only 11 reads demonstrated methylation levels below 60%. This observation indicates that the methylation levels of divergent D4Z4 repeats were uniformly high in the peripheral blood DNA of both FSHD1 and FSHD2 affected participants, as well as unaffected individuals.
Methylation gradients are seen in other high CpG ‘continents’
To identify whether the methylation gradients identified in the 4q and 10q D4Z4 arrays were unique, we conducted a genome-wide search for other regions of comparable CpG density to assess whether these areas harbored similar methylation gradients. A 33 kb sliding window, equivalent in size to 10 D4Z4 repeat units, was scanned across the T2T reference genome using a step size of 3.3 kb (~1 repeat unit) to rank the genome-wide CpG densities in 33 kb segments. CpG density for each individual 33kb segment was plotted against the location of the segment in a Manhattan plot with segments showing >5% CpG density highlighted for further analysis (Fig 6A). The 4q and 10q D4Z4 arrays were the two most CpG-dense regions across the genome for this window length, with densities of approximately 9.8% for the 33 kb window. There were 8 additional regions with 33 kb windows of >7.0% CpG density (Table S4). Five peaks were from 45S ribosomal DNA (rDNA) array clusters, and unique peaks were seen for the chr1 LM-tRNA cluster, the chr1 5S rDNA cluster, and the chr19 SST1 cluster. Methylation plots from the HG002 cell line showed evidence of methylation gradients in the rDNA arrays, the chr1 tRNA cluster, and the chr1 5S rDNA cluster (Fig. S3).
Figure 6. Methylation gradients in other CpG-dense repeat regions.
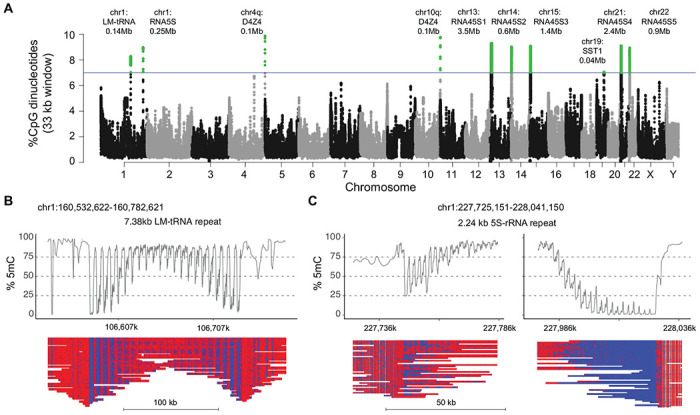
(A) Manhattan plot of CpG density in 33 kb sliding windows (step size = 3.3 kb) across the T2T CHM13 v2.0 reference genome. Windows above 7% CpG density are marked as green dots. Alignment and methylation plots of untargeted reads from the C4 control (R10.4.1_e8.2 nanopores) mapped to (B) the tRNA cluster on chromosome 1 and (C) the 5S rRNA cluster on chromosome 1. Mapped reads are anchored by unique flanking sequence and the two ends of the 5S rRNA cluster are shown separately.
Since long-range assembly across the 45kb rRNA arrays remains challenging, we focused on assembly and methylation analysis from the chr1 tRNA and 5S rDNA cluster using the peripheral blood reads from control C4. The chr1 tRNA has a 7.4 kb LM-tRNA repeat and the 5S region has a 2.2 kb 5S-rRNA repeat, so we used only reads anchored uniquely into flanking sequence for methylation analysis. The LM-tRNA region had a symmetrical gradient on both ends of the repeat array (Fig. 6B), while the 5S-rRNA region had asymmetrical gradients (Fig. 6C), confirming the presence of methylation gradients in repeat segments outside of the 4q and 10q D4Z4 arrays. Notably, the chr1 tRNA and 5S rRNA clusters were previously identified as hypomethylated regions in FSHD2 patients 37.
DISCUSSION
FSHD is one of the most common muscular dystrophies, yet genetic analysis is among the most difficult of any monogenetic disorder, due to the complex mechanism combining genetic and epigenetic factors with long stretches of repetitive DNA that have close paralogs elsewhere in the genome. Southern blot techniques have been the gold standard for diagnosis and research analysis for many years, but recent development of optical mapping, molecular combing, and next-generation sequencing based bisulfite sequencing techniques are rapidly enhancing our understanding of the genetics of FSHD 21–26.
