

Abstract
Selinexor is a first-in-class inhibitor of the nuclear exportin XPO1 that was recently FDA-approved for the treatment of multiple myeloma and diffuse large B-cell lymphoma. In relapsed/refractory acute myeloid leukemia (AML), selinexor has shown promising activity, suggesting that selinexor-based combination therapies may have clinical potential. Here, motivated by the hypothesis that selinexor’s nuclear sequestration of diverse substrates imposes pleiotropic fitness effects on AML cells, we systematically catalogue the pro- and anti-fitness consequences of selinexor treatment. We discover that selinexor activates PI3Kγ-dependent AKT signaling in AML by upregulating the purinergic receptor P2RY2. Inhibiting this axis potentiates the anti-leukemic effects of selinexor in AML cell lines, patient-derived primary cultures, and multiple mouse models of AML. In a syngeneic, MLL-AF9-driven mouse model of AML, treatment with selinexor and ipatasertib outperforms both standard-of-care chemotherapy and chemotherapy with selinexor. Together, these findings establish drug-induced P2RY2-AKT signaling as an actionable consequence of XPO1 inhibition in AML.
Introduction
Selinexor is an inhibitor of nuclear export that recently received FDA approval for the treatment of multiple myeloma (1, 2) and diffuse large B-cell lymphoma (3), and is in early clinical trials for advanced solid and hematologic malignancies (4, 5). There is interest in selinexor’s potential for treating acute myeloid leukemia (AML), supported by preclinical evidence showing that inhibition of nuclear export promotes cell cycle arrest and apoptosis in AML cells (6–8). These therapeutic effects are evidenced in bulk leukemic populations and in leukemia-initiating cells (LICs) (9, 10), and can be enhanced by combining selinexor with existing chemotherapies (11–15). In patients with relapsed or refractory AML, selinexor was tolerable and active, producing complete responses in a substantial fraction of patients as monotherapy or in combination with chemotherapy (11, 15–20). Therefore, there is an increasing need to understand the largely undefined determinants of response to XPO1 inhibition in hematologic malignancies.
Mechanistically, selinexor blocks nuclear-cytoplasmic export by directly inhibiting the nuclear export protein XPO1, which facilitates RanGTP-dependent transport of nuclear export sequence (NES)-bearing cargos from the nucleus to the cytoplasm (21). Inhibition of XPO1 results in nuclear accumulation of its substrates, among them: tumor suppressor proteins, cell cycle regulators, and DNA damage response proteins such as Rb, p53, p21, p27, FOXO3, BRCA1, CHK1, and RAD51 (21). In cancer, these proteins, which require nuclear localization to function, are dislocated to the cytoplasm due to the activation of oncogenic signaling (22–25) or frank upregulation of XPO1 (26, 27); returning them to the nucleus restores their activity, providing an anti-cancer effect. In AML, mutations in NPM1 promote its pro-leukemic, cytoplasmic localization (NPM1c) (28, 29). XPO1 inhibition enforces nuclear relocalization of NPM1c, promoting leukemic differentiation and growth arrest (30). However, XPO1 is not specific for protein clients with tumor suppressive activity. Unbiased proteomic studies have identified hundreds of XPO1 substrates in human cells (31, 32). This suggests that XPO1 inhibition likely engages numerous cellular programs simultaneously, creating the possibility that selinexor treatment may paradoxically activate pro-oncogenic processes.
Here, we use unbiased functional genomics and proteomics to identify activation of a P2RY2-PI3Kγ-AKT signaling pathway as a deterministic, pro-fitness consequence of selinexor treatment in AML cells. We find that inhibition of this pathway potentiates the anti-leukemic effect of selinexor in vitro and in vivo. We demonstrate that XPO1 inhibition activates pro-fitness P2RY2-AKT signaling in AML cells and that combination therapies that anticipate this pro-fitness activation may unlock selinexor’s clinical potential.
Results
Parallel profiling identifies selinexor-induced AKT activation.
To identify direct, cell-beneficial sequelae of selinexor treatment that could be therapeutically targeted, we searched for pathways that satisfied two requirements: (1) treatment of AML cells with selinexor modulated the pathway, and (2) genetic or pharmacological modulation of the pathway sensitized AML cells to selinexor treatment. We undertook phenotypic screens designed to address these requirements (Fig. 1a).
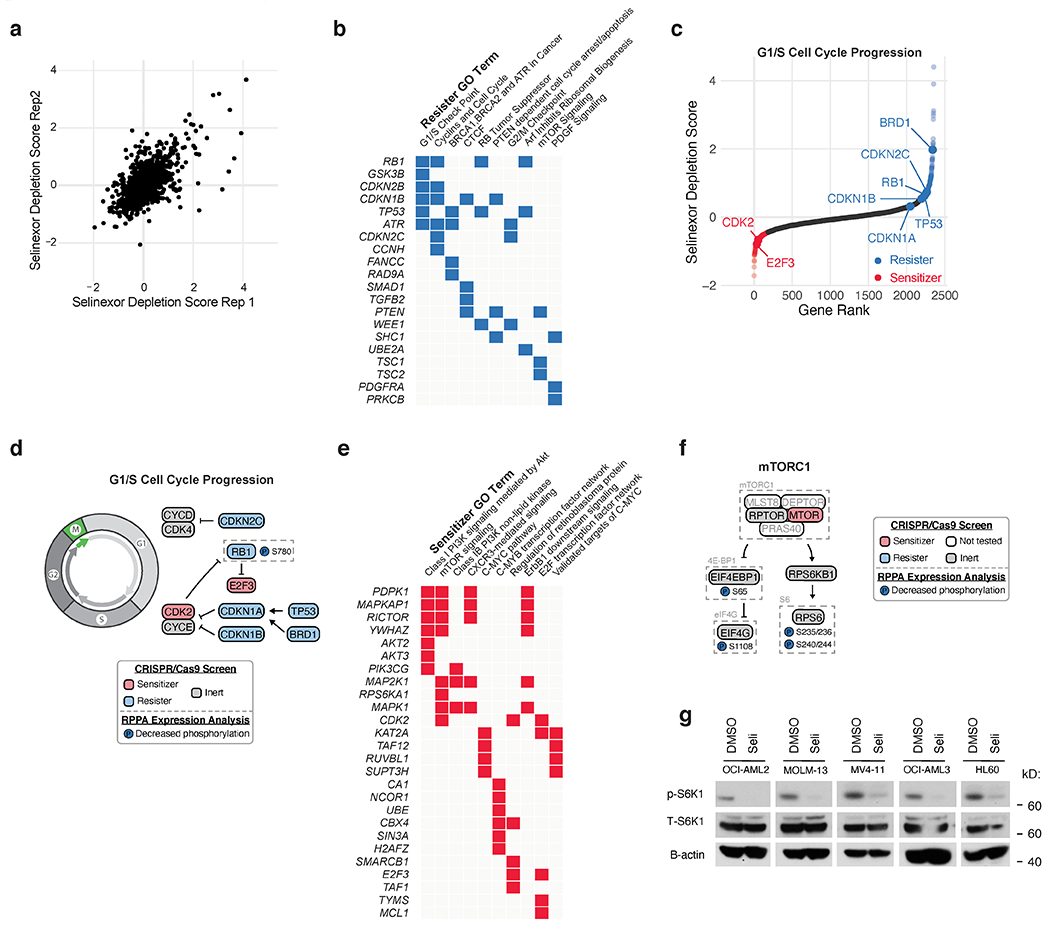
Figure 1. Orthogonal Functional Genomic and Proteomic Analyses Nominate AKT Activation as an Actionable Consequence of Selinexor Treatment.
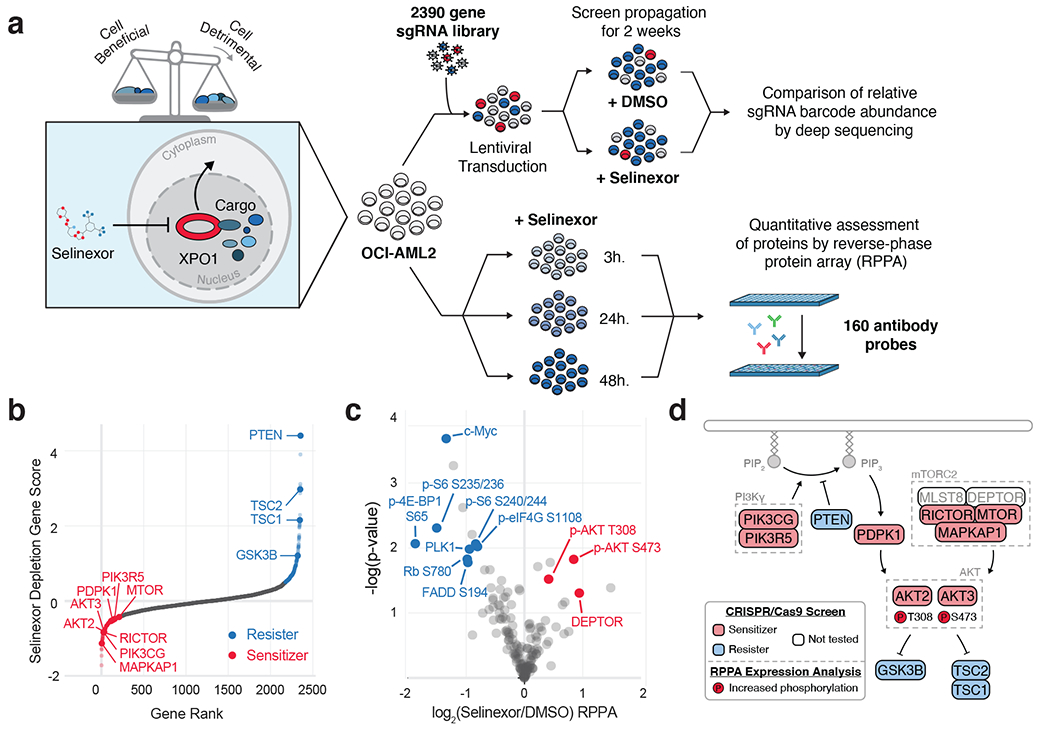
a) Experimental strategy for parallel assessment of cell-beneficial and cell-detrimental effects of nuclear export inhibition with the XPO1 inhibitor, selinexor. Pooled CRISPR-Cas9 screening in OCI-AML2 cells treated with selinexor reveals genetic modifiers of drug sensitivity. Reverse phase protein array (RPPA) analysis in selinexor treated OCI-AML2 cells reveals drug-responsive protein and phosho-protein expression.
b) Selinexor depletion gene scores ranked from most depleted to most enriched in the selinexor versus vehicle treated populations. Scoring genes in the PI3K/AKT pathway are annotated as sensitizers (orange, depleted in the selinexor population) or resisters (blue, enriched in the selinexor population). Screens conducted as n = 2 independent replicates with n = 5 sgRNAs per gene.
c) Volcano plot depicting differential expression for 160 RPPA probes following 48 hours of selinexor treatment relative to statistical significance in dataset. Annotated probes comprise pathways with decreased expression (blue) or increased expression (orange). RPPA expression analysis conducted as n = 3 independent experiments. P-values computed by multiple unpaired (two-sided) t-tests.
d) Schematic relating PI3K/AKT pathway members to selinexor depletion gene scores and RPPA expression. Genes scoring as selinexor sensitizers are shaded in orange; genes scoring as selinexor resisters are shaded in blue; genes absent from library not shaded. Phosphorylated proteins with selinexor-induced increased (orange) RPPA expression are indicated.
First, we used a CRISPR/Cas9-based loss-of-function screen to identify genetic sensitizers to selinexor. In this assay, OCI-AML2 cells were transduced with a CRISPR/Cas9-knockout library and cultured with or without selinexor for two weeks. Samples from zero- and two-week time points were deconvoluted using deep sequencing to identify potential genetic sensitizers, genes whose ablation reduced their representation within the population of selinexor-treated cells. We used a CRISPR/Cas9 library focused on oncogenic, proliferative, and survival pathways totaling 11,950 sgRNAs targeting 2390 genes plus 50 non-targeting sgRNAs (Extended Data Fig. 1a, Supplementary Table 1). Data from this effort was included in a prior study (33), but a full dissection of selinexor’s sensitizer interactions has been reserved for this study.
The screen identified many selinexor sensitivity modifiers. Among resisters, genes whose loss conferred selinexor resistance and whose representation was enriched with drug, gene ontology (GO) pathway analysis identified cell cycle modulators (Extended Data Fig. 1b), tumor suppressors, and known XPO1 substrates p21 (CDKN1A), p27 (CDKN1B), RB (RB1), and p53 (TP53) (Extended Data Fig. 1c,d). These findings cohered with selinexor’s known ability to induce G1 arrest and apoptosis, given the roles that p21, p27, RB, and p53 play in restricting G1/S progression (34) and the role of p53 in apoptosis (35). p53 loss is a negative predictor of response to XPO1 inhibition in AML (7). BRD1, which indirectly modulates the cell cycle through CDKN1A and CDK1 (36), scored as a resister (Extended Data Fig. 1c,d). The screen also identified sensitizers, genes whose loss potentiated the effects of selinexor, depleting their representation in the presence of drug (Extended Data Fig. 1e). Multiple sensitizers suggested that interference with cell cycle progression (CDK2, E2F3) and c-MYC targets (KAT2A, TAF12, RUVBL1, SUPT3H) could sensitize AML cells to selinexor (Extended Data Fig. 1c,d,e). Cell cycle genes that scored as sensitizers were directionally consistent with those that scored as resisters: RB receives inhibitory phosphorylation from CDK2 and represses E2F3 (Extended Data Fig. 1d) (37).
The strongest phenotype identified in the loss-of-function CRISPR screen belonged to PTEN, which was enriched 16-fold in the selinexor-treated versus control-treated populations, identifying it as a resister (Fig. 1b). PTEN catalyzes dephosphorylation of PIP3 to PIP2, and plays a tumor-suppressive role within the PI3K/AKT pathway (38). Many nodes in this pathway scored; accordingly, PI3K/AKT signaling and PTEN signaling were identified by GO pathway analysis as enriched signatures (Extended Data Fig. 1b,e). PIK3CG and PIK3R5 respectively encode p110γ and p101, complementary catalytic and regulatory subunits of PI3-kinase (39), and were identified as sensitizers (Fig. 1b). PDPK1, which encodes PDK1, scored as a sensitizer, as did AKT2 and AKT3, encoding isoforms of the PDK1 substrate AKT. AKT also receives activating phosphorylation from mTORC2, a multi-subunit complex whose components, encoded by RICTOR, MTOR, and MAPKAP1, scored as sensitizers (Fig. 1b). AKT provides inhibitory phosphorylation to GSK3b, TSC1, and TSC2 (38), which all scored as resisters (Fig. 1b). These data implicated the PI3K/AKT pathway as a modifier of selinexor sensitivity.
Next, to identify pathways activated by selinexor, we used a reverse phase protein array (RPPA) to analyze AML cells treated with DMSO or selinexor for 48 hours. Our RPPA platform, which assesses 160 epitopes and phospho-epitopes representing cellular growth, proliferation, and signaling pathways, identified (Supplementary Table 2) several drug-induced effects supporting described mechanisms of selinexor. Selinexor treatment suppressed proteins or phosphorylation marks implicated in G1/S progression such as PLK1, phospho-Rb at Ser780, and phospho-FADD at Ser 194 (6, 40) (Fig. 1c). Selinexor-induced downregulations of phospho-S6 at Ser235/236 and Ser240/244, phospho-eIF4G at Ser1108, and phospho-4E-BP1 at Ser65 (Fig. 1c, Extended Data Fig. 1f) are consistent with work showing that disruption of nuclear-cytoplasmic shuttling diminishes S6K activation and 4E-BP1 phosphorylation (41). Similar observations have been made in multiple myeloma (42), and are supported here by reciprocal upregulation of DEPTOR, a negative regulator of mTOR . Across five AML cell lines, we confirmed that treatment with selinexor reduced phospho-S6K1 (Extended Data Fig. 1g), suggesting that selinexor treatment suppresses mTORC1-dependent signaling. Notably, two of the strongest upregulations identified by RPPA were in AKT (both Thr308 and Ser473) (Fig. 1c), aligning the results from the RPPA and the selinexor sensitizer screen. Together, they suggest that selinexor treatment activates PI3K/AKT, and that targeting this pathway could sensitize AML cells to selinexor (Fig. 1d).
Selinexor treatment activates PI3K/AKT signaling.
To validate selinexor’s ability to activate PI3K/AKT signaling, we treated five AML cell lines with selinexor for 24 hours. Across all five lines, western blots of selinexor-treated cells revealed increased AKT phosphorylation at Thr308 and Ser473, and phosphorylation of AKT substrates GSK3b Ser9 and BAD Ser136, (Fig. 2a). Additional AKT-dependent phospho-sites on PRAS40, FOXO3a, and TSC2 were assessed in OCI-AML2 cells and were increased (Extended Data Fig. 2a). Eltanexor, a second-generation inhibitor of XPO1 also prompted phosphorylation of AKT (Extended Data Fig. 2b). To characterize the kinetics of selinexor-induced AKT activation, we sampled selinexor-treated AML cells through 48 hours and observed activation of AKT signaling starting by 24 hours (Fig. 2b).
Figure 2. Selinexor Activates PI3K/AKT Signaling in AML cells.
