

Abstract
Background
Known risk alleles for epithelial ovarian cancer (EOC) account for approximately 40% of the heritability for EOC. Copy number variants (CNVs) have not been investigated as EOC risk alleles in a large population cohort.
Methods
Single nucleotide polymorphism array data from 13 071 EOC cases and 17 306 controls of White European ancestry were used to identify CNVs associated with EOC risk using a rare admixture maximum likelihood test for gene burden and a by-probe ratio test. We performed enrichment analysis of CNVs at known EOC risk loci and functional biofeatures in ovarian cancer–related cell types.
Results
We identified statistically significant risk associations with CNVs at known EOC risk genes; BRCA1 (PEOC = 1.60E-21; OREOC = 8.24), RAD51C (Phigh-grade serous ovarian cancer [HGSOC] = 5.5E-4; odds ratio [OR]HGSOC = 5.74 del), and BRCA2 (PHGSOC = 7.0E-4; ORHGSOC = 3.31 deletion). Four suggestive associations (P < .001) were identified for rare CNVs. Risk-associated CNVs were enriched (P < .05) at known EOC risk loci identified by genome-wide association study. Noncoding CNVs were enriched in active promoters and insulators in EOC-related cell types.
Conclusions
CNVs in BRCA1 have been previously reported in smaller studies, but their observed frequency in this large population-based cohort, along with the CNVs observed at BRCA2 and RAD51C gene loci in EOC cases, suggests that these CNVs are potentially pathogenic and may contribute to the spectrum of disease-causing mutations in these genes. CNVs are likely to occur in a wider set of susceptibility regions, with potential implications for clinical genetic testing and disease prevention.
Epithelial ovarian cancer (EOC) has a complex genetic architecture. Genetic risk alleles include highly penetrant pathogenic mutations in the BRCA1 and BRCA2 genes (1); rare mutations in moderately penetrant risk genes including BRIP1, RAD51D, RAD51C, FANCM, and PALB2 (2-5); and many common, low-risk polymorphisms identified by genome-wide association studies (GWAS) (6–17). The lifetime risk for ovarian cancer is 1.4% in the general population of the United States, however, this is greatly increased in carriers of deleterious mutations in BRCA1 and BRCA2 (44% and 17% lifetime risk, respectively) (18). The presence of a BRCA1 or BRCA2 mutation remains the strongest genetic risk factor for predicting a woman’s risk of EOC and is now routinely used to guide clinical interventions, including highly effective prevention by risk-reducing surgery. The genetic risk alleles for EOC identified so far account for approximately 40% of the heritability, suggesting there are many genetic risk alleles yet to be discovered (19).
The human genome harbors approximately 5000 to 10 000 structural variants (SVs), including deletions, duplications, insertions, and inversions, estimated to impact up to 13% of the human genome (20–22). By comparison, single nucleotide polymorphisms (SNPs) are estimated to affect approximately 0.1% of the human genome; thus, the estimated proportion of the human genome under structural variation is far higher than that due to SNPs. Despite this, copy number variants (CNVs; deletions and duplications) have not been analyzed at a similar scale as SNP variation, because of the cost of whole genome sequencing and technical challenges calling CNVs from genotyping arrays.
Previous studies have reported CNVs that contribute to the disease risk of other complex diseases such as breast cancer, pancreatic cancer, and diabetes (23–32). Similar extensive studies have not been performed in EOC cases, in part because of difficulty identifying large genotyped EOC case-control populations that can detect rare CNVs with sufficient power (33). Two previous genome-wide CNV analyses in approximately 1000 EOC cases and approximately 3000 unaffected controls failed to identify CNVs associated with disease risk or survival after multiple testing correction (34,35). In the current study, we have used genome-wide genotyping data from 13 071 EOC cases, including 8679 high-grade serous ovarian cancer (HGSOC) cases and 17 306 controls to identify CNVs throughout the genome and evaluate their associations with EOC risk.
Methods
Participants
The Ovarian Cancer Association Consortium (OCAC) collated and genotyped blood-derived DNA on the Illumina Infinium OncoArray as previously described (6). We selected 13 071 cases and 17 306 controls of White European ancestry from OCAC studies within countries with both cases and controls that passed genotyping quality control measures previously described (6). All participants signed an informed consent approved by the institutional review board of the recruiting institution. Demographics for these participants and their CNV distributions are listed in Supplementary Table 1 (available online) and Table 1.
Table 1.
Study participant age and copy number variant distributionsa
| Statistical category | Controls | EOC cases | HGSOC cases | P EOC | P HGSOC |
|---|---|---|---|---|---|
| (n = 17 306) | (n = 13 071) | (n = 8679) | |||
| Mean age (range) | 56.1 (18-97) | 58.9 (16-93) | 60.1 (18-93) | <.001 | <.001 |
| CNV segments, No. (%) | |||||
| All | 91 674 | 69 056 | 46 831 | — | — |
| Deletions | 46 637 (50.9) | 35 165 (50.9) | 23 900 (51.0) | — | — |
| Duplications | 45 037 (49.1) | 33 891 (49.1) | 22 931 (49.0) | — | — |
| CNV segments, mean | |||||
| All | 5.3 | 5.3 | 5.3 | .59 | .68 |
| Deletions | 2.7 | 2.7 | 2.7 | .78 | .83 |
| Duplications | 2.6 | 2.6 | 2.6 | .63 | .44 |
| Median CNV length, kb | |||||
| All | 22.2 | 21.9 | 21.4 | .57 | .91 |
| Deletions | 13.7 | 13.4 | 13.5 | .68 | .77 |
| Duplications | 37.1 | 36.7 | 36.1 | .64 | .87 |
| Mean CNV length, kb | |||||
| All | 67.4 | 67.8 | 67.3 | .57 | .91 |
| Deletions | 44.6 | 44.9 | 44.8 | .68 | .77 |
| Duplications | 91.0 | 91.6 | 90.8 | .64 | .87 |
CNV = copy number variant; EOC = epithelial ovarian cancer; HGSOC = high-grade serous ovarian cancer.
CNV Calling Method
The CamCNV pipeline was used to call rare CNVs from the log R ratio (LRR) intensity measurements for each OncoArray probe (36). Principal component analysis adjustment was applied to the LRR for each OCAC study to mitigate the impact of technical batch effects. We excluded outlier probes based on LRR residual. Remaining CNV calls after additional quality control exclusions (see Supplementary Methods, available online) were lifted into hg38 from hg19 for downstream analysis, using University of California, Santa Cruz, Genome Browser liftOver.
Rare CNVs Association Analysis
A likelihood ratio test was performed to test for association with deletions or duplications at each probe where CNVs were observed in at least 0.05% of samples. For downstream enrichment analysis, we used probes covered by at least 5 CNVs (20 981 probes with deletions only, 30 917 with duplications only, and 5515 with deletions and duplications). Individual copy number variants were assigned the minimum P value of any probes they overlapped.
Gene burden analysis was performed by assigning probes to a single protein coding gene in the University of California, Santa Cruz, Genome Browser’s knownGene table. Gene burden analyses were performed using the rare admixture maximum likelihood test (RAML) on genes with at least 5 samples carrying a CNV (37). The Bonferroni correction based on the number of genes tested in RAML for deletions, duplications, and both types of CNVs combined was a P value less than 6.37E-6 for all EOC and a P value less than 7.07E-6 for HGSOC. Because the smallest exact P value in our RAML analysis was a P value less than 1.0E-6, we additionally performed a binomial test using the frequencies of CNVs in cases and controls within BRCA1 to obtain a more precise P value.
GWAS and Transcriptome-Wide Association Study Enrichment Analysis
Enrichment of CNVs at known EOC risk loci was performed at known genome-wide statistically significant loci from the most recent GWAS of EOC and HGSOC (Coetzee S, Dareng EO, Peng P-C, Rosenow W, Tyrer JP, Chen S, et al, In Review) and genes identified by transcriptome-wide association (TWAS) studies by Gusev et al. (38) and Lu et al. (39). Analysis was performed twice; with CNVs overlapping BRCA1 included and then excluded. Genes at GWAS genome-wide statistically significant loci and TWAS genes (n = 37) were mapped to linkage disequilibrium (LD) blocks from the 1000 Genomes (1000G) European subpopulation (40) and CNVs intersecting these LD blocks retained for analysis. Enrichment was performed using a foreground of CNVs associated with EOC containing 1 or more probes with a P value less than .05 in association analyses. The foreground was used to generate a 1000-fold randomly selected background. Enrichment analysis was performed in R using FunciVar (41,42).
Functional Annotation and Noncoding Enrichment Analysis
Functional biofeatures for 18 cell lines related to ovarian cancer or with shared biological features of candidate precursor cell types (Supplementary Table 3, available online) were collated for enrichment analysis. Individual samples were processed and analyzed as previously described (42,43) and are described in detail in the Supplementary Methods (available online). Enrichment was performed with FunciVar (42), using a foreground of noncoding CNVs associated with EOC and/or HGSOC and a 1000-fold randomly selected set of regions as a background. Using these 2 lists, FunciVar then intersects each variant with functional annotations, which in this analysis were our ChromHMM states lifted into hg38. The significance of results is reported as probability that foreground variants have more overlaps with the functional annotation than background regions.
Common CNVs Tagged by SNPs
A list of SNPs in high LD (≥0.8) with common CNVs identified in 1000G (44) were looked up in the most recent EOC and HGSOC GWAS (Coetzee S, Dareng EO, Peng P-C, Rosenow W, Tyrer JP, Chen S, et al, In Review). We applied a Bonferroni threshold to identify CNV-tagging SNPs that were statistically significantly associated within the nonmucinous EOC GWAS analysis, which includes all invasive subtypes except for the mucinous histotype. SNPs were considered statistically significant with a P value less than 2.09E-6.
Results
Rare CNVs at Known EOC Susceptibility Gene Loci
We identified 160 730 CNV segments, with an average of 5.3 CNVs detected in each study participant. The median deletion size was 13.6 kb, and the median duplication size was 37.0 kb (Table 1). Rare CNVs retained for analysis ranged from 0.003% to 2.95% frequency (Table 1). More than 49% of deletions and 30% of duplications in our dataset overlapped (≥90% of length) with a rare CNV identified in women of European descent in the 1000G. Gene burden analysis was performed for all EOC cases and in HGSOC cases separately (Bonferroni corrected significance thresholds P ≤ 6.37E-6 and P ≤ 7.07E-6, respectively). In both analyses, the most statistically significant risk gene was BRCA1 (PEOC < 1.0E-6, odds ratio [OR]EOC = 8.24; PHGSOC < 1.0E-6, ORHGSOC = 7.29; Table 2; Supplementary Tables 4 and 5, available online). We identified 65 cases and 5 controls predicted to be hemizygous for a deletion, and 40 cases and 12 control participants with predicted duplications; 105 of 13 071 (0.80%) EOC cases, 93 of 8679 (1.1%) HGSOC cases, and 17 of 17 306 (0.098%) controls harbored a predicted deletion or duplication of BRCA1 (P = 1.60E-21). Deletions and duplications at the BRCA1 locus are illustrated in Figure 1, A. The most common CNV we found in BRCA1 is a duplication at exon 13, a known relatively common CNV also called BRCA1-ins6kbEx13 described in Mazoyer et al. (45). This duplication is found in 20 cases and 0 controls. The most common deletion in BRCA1 in our data is found in exon 22, where a common deletion is known in families from the Netherlands (46). This was found in 10 cases, 5 of which are from the Netherlands (2.3% of all Netherlands cases have this specific CNV). The most common CNV in BRCA2 was a previously reported (47) deletion of exons 14-16, found in 4 cases in our study.