Recently developed long read sequencing techniques such as nanopore sequencing, have opened the door to analysis of highly repetitive DNA in areas of the genome that were previously unmappable. Using these techniques, researchers completed sequencing of a full reference human genome for the first time, including mapping of repetitive DNA such as the D4Z4 arrays important in FSHD 28. These long-read sequencing technologies, have made sequencing analysis for FSHD feasible for the first time. Initial efforts to use nanopore sequencing for FSHD, struggled due to lack of read-depth in 4q derived D4Z4 arrays from genomic analyses 30. Development of strategies to enrich sequences of interest using Cas9-targeting has led to improved read-depth for regions of interest (Gilpatrick et al. 2020), including the D4Z4 array associated with FSHD 31, 32. Nanopore sequencing not only opens the door to the full sequencing of the D4Z4 array, but also allows determination of CpG methylation in the array, a critical aspect of genetic analysis for FSHD.
Here we described a refinement of the Cas9 targeting technique that can successfully enrich 4q and 10q D4Z4 arrays and provide a comprehensive tool for genetic analysis of FSHD. We demonstrated on-target p13E-11-to-pLAM reads enriched to 0.5-1.1% of all mapped reads, which provides sufficient read depth for analysis of 4q vs 10q sequences, determination of A/B haplotype, and size and methylation status of the array. Surprisingly, the bulk of off-target reads were near-target reads that arise from divergent D4Z4 arrays scattered throughout the genome, and accounted for 25-57% of all mapped reads. Exclusion of pLAM guide RNAs from the assay removed nearly all near-target reads, but did not increase recovery of full-length reads. The high read-depth of near-target sequences from pLAM gRNAs suggested high efficiency Cas9 cleavage per genome equivalent.
Our investigation revealed that the use of a single gRNA within the D4Z4 repeat led to a substantial retrieval of D4Z4 repeat reads, with up to 30% on-target reads, the majority of which (>95%) were 3.3 kb monomer, single D4Z4 reads. These findings offer additional support for the high efficiency of Cas9 cleavage. While the order of the individual repeats is lost using D4Z4 targeted guide RNAs, this assay adds read-depth and maintains the ability to separate 4qA, 4qB, and 10q reads, quantify methylation, and can be done on lower cost Flongle flow cells, providing a quick screen to differentiate FSHD and healthy controls. Furthermore, the retrieval of uniform 3.3 kb reads offers a semi-quantitative measure of D4Z4 copy number, as they are not affected by the length-dependent bias commonly seen in nanopore sequencing. A combined approach using libraries with p13E-11 and pLAM guide RNAs and libraries with D4Z4 guide RNAs on the same MinION flowcell provided adequate read-depth for comprehensive analysis and maintained the advantages of both assays. The combined assay provided read-depth comparable to non-targeted reads obtained from whole genome sequencing in a healthy control based on multiple PromethION flow cells obtained at a vastly higher cost.
The ability of nanopore sequencing to provide the methylation status of individual CpG sites on individual molecules allowed us to observe asymmetric methylation gradients that form at the proximal end of D4Z4 repeat arrays and reach a saturation point at approximately the 10th repeat. This finding provides a biophysical correlate for why contractions of less than 10 repeats are associated with FSHD. We postulated a simple model for directional barriers to methylation spreading across the 4q and 10q arrays that would establish the observed length-dependent, asymmetric gradients. While ad hoc, the model predicts the presence of asymmetric methylation gradients and the length-dependent change. The absence of methylation gradients and the uniform hypermethylation observed in paralogous D4Z4 clusters may provide insight to elements that restrict or promote methylation spreading.
It has been shown that methylation at the single FseI site in the proximal (centromeric) D4Z4 repeat is correlated with repeat array size 38. The authors proposed a metric (Delta1 score) based on proximal FseI methylation and a non-linear relationship between the cumulative array size, and used this as a marker for disease onset and progression. The length-dependent effect modeled by the Delta1 score was confirmed in our study by the use of a linear mixed-effect model to estimate the degree of change in the intercept value of the methylation gradient with increasing array size. The Delta1 score is particularly sensitive to detecting hypomethylation due to SMCHD1 mutations in FSHD2 individuals due to hypomethylation of both 4q and 10q alleles. The decreased slope of the methylation gradient we observed in FSHD2 individuals provides an alternative method for quantifying D4Z4 methylation that can be combined with single D4Z4 guide RNA libraries which capture and quantitate methylation from all 4q and 10q repeats. Although fitting the intercept and slope using a linear mixed-effect model allowed for the quantification of haplotype-specific methylation levels, this approach only accounted for a part of the considerable variability in read-level methylation observed with different haplotypes. Alternative approaches that capture this read-to-read variation may be useful as metrics for association with clinical markers of disease.