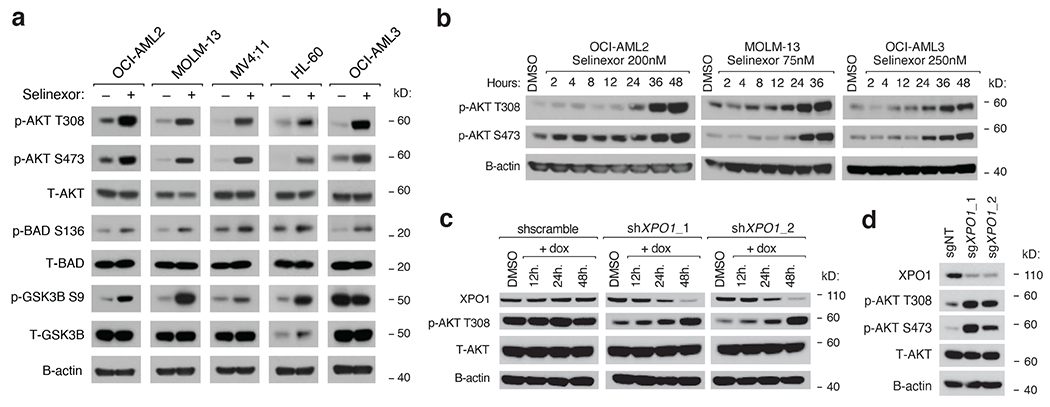
a) Immunoblot depicting protein levels of phosphorylated and total PI3K/AKT pathway members following treatment of a panel of AML cell lines with selinexor. Selinexor was dosed at the following concentrations for each cell line: OCI-AML2 (200nM), MOLM-13 (75nM), MV;411 (50nM), HL-60 (300nM), OCI-AML3 (250nM).
b) Immunoblot depicting protein levels of phosphorylated AKT at T308 and S473 in OCI-AML2, OCI-AML3, and MOLM-13 cells treated with Selinexor for indicated duration.
c) Immunoblot depicting protein levels of XPO1 and total and phosphorylated AKT at T308 in OCI-AML2 cells with doxycycline (dox) inducible shRNAs targeting XPO1 versus scrambled shRNA control. Cells were exposed to dox (75ng/mL) for indicated durations prior to collection.
d) Immunoblot depicting protein levels of XPO1, total and phosphorylated AKT at T308 and S473 in OCI-AML2 cells with sgRNAs targeting XPO1 versus non-targeting controls.
Representative immunoblots of n=3-5 biologically independent experiments yielding similar results. B-actin included as loading control.
Next, we used two doxycycline-inducible shRNAs targeting XPO1 to induce progressive knockdown of XPO1 over 48 hours and reciprocal induction of AKT phosphorylation at Thr308 (Fig. 2c). We also used two sgRNAs to target XPO1 via CRISPR/Cas9, which increased phosphorylation at AKT Thr308 and Ser473 (Fig. 2d). These experiments genetically phenocopied the effects of selinexor treatment on AKT signaling, indicating that AKT activation is an on-target effect of XPO1 inhibition by selinexor. To assess the exclusivity of these findings for selinexor, we treated cells with eight different anti-leukemic drugs for 24 hours. None activated AKT, suggesting that drug-induced activation of AKT is uncommon (Extended Data Fig. 2c).
FACS-based screens identify modifiers of AKT activation.
To define the mechanism of selinexor-induced AKT activation, we sought to identify genes that were both necessary for selinexor-induced AKT activation and upregulated in response to selinexor. Finding genes that satisfied these conditions required two separate experiments: a functional screen to identify the genetic determinants of selinexor-induced AKT activation, and a transcriptomic analysis of selinexor-treated versus vehicle-treated cells.
To identify genes required for selinexor-induced AKT activation, we performed a full-genome, loss-of-function CRISPR screen (43) in selinexor-treated cells, using fluorescence-activated cell sorting (FACS) to quantify drug-induced shifts in phosphorylated AKT (Fig. 3a). We selected phospho-Thr308 as the measured phosphoepitope because its phosphorylation by PDK1 represents upstream phosphoinositide mobilization (mTORC2-depent Ser473 phosphorylation stabilizes Thr308 phosphorylation). In library-non-expressing OCI-AML2 cells, selinexor treatment produced a rightward, positive shift in the population distribution of phospho-Thr308 (Fig. 3b, Extended Data Fig. 3a,b). Interestingly, in library-expressing cells, selinexor treatment produced a bimodal distribution, with a larger, right-shifted peak and a smaller, left-shifted peak (Fig. 3c). From the full distribution, two subpopulations were collected: the top fraction, comprised of cells from the top 10%, and the bottom fraction, cells whose AKT activation was not induced by selinexor (roughly 18%). The compositional abundance of sgRNAs in the bottom and top fractions was deconvoluted through deep sequencing.
Figure 3. FACS-Based CRISPR/Cas9 Screening Identifies Genetic Determinants of Selinexor-Induced AKT Activation.
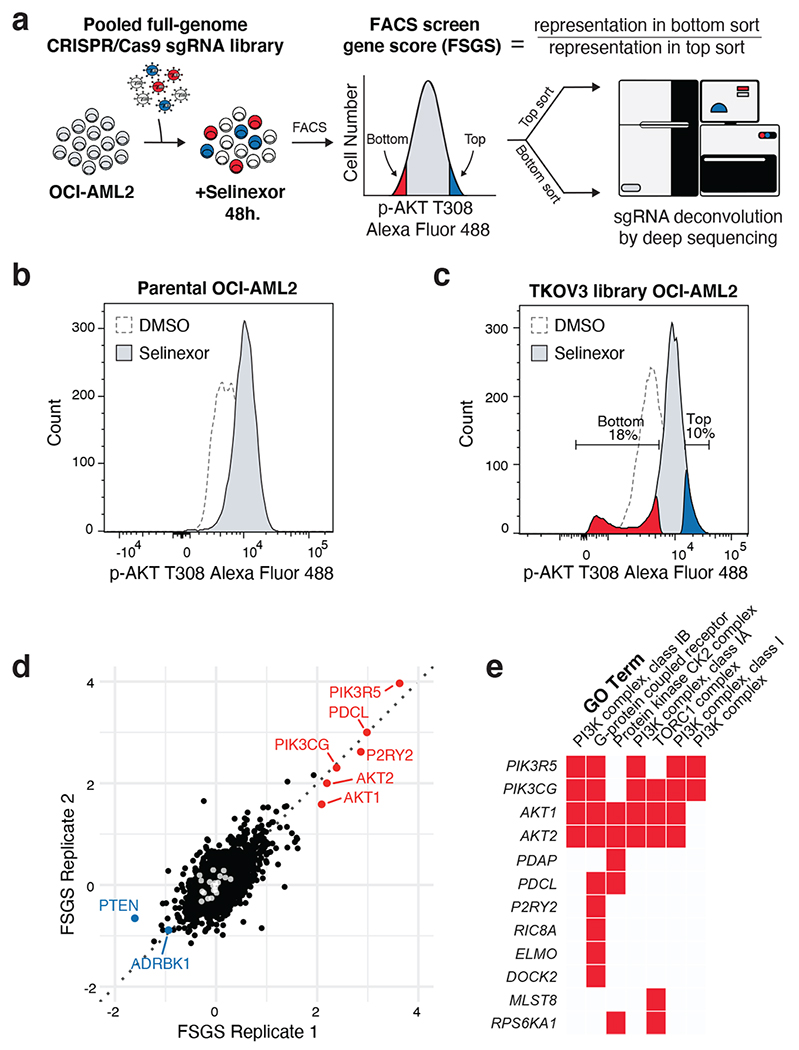
a) FACS-based CRISPR/Cas9 screening strategy to identify genetic modifiers of AKT phosphorylation in selinexor-treated AML cells. sgRNA library transduced OCI-AML2 cells were treated with selinexor for 48 hours, fixed/permeabilized and stained with phosphorylated AKT T308 primary antibody followed by Alexa Flour 488 conjugated secondary antibody. Stained cells were then sorted according to phosphorylated AKT T308 expression into high-expressing cells (top sort) and low-expressing cells (bottom sort). Genomic DNA was extracted and sgRNA barcodes were amplified and indexed prior to deep sequencing. The FACS screen gene score (FSGS) enumerates genes whose sgRNA representatives were enriched in the top or bottom sorted populations.
b) Histogram depicting distribution of phosphorylated AKT T308 expression in parental OCI-AML2 cells treated with vehicle or selinexor for 48 hours.
c) Histogram depicting distribution of phosphorylated AKT T308 expression in sgRNA library transduced OCI-AML2 cells treated with vehicle or selinexor for 48 hours. Gates defining top (blue) and bottom (orange) sorted population in selinexor treated cells are indicated.
d) Scatterplot depicting replicate FSGS values. Scoring genes enriched in the bottom sort (orange) or the top sort (blue) are annotated. LacZ, EGFP and luciferase targeting controls indicated in white. Screens conducted as n = 2 independent replicates with n = 4 sgRNAs per gene.
e) Gene ontology (GO) analysis of scoring genes enriched in the bottom sort with a p-value < 5e-4. GO performed using Enrichr.
To stratify genes in the FACS screen, we calculated the gene-wise ratio of representation in the bottom fraction over the top fraction, the ‘FACS screen gene score’, or FSGS. Genes that positively affect selinexor-induced AKT activation should be increased in the numerator and suppressed in the denominator of the quotient. Using this approach, we identified candidate positive and negative modifiers of AKT phosphorylation (Supplementary Table 3). Among them, AKT1 and AKT2 scored as positive modifiers of AKT activation and PTEN scored as a negative modifier, suggesting that the FACS screen can accurately identify determinants of selinexor-induced AKT activation (Fig. 3d). The screen also identified PIK3CG and PIK3R5 as positive modifiers of AKT activation (Fig. 3d). These themes were corroborated by GO pathway analysis, which pulled out PI3K/AKT-related pathways and highlighted G protein-coupled receptor (GPCR) signaling (Fig. 3e, Extended Data Fig. 3c).
P2RY2 is upregulated by selinexor treatment.
Next, we performed RNA-seq on OCI-AML2 and MOLM13 cells treated with selinexor or vehicle for 36 hours. GSEA analysis highlighted cell cycle-related processes (Extended Data Fig. 4a,b), consistent with XPO1’s localization at kinetochore assemblies and regulation of mitotic progression (44). There was concordance among differentially-expressed genes between the two cell lines (Extended Data Fig. 4c). Among all genes, there were 185 whose expression increased at least two-fold with selinexor treatment (Supplementary Table 4), including many XPO1 response genes (10, 45). One such gene, P2RY2, was also among the highest-scoring in the FACS screen, suggesting that its selinexor-induced upregulation could contribute to selinexor-induced AKT activation (Fig. 4a). We validated the selinexor-induced transcriptional upregulation of P2RY2 in five AML cell lines by qRT-PCR (Fig. 4b). If transcriptional induction of P2RY2 was a determinant of selinexor-induced AKT activation, then overexpression of P2RY2 in AML should predict enrichment for our selinexor gene signature. To test this, we reanalyzed two gene expression datasets of de novo AML (46, 47), dividing samples into high and low P2RY2 expressors. In both datasets, AML samples expressing high P2RY2 were enriched for our selinexor-induced signature than samples expressing low P2RY2 (Extended Data Fig. 4d). These results independently associate P2RY2 expression with our selinexor-induced signature, corroborating a potential relationship between P2RY2 expression, AKT activation, and selinexor treatment.
Figure 4. PI3Kγ Promotes AKT Activation Upon Selinexor-Induced P2RY2 Upregulation.
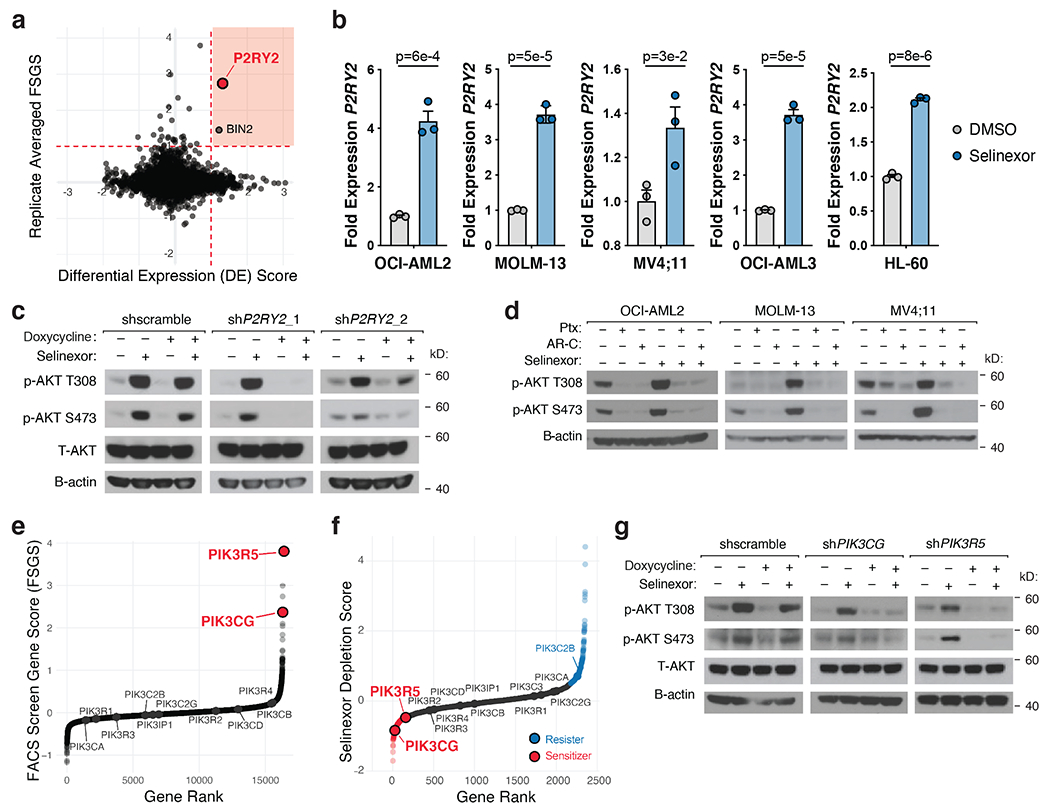
a) Scatterplot depicting replicate-averaged FSGS compared to differential gene expression (DE) score with selinexor treatment. RNA-seq analysis of selinexor versus vehicle treated OCI-AML2 and MOLM-13 cells yielded DE for each cell line. Plotted DE score is the average DE across the two cell lines. Orange shaded region denotes genes that were both transcriptionally upregulated by selinexor treatment and yielded a high FSGS score in the FACS-based CRISPR/Cas9 screen. RNA-seq conducted as n = 3 biologically independent replicates.
b) Relative expression of P2RY2 across a panel of selinexor-treated AML cell lines compared to DMSO control. P-values computed using multiple unpaired (two-sided) t-tests. Data are presented as mean +/−s.e.m. for n = 3 biologically independent replicates.
c) Immunoblot depicting protein levels of total and phosphorylated AKT at T308 and S473 in OCI-AML2 cells with doxycycline-inducible shRNAs targeting P2RY2 versus scrambled shRNA control. Cells were exposed to doxycycline (75ng/mL) for 48 hours and treated with either vehicle or selinexor for 36 hours.
d) Immunoblot depicting protein levels of phosphorylated AKT at T308 and S473 in OCI-AML2, MOLM-13, and MV4;11 cells following treatment of pertussis toxin (Ptx 100ng/mL), AR-C 118925XX (AR-C 2.5μM) or selinexor alone and in combination for 36 hours.
e) Replicate-averaged FSGS ranked from most depleted to most enriched in bottom sort. Isoforms of PI3K are annotated.
f) Selinexor depletion gene scores ranked from most depleted to most enriched in the selinexor versus vehicle treated populations. Isoforms of PI3K are annotated.
g) Immunoblot depicting protein levels of total and phosphorylated AKT at T308 and S473 in OCI-AML2 cells with doxycycline-inducible shRNAs against PIK3CG and PIK3R5 versus scrambled shRNA control. Cells were exposed to doxycycline (75ng/mL) for 48 hours and treated with either vehicle or 200nM selinexor for 36 hours.
Figure 4c,d,g Representative immunoblots of n=2-3 biologically independent experiments yielding similar results. B-actin included as loading control.
P2RY2 encodes a purinergic GPCR that acts as an extracellular ATP/UTP sensor (48). The notion that P2RY2 might be responsible for selinexor-induced AKT signaling was intriguing for a few reasons. First, our FACS screen highlighted several GPCR-related genes. The aforementioned PIK3CG and PIK3R5 encode catalytic and regulatory subunits of PI3Kγ. In particular, PIK3R5 encodes p101, a regulatory adaptor of PI3Kγ that facilitates signaling inputs from GPCRs via Gβγ. PDCL, which encodes a Gβγ modulator, phosducin-like protein, scored as the second-highest positive modifier of AKT activation (Fig. 3d). ADRBK1, which scored as a negative modifier of AKT activation, encodes GRK2, which phosphorylates phosducin-like protein, inhibiting its capacity to bind Gβγ (Fig. 3d) (49). Additionally, P2RY2 itself can activate AKT signaling in cancer (48, 50); it is overexpressed in AML (51); it is upregulated in AML cells co-cultured with bone marrow adipocytes (52).
Before assessing the role of P2RY2 in selinexor-induced AKT activation, we tested whether ATP or UTP could activate AKT in AML cell lines. Cells cultured with exogenous ATP activated AKT signaling (Extended Data Fig. 4e) while cells cultured with exogenous UTP did not (Extended Data Fig. 4f), implying that selinexor may activate AKT through induced release of extracellular ATP. We tested this by treating cells with selinexor versus vehicle in the presence of ectonucleotidase inhibitor ARL67156 and quantifying the extracellular ATP released after 36 hours. After normalizing for cell quantity, there was no increase in extracellular ATP in cells treated with selinexor, suggesting that AKT activation is predominantly driven by upregulation of P2RY2, not an increased abundance of ligand (Extended Data Fig. 4g).
P2RY2 is required for selinexor-induced activation of AKT.
To verify the role of P2RY2 in selinexor-induced AKT activation, we used two doxycycline-inducible shRNAs to knock down P2RY2. P2RY2 knockdown blunted selinexor-induced AKT activation (Fig. 4c, Extended Data Fig. 4h). CRISPR/Cas9-mediated knockout of P2RY2 with three sgRNAs similarly abrogated AKT signaling (Extended Data Fig. 4i,j). We phenocopied this effect using AR-C118925XX (AR-C), a P2RY2 antagonist (53), and pertussis toxin (PTX), which uncouples GPCR signaling through ADP-ribosylation of Gi and Go (Fig. 4d). Conversely, ectopic P2RY2 overexpression promoted AKT activation (Extended Data Fig. 4k,l). These data demonstrated that increased P2RY2 was sufficient to activate AKT, and that selinexor’s activation of AKT was P2RY2-dependent.