Table 2.
Gene burden testing results for rare CNVs in all EOC or HGSOC cases with a P value less than .002
| Gene | No. of cases | No. of HGSOC | No. of controls | OR EOC | ORHGSOC | CNV typea | All cases | HGSOC cases |
|---|---|---|---|---|---|---|---|---|
| P | P | |||||||
| BRCA1 | 40 | 35 | 12 | 4.42 | 3.87 | Duplication | <1.00E-06 | <1.00E-06 |
| BRCA1 | 65 | 58 | 5 | 17.29 | 15.42 | Deletion | <1.00E-06 | <1.00E-06 |
| RAD51C | 17 | 14 | 4 | 5.63 | 4.64 | Both | 7.00E-04 | 4.33E-04 |
| RAD51C | 14 | 13 | 3 | 6.18 | 5.74 | Deletion | 6.00E-03 | 5.50E-04 |
| PRKACG | 0 | 0 | 15 | 0.00 | 0.00 | Duplication | 5.67E-04 | 9.00E-03 |
| BRCA2 | 13 | 10 | 4 | 4.31 | 3.31 | Deletion | 6.20E-03 | 7.00E-04 |
| FBLIM1 | 11 | 6 | 0 | NA | NA | Deletion | 8.50E-04 | 4.80E-03 |
| HAS3 | 12 | 10 | 5 | 3.18 | 2.65 | Duplication | 1.13E-02 | 1.20E-03 |
| ARHGAP24 | 12 | 9 | 3 | 5.30 | 3.97 | Both | 1.39E-02 | 1.40E-03 |
| LSP1 | 35 | 27 | 20 | 2.32 | 1.79 | Both | 2.41E-02 | 1.40E-03 |
| SNX29 | 8 | 5 | 1 | 10.60 | 6.62 | Duplication | 1.40E-03 | 7.70E-03 |
| PIP5K1B | 1 | 0 | 20 | 0.07 | 0.00 | Both | 1.53E-03 | 2.73E-02 |
| ALKBH4 | 6 | 2 | 0 | NA | NA | Duplication | 1.65E-03 | NA |
| LRWD1 | 6 | 2 | 0 | NA | NA | Duplication | 1.65E-03 | NA |
| LSP1 | 33 | 26 | 17 | 2.57 | 2.03 | Duplication | 2.31E-02 | 1.70E-03 |
| TTLL2 | 8 | 3 | 1 | 10.60 | 3.97 | Duplication | 1.80E-03 | 3.29E-02 |
| NAT1 | 8 | 6 | 2 | 5.30 | 3.97 | Deletion | 1.90E-03 | 6.00E-03 |
Combined duplications and deletions P value result included only if it was more statistically significant than deletions or duplications alone. CNV = copy number variant; EOC = epithelial ovarian cancer; HGSOC = high-grade serous ovarian cancer; OR = odds ratio; NA = Not Available.
Figure 1.

CNVs identified at the BRCA1, BRCA2, and RAD51C susceptibility gene risk loci in EOC cases and controls. CNVs of varying size predicting deletions (horizontal red bars) and duplications (horizontal blue bars) in EOC cases (solid bars) and controls (faint bars) at the (A) BRCA1, (B) BRCA2, and (C) RAD51C gene loci. The location of all probes genotyped on the Illumina OncoArray and used to “call” copy number variations are shown as vertical blue lines. CNV = copy number variants.
We found evidence of CNV EOC risk associations spanning 2 additional known ovarian cancer susceptibility gene regions: RAD51C (PEOC = 7.0E-4, OREOC = 5.63; PHGSOC = 4.33E-4, ORHGSOC = 4.64) and BRCA2 (deletions only; PEOC = 0.0062, OREOC = 4.31; PHGSOC = 7.0E-4, ORHGSOC = 3.31; Table 2). Risk associations were stronger in HGSOC, consistent with previous studies of these genes (Table 2; Figure 1, B and C) (48,49). In addition, we found evidence of association for 12 genes not previously associated with EOC risk (P < .002; Table 2) including PRKACG, a cAMP-dependent protein kinase catalytic subunit gamma at 9q21.11 associated with a decreased risk in all EOC cases (P < EOC = 5.67E-4, OREOC = 0); the filamin-binding LIM protein 1 (FBLIM1) gene locus at 1p36.21 associated with increased risk in all EOC cases (PEOC = 8.50E-4, OREOC = Not Available [NA]); and ARHGAP24 at 4q21.23, where both deletions and duplication were associated with an increased risk for HGSOC (P = .00140, ORHGSOC = 3.97; Table 2).
Rare CNV Association Analysis
To detect associations with individual CNVs, we restricted analyses to probes intersecting deletions or duplications with a frequency of at least 0.05% of samples (n = 16 for EOC, n = 13 for HGSOC). There were 6882 probes with deletions, and 9778 probes with duplications were analyzed. We identified 16 CNVs associated with risk for EOC or HGSOC (Table 3; Figure 2). Some individual deletions and duplications within BRCA1 are frequent enough to appear in this analysis, and they are the only CNVs with P values below a significance threshold corrected for multiple testing. Outside of the BRCA1 locus, the most statistically significant deletion falls within the long noncoding LINC01194 also known as Cancer Testis Antigen 49 (n = 137; P = .0007). The strongest novel duplication result (n = 90; P = .0003) falls within the seventh intron of the DCDC2 gene.
Table 3.
CNVs statistically significantly associated with EOC and HGSOC with a P value less than .005
| Chr | CNV region start | CNV region end | Type | No. sig probes | Probe location | OREOC | P EOC | EOC Carrier count | ORHGSOC | P HGSOC | HGSOC carrier count |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 17 | 43 080 276 | 43 082 575 | Duplication | 7 | BRCA1 coding | NAa | 9.72E-10 | 26 | NAa | 1.02E-11 | 23 |
| 17 | 43 049 093 | 43 125 836 | Deletion | 6 | BRCA1 coding | 28.6 | 2.57E-07 | 20 | 33.22 | 5.38E-07 | 16 |
| 6 | 24 221 271 | 24 221 660 | Duplication | 3 | DCDC2 intronic | 0.45 | 3.65E-04 | 90 | 0.53 | 9.69E-03 | 84 |
| 5 | 12 692 574 | 12 726 378 | Deletion | 7 | LINC01194 LncRNA | 0.53 | 7.22E-04 | 137 | 0.59 | 9.90E-03 | 126 |
| 9 | 69 064 550 | 69 225 129 | Duplication | 6 | FXN, TJP2 coding | 0.19 | 1.48E-03 | 24 | 0.19 | 5.51E-03 | 23 |
| 17 | 1 197 175 | 1 198 288 | Deletion | 4 | ABR intronic | 0.62 | 2.50E-03 | 188 | 0.63 | 9.71E-03 | 168 |
| 9 | 116 713 991 | 116 729 732 | Deletion | 4 | ASTN2 coding | 3.53 | 2.55E-03 | 26 | 3.36 | 9.74E-03 | 19 |
| 23 | 67 910 806 | 67 923 215 | Duplication | 3 | Intergenic | 2.7 | 3.38E-03 | 40 | 2.32 | 3.07E-02 | 30 |
| 11 | 1 841 637 | 1 886 457 | Duplication | 5 | LSP1 intronic | 2.46 | 4.66E-03 | 44 | 3.1 | 7.68E-04 | 31 |
| 2 | 50 669 275 | 50 697 220 | Deletion | 5 | NRXN1 intronic | 2.93 | 5.14E-03 | 30 | 3.32 | 3.74E-03 | 25 |
| 12 | 115 341 220 | 115 341 234 | Deletion | 3 | Intergenic | 0.3 | 6.58E-03 | 28 | 0.18 | 3.11E-03 | 25 |
| 10 | 82 776 430 | 82 797 323 | Deletion | 4 | NRG3 intronic | 3.91 | 7.09E-03 | 21 | 4.44 | 4.33E-03 | 18 |
| 9 | 12 350 523 | 12 443 586 | Deletion | 11 | Intergenic | 2.33 | 1.14E-02 | 40 | 3.01 | 1.68E-03 | 36 |
| 23 | 36 457 791 | 36 541 981 | Duplication | 4 | Intergenic | 0.38 | 1.23E-02 | 34 | 0.15 | 8.57E-04 | 28 |
| 6 | 168 031 413 | 168 196 562 | Duplication | 37 | KIF25, FRMD1 | 0.85 | 3.81E-02 | 753 | 0.76 | 1.93E-03 | 628 |
| 1 | 195 862 228 | 195 905 802 | Deletion | 10 | Intergenic | 1.43 | 5.66E-02 | 119 | 1.83 | 2.18E-03 | 110 |
NA: This deletion was only observed in cases; odds ratio (OR) could not be calculated. CNV = copy number variant; EOC = epithelial ovarian cancer; HGSOC = high-grade serous ovarian cancer.
Figure 2.

A) Manhattan plots showing the results of single-probe CNV association testing. At a Bonferroni P value cutoff (blue line) of P < 8.71E-7 for the all EOC cases (based on 57 432 tests) and P < 8.56E-7 for HGSOC cases only (based on 58 382 tests) identified statistically significant probes at the BRCA1 gene locus. Evidence of several additional risk associations with a Bonferroni P value cutoff of P < 5E-4, including associations at intergenic sites, are also shown. B) Manhattan plot displaying results of common CNV analysis. At a Bonferroni P value cutoff of P < 2.09E-6 based on 23 960 tag SNPs included in the lookup, we identified statistically significant SNPs at 4 loci. At these loci, there are common CNVs in high linkage disequilibrium with GWAS SNPs that may account for some of the variation leading to differences in risk at that SNP. CNV = copy number variants; EOC = epithelial ovarian cancer; GWAS = genome-wide association studies; HGSOC = high-grade serous ovarian cancer; SNP = single nucleotide variants.
CNV Enrichment at Risk Loci Identified by GWAS and TWAS
Statistically significant CNVs intersected LD blocks at EOC GWAS risk regions at 7 of 27 EOC risk loci, even when BRCA1 CNVs were excluded (P < .05; Table 4; Supplementary Tables 8 and 9, available online). HGSOC histotype-specific GWAS regions were also statistically significantly enriched for risk CNVs; statistically significant CNVs intersected 12 of 30 loci (P < .05; Table 4; Supplementary Table 8, available online). GWAS risk regions with a statistically significant enrichment for CNVs intersected both known and potentially novel causal genes. CNVs identified in all EOC cases were statistically significantly enriched within the bodies of TWAS genes in EOC but not HGSOC (Supplementary Table 9, available online), and the regions defined by LD blocks around the same TWAS genes were not statistically significantly enriched for CNVs in either EOC or HGSOC.