Bisulfite sequencing and methylated DNA immunoprecipitation analysis have been widely used for high resolution methyl-CpG analysis of the D4Z4 repeat 9, 34, 39, 40. These studies have been recently refined with improvements in amplicon specificity for discriminating 4q and 10q alleles, and by NGS-based analysis of bisulfite converted amplicons 25, 26, 41. The methyl-CpG patterns observed in our study confirmed several aspects of these prior studies, including extended hypomethylation at the 5’ end of the D4Z4 repeat including the CTCF/insulator region and the DR1 region. In a new finding, we identified preferential methylation of certain CpGs in close proximity to the DUX4 transcription start site, and we observed a consistent pattern of methylation across all ~320 individual CpGs within each 3.3 kb repeat.
This recurrent methylation pattern was independent of 4q or 10q haplotypes, FSHD1, FSHD2, or unaffected status, and was evident in both averaged single-CpG methylation estimates and kernel-smoothing analysis. Several aspects of this fine-scale methylation pattern suggested that it may be related to the chromatin-state of the D4Z4 repeat. Two regions of relative hypomethylation were consistently observed at around positions 100 to 700, overlapping with the CTCF/insulator region 42, and from 1400 to 1700, corresponding to the D4Z4 binding element (DBE) site for YY1/HMGB2/NCL complexes situated in the DUX4 proximal promoter region 43. Recent affinity capture of protein complexes bound to D4Z4 repeats suggest that these regions may also participate in binding the Nucleosome Remodeling Deacetylase (NuRD) and Chromatin Assembly Factor 1 (CAF-1) complexes 44.
A fine-scale methylation pattern with a periodicity of approximately 180 nts was observed starting near the methylation peak at position 1700. This pattern was also detected using long-read sequencing data from the HG002 lymphoblastoid cell line, which included companion NOMe-seq data to concurrently investigate nucleosome positioning and CpG methylation. NOMe-seq data from a proximal D4Z4 repeat with an intermediate level of CpG methylation suggested that the phasic CpG methylation signal was consistent with a phased nucleosome array in which the more highly methylated CpG peaks were wrapped around nucleosomes. Although it is unclear how phased nucleosomes may contribute to DUX4 repression, we can speculate that the preferentially methylated CpGs near position 1700 may participate in positioning of nucleosomes by localizing NuRD complexes through their methyl-CpG-binding protein domain 2 (MBD2) subunits to specific methyl-CpGs. Recent studies indicate that MBD2 has high affinity for large and densely methylated CpG islands, allowing recruitment of NuRD to facilitate nucleosome positioning, histone deacetylation, and chromatin compaction 45. Further investigation is required to determine whether the fine-scale methylation pattern observed within each D4Z4 repeat is established during development or whether it arises from the dynamic turnover of methyl-CpGs, reflecting changes in the underlying chromatin state.
The T2T reference genome enabled us to map near-target reads from the pLAM gRNAs to the repetitive regions of acrocentric chromosomes and analyze the methylation state of paralogous D4Z4 copies. Our findings were consistent with previous studies, which suggest that the D4Z4-4qAS-pLAM sequence is the ancestral form of the D4Z4 retroelement that radiated and diverged during primate and human evolution 46. The paralogous D4Z4 sequences we catalogued in the T2T genome uniformly share ~90% nucleotide identity with the D4Z4-4qAS-pLAM sequence but with reduced CpG densities of approximately 5.4%. Notably, all these paralogous sequences, regardless of length or location in the genome, and irrespective of whether the individual had FSHD1, FSHD2, or was unaffected, showed high levels of methylation. This hypermethylation was also observed in a large 380 kb cluster on chromosome 22, which has features similar to the 4q D4Z4 region, including an inverted DUX4c paralog located before a FRG2 paralog at one end, and a full-length D4Z4-4qAS-pLAM paralog at the other end.
One of the more striking aspects of the 4q D4Z4 array is its extreme CpG density. We used this property to scan the genome with a window size of 33 kb for other large regions of extreme density. The 4q and 10q arrays have the highest CpG density in the genome for this window size, with an median of 9.8 and 9.7%, respectively, and we identified 8 other genomic regions with densities >7.0%. The next densest region was a chromosome 1 tRNA cluster at 7.9% CpG with a 7.4 kb LM-tRNA repeating unit. This region also displayed methylation gradients that stretch over a ~50 kb interval. Unlike the D4Z4 arrays, these gradients appear symmetrical on both ends of the cluster. This tRNA tandem repeat has also been shown to contain a conserved CTCF binding site and the repeat unit has features of a genomic boundary element 47. We also observed methylation gradients at the two ends of the chr1 5S rDNA cluster with of repeating 2.2 kb 5S-rRNA repeat, although in this case, the gradients were asymmetric. Several regions of the 45S rDNA clusters were in this extreme CpG density range and there was suggestive evidence of methylation gradients at their borders. Notably, both the chr1 LM-tRNA and 5S rDNA cluster have been identified as one of the few genomic regions that are hypomethylated in the presence of heterozygous SMCHD1 mutations 37. This suggests that shared features among these repetitive clusters with extreme CpG densities may be utilizing common processes to link CpG methylation to the formation of repressive chromatin domains.