Because PIK3CG and PIK3R5 were the only PI3-kinase subunits that scored in both CRISPR screens, we reasoned that PI3Kγ may be responsible for P2RY2-driven activation of AKT (Fig. 4e, 4f). Using inducible shRNA constructs, we showed that, like knockdown of P2RY2, knockdown of PIK3CG or PIK3R5 blunted AKT activation (Fig. 4g, Extended Data Fig. 5a,b,c). This effect was phenocopied using the PI3Kγ-specific inhibitor IPI-549 but not inhibitors of other catalytic PI3-kinase isoforms (Extended Data Fig. 5d). None of the catalytic PI3-kinase isoforms were upregulated with selinexor treatment (Extended Data Fig. 5e). However, because purinergic signaling involves more than PI3-kinase, we reanalyzed data from our CRISPR screens and RNAseq experiments to look for canonical, downstream purinergic effectors. These searches did not implicate any leads; western blot analysis of phospho-substrates of downstream kinases PKA/PKC were not increased in selinexor-treated cells (Extended Data Fig. 5f).
Selinexor-induced activation of AKT requires RAS activation.
In order for PI3Kγ to be fully responsive to Gβγ, p110γ must associate with both p101 and activated RAS (GTP-RAS) (54, 55). We wondered whether selinexor-induced P2RY2 signaling activated RAS as an accessory to full PI3Kγ activation. Several lines of experimental evidence support this model. First, RAF1-RAS-binding-domain (RAF1-RBD) pulldowns revealed increased GTP-RAS in selinexor- versus vehicle-treated samples, evidence that selinexor-treatment activated RAS (Extended Data Fig. 6a). AR-C reversed the increased GTP-RAS observed upon RAF1-RBD pulldown following selinexor treatment (Extended Data Fig. 6b), indicating that P2RY2 was required for selinexor-induced RAS activation. Likewise, doxycycline-inducible shRNAs targeting P2RY2 phenocopied the effect of P2RY2 inhibition on selinexor-mediated GTP-RAS loading (Extended Data Fig. 6c). These data suggest that selinexor treatment activates RAS in a P2RY2-dependent manner.
To test the necessity of RAS for selinexor-induced AKT activation, we used doxycycline-inducible shRNAs to knockdown KRAS and NRAS. Cells with simultaneous KRAS and NRAS knockdown retained their viability but could not mount an AKT-activating response to selinexor, suggesting that RAS is required for selinexor-induced activation of AKT (Extended Data Fig. 6d). Singular knockdowns of KRAS and NRAS completely suppressed selinexor-induced AKT activation (Extended Data Fig. 6e). This may explain why RAS isoforms did not score in either CRISPR screen, although we cannot rule out isoform-specific, compensatory upregulation (56, 57). Next, we posited that selinexor treatment might also activate mitogen-activated protein kinase signaling. Indeed, RPPA analysis of selinexor- versus vehicle-treated cells confirmed an increase in phospho-ERK at Thr202/Tyr204 (Supplementary Table 2). Unlike non-small cell lung cancer, where XPO1 is a mutant KRAS-associated dependency (58), our findings suggest that in AML, selinexor-provoked RAS activation promotes resistance to selinexor through AKT activation. This is supported by experiments demonstrating that ectopic expression of oncogenic RAS and AKT mutants confer resistance to selinexor (Extended Data Fig. 6f), and by publicly-available genetic dependency data, which reveal no relationship between RAS status and sensitivity to XPO1 knockout (Extended Data Fig. 6g). Together, these results support a model where selinexor treatment promotes P2RY2 upregulation, coordinately activating PI3Kγ and RAS, and empowering PI3Kγ to activate AKT.
AKT inhibition potentiates selinexor-induced apoptosis in AML.
Having determined that selinexor treatment activates AKT signaling in a P2RY2-, RAS-, and p110γ-dependent manner, we sought to explore AKT inhibition as a potentiator of selinexor’s anti-leukemic effects. First, we treated a panel of eight AML cell lines with selinexor in combination with three AKT inhibitors: MK2206, ipatasertib, and GSK690693, and showed that each could sensitize cells to selinexor (Fig. 5a). This was observed in both NPM1 wild type and mutant cell lines, suggesting that it stands apart from the dependence of NPM1-mutant AML on AKT (59). Because both allosteric and ATP-competitive AKT inhibitors sensitize cells to selinexor, the sensitization effect is likely on target to AKT. MK-2206 was selected for in vitro follow-up studies. Bliss criteria confirmed synergy between MK-2206 and selinexor across a range of drug doses (Extended Data Fig. 7a). This is consistent with the ability of shRNA-mediated XPO1 knockdown to sensitize cells to treatment with MK-2206 (Extended Data Fig. 7b). This combination forestalled the outgrowth of selinexor resistance over 8 weeks (Fig. 5b, Extended Data Fig. 7c). In contrast, pairing selinexor with the mTORC1 inhibitor everolimus did not elicit a synergistic response (Extended Data Fig. 7d). Separately, doxycycline-induced, shRNA knockdown of P2RY2 sensitized cells to selinexor (Extended Data Fig. 7e). This effect was phenocopied using AR-C (Extended Data Fig. 7f). In addition, we treated cells with selinexor in combination with: PI3Kα-specific inhibitor BYL-719, PI3Kβ-specific inhibitor TGX-221, PI3Kδ-specific inhibitor Cal-101, and IPI-549. IPI-549 could sensitize each AML cell line to selinexor more than the PI3K-α/β/δ-specific inhibitors (Extended Data Fig. 7g).
Figure 5. Inhibition of AKT Potentiates the Anti-Leukemic Effect of Selinexor.
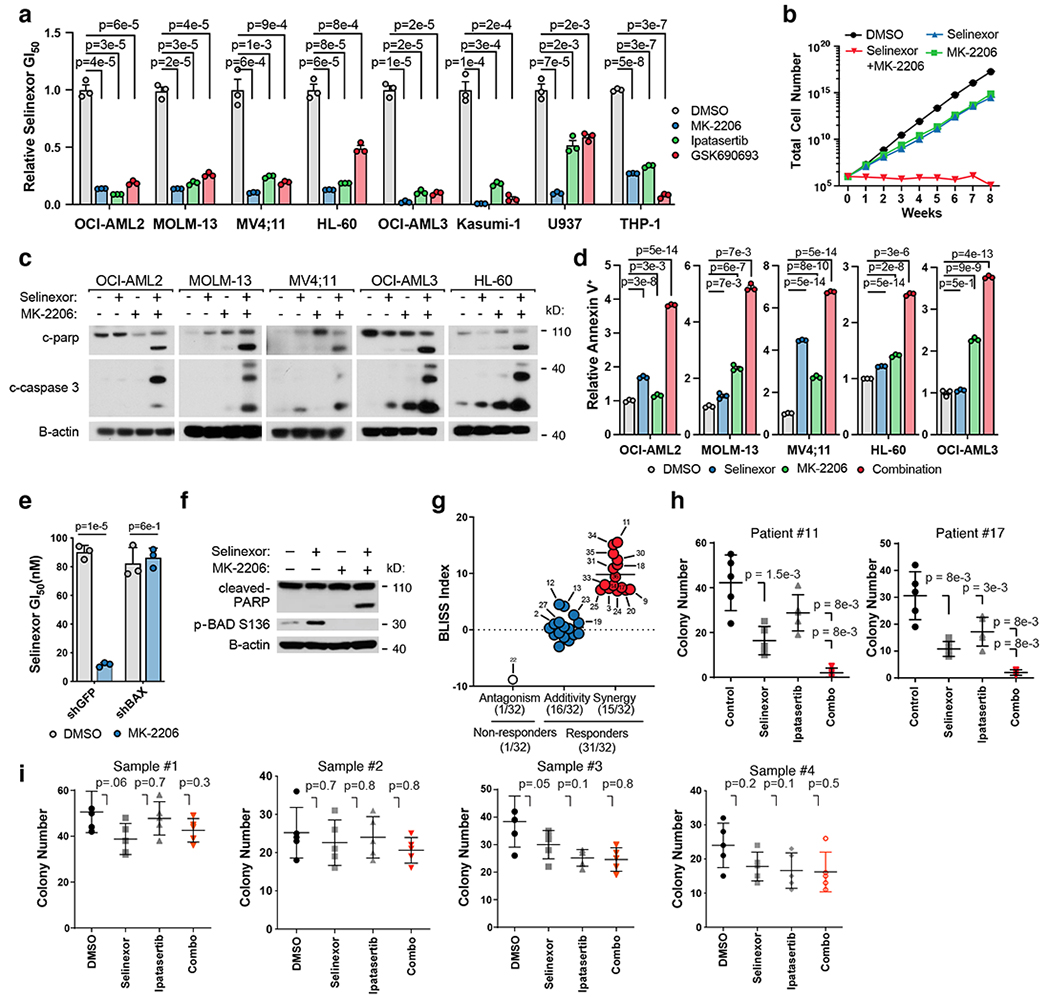
a) Relative GI50 values (GI50 selinexor + AKT inhibitor / GI50 selinexor alone) of selinexor in combination with AKT inhibitors across a panel of AML cell lines. Background AKT inhibitors dosed by cell line (OCI-AML2, 5μM; MOLM-13-3μM; MV4;11-3μM; HL-60-5μM; OCI-AML3-3μM; Kasumi-1-3μM; U937-3μM; THP-1,-5μM).
b) Time-to-progression assay of OCI-AML2 cells treated with 200nM selinexor, 5μM MK-2206, or the two drugs in combination. Data are presented as mean for n = 3 biologically independent cell populations.
c) Immunoblot depicting protein levels of cleaved CASPASE 3 and cleaved PARP across a panel of AML cell lines treated with selinexor (OCI-AML2, 200nM; MOLM-13, 75nM; MV4;11, 50nM; OCI-AML3, 250nM; HL-60, 300nM), MK-2206 dosed as in (a) or the combination.
d) Percentage of cells staining annexin V+ across a panel of AML cell lines treated with selinexor, MK-2206 or the combination; relative to DMSO control. P-values computed using one-way ANOVA with Tukey’s method for multiple comparisons; data are presented as mean +/−s.e.m. for n = 3 biological replicates.
e) Absolute selinexor GI50 values in OCI-AML2 cells harboring shRNAs targeting BAX (shBAX_71) versus GFP control and treated with selinexor + DMSO or selinexor + MK-2206.
f) Immunoblot depicting protein levels of cleaved PARP and phospho-BAD S136 in OCI-AML2 cells treated with selinexor (200nM), MK-2206 (5μM), or the combination.
g) Bliss synergy scores for 32 primary patient samples treated with a drug-dilution matrix. Bliss values ≤ −5, > −5 and < 5, and ≥ 5 denote antagonism, additivity, and synergy, respectively.
h) Methylcellulose colony formation of primary AML patient samples treated with selinexor (#11: 5nM, #17: 5nM) and/or ipatasertib (#11: 0.75μM, #17: 5μM) as indicated.
i) Methylcellulose colony formation of normal cord-blood-derived CD34+ cells treated with selinexor (5nM) and/or ipatasertib (Sample #1-2: 0.75μM, Sample #3-4: 5μM) as indicated.
Figure 5 a,e P-values computed using multiple unpaired (two-sided) t-tests; data are presented as mean +/−s.e.m. for n = 3 biologically independent replicates.
Figure h,i 5 p-values calculated using nonparametric Mann-Whitney test; data are presented as mean +/− s.d. for n = 5 biologically independent replicates. Colonies were stained and counted at 14 days.
Figure 5 c,f Representative immunoblots of n=2-3 biologically independent experiments yielding similar results. B-actin included as loading control.
Finally, we assessed the ability of AKT and XPO1 co-inhibition to provoke apoptosis. Western blot analysis revealed that AKT inhibition enhanced selinexor-induced cleavage of PARP and caspase 3 across cell lines (Fig. 5c, Extended Data Fig. 7h). This was corroborated by annexin staining and suggested that the observed synergy was from potentiation of drug-induced apoptosis (Fig. 5d, Extended Data Fig. 7i). To confirm this, we knocked down the pro-apoptotic protein BAX using two BAX-directed shRNAs before treating cells with the AKT inhibitor and selinexor combination (Extended Data Fig. 7j). BAX knockdown rescued the synergistic effect of AKT inhibition on selinexor activity (Fig. 5e). Moreover, treatment with selinexor promoted inhibitory phosphorylation of the pro-apoptotic protein BAD at Serine 136, an AKT substrate residue. This anti-apoptotic phosphorylation event is blocked by AKT inhibition, promoting increased PARP cleavage (Fig. 5f). These findings are consistent with our screen and propose that increased apoptosis, rather than growth inhibition, drives the synergy between selinexor and AKT inhibition.
Next, we tested the combination of AKT inhibition and selinexor in primary patient-derived AML samples, choosing the AKT inhibitor ipatasertib for its relative clinical maturity (60). We treated a panel of 32 AML patient-derived samples with a drug-dilution matrix comprised of 88 selinexor and ipatasertib dose combinations for 5 days before quantifying cell viability for each combination. We analyzed each viability matrix for Bliss synergy: 31 of 32 patient samples exhibited a stronger response to the combination of selinexor and ipatasertib than either agent alone (Supplementary Table 5). Among these 31 samples, 15 exhibited synergistic sensitivity to the combination (Bliss score above 5) while 16 displayed an additive sensitivity (Bliss scores between −5 and 5) (Fig. 5g). A 60-gene next generation sequencing panel revealed that patient cells with mutations in cohesin factor genes (STAG2, SMC1A, SMC3, CTCF) may exhibit heightened sensitivity to the combination of selinexor and ipatasertib (Fisher test p-value = 0.038, Supplementary Table 5). These results suggest that this combination may be active across a spectrum of AML patients. To further validate these findings, two patient samples with sufficient cell material were cultured in methylcellulose and treated with selinexor, ipatasertib, or the combination, at concentrations identified by the drug-drug interaction assay as most synergistic. In both samples, fewer colonies formed when treated with the combination than when treated with either agent alone, consistent with the synergy previously observed in liquid culture (Fig. 5h). At these concentrations, we did not observe synergistically deleterious effects on the colony-forming capacity or growth of normal cord-blood-derived CD34+ cells (Fig. 5i, Extended Data Fig. 7k).
AKT inhibition sensitizes mouse models of AML to selinexor.
Before assessing combined AKT inhibition and selinexor treatment in vivo, we established the maximally-tolerated combination dose in naive C57BL/6 mice at 65mg/kg ipatasertib plus 15mg/kg selinexor, every other day (Extended Data Fig. 8a,b). This dose of selinexor elicited an on-target response, evidenced by induction of p53 (Extended Data Fig. 8c) (7, 61). We did not observe significant changes in the abundance of hematopoietic stem cells (LinLow/Kit+/Sca-1+) in mice treated with the selinexor and ipatasertib combination compared to vehicle-treated mice (Extended Data Fig. 8d). In contrast, the myeloid progenitor subpopulation (LinLow/Kit+/Sca-1−) was increased by an average of 1.5-fold in mice treated with the selinexor and ipatasertib combination; this was also observed in mice treated with cytarabine and anthracycline-based chemotherapy (Extended Data Fig. 8e). While the selinexor and ipatasertib combination did not impose noticeable effects on the LinLow/Sca-1−/Kit+/Cd16/32−/Cd34− MEP cell compartment, it promoted slight expansion of the LinLow/Sca-1−/Kit+/Cd16/32−/Cd34+ CMP fraction and a reduction in the LinLow/Sca-1−/Kit+/Cd16/32+/Cd34+ GMP cell compartment (Extended Data Fig. 8f,g,h). In addition, evaluation of more mature hematopoietic cell populations revealed that ipatasertib combined with selinexor prompted no significant alterations to the proportion of Cd3+ T-cells, Ter-119+ erythrocytes, and Mac-1+/Gr-1+ granulocytes compared to vehicle, and decreased the proportion of B220+ B-cells and Mac-1+/Gr-1− monocytes to the same extent as chemotherapy. An exception here involves Cd41+ megakaryocytes, whose proportion remained unchanged with chemotherapy but was decreased with ipatasertib and selinexor treatment (Extended Data Fig. 8i,j,k,l,m,n).
We followed up our preclinical investigation of ipatasertib and selinexor using this dosing regimen, selecting an aggressive, MLL-AF9-driven syngeneic mouse model of AML whose median time to disease progression was 14 days from injection. This model was treated every other day with five cycles of either vehicle, 15mg/kg selinexor, 65mg/kg ipatasertib, or the combination. While animals that received selinexor or ipatasertib survived, on average, no more than 10 days longer than those that received vehicle, animals that received both selinexor and ipatasertib survived nearly 30 days longer (Fig. 6a). Treatment with both agents was also associated with reduction of the marrow MLL-AF9 blast fraction (Fig. 6b).
Figure 6. Preclinical Efficacy of Combined XPO1 and AKT Inhibition in AML.