Table 4.
GWAS loci with CNVs associated with EOC or HGSOC risk (P < .05)
| Cytoband | rsID | GWAS P | LD block start | LD block end |
|---|---|---|---|---|
| NMOC loci | ||||
| 3q28 | rs9869209 | 6.61E-09 | 190508818 | 192626025 |
| 5p15.33 | rs4449583 | 2.76E-21 | 982137 | 2132328 |
| 10p12.31 | rs7084454 | 1.86E-12 | 19427949 | 22483354 |
| 17q21.31a | rs146596949 | 1.26E-51 | 41743558 | 43694719 |
| 17q21.31a | rs575499584 | 4.12E-18 | 44979537 | 47798656 |
| 17q21.32 | rs12946636 | 9.92E-25 | 47798656 | 49440038 |
| 19p13.11 | rs4808075 | 5.76E-26 | 16263605 | 18299052 |
| HGSOC associated loci | ||||
| 4q13.2/4q13.3 | rs4149419 | 2.66E-08 | 67989047 | 70183435 |
| 5p15.33 | rs4449583 | 1.09E-19 | 982137 | 2132328 |
| 9q31.1 | rs2122577 | 2.94E-09 | 103205694 | 104819468 |
| 9p22.1/9p21.3 | rs7851336 | 2.54E-10 | 18661053 | 20463536 |
| 10p12.31 | rs7084454 | 1.48E-09 | 19427949 | 22483354 |
| 13q13.1 | rs11571815 | 4.32E-09 | 31727678 | 33202766 |
| 15q26.1 | rs76119208 | 3.36E-09 | 89932319 | 91621162 |
| 17q12 | rs11657964 | 2.63E-12 | 36141651 | 38653091 |
| 17q21.31 | rs146596949 | 3.11E-56 | 41743558 | 43694719 |
| 17q21.31 | rs575499584 | 1.69E-19 | 44979537 | 47798656 |
| 17q21.32 | rs12946636 | 5.54E-19 | 47798656 | 49440038 |
| 19p13.11 | rs56069439 | 8.41E-38 | 16263605 | 18299052 |
BRCA1 locus. CNV = copy number variants; GWAS = genome-wide association analysis; LD = linkage disequilibrium; HGSOC = high-grade serous ovarian cancer; NMOC = all nonmucinous ovarian cancer.
EOC Risk Associations for Common CNVs
To identify common CNVs associated with EOC and HGSOC risk, we used tag SNPs (r2 > 0.8) for common CNVs in participants of European descent (>1% frequency). We evaluated 23 960 SNPs tagging 3681 CNVs in the largest GWAS dataset for EOC and HGSOC risk (Supplementary Table 10, available online). We identified 4 statistically significant SNPs tagging 4 CNVs at 2 loci (P < 2.09E-6) in both the EOC (excludes mucinous EOC) and the HGSOC histotype–specific GWAS (Figure 2, Table 5). At 9p22.2, the risk-associated CNV lies between BNC2 and CNTLN, intersecting the promoter of a long noncoding RNA and the previously identified risk SNPs for EOC (50). The CNVs at 17q21.31 are within a common inversion polymorphism also associated with a microdeletion syndrome and predicted to disrupt LINC02210-CRHR1, MAPT, and KANSL1 (51,52).
Table 5.
HRC tagSNPs in LD with known common CNVs from 1000G that are statistically significantly associated with EOC risk (hg38)a
| tagSNP | Chr | Position | Effect allele | Noneffect allele | CNV start | CNV end | CNV type | Length (bp) | P NMOC | No. sig tagSNPs | P HGSOC | No. sig tagSNPs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs10962691 | 9 | 16915107 | G | C | 16905594 | 16905924 | Deletion | 330 | 1.48E-50 | 3 | 4.38E-68 | 3 |
| rs17689104 | 17 | 45705126 | G | A | 45753354 | 45753478 | Deletion | 124 | 3.12E-12 | 152 | 9.59E-15 | 152 |
| rs17688922 | 17 | 45701985 | A | G | 45753354 | 45753478 | Deletion | 124 | 3.17E-12 | 152 | 9.55E-15 | 152 |
| rs8080583 | 17 | 46085231 | A | C | 46009357 | 46009595 | Deletion | 238 | 1.85E-12 | 179 | 2.32E-15 | 181 |
| rs8080583 | 17 | 46085231 | A | C | 46146541 | 46146855 | Deletion | 314 | 1.85E-12 | 179 | 2.32E-15 | 181 |
1000G = 1000 Genomes project; CNV = copy number variant; EOC = epithelial ovarian cancer; HGSOC = high-grade serous ovarian cancer; HRC = Haplotype Reference Consortium; LD = linkage disequilibrium; NMOC = all nonmucinous ovarian cancer; SNP = single nucleotide polymorphism.
CNVs Are Enriched in Active Regulatory Elements in Ovarian Cancer–Related Cell Types
We identified 1707 and 1948 CNVs within nonprotein-coding DNA regions associated with EOC or HGSOC risk, respectively (P < .05; Supplementary Tables 6 and 7, available online). We evaluated the enrichment of these CNVs in chromatin states (weak promoter, active promoter, active region, active enhancer, weak enhancer, insulator, and transcribed) mapped in 18 ovarian cancer–related cell types (Supplementary Table 3, available online) (Plummer JT, Dezem FS, Davis B, Chen S, Seo J-H, Giambartolomei C et al, In Review). We identified statistically significant enrichment of EOC risk CNVs in insulators and modest enrichment at weak promoters (Figure 3); depletion in active promoters; and enhancers (Supplementary Figure 1, Supplementary Table 11, available online). Restricting the analysis to HGSOC risk CNVs to HGSOC showed a similar pattern of enrichment (Supplementary Figure 2, available online).
Figure 3.
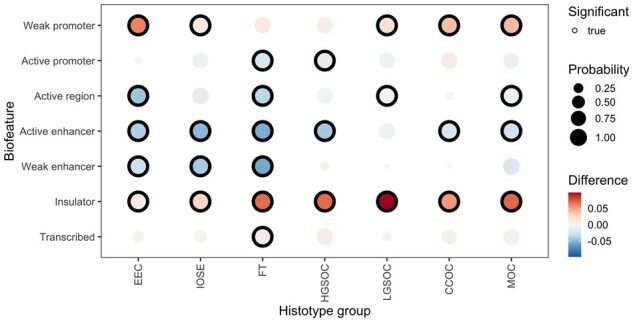
Enrichment of EOC statistically significant CNVs (P < .05) in functional biofeatures in ovarian cancer–related cell types. EOC risk CNVs are statistically significantly enriched in insulators across all ovarian cancer–relevant histotype consensus groups. The total number of risk CNVs in each biofeature per histotype grouping can be found in Supplementary Table 11 (available online). Abbreviations for histotypes are as follows: CCOC = clear cell ovarian cancer; EEC = endometriosis (precursor cell type); FT = fallopian tube secretory epithelial cells (precursor cell type); HGSOC = high-grade serous ovarian cancer; IOSE = immortalized ovarian surface epithelium (precursor cell type); LGSOC = low-grade serous ovarian cancer; MOC = mucinous ovarian cancer. CNV = copy number variants; EOC = epithelial ovarian cancer.
Discussion
In this study, we used genome-wide genotype array probe signal intensity data for more than 13 000 EOC cases and more than 17 000 controls to characterize CNVs and evaluate their associations with EOC and HGSOC risk. This study represents the largest to evaluate the contribution of CNVs to ovarian cancer risk performed to date. Two previous studies failed to find strong evidence of CNVs associated with EOC risk (34,35). Both prior studies focused on common CNVs (>1% frequency), whereas we focused on rare CNVs, and this, along with the large difference in sample size, likely contributed to the lack of replication. Using gene burden analyses, we identified highly statistically significant deletions and duplications at the BRCA1 gene locus and confirmed these findings using single probe association testing. We also found evidence of CNV risk associations at 2 other EOC susceptibility loci: RAD51C and BRCA2. A subset of EOC cases and controls included in this study have been previously sequenced to identify germline BRCA1 (n = 89), BRCA2 (n = 106), and RAD51C (n = 8) coding variants (48,49,53), and none of the 203 patients carrying a pathogenic mutation in any of these genes also harbored a predicted CNV in these genes. As single nucleotide variants (SNVs) and CNVs are rare in these genes, we expect patients with concurrent pathogenic SNVs and CNVs to be extremely rare. For all 3 loci, EOC risk estimates were stronger when we restricted the analyses to HGSOC cases only, consistent with previous studies indicating that mutations in these genes are more strongly associated with HGSOC.
Prior studies report pathogenic BRCA1 coding sequence mutations at a frequency of 5.3% in HGSOC (48), and we identified CNVs at the BRCA1 gene locus in 1.1% of HGSOC cases, suggesting CNVs represent a substantial contribution to the overall prevalence of BRCA1 mutations in HGSOC cases. Previous candidate studies identified pathogenic deletions and rearrangements involving BRCA1, BRCA2, and moderate-risk CNVs in high-risk hereditary breast and ovarian cancer (HBOC) families where a mutation was not identified in clinical testing (54–61), and we identified deletions and duplications overlapping previously reported CNVs, such as deletions in exon 2-9 of RAD51C or deletions in exons 14-16 of BRCA2 (55,57). BRCA2 CNV mutations are rarer than BRCA1 CNVs, however, they are still estimated to account for up to 8% of germline BRCA2 mutations (47,62–67). The contribution of CNVs to BRCA1 varies greatly depending on population, with CNVs being 3% of BRCA1 mutations in South African HBOC families (68) and 27%-36% of BRCA1 mutations in Dutch HBOC families (46,69). CNVs account for a smaller proportion of BRCA2 carriers comparatively, with a Danish study of HBOC families finding BRCA1 CNVs in 12.5% of all BRCA1 carriers but only 2% of BRCA2 carriers (62). Most estimates of contribution are from screening individuals in hereditary breast and ovarian cancer families rather than all ovarian cancer cases, as in our study, which may partially account for the fewer CNVs seen in our data. It is likely that BRCA2 and RAD51C contain clinically relevant CNVs but also that other moderate-risk genes with CNVs or structural variants would be found in a cohort with sufficient sample size and a sensitive detection method. It is more difficult to find estimates of CNV contribution to these genes in nonfamilial studies. In a study of 376 000 participants undergoing genetic testing, 12.7% of pathogenic variants in BRCA1, 1.9% of pathogenic variants in BRCA2, and 21.1% of pathogenic variants in RAD51C were large rearrangements (70). The percent of all ovarian cancer patients with a CNV vs SNV as their pathogenic mutation in these genes is not currently available.