Several limitations are associated with our analysis of targeted sequence and methylation analysis of D4Z4 arrays. Our technique improves on the yield of targeted D4Z4 reads from prior studies using nanopore sequencing, however, the yield of full-length reads covering the entire D4Z4 array needs further improvement. Our analysis of near-target reads and the D4Z4 gRNA targeted reads revealed high Cas9 cutting efficiency, so further design of guide RNAs is not likely to improve yield. The presence of near-target reads from divergent D4Z4 reads throughout the genome did not diminish the yield of on-target reads, since the flow cells were still operating at reduced levels of pore occupancy due to a sub-optimal ratio of adaptered molecules to available pores, even with high DNA loads. Because of the complex and repetitive nature of the target sequences, our technique requires a high level of expertise in mapping of the target reads to appropriate reference sequences. Furthermore, cost for the MinION flowcells is relatively high. Improved pore chemistry, basecalling, and base modification accuracy are likely to provide additional precision, including sequencing the complementary strand from the same molecule. Optimization for lower cost flowcells such as the Flongle, may also help minimize cost and maximize yield. Our analysis for this study relied on genomic DNA from peripheral blood, it remains to be seen whether DNA samples from other tissues will have methylation patterns consistent with those observed here.
Our Cas9 targeted nanopore sequencing assay provides an important tool for genetic analysis of FSHD related D4Z4 arrays. This technique offers an additional tool for clinical diagnosis that can better quantify array length and methylation status, but can also help to provide additional genomic information for unusual cases including somatic mosaicism, 4q-to-10q translocations, and cis D4Z4 duplications which are not uncommon in FSHD clinics 11, 23, 48, 49. Furthermore, this tool opens the door to a more detailed methylation analysis, including analysis of the methylation gradient across the array, and methylation patterns within the individual repeats. Here, we show that the chromosome 4q and 10q D4Z4 arrays share a high CpG density and methylation gradients that are seen in only a few sites in the genome including tRNA and rRNA clusters, each with unique regulation of copy number and gene expression 50, 51. Understanding these unique structures and regulatory mechanisms is especially important in light of emerging therapies including antisense knockdown, targeted re-methylation, and other approaches of DUX4 transcriptional regulation 52–57.
METHODS
Subjects and samples.
Subjects with clinical history of FSHD and healthy controls were enrolled after consent to participation under a University of Utah Institution Review Board approved protocol. FSHD1 participants had either confirmed clinical genetic testing or positive family history of FSHD with clinical symptoms consistent with FSHD. FSHD2 participants had pathogenic variants in SMCHD1 identified by clinical genetic testing. Publicly available Oxford Nanopore files were downloaded from s3://ont-open-data/cliveome_kit14_2022.05/ for the peripheral blood library prepared from a control subject (C4). Genomic DNA was purified from whole blood using standard techniques provided by Gentra Puregene Blood Kits (Qiagen, cat # 158389).
Targeted nanopore sequencing
Genomic DNA libraries were generated using the Cas9 Sequencing Kit (Oxford Nanopore Technologies, cat # SQK-CS9109). crRNAs are annealed with tracrRNA (IDT, cat # 1072532) by combining: 1 μL of 100μM of crRNA pooled oligonucleotides (see Table S5 for sequences), 1 μL of 100μM tracrRNA, 8 μL Nuclease-free Duplex buffer (IDT, cat # 1072570) and heated to 95°C for 5 min. Cas9 ribonucleoprotein complexes (RNPs) were assembled in a 1.5 mL microfuge tube by mixing: 79.2 μL Nuclease-free water, 10μL Reaction Buffer, 10μL annealed crRNA/tracrRNAs, 0.8 μL HiFi Cas9 Nuclease v.3 (IDT, cat # 1081060) and incubating at room temperature for 30 min. Genomic DNA was dephosphorylated by incubating 3 μL Reaction Buffer, 6 μg genomic DNA and 3 μL of Phosphatase at 37°C for 10 min in a total volume of 30 uL, followed by 80°C for 2 min. to denature. Cleavage and dATP-tailing of the dephosphorylated DNA was performed by adding 10 μL of the Cas9 RNPs, 1 μL 10 mM dATP, 1 μL of Taq Polymerase and incubating at 37°C for 15 min. in a total volume of 42 μL, followed by 72°C for 5 min. to inactivate the enzyme.