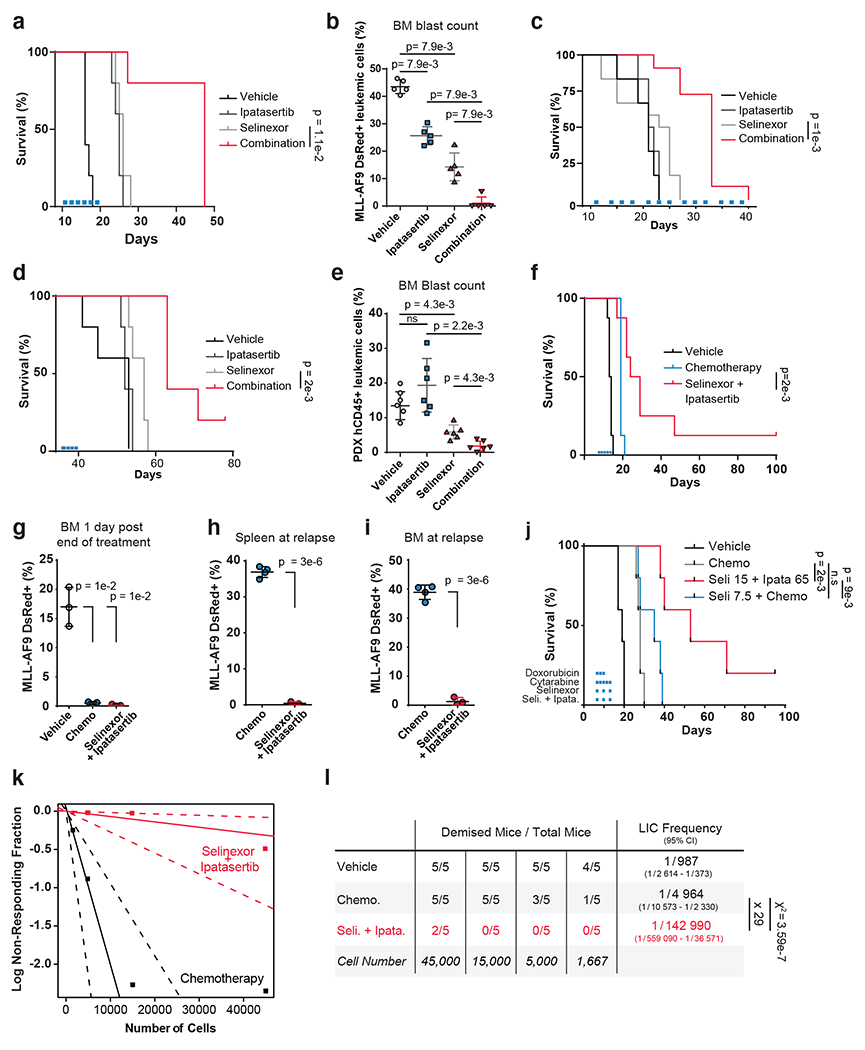
a, c-d) Kaplan–Meier survival curves of MLL-AF9 syngeneic (a), OCI-AML2 cell-line NSG xenograft (c) or patient-derived AML-engrafted NOG-EXL (d) mouse models treated with vehicle, 65mg/kg ipatasertib, 15mg/kg selinexor or the drug combination; n = 5 (a,d),n=10 (c), mice per cohort.
b) FACS quantification of MLL-AF9 dsRed+ leukemic blast cells mouse bone marrow aspirates (n=5 biologically independent mice per group) following treatment with conditions indicated in (a).
e) FACS quantification of human CD45+ leukemic blast cells from NOG-EXL mouse bone marrow aspirates (n=6 biologically independent mice per group) on day 28. Human CD45+ cells were injected on day 0; engraftment was confirmed on day 12; mice were treated day 13 through day 21
f) Kaplan–Meier survival curves of MLL-AF9 syngeneic mouse model of AML treated with vehicle, standard-of-care chemotherapy (1mg/kg doxorubicin and 100mg/kg cytarabine) or the combination of 65mg/kg ipatasertib plus 15mg/kg selinexor; n = 8 mice per cohort.
g) FACS quantification of MLL-AF9 dsRed+ leukemic blast cells from mouse bone marrow aspirates following treatment with conditions indicated in (f); n = 3 biologically independent mice.
h) FACS quantification of MLL-AF9 dsRed+ leukemic blast cells in spleen upon disease relapse following standard-of-care chemotherapy (1mg/kg doxorubicin and 100mg/kg cytarabine) versus the 65mg/kg ipatasertib plus 15mg/kg selinexor drug combination; n = 4 biologically independent mice in control and n= 3 selinexor + ipatasertib.
i) FACS quantification of MLL-AF9 dsRed+ leukemic blast cells in bone marrow with conditions indicated in (h); n = 4 biologically independent mice in control and n= 3 selinexor + ipatasertib.
j) Kaplan–Meier survival curves of MLL-AF9 syngeneic mouse model of AML treated with vehicle, standard-of-care chemotherapy (1mg/kg doxorubicin and 100mg/kg cytarabine), the combination of chemotherapy plus 7.5mg/kg selinexor, or the combination of 65mg/kg ipatasertib plus 15mg/kg Selinexor conducted with n = 5 mice per cohort.
k) Limiting dilution assay performed on MLL-AF9 cells isolated from primary mice treated with either standard chemotherapy (100mg/kg cytarabine and 1mg/kg doxorubicin) or the combination of 15mg/kg selinexor and 65mg/kg ipatasertib for 24hrs and reinjected into secondary recipient mice with n=5 biologically independent replicates per cohort.
l) Determination of leukemia-initiating cell (LIC) frequency with a 95% confidence interval in each group using extreme limiting dilution analysis (ELDA). Statistical significance determined by a two-sided chi-squared test.
Figure 6 b,e, g–i Data are presented +/− s.d. P-values calculated using nonparametric Mann-Whitney test (b, e) or two-tailed Welch’s t-test (g-i).
Figure 6 a,d,f,j Statistical significance determined by log-rank (Mantel-Cox) test. Duration treatments conditions depicted as colored bars.
We further validated our combination using an orthotopic xenograft model of AML, established by introducing OCI-AML2 cells into nonobese diabetic (NOD)-severe combined immunodeficiency (SCID) IL2R-γnull mice (NSG mice). Consistent with results from the MLL-AF9-driven syngeneic AML mouse model, the combination of selinexor and ipatasertib significantly prolonged survival in our human xenograft model more than either drug alone (Fig. 6c). An analogous experiment was subsequently conducted using a patient-derived xenograft (PDX) model, established by transplanting primary cells into NOD/shi-SCID/IL2R-γnull (NOG)-Tg(SV40/HTLV-IL3,CSF2)(EXL) mice (NOG-EXL mice) (Extended Data Fig. 9a). The combination of selinexor and ipatasertib significantly prolonged mouse survival compared to mice treated with either agent individually (Fig. 6d). The group treated with both agents exhibited a lower percentage of human CD45+ leukemic blasts in bone marrow than animals treated with either drug alone (Fig. 6e). Synergy between selinexor and ipatasertib was confirmed across doses in cells from this PDX model in vitro (Extended Data Fig. 9b). Together, these data demonstrate that ipatasertib enhances the therapeutic benefit of selinexor treatment in preclinical mouse models of AML.
Selinexor plus AKT inhibition outperforms chemotherapy.
To compare selinexor and ipatasertib against standard-of-care induction chemotherapies, we treated MLL-AF9 AML-bearing mice with either vehicle, cytarabine (100mg/kg) and doxorubicin (1mg/kg), or selinexor (15mg/kg) and ipatasertib (65mg/kg), every other day for five days. Combining selinexor and ipatasertib conferred a survival advantage over maximally-tolerated doses of chemotherapy (Fig. 6f). Bone marrows of treated mice, sampled one day after treatment completion, revealed decreased leukemic cells, suggesting that both combinations can achieve marrow penetrance (Fig. 6g). Subsequently, analysis of bone marrow and splenic samples taken upon first relapse in either treatment group revealed that the combination of selinexor and ipatasertib could provide durable responses while cytarabine and doxorubicin could not (Fig. 6h,i).
Since selinexor has been combined with standard-of-care chemotherapy in early clinical trials (16, 17), we sought to benchmark the selinexor and ipatasertib combination against selinexor and chemotherapy. We treated cohorts of MLL-AF9 AML-bearing mice with selinexor plus ipatasertib, chemotherapy (cytarabine and doxorubicin), or the combination of selinexor and chemotherapy, compared against vehicle. Mice treated with ipatasertib could tolerate selinexor at 15mg/kg; no mice treated with ipatasertib and selinexor suffered treatment-related mortality and weight loss was below 10% (Extended Data Fig. 9c). In contrast, while mice could tolerate chemotherapy alone (Extended Data Fig. 9d), mice treated with chemotherapy were unable to tolerate the additional 15mg/kg selinexor; all five mice treated with that combination sustained weight loss exceeding 20% and suffered drug-related mortality by day 16 (Extended Data Fig. 9e). To address this, we reduced the selinexor dose to 7.5mg/kg when paired with chemotherapy. Even still, the selinexor and chemotherapy-treated mice exceeded 15% weight loss and one out of five mice was lost to mortality at day 16 (Extended Data Fig. 9f).
Flow cytometry performed two days after treatment termination revealed that the leukemia burden was reduced in all groups versus controls (Extended Data Fig. 9g). Strikingly, MLL-AF9-driven leukemia in animals treated with selinexor and chemotherapy relapsed faster than in mice treated with ipatasertib and selinexor (Fig. 6j, Extended Data Fig. 9h), indicating that ipatasertib is a more tolerable and effective partner for selinexor than standard chemotherapy. This aligns with in vitro drug sensitivity data suggesting that selinexor does not synergize with either cytarabine or daunorubicin (Extended Data Fig. 9i).
Last, studies have suggested that drug resistance in AML is driven by leukemia-initiating cells (LICs), cells capable of seeding leukemia (62). Therapies that engender durable responses require drugs that eradicate the LIC fraction (63). To assess the ability of selinexor and ipatasertib to target LICs, we harvested MLL-AF9 leukemic cells from mice treated with either vehicle, selinexor and ipatasertib, or cytarabine and doxorubicin. Twenty-four hours after treatment, the leukemic burdens in treated mice were assessed by flow cytometry (Extended Data Fig. 10a,b). Leukemic cells were sorted for DsRed+, diluted to establish cell concentrations, and reinjected into sublethally-irradiated recipient mice. Using extreme limiting dilution analysis, we observed a 29-fold decrease in LIC frequency in secondary recipients injected with blasts harvested from selinexor plus ipatasertib-treated donor mice compared to those engrafted with blasts harvested from cytarabine plus doxorubicin-treated donor animals (Fig. 6k,l). Across cell concentrations, the survival of mice injected with blasts pretreated with selinexor and ipatasertib was substantially longer than the chemotherapy-pretreated group (Fig. 6l, Extended Data Fig. 10c).
Discussion
Here, we report that treating AML cells with the XPO1 inhibitor selinexor activates the PI3K/AKT pathway through transcriptional upregulation of P2RY2. Using in vitro, in vivo, and patient-derived model systems, we demonstrate that co-inhibition of AKT potentiates the anti-leukemic effects of selinexor.
Principally, our data nominate the combination of selinexor and AKT inhibition for treating AML. This drug combination was widely synergistic across multiple cell lines, AKT inhibitors, and assays. Notably, in three mouse models of AML, treatment with selinexor and an AKT inhibitor prolonged survival versus selinexor alone. Further, the selinexor plus ipatasertib combination conferred a survival advantage and reduced the LIC burden over maximally-tolerated standard-of-care chemotherapy. These results are particularly striking in light of selinexor’s recent clinical developments, suggesting that selinexor-based regimens could be augmented through AKT inhibition (16–18, 61).
More broadly, this work addresses contrasting notions of how anticancer therapies affect cancer cell fitness: one which holds that therapies monotonically restrict cell fitness, and another, which reserves the possibility that anticancer drugs may elicit pro-fitness effects separable from anti-fitness effects. Our study reveals that selinexor treatment compels the activation of signaling cascades that simultaneously restrict and support AML cell fitness. These effects are not compensatory, and are set apart from cellular adaptations to drug-induced stress; they represent on-target sequelae of XPO1 inhibition, not the off-target effects of imperfect inhibitors; they exhibit determinism at the cellular level, and do not represent rare, subclonal events that are selected for over time. We present selinexor as an archetype, but we suspect that many small-molecule inhibitors analogously activate pro-fitness effects, particularly those targeting general cellular processes such as chromatin regulation, transcription, translation, and protein degradation. These processes affect many features of the cell, and drugs targeting them may trigger unforeseen effects.
The principles discussed here lay out two avenues for future study. First, since discovering that AKT inhibition synergizes with selinexor in AML, our group has been working to initiate clinical trials that exploit this relationship. Second, our work supports the idea that the efficacy of any targeted therapy is a summation of anti-fitness and pro-fitness effects, emphasizing the importance of cataloguing therapy-induced, pro-fitness effects. The techniques modeled in this study could be applied at scale to explore the pro-fitness effects brought upon by other therapies, complementing our knowledge of how those therapies exert their anti-fitness effects. At a minimum, this would provide another means for taxonomizing our compendium of cancer treatments. In theory, it could provide a rational framework for designing effective therapeutic combinations.
Materials and Methods
Our research complies with all relevant ethical regulations. In vivo experiments were approved by and performed according to guidance from the American Association for Laboratory Animal Science and the French National Committee on Animal Care. Primary AML patient samples were handled in accordance with a protocol approved by the INSERM IRB.
Cell lines and reagents
Cell lines were maintained in a humidified incubator at 37 °C with 5% CO2. OCI-AML2, MOLM13, MV4;11, HL-60, OCI-AML3, Kasumi-1, U937, THP-1 cells were cultured in RPMI-1640 medium with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. 293FT cells were cultured in DMEM high glucose medium with 10% FBS, 1% penicillin/streptomycin, 1% sodium pyruvate, 1% non-essential amino acids, and 1% GlutaMax. MV4;11 (CRL-9591), HL-60 (CCL-240), Kasumi-1 (CRL-2724), U937 (CRL-1593.2), and THP-1 (TIB-202) cell lines were purchased from Duke University Cell Culture Facility. OCI-AML2, MOLM13, and OCI-AML3 cell lines were received as a gift from the lab of Dr. Anthony Letai. All cell lines were authenticated by STR profiling prior to use. All experiments performed using aliquots of positively identified cell lines. Drugs were purchased from ApexBio (MK-2206, BYL-719), Tocris (AR-C118925XX, ARL67156), Sigma-Aldrich (Pertussis toxin) and SelleckChem (Selinexor, IPI-549, ipatasertib, GSK690693, everolimus, cytarabine, daunorubicin).
Short-term drug sensitivity assay (GI50)
Cell viability assays conducted as described previously (33). Briefly, AML cells were seeded at 7,500 cells/well, treated with vehicle or a serial dilution of selinexor (individually or combination with fixed-concentration background drug) and assessed for viability after 72-hours using Cell Titer Glo (Promega). Raw luminescence values for each treatment condition normalized to either the vehicle-treated well (selinexor individually) or the background drug only well (selinexor combinations). GI50 values interpolated from dose-response curves plotted using GraphPad Prism v7, v8.0.2, v9.0.1 software.
Time-to-progression assay
Performed as previously described (64). Briefly, cells plated into 10cm plates at 1E6 cells per plate, treated with drug or vehicle, counted weekly, replated (up to 1E6) and treated for 8 weeks. Weekly growth rates (μ) were calculated from the number of cells plated the prior week (N0) and the number counted the current week (N) using the formula ln(N) = ln(N0) + μ*t; where t is elapsed time in hours. Virtual cell number extrapolated from growth rate.
Western immunoblotting
Immunoblotting:
Immunoblotting performed as previously described (33), with slight modification. Protein lysates were prepared with 1X CST Lysis Buffer (CST #9803) with 1X cOmplete protease inhibitor cocktail (Roche #04693124001) and 1X PhosSTOP phosphatase inhibitor (Roche #04906837001), rotated for 15 minutes, cleared by centrifugation at 20,000g for 10 minutes at 4 °C and normalized by total protein content using Bradford analysis. Membranes were probed with the following primary antibodies (where available: clone, catalogue number, dilution): β-actin (13E5) (CST #4970 diluted 1:5000 in 5% BSA), p-AKT T308 (244F9) (CST #4056 diluted 1:1000 in 5% BSA), p-AKT S473 (D9E) (CST #4060 diluted 1:1000 in 5% BSA), T-AKT (C67E7) (CST #4691 diluted 1:3000 in 5% BSA), p-GSK3β S9 (D85E12) (CST #5558 diluted 1:1000 in 5% BSA), p-BAD S136 (D25H8) (CST #4366 diluted 1:1000 in 5% BSA), XPO1 (C-1) (sc # 74454 diluted 1:100 in 5% BSA), cleaved-PARP (D64E10) (CST #5625 diluted 1:1000 in 5% BSA), cleaved-Caspase3 D175 (CST #9661 diluted 1:500 in 5% BSA), p110-g (D55D5) (CST #5405 diluted 1:1000 in 5% BSA), p110α (C73F8) (CST #4249 diluted 1:1000 in 5% BSA), p110β (C33D4) (CST #3011 diluted 1:1000 in 5% BSA), p110δ (D1Q7R) (CST #34050 diluted 1:1000 in 5% BSA), p101 (D32A5) (CST #5569 diluted 1:1000 in 5% BSA), Phospho-PKC Substrate Motif [(R/K)XpSX(R/K)] MultiMab™ Rabbit mAb mix (CST #6967 diluted 1:1000 in 5% BSA), Phospho-PKA Substrate (RRXS*/T*) (100G7E) Rabbit (CST #5569 diluted 1:1000 in 5% BS, T-S6K1 (CST#9202 1:1000 in 5% BSA) or p-S6K1 (CST#9205 1:500 in 5% BSA) overnight (16 hours). Following incubation with HRP-conjugated secondary antibody, blots were developed with SuperSignal West Pico PLUS Chemiluminescent Substrate (ThermoFisher) or ECL Western Blotting Substrate (ThermoFisher).
RT-qPCR analysis
RT-qPCR analysis was performed as previously described (33). The primers used in our analysis are provided (Supplementary Table 6).
Lentivirus production
293FT cells were grown to 70-80% confluency in a 10cM and transfected using Lipofectamine 2000 (Invitrogen), PLUS Reagent (Invitrogen), 8.164μg of psPAX2, 5.336μg of pVSVg, and 10.667 μg of plasmid DNA diluted in Opti-MEM according to manufacturer’s instructions. Briefly, psPAX2, pVSVg and plasmid DNA were mixed with 785μL Opti-MEM. 103.2μL PLUS Reagent was diluted in 785μL Opti-MEM and gently pipetted onto DNA mixture. After a 5-minute room temperature incubation, 94.6μL Lipofectamine 2000 in 1.570mL Opti-MEM was pipetted onto DNA/PLUS Reagent mixture. After another 5-minute room temperature incubation, the mixture was added to 293FT cells. After a 5-hour 37 °C incubation of 293FT cells with transfection mixture, media was aspirated and exchanged for virus harvest media (30% FBS in described 293FT media). After 48 hours, media containing virus was harvested, filtered with a 0.45μM filter and stored at −80°C.
Doxycycline-inducible shRNA constructs
Controlled expression of shRNAs achieved using a doxycycline-inducible pLKO-Tet-On lentiviral system (65, 66) using shRNA sequences from the LEGACY shRNA inventory (Supplementary Table 7). Top and bottom oligos were annealed, ligated with AgeI/EcoRI digested gel-purified pLKO-Tet-On vector, transformed into competent One Shot Stabl3 E. coli cells (Invitrogen #C737303) and spread onto LB/Amp plates. Individual colonies were selected and plasmid DNA was purified (Qiagen) and sequence validated.