CNV association analyses also identified novel candidate ovarian cancer susceptibility genes, including FBLIM1, HAS3, and LSP1. Germline whole-exome sequencing studies have previously implicated FBLIM1 as a putative susceptibility gene in HGSOC (71). The gene is differentially expressed between benign and malignant murine ovarian surface epithelial cells and is dysregulated in ovarian cancers (72,73). LSP1 is a candidate breast cancer susceptibility gene and may interact multiplicatively to increase breast cancer risk for BRCA2 mutation carriers (16,74–76), and the HAS3 gene may also be associated with the development of chemoresistant ovarian cancer (77,78).
Rare variant association analysis detected a number of suggestive associations for individual variants. Only the BRCA1 variants with large effect sizes (OR > 10) passed the multiple-testing P value threshold. If associations for rare CNVs are to be confirmed, a key question is the magnitude of effect sizes we should expect for CNVs outside the known genes. Sample size requirements scale linearly with decreasing minor allele frequence but quadratically for decreasing odds ratios (1/|[OR - 1]) (79). When compared with associations for common noncoding SNPs, the possible associations in this analysis have large odds ratios ranging from 0.15 to 0.76 and from 1.83 to 4.44. It is plausible that evolutionary younger rare variants not yet removed by negative selection can have a stronger biological effect than older common variants. There is also some evidence from sequencing studies that noncoding SVs such as CNVs are more likely to have a stronger biological effect than SNVs. For example, Abel et al. (80) calculated that each individual carried 122 rare variants (63% SNVs, 20% indels, 17% SVs) predicted to be deleterious, and given their relative frequency, SVs are 841-fold more likely to be deleterious than rare SNVs and 341-fold more than rare indels. We estimate that the probe coverage on the OncoArray allows us to detect up to 10% of the deletions and 25% of the duplications identified by the 1000G in the 0.05% to 1% frequency range in the European population.
The most statistically significantly risk-associated deletion impacts part of the long noncoding RNA LINC01194 (n = 137, OR = 0.53; P = .0007). There is some evidence for an oncogenic role for LINC01194 from expression analyses in colorectal tumors (81) and prostate tumors and cancer cell lines (82).The strongest duplication association was observed in an intron at the start of the DCDC2 gene (n = 90, OR = 0.45; P = .0004). Interestingly, the reverse strand of this gene encodes KAAG1, which has been identified as an antigen expressed on the surface of cancer cells in a high proportion of ovarian tumors (83). The strongest result in the HGSOC analysis is for a duplication covering the first exon of the LSP1 gene (n = 37, OR = 3.10; P = .0008) (71).
We observed enrichment of risk-associated CNVs at EOC risk loci identified by GWAS. A wide variety of genetic variation, including SNVs and CNVs at GWAS loci, may contribute cumulatively to observed signal through aggregation by LD, and CNV analysis may implicate candidate genes for further functional analysis. Most EOC-risk associated variants identified by GWAS lie in noncoding DNA regions. In our study, noncoding risk-associated CNVs were enriched in weak promoters and insulators (bound CTCF motifs), suggesting they mediate gene expression through their interaction with regulatory elements and the 3-dimensional structure of the genome. Studies have shown germline risk variants and CNVs altering CTCF sites underlie some human diseases (84,85).
We have used genome-wide analysis to identify rare CNVs associated with ovarian cancer risk, including at known EOC susceptibility gene loci BRCA1, BRCA2, and RAD51C. Given the frequency at which we detected these CNVs, it may be appropriate to expand the content of genetic risk assessment panels for breast and ovarian cancer to universally include coverage of CNVs at BRCA1, BRCA2, and RAD51C as likely pathogenic variants where such testing is not already standard (86). CNVs likely represent a missing fraction of heritability for ovarian cancer at known susceptibility genes and as independent risk variants. Evaluating the frequency of CNVs in larger EOC case-control populations and with whole genome sequencing on a population scale is warranted to improve our understanding of the genetic architecture for EOC.
Funding
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health (5T32GM118288-03); the CSMC Precision Health Initiative; and the Tell Every Amazing Lady About Ovarian Cancer Louisa M. McGregor Ovarian Cancer Foundation. Supported in part by the Ovarian Cancer Research Fund thanks to donations by the family and friends of Kathryn Sladek Smith (PPD/RPCI.07); the US National Cancer Institute GAME-ON Post-GWAS Initiative (U19-CA148112); the Wellcome Trust (076113; the National Cancer Institute and National Human Genome Research Institute (dbGap accession number phs000178.v8.p7); the U.S. National Institutes of Health (CA1X01HG007491-01 (CIA), U19-CA148112 (TAS), R01-CA149429 (CMP) and R01-CA058598 (MTG); Canadian Institutes of Health Research (MOP-86727 (LEK); the Ovarian Cancer Research Fund (AB); a European Commission’s Seventh Framework Programme grant (agreement number 223175 - HEALTH-F2-2009-223175); the U.S. Army Medical Research and Materiel Command (DAMD17-01-1-0729); National Health & Medical Research Council of Australia (199600, 400413 and 400281); Cancer Councils of New South Wales, Victoria, Queensland, South Australia and Tasmania and Cancer Foundation of Western Australia (Multi-State Applications 191, 211 and 182); Ovarian Cancer Australia and the Peter MacCallum Foundation; ELAN Funds of the University of Erlangen-Nuremberg; National Kankerplan; Breast Cancer Now, Institute of Cancer Research; the National Institutes of Health (NIH)/National Center for Advancing Translational Sciences (NCATS) (ULTR000445 - the 1S10RR025141-01 instrumentation award and Vanderbilt CTSA grant); the European Commission (DG-SANCO); the International Agency for Research on Cancer; Danish Cancer Society (Denmark) (EMC 2014-6699); Ligue Contre le Cancer, Institut Gustave Roussy, Mutuelle Générale de l’Education Nationale; Institut National de la Santé et de la Recherche Médicale (INSERM) (France); German Cancer Aid; German Cancer Research Center (DKFZ); Federal Ministry of Education and Research (BMBF) (Germany); the Hellenic Health Foundation (Greece); Associazione Italiana per la Ricerca sul Cancro-AIRC-Italy and National Research Council (Italy); Dutch Ministry of Public Health, Welfare and Sports (VWS); Netherlands Cancer Registry (NKR); LK Research Funds; Dutch Prevention Funds; Dutch ZON (Zorg Onderzoek Nederland); World Cancer Research Fund (WCRF); Statistics Netherlands (the Netherlands); ERC-2009-AdG 232997 and Nordforsk, Nordic Centre of Excellence programme on Food, Nutrition and Health (Norway); Health Research Fund (FIS), PI13/00061 to Granada, PI13/01162 to EPIC-Murcia; Regional Governments of Andalucía, Asturias, Basque Country, Murcia and Navarra, ISCIII RETIC (RD06/0020) (Spain); Swedish Cancer Society, Swedish Research Council and County Councils of Skåne and Västerbotten (Sweden); Cancer Research UK (14136 to EPIC-Norfolk; C570/A16491 and C8221/A19170 to EPIC-Oxford), Medical Research Council (1000143 to EPIC-Norfolk, MR/M012190/1 to EPIC-Oxford) (United Kingdom); German Federal Ministry of Education and Research, Programme of Clinical Biomedical Research (01 GB 9401) and the German Cancer Research Center (DKFZ); U.S. National Institutes of Health (R01-CA58598, N01-CN-55424 and N01-PC-67001); intramural funding; Rudolf-Bartling Foundation; Helsinki University Hospital Research Fund; University of Pittsburgh School of Medicine Dean’s Faculty Advancement Award (F. Modugno); Department of Defense (DAMD17-02-1-0669); NCI (K07-CA080668, R01-CA95023, P50-CA159981 MO1-RR000056 R01-CA126841); intramural funding from the Rudolf-Bartling Foundation; ERC-2011-AdG 294576-risk factors cancer, Swedish Cancer Society, Swedish Research Council, Beta Kamprad Foundation; the National Cancer Institute (R01- CA61107), the Danish Cancer Society, Copenhagen, Denmark (94 222 52); the Mermaid I project; National Institutes of Health (R01-CA122443, P30-CA15083, P50-CA136393); Mayo Foundation; Minnesota Ovarian Cancer Alliance; Fred C. and Katherine B. Andersen Foundation; VicHealth and Cancer Council Victoria, Cancer Council Victoria, National Health and Medical Research Council of Australia (NHMRC) (209057, 251533, 396414, and 504715); DOD Ovarian Cancer Research Program (W81XWH-07-0449); Moffitt Cancer Center; Merck Pharmaceuticals; the state of Florida; Hillsborough County; the city of Tampa; National Institutes of Health (R01-CA76016); the Department of Defense (DAMD17-02-1-0666); National Institutes of Health (R01-CA54419 and P50-CA105009); Department of Defense (W81XWH-10-1-02802. UM1 CA186107, P01 CA87969, R01 CA49449, R01-CA67262, UM1 CA176726); Radboud University Medical Centre; Canadian Institutes of Health Research grant (MOP-86727); National Institutes of Health/National Cancer Institute 1 (R01CA160669-01A1); Intramural Research Program of the National Cancer Institute; Pomeranian Medical University; Cancer Research UK (C490/A10119 C490/A10124); UK National Institute for Health Research Biomedical Research Centres at the University of Cambridge; Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (Z01-ES044005 and Z01-ES049033); The Swedish Cancer Foundation and the Swedish Research Council (VR 2017-00644); National Institutes of Health (R01-CA106414-A2); American Cancer Society (CRTG-00-196-01-CCE); Department of Defense (DAMD17-98-1-8659); Celma Mastry Ovarian Cancer Foundation; National Institutes of Health (R01-CA058860); the Lon V Smith Foundation (LVS-39420); The Eve Appeal (The Oak Foundation); National Institute for Health Research University College London Hospitals Biomedical Research Centre and MRC core funding (MR_UU_12023); P01CA17054, P30CA14089, R01CA61132, N01PC67010, R03CA113148, R03CA115195, N01CN025403, and California Cancer Research Program (00-01389 V-20170, 2II0200); National Science Centre (N N301 5645 40); The Maria Sklodowska-Curie Memorial Cancer Centre; and the Institute of Oncology, Warsaw, Poland. The Nurses’ Health Study (NHS) was supported by the National Institutes of Health (UM1 CA186107, P01 CA87969, R01 CA49449) and the NHS II was supported by the National Institutes of Health (U01 CA176726, R01 CA67262). Joe Dennis is supported by the CanRisk Cancer Research UK programme grant: PPRPGM-Nov20\100002 and by the Confluence project which is funded with intramural funds from the National Cancer Institute Intramural Research Programme, National Institutes of Health.
Notes
Role of the funder: The funding agencies had no role in the conceptualization of the study; collection or analysis of the data; or drafting and editing of the manuscript, or decision to submit it for publication.
Disclosures: Matthias W. Beckmann conducts research funded by Amgen, Novartis, and Pfizer. Peter A. Fasching conducts research funded by Amgen, Novartis, and Pfizer, and he received Honoraria from Roche, Novartis, and Pfizer. Usha Menon has stock ownership in Abcodia Ltd. AHE, who is a JNCI Associate Editor and co-author on this article, was not involved in the editorial review or decision to publish the manuscript. The remaining authors have no conflicts of interest to disclose.
Author contributions: Conceptualization: AAD, JD, JPY, PDPP, DFE, SAG, MRJ. Data curation: AAD, JD, JPY, PCP, SGC, ALR, JTP, BDD, SC, FSD. Formal Analysis: AAD, JD, JPY. Funding acquisition: HAC, AB, RB, GCT, LSC, TD, ELG, MTG, JG, LAK, SKK, JK, DL, NDL, UM, RLM, FM, ANM, HO, CLP, SJR, ER, MJR, IR, DPS, JMS, RS, KLT, SST, DVE, PMW, NW, AW, AHW, PDPP, SAG. Investigation: AAD, JD, JPY, PDPP, DFE, SAG, MRJ. Methodology: AAD, JD, JPY, PDPP, DFE, SAG, MRJ. Project administration: MJR, PDPP, DFE, SAG, MRJ. Resources: AADV, JD, JPT, PCP, SGC, ALR, JTP, BDD, SC, FSD, KKHA, HAC, NNA, MWB, ABF, AB, NVB, JDB, RB, IC, JCC, GCT, LSC, ADF, JAD, TD, DME, AHE, PAF, RTF, GGG, ELG, MTG, JG, NH, MATH, CH, DGH, AJ, PK, SK, BYK, EKK, LAK, SKK, JK, ML, DL, NDL, JL, TM, UM, RLM, FM, ANM, KBM, HN, KO, HO, CLP, TP, SJR, ER, MJR, IR, DPS, JMS, VWS, WS, HS, RS, KLT, PJT, LT, SST, EVN, DVE, PMW, NW, ASW, AW, AHW, AZ, MLF, KL, PDPP, DFE, SAG, MRJ. Software: AADV, JD, JPT, PCP, SGC. Supervision: PDPP, DFE, SAG, MRJ. Validation: AAD, JD, JPY, MRJ. Visualization: AAD, JD, MRJ. Writing—original draft: AAD, JD, JPY, PDPP, DFE, SAG, MRJ. Writing—review & editing: AADV, JD, JPT, PCP, SGC, ALR, JTP, BDD, SC, FSD, KKHA, HAC, NNA, MWB, ABF, AB, NVB, JDB, RB, IC, JCC, GCT, LSC, ADF, JAD, TD, DME, AHE, PAF, RTF, GGG, ELG, MTG, JG, NH, MATH, CH, DGH, AJ, PK, SK, BYK, EKK, LAK, SKK, JK, ML, DL, NDL, JL, TM, UM, RLM, FM, ANM, KBM, HN, KO, HO, CLP, TP, SJR, ER, MJR, IR, DPS, JMS, VWS, WS, HS, RS, KLT, PJT, LT, SST, EVN, DVE, PMW, NW, ASW, AW, AHW, AZ, MLF, KL, PDPP, DFE, SAG, MRJ.
Acknowledgements: We thank the study participants, doctors, nurses, clinical and scientific collaborators, health-care providers, and health information sources who have contributed to the many studies contributing to this manuscript. The Australian Ovarian Cancer Study (AOCS) also acknowledges the cooperation of the participating institutions in Australia and the contribution of the study nurses, research assistants, and all clinical and scientific collaborators. The complete AOCS Group can be found at www.aocstudy.org. We would like to thank all of the women who participated in this research program. The datasets used for the analyses described were in part obtained from Vanderbilt University Medical Center’s BioVU. The authors would like to thank all members and investigators of the Rotterdam Ovarian Cancer Study. Some cases and their vital status were ascertained through the Victorian Cancer Registry (VCR) and the Australian Institute of Health and Welfare (AIHW), including the National Death Index and the Australian Cancer Database. The authors would like to thank The Total Cancer Care Protocol and the Collaborative Data Services and Tissue Core Facilities at the H. Lee Moffitt Cancer Center & Research Institute, a National Cancer Institute–designated Comprehensive Cancer Center (P30-CA076292), Merck Pharmaceuticals, and the state of Florida. The Nurses’ Health Study and Nurses’ Health Study II thank the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, and WY.
Disclaimers: The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Prior presentations: Some of the content has been presented publicly as a poster at the 69th Annual Meeting of the American Society for Human Genetics in 2019 in Houston on October 17, 2019 (Abstract #985). The abstract can be accessed here: https://www.ashg.org/wp-content/uploads/2019/10/ASHG-2019-poster-abstracts.pdf.
Supplementary Material
Contributor Information
Amber A DeVries, Center for Bioinformatics and Functional Genomics, Department of Biomedical Sciences, Cedars-Sinai Medical Center, Los Angeles, CA, USA.
Joe Dennis, Centre for Cancer Genetic Epidemiology, Department of Public Health and Primary Care, University of Cambridge, Cambridge, UK.
Jonathan P Tyrer, Centre for Cancer Genetic Epidemiology, Department of Oncology, University of Cambridge, Cambridge, UK.
Pei-Chen Peng, Center for Bioinformatics and Functional Genomics, Department of Biomedical Sciences, Cedars-Sinai Medical Center, Los Angeles, CA, USA.
Simon G Coetzee, Center for Bioinformatics and Functional Genomics, Department of Biomedical Sciences, Cedars-Sinai Medical Center, Los Angeles, CA, USA.
Alberto L Reyes, Center for Bioinformatics and Functional Genomics, Department of Biomedical Sciences, Cedars-Sinai Medical Center, Los Angeles, CA, USA.
Jasmine T Plummer, Center for Bioinformatics and Functional Genomics, Department of Biomedical Sciences, Cedars-Sinai Medical Center, Los Angeles, CA, USA; Applied Genomics, Computation and Translational Core, Cedars-Sinai Medical Center, Los Angeles, CA, USA.
Brian D Davis, Center for Bioinformatics and Functional Genomics, Department of Biomedical Sciences, Cedars-Sinai Medical Center, Los Angeles, CA, USA; Applied Genomics, Computation and Translational Core, Cedars-Sinai Medical Center, Los Angeles, CA, USA.
Stephanie S Chen, Center for Bioinformatics and Functional Genomics, Department of Biomedical Sciences, Cedars-Sinai Medical Center, Los Angeles, CA, USA; Applied Genomics, Computation and Translational Core, Cedars-Sinai Medical Center, Los Angeles, CA, USA.
Felipe Segato Dezem, Center for Bioinformatics and Functional Genomics, Department of Biomedical Sciences, Cedars-Sinai Medical Center, Los Angeles, CA, USA.
Katja K H Aben, Radboud Institute for Health Sciences, Radboud University Medical Center, Nijmegen, The Netherlands; Netherlands Comprehensive Cancer Organisation, Utrecht, The Netherlands.
Hoda Anton-Culver, Department of Medicine, Genetic Epidemiology Research Institute, University of California Irvine, Irvine, CA, USA.
Natalia N Antonenkova, N.N. Alexandrov Research Institute of Oncology and Medical Radiology, Minsk, Belarus.
Matthias W Beckmann, Department of Gynecology and Obstetrics, Comprehensive Cancer Center Erlangen-European Metropolitan Region of Nuremberg, Friedrich-Alexander University Erlangen-Nuremberg, University Hospital Erlangen, Erlangen, Germany.
Alicia Beeghly-Fadiel, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, Nashville, TN, USA.
Andrew Berchuck, Department of Gynecologic Oncology, Duke University Hospital, Durham, NC, USA.
Natalia V Bogdanova, N.N. Alexandrov Research Institute of Oncology and Medical Radiology, Minsk, Belarus; Department of Radiation Oncology, Hannover Medical School, Hannover, Germany; Gynaecology Research Unit, Hannover Medical School, Hannover, Germany.
Nadja Bogdanova-Markov, Institute of Human Genetics, University of Münster, Münster, Germany.
James D Brenton, Cancer Research UK Cambridge Institute, University of Cambridge, Cambridge, UK.
Ralf Butzow, Department of Pathology, Helsinki University Hospital, University of Helsinki, Helsinki, Finland.
Ian Campbell, Cancer Genetics Laboratory, Research Division, Peter MacCallum Cancer Center, Melbourne, Victoria, Australia; Sir Peter MacCallum Department of Oncology, The University of Melbourne, Melbourne, Victoria, Australia.
Jenny Chang-Claude, Division of Cancer Epidemiology, German Cancer Research Center (DKFZ), Heidelberg, Germany; Cancer Epidemiology Group, University Cancer Center Hamburg (UCCH), University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
Georgia Chenevix-Trench, Department of Genetics and Computational Biology, QIMR Berghofer Medical Research Institute, Brisbane, Queensland, Australia.
Linda S Cook, Epidemiology, School of Public Health, University of Colorado, Aurora, CO, USA; Community Health Sciences, University of Calgary, Calgary, AB, Canada.
Anna DeFazio, Centre for Cancer Research, The Westmead Institute for Medical Research, Sydney, New South Wales, Australia; Department of Gynaecological Oncology, Westmead Hospital, Sydney, New South Wales, Australia; The Daffodil Centre, a joint venture with Cancer Council NSW, The University of Sydney, Sydney, New South Wales, Australia.
Jennifer A Doherty, Huntsman Cancer Institute, Department of Population Health Sciences, University of Utah, Salt Lake City, UT, USA.
Thilo Dörk, Gynaecology Research Unit, Hannover Medical School, Hannover, Germany.
Diana M Eccles, Faculty of Medicine, University of Southampton, Southampton, UK.
A Heather Eliassen, Channing Division of Network Medicine, Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, USA; Department of Epidemiology, Harvard T.H. Chan School of Public Health, Boston, MA, USA; Department of Nutrition, Harvard T.H. Chan School of Public Health, Boston, MA, USA.
Peter A Fasching, Department of Gynecology and Obstetrics, Comprehensive Cancer Center Erlangen-European Metropolitan Region of Nuremberg, Friedrich-Alexander University Erlangen-Nuremberg, University Hospital Erlangen, Erlangen, Germany; David Geffen School of Medicine, Department of Medicine Division of Hematology and Oncology, University of California at Los Angeles, Los Angeles, CA, USA.
Renée T Fortner, Division of Cancer Epidemiology, German Cancer Research Center (DKFZ), Heidelberg, Germany.
Graham G Giles, Cancer Epidemiology Division, Cancer Council Victoria, Melbourne, Victoria, Australia; Centre for Epidemiology and Biostatistics, Melbourne School of Population and Global Health, The University of Melbourne, Melbourne, Victoria, Australia; Precision Medicine, School of Clinical Sciences at Monash Health, Monash University, Clayton, Victoria, Australia.
Ellen L Goode, Department of Quantitative Health Sciences, Division of Epidemiology, Mayo Clinic, Rochester, MN, USA.
Marc T Goodman, Samuel Oschin Comprehensive Cancer Institute, Cancer Prevention and Genetics Program, Cedars-Sinai Medical Center, Los Angeles, CA, USA.
Jacek Gronwald, Department of Genetics and Pathology, Pomeranian Medical University, Szczecin, Poland.
Niclas Håkansson, Institute of Environmental Medicine, Karolinska Institutet, Stockholm, Sweden.
Michelle A T Hildebrandt, Department of Epidemiology, University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Chad Huff, Department of Epidemiology, University of Texas MD Anderson Cancer Center, Houston, TX, USA.
David G Huntsman, Department of Obstetrics and Gynecology, University of British Columbia, Vancouver, BC, Canada; Department of Molecular Oncology, BC Cancer Research Centre, Vancouver, BC, Canada.
Allan Jensen, Department of Lifestyle, Reproduction and Cancer, Danish Cancer Society Research Center, Copenhagen, Denmark.
Siddhartha Kar, Medical Research Council Integrative Epidemiology Unit, University of Bristol, Bristol, UK; Section of Translational Epidemiology, Division of Population Health Sciences, Bristol Medical School, University of Bristol, Bristol, UK.
Beth Y Karlan, David Geffen School of Medicine, Department of Obstetrics and Gynecology, University of California at Los Angeles, Los Angeles, CA, USA.
Elza K Khusnutdinova, Institute of Biochemistry and Genetics, Ufa Federal Research Centre of the Russian Academy of Sciences, Ufa, Russia; Saint Petersburg State University, Saint Petersburg, Russia.
Lambertus A Kiemeney, Radboud Institute for Health Sciences, Radboud University Medical Center, Nijmegen, The Netherlands.
Susanne K Kjaer, Department of Lifestyle, Reproduction and Cancer, Danish Cancer Society Research Center, Copenhagen, Denmark; Department of Gynaecology, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark.
Jolanta Kupryjanczyk, Department of Pathology and Laboratory Diagnostics, Maria Sklodowska-Curie National Research Institute of Oncology, Warsaw, Poland.
Marilyne Labrie, Knight Cancer Institute, Oregon Health & Science University, Portland, OR, USA.
Diether Lambrechts, VIB Center for Cancer Biology, VIB, Leuven, Belgium; Laboratory for Translational Genetics, Department of Human Genetics, KU Leuven, Leuven, Belgium.
Nhu D Le, Cancer Control Research, BC Cancer, Vancouver, BC, Canada.
Jan Lubiński, Department of Genetics and Pathology, Pomeranian Medical University, Szczecin, Poland.
Taymaa May, Division of Gynecologic Oncology, University Health Network, Princess Margaret Hospital, Toronto, Ontario, Canada.
Usha Menon, MRC Clinical Trials Unit, Institute of Clinical Trials & Methodology, University College London, London, UK.
Roger L Milne, Cancer Epidemiology Division, Cancer Council Victoria, Melbourne, Victoria, Australia; Centre for Epidemiology and Biostatistics, Melbourne School of Population and Global Health, The University of Melbourne, Melbourne, Victoria, Australia; Precision Medicine, School of Clinical Sciences at Monash Health, Monash University, Clayton, Victoria, Australia.
Francesmary Modugno, Women’s Cancer Research Center, Magee-Womens Research Institute and Hillman Cancer Center, Pittsburgh, PA, USA; Department of Obstetrics, Gynecology and Reproductive Sciences, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA.
Alvaro N Monteiro, Department of Cancer Epidemiology, Moffitt Cancer Center, Tampa, FL, USA.
Kirsten B Moysich, Department of Cancer Prevention and Control, Roswell Park Cancer Institute, Buffalo, NY, USA.
Kunle Odunsi, Department of Oncology, University of Chicago Medicine Comprehensive Cancer Center, Chicago, IL, USA; Department of Obstetrics and Gynecology, University of Chicago Medicine Comprehensive Cancer Center, Chicago, IL, USA.
Håkan Olsson, Oncology, Department of Clinical Sciences, Lund University, Lund, Sweden.
Celeste L Pearce, Department of Epidemiology, University of Michigan School of Public Health, Ann Arbor, MI, USA; Department of Preventive Medicine, Keck School of Medicine, University of Southern California Norris Comprehensive Cancer Center, Los Angeles, CA, USA.
Tanja Pejovic, Laboratory for Translational Genetics, Department of Human Genetics, KU Leuven, Leuven, Belgium; Department of Obstetrics and Gynecology, Oregon Health & Science University, Portland, OR, USA.
Susan J Ramus, School of Women’s and Children’s Health, Faculty of Medicine and Health, University of NSW Sydney, Sydney, New South Wales, Australia; Adult Cancer Program, Lowy Cancer Research Centre, University of NSW Sydney, Sydney, New South Wales, Australia.
Elio Riboli, Imperial College London, London, UK.
Marjorie J Riggan, Department of Gynecologic Oncology, Duke University Hospital, Durham, NC, USA.
Isabelle Romieu, Nutrition and Metabolism Section, International Agency for Research on Cancer (IARC-WHO), Lyon, France.
Dale P Sandler, Epidemiology Branch, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC, USA.
Joellen M Schildkraut, Department of Epidemiology, Rollins School of Public Health, Emory University, Atlanta, GA, USA.
V Wendy Setiawan, Department of Preventive Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA, USA.
Weiva Sieh, Department of Population Health Science and Policy, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, USA.
Honglin Song, Department of Public Health and Primary Care, University of Cambridge, Cambridge, UK.
Rebecca Sutphen, Epidemiology Center, College of Medicine, University of South Florida, Tampa, FL, USA.
Kathryn L Terry, Department of Epidemiology, Harvard T.H. Chan School of Public Health, Boston, MA, USA; Obstetrics and Gynecology Epidemiology Center, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, USA.
Pamela J Thompson, Samuel Oschin Comprehensive Cancer Institute, Cancer Prevention and Genetics Program, Cedars-Sinai Medical Center, Los Angeles, CA, USA.
Linda Titus, Muskie School of Public Policy, Public Health, Portland, ME, USA.
Shelley S Tworoger, Department of Cancer Epidemiology, Moffitt Cancer Center, Tampa, FL, USA.
Els Van Nieuwenhuysen, Division of Gynecologic Oncology, Department of Gynecology and Obstetrics, Leuven Cancer Institute, Leuven, Belgium.
Digna Velez Edwards, Division of Quantitative Sciences, Department of Obstetrics and Gynecology, Department of Biomedical Sciences, Women’s Health Research, Vanderbilt University Medical Center, Nashville, TN, USA.
Penelope M Webb, Population Health Department, QIMR Berghofer Medical Research Institute, Brisbane, Queensland, Australia.
Nicolas Wentzensen, Division of Cancer Epidemiology and Genetics, National Cancer Institute, Bethesda, MD, USA.
Alice S Whittemore, Department of Epidemiology and Population Health, Stanford University School of Medicine, Stanford, CA, USA; Department of Biomedical Data Science, Stanford University School of Medicine, Stanford, CA, USA.
Alicja Wolk, Institute of Environmental Medicine, Karolinska Institutet, Stockholm, Sweden; Department of Surgical Sciences, Uppsala University, Uppsala, Sweden.
Anna H Wu, Department of Preventive Medicine, Keck School of Medicine, University of Southern California Norris Comprehensive Cancer Center, Los Angeles, CA, USA.
Argyrios Ziogas, Department of Medicine, Genetic Epidemiology Research Institute, University of California Irvine, Irvine, CA, USA.
Matthew L Freedman, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA.
Kate Lawrenson, Division of Gynecologic Oncology, Department of Obstetrics and Gynecology, Women’s Cancer Program at the Samuel Oschin Cancer Institute Cedars-Sinai Medical Center, Los Angeles, CA, USA.
Paul D P Pharoah, Centre for Cancer Genetic Epidemiology, Department of Public Health and Primary Care, University of Cambridge, Cambridge, UK; Centre for Cancer Genetic Epidemiology, Department of Oncology, University of Cambridge, Cambridge, UK.
Douglas F Easton, Centre for Cancer Genetic Epidemiology, Department of Public Health and Primary Care, University of Cambridge, Cambridge, UK; Centre for Cancer Genetic Epidemiology, Department of Oncology, University of Cambridge, Cambridge, UK.
Simon A Gayther, Center for Bioinformatics and Functional Genomics, Department of Biomedical Sciences, Cedars-Sinai Medical Center, Los Angeles, CA, USA.
Michelle R Jones, Center for Bioinformatics and Functional Genomics, Department of Biomedical Sciences, Cedars-Sinai Medical Center, Los Angeles, CA, USA.
OPAL Study Group:
P Webb, A DeFazio, M Friedlander, A Obermair, P Grant, C Nagle, V Beesley, G Chevenix-Trench, D Bowtell, P Blomfield, A Brand, A Davis, Y Leung, J Nicklin, M Quinn, K Livingstone, H O'Neill, M Williams, A Black, A Hadley, A Glasgow, A Garrett, A Rao, C Shannon, C Steer, D Allen, D Neesham, G Otton, G Au-Yeung, G Goss, G Wain, G Gard, G Robertson, J Lombard, J Tan, J McNeilage, J Power, J Coward, J Miller, J Carter, J Lamont, K M Wong, K Reid, L Perrin, L Milishkin, M Nascimento, M Buck, M Bunting, M Harrison, N Chetty, N Hacker, O McNally, P Harnett, P Beale, R Awad, R Mohan, R Farrell, R McIntosh, R Rome, R Sayer, R Houghton, R Hogg, R Land, S Baron-Hay, S Paramasivum, S Pather, S Hyde, S Salfinger, S Valmadre, T Jobling, T Manolitsas, T Bonaventura, and V Arora
AOCS Group:
D Bowtell, G Chenevix-Trench, A Green, P Webb, A DeFazio, D Gertig, N Traficante, S Fereday, S Moore, J Hung, K Harrap, T Sadkowsky, N Pandeya, M Malt, R Robertson, T Vanden Bergh, M Jones, P McKenzie, J Maidens, K Nattress, Y E Chiew, A Stenlake, H Sullivan, B Alexander, P Ashover, S Brown, T Corrish, L Green, L Jackman, K Ferguson, K Martin, A Martyn, B Ranieri, J White, V Jayde, L Bowes, P Mamers, L Galletta, D Giles, J Hendley, K Alsop, T Schmidt, H Shirley, C Ball, C Young, S Viduka, H Tran, S Bilic, L Glavinas, J Brooks, R Stuart-Harris, F Kirsten, J Rutovitz, P Clingan, A Glasgow, A Proietto, S Braye, G Otton, J Shannon, T Bonaventura, J Stewart, and S Begbie
Data Availability
All data used and code developed for these analyses are available on Github at https://github.com/Jones-Lab-CSMC/OCAC-Oncoarray-CamCNV.
References
- 1. Ramus SJ, Gayther SA. The contribution of BRCA1 and BRCA2 to ovarian cancer. Mol Oncol. 2009;3(2):138-150. doi: 10.1016/j.molonc.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ramus SJ, Song H, Dicks E, et al. Germline mutations in the BRIP1, BARD1, PALB2, and NBN genes in women with ovarian cancer. J Natl Cancer Inst. 2015;107(11):djv214. doi: 10.1093/jnci/djv214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Norquist BM, Harrell MI, Brady MF, et al. Inherited mutations in women with ovarian carcinoma. JAMA Oncol. 2016;2(4):482-490. doi: 10.1001/jamaoncol.2015.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Walsh T, Casadei S, Lee MK, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108(44):18032-18037. doi: 10.1073/pnas.1115052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rafnar T, Gudbjartsson DF, Sulem P, et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet. 2011;43(11):1104-1107. doi: 10.1038/ng.955. [DOI] [PubMed] [Google Scholar]
- 6. Phelan CM, Kuchenbaecker KB, Tyrer JP, et al. ; for the OPAL study group. Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat Genet. 2017;49(5):680-691. doi: 10.1038/ng.3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pharoah PDP, Tsai Y-Y, Ramus SJ, et al. ; for the Australian Ovarian Cancer Study Group. GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat Genet. 2013;45(4):362-370.e1. doi: 10.1038/ng.2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Song H, Ramus SJ, Tyrer J, et al. ; for the Australian Cancer (Ovarian) Study. A genome-wide association study identifies a new ovarian cancer susceptibility locus on 9p22.2. Nat Genet. 2009;41(9):996-1000. doi: 10.1038/ng.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goode EL, Chenevix-Trench G, Song H, et al. ; for the Ovarian Cancer Association Consortium (OCAC). A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat Genet. 2010;42(10):874-879. doi: 10.1038/ng.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Permuth-Wey J, Lawrenson K, Shen HC, et al. ; for the Consortium of Investigators of Modifiers of BRCA1/2. Identification and molecular characterization of a new ovarian cancer susceptibility locus at 17q21.31. Nat Commun. 2013;4:1627. doi: 10.1038/ncomms2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shen H, Fridley BL, Song H, et al. ; for the Australian Cancer Study. Epigenetic analysis leads to identification of HNF1B as a subtype-specific susceptibility gene for ovarian cancer. Nat Commun. 2013;4:1628. doi: 10.1038/ncomms2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bojesen SE, Pooley KA, Johnatty SE, et al. ; for the Genetic Modifiers of Cancer Risk in BRCA1/2 Mutation Carriers (GEMO). Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45(4):371-384.e1. doi: 10.1038/ng.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kuchenbaecker KB, Ramus SJ, Tyrer J, et al. ; for the EMBRACE. Identification of six new susceptibility loci for invasive epithelial ovarian cancer. Nat Genet. 2015;47(2):164-171. doi: 10.1038/ng.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kelemen LE, Lawrenson K, Tyrer J, et al. ; for the Ovarian Cancer Association Consortium. Genome-wide significant risk associations for mucinous ovarian carcinoma. Nat Genet. 2015;47(8):888-897. doi: 10.1038/ng.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kar SP, Beesley J, Amin Al Olama A, et al. ; for the ABCTB Investigators. Genome-wide meta-analyses of breast, ovarian, and prostate cancer association studies identify multiple new susceptibility loci shared by at least two cancer types. Cancer Discov. 2016;6(9):1052-1067. doi: 10.1158/2159-8290.CD-15-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Couch FJ, Wang X, McGuffog L, et al. ; for the CIMBA. Genome-wide association study in BRCA1 mutation carriers identifies novel loci associated with breast and ovarian cancer risk. PLoS Genet. 2013;9(3):e1003212. doi: 10.1371/journal.pgen.1003212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bolton KL, Tyrer J, Song H, et al. ; for the Ovarian Cancer Association Consortium. Common variants at 19p13 are associated with susceptibility to ovarian cancer. Nat Genet. 2010;42(10):880-884. doi: 10.1038/ng.666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pearce CL, Stram DO, Ness RB, et al. Population distribution of lifetime risk of ovarian cancer in the United States. Cancer Epidemiol Biomarkers Prev. 2015;24(4):671-676. doi: 10.1158/1055-9965.EPI-14-1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lu Y, Ek WE, Whiteman D, et al. Most common “sporadic” cancers have a significant germline genetic component. Hum Mol Genet. 2014;23(22):6112-6118. doi: 10.1093/hmg/ddu312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brandler WM, Antaki D, Gujral M, et al. Frequency and complexity of de novo structural mutation in autism. Am J Hum Genet. 2016;98(4):667-679. doi: 10.1016/j.ajhg.2016.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature. 2006;444(7118):444-454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods. 2012;9(3):215-216. doi: 10.1038/nmeth.1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Long J, Delahanty RJ, Li G, et al. A common deletion in the APOBEC3 genes and breast cancer risk. J Natl Cancer Inst. 2013;105(8):573-579. doi: 10.1093/jnci/djt018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kuusisto KM, Akinrinade O, Vihinen M, Kankuri-Tammilehto M, Laasanen S-L, Schleutker J. Copy number variation analysis in familial BRCA1/2-negative Finnish breast and ovarian cancer. PLoS One. 2013;8(8):e71802. doi: 10.1371/journal.pone.0071802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kumaran M, Cass CE, Graham K, et al. Germline copy number variations are associated with breast cancer risk and prognosis. Sci Rep. 2017;7(1):14621. doi: 10.1038/s41598-017-14799-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Komatsu A, Nagasaki K, Fujimori M, Amano J, Miki Y. Identification of novel deletion polymorphisms in breast cancer. Int J Oncol . 2008;33(2):261-270. doi: 10.3892/ijo_00000005. [DOI] [PubMed] [Google Scholar]
- 27. Walker LC, Pearson JF, Wiggins GAR, Giles GG, Hopper JL, Southey MC. Increased genomic burden of germline copy number variants is associated with early onset breast cancer: Australian Breast Cancer Family Registry. Breast Cancer Res. 2017;19(1):30. doi: 10.1186/s13058-017-0825-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pylkäs K, Vuorela M, Otsukka M, Kallioniemi A, Jukkola-Vuorinen A, Winqvist R. Rare copy number variants observed in hereditary breast cancer cases disrupt genes in estrogen signaling and TP53 tumor suppression network. PLoS Genet. 2012;8(6):e1002734. doi: 10.1371/journal.pgen.1002734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. de Jesús Ascencio-Montiel I, Pinto D, Parra EJ, Valladares-Salgado A, Cruz M, Scherer SW. Characterization of large copy number variation in Mexican type 2 diabetes subjects. Sci Rep. 2017;7(1):17105. doi: 10.1038/s41598-017-17361-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bae JS, Cheong HS, Kim J-H, et al. The genetic effect of copy number variations on the risk of type 2 diabetes in a Korean population. PLoS One. 2011;6(4):e19091. doi: 10.1371/journal.pone.0019091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Craddock N, Hurles ME, Cardin N, et al. ; for the Wellcome Trust Case Control Consortium. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464(7289):713-720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Al-Sukhni W, Joe S, Lionel AC, et al. Identification of germline genomic copy number variation in familial pancreatic cancer. Hum Genet. 2012;131(9):1481-1494. doi: 10.1007/s00439-012-1183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Collins RL, Brand H, Karczewski KJ, et al. ; for the Genome Aggregation Database Consortium. A structural variation reference for medical and population genetics. Nature. 2020;581(7809):444-451. doi: 10.1038/s41586-020-2287-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reid BM, Permuth JB, Chen YA, et al. Genome-wide analysis of common copy number variation and epithelial ovarian cancer risk. Cancer Epidemiol Biomarkers Prev. 2019;28(7):1117-1126. doi: 10.1158/1055-9965.EPI-18-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fridley BL, Chalise P, Tsai Y-Y, et al. Germline copy number variation and ovarian cancer survival. Front Genet. 2012;3:142. doi: 10.3389/fgene.2012.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dennis J, Walker L, Tyrer J, Michailidou K, Easton DF. Detecting rare copy number variants from Illumina genotyping arrays with the CamCNV pipeline: segmentation of z-scores improves detection and reliability. Genet Epidemiol. 2021;45(3):237-248. doi: 10.1002/gepi.22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tyrer JP, Guo Q, Easton DF, Pharoah PDP. The admixture maximum likelihood test to test for association between rare variants and disease phenotypes. BMC Bioinformatics. 2013;14:177. doi: 10.1186/1471-2105-14-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gusev A, Lawrenson K, Lin X, et al. ; for the Ovarian Cancer Association Consortium. A transcriptome-wide association study of high-grade serous epithelial ovarian cancer identifies new susceptibility genes and splice variants. Nat Genet. 2019;51(5):815-823. doi: 10.1038/s41588-019-0395-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lu Y, Beeghly-Fadiel A, Wu L, et al. A transcriptome-wide association study among 97,898 women to identify candidate susceptibility genes for epithelial ovarian cancer risk. Cancer Res. 2018;78(18):5419-5430. doi: 10.1158/0008-5472.CAN-18-0951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Berisa T, Pickrell JK. Approximately independent linkage disequilibrium blocks in human populations. Bioinformatics. 2016;32(2):283-285. doi: 10.1093/bioinformatics/btv546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Coetzee S. Simon-Coetzee/Funcivar; 2018. https://github.com/Simon-Coetzee/funcivar.
- 42. Jones MR, Peng P-C, Coetzee SG, et al. ; for the Ovarian Cancer Association Consortium. Ovarian cancer risk variants are enriched in histotype-specific enhancers and disrupt transcription factor binding sites. Am J Hum Genet. 2020;107(4):622-635. doi: 10.1016/j.ajhg.2020.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reyes ALP, Silva TC, Coetzee SG, et al. GENAVi: a shiny web application for gene expression normalization, analysis and visualization. BMC Genomics. 2019;20(1):745. doi: 10.1186/s12864-019-6073-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Handsaker RE, Korn JM, Nemesh J, McCarroll SA. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat Genet. 2011;43(3):269-276. doi: 10.1038/ng.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mazoyer S. The exon 13 duplication in the BRCA1 gene is a founder mutation present in geographically diverse populations. Am J Hum Genet. 2000;67(1):207-212. doi: 10.1086/302974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Petrij-Bosch A, Peelen T, van Vliet M, et al. BRCA1 genomic deletions are major founder mutations in Dutch breast cancer patients. Nat Genet. 1997;17(3):341-345. doi: 10.1038/ng1197-341. [DOI] [PubMed] [Google Scholar]
- 47. Woodward AM, Davis TA, Silva AGS, Kirk JA, Leary JA; for the kConFab Investigators. Large genomic rearrangements of both BRCA2 and BRCA1 are a feature of the inherited breast/ovarian cancer phenotype in selected families. J Med Genet. 2005;42(5):e31. doi: 10.1136/jmg.2004.027961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Song H, Cicek MS, Dicks E, et al. The contribution of deleterious germline mutations in BRCA1, BRCA2 and the mismatch repair genes to ovarian cancer in the population. Hum Mol Genet. 2014;23(17):4703-4709. doi: 10.1093/hmg/ddu172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Song H, Dicks E, Ramus SJ, et al. Contribution of germline mutations in the RAD51B, RAD51C, and RAD51D genes to ovarian cancer in the population. J Clin Oncol. 2015;33(26):2901-2907. doi:10.1200/J Clin Oncol.2015.61.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Buckley MA, Woods NT, Tyrer JP, et al. ; for the Ovarian Cancer Association Consortium. Functional analysis and fine mapping of the 9p22.2 ovarian cancer susceptibility locus. Cancer Res. 2019;79(3):467-481. doi: 10.1158/0008-5472.CAN-17-3864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. de Jong S, Chepelev I, Janson E, et al. Common inversion polymorphism at 17q21.31 affects expression of multiple genes in tissue-specific manner. BMC Genomics. 2012;13:458. doi: 10.1186/1471-2164-13-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Koolen DA, Vissers LELM, Pfundt R, et al. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet. 2006;38(9):999-1001. doi: 10.1038/ng1853. [DOI] [PubMed] [Google Scholar]
- 53. Gayther SA, Russell P, Harrington P, Antoniou AC, Easton DF, Ponder BA. The contribution of germline BRCA1 and BRCA2 mutations to familial ovarian cancer: no evidence for other ovarian cancer-susceptibility genes. Am J Hum Genet. 1999;65(4):1021-1029. doi: 10.1086/302583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kerkhof J, Schenkel LC, Reilly J, et al. Clinical validation of copy number variant detection from targeted next-generation sequencing panels. J Mol Diagn. 2017;19(6):905-920. doi: 10.1016/j.jmoldx.2017.07.004. [DOI] [PubMed] [Google Scholar]
- 55. Boone PM, Soens ZT, Campbell IM, et al. Incidental copy-number variants identified by routine genome testing in a clinical population. Genet Med. 2013;15(1):45-54. doi: 10.1038/gim.2012.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Scaglione GL, Concolino P, De Bonis M, et al. A whole germline BRCA2 gene deletion: how to learn from CNV in Silico analysis. Int J Mol Sci. 2018;19(4):961. doi: 10.3390/ijms19040961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schmidt AY, Hansen TVO, Ahlborn LB, Jønson L, Yde CW, Nielsen FC. Next-generation sequencing-based detection of germline copy number variations in BRCA1/BRCA2: validation of a one-step diagnostic workflow. J Mol Diagn. 2017;19(6):809-816. doi: 10.1016/j.jmoldx.2017.07.003. [DOI] [PubMed] [Google Scholar]
- 58. Truty R, Paul J, Kennemer M, et al. Prevalence and properties of intragenic copy-number variation in Mendelian disease genes. Genet Med. 2019;21(1):114-123. doi: 10.1038/s41436-018-0033-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bozsik A, Pócza T, Papp J, et al. Complex characterization of germline large genomic rearrangements of the BRCA1 and BRCA2 Genes in high-risk breast cancer patients-novel variants from a large national center. Int J Mol Sci. 2020;21(13):4650. doi: 10.3390/ijms21134650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hirotsu Y, Ooka Y, Sakamoto I, Nakagomi H, Omata M. Simultaneous detection of genetic and copy number alterations in BRCA1/2 genes. Oncotarget. 2017;8(70):114463-114473. doi: 10.18632/oncotarget.22962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nunziato M, Starnone F, Lombardo B, et al. Fast detection of a BRCA2 large genomic duplication by next generation sequencing as a single procedure: a case report. Int J Mol Sci. 2017;18(11):2487. doi: 10.3390/ijms18112487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hansen TO, Jønson L, Albrechtsen A, Andersen MK, Ejlertsen B, Nielsen FC. Large BRCA1 and BRCA2 genomic rearrangements in Danish high risk breast-ovarian cancer families. Breast Cancer Res Treat. 2009;115(2):315-323. doi: 10.1007/s10549-008-0088-0. [DOI] [PubMed] [Google Scholar]
- 63. Gad S, Klinger M, Caux-Moncoutier V, et al. Bar code screening on combed DNA for large rearrangements of the BRCA1 and BRCA2 genes in French breast cancer families. J Med Genet. 2002;39(11):817-821. doi: 10.1136/jmg.39.11.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lahti-Domenici J, Rapakko K, Pääkkönen K, et al. Exclusion of large deletions and other rearrangements in BRCA1 and BRCA2 in Finnish breast and ovarian cancer families. Cancer Genet Cytogenet. 2001;129(2):120-123. doi: 10.1016/s0165-4608(01)00437-x. [DOI] [PubMed] [Google Scholar]
- 65. Peelen T, van Vliet M, Bosch A, et al. Screening for BRCA2 mutations in 81 Dutch breast-ovarian cancer families. Br J Cancer. 2000;82(1):151-156. doi: 10.1054/bjoc.1999.0892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Agata S, Dalla Palma M, Callegaro M, et al. Large genomic deletions inactivate the BRCA2 gene in breast cancer families. J Med Genet. 2005;42(10):e64. doi: 10.1136/jmg.2005.032789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Peixoto A, Santos C, Rocha P, et al. The c.156_157insAlu BRCA2 rearrangement accounts for more than one-fourth of deleterious BRCA mutations in northern/central Portugal. Breast Cancer Res Treat. 2009;114(1):31-38. doi: 10.1007/s10549-008-9978-4. [DOI] [PubMed] [Google Scholar]
- 68. Sluiter MD, van Rensburg EJ. Large genomic rearrangements of the BRCA1 and BRCA2 genes: review of the literature and report of a novel BRCA1 mutation. Breast Cancer Res Treat. 2011;125(2):325-349. doi: 10.1007/s10549-010-0817-z. [DOI] [PubMed] [Google Scholar]
- 69. Hogervorst FBL, Nederlof PM, Gille JJP, et al. Large genomic deletions and duplications in the BRCA1 gene identified by a novel quantitative method. Cancer Res. 2003;63(7):1449-1453. [PubMed] [Google Scholar]
- 70. Mancini-DiNardo D, Judkins T, Kidd J, et al. Detection of large rearrangements in a hereditary pan-cancer panel using next-generation sequencing. BMC Med Genomics. 2019;12(1):138. doi: 10.1186/s12920-019-0587-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Subramanian DN, Zethoven M, McInerny S, et al. Exome sequencing of familial high-grade serous ovarian carcinoma reveals heterogeneity for rare candidate susceptibility genes. Nat Commun. 2020;11(1):1640. doi: 10.1038/s41467-020-15461-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Creekmore AL, Silkworth WT, Cimini D, Jensen RV, Roberts PC, Schmelz EM. Changes in gene expression and cellular architecture in an ovarian cancer progression model. PLoS One. 2011;6(3):e17676. doi: 10.1371/journal.pone.0017676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Emmanuel C, Gava N, Kennedy C, et al. ; for the Australian Ovarian Cancer Study Group. Comparison of expression profiles in ovarian epithelium in vivo and ovarian cancer identifies novel candidate genes involved in disease pathogenesis. PLoS One. 2011;6(3):e17617. doi: 10.1371/journal.pone.0017617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Huijts PEA, Vreeswijk MPG, Kroeze-Jansema KHG, et al. Clinical correlates of low-risk variants in FGFR2, TNRC9, MAP3K1, LSP1 and 8q24 in a Dutch cohort of incident breast cancer cases. Breast Cancer Res. 2007;9(6):R78. doi: 10.1186/bcr1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gates MA, Tworoger SS, Terry KL, et al. Breast cancer susceptibility alleles and ovarian cancer risk in 2 study populations. Int J Cancer. 2009;124(3):729-733. doi: 10.1002/ijc.23924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Antoniou AC, Sinilnikova OM, McGuffog L, et al. ; for the CIMBA. Common variants in LSP1, 2q35 and 8q24 and breast cancer risk for BRCA1 and BRCA2 mutation carriers. Hum Mol Genet. 2009;18(22):4442-4456. doi: 10.1093/hmg/ddp372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lokman NA, Price ZK, Hawkins EK, Macpherson AM, Oehler MK, Ricciardelli C. 4-Methylumbelliferone inhibits cancer stem cell activation and overcomes chemoresistance in ovarian cancer. Cancers (Basel). 2019;11(8):1187., doi:10.3390/cancers11081187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ricciardelli C, Ween MP, Lokman NA, Tan IA, Pyragius CE, Oehler MK. Chemotherapy-induced hyaluronan production: a novel chemoresistance mechanism in ovarian cancer. BMC Cancer. 2013;13:476. doi: 10.1186/1471-2407-13-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747-753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Abel HJ, Larson DE, Regier AA, et al. ; for the NHGRI Centers for Common Disease Genomics. Mapping and characterization of structural variation in 17,795 human genomes. Nature. 2020;583(7814):83-89. doi: 10.1038/s41586-020-2371-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang X, Liu Z, Tong H, et al. Linc01194 acts as an oncogene in colorectal carcinoma and is associated with poor survival outcome. Cancer Manag Res. 2019;11:2349-2362. doi: 10.2147/CMAR.S189189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Song HR, Guo XB, Duan Y, Meng HY, Wang ZY. PAX5-induced upregulation of LINC01194 exerts oncogenic properties by regulating GOLPH3 expression via miR-486-5p in prostate cancer. Eur Rev Med Pharmacol Sci. 2021;25(6):2528-2541. doi: 10.26355/eurrev_202103_25416. [DOI] [PubMed] [Google Scholar]
- 83. Zammarchi F, Bertelli F, Havenith K, Kirby I, Chivers S, Berkel P. V. Abstract 234: Pre-clinical characterization of 3A4-PL1601, a novel pyrrolobenzodiazepine (PBD) dimer-based antibody-drug conjugate (ADC) directed against KAAG1-expressing malignancies. In: Experimental and Molecular Therapeutics. Am Assoc Cancer Res. 2019;79(suppl 13):234-234. doi: 10.1158/1538-7445.AM2019-234. [DOI] [Google Scholar]
- 84. Han L, Zhao X, Benton ML, et al. ; for the CommonMind Consortium. Functional annotation of rare structural variation in the human brain. Nat Commun. 2020;11(1):2990. doi: 10.1038/s41467-020-16736-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Laplana M, Royo JL, García LF, Aluja A, Gomez-Skarmeta JL, Fibla J. SIRPB1 copy-number polymorphism as candidate quantitative trait locus for impulsive-disinhibited personality. Genes Brain Behav. 2014;13(7):653-662. doi: 10.1111/gbb.12154. [DOI] [PubMed] [Google Scholar]
- 86. Casilli F, Di Rocco ZC, Gad S, et al. Rapid detection of novel BRCA1 rearrangements in high-risk breast-ovarian cancer families using multiplex PCR of short fluorescent fragments. Hum Mutat. 2002;20(3):218-226. doi: 10.1002/humu.10108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used and code developed for these analyses are available on Github at https://github.com/Jones-Lab-CSMC/OCAC-Oncoarray-CamCNV.