

Abstract
Angioleiomyoma is a benign, pericytic (perivascular) neoplasm that primarily occurs in the subcutis or dermis of the extremities. The lesion typically presents as a small, firm, slow-growing, painful nodule. Magnetic resonance imaging reveals the lesion to be a well-defined, round to oval mass with signal intensity similar to or slightly hyperintense to that of skeletal muscle on T1-weightwed sequences. A dark reticular sign on T2-weighted sequences appears to be a characteristic feature of angioleiomyoma. Prominent enhancement is usually seen after intravenous contrast. Histologically, the lesion consists of well-differentiated smooth muscle cells with many vascular channels. Based on vascular morphologies, angioleiomyoma is classified into three subtypes: solid, venous, and cavernous. By immunohistochemistry, angioleiomyoma is diffusely positive for smooth muscle actin and calponin and variably for h-caldesmon and desmin. Conventional cytogenetic studies have demonstrated relatively simple karyotypes characterized by one or few structural rearrangements or numerical aberrations. In addition, metaphase comparative genomic hybridization analyses have revealed recurrent loss of 22q and gain of Xq. Angioleiomyoma can be successfully treated with simple excision, with a very low recurrence rate. Knowledge of this peculiar neoplasm is important because it can mimic a variety of benign and malignant soft-tissue tumors. This review provides an updated overview of the clinical, radiological, histopathological, cytogenetic, and molecular genetic features of angioleiomyoma.
Keywords: Angioleiomyoma, vascular leiomyoma, cytogenetics, review
Angioleiomyoma, also known as vascular leiomyoma, is a distinctive, relatively common, benign soft-tissue tumor originating from the smooth muscle layer of blood vessels. It belongs to the pericytic (perivascular) tumor group according to the 2020 World Health Organization Classification of Soft Tissue Tumors (1). The etiology of angioleiomyoma remains unknown. Angioleiomyoma can show a morphological overlap with a variety of benign and malignant soft-tissue tumors, including angiomyolipoma, myopericytoma and leiomyosarcoma. In our extensive experience, it is often difficult to diagnose angioleiomyoma preoperatively and this lesion may not be recognized as the cause of pain for some time. Advances in knowledge of the imaging, histopathology, and genetics of angioleiomyoma are leading to more accurate diagnosis and appropriate treatment. This review highlights the clinical, radiological, histological, immunohistochemical, cytogenetic and molecular genetic features of angioleiomyoma.
Clinical Features
Angioleiomyoma has a peak incidence in the fourth to sixth decades of life. The solid type is more common in females, whereas the venous and cavernous types show a slight male predominance (1). It typically presents as a solitary, firm, well-circumscribed, slow-growing, subcutaneous nodule, which is often painful. Angioleiomyoma shows a wide anatomic distribution but most frequently occurs in the extremities, particularly the lower leg. The diameter ranges from 0.2 to 4.3 cm (usually less than 2 cm) (2). Simple excision is the treatment of choice and prognosis is excellent. It is recommended that patients are followed up for one year after excision (3).
Radiologic Features
Various imaging modalities have been applied for the detection of angioleiomyoma. The radiographic and computed tomographic (CT) features of this neoplasm are not specific but ultrasonographic and magnetic resonance imaging (MRI) features are relatively characteristic. It is important to be familiar with the key imaging features of angioleiomyoma for its appropriate management.
Radiographs may be normal or reveal only a nonspecific soft-tissue mass. In such cases, it is often not possible to establish a meaningful differential diagnosis. Calcification may be seen in acral regions (4-6) and has been interpreted as degenerative in nature, probably related to repetitive minor trauma (6). CT reveals a well-defined soft-tissue mass with tissue attenuation similar to that of skeletal muscle. Ultrasound shows a solid, ovoid, well-defined mass with a relatively homogeneous, hypoechoic echotexture (7). Color doppler examination demonstrates prominent hyper-vascularity and may display associated feeding vessels. However, echogenicity and vascularity can be variable for angioleiomyoma based on the histological subtypes (8). On MRI, the lesion is well-defined and displays isointense or slightly hyperintense relative to skeletal muscle on T1-weighted images (Figure 1A) and variable heterogeneous signal intensity on T2-weighted images (9-12). A peripheral rim of low signal intensity can be seen on both T1- and T2-weighted images (10). A dark reticular sign on T2-weighted images appears to be a characteristic feature of angioleiomyoma (Figure 1B) (11). Contrast-enhanced MRI demonstrates prominent homogeneous and heterogeneous enhancement (Figure 1C). The presence of an adjacent tortuous vascular structure has also been reported after intravenous contrast (10). The vascular nature of the lesion may be identified on Tc-99m red blood cell perfusion and blood-pool scintigraphy (13).