CRISPR/Cas9 selinexor sensitizer screen
Custom sgRNA library generation:
Our sgRNA library was designed, cloned and amplified as previously described (33, 67). Each unique 20 base pair sgRNA was appended/prepended and synthesized as an oligo pool by Custom Array Inc. The pooled inserts were PCR amplified using NEB Phusion Hotstart enzyme mix and cleaned up with Axygen magnetic PCR beads (Fisher Scientific). Gibson assembly was performed using 100ng of FastDigest BsmBI digested lentiCRISPRv2 (Addgene plasmid #52961), 40ng of prepped sgRNA insert and 10μL of Gibson assembly master mix (NEB). 1μL of the Gibson reaction product was transformed into electrocompetent cells (E. cloni 10G ELITE, Lucigen #60052-2), spread onto LB-ampicillin plates and incubated at 37 °C for 16 hours. Colonies counted to ensure > 40X coverage of library, scraped and plasmid DNA was isolated using a Maxiprep kit (Qiagen). The library used to perform the selinexor sensitizer screen has previously been published (33).
Individual sgNT and sgXPO1 oligonucleotides were similarly prepared, cleaned-up, cloned, transformed, isolated, and sequence validated.
Pooled CRISPR screening:
Virus production, titering and transduction performed as previously described (33). Briefly, viral titer (Infectious Units (IFU)/mL) was found by transducing OCI-AML2 cells with a 1:3 dilution series of library virus, selecting cells with puromycin for 2 days and determining multiplicity of infection (MOI) from % of cells infected. Viral titer is equal to (# cells seeded * MOI * Virus dilution factor) / (Virus volume added to each well). OCI-AML2 cells were transduced at an MOI of 0.2 at 1000X coverage of the library and puromycin selected for 7 days prior to dividing into selinexor-treated versus vehicle-treated populations. For the selinexor sensitizer screen, selinexor was dosed at 100nM, a concentration that yielded sufficient selective pressure without excessive cell death (approximately the GI50 concentration) over the two-week screen period. Each drug/vehicle condition was conducted in biologically independent replicate and carried at >1000X coverage for 2 weeks. Genomic DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen) from 25E6 cell samples taken prior to dividing cells into treatment conditions (time zero) and after completion of the two-week screen. Amplification of the sgRNA barcodes and indexing of each sample was performed via 2-step PCR as previously described (33, 67).
Screen processing and analysis:
To determine differences in sgRNA composition between samples, deep sequencing was performed by Hudson Alpha Institute for Biotechnology using the Illumina Nextseq platform (single-ended 75 bp). As previously described (33), barcoded reads were converted to guide-level counts and the fractional representation (FR) of each sgRNA construct was found by dividing the count of each sgRNA in a sample by the sum of all sgRNA counts in that sample. The selinexor construct-level depletion score was found by comparing the 2-week drug-treated population to the 2-week vehicle-treated population (Selinexortime = 2weeks / DMSOtime = 2weeks). Construct-level depletion scores were collapsed to gene-level depletion scores by taking the average depletion score across 5 sgRNA constructs. All depletion/enrichment effects reported as log2 ratios. All described analyses conducted using the R Statistical Environment, see Code Availability section below.
The results of this sensitizer screen were included in a larger effort (33), although a full dissection of selinexor’s sensitizer interactions has been reserved for this study.
Reverse-phase protein array (RPPA) analysis sample preparation
OCI-AML2 cells were treated with DMSO or selinexor for 3, 24 and 48-hours at which point 3E6 cells were pelleted and frozen at −80 °C. RPPA analysis was performed as previously described (68, 69). All samples were conducted in biological triplicate and normalized to DMSO control at each respective timepoint.
p-AKT T308 FACS-based CRISPR/Cas9 screen in selinexor treated OCI-AML2 cells
sgRNA library amplification:
The Toronto Knockout CRISPR Library – Version 3 (TKOv3) was obtained from Addgene (Pooled Libraries #90294, #125517) and amplified according to provided published protocol. The TKOv3 library contains 70,948 sgRNAs targeting 18,053 protein coding genes (4 sgRNAs targeting each gene) and 142 non-targeting control sgRNAs against LacZ, EGFP and luciferase. Briefly, TKOv3 pooled plasmid library DNA was diluted 1:10 in TE and electroporated into Endura electrocompetent cells (Lucigen, #60242) at a coverage of >25X of the library. The library plasmid pool was purified using a Maxiprep kit (Qiagen). Virus generation and library expression performed as described at minimum of 1000x coverage.
Fixation and intracellular p-AKT T308 staining:
Ten days after transduction, library-expressing OCI-AML2 cells were treated with DMSO or 200nM selinexor. After 48-hours, cells were pelleted, washed with 1X PBS and fixed/permeabilized using the Thermo IC Fixation Kit. Fixation, permeabilization and staining was performed according to manufacturer’s instruction with slight modification; fixed 35E6 cells/15mL tube (buffers scaled) at R.T. for 15 minutes , washed with 1X Permeabilization Buffer followed by wash with FACS buffer (1X PBS, 2% FBS, 0.1% Sodium Azide, 2mM EDTA). Cells were stained in 1X Permeabilzation Buffer with a 1:200 dilution of p-AKT T308 (244F9) (CST #4056) primary antibody (1mL of Permeabilzation Buffer/primary antibody solution per 35E6 cells) at R.T. for 2 hours with gentle rocking. Cells were again washed with 1X Permeabilization Buffer followed by FACS buffer and stained in 1X Permeabilzation Buffer with 1:200 dilution of Alexa Fluor 488 Conjugate (CST #4412) secondary antibody at R.T. for 1 hour with gentle rocking. Cells were washed with 1X Permeabilization Buffer followed by FACS buffer and resuspended at 25E6cells/mL in FACS buffer in preparation for FACS analysis. All centrifugations performed at 700g.
FACS analysis and sorting:
To achieve sufficient coverage of the library in the sorted cell populations, 144E6 (2000X coverage) selinexor-treated, fixed and stained cells were sorted in each replicate. This ensured that both the top and bottom sort populations retained at least 200X coverage of the library. Cells were strained with a 0.3μM filter, FACS analyzed and sorted using the Astrious Cell Sorter (Beckman Coulter). Cells were gated for live cells based on FSC/SSC and singlets based on FSC (Extended Data Fig. 3a,b). The bottom 18% and top 10% p-AKT T308 expressing cells were collected into 1X PBS in separate collection tubes. The bottom 18% gate represents selinexor-treated cells that were unable to activate AKT relative to DMSO-treated cells. To prevent the fixed cells from sticking to the collection tubes, 2% FBS was spiked into sorted cell populations. Cells were distributed into 1.5E6 cell aliquots, pelleted at 700g, followed by genomic DNA extraction using Arcturus PicoPure DNA Extraction Kit (ThermoFisher #KIT0103) according to manufacturer’s instructions. Amplification of the sgRNA barcodes and indexing of each sample was performed via 2-step PCR as previously described (Supplementary Table 8) (43).
Screen processing and analysis:
To determine the sgRNA composition in the bottom and top-sorted cell populations, deep sequencing was performed by the Duke Sequencing and Genomic Technologies core using the Illumina NovaSeq 6000 platform (single-ended 75 base pair reads). Barcoded reads were converted to fractional representation as described in analysis of CRISPR/Cas9 selinexor sensitizer screen. Construct-level scores were found by comparing the fractional representation of each sgRNA in the bottom sort population to that of the top sort population (SelinexorBottom sort / SelinexorTop sort). sgRNA constructs that registered fewer than 100 counts were excluded from downstream analysis. Construct-level data was collapsed to a gene-level FACS screen gene score (FSGS) by taking the average score across the (up to 4) sgRNA constructs. Genes without a minimum of two sgRNA constructs were excluded. All FSGSs were reported as log2 ratios. All described manipulations were performed in R, see Code Availability section below.
RNA-seq gene expression analysis:
OCI-AML2 and MOLM-13 cells were treated for 36 hours with vehicle or selinexor (200nM for OCI-AML2 and 75nM for MOLM-13) in biologically independent triplicate. RNA was isolated from whole cells with the RNEasy Mini kit (Qiagen) and sent for paired-end non-stranded RNA-sequencing by Novogene. Reads uniquely mapped to annotated genes were analyzed using DEseq2 (70) and assessed for differential expression in selinexor versus vehicle-treated conditions. To generate the selinexor signature, the top 300 differentially up and downregulated genes between DMSO and Selinexor conditions (defined based on the lowest adjusted p-value and log2 fold change) were derived from each cell lines, and at the intersection of these two gene lists, we identified a common signature of 166 and 122 genes and up and downregulated, respectively, in both AML cell lines.
P2RY2 expression patient stratification:
Gene expression data of AML patients whose transcriptomic profiling was available from TCGA-LAML (n=198 AML samples) or GSE14468 (n = 526 AML samples), were z-score normalized and high versus low P2RY2 levels were evaluated based on the absolute z-score cut-off of 0.75. On versus off selinexor signatures were assigned for each patient based on the ES z-score > 1 or < −1, respectively. The significance of the differences between the proportions of each subgroup of patients was evaluated by applying the two tailed Fisher’s Exact Test implemented in the function fisher.test (stats package, R 2.14, http://cran.r-project.org/).
Gene Ontology analysis:
Gene Ontology analysis was performed on genes enriched in the bottom sort versus top sort of p-AKT T308 FACS screen (FSGS > 1.5) using Enrichr web-based tool (https://amp.pharm.mssm.edu/Enrichr/) (71, 72). Gene Ontology on selinexor sensitizer and resister genes was performed independently on genes with a depletion score of < −0.75 and > 0.75, respectively.
Patient samples:
Synergy assessment:
Upon consent of informed donors without history of hematological disorders, umbilical cord blood samples were supplied by the EFS (Bourgogne Franche-Comté, France) and CD34+ cells were purified (Ficoll-Paque PLUS 17-1440-02 GE Healthcare) using the CD34+ cells magnetic isolation kit and AutoMACS™ separator according to manufacturer instructions (Miltenyi Biotec, Bergish Gladbach, Germany). Blood or bone marrow samples collected from informed patients under consent from a St Louis Hospital IRB-approved protocol were collected; Ficoll gradient centrifugation was performed to isolate mononucleated cells. Red blood cells were lysed (Red Blood Cell Lysing Buffer Hybri-Max R7757 Sigma Life Science) and either the normal cord-blood-derived CD34+ cells or the leukemic blasts were resuspended in patient medium (RPMI 10% FBS 1%Pen/Strept with the cytokines TPO, EPO, SCF, FLT3, IL3, IL6, G-CSF, GM-CSF). Cells were seeded into 384 well plates at 5000 cells per well and treated in quadruplicate with top doses of 1μM for selinexor and 50μM for ipatasertib with 1:2 dilutions between doses. After 120 hours, CellTiter-Glo® Luminescent Cell Viability Assay was used as a readout of viability. Synergy across the matrix was assessed by calculating the average Bliss score across all doses using synergyfinder (73). Negative scores indicate antagonism while positive scores indicate synergy. For this study, combinations where doses of either drug alone produced greater than 90% loss of viability, precluding observable synergy, were excluded; we used a Bliss cutoff of 1 to denote the presence of strong synergy.
Patient profiling:
Primary patient samples were collected as part of an ongoing clinical registry at St Louis Hospital (THEMA, IRB approval: IDRCB 2021-A00940-41) and stored at the St Louis Hospital tumor biobank. Informed consent was obtained prior to collection; no compensation was provided. Samples retrieved from these tumor banks were anonymized and their storage in our laboratory was declared to the Ministry of Higher Education, Research and Innovation. Secondary use of primary patient cells derived from clinical practice (bone marrow biopsies or blood samples) was approved by the INSERM IRB. No identifying information and no personal data were made available to the research teams, and only anonymized clinical data were accessible, in compliance with French protection of personal data law and in accordance with the Declaration of Helsinki. Cytogenetic analyses were carried out using karyotyping and FISH studies guided by karyotype; genetic profiling consisted of fragment analysis for NPM1, FLT3, and IDH1/2 mutational status and by targeted-sequencing of 60 recurrently mutated genes in AML at > 500X coverage (Agilent SureSelect, Illumina). This information, along with age and sex for all samples, has been provided (Supplementary Table 5).
Methylcellulose assay:
Cord-blood-derived CD34+ cells and additional primary AML cell aliquots from patient samples 11 and 17 were obtained from the bone marrow aspirate, seeded in methylcellulose-based medium MethoCult H4435 (Stem Cell Technologies) at a concentration of 20E3 cells/plate in triplicate, and treated with the indicated concentrations of Selinexor, Ipatasertib, or the combination of both compounds. Plates were quantified 14 days later with MTT staining.
In vivo transplantation
OCI-AML2 cell line xenograft:
The Duke University Institutional Animal Care & Use Committee (IACUC) reviewed and approved the cell line xenograft transplantation and treatment protocol described in this study. OCI-AML2 cells were IMPACT tested and confirmed mycoplasma negative prior to engraftment. Approximately 1 × 106 luciferase-expressing OCI-AML2 cells suspended in 0.1mL sterile 1X PBS were tail vein injected into 5-6 week old male nonobese diabetic (NOD)-severe combined immunodeficiency (SCID) IL2R-γnull mice. Two weeks after injection, mice were assessed for successful engraftment by IVIS bioluminescence imaging and analysis using Living Image software. Mice were sorted by bioluminescence, treated M/W/F with selinexor (10mg/kg) by oral gavage, ipatasertib (75mg/kg) by oral gavage, or both selinexor and ipatasertib. Drugs were formulated in OraPlus suspending vehicle. Mice were monitored daily for signs of distress, such as weakness, weight loss, ruffled coat, lethargy or bruising. Observance of humane endpoints as described in our protocol—loss of body weight > 15% free-feeding body weight, inability to rise or ambulate, presence of labored respiration, wound ulceration, or other signs of active infection—resulted in euthanization. As this study pertains to leukemia, a maximal tumor size was not enforced. Drug treatments and routine monitoring continued for 6 weeks or until a humane endpoint was reached.
PDX models:
The French National Committee on Animal Care reviewed and approved all experiments using the PDX models described in this study.
PDX engraftment performed as previously described (33).
Twelve days after injection, mice were assessed for engraftment: peripheral blood samples and bone marrow biopsies were resuspended in PBS, 0.5% BSA, 2mM EDTA prior to staining with anti-human PE-Vio770-coupled CD45 (hCD45) antibody (130-113-119, Miltenyi). Cells were washed three times in PBS 2mM EDTA and hCD45-positive cells were assessed using a FACScanto II. Following engraftment, mice were randomized and treated every other day for one week either by oral gavage with 65 mg kg–1 ipatasertib (OraPlus) or 15 mg kg–1 selinexor (OraPlus) or with these drugs combined as indicated (day 13 through day 21). Mice were monitored for signs of distress: rough fur, tremors, isolation from cage mates, weight loss >15% of original body weight, and temperature decrease; no limit points were exceeded in our experiments. After one week, mice were euthanized and bone marrow was harvested to analyze the leukemic cells in each group (day 28). Samples were washed in PBS and resuspended in 0.5% BSA, 2 mM EDTA–PBS before staining with the PE-Vio770-coupled CD45 antibody and flow cytometry analysis.
PDX patient characteristics:
The PDX sample was derived from a 69-year-old female who was diagnosed with secondary AML with MDS-related changes; patient was previously treated with mitoxantrone/ etoposide/ cytarabine + lenalidomide; genetic profiling revealed mutations in CEBPA/ ASXL1/ RUNX1/ EZH2/ JAK2/ TET2; patient karyotype is 46,XX,t(6;7)(q23;q11.2)[1]/46,XX[cp19].
MLL-AF9 model:
The French National Committee on Animal Care reviewed and approved all experiments described. The Sca-1− /c-Kit+ myeloid progenitor fraction was sorted from total bone marrow following exclusion of Cd5+, Cd127+, Cd45R+, and Ter-119+ cell populations using FACS Aria (BD Biosciences) and were transduced with an MLL-AF9-IRES-DsRed retroviral vector (gift from Dr. Scott Armstrong’s laboratory, DFCI, Boston, USA) (74). Two months after transplantation, sick mice were euthanized, their bone marrow harvested, flow-sorted for the MLL-AF9-positive granulo-monocytic bone marrow progenitor population (DsRed+/Sca-1−/c-Kit+/Cd16/32+/Cd34+) and subsequently reinjected into sublethally-irradiated recipient C57BL/6 male donor mice (The Jackson Laboratory). MLL-AF9 dsRed+ cells were harvested and sorted from whole bone marrow of recipient mice and approximately 0.1 × 106 dsRed+ sorted cells were tail-vein-injected into sublethally irradiated (350 cGy) 6–8-week-old male C57BL/6 mice. Ten days later, mice were randomized and treated every other day for 5 or 10 days either by oral gavage with ipatasertib (65 mg/kg) (OraPlus), selinexor (15 mg/kg) (OraPlus), or both. Doxorubicin (1 mg/kg) and cytarabine (100 mg/kg) were resuspended (HBSS) and delivered intraperitoneally on days 1-3, and cytarabine alone on days 4-5. For combinations of selinexor and chemotherapy, 7.5 mg/kg selinexor was used on days 1, 3, 5. Mice were monitored for signs of distress: rough fur, tremors, cage isolation, weight loss >15%, and hypothermia; none of these limit points were exceeded over the course of our experiments. As this study pertains to leukemia, a maximal tumor size was not enforced. Bone marrow biopsies were performed on anesthetized animals 24 or 48 hours after treatment completion. Biopsies were washed in PBS and resuspended in 2 mM EDTA–PBS before flow cytometry analysis. Upon relapse, mice were sacrificed and bone marrow and spleen were collected, washed with PBS, and resuspended in 0.5% BSA, 2 mM EDTA–PBS before flow cytometry analysis.
MLL-AF9 limiting dilution assay:
20 × 106 viable MLL-AF9-positive leukemic cells were harvested and sorted from sick mice treated with either the combination of Selinexor (15mg/kg) and Ipatasertib (65mg/kg) or the combination of the chemotherapeutic agents Cytarabine (100mg/kg) and Doxorubicin (1mg/kg) for 16 hrs. Sorted MLL-AF9 cells serially diluted to appropriate cell concentrations for reinjection into sublethally-irradiated secondary recipient mice (either 45,000, 15,000, 5,000, or 1,667 cells per mouse in a total 5 mice per group). Demised mice were counted and limiting dilution analyses performed using the Extreme Limiting Dilution Analysis (ELDA) function of the ‘StatMod’ package (http://bioinf.wehi.edu.au/software/elda/index.html). Leukemia-Initiating Cell (LIC) frequencies between groups of secondary recipient animals were compared using the likelihood ratio chi-squared test (75).
Statistics and Reproducibility
Results shown as mean ± s.e.m. unless specified; two-sample hypotheses were tested using the unpaired Student’s t-test at unadjusted two-sided level of 0.05. Welch correction applied in cases where distribution variances determined group dependent. P-values provided as exact values whenever significant. Unless noted, data distributions were assumed normal, not formally tested. Where an underlying Gaussian distribution could not be assumed, nonparametric Mann-Whitney test used to test two-sample hypotheses at an unadjusted two-sided type I level of 0.05. Unless otherwise noted, all measurements were from distinct samples. Box plot elements defined as: box extending 25th to 75th percentile; whiskers extending minimum to maximum values; median indicated by traversing line. No statistical methods used to pre-determine murine cohort sizes but cohorts are similar to those reported in previous publications (33, 74, 76). Mice were only excluded from study when leukemic engraftment failed; this exclusion occurred prior to randomization/cohorting and treatment. Otherwise, no animals or data points excluded unless noted. Where noted, mice were randomized according to their disease burden in blood or bone marrow prior to treatment. The investigators were not blinded to allocation during experiments or analysis. Unless otherwise indicated, representative immunoblots are reflective of n=3-5 biologically independent experiments yielding similar results. B-actin is included as a protein loading control. Computing assistance for statistical analysis and/or data visualization was performed using GraphPad Prism v7, v8.0.2, v9.0.1; R v3.5.1, v4.0.2 (https://www.r-project.org/); Adobe Illustrator CC 2017, 2020; FlowJo 2019 and Microsoft Excel 2016-2021.
Data Availability
All data associated with this study are available in the main text or the supplementary materials, or can otherwise be made available from the corresponding author on reasonable request. Raw counts table for both the CRISPR/Cas9 sensitizer and p-AKT T308 FACS based screen are included as Supplementary Tables 9 and 10, and on Github as below. RNA-seq data from OCI-AML2 and MOLM-13 cells treated with selinexor has been deposited in the Gene Expression Omnibus (GEO) under accession code GSE181003. Source data for Fig. 1, 2, 3, 4, 5, 6 and Extended Data Fig. 1, 2, 4, 5, 6, 7, 8, 9, 10 have been provided as Source Data files.
Code Availability
Script and associated raw data for reanalyzing sensitizer and FACS-based CRISPR–Cas9 screens are available on Github (https://github.com/linkvein/selinexor_p2ry2_akt).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1. CRISPR/Cas9 and RPPA analyses reveal signaling pathways modulated by Selinexor treatment.
a) Scatterplot depicting replicate selinexor depletion gene scores from CRISPR/Cas9 drug-modifier screen. Screens conducted as n = 2 independent replicates with n = 5 sgRNAs per gene.
b) Gene ontology (GO) analysis of selinexor “resister” genes; performed using Enrichr.
c) Selinexor depletion gene scores ranked from most depleted to most enriched in the selinexor versus vehicle treated populations. Predicted genetic modifiers of selinexor sensitivity involved in G1/S cell cycle progression are annotated.
d) Schematic relating G1/S cell cycle regulators to selinexor depletion gene scores and RPPA expression. Annotated as in Fig. 1d.
e) GO analysis of selinexor “sensitizer” genes; performed using Enrichr.
f) Schematic relating mTORC1 signaling to selinexor depletion gene scores and RPPA expression. Annotated as in Fig. 1d.
g) Immunoblot depicting protein levels of phosphorylated and total S6K1 in five AML cell lines treated with DMSO or selinexor. B-actin included as loading control. Representative immunoblots of n = 2 independent experiments yielding similar results.
Extended Data Fig. 2. Activation of AKT signaling is a specific consequence of XPO1 inhibition.
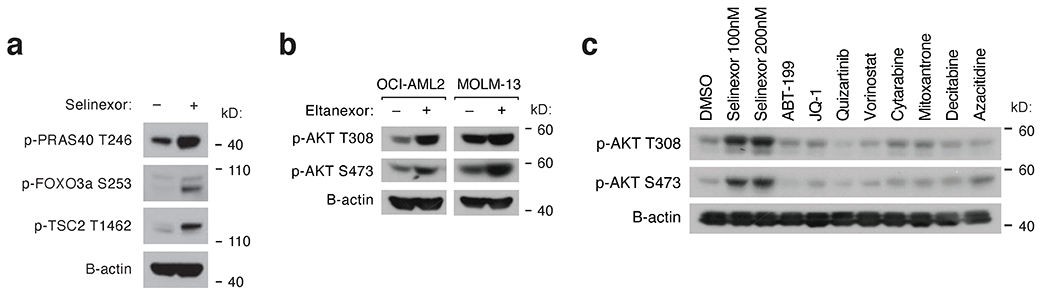
a) Immunoblot depicting protein levels of phosphorylated PRAS40, FOXO3a, and TSC2 following 24-hour treatment of OCI-AML2 cells with selinexor.
b) Immunoblot depicting protein levels of phosphorylated AKT at T308 and S473 following 24-hour treatment of OCI-AML2 and MOLM-13 cells with eltanexor.
c) Immunoblot depicting protein levels of phosphorylated AKT at T308 and S473 in OCI-AML2 cells treated with a panel of standard-of-care therapies for 24 hours.
Representative immunoblots of n = 2-3 independent experiments yielding similar results. B-actin included as loading control.
Extended Data Fig. 3. Gating strategy and Gene Ontology analysis of FACS-based CRISPR/Cas9 screen.
a) Scatterplot depicting gating strategy to isolate live sgRNA library transduced OCI-AML2 cells based on forward-scatter (FSC) and side-scatter (SSC).
b) Scatterplot depicting gating strategy to isolate singlet sgRNA library transduced OCI-AML2 cells based on FSC.
c) Gene ontology (GO) analysis table of scoring genes enriched in the bottom sort (FSGS of > 1.5). P-values calculated by Enrichr using Benjamini-Hochberg correction for multiple hypothesis testing.
Extended Data Fig. 4. Selinexor-induced AKT activation requires the P2RY2 purinergic receptor.
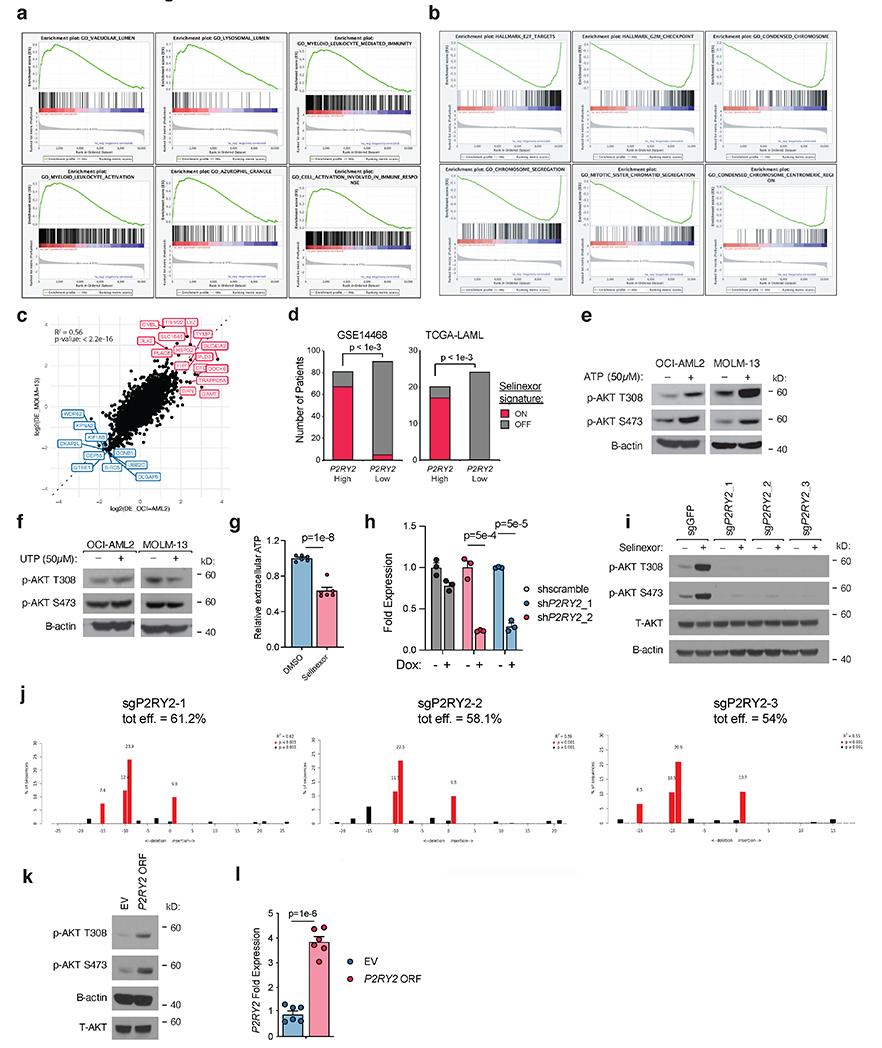
a) GSEA plots for gene ontologies enriched in RNA-seq datasets upon selinexor treatment.
b) GSEA plots for gene ontologies depleted in RNA-seq datasets upon selinexor treatment.
c) Comparison of differential gene expression analysis in OCI-AML2 and MOLM-13 cells treated +/− selinexor. The genes in red are upregulated in both cell lines when treated with selinexor whereas the genes in blue are downregulated in both cell lines.
d) Selinexor gene signature representation in AML patients with low versus high P2RY2 expression. ON versus OFF selinexor signatures were assigned for each patient based on the ES z-score > 1 or < −1, respectively. The number of selinexor-ON patients in the P2RY2 high subset versus the P2RY2 low subset was compared; P-values computed using two-sided Fisher’s t-test.
e) Immunoblot depicting protein levels of phosphorylated AKT at T308 and S473 in OCI-AML2 and MOLM-13 cells treated with 50μM ATP for 48 hours.
f) As in (e) but cells were treated with UTP.
g) Relative extracellular ATP concentration in OCI-AML2 cells treated with selinexor versus DMSO control for 36 hours. P-values computed using Welch’s unpaired (two-sided) t-tests; data are presented as mean +/− s.e.m. for n = 6 biological replicates.
h) Relative expression of P2RY2 in OCI-AML2 cells stably expressing indicated TetOn shRNA constructs following 48 hours of doxycycline treatment.
i) Immunoblot depicting protein levels of total and phosphorylated AKT at T308 and S473 in OCI-AML2 cells expressing Cas9 and sgRNAs targeting GFP control or P2RY2 treated with selinexor or DMSO. B-actin included as loading control.
j) Tide analysis of OCI-AML2 cells expressing sgP2RY2 to access knockout efficiency. .
k) Immunoblot depicting protein levels of total and phosphorylated AKT at T308 and S473 in OCI-AML2 cells overexpressing P2RY2 or empty vector in OCI-AML2 cells. B-actin included as loading control.
l) Relative expression of P2RY2 in OCI-AML2 cells stably expressing either empty vector or P2RY2 ORF.
Extended Data Figure 4h, l P-values computed using multiple unpaired (two-sided) t-tests; data presented as mean +/−s.e.m. for n = 3 biological replicates.
Extended Data Figure 4e,f,i,k Representative immunoblots of n=2-3 biologically independent experiments yielding similar results. B-actin included as loading control.
Extended Data Fig. 5. Isoform specific dependency on PI3Kγ for Selinexor-induced AKT activation.
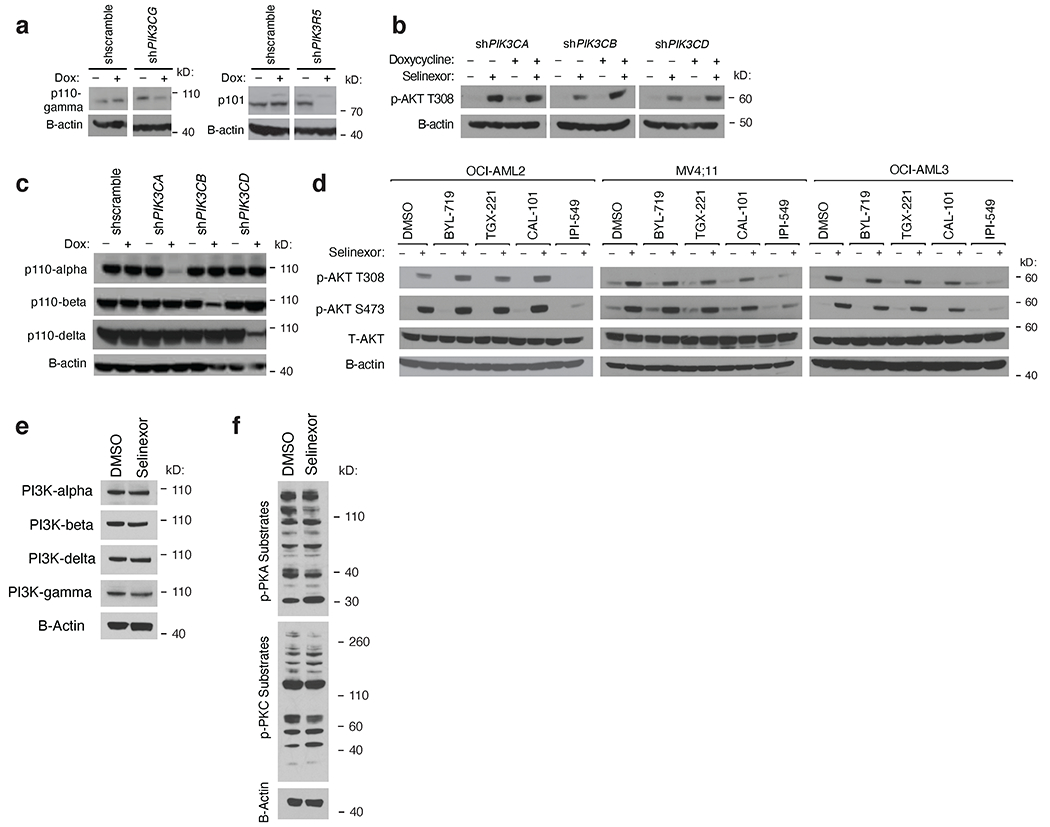
a) Immunoblot depicting protein levels of p110-gamma and p101 in OCI-AML2 cells harboring doxycycline-inducible shRNAs targeting PIK3CG (encoding for p110-gamma) and PIK3R5 (encoding for p101) versus scrambled control. B-actin included as loading control.
b) Immunoblot depicting protein levels of phosphorylated AKT at T308 in OCI-AML2 cells harboring doxycycline-inducible shRNAs targeting PIK3CA, PIK3CB and PIK3CD versus scrambled control following treatment with selinexor for 36 hours. B-actin included as loading control.
c) Immunoblot depicting protein levels of p110α, p110β and p110δ in OCI-AML2 cells harboring doxycycline-inducible shRNAs targeting PIK3CA, PIK3CB and PIK3CD versus scrambled control. B-actin included as loading control.
d) Immunoblot depicting protein levels of total and phosphorylated AKT at T308 and S473 in OCI-AML2, MV4;11 and OCI-AML3 cells treated with BYL-719 (PI3K-alpha inhibitor), TGX-221 (PI3K-beta inhibitor), CAL-101 (PI3K-delta inhibitor) or IPI-549 (PI3K-gamma inhibitor) with or without selinexor for 36 hours. OCI-AML2 cells were treated with 500nM of PI3K inhibitors and MV;411 and OCI-AML3 cells were treated with 100nM of PI3K inhibitors. B-actin included as loading control.
e) Immunoblot depicting protein levels of catalytic PI3K isoforms in OCI-AML2 cells treated with selinexor or DMSO. B-actin included as loading control.
f) Immunoblot depicting protein levels of phospho-substrates of PKA or PKC in OCI-AML2 cells treated with selinexor or DMSO. B-actin included as loading control.
Representative immunoblots of n=2-4 biologically independent experiments yielding similar results. B-actin included as loading control.
Extended Data Fig. 6. The activity and requirement of Ras for complete Selinexor-induced AKT activation.
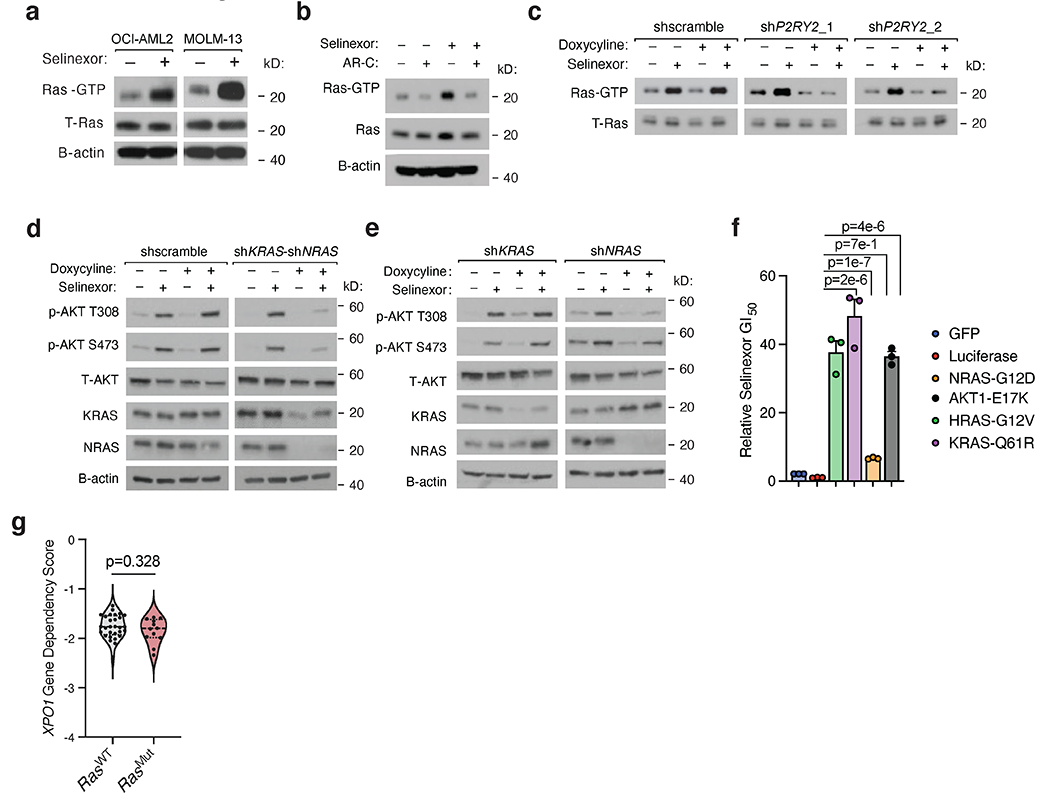
a) Immunoblot depicting active Ras following co-immunoprecipitation of Ras-GTP with GST-Raf1-Ras-binding domain (RBD) fusion proteins in a panel of selinexor-treated AML cell lines. Total Ras in input shown as control.
b) Immunoblot depicting active Ras as in (c) in OCI-AML2 cells treated with AR-C 118925XX (2.5μM) and Selinexor (200nM), alone and in combination. Total Ras in input shown as control.
c) Immunoblot depicting protein levels of immunoprecipitated GTP-bound Ras in OCI-AML2 cells with doxycycline-inducible shRNAs targeting P2RY2 versus scrambled shRNA control. Cells were exposed to doxycycline (75ng/mL) for 48 hours and treated with either vehicle or selinexor for 36 hours. Total Ras in input shown as control.
d) Immunoblot depicting protein levels of phosphorylated AKT at T308 and S473 in OCI-AML2 cells co-expressing doxycycline-inducible shRNAs against NRAS and KRAS versus scrambled shRNA control. Cells were exposed to doxycycline (75ng/mL) for 48 hours and treated with either vehicle or selinexor for 36 hours.
e) Immunoblot depicting protein levels of phosphorylated AKT at T308 and S473 in OCI-AML2 cells expressing doxycycline-inducible shRNAs against NRAS or KRAS. Cells were exposed to doxycycline (75ng/mL) for 48 hours and treated with either vehicle or selinexor for 36 hours.
f) Relative 72h selinexor GI50 and dose-response curves in OCI-AML2 cells expressing GFP or Luciferase control or activating constructs of Ras or AKT. P-values computed using one-way ANOVA with Tukey’s method for multiple comparisons; data are presented as mean +/−s.e.m. for n = 3 biological replicates.
g) Genetic dependency of AML cell lines on XPO1 as defined by the DepMap dataset. P-values computed using unpaired (two-sided) t-test.
Extended Data Figure 6a–e Representative immunoblots of n=2-4 biologically independent experiments yielding similar results. B-actin included as loading control.
Extended Data Fig. 7. The anti-leukemic effect of AKT inhibition combined with Selinexor in cell line models of AML.
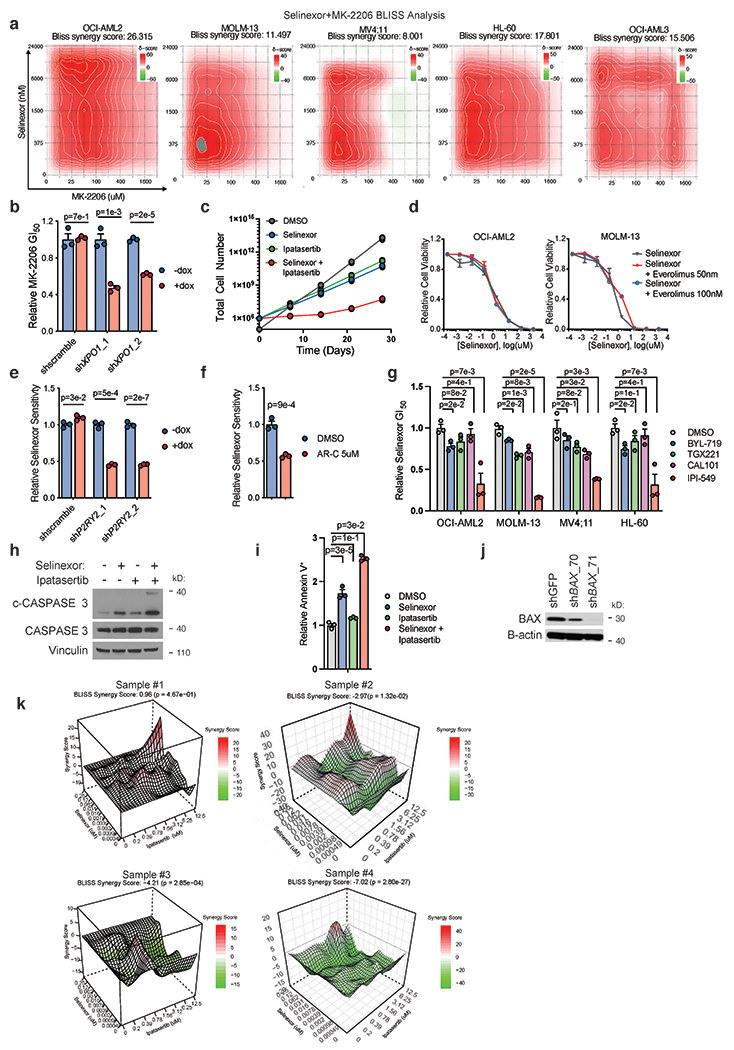
a) Bliss synergy analysis 2D plots for a panel of AML cell lines treated with a dilution series of selinexor and MK-2206. Delta scores indicate synergy (red) and antagonism (green) across the drug dilution matrix.
b) Relative MK-2206 GI50 values in OCI-AML2 cells harboring doxycycline (dox)-inducible shRNAs targeting XPO1 versus scrambled shRNA control. Cells treated with dox for 48 hours prior to incubation with MK-2206 drug-dilution series. Relative MK-2206 GI50 value defined as (GI50 MK-2206 -dox) / (GI50 MK-2206 + dox).
c) Time-to-progression assay of OCI-AML2 cells treated with 200nM selinexor, 5μM ipatasertib, or the two drugs in combination.
d) Selinexor dose response curves in OCI-AML2 and MOLM-13 cell lines treated with everolimus (50nM or 100nM) in the background.
e) Relative selinexor GI50 values in OCI-AML2 cells harboring doxycycline-inducible shRNAs targeting P2RY2 versus scrambled shRNA control. Cells were pre-treated with dox for 48 hours prior to incubation with selinexor drug-dilution series. Relative selinexor sensitivity values defined as (GI50 selinexor-dox) / (GI50 selinexor + dox). .
f) Relative selinexor GI50 values in OCI-AML2 cells co-treated with 5μM AR-C or DMSO.
g) Relative GI50 values of selinexor in combination with PI3K-α/β/δ/γ-specific inhibitors across a panel of AML cell lines. Relative selinexor GI50 value defined as (GI50 selinexor + PI3K inhibitor) / (GI50 selinexor alone). Background PI3K inhibitors dosed by cell line (OCI-AML2, 1μM; HL-60, 4 μM; MOLM-13, 2μM; MV4;11, 2μM; THP-1, 1μM).
h) Immunoblot depicting protein levels of CASPASE 3 and cleaved CASPASE 3 in OCI-AML2 cells treated with selinexor (200nM), ipatasertib (5μM), or the combination. Vinculin shown as control.
i) Drug-treated induction of annexin positivity in OCI-AML2 cells, as measured by flow cytometry, relative to baseline annexin positivity elicited with DMSO treatment.
j) Immunoblot depicting protein levels of BAX in OCI-AML2 cells expressing hairpins targeting BAX or GFP control. B-actin shown as loading control.
k) Bliss synergy landscape for two healthy donor cord blood-derived CD34+ cells treated with selinexor versus ipatasertib across a drug-dilution matrix.
Extended Data Figure 7b–g, i data are presented as mean +/− s.e.m. for n = 3 biologically independent replicates.
Extended Data Figure 7b,e–h,i P-values computed using multiple unpaired (two-sided) t-tests.
Extended Data Figure 7h,j Representative immunoblots of n=2 biologically independent experiments yielding similar results
Extended Data Fig. 8. The effect of Selinexor combined with AKT inhibition on mouse weight and hematopoietic cell compartment.
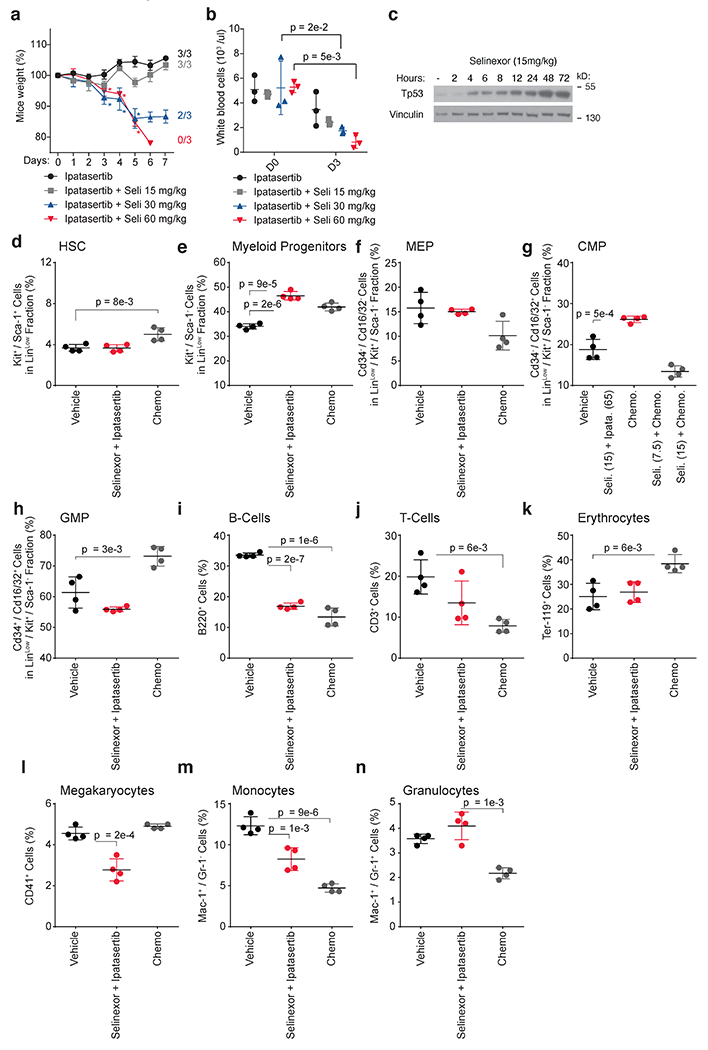
a) Dose-escalation study assessing mouse weight in disease naïve C57BL/6 mice. Mice were treated until Day 4 with indicated treatment regimens (n=3 biologically independent mice). P-values calculated using a two-way ANOVA; data are presented as +/− mean s.d.
b) Dose-escalation study assessing white blood cell count in disease naïve C57BL/6 mice. Mice were treated until Day 4 with indicated treatment regimens (n=3 biologically independent mice). P-values calculated using a two-way ANOVA; data are presented as +/− mean s.d.
c) Immunoblot depicting protein level of Tp53 in DsRed+ MLL-AF9 cells from mice treated in vivo with Selinexor for indicated duration. Each timepoint representants an independent biologic replicate. Representative immunoblots of n=2 biologically independent experiments yielding similar results. Vinculin shown as loading control.
d-n) Flow cytometric measurement of various hematopoietic cell types, each defined by gating strategies labeled on respective y-axes, in C57BL/6 mice treated with vehicle, chemotherapy (1mg/kg doxorubicin and 100mg/kg cytarabine), or the combination of 65mg/kg ipatasertib and 15mg/kg selinexor. Data are presented as mean +/− s.d. for n = 4 biologically independent replicates. Where indicated, P-values computed using unpaired (two-sided) t-test
Extended Data Fig. 9. Analysis of engraftment, tolerability, and efficacy of Selinexor combined with AKT inhibition in mouse models of AML.
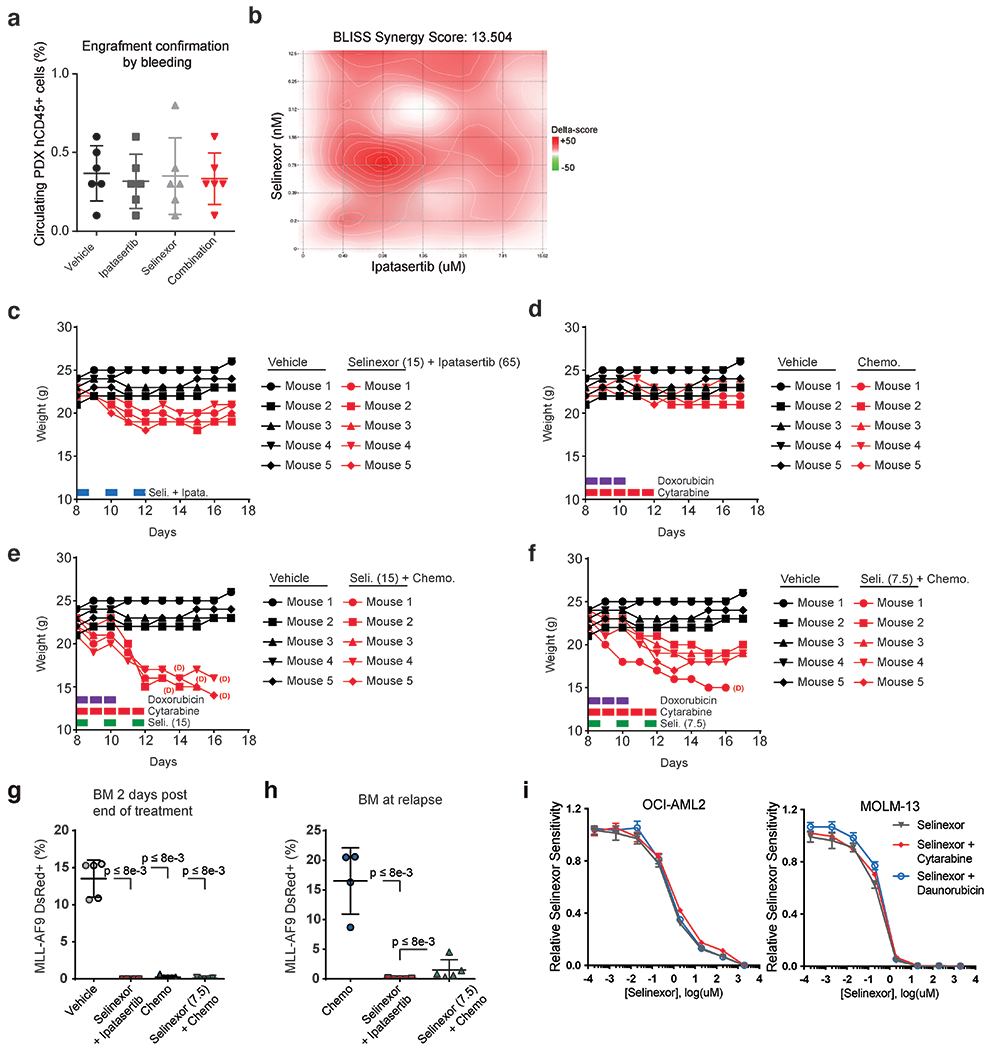
a) Confirmation of PDX engraftment by measurement of circulating human CD45+ cells transplanted into NOG-EXL mice (n=6 biologically independent mice per group); data are presented as mean +/− s.d.
b) Bliss synergy analysis 2D plots for PDX model of AML treated with a dilution series of selinexor and ipatasertib. Delta scores indicate synergy (red) and antagonism (green) across drug dilution matrix.
c) Weight of C57BL/6 mice treated with vehicle or selinexor (15mg/kg) plus ipatasertib (65mg/kg).
d) Weight of C57BL/6 mice treated with vehicle or chemotherapy (doxorubicin 1mg/kg, cytarabine 100mg/kg).
e) Weight of C57BL/6 mice treated with vehicle or selinexor (15mg/kg) plus chemotherapy (doxorubicin 1mg/kg, cytarabine 100mg/kg).
f) Weight of C57BL/6 mice treated with vehicle or selinexor (7.5mg/kg) plus chemotherapy (doxorubicin 1mg/kg, cytarabine 100mg/kg).
g) Measurement by flow cytometry of the proportion of DsRed+ MLL-AF9 cells taken from bone marrows (n=5 biologically independent mice per group) two days following treatment with vehicle, 15mg/kg selinexor plus 65mg/kg ipatasertib, chemotherapy (1mg/kg doxorubicin and 100mg/kg cytarabine), or 7.5mg/kg selinexor plus chemotherapy; P-values calculated using unpaired Mann-Whitney test; data are presented as mean +/− s.d.
h) Measurement by flow cytometry of DsRed+ MLL-AF9 cells taken from bone marrows at the point of relapse observed in chemotherapy-treated mice (n=5 biologically independent mice per group except in the chemotherapy-treated subgroup in which n=4). P-values calculated using unpaired Mann-Whitney test; data are presented as mean +/− s.d.
i) Selinexor dose response in OCI-AML2 and MOLM-13 cells treated with cytarabine (50nM) or daunorubicin (5nM) in the background; data are presented as mean +/− s.e.m. for n = 3 biologically independent experiments.
Extended Data Figure 9c–f Dosing schedule shown at bottom left. (D) = mouse demise.
Extended Data Fig. 10. Efficacy of Selinexor combined with AKT inhibition on leukemia initiating cells.
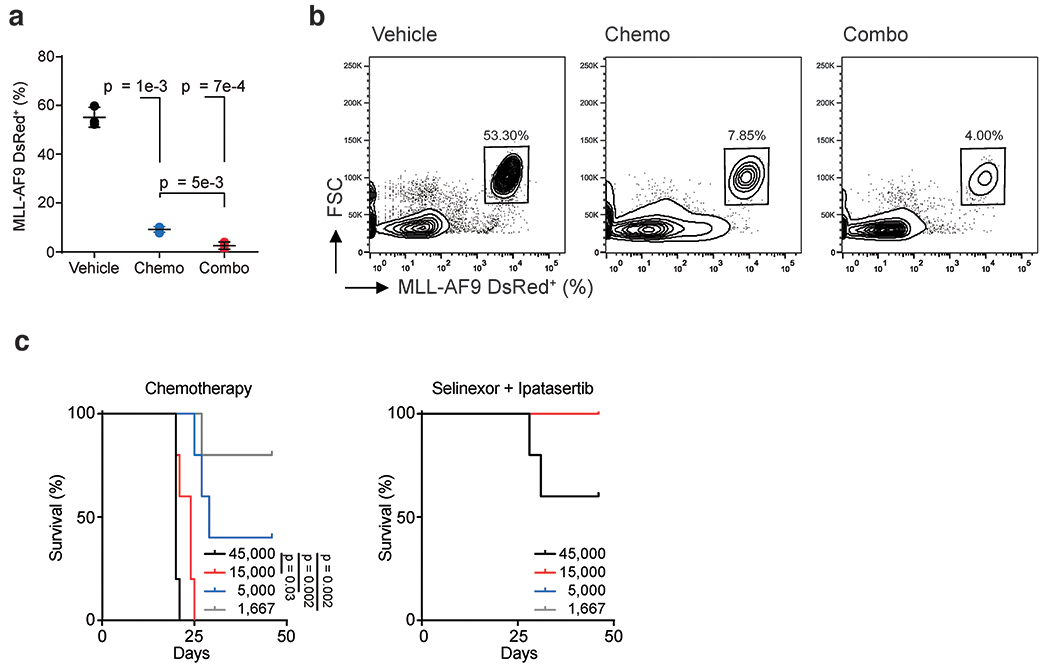
a) Leukemic burden of mice (n=3 biologically independent mice per group) prior to sorting and engraftment for extreme limiting dilution assay. P-values calculated using unpaired two-sided Welch’s test; data are presented as mean +/−s .d.
b) FACS plots depicting gating strategy to isolate dsRed+ MLL-AF9 cells prior to engraftment for extreme limiting-dilution assay.
c) Kaplan-Meier curves showing overall survival of secondary recipient mice (n=5 biologically independent mice) from each group in limiting dilution assay for determining LIC frequency. Statistical significance determined by log-rank (Mantel-Cox) test. n.s, not significant.
Supplementary Material
Supplementary Table 1. CRISPR/Cas9 selinexor sensitizer screen in OCI-AML2 cells
Supplementary Table 2. RPPA protein expression analysis at three selinexor treatment timepoints and normalized to DMSO control
Supplementary Table 3. FACS screen gene score (FSGS) indicating enrichment in bottom sort vs. top sort of selinexor treated, full-genome sgRNA library containing OCI-AML2 cells FACS sorted by p-AKT T308 expression
Supplementary Table 4. RNA-seq analysis showing differential gene expression in selinexor vs. vehicle treated OCI-AML2 and MOLM-13 cells
Supplementary Table 5. AML patients’ clinical features
Supplementary Table 6. qRT-PCR Primers
Supplementary Table 7. shRNA sequences
Supplementary Table 8. Sequencing Primers
Supplementary Table 9. CRISPR/Cas9 selinexor sensitizer screen in OCI-AML2 cells; Raw Counts
Supplementary Table 10. CRISPR/Cas9 selinexor FACS screen OCI-AML2 cells; Raw Counts
Acknowledgements
We would like to thank the members of the Kris Wood and Alexandre Puissant laboratories for their scientific input. In particular, we thank Colin A. Martz, Daniel P. Nussbaum, Grace R. Anderson, and Peter S. Winter for their insightful comments.
Funding
This work was supported by Duke University School of Medicine start-up funds and support from the Duke Cancer Institute (K.C.W.), NIH awards (R01CA207083 to K.C.W., F30CA206348 to K.H.L., K00CA245732-04 to J.H., F30CA247323 to C.C.S.), the Duke Medical Scientist Training Program (T32 GM007171 to K.H.L., C.F.B., C.C.S. and S.T.K.), the Duke Undergraduate Research Support Office (to J.C.R. and A.X.), the National Science Scholarship for PhD studies from the Agency for Science, Technology and Research, Singapore (H.X.A), the ATIP/AVENIR French research program (to A.P. and G.S.), and the EHA research grant for Non-Clinical Advanced Fellow (to A.P.). A.P. is a recipient of support from the ERC Starting program (758848), and supported by the St Louis Association for leukemia research and the FSER association. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors(s) and do not necessarily reflect the views of the National Science Foundation or the NIH.
Competing Interests
K.C.W. is a founder, consultant, and equity holder at Tavros Therapeutics and Celldom and has performed consulting work for Guidepoint Global, Bantam Pharmaceuticals, and Apple Tree Partners. T.S. Pardee has received research funding from Karyopharm and has served as a paid advisor to Karyopharm. R. Itzykson has received honoraria from Karyopharm for consulting. E.P. and M.P. are inventors on patent applications that cover aspects of the reverse phase protein microarray; as inventors, they are entitled to receive royalties as provided by US Law and George Mason University policy. E.P. and M.P. receive royalties from and are consultants of TheraLink Technologies, Inc. E.P. is a shareholder of TheraLink Technologies Inc., and shareholder and consultant of Perthera, Inc. The remaining authors have no competing interests to declare.
References
- 1.Chari A et al. Oral Selinexor-Dexamethasone for Triple-Class Refractory Multiple Myeloma. N Engl J Med 381, 727–738 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Gavriatopoulou M et al. Integrated safety profile of selinexor in multiple myeloma: experience from 437 patients enrolled in clinical trials. Leukemia, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalakonda N et al. Selinexor in patients with relapsed or refractory diffuse large B-cell lymphoma (SADAL): a single-arm, multinational, multicentre, open-label, phase 2 trial. Lancet Haematol 7, e511–e522 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Mahipal A, Malafa M, Importins and exportins as therapeutic targets in cancer. Pharmacol Ther 164, 135–143 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Senapedis WT, Baloglu E, Landesman Y, Clinical translation of nuclear export inhibitors in cancer. Semin Cancer Biol 27, 74–86 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Ranganathan P et al. Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood 120, 1765–1773 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kojima K et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood 121, 4166–4174 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Etchin J et al. Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia 27, 66–74 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Etchin J et al. KPT-8602, a second-generation inhibitor of XPO1-mediated nuclear export, is well tolerated and highly active against AML blasts and leukemia-initiating cells. Leukemia 31, 143–150 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Etchin J et al. Activity of a selective inhibitor of nuclear export, selinexor (KPT-330), against AML-initiating cells engrafted into immunosuppressed NSG mice. Leukemia 30, 190–199 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang W et al. Combinatorial targeting of XPO1 and FLT3 exerts synergistic anti-leukemia effects through induction of differentiation and apoptosis in FLT3-mutated acute myeloid leukemias: from concept to clinical trial. Haematologica 103, 1642–1653 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ranganathan P et al. XPO1 Inhibition using Selinexor Synergizes with Chemotherapy in Acute Myeloid Leukemia by Targeting DNA Repair and Restoring Topoisomerase IIalpha to the Nucleus. Clin Cancer Res 22, 6142–6152 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ranganathan P et al. Decitabine priming enhances the antileukemic effects of exportin 1 (XPO1) selective inhibitor selinexor in acute myeloid leukemia. Blood 125, 2689–2692 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fischer MA et al. Venetoclax response is enhanced by selective inhibitor of nuclear export compounds in hematologic malignancies. Blood Adv 4, 586–598 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abboud R et al. Selinexor combined with cladribine, cytarabine, and filgrastim in relapsed or refractory acute myeloid leukemia. Haematologica, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sweet K et al. Phase I Clinical Trial of Selinexor in Combination with Daunorubicin and Cytarabine in Previously Untreated Poor-Risk Acute Myeloid Leukemia. Clin Cancer Res 26, 54–60 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang AY et al. A phase I study of selinexor in combination with high-dose cytarabine and mitoxantrone for remission induction in patients with acute myeloid leukemia. J Hematol Oncol 11, 4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walter Fiedler JC, Amberg Stefanie, Kebenko Maxim, Thol Felicitas, Schlipfenbacher Vera, Wilke Anne Christine, Modemann Franziska, Janning Melanie, Serve Hubert, Ganser Arnold, Bokemeyer Carsten, Theile Susann, Deppermann Ute, Kranich Anne, Heuser Michael, in European Hematology Association. (Elicium 2, 2019). [Google Scholar]
- 19.Bhatnagar B et al. Selinexor in combination with decitabine in patients with acute myeloid leukemia: results from a phase 1 study. Leuk Lymphoma 61, 387–396 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Timothy M, Pardee S et al. paper presented at the ASH 2020, November 5, 2020. 2020. [Google Scholar]
- 21.Tan DS, Bedard PL, Kuruvilla J, Siu LL, Razak AR, Promising SINEs for embargoing nuclear-cytoplasmic export as an anticancer strategy. Cancer Discov 4, 527–537 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Shin I et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med 8, 1145–1152 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Liang J et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med 8, 1153–1160 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Plo I et al. AKT1 inhibits homologous recombination by inducing cytoplasmic retention of BRCA1 and RAD51. Cancer Res 68, 9404–9412 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Feng Z, Kachnic L, Zhang J, Powell SN, Xia F, DNA damage induces p53-dependent BRCA1 nuclear export. J Biol Chem 279, 28574–28584 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Huang WY et al. Prognostic value of CRM1 in pancreas cancer. Clin Invest Med 32, E315 (2009). [PubMed] [Google Scholar]
- 27.van der Watt PJ et al. The Karyopherin proteins, Crm1 and Karyopherin beta1, are overexpressed in cervical cancer and are critical for cancer cell survival and proliferation. Int J Cancer 124, 1829–1840 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bolli N et al. Born to be exported: COOH-terminal nuclear export signals of different strength ensure cytoplasmic accumulation of nucleophosmin leukemic mutants. Cancer Res 67, 6230–6237 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Falini B et al. Altered nucleophosmin transport in acute myeloid leukaemia with mutated NPM1: molecular basis and clinical implications. Leukemia 23, 1731–1743 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Brunetti L et al. Mutant NPM1 Maintains the Leukemic State through HOX Expression. Cancer Cell 34, 499–512 e499 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirli K et al. A deep proteomics perspective on CRM1-mediated nuclear export and nucleocytoplasmic partitioning. Elife 4, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thakar K, Karaca S, Port SA, Urlaub H, Kehlenbach RH, Identification of CRM1-dependent Nuclear Export Cargos Using Quantitative Mass Spectrometry. Mol Cell Proteomics 12, 664–678 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin KH et al. Using antagonistic pleiotropy to design a chemotherapy-induced evolutionary trap to target drug resistance in cancer. Nat Genet, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vermeulen K, Van Bockstaele DR, Berneman ZN, The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif 36, 131–149 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fridman JS, Lowe SW, Control of apoptosis by p53. Oncogene 22, 9030–9040 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Klein K et al. Evaluating the bromodomain protein BRD1 as a therapeutic target in rheumatoid arthritis. Sci Rep 8, 11125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henley SA, Dick FA, The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div 7, 10 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manning BD, Toker A, AKT/PKB Signaling: Navigating the Network. Cell 169, 381–405 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fruman DA, Rommel C, PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 13, 140–156 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marcus JM, Burke RT, DeSisto JA, Landesman Y, Orth JD, Longitudinal tracking of single live cancer cells to understand cell cycle effects of the nuclear export inhibitor, selinexor. Sci Rep 5, 14391 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim JE, Chen J, Cytoplasmic-nuclear shuttling of FKBP12-rapamycin-associated protein is involved in rapamycin-sensitive signaling and translation initiation. Proc Natl Acad Sci U S A 97, 14340–14345 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Argueta C et al. Selinexor synergizes with dexamethasone to repress mTORC1 signaling and induce multiple myeloma cell death. Oncotarget 9, 25529–25544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hart T et al. Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3 (Bethesda) 7, 2719–2727 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arnaoutov A et al. Crm1 is a mitotic effector of Ran-GTP in somatic cells. Nat Cell Biol 7, 626–632 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Crochiere M et al. Deciphering mechanisms of drug sensitivity and resistance to Selective Inhibitor of Nuclear Export (SINE) compounds. BMC Cancer 15, 910 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.N. Cancer Genome Atlas Research et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368, 2059–2074 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wouters BJ et al. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood 113, 3088–3091 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E, Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer 18, 601–618 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Ruiz-Gomez A et al. Phosphorylation of phosducin and phosducin-like protein by G protein-coupled receptor kinase 2. J Biol Chem 275, 29724–29730 (2000). [DOI] [PubMed] [Google Scholar]
- 50.Hu LP et al. Targeting Purinergic Receptor P2Y2 Prevents the Growth of Pancreatic Ductal Adenocarcinoma by Inhibiting Cancer Cell Glycolysis. Clin Cancer Res 25, 1318–1330 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Maiga A et al. Transcriptome analysis of G protein-coupled receptors in distinct genetic subgroups of acute myeloid leukemia: identification of potential disease-specific targets. Blood Cancer J 6, e431 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tabe Y et al. Bone Marrow Adipocytes Facilitate Fatty Acid Oxidation Activating AMPK and a Transcriptional Network Supporting Survival of Acute Monocytic Leukemia Cells. Cancer Res 77, 1453–1464 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muoboghare MO, Drummond RM, Kennedy C, Characterisation of P2Y2 receptors in human vascular endothelial cells using AR-C118925XX, a competitive and selective P2Y2 antagonist. Br J Pharmacol 176, 2894–2904 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pacold ME et al. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell 103, 931–943 (2000). [DOI] [PubMed] [Google Scholar]
- 55.Suire S et al. Gbetagammas and the Ras binding domain of p110gamma are both important regulators of PI(3)Kgamma signalling in neutrophils. Nat Cell Biol 8, 1303–1309 (2006). [DOI] [PubMed] [Google Scholar]
- 56.El-Brolosy MA et al. Genetic compensation triggered by mutant mRNA degradation. Nature 568, 193–197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ma Z et al. PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature 568, 259–263 (2019). [DOI] [PubMed] [Google Scholar]
- 58.Kim J et al. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature 538, 114–117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ren Z et al. Opposing effects of NPM1wt and NPM1c mutants on AKT signaling in AML. Leukemia 34, 1172–1176 (2020). [DOI] [PubMed] [Google Scholar]
- 60.Saura C et al. A First-in-Human Phase I Study of the ATP-Competitive AKT Inhibitor Ipatasertib Demonstrates Robust and Safe Targeting of AKT in Patients with Solid Tumors. Cancer Discov 7, 102–113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garzon R et al. A phase 1 clinical trial of single-agent selinexor in acute myeloid leukemia. Blood 129, 3165–3174 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thomas D, Majeti R, Biology and relevance of human acute myeloid leukemia stem cells. Blood 129, 1577–1585 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pollyea DA, Jordan CT, Therapeutic targeting of acute myeloid leukemia stem cells. Blood 129, 1627–1635 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Lin KH et al. Systematic Dissection of the Metabolic-Apoptotic Interface in AML Reveals Heme Biosynthesis to Be a Regulator of Drug Sensitivity. Cell Metab 29, 1217–1231 e1217 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wiederschain D et al. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 8, 498–504 (2009). [DOI] [PubMed] [Google Scholar]
- 66.Wee S et al. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A 105, 13057–13062 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shalem O et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pierobon M et al. Enrichment of PI3K-AKT-mTOR Pathway Activation in Hepatic Metastases from Breast Cancer. Clin Cancer Res 23, 4919–4928 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baldelli E et al. Reverse Phase Protein Microarrays. Methods Mol Biol 1606, 149–169 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen EY et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ianevski A, Giri AK, Aittokallio T, SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res 48, W488–W493 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Su A et al. The Folate Cycle Enzyme MTHFR Is a Critical Regulator of Cell Response to MYC-Targeting Therapies. Cancer Discov 10, 1894–1911 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hu Y, Smyth GK, ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 347, 70–78 (2009). [DOI] [PubMed] [Google Scholar]
- 76.Fenouille N et al. The creatine kinase pathway is a metabolic vulnerability in EVI1-positive acute myeloid leukemia. Nat Med 23, 301–313 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. CRISPR/Cas9 selinexor sensitizer screen in OCI-AML2 cells
Supplementary Table 2. RPPA protein expression analysis at three selinexor treatment timepoints and normalized to DMSO control
Supplementary Table 3. FACS screen gene score (FSGS) indicating enrichment in bottom sort vs. top sort of selinexor treated, full-genome sgRNA library containing OCI-AML2 cells FACS sorted by p-AKT T308 expression
Supplementary Table 4. RNA-seq analysis showing differential gene expression in selinexor vs. vehicle treated OCI-AML2 and MOLM-13 cells
Supplementary Table 5. AML patients’ clinical features
Supplementary Table 6. qRT-PCR Primers
Supplementary Table 7. shRNA sequences
Supplementary Table 8. Sequencing Primers
Supplementary Table 9. CRISPR/Cas9 selinexor sensitizer screen in OCI-AML2 cells; Raw Counts
Supplementary Table 10. CRISPR/Cas9 selinexor FACS screen OCI-AML2 cells; Raw Counts
Data Availability Statement
All data associated with this study are available in the main text or the supplementary materials, or can otherwise be made available from the corresponding author on reasonable request. Raw counts table for both the CRISPR/Cas9 sensitizer and p-AKT T308 FACS based screen are included as Supplementary Tables 9 and 10, and on Github as below. RNA-seq data from OCI-AML2 and MOLM-13 cells treated with selinexor has been deposited in the Gene Expression Omnibus (GEO) under accession code GSE181003. Source data for Fig. 1, 2, 3, 4, 5, 6 and Extended Data Fig. 1, 2, 4, 5, 6, 7, 8, 9, 10 have been provided as Source Data files.