Ligation of sequencing adaptors to the genomic DNA was performed by combining 20 μL Ligation Buffer (LNB), 3 μL Nuclease-free water, 10 μL T4 Ligase and 5 μL Adapter mix (AMX) in a separate microfuge tube. The adapter mixture was then gently added in 2 successive aliquots to the sample and incubated at room temp. for 10 min. The Cas9 library was purified using AMPure XP (Beckman Coulter, cat # A63881) beads. 80 μL of SPRI dilution buffer was added to the library and bound to 48 μL of the beads at room temp. for 10 min. The beads were washed 2x with 250 μL of either Long Fragment Buffer (p13E-11 and pLAM guide RNAs) or Short Fragment Buffer (D4Z4 guide RNA). After a brief drying the beads were resuspended in either 13 μL (MinION samples) or 7 μL (Flongle samples) of Elution Buffer (EB), incubated at room temp. for 10 min. and transferred to a new microfuge tube.
Cas9 libraries were loaded onto either MinION Flow Cells (Oxford Nanopore Technologies, cat # FLO-MIN106D, version R9.4.1 or FLO-MIN114, version R10.4.1) and run on a MinION Sequencing Device (Oxford Nanopore Technologies, cat # MIN-101B) or loaded onto Flongle Flow Cells (Oxford Nanopore Technologies, cat # FLO-FLG001) and run on a Flongle adapter (Oxford Nanopore Technologies, cat # ADP-FLG001) inserted into the MinION Sequencing Device. The MinION Flow Cells were primed with 1000 μL Flush Buffer (FB) + FLT (Oxford Nanopore Technologies, cat # EXP-FLP002). 12 μL of the Cas9 libraries were combined with 37.5 μL Sequencing Buffer (SQB) + 25.5 μL Loading Beads (LB) and loaded onto the MinION Flow Cell. The Flongle Flow Cells were primed with 117 μL Flush Buffer II (FBII) + 3 μL FLT (Oxford Nanopore Technologies, cat # EXP-FSE001). 5 μL of the Cas9 libraries were combined with 15 μL Sequencing Buffer II (SBII) + 10 μL Loading Buffer II (LBII) and loaded into the Flongle Flow Cell. Data collection was controlled using the MinKNOW software on a Linux desktop computer through a USB 3.0 port as described for the MinION Mk1B IT requirements (Oxford Nanopore Technologies).
Modified basecalling and alignment
Read-level modified basecalling was performed on fast5 files producing FASTQ and bam output files using the GPU-enabled ONT guppy_basecaller v6.3.8 with the options “--trim_adapters --trim_strategy dna --do_read_splitting” and the dna_r9.4.1_450bps_modbases_5mc_cg_sup.cfg or dna_r10.4.1_e8.2_400bps_modbases_5mc_cg_sup.cfg config files, unless otherwise noted. FASTQ/A sequences were aligned to the T2T CHM13v2.0 reference genome that included a 200 kb accessory sequence for the 4qB D4Z4 region from the HG002 chromosome 4 sequence. Minimap2 (v2.22) and samtools (v1.12) 58 was used to generate and process alignments using the options “-ax map-ont” and parsing the output with the samtools -F 2304 flag to remove secondary and supplementary alignments. Alignments were processed with custom scripts that regionally extracted FASTA reads for secondary alignments to 4q, 10q and 4qB reference sequence for scoring mismatches and assigning reads to haplotypes.