Figure 1. Magnetic resonance images of angioleiomyoma of the ankle in a 60-year-old man. (A) T1-weighted sequence shows a well-defined subcutaneous mass (yellow arrow) with signal intensity similar to that of skeletal muscle. (B) T2-weighted sequence shows that the mass has high signal intensity. A dark reticular sign (yellow arrows) can be seen. (C) Contrast-enhanced fat-suppressed T1-weighted sequence reveals prominent homogeneous enhancement throughout the mass (yellow arrow).

Histological and Immunohistochemical Characteristics
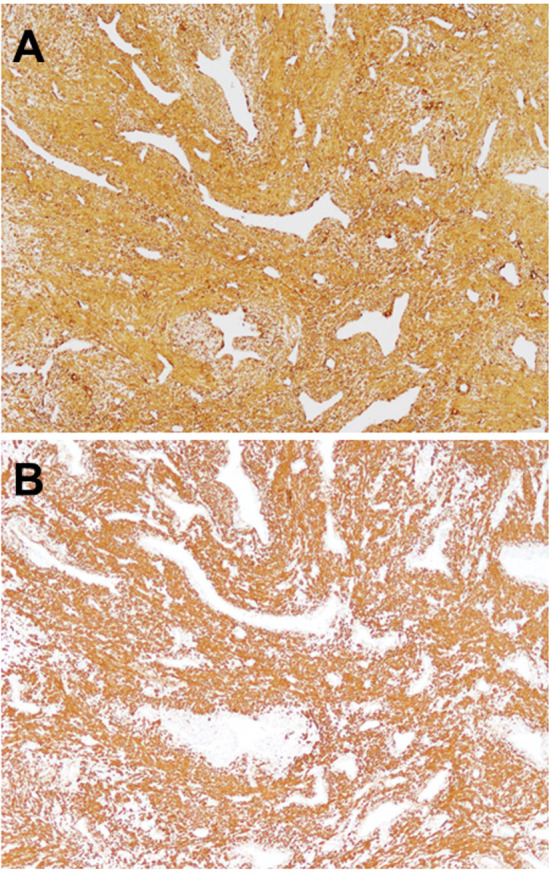
Grossly, angioleiomyoma is an ovoid, well-circumscribed, rubbery nodule with a gray-white or brown cut surface. The lesion with calcification can appear gritty on sectioning (Figure 2). An attached blood vessel wall may be identified (1). Histologically, the lesion is composed of well-differentiated smooth muscle cells with intervening vascular channels (Figure 3A). In addition, adipocytic metaplasia, hyalinization and calcification may be seen (Figure 3B). Mitotic figures are absent. Based on vascular morphologies, angioleiomyoma is subclassified into the three histological subtypes: solid, venous, and cavernous (1). The solid type has closely compacted smooth muscle cells with intervening thin-walled, slit-like vascular channels. The venous type shows thick muscle-coated blood vessels with intervascular smooth muscle bundles. The cavernous type is characterized by delated vascular channels with attenuated walls of smooth muscle cells. In the largest series of 562 cases of angioleiomyoma, Hachisuga et al. reported that the solid type was the most common (66%), followed by the venous (23%) and cavernous (11%) types (2). Immunohistochemically, the tumor cells are diffusely positive for smooth muscle actin (SMA) (Figure 4A), muscle-specific actin (MSA) and calponin and variably for h-caldesmon (Figure 4B) and desmin (14). Immunostainings for S-100 protein and HMB45 are typically negative.
Figure 2. Gross appearance of angioleiomyoma showing a wellcircumscribed mass with a gray-white, gritty cut surface.
Figure 3. Histological features of angioleiomyoma. (A) The tumor is composed of well-differentiated smooth muscle cells with intervening vascular channels. (B) Calcification can be seen. (hematoxylin and eosin staining, original magnification ×100).
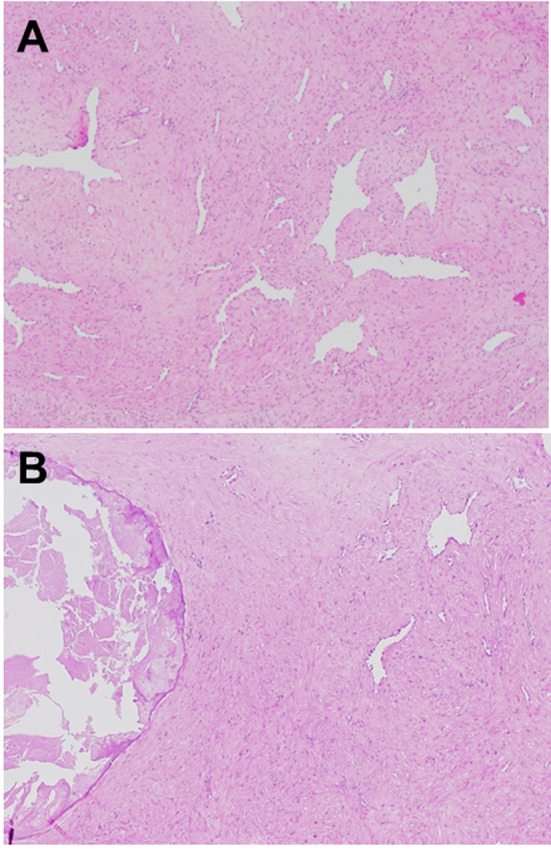
Figure 4. Immunohistochemical features of angioleiomyoma. The tumor cells are diffusely positive for smooth muscle actin (A) and h-caldesmon (B) (original magnification ×100).
Cytogenetic and Molecular Genetic Features
Only six cases of angioleiomyoma have been cytogenetically characterized in the literature (15-19) (Table I). Angio-leiomyoma displays mostly simple karyotypes characterized by one or few chromosomal rearrangements or numerical aberrations. Previously, we identified the presence of DNA copy number changes involving one or two chromosomes in 8 (35%) of 23 cases of angioleiomyoma (20). The most common recurrent change was a loss of 22q11. Recurrent gain of Xq was also observed. Recently, Notch Receptor 2 (NOTCH2) gene rearrangements have been detected in a very small subset of angioleiomyomas (21). Further studies are needed to better understand the correlation between certain genomic alterations and distinct biological behavior.
Table I. Chromosomal changes in angioleiomyoma.

F: Female; M: male.
Differential Diagnosis
The differential diagnosis for angioleiomyoma is broad and includes both benign and malignant entities such as schwannoma, glomus tumor and synovial sarcoma (11,22). In our opinion and experience, the most significant differential diagnosis is synovial sarcoma, and its small, well-circumscribed mass may give the wrong impression of a benign lesion by clinical examination and imaging.
Schwannoma is an almost invariably slow-growing neoplasm that may present as a painful mass, often less than 5 cm in size. It can occur at any age but has a peak incidence in the fourth and sixth decades of life (23). Radiographs are frequently normal. In rare cases, a fusiform soft-tissue mass with surrounding fat may be seen. Calcification is uncommon. Ultrasound typically shows a well-defined hypoechoic mass with variable posterior acoustic enhancement (24). An entering and exiting nerve sign and a polar blood supply sign on color doppler flow imaging can be seen (25). Ogata et al. reported that several ultrasonographic features including maximum diameter, tumor localization and continuity with the nerve were significantly different between schwannoma and angioleiomyoma (26). On MRI, the lesion usually displays intermediate or low signal intensity on T1-weighted images and high signal intensity on T2-weighted images. A target sign is one of the characteristic imaging features of schwannoma (27) and may be helpful to distinguish it from angioleiomyoma (28). A fascicular sign and a split-fat sign may also be observed. The histological hallmark of schwannoma is the pattern of alternating Antoni A and B areas. Immunohistochemically, the tumor cells are diffusely positive for S-100 protein (23).
Glomus tumor is a rare pericytic neoplasm that typically presents as a small, red-blue, painful nodule in the distal extremities, particularly the subungual region. It occurs in all age groups but more often in young adults (29). Radiographs may be normal or reveal a small soft-tissue mass related to the nail bed. Extrinsic erosion of the bone, often with a sclerotic margin, may be seen in subungual glomus tumor (30). Ultrasound usually reveals a solid, well-defined hypoechoic mass with prominent vascularity on color doppler examination (31,32). On MRI, the lesion displays intermediate or low signal intensity on T1-weighted images and high signal intensity on T2-weighted images (32). Contrast-enhanced MRI demonstrates prominent homogeneous enhancement (32,33). Histologically, the lesion consists of uniform, round cells with sharp cellular borders. Immunohistochemically, the tumor cells are positive for SMA, MSA and h-caldesmon (29). In 2015, we reported the first case of glomus tumor with a t(1;5)(p13;q32) translocation (33). The discovery of NOTCH1/2/3-MicroRNA 143 (MIR143) fusion genes has recently led to more precise diagnosis of glomus tumor (21).
Synovial sarcoma is a malignant mesenchymal neoplasm that usually presents as a slow-growing, painful mass in the extremities, particularly around the knee. The duration of symptoms is quite variable. It most commonly affects adolescents and young adults (34). Radiographs may be normal or reveal a well-defined or lobulated soft-tissue mass. Calcification, often eccentrical or at the periphery of the lesion, is seen in one-third of cases (35). Adjacent bone involvement (periosteal reaction, osseous remodeling, or frank bony invasion) may be seen. CT shows a heterogeneous soft-tissue mass with tissue attenuation similar to or slightly lower than that of skeletal muscle. Contrast-enhanced CT reveals heterogeneous or nodular enhancement in almost all cases (36). Ultrasound reveals a focal, nodular, round, or lobulated, solid but hypoechoic soft-tissue mass suggestive of a more indolent, less aggressive process (37). On MRI, the lesion is relatively well-defined and appears isointense or slightly hyperintense relative to skeletal muscle on T1-weighted images and displays prominent heterogeneous signal intensity on T2-weighted images. A triple signal pattern on T2-weighted images is often seen (38). Contrast-enhanced MRI typically demonstrates prominent heterogeneous enhancement (36). However, small lesions (less than 5 cm) may have a predominantly homogeneous appearance on all MR images (39). Histologically, synovial sarcoma can be classified as monophasic or biphasic. Monophasic synovial sarcoma is composed of fascicles of relatively uniform spindle cells with sparse cytoplasm and hyperchromatic nuclei with regular granular chromatin and inconspicuous nucleoli. Biphasic synovial sarcoma has epithelial and spindle cell components (40). Immunohistochemically, the tumor cells are positive for epithelial membrane antigen (EMA) and cytokeratins. A large majority of synovial sarcomas are positive for BCL2 and CD99. Notably, transducin-like enhancer of split 1 (TLE1) staining is highly sensitive for synovial sarcoma (34). Synovial sarcoma is characterized by the presence of a unique t(X;18)(p11;q11) translocation, resulting in a fusion of SS18 subunit of BAF chromatin remodeling complex (SS18) (18q11) and SSX family member (SSX) (Xp11) (34). Identification of the SS18-SSX fusion would be helpful diagnostically for synovial sarcoma in more difficult cases.
Conclusion
Angioleiomyoma is a distinct, benign, pericytic neoplasm and simple excision is usually curative. A diagnosis of angioleiomyoma can be formed on a routine microscopic assessment in a typical clinical setting. Angioleiomyoma should be considered a possible diagnosis when a well-demarcated oval or round subcutaneous mass with T1-isointense–to–slightly high signal, T2-high signal intensity, hypointense rim and intense enhancement is seen in the soft tissue of the extremities. Rearrangements of 22q and Xq are prominent in this peculiar neoplasm. Further studies are required to determine the biological consequences of these genomic alterations in angioleiomyoma.
Conflicts of Interest
The Authors declare no conflicts of interest associated with this article.
Authors’ Contributions
MK collected the data, searched the literature, and drafted the article. JN was a major contributor to writing the article. KK performed the histopathological evaluations. TK, SN and TY reviewed the article. All Authors read and approved the final article.
Acknowledgements
This study was supported in part by the Japan Society for the Promotion of Science KAKENHI (21K09336).
References
- 1.Matsuyama A. Lyon, IARC Press. 2020. Angioleiomyoma. In: World Health Organization Classification of Tumours: Soft Tissue and Bone Tumours. p. pp. 186. [Google Scholar]
- 2.Hachisuga T, Hashimoto H, Enjoji M. Angioleiomyoma. A clinicopathologic reappraisal of 562 cases. Cancer. 1984;54(1):126–130. doi: 10.1002/1097-0142(19840701)54:1<126::aid-cncr2820540125>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 3.Yeung CM, Moore L, Lans J, Lozano-Calderón L. Angioleiomyoma of the hand: a case series and review of the literature. Arch Bone Jt Surg. 2020;8(3):373–377. doi: 10.22038/ABJS.2019.14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kacerovska D, Michal M, Kreuzberg B, Mukensnabl P, Kazakov DV. Acral calcified vascular leiomyoma of the skin: a rare clinicopathological variant of cutaneous vascular leiomyomas: report of 3 cases. J Am Acad Dermatol. 2008;59(6):1000–1004. doi: 10.1016/j.jaad.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 5.Blalock TW, Kerr C, DeRienzo DP, Greenway HT. Rare case of acral calcified angioleiomyoma with macroscopic calcium extrusion. Foot Ankle Surg. 2015;21(2):e36–e39. doi: 10.1016/j.fas.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 6.Suárez-Peñaranda JM, Pita da Veiga G, Pérez-Muñoz N, Fernández-Figueras MT. Acral calcified vascular leiomyoma: Report of 3 cases and literature review. Am J Dermatopathol. 2021;43(10):732–735. doi: 10.1097/DAD.0000000000001773. [DOI] [PubMed] [Google Scholar]
- 7.Kang BS, Shim HS, Kim JH, Kim YM, Bang M, Lim S, Park GM, Lee TY, Ha ND, Kwon WJ. Angioleiomyoma of the extremities: Findings on ultrasonography and magnetic resonance imaging. J Ultrasound Med. 2019;38(5):1201–1208. doi: 10.1002/jum.14798. [DOI] [PubMed] [Google Scholar]
- 8.Kim DG, Lee SJ, Choo HJ, Kim SK, Cha JG, Park HJ, Kwon JW, Kim TE, Jung SJ. Ultrasonographic findings of subcutaneous angioleiomyomas in the extremities based on pathologic subtypes. Korean J Radiol. 2018;19(4):752–757. doi: 10.3348/kjr.2018.19.4.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gupte C, Butt SH, Tirabosco R, Saifuddin A. Angioleiomyoma: magnetic resonance imaging features in ten cases. Skeletal Radiol. 2008;37(11):1003–1009. doi: 10.1007/s00256-008-0518-4. [DOI] [PubMed] [Google Scholar]
- 10.Yoo HJ, Choi JA, Chung JH, Oh JH, Lee GK, Choi JY, Hong SH, Kang HS. Angioleiomyoma in soft tissue of extremities: MRI findings. AJR Am J Roentgenol. 2009;192(6):W291–W294. doi: 10.2214/AJR.07.3952. [DOI] [PubMed] [Google Scholar]
- 11.Edo H, Matsunaga A, Matsukuma S, Mikoshi A, Susa M, Horiuchi K, Shinmoto H. Angioleiomyoma of the extremities: correlation of magnetic resonance imaging with histopathological findings in 25 cases. Skeletal Radiol. 2022;51(4):837–848. doi: 10.1007/s00256-021-03888-4. [DOI] [PubMed] [Google Scholar]
- 12.Nishio J, Aoki M, Tanaka Y, Iwasaki H, Naito M. Painless angioleiomyoma of the first web space of the hand. In Vivo. 2013;27(4):519–522. [PubMed] [Google Scholar]
- 13.Lim ST, Kim MW, Sohn MH. Tc-99m RBC perfusion and blood-pool scintigraphy in the evaluation of vascular leiomyoma of the hand. Ann Nucl Med. 2002;16(4):293–296. doi: 10.1007/BF03000110. [DOI] [PubMed] [Google Scholar]
- 14.Matsuyama A, Hisaoka M, Hashimoto H. Angioleiomyoma: a clinicopathologic and immunohistochemical reappraisal with special reference to the correlation with myopericytoma. Hum Pathol. 2007;38(4):645–651. doi: 10.1016/j.humpath.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 15.Heim S, Mandahl N, Kristoffersson U, Mitelman F, Rööser B, Rydholm A, Willén H. Structural chromosome aberrations in a case of angioleiomyoma. Cancer Genet Cytogenet. 1986;20(3-4):325–330. doi: 10.1016/0165-4608(86)90091-9. [DOI] [PubMed] [Google Scholar]
- 16.Nilbert M, Mandahl N, Heim S, Rydholm A, Willén H, Mitelman F. Cytogenetic abnormalities in an angioleiomyoma. Cancer Genet Cytogenet. 1989;37(1):61–64. doi: 10.1016/0165-4608(89)90075-7. [DOI] [PubMed] [Google Scholar]
- 17.Sonobe H, Ohtsuki Y, Mizobuchi H, Toda M, Shimizu K. An angiomyoma with t(X;10)(q22;q23.2) Cancer Genet Cytogenet. 1996;90(1):54–56. doi: 10.1016/0165-4608(96)00070-2. [DOI] [PubMed] [Google Scholar]
- 18.Hennig Y, Caselitz J, Stern C, Bartnitzke S, Bullerdiek J. Karyotype evolution in a case of uterine angioleiomyoma. Cancer Genet Cytogenet. 1999;108(1):79–80. doi: 10.1016/s0165-4608(98)00123-x. [DOI] [PubMed] [Google Scholar]
- 19.Welborn J, Fenner S, Parks R. Angioleiomyoma: a benign tumor with karyotypic aberrations. Cancer Genet Cytogenet. 2010;199(2):147–148. doi: 10.1016/j.cancergencyto.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 20.Nishio J, Iwasaki H, Ohjimi Y, Ishiguro M, Kobayashi K, Nabeshima K, Naito M, Kikuchi M. Chromosomal imbalances in angioleiomyomas by comparative genomic hybridization. Int J Mol Med. 2004;13(1):13–16. [PubMed] [Google Scholar]
- 21.Agaram NP, Zhang L, Jungbluth AA, Dickson BC, Antonescu CR. A molecular reappraisal of glomus tumors and related pericytic neoplasms with emphasis on NOTCH-gene fusions. Am J Surg Pathol. 2020;44(11):1556–1562. doi: 10.1097/PAS.0000000000001531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pham K, Ezuddin NS, Pretell-Mazzini J, Subhawong TK. Small soft tissue masses indeterminate at imaging: histological diagnoses at a tertiary orthopedic oncology clinic. Skeletal Radiol. 2019;48(10):1555–1563. doi: 10.1007/s00256-019-03205-0. [DOI] [PubMed] [Google Scholar]
- 23.Perry A, Jo VY. Lyon, IARC Press. 2020. Schwannoma. In: World Health Organization Classification of Tumours: Soft Tissue and Bone Tumours; p. pp. 226. [Google Scholar]
- 24.Reynolds DL Jr, Jacobson JA, Inampudi P, Jamadar DA, Ebrahim FS, Hayes CW. Sonographic characteristics of peripheral nerve sheath tumors. AJR Am J Roentgenol. 2004;182(3):741–744. doi: 10.2214/ajr.182.3.1820741. [DOI] [PubMed] [Google Scholar]
- 25.Yuan Y, Gao J, Xiong G, Guo L. Diagnostic accuracy of multiparametric ultrasound for peripheral nerve schwannoma. Acta. 2022;Radiol:2841851221125109. doi: 10.1177/02841851221125109. [DOI] [PubMed] [Google Scholar]
- 26.Ogata D, Takeji M, Murakami T, Yanagisawa H, Kuramochi A, Tsuchida T. Comparison of ultrasonographic findings of schwannomas and angioleiomyomas. J Dermatol. 2018;45(7):837–843. doi: 10.1111/1346-8138.14358. [DOI] [PubMed] [Google Scholar]
- 27.Koga H, Matsumoto S, Manabe J, Tanizawa T, Kawaguchi N. Definition of the target sign and its use for the diagnosis of schwannomas. Clin Orthop Relat Res. 2007;464:224–229. doi: 10.1097/BLO.0b013e3181583422. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto K, Nishio J, Yano S, Naito M. Solitary schwannoma of the sural nerve: An unusual clinical presentation. Exp Ther Med. 2014;7(1):90–92. doi: 10.3892/etm.2013.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Specht K, Antonescu CR. Lyon, IARC Press. 2020. Glomus tumor. In: World Health Organization Classification of Tumours: Soft Tissue and Bone Tumours; p. pp. 179. [Google Scholar]
- 30.Vandenberghe L, De Smet L. Subungual glomus tumours: a technical tip towards diagnosis on plain radiographs. Acta Orthop Belg. 2010;76(3):396–397. [PubMed] [Google Scholar]
- 31.Park HJ, Jeon YH, Kim SS, Lee SM, Kim WT, Park NH, Park SI, Hong HP, Rho MH. Gray-scale and color Doppler sonographic appearances of nonsubungual soft-tissue glomus tumors. J Clin Ultrasound. 2011;39(6):305–309. doi: 10.1002/jcu.20830. [DOI] [PubMed] [Google Scholar]
- 32.Glazebrook KN, Laundre BJ, Schiefer TK, Inwards CY. Imaging features of glomus tumors. Skeletal Radiol. 2011;40(7):855–862. doi: 10.1007/s00256-010-1067-1. [DOI] [PubMed] [Google Scholar]
- 33.Nishio J, Nabeshima K, Mori S, Naito M. Translocation (1;5) in a glomus tumor. Anticancer Res. 2015;35(11):6167–6170. [PubMed] [Google Scholar]
- 34.Suurmeijer AJH, Ladanyi M, Nielsen TO. Lyon, IARC Press. 2020. Synovial sarcoma. In: World Health Organization Classification of Tumours: Soft Tissue and Bone Tumours; p. pp. 290. [Google Scholar]
- 35.Gazendam AM, Popovic S, Munir S, Parasu N, Wilson D, Ghert M. Synovial sarcoma: a clinical review. Curr Oncol. 2021;28(3):1909–1920. doi: 10.3390/curroncol28030177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tateishi U, Hasegawa T, Beppu Y, Satake M, Moriyama N. Synovial sarcoma of the soft tissues: prognostic significance of imaging features. J Comput Assist Tomogr. 2004;28(1):140–148. doi: 10.1097/00004728-200401000-00024. [DOI] [PubMed] [Google Scholar]
- 37.Marzano L, Failoni S, Gallazzi M, Garbagna P. The role of diagnostic imaging in synovial sarcoma. Our experience. Radiol Med. 2004;107(5-6):533–540. [PubMed] [Google Scholar]
- 38.Jones BC, Sundaram M, Kransdorf MJ. Synovial sarcoma: MR imaging findings in 34 patients. AJR Am J Roentgenol. 1993;161(4):827–830. doi: 10.2214/ajr.161.4.8396848. [DOI] [PubMed] [Google Scholar]
- 39.Blacksin MF, Siegel JR, Benevenia J, Aisner SC. Synovial sarcoma: frequency of nonaggressive MR characteristics. J Comput Assist Tomogr. 1997;21(5):785–789. doi: 10.1097/00004728-199709000-00025. [DOI] [PubMed] [Google Scholar]
- 40.Nishio J, Iwasaki H, Ishiguro M, Ohjimi Y, Isayama T, Naito M, Kikuchi M. Identification of syt-ssx fusion transcripts in both epithelial and spindle cell components of biphasic synovial sarcoma in small tissue samples isolated by membrane-based laser microdissection. Virchows Arch. 2001;439(2):152–157. doi: 10.1007/s004280100428. [DOI] [PubMed] [Google Scholar]