CpG methylation analysis
Reference-based modified basecalling was performed using Megalodon v2.5.0 (https://github.com/nanoporetech/megalodon) with guppy v.6.4.2 using the Remora modified base model. The megalodon options included the remora settings as “--remora-modified-bases dna_r9.4.1_e8 sup 0.0.0 5mc CG 1 --mod-map-base-conv m C”, the guppy settings “--guppy-config dna_r9.4.1_450bps_modbases_5mc_cg_sup.cfg”, and the output settings as “--outputs basecalls mappings mod_mappings mods”. Single-read methylation plots were generated with modbamtools (https://www.biorxiv.org/content/10.1101/2022.07.07.499188v1) and averaged methylation levels from the reference anchored base calls were parsed from the Megalodon per_read_modified_base_calls.txt output file. Aggregate modified base counts stored in a modified-base BAM files were converted to bedMethyl files using the modbam2bed tool (https://github.com/epi2me-labs/modbam2bed). All statistical analyses of methylation levels were performed with R version 4.2.1. The linear mixed-effect model analysis used the lme4, lmerTest, and broom.mixed packages with the model equation: fitN <- lmer(met_freq ~ (RepeatN * Sample) + (1+RepeatN | read), REML = F), where met_freq was the repeat-level methylation value, RepeatN was D4Z4 repeat number (centromeric repeat numbering beginning at 0) and read as the nanopore read name. Kernel smoothing of bedMethyl files was performed with the R package smoothr using the smooth_ksmooth function and plotted with ggplot2.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Jamshid Arjomand from the FSHD Society and Charles Emerson, Lou Kunkel, and the UMass Chan Wellstone Center for FSHD team for helpful discussions in developing the study. We are grateful for the long history of engagement of FSHD families in Utah and for the work of Mark Leppert and Kevin Flanigan and other Utah FSHD investigators over the past 7 decades. This work was supported by the National Institutes of Health [Wellstone P50 HD060848, NINDS K08 NS097631] and the FSHD Society.
COMPETING INTERESTS
RJB is receiving funding via contracts for research and clinical trials from Avexis, PTC Therapeutics, Sarepta Therapeutics, Pfizer, Biogen and Ionis Pharmaceticals. He serves on scientific advisory boards for Sarepta Therapeutics, Biogen, Avexis and Pfizer. DMD, BD, SM, and RBW have no competing interest.
REFERENCES
- 1.Flanigan KM, Coffeen CM, Sexton L, Stauffer D, Brunner S, Leppert MF. Genetic characterization of a large, historically significant Utah kindred with facioscapulohumeral dystrophy. Neuromuscul Disord 2001;11:525–529. [DOI] [PubMed] [Google Scholar]
- 2.Mostacciuolo ML, Pastorello E, Vazza G, et al. Facioscapulohumeral muscular dystrophy: epidemiological and molecular study in a north-east Italian population sample. Clinical genetics 2009;75:550–555. [DOI] [PubMed] [Google Scholar]
- 3.Padberg G. Facioscapulohumeral Disease. University of Leiden. Leiden; The Netherlands: 1982. [Google Scholar]
- 4.Tyler FH, Stephens FE. Studies in disorders of muscle. II Clinical manifestations and inheritance of facioscapulohumeral dystrophy in a large family. Annals of internal medicine 1950;32:640–660. [DOI] [PubMed] [Google Scholar]
- 5.Scully MA, Eichinger KJ, Donlin-Smith CM, Tawil R, Statland JM. Restrictive lung involvement in facioscapulohumeral muscular dystrophy. Muscle Nerve 2014;50:739–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Statland JM, Tawil R. Risk of functional impairment in Facioscapulohumeral muscular dystrophy. Muscle Nerve 2014;49:520–527. [DOI] [PubMed] [Google Scholar]
- 7.Hendrickson PG, Dorais JA, Grow EJ, et al. Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat Genet 2017;49:925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lemmers RJ, van der Vliet PJ, Klooster R, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010;329:1650–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Greef JC, Lemmers RJ, van Engelen BG, et al. Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum Mutat 2009;30:1449–1459. [DOI] [PubMed] [Google Scholar]
- 10.Lemmers RJ, Wohlgemuth M, van der Gaag KJ, et al. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am J Hum Genet 2007;81:884–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rieken A, Bossler AD, Mathews KD, Moore SA. CLIA Laboratory Testing for Facioscapulohumeral Dystrophy: A Retrospective Analysis. Neurology 2021;96:e1054–e1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salort-Campana E, Nguyen K, Bernard R, et al. Low penetrance in facioscapulohumeral muscular dystrophy type 1 with large pathological D4Z4 alleles: a cross-sectional multicenter study. Orphanet J Rare Dis 2012;10:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Statland JM, Donlin-Smith CM, Tapscott SJ, Lemmers RJLF, van der Maarel SM, Tawil R. Milder phenotype in facioscapulohumeral dystrophy with 7-10 residual D4Z4 repeats. Neurology 2015;85:2147–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamanaka K, Šikrová D, Mitsuhashi S, et al. Homozygous nonsense variant in LRIF1 associated with facioscapulohumeral muscular dystrophy. Neurology 2020;94:e2441–e2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lemmers RJ, Tawil R, Petek LM, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet 2012;44:1370–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van den Boogaard ML, Lemmers R, Balog J, et al. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am J Hum Genet 2016;98:1020–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sacconi S, Briand-Suleau A, Gros M, et al. FSHD1 and FSHD2 form a disease continuum. Neurology 2019;92:e2273–e2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Winokur ST, Bengtsson U, Feddersen J, et al. The DNA rearrangement associated with facioscapulohumeral muscular dystrophy involves a heterochromatin-associated repetitive element: implications for a role of chromatin structure in the pathogenesis of the disease. Chromosome Res 1994;2:225–234. [DOI] [PubMed] [Google Scholar]
- 19.Lyle R, Wright TJ, Clark LN, Hewitt JE. The FSHD-associated repeat, D4Z4, is a member of a dispersed family of homeobox-containing repeats, subsets of which are clustered on the short arms of the acrocentric chromosomes. Genomics 1995;28:389–397. [DOI] [PubMed] [Google Scholar]
- 20.Leidenroth A, Clapp J, Mitchell LM, et al. Evolution of DUX gene macrosatellites in placental mammals. Chromosoma 2012;121:489–497. [DOI] [PubMed] [Google Scholar]
- 21.Zampatti S, Colantoni L, Strafella C, et al. Facioscapulohumeral muscular dystrophy (FSHD) molecular diagnosis: from traditional technology to the NGS era. Neurogenetics 2019;20:57–64. [DOI] [PubMed] [Google Scholar]
- 22.Dai Y, Li P, Wang Z, et al. Single-molecule optical mapping enables quantitative measurement of D4Z4 repeats in facioscapulohumeral muscular dystrophy (FSHD). J Med Genet 2020;57:109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen K, Puppo F, Roche S, et al. Molecular combing reveals complex 4q35 rearrangements in Facioscapulohumeral dystrophy. Hum Mutat 2017;38:1432–1441. [DOI] [PubMed] [Google Scholar]
- 24.Vasale J, Boyar F, Jocson M, et al. Molecular combing compared to Southern blot for measuring D4Z4 contractions in FSHD. Neuromuscul Disord 2015;25:945–951. [DOI] [PubMed] [Google Scholar]
- 25.Gould T, Jones TI, Jones PL. Precise Epigenetic Analysis Using Targeted Bisulfite Genomic Sequencing Distinguishes FSHD1, FSHD2, and Healthy Subjects. Diagnostics (Basel) 2021;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roche S, Dion C, Broucqsault N, et al. Methylation hotspots evidenced by deep sequencing in patients with facioscapulohumeral dystrophy and mosaicism. Neurol Genet 2019;5:e372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lemmers R, van der Vliet PJ, Granado DSL, et al. High-resolution breakpoint junction mapping of proximally extended D4Z4 deletions in FSHD1 reveals evidence for a founder effect. Hum Mol Genet 2022;31:748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nurk S, Koren S, Rhie A, et al. The complete sequence of a human genome. Science 2022;376:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deamer D, Akeson M, Branton D. Three decades of nanopore sequencing. Nat Biotechnol 2016;34:518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mitsuhashi S, Nakagawa S, Takahashi Ueda M, Imanishi T, Frith MC, Mitsuhashi H. Nanopore-based single molecule sequencing of the D4Z4 array responsible for facioscapulohumeral muscular dystrophy. Sci Rep 2017;7:14789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilpatrick T, Lee I, Graham JE, et al. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat Biotechnol 2020;38:433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hiramuki Y, Kure Y, Saito Y, et al. Simultaneous measurement of the size and methylation of chromosome 4qA-D4Z4 repeats in facioscapulohumeral muscular dystrophy by long-read sequencing. J Transl Med 2022;20:517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoyt SJ, Storer JM, Hartley GA, et al. From telomere to telomere: The transcriptional and epigenetic state of human repeat elements. Science 2022;376:eabk3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartweck LM, Anderson LJ, Lemmers RJ, et al. A focal domain of extreme demethylation within D4Z4 in FSHD2. Neurology 2013;80:392–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gershman A, Sauria MEG, Guitart X, et al. Epigenetic patterns in a complete human genome. Science 2022;376:eabj5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kelly TK, Liu Y, Lay FD, Liang G, Berman BP, Jones PA. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res 2012;22:2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mason AG, Slieker RC, Balog J, et al. SMCHD1 regulates a limited set of gene clusters on autosomal chromosomes. Skelet Muscle 2017;7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lemmers RJ, Goeman JJ, van der Vliet PJ, et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum Mol Genet 2015;24:659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones TI, Yan C, Sapp PC, et al. Identifying diagnostic DNA methylation profiles for facioscapulohumeral muscular dystrophy in blood and saliva using bisulfite sequencing. Clin Epigenetics 2014;6:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaillard MC, Roche S, Dion C, et al. Differential DNA methylation of the D4Z4 repeat in patients with FSHD and asymptomatic carriers. Neurology 2014;83:733–742. [DOI] [PubMed] [Google Scholar]
- 41.Huichalaf C, Micheloni S, Ferri G, Caccia R, Gabellini D. DNA methylation analysis of the macrosatellite repeat associated with FSHD muscular dystrophy at single nucleotide level. PloS one 2014;9:e115278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ottaviani A, Rival-Gervier S, Boussouar A, et al. The D4Z4 macrosatellite repeat acts as a CTCF and A-type lamins-dependent insulator in facio-scapulo-humeral dystrophy. PLoS Genet 2009;5:e1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gabellini D, Green MR, Tupler R. Inappropriate gene activation in FSHD: a repressor complex binds a chromosomal repeat deleted in dystrophic muscle. Cell 2002;110:339–348. [DOI] [PubMed] [Google Scholar]
- 44.Campbell AE, Shadle SC, Jagannathan S, et al. NuRD and CAF-1-mediated silencing of the D4Z4 array is modulated by DUX4-induced MBD3L proteins. Elife 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leighton GO, Irvin EM, Kaur P, et al. Densely methylated DNA traps Methyl-CpG-binding domain protein 2 but permits free diffusion by Methyl-CpG-binding domain protein 3. J Biol Chem 2022;298:102428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lemmers RJ, van der Vliet PJ, van der Gaag KJ, et al. Worldwide population analysis of the 4q and 10q subtelomeres identifies only four discrete interchromosomal sequence transfers in human evolution. Am J Hum Genet 2010;86:364–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Darrow EM, Chadwick BP. A novel tRNA variable number tandem repeat at human chromosome 1q23.3 is implicated as a boundary element based on conservation of a CTCF motif in mouse. Nucleic Acids Res 2014;42:6421–6435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qiu L, Ye Z, Lin L, et al. Clinical and genetic features of somatic mosaicism in facioscapulohumeral dystrophy. J Med Genet 2020;57:777–785. [DOI] [PubMed] [Google Scholar]
- 49.Lemmers R, van der Vliet PJ, Vreijling JP, et al. Cis D4Z4 repeat duplications associated with facioscapulohumeral muscular dystrophy type 2. Hum Mol Genet 2018;27:3488–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hummel G, Warren J, Drouard L. The multi-faceted regulation of nuclear tRNA gene transcription. IUBMB Life 2019;71:1099–1108. [DOI] [PubMed] [Google Scholar]
- 51.Hori Y, Engel C, Kobayashi T. Regulation of ribosomal RNA gene copy number, transcription and nucleolus organization in eukaryotes. Nat Rev Mol Cell Biol 2023. [DOI] [PubMed] [Google Scholar]
- 52.Chen JC, King OD, Zhang Y, et al. Morpholino-mediated Knockdown of DUX4 Toward Facioscapulohumeral Muscular Dystrophy Therapeutics. Molecular therapy : the journal of the American Society of Gene Therapy 2016;24:1405–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marsollier AC, Ciszewski L, Mariot V, et al. Antisense targeting of 3’ end elements involved in DUX4 mRNA processing is an efficient therapeutic strategy for facioscapulohumeral dystrophy: a new gene-silencing approach. Hum Mol Genet 2016;25:1468–1478. [DOI] [PubMed] [Google Scholar]
- 54.Bouwman LF, den Hamer B, van den Heuvel A, et al. Systemic delivery of a DUX4-targeting antisense oligonucleotide to treat facioscapulohumeral muscular dystrophy. Mol Ther Nucleic Acids 2021;26:813–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rashnonejad A, Amini-Chermahini G, Taylor NK, Wein N, Harper SQ. Designed U7 snRNAs inhibit DUX4 expression and improve FSHD-associated outcomes in DUX4 overexpressing cells and FSHD patient myotubes. Mol Ther Nucleic Acids 2021;23:476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cohen J, DeSimone A, Lek M, Lek A. Therapeutic Approaches in Facioscapulohumeral Muscular Dystrophy. Trends Mol Med 2021;27:123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Himeda CL, Jones TI, Jones PL. Targeted epigenetic repression by CRISPR/dSaCas9 suppresses pathogenic DUX4-fl expression in FSHD. Mol Ther Methods Clin Dev 2021;20:298–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 2021;37:4572–